X-ray Crystallography I. James Fraser Macromolecluar Interactions BP204
|
|
|
- Ethan Gardner
- 6 years ago
- Views:
Transcription
1 X-ray Crystallography I James Fraser Macromolecluar Interactions BP204
2 Key take-aways 1. X-ray crystallography results from an ensemble of Billions and Billions of molecules in the crystal 2. Models in the PDB are often sub-optimal and can contain errors 3. Intensity of spots relates to the electron density (which relates to the molecules) in the unit cell 4. Positions of spots relates to the arrangement of unit cells in the crystal 5. Every spot contains contributions from every part of the crystal. Every part of the map contains contributions from every spot
3 Key outcomes Understand Table 1 in X-ray Papers (now often Table S1 ) Understand the basic workflow of determining a crystal structure Embrace the beauty and challenge of disorder at high and low resolution



4 Today we are going to tackle crystallography in reverse Texts begin with diffraction theory from a series of point atoms (e.g. Biomolecular Crystallography, Rupp; Principles of Protein X-ray Crystallography, Drenth; Crystallography Made Crystal Clear, Rhoades) Bob teaches mini-course in Spring with this level of detail Today - model to reflections;tomorrow - phasing
5
6 What is a protein structure?

7 What is a protein structure Is it a: pretty cartoon... space-filling set of spheres... picture of the protein in the crystal... computational picture of the protein... representation of atoms that satisfies experimental constraints... PDB formatted text file... model!!!

8 Moreover... a model of the crystal lattice...
9 ProteinDataBank Files are text: chemistry, sequence, position, certainty HEADER HYDROLASE 10-DEC-06 2O7A TITLE T4 LYSOZYME C-TERMINAL FRAGMENT COMPND MOL_ID: 1; COMPND 2 MOLECULE: LYSOZYME; REMARK 3 FIT TO DATA USED IN REFINEMENT (NO CUTOFF). REMARK 3 R VALUE (WORKING + TEST SET, NO CUTOFF) : NULL REMARK 3 R VALUE (WORKING SET, NO CUTOFF) : REMARK 3 FREE R VALUE (NO CUTOFF) : ATOM 1 N VAL A N ATOM 2 CA VAL A C ATOM 3 C VAL A C ATOM 4 O VAL A O ATOM 5 CB VAL A C... MASTER END
' XPray' Robert'M.")
10 The$universe$of$protein$structures:$$ Our$knowledge$about$protein$structures$is$increasing..$ 65,271' protein' structures' are' deposited' in' PDB' (2/15/2010).' This'number'is'growing'by'>'~7000'a'year'' Growing'input'from'Structural'Genomics'HT'structure' determinajon'(>1000'structures'a'year)' XPray' Robert'M.'Stroud'2012' 9'
11 How do we tell if a model is good? physically (packing, contacts) chemically (bond lengths, bond angles, chirality, planarity, torsions) crystallographically (real space fits - B- factors, R-factor) statistically (R-free, CC1/2) Most of these stats appear in Table I

12 Physical Checks Bad Steric clashes Good Overall clash score (number of bad overlaps per 1000 atoms) A clash: disallowed atom pair overlap 0.4 Å MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Davis et al, Nucleic Acids Research, 2007, Vol. 35
13 Chemical checks Bond lengths and angles - Typical values (resolutions ~1.5-2Å): rmsd(bonds)~0.02å, rmsd(angles)~2 o These values can be smaller at lower resolution (~2.5-3Å), approaching 0 at ~3Å and lower resolution, and they can be larger at higher resolution (~1.5Å and higher). Engh and Huber, Acta Cryst, 1991

14 Chemical checks backbone and side chain torsion angles Ramachandran plot Rotamers Rotamers: a set of conformers arising from restricted rotation about one single bond χ 2 χ 1 Typically 1-3% outliers
15 Are all outliers bad?

16 ...not if justified by fit to electron density map...what forces might cause this?
17 ...similarly for side chains Flagged as rotamer outlier Correct rotamer
18 ... each outlier should be explainable by examining the electron density AND by forces acting in context of the whole protein





19 Phenix offers handy tools for looking at outliers PHENIX tools for model validation outliers in graphs also recenter Coot
20 What the Fobs-Fcalc are electron density maps?
21

22 Density maps can offer a model free view 2mFo-DFc (blue) mfo-dfc (red, green) m and D are de-biasing coefficients Fobs = Observed Amplitude Fcalc = Model-based Amplitude!
 - Include reflection F(000)")
23 Maps are contoured in units of SIGMA (rmsd) Typically 1.0 for 2Fo-Fc, +/-3 for Fo-Fc Two ways of bringing a map into some scale: - Divide it by standard deviation (map in sigmas) - Include reflection F(000) and divide map by the unit cell volume. Model should be complete to estimate F(000). Map in e/å 3.
24 to Coot!!!


25 Computationally it is very beneficial to approximate the electron density arising from each atom as a Gaussian function - Electron density at the point r of an atom located at position r 0 and having B-factor B and occupancy q: 5 3 / 2 4π ρ atom (r,r 0,B,q) = q a k exp 4π 2 r r 0 k=1 b k + B b k + B - Number of terms in the above formula depends on how accurately we want to model an atom 2 q and B are hard to separate (even at very high resolution)


26 looks very nice: However in practice we see densities more like:
27 What are B-factors? ATOM 1 N VAL A N ATOM 2 CA VAL A C ATOM 3 C VAL A C ATOM 4 O VAL A O ATOM 5 CB VAL A C F calc ( ~ h)= X j f j exp 1 4 B j ~ h t ~ h exp 2 i ~ h t ~x j, (1) i.e. three coordinates ~x j =(x j,y j,z j ) and one isotropic B-Factor B j are refined for each atom j (f j is the respective scattering factor, ~ h a reciprocal lattice vector). The overall mean displacement of an atom originates from several sources: di erent conformations in di erent unit cells ( internal static disorder ) vibration or dynamic transitions within molecules ( internal dynamic disorder ) lattice defects lattice vibrations (acoustical phonons) +restraints, +model errors!
28 Going anisotropic means 6 parameters instead of 1 F calc ( ~ h)= X j f j exp 2 2 ~ h t U j ~ h exp 2 i ~ h t ~x j, When is this decision justified?
00476-x available online at http://www.")
 320, 783 799 Structural Basis for Mobility in the 1.")

29 What is q? doi: /s (02)00476-x available online at on Bw J. Mol. Biol. (2002) 320, Structural Basis for Mobility in the 1.1 Å Crystal Structure of the NG Domain of Thermus aquaticus Ffh Ursula D. Ramirez 1, George Minasov 1, Pamela J. Focia 1 Robert M. Stroud 2, Peter Walter 2, Peter Kuhn 3 and Douglas M. Freymann 1 *


30 Difference Maps: Fo-Fc Error in position Error in occupancy Error in B-factor Should be green, not blue...
31 Model anisotropic atom with isotropic Add positional error

32 Ser residue needs a different rotamer
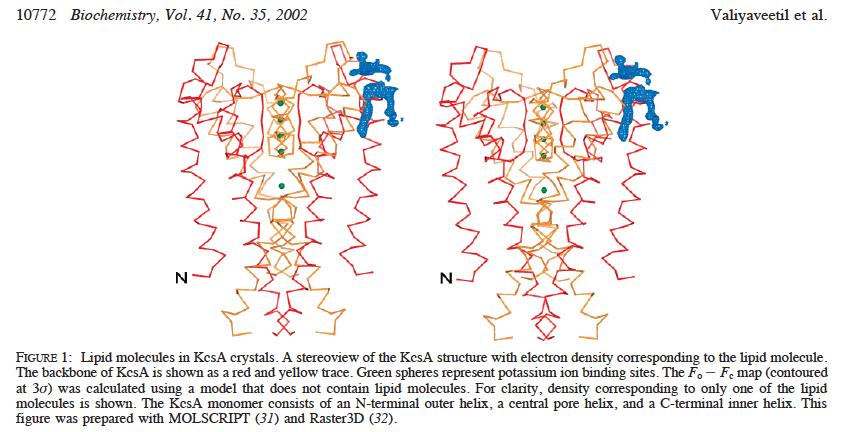
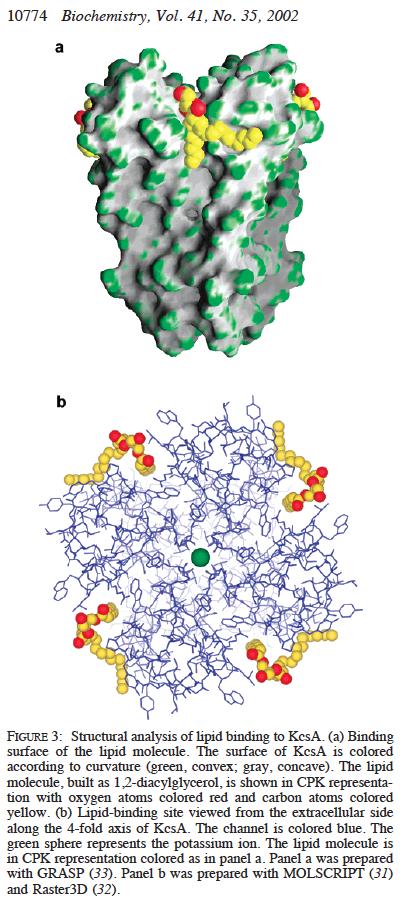
33 Fo#Fc%maps%iden.fy%everything%ordered%that%is%'missing'% mapmap% #Eliminate%Bias% #Half%electron%content% #See%electrons% Robert%M.%Stroud%2012% 70%

34 Also useful in dynamic crystallography
35 Refinement is the process of minimizing Fo-Fc...need to balance prior knowledge and data...an iterative process, difference maps minimized, and 2Fo-Fc maps improve (phases... we are coming to this)
36 Structure refinement is a process of changing a model parameters in order to optimize a goal (target) function: T = F(Experimental data, Model parameters, A priori knowledge) - Experimental data a set of diffraction amplitudes Fobs (and phases, if available). - Model parameters: coordinates, ADP, occupancies, bulk-solvent, - A priori knowledge (restraints or constraints) additional information that may be introduced to compensate for the insufficiency of experimental data (finite resolution, poor data-to-parameters ratio) Typically: T = T DATA + w*t RESTRAINTS - E DATA relates model to experimental data - E RESTRAINTS represents a priori knowledge - w is a weight to balance the relative contribution of E DATA and E RESTRAINTS
 Hands & eyes (Via Coot) X MIN solution XMAX Target function profile Local minima Global")
37 Gradient-driven minimization Target function profile Simulated annealing (SA) Target function profile Local minimum Global minimum Deeper local minimum Global minimum Grid search (Sample parameter space within known range [X MIN, X MAX ]) Hands & eyes (Via Coot) X MIN solution XMAX Target function profile Local minima Global minimum
38 How do we tell if a model is good? physically (packing, contacts) chemically (bond lengths, bond angles, chirality, planarity, torsions) crystallographically (real space fits - B- factors, R-factor) statistically (R-free, CC1/2) Most of these stats appear in Table I

39 Hands and Eyes are still important! Minimization Simulated Annealing Real-space grid search Both minimization and SA can fix it This is beyond the convergence radius for minimization This is beyond the convergence radius for minimization and SA
40 CC = % ' & $ " OBS # " OBS $ " CALC # " CALC grid points $ grid points " OBS # " OBS 2 grid points $ grid points " CALC # " CALC 2 ( * ) 1/ 2 Scale independent Can be computed for the whole structure (not really interesting you already have R-factor) or locally (most interesting; typically computed per residue) Values greater than ~0.8 indicate good correlation May give high correlation for weak densities Map CC is correlated with B-factor: poorly defined regions typically have low map CC and high B-factors
41 although this emphasizes local adjustments, refinement is global Every&X(ray&reflec,on&(h,k,l)&has&a&contribu,ng&wave&from&all&atoms&.& & & & ρ(x,y,z)&=&σ & F (h,k,l) &e ((2πi(hx+ky+lz))& & & F (h,k,l) &=&Σ j &f j &e (2πi & (hx+ky+lz))& or ρ(x,y,z)&=&σ F (h,k,l) &e ((2πi(hx+ky+lz)&+&φ hkl )& & Every&point&in&the&density&map&has&contribu,ons&from&every&reflec,on& & &
42 R-factor formula R-factor values: - Expected value for a random model R~59% - You can see some model in 2mFo-DFc map, R~30% - You can see most of the model in 2mFo-DFc map, R<20% - Perfect model R~0% R = # reflections # F OBS reflections F OBS " F MODEL F MODEL = k OVERALL e "su CRYSTAL s t F CALC_ATOMS + k SOL e " B 2 # SOL s & 4 % F MASK ( $ ' Sometimes the R-factor looks very good (you would expect a good model) but the model-to-map fit is terrible Overfitting.
43 Let s suppose: (red, blue or green) is the model: y = ax + b (2 parameters: a and b) is the data. Lot s of data one single correct model Less data more ambiguity, less certainty: a bunch of models Little data variety of models: from good to completely wrong R-factor is good R-factor may be good too R-factor = 0 for all models (including wrong ones)
44 Let s suppose: model: y = ax + b (2 parameters: a and b) data model described using more parameters: y=ax 2 +bx+c model described using even more parameters: y=a 1 x n +a 2 x n-1 + Less parameters More parameters Much more parameters R-factor is good R-factor is better R=0
 - Ignoring data (cutting by resolution, sigma, anisotropy correction) - Inoptimal parameterization - Excess of")
45 What leads to overfitting? - Insufficient amount of data (low resolution, poor completeness) - Ignoring data (cutting by resolution, sigma, anisotropy correction) - Inoptimal parameterization - Excess of imagination - Bad weights Choice for model parameterization depends on amount of available data and its resolution Key resolution limits and corresponding features
46 Solution: cross-validation (R-free factor): - At the beginning of structure solution split the data into two sets: test set (~5-10% of randomly selected data), and work set (the rest). - From this point on you look at two R-factors: R-work (computed using work set), and R-free (computed using test set) Dataset (F OBS ) work test Work set reflections are used for everything: model building, refinement, map calculation, Test set reflections are never used for any model optimization, expect Rfree factor calculation Rationale: the model that fits well ~90% of work set should fit well 10% of excluded data (test set). Since test set data does not participate in refinement, Rfree > Rwork. The gap Rfree Rwork depends on resolution and ranges from 5-7% (at medium to low resolution) to ~0.5A 1% (at ultra-high resolution)
47 Why does Rfree work so well? Every&X(ray&reflec,on&(h,k,l)&has&a&contribu,ng&wave&from&all&atoms&.& & & & ρ(x,y,z)&=&σ & F (h,k,l) &e ((2πi(hx+ky+lz))& & & F (h,k,l) &=&Σ j &f j &e (2πi & (hx+ky+lz))& or ρ(x,y,z)&=&σ F (h,k,l) &e ((2πi(hx+ky+lz)&+&φ hkl )& & Every&point&in&the&density&map&has&contribu,ons&from&every&reflec,on& & &
48 What the are Fhkl reflections, structure factors, amplitudes, spots?

49
50 We rotate the crystal to place a different set of reflections on the detector Robert(M.(Stroud(2012( 10(
51 Ewald sphere construction given: wavelength angle lattice distance from detector orientation of lattice relative to detector predicts: which diffracted waves satisfy Bragg s law
52 Each reflection is measured multiple times F = sqrt(intensity) SigI = error in Intensity (resulting from multiple observations)
53 Where to cut the data? I/sigma - background Rmerge - consistency CC1/2, CC* - effect on refinement (Karplus and Diederichs, Science, 2012) Robert(M.(Stroud(2012( 10(
54 If you have too many overloads
")
55 if you throw out weak (low res) data

56 if you randomly miss data (like Rfree)
 - why you need a whole")
57 if you miss slices of data (bad strategy) - why you need a whole dataset

# ## Structure#is#the# inverse #")
58 Sca-ering#pa-ern#is#the## Fourier#transform#of#the#structure## FT# FT =1# F(S)#=#Σ j #f j #e (2πirj.S)# ## Structure#is#the# inverse # Fourier#transform#of#the## Sca-ering#pa-ern### ρ(r)"="σ " F(S)"e (&2πir.S)"

59 A crystal only samples the parts of the transform that satisfy Bragg s Law a% b% FT% FT #1% 1/b%
60 F(h,k,l)&=&Σj&fj&e(2πi&(hx+ky+lz))& Every&X(ray&reflec,on&(h,k,l)&has&a&contribu,ng&wave&from&all&atoms&.& & & & ρ(x,y,z)&=&σ&f(h,k,l)&e((2πi(hx+ky+lz))& & & or ρ(x,y,z)&=&σ F(h,k,l) &e((2πi(hx+ky+lz)&+&φhkl)& & Every&point&in&the&density&map&has&contribu,ons&from&every&reflec,on& & & Fourier Transform
61 Waves have phase too... next lecture - and paper... how model phases bias our maps and how to solve the phase problem & & or ρ(x,y,z)&=&σ F (h,k,l) &e ((2πi(hx+ky+lz)&+&φ hkl )& & Every&point&in&the&density&map&has&contribu,ons&from&every&reflec,on& & &
62 Key take-aways 1. X-ray crystallography results from an ensemble of Billions and Billions of molecules in the crystal 2. Models in the PDB are often sub-optimal and can contain errors 3. Intensity of spots relates to the electron density (which relates to the molecules) in the unit cell 4. Positions of spots relates to the arrangement of unit cells in the crystal 5. Every spot contains contributions from every part of the crystal. Every part of the map contains contributions from every spot
63 Key outcomes Understand Table 1 in X-ray Papers (now often Table S1 ) Understand the basic workflow of determining a crystal structure Embrace the beauty and challenge of disorder at high and low resolution
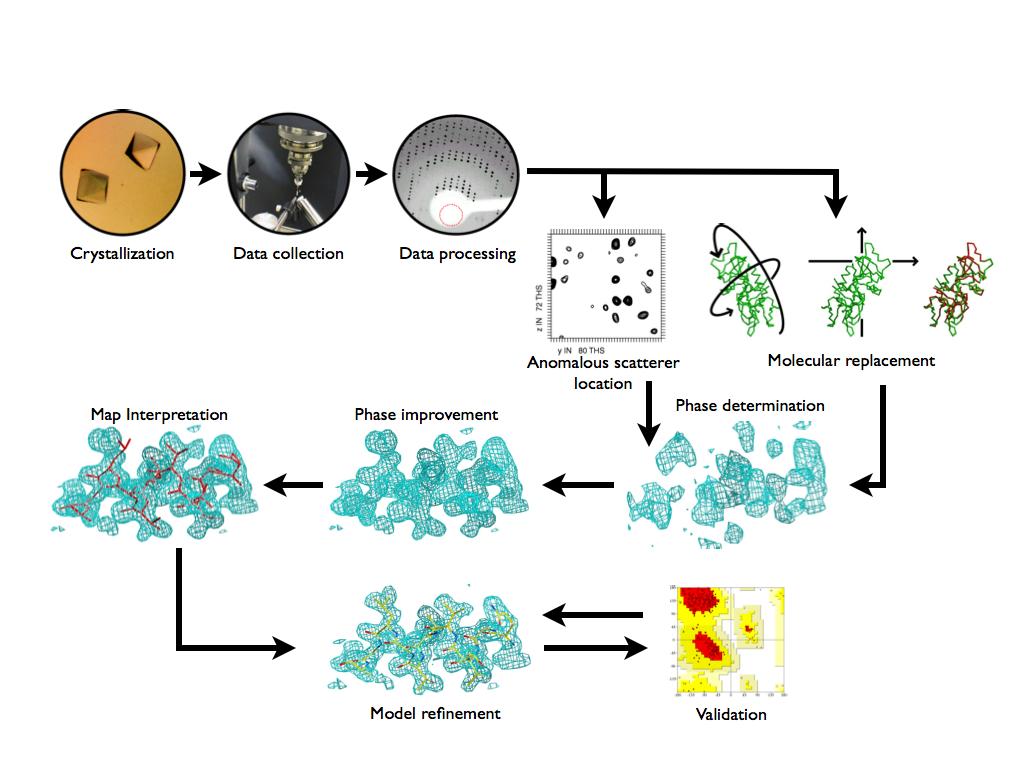
Summary of Experimental Protein Structure Determination. Key Elements
 Programme 8.00-8.20 Summary of last week s lecture and quiz 8.20-9.00 Structure validation 9.00-9.15 Break 9.15-11.00 Exercise: Structure validation tutorial 11.00-11.10 Break 11.10-11.40 Summary & discussion
Programme 8.00-8.20 Summary of last week s lecture and quiz 8.20-9.00 Structure validation 9.00-9.15 Break 9.15-11.00 Exercise: Structure validation tutorial 11.00-11.10 Break 11.10-11.40 Summary & discussion
Direct Method. Very few protein diffraction data meet the 2nd condition
 Direct Method Two conditions: -atoms in the structure are equal-weighted -resolution of data are higher than the distance between the atoms in the structure Very few protein diffraction data meet the 2nd
Direct Method Two conditions: -atoms in the structure are equal-weighted -resolution of data are higher than the distance between the atoms in the structure Very few protein diffraction data meet the 2nd
X-ray Crystallography
 2009/11/25 [ 1 ] X-ray Crystallography Andrew Torda, wintersemester 2009 / 2010 X-ray numerically most important more than 4/5 structures Goal a set of x, y, z coordinates different properties to NMR History
2009/11/25 [ 1 ] X-ray Crystallography Andrew Torda, wintersemester 2009 / 2010 X-ray numerically most important more than 4/5 structures Goal a set of x, y, z coordinates different properties to NMR History
Linking data and model quality in macromolecular crystallography. Kay Diederichs
 Linking data and model quality in macromolecular crystallography Kay Diederichs Crystallography is highly successful Can we do better? Error in experimental data Error = random + systematic Multiplicity
Linking data and model quality in macromolecular crystallography Kay Diederichs Crystallography is highly successful Can we do better? Error in experimental data Error = random + systematic Multiplicity
Molecular Biology Course 2006 Protein Crystallography Part II
 Molecular Biology Course 2006 Protein Crystallography Part II Tim Grüne University of Göttingen Dept. of Structural Chemistry December 2006 http://shelx.uni-ac.gwdg.de tg@shelx.uni-ac.gwdg.de Overview
Molecular Biology Course 2006 Protein Crystallography Part II Tim Grüne University of Göttingen Dept. of Structural Chemistry December 2006 http://shelx.uni-ac.gwdg.de tg@shelx.uni-ac.gwdg.de Overview
Electronic Supplementary Information (ESI) for Chem. Commun. Unveiling the three- dimensional structure of the green pigment of nitrite- cured meat
 for Chem. Commun. Unveiling the three- dimensional structure of the green pigment of nitrite- cured meat") Electronic Supplementary Information (ESI) for Chem. Commun. Unveiling the three- dimensional structure of the green pigment of nitrite- cured meat Jun Yi* and George B. Richter- Addo* Department of Chemistry
Electronic Supplementary Information (ESI) for Chem. Commun. Unveiling the three- dimensional structure of the green pigment of nitrite- cured meat Jun Yi* and George B. Richter- Addo* Department of Chemistry
Data quality indicators. Kay Diederichs
 Data quality indicators Kay Diederichs Crystallography has been highly successful Now 105839 Could it be any better? 2 Confusion what do these mean? CC1/2 Rmerge Rsym Mn(I/sd) I/σ Rmeas CCanom Rpim Rcum
Data quality indicators Kay Diederichs Crystallography has been highly successful Now 105839 Could it be any better? 2 Confusion what do these mean? CC1/2 Rmerge Rsym Mn(I/sd) I/σ Rmeas CCanom Rpim Rcum
Ensemble refinement of protein crystal structures in PHENIX. Tom Burnley Piet Gros
 Ensemble refinement of protein crystal structures in PHENIX Tom Burnley Piet Gros Incomplete modelling of disorder contributes to R factor gap Only ~5% of residues in the PDB are modelled with more than
Ensemble refinement of protein crystal structures in PHENIX Tom Burnley Piet Gros Incomplete modelling of disorder contributes to R factor gap Only ~5% of residues in the PDB are modelled with more than
Crystal lattice Real Space. Reflections Reciprocal Space. I. Solving Phases II. Model Building for CHEM 645. Purified Protein. Build model.
 I. Solving Phases II. Model Building for CHEM 645 Purified Protein Solve Phase Build model and refine Crystal lattice Real Space Reflections Reciprocal Space ρ (x, y, z) pronounced rho F hkl 2 I F (h,
I. Solving Phases II. Model Building for CHEM 645 Purified Protein Solve Phase Build model and refine Crystal lattice Real Space Reflections Reciprocal Space ρ (x, y, z) pronounced rho F hkl 2 I F (h,
Protein Crystallography Part II
 Molecular Biology Course 2007 Protein Crystallography Part II Tim Grüne University of Göttingen Dept. of Structural Chemistry November 2007 http://shelx.uni-ac.gwdg.de tg@shelx.uni-ac.gwdg.de Overview
Molecular Biology Course 2007 Protein Crystallography Part II Tim Grüne University of Göttingen Dept. of Structural Chemistry November 2007 http://shelx.uni-ac.gwdg.de tg@shelx.uni-ac.gwdg.de Overview
Macromolecular Crystallography Part II
 Molecular Biology Course 2009 Macromolecular Crystallography Part II Tim Grüne University of Göttingen Dept. of Structural Chemistry November 2009 http://shelx.uni-ac.gwdg.de tg@shelx.uni-ac.gwdg.de From
Molecular Biology Course 2009 Macromolecular Crystallography Part II Tim Grüne University of Göttingen Dept. of Structural Chemistry November 2009 http://shelx.uni-ac.gwdg.de tg@shelx.uni-ac.gwdg.de From
SHELXC/D/E. Andrea Thorn
 SHELXC/D/E Andrea Thorn What is experimental phasing? Experimental phasing is what you do if MR doesn t work. What is experimental phasing? Experimental phasing methods depend on intensity differences.
SHELXC/D/E Andrea Thorn What is experimental phasing? Experimental phasing is what you do if MR doesn t work. What is experimental phasing? Experimental phasing methods depend on intensity differences.
Full wwpdb X-ray Structure Validation Report i
 Full wwpdb X-ray Structure Validation Report i Mar 10, 2018 01:44 am GMT PDB ID : 1MWP Title : N-TERMINAL DOMAIN OF THE AMYLOID PRECURSOR PROTEIN Authors : Rossjohn, J.; Cappai, R.; Feil, S.C.; Henry,
Full wwpdb X-ray Structure Validation Report i Mar 10, 2018 01:44 am GMT PDB ID : 1MWP Title : N-TERMINAL DOMAIN OF THE AMYLOID PRECURSOR PROTEIN Authors : Rossjohn, J.; Cappai, R.; Feil, S.C.; Henry,
Resolution: maximum limit of diffraction (asymmetric)
") Resolution: maximum limit of diffraction (asymmetric) crystal Y X-ray source 2θ X direct beam tan 2θ = Y X d = resolution 2d sinθ = λ detector 1 Unit Cell: two vectors in plane of image c* Observe: b*
Resolution: maximum limit of diffraction (asymmetric) crystal Y X-ray source 2θ X direct beam tan 2θ = Y X d = resolution 2d sinθ = λ detector 1 Unit Cell: two vectors in plane of image c* Observe: b*
Full wwpdb X-ray Structure Validation Report i
 Full wwpdb X-ray Structure Validation Report i Mar 8, 2018 10:24 pm GMT PDB ID : 1A30 Title : HIV-1 PROTEASE COMPLEXED WITH A TRIPEPTIDE INHIBITOR Authors : Louis, J.M.; Dyda, F.; Nashed, N.T.; Kimmel,
Full wwpdb X-ray Structure Validation Report i Mar 8, 2018 10:24 pm GMT PDB ID : 1A30 Title : HIV-1 PROTEASE COMPLEXED WITH A TRIPEPTIDE INHIBITOR Authors : Louis, J.M.; Dyda, F.; Nashed, N.T.; Kimmel,
Full wwpdb X-ray Structure Validation Report i
 Full wwpdb X-ray Structure Validation Report i Mar 8, 2018 06:13 pm GMT PDB ID : 5G5C Title : Structure of the Pyrococcus furiosus Esterase Pf2001 with space group C2221 Authors : Varejao, N.; Reverter,
Full wwpdb X-ray Structure Validation Report i Mar 8, 2018 06:13 pm GMT PDB ID : 5G5C Title : Structure of the Pyrococcus furiosus Esterase Pf2001 with space group C2221 Authors : Varejao, N.; Reverter,
Full wwpdb X-ray Structure Validation Report i
 Full wwpdb X-ray Structure Validation Report i Mar 14, 2018 02:00 pm GMT PDB ID : 3RRQ Title : Crystal structure of the extracellular domain of human PD-1 Authors : Lazar-Molnar, E.; Ramagopal, U.A.; Nathenson,
Full wwpdb X-ray Structure Validation Report i Mar 14, 2018 02:00 pm GMT PDB ID : 3RRQ Title : Crystal structure of the extracellular domain of human PD-1 Authors : Lazar-Molnar, E.; Ramagopal, U.A.; Nathenson,
ABSTRACT. Chong Zhang
 ABSTRACT Improved Biomolecular Crystallography at Low Resolution with the Deformable Complex Network Approach by Chong Zhang It is often a challenge to atomically determine the structure of large macromolecular
ABSTRACT Improved Biomolecular Crystallography at Low Resolution with the Deformable Complex Network Approach by Chong Zhang It is often a challenge to atomically determine the structure of large macromolecular
Macromolecular Crystallography Part II
 Molecular Biology Course 2010 Macromolecular Crystallography Part II University of Göttingen Dept. of Structural Chemistry November 2010 http://shelx.uni-ac.gwdg.de tg@shelx.uni-ac.gwdg.de Crystallography
Molecular Biology Course 2010 Macromolecular Crystallography Part II University of Göttingen Dept. of Structural Chemistry November 2010 http://shelx.uni-ac.gwdg.de tg@shelx.uni-ac.gwdg.de Crystallography
Resolution and data formats. Andrea Thorn
 Resolution and data formats Andrea Thorn RESOLUTION Motivation Courtesy of M. Sawaya Map resolution http://www.bmsc.washington.edu/people/verlinde/experiment.html Data quality indicators Resolution accounts
Resolution and data formats Andrea Thorn RESOLUTION Motivation Courtesy of M. Sawaya Map resolution http://www.bmsc.washington.edu/people/verlinde/experiment.html Data quality indicators Resolution accounts
CCP4 Diamond 2014 SHELXC/D/E. Andrea Thorn
 CCP4 Diamond 2014 SHELXC/D/E Andrea Thorn SHELXC/D/E workflow SHELXC: α calculation, file preparation SHELXD: Marker atom search = substructure search SHELXE: density modification Maps and coordinate files
CCP4 Diamond 2014 SHELXC/D/E Andrea Thorn SHELXC/D/E workflow SHELXC: α calculation, file preparation SHELXD: Marker atom search = substructure search SHELXE: density modification Maps and coordinate files
Full wwpdb X-ray Structure Validation Report i
 Full wwpdb X-ray Structure Validation Report i Jan 14, 2019 11:10 AM EST PDB ID : 6GYW Title : Crystal structure of DacA from Staphylococcus aureus Authors : Tosi, T.; Freemont, P.S.; Grundling, A. Deposited
Full wwpdb X-ray Structure Validation Report i Jan 14, 2019 11:10 AM EST PDB ID : 6GYW Title : Crystal structure of DacA from Staphylococcus aureus Authors : Tosi, T.; Freemont, P.S.; Grundling, A. Deposited
Full wwpdb X-ray Structure Validation Report i
 Full wwpdb X-ray Structure Validation Report i Jan 28, 2019 11:10 AM EST PDB ID : 6A5H Title : The structure of [4+2] and [6+4] cyclase in the biosynthetic pathway of unidentified natural product Authors
Full wwpdb X-ray Structure Validation Report i Jan 28, 2019 11:10 AM EST PDB ID : 6A5H Title : The structure of [4+2] and [6+4] cyclase in the biosynthetic pathway of unidentified natural product Authors
Full wwpdb X-ray Structure Validation Report i
 Full wwpdb X-ray Structure Validation Report i Jan 17, 2019 09:42 AM EST PDB ID : 6D3Z Title : Protease SFTI complex Authors : Law, R.H.P.; Wu, G. Deposited on : 2018-04-17 Resolution : 2.00 Å(reported)
Full wwpdb X-ray Structure Validation Report i Jan 17, 2019 09:42 AM EST PDB ID : 6D3Z Title : Protease SFTI complex Authors : Law, R.H.P.; Wu, G. Deposited on : 2018-04-17 Resolution : 2.00 Å(reported)
Structure factors again
 Structure factors again Remember 1D, structure factor for order h F h = F h exp[iα h ] = I 01 ρ(x)exp[2πihx]dx Where x is fractional position along unit cell distance (repeating distance, origin arbitrary)
Structure factors again Remember 1D, structure factor for order h F h = F h exp[iα h ] = I 01 ρ(x)exp[2πihx]dx Where x is fractional position along unit cell distance (repeating distance, origin arbitrary)
SUPPLEMENTARY INFORMATION
 Table of Contents Page Supplementary Table 1. Diffraction data collection statistics 2 Supplementary Table 2. Crystallographic refinement statistics 3 Supplementary Fig. 1. casic1mfc packing in the R3
Table of Contents Page Supplementary Table 1. Diffraction data collection statistics 2 Supplementary Table 2. Crystallographic refinement statistics 3 Supplementary Fig. 1. casic1mfc packing in the R3
Protein Crystallography
 Protein Crystallography Part II Tim Grüne Dept. of Structural Chemistry Prof. G. Sheldrick University of Göttingen http://shelx.uni-ac.gwdg.de tg@shelx.uni-ac.gwdg.de Overview The Reciprocal Lattice The
Protein Crystallography Part II Tim Grüne Dept. of Structural Chemistry Prof. G. Sheldrick University of Göttingen http://shelx.uni-ac.gwdg.de tg@shelx.uni-ac.gwdg.de Overview The Reciprocal Lattice The
Anisotropy in macromolecular crystal structures. Andrea Thorn July 19 th, 2012
 Anisotropy in macromolecular crystal structures Andrea Thorn July 19 th, 2012 Motivation Courtesy of M. Sawaya Motivation Crystal structures are inherently anisotropic. X-ray diffraction reflects this
Anisotropy in macromolecular crystal structures Andrea Thorn July 19 th, 2012 Motivation Courtesy of M. Sawaya Motivation Crystal structures are inherently anisotropic. X-ray diffraction reflects this
1.b What are current best practices for selecting an initial target ligand atomic model(s) for structure refinement from X-ray diffraction data?!
 for structure refinement from X-ray diffraction data?!") 1.b What are current best practices for selecting an initial target ligand atomic model(s) for structure refinement from X-ray diffraction data?! Visual analysis: Identification of ligand density from
1.b What are current best practices for selecting an initial target ligand atomic model(s) for structure refinement from X-ray diffraction data?! Visual analysis: Identification of ligand density from
Protein crystallography. Garry Taylor
 Protein crystallography Garry Taylor X-ray Crystallography - the Basics Grow crystals Collect X-ray data Determine phases Calculate ρ-map Interpret map Refine coordinates Do the biology. Nitrogen at -180
Protein crystallography Garry Taylor X-ray Crystallography - the Basics Grow crystals Collect X-ray data Determine phases Calculate ρ-map Interpret map Refine coordinates Do the biology. Nitrogen at -180
Full wwpdb X-ray Structure Validation Report i
 Full wwpdb X-ray Structure Validation Report i Mar 13, 2018 04:03 pm GMT PDB ID : 5NMJ Title : Chicken GRIFIN (crystallisation ph: 6.5) Authors : Ruiz, F.M.; Romero, A. Deposited on : 2017-04-06 Resolution
Full wwpdb X-ray Structure Validation Report i Mar 13, 2018 04:03 pm GMT PDB ID : 5NMJ Title : Chicken GRIFIN (crystallisation ph: 6.5) Authors : Ruiz, F.M.; Romero, A. Deposited on : 2017-04-06 Resolution
wwpdb X-ray Structure Validation Summary Report
 wwpdb X-ray Structure Validation Summary Report io Jan 31, 2016 06:45 PM GMT PDB ID : 1CBS Title : CRYSTAL STRUCTURE OF CELLULAR RETINOIC-ACID-BINDING PROTEINS I AND II IN COMPLEX WITH ALL-TRANS-RETINOIC
wwpdb X-ray Structure Validation Summary Report io Jan 31, 2016 06:45 PM GMT PDB ID : 1CBS Title : CRYSTAL STRUCTURE OF CELLULAR RETINOIC-ACID-BINDING PROTEINS I AND II IN COMPLEX WITH ALL-TRANS-RETINOIC
Full wwpdb X-ray Structure Validation Report i
 Full wwpdb X-ray Structure Validation Report i Mar 8, 2018 08:34 pm GMT PDB ID : 1RUT Title : Complex of LMO4 LIM domains 1 and 2 with the ldb1 LID domain Authors : Deane, J.E.; Ryan, D.P.; Maher, M.J.;
Full wwpdb X-ray Structure Validation Report i Mar 8, 2018 08:34 pm GMT PDB ID : 1RUT Title : Complex of LMO4 LIM domains 1 and 2 with the ldb1 LID domain Authors : Deane, J.E.; Ryan, D.P.; Maher, M.J.;
Ultra-high resolution structures in validation
 Ultra-high resolution structures in validation (and not only...) Mariusz Jaskolski Department of Crystallography,, A. Mickiewicz University Center for Biocrystallographic Research, Polish Academy of Sciences,
Ultra-high resolution structures in validation (and not only...) Mariusz Jaskolski Department of Crystallography,, A. Mickiewicz University Center for Biocrystallographic Research, Polish Academy of Sciences,
X-ray Crystallography. Kalyan Das
 X-ray Crystallography Kalyan Das Electromagnetic Spectrum NMR 10 um - 10 mm 700 to 10 4 nm 400 to 700 nm 10 to 400 nm 10-1 to 10 nm 10-4 to 10-1 nm X-ray radiation was discovered by Roentgen in 1895. X-rays
X-ray Crystallography Kalyan Das Electromagnetic Spectrum NMR 10 um - 10 mm 700 to 10 4 nm 400 to 700 nm 10 to 400 nm 10-1 to 10 nm 10-4 to 10-1 nm X-ray radiation was discovered by Roentgen in 1895. X-rays
The structure of Aquifex aeolicus FtsH in the ADP-bound state reveals a C2-symmetric hexamer
 Volume 71 (2015) Supporting information for article: The structure of Aquifex aeolicus FtsH in the ADP-bound state reveals a C2-symmetric hexamer Marina Vostrukhina, Alexander Popov, Elena Brunstein, Martin
Volume 71 (2015) Supporting information for article: The structure of Aquifex aeolicus FtsH in the ADP-bound state reveals a C2-symmetric hexamer Marina Vostrukhina, Alexander Popov, Elena Brunstein, Martin
Handout 12 Structure refinement. Completing the structure and evaluating how good your data and model agree
 Handout 1 Structure refinement Completing the structure and evaluating how good your data and model agree Why you should refine a structure We have considered how atoms are located by Patterson, direct
Handout 1 Structure refinement Completing the structure and evaluating how good your data and model agree Why you should refine a structure We have considered how atoms are located by Patterson, direct
7.91 Amy Keating. Solving structures using X-ray crystallography & NMR spectroscopy
 7.91 Amy Keating Solving structures using X-ray crystallography & NMR spectroscopy How are X-ray crystal structures determined? 1. Grow crystals - structure determination by X-ray crystallography relies
7.91 Amy Keating Solving structures using X-ray crystallography & NMR spectroscopy How are X-ray crystal structures determined? 1. Grow crystals - structure determination by X-ray crystallography relies
Proteins. Central Dogma : DNA RNA protein Amino acid polymers - defined composition & order. Perform nearly all cellular functions Drug Targets
 Proteins Central Dogma : DNA RNA protein Amino acid polymers - defined composition & order Perform nearly all cellular functions Drug Targets Fold into discrete shapes. Proteins - cont. Specific shapes
Proteins Central Dogma : DNA RNA protein Amino acid polymers - defined composition & order Perform nearly all cellular functions Drug Targets Fold into discrete shapes. Proteins - cont. Specific shapes
TLS and all that. Ethan A Merritt. CCP4 Summer School 2011 (Argonne, IL) Abstract
 Abstract") TLS and all that Ethan A Merritt CCP4 Summer School 2011 (Argonne, IL) Abstract We can never know the position of every atom in a crystal structure perfectly. Each atom has an associated positional uncertainty.
TLS and all that Ethan A Merritt CCP4 Summer School 2011 (Argonne, IL) Abstract We can never know the position of every atom in a crystal structure perfectly. Each atom has an associated positional uncertainty.
Fourier Syntheses, Analyses, and Transforms
 Fourier Syntheses, Analyses, and Transforms http://homepages.utoledo.edu/clind/ The electron density The electron density in a crystal can be described as a periodic function - same contents in each unit
Fourier Syntheses, Analyses, and Transforms http://homepages.utoledo.edu/clind/ The electron density The electron density in a crystal can be described as a periodic function - same contents in each unit
RNA protects a nucleoprotein complex against radiation damage
 Supporting information Volume 72 (2016) Supporting information for article: RNA protects a nucleoprotein complex against radiation damage Charles S. Bury, John E. McGeehan, Alfred A. Antson, Ian Carmichael,
Supporting information Volume 72 (2016) Supporting information for article: RNA protects a nucleoprotein complex against radiation damage Charles S. Bury, John E. McGeehan, Alfred A. Antson, Ian Carmichael,
Data quality noise, errors, mistakes
 Data quality noise, errors, mistakes Kay Diederichs Protein Crystallography / Molecular Bioinformatics University of Konstanz, Germany Crystallography has been extremely successful Protein Data Bank on
Data quality noise, errors, mistakes Kay Diederichs Protein Crystallography / Molecular Bioinformatics University of Konstanz, Germany Crystallography has been extremely successful Protein Data Bank on
X-ray Diffraction. Diffraction. X-ray Generation. X-ray Generation. X-ray Generation. X-ray Spectrum from Tube
 X-ray Diffraction Mineral identification Mode analysis Structure Studies X-ray Generation X-ray tube (sealed) Pure metal target (Cu) Electrons remover inner-shell electrons from target. Other electrons
X-ray Diffraction Mineral identification Mode analysis Structure Studies X-ray Generation X-ray tube (sealed) Pure metal target (Cu) Electrons remover inner-shell electrons from target. Other electrons
Full wwpdb X-ray Structure Validation Report i
 Full wwpdb X-ray Structure Validation Report i Feb 17, 2018 01:16 am GMT PDB ID : 1IFT Title : RICIN A-CHAIN (RECOMBINANT) Authors : Weston, S.A.; Tucker, A.D.; Thatcher, D.R.; Derbyshire, D.J.; Pauptit,
Full wwpdb X-ray Structure Validation Report i Feb 17, 2018 01:16 am GMT PDB ID : 1IFT Title : RICIN A-CHAIN (RECOMBINANT) Authors : Weston, S.A.; Tucker, A.D.; Thatcher, D.R.; Derbyshire, D.J.; Pauptit,
Crystals, X-rays and Proteins
 Crystals, X-rays and Proteins Comprehensive Protein Crystallography Dennis Sherwood MA (Hons), MPhil, PhD Jon Cooper BA (Hons), PhD OXFORD UNIVERSITY PRESS Contents List of symbols xiv PART I FUNDAMENTALS
Crystals, X-rays and Proteins Comprehensive Protein Crystallography Dennis Sherwood MA (Hons), MPhil, PhD Jon Cooper BA (Hons), PhD OXFORD UNIVERSITY PRESS Contents List of symbols xiv PART I FUNDAMENTALS
Electron Density at various resolutions, and fitting a model as accurately as possible.
 Section 9, Electron Density Maps 900 Electron Density at various resolutions, and fitting a model as accurately as possible. ρ xyz = (Vol) -1 h k l m hkl F hkl e iφ hkl e-i2π( hx + ky + lz ) Amplitude
Section 9, Electron Density Maps 900 Electron Density at various resolutions, and fitting a model as accurately as possible. ρ xyz = (Vol) -1 h k l m hkl F hkl e iφ hkl e-i2π( hx + ky + lz ) Amplitude
Refinement of Disorder with SHELXL
 Refinement of Disorder with SHELXL Peter Müller MIT pmueller@mit.edu Single Crystal Structure Determination A crystal is a potentially endless, three-dimensional, periodic discontinuum built up by atoms,
Refinement of Disorder with SHELXL Peter Müller MIT pmueller@mit.edu Single Crystal Structure Determination A crystal is a potentially endless, three-dimensional, periodic discontinuum built up by atoms,
Nitrogenase MoFe protein from Clostridium pasteurianum at 1.08 Å resolution: comparison with the Azotobacter vinelandii MoFe protein
 Acta Cryst. (2015). D71, 274-282, doi:10.1107/s1399004714025243 Supporting information Volume 71 (2015) Supporting information for article: Nitrogenase MoFe protein from Clostridium pasteurianum at 1.08
Acta Cryst. (2015). D71, 274-282, doi:10.1107/s1399004714025243 Supporting information Volume 71 (2015) Supporting information for article: Nitrogenase MoFe protein from Clostridium pasteurianum at 1.08
X-Ray structure analysis
 X-Ray structure analysis Kay Diederichs kay.diederichs@uni-konstanz.de Analysis of what? Proteins ( /ˈproʊˌtiːnz/ or /ˈproʊti.ɨnz/) are biochemical compounds consisting of one or more polypeptides typically
X-Ray structure analysis Kay Diederichs kay.diederichs@uni-konstanz.de Analysis of what? Proteins ( /ˈproʊˌtiːnz/ or /ˈproʊti.ɨnz/) are biochemical compounds consisting of one or more polypeptides typically
HOMOLOGY MODELING. The sequence alignment and template structure are then used to produce a structural model of the target.
 HOMOLOGY MODELING Homology modeling, also known as comparative modeling of protein refers to constructing an atomic-resolution model of the "target" protein from its amino acid sequence and an experimental
HOMOLOGY MODELING Homology modeling, also known as comparative modeling of protein refers to constructing an atomic-resolution model of the "target" protein from its amino acid sequence and an experimental
X-ray Data Collection. Bio5325 Spring 2006
 X-ray Data Collection Bio535 Spring 006 Obtaining I hkl and α (Ihkl) from Frame Images Braggs Law -predicts conditions for in-phase scattering by equivalent atoms lying in planes that transect a crystal.
X-ray Data Collection Bio535 Spring 006 Obtaining I hkl and α (Ihkl) from Frame Images Braggs Law -predicts conditions for in-phase scattering by equivalent atoms lying in planes that transect a crystal.
Phase problem: Determining an initial phase angle α hkl for each recorded reflection. 1 ρ(x,y,z) = F hkl cos 2π (hx+ky+ lz - α hkl ) V h k l
 = F hkl cos 2π (hx+ky+ lz - α hkl ) V h k l") Phase problem: Determining an initial phase angle α hkl for each recorded reflection 1 ρ(x,y,z) = F hkl cos 2π (hx+ky+ lz - α hkl ) V h k l Methods: Heavy atom methods (isomorphous replacement Hg, Pt)
Phase problem: Determining an initial phase angle α hkl for each recorded reflection 1 ρ(x,y,z) = F hkl cos 2π (hx+ky+ lz - α hkl ) V h k l Methods: Heavy atom methods (isomorphous replacement Hg, Pt)
Molecular Modeling lecture 2
 Molecular Modeling 2018 -- lecture 2 Topics 1. Secondary structure 3. Sequence similarity and homology 2. Secondary structure prediction 4. Where do protein structures come from? X-ray crystallography
Molecular Modeling 2018 -- lecture 2 Topics 1. Secondary structure 3. Sequence similarity and homology 2. Secondary structure prediction 4. Where do protein structures come from? X-ray crystallography
Schematic representation of relation between disorder and scattering
 Crystal lattice Reciprocal lattice FT Schematic representation of relation between disorder and scattering ρ = Δρ + Occupational disorder Diffuse scattering Bragg scattering ρ = Δρ + Positional
Crystal lattice Reciprocal lattice FT Schematic representation of relation between disorder and scattering ρ = Δρ + Occupational disorder Diffuse scattering Bragg scattering ρ = Δρ + Positional
PSD '18 -- Xray lecture 4. Laue conditions Fourier Transform The reciprocal lattice data collection
 PSD '18 -- Xray lecture 4 Laue conditions Fourier Transform The reciprocal lattice data collection 1 Fourier Transform The Fourier Transform is a conversion of one space into another space with reciprocal
PSD '18 -- Xray lecture 4 Laue conditions Fourier Transform The reciprocal lattice data collection 1 Fourier Transform The Fourier Transform is a conversion of one space into another space with reciprocal
SOLID STATE 18. Reciprocal Space
 SOLID STATE 8 Reciprocal Space Wave vectors and the concept of K-space can simplify the explanation of several properties of the solid state. They will be introduced to provide more information on diffraction
SOLID STATE 8 Reciprocal Space Wave vectors and the concept of K-space can simplify the explanation of several properties of the solid state. They will be introduced to provide more information on diffraction
Acta Crystallographica Section F
 Supporting information Acta Crystallographica Section F Volume 70 (2014) Supporting information for article: Chemical conversion of cisplatin and carboplatin with histidine in a model protein crystallised
Supporting information Acta Crystallographica Section F Volume 70 (2014) Supporting information for article: Chemical conversion of cisplatin and carboplatin with histidine in a model protein crystallised
3.012 Structure An Introduction to X-ray Diffraction
 3.012 Structure An Introduction to X-ray Diffraction This handout summarizes some topics that are important for understanding x-ray diffraction. The following references provide a thorough explanation
3.012 Structure An Introduction to X-ray Diffraction This handout summarizes some topics that are important for understanding x-ray diffraction. The following references provide a thorough explanation
X- ray crystallography. CS/CME/Biophys/BMI 279 Nov. 12, 2015 Ron Dror
 X- ray crystallography CS/CME/Biophys/BMI 279 Nov. 12, 2015 Ron Dror 1 Outline Overview of x-ray crystallography Crystals Electron density Diffraction patterns The computational problem: determining structure
X- ray crystallography CS/CME/Biophys/BMI 279 Nov. 12, 2015 Ron Dror 1 Outline Overview of x-ray crystallography Crystals Electron density Diffraction patterns The computational problem: determining structure
Model and data. An X-ray structure solution requires a model.
 Model and data An X-ray structure solution requires a model. This model has to be consistent with: The findings of Chemistry Reflection positions and intensities Structure refinement = Model fitting by
Model and data An X-ray structure solution requires a model. This model has to be consistent with: The findings of Chemistry Reflection positions and intensities Structure refinement = Model fitting by
Introduction to Comparative Protein Modeling. Chapter 4 Part I
 Introduction to Comparative Protein Modeling Chapter 4 Part I 1 Information on Proteins Each modeling study depends on the quality of the known experimental data. Basis of the model Search in the literature
Introduction to Comparative Protein Modeling Chapter 4 Part I 1 Information on Proteins Each modeling study depends on the quality of the known experimental data. Basis of the model Search in the literature
CS273: Algorithms for Structure Handout # 13 and Motion in Biology Stanford University Tuesday, 11 May 2003
 CS273: Algorithms for Structure Handout # 13 and Motion in Biology Stanford University Tuesday, 11 May 2003 Lecture #13: 11 May 2004 Topics: Protein Structure Determination Scribe: Minli Zhu We acknowledge
CS273: Algorithms for Structure Handout # 13 and Motion in Biology Stanford University Tuesday, 11 May 2003 Lecture #13: 11 May 2004 Topics: Protein Structure Determination Scribe: Minli Zhu We acknowledge
Chapter 2. X-ray X. Diffraction and Reciprocal Lattice. Scattering from Lattices
 Chapter. X-ray X Diffraction and Reciprocal Lattice Diffraction of waves by crystals Reciprocal Lattice Diffraction of X-rays Powder diffraction Single crystal X-ray diffraction Scattering from Lattices
Chapter. X-ray X Diffraction and Reciprocal Lattice Diffraction of waves by crystals Reciprocal Lattice Diffraction of X-rays Powder diffraction Single crystal X-ray diffraction Scattering from Lattices
Small Molecule Crystallography Lab Department of Chemistry and Biochemistry University of Oklahoma 101 Stephenson Parkway Norman, OK
 Small Molecule Crystallography Lab Department of Chemistry and Biochemistry University of Oklahoma 101 Stephenson Parkway Norman, OK 73019-5251 Sample: KP-XI-cinnamyl-chiral alcohol Lab ID: 12040 User:
Small Molecule Crystallography Lab Department of Chemistry and Biochemistry University of Oklahoma 101 Stephenson Parkway Norman, OK 73019-5251 Sample: KP-XI-cinnamyl-chiral alcohol Lab ID: 12040 User:
SUPPLEMENTARY INFORMATION
 SUPPLEMENTARY INFORMATION doi:10.1038/nature11539 Supplementary Figure 1 Schematic representation of plant (A) and mammalian (B) P 2B -ATPase domain organization. Actuator (A-), nucleotide binding (N-),
SUPPLEMENTARY INFORMATION doi:10.1038/nature11539 Supplementary Figure 1 Schematic representation of plant (A) and mammalian (B) P 2B -ATPase domain organization. Actuator (A-), nucleotide binding (N-),
Plasmid Relevant features Source. W18N_D20N and TrXE-W18N_D20N-anti
 Table S1. E. coli plasmids Plasmid Relevant features Source pdg680 T. reesei XynII AA 2-190 with C-terminal His 6 tag optimized for E. coli expression in pjexpress401 Wan et al. (in press) psbn44d psbn44h
Table S1. E. coli plasmids Plasmid Relevant features Source pdg680 T. reesei XynII AA 2-190 with C-terminal His 6 tag optimized for E. coli expression in pjexpress401 Wan et al. (in press) psbn44d psbn44h
SOLID STATE 9. Determination of Crystal Structures
 SOLID STATE 9 Determination of Crystal Structures In the diffraction experiment, we measure intensities as a function of d hkl. Intensities are the sum of the x-rays scattered by all the atoms in a crystal.
SOLID STATE 9 Determination of Crystal Structures In the diffraction experiment, we measure intensities as a function of d hkl. Intensities are the sum of the x-rays scattered by all the atoms in a crystal.
Molecular Replacement (Alexei Vagin s lecture)
") Molecular Replacement (Alexei Vagin s lecture) Contents What is Molecular Replacement Functions in Molecular Replacement Weighting scheme Information from data and model Some special techniques of Molecular
Molecular Replacement (Alexei Vagin s lecture) Contents What is Molecular Replacement Functions in Molecular Replacement Weighting scheme Information from data and model Some special techniques of Molecular
Experimental phasing, Pattersons and SHELX Andrea Thorn
 Experimental phasing, Pattersons and SHELX Andrea Thorn What is experimental phasing? Experimental phasing is what you do if MR doesn t work. What is experimental phasing? Experimental phasing methods
Experimental phasing, Pattersons and SHELX Andrea Thorn What is experimental phasing? Experimental phasing is what you do if MR doesn t work. What is experimental phasing? Experimental phasing methods
This is an author produced version of Privateer: : software for the conformational validation of carbohydrate structures.
 This is an author produced version of Privateer: : software for the conformational validation of carbohydrate structures. White Rose Research Online URL for this paper: http://eprints.whiterose.ac.uk/95794/
This is an author produced version of Privateer: : software for the conformational validation of carbohydrate structures. White Rose Research Online URL for this paper: http://eprints.whiterose.ac.uk/95794/
HTCondor and macromolecular structure validation
 HTCondor and macromolecular structure validation Vincent Chen John Markley/Eldon Ulrich, NMRFAM/BMRB, UW@Madison David & Jane Richardson, Duke University Macromolecules David S. Goodsell 1999 Two questions
HTCondor and macromolecular structure validation Vincent Chen John Markley/Eldon Ulrich, NMRFAM/BMRB, UW@Madison David & Jane Richardson, Duke University Macromolecules David S. Goodsell 1999 Two questions
Rigid body Rigid body approach
 Rigid body Rigid body approach Model molecule is a fragment, which is placed arbitrarily and does not contribute to structure factors Model molecule is transformed to Actual positions by translation vector
Rigid body Rigid body approach Model molecule is a fragment, which is placed arbitrarily and does not contribute to structure factors Model molecule is transformed to Actual positions by translation vector
NMR, X-ray Diffraction, Protein Structure, and RasMol
 NMR, X-ray Diffraction, Protein Structure, and RasMol Introduction So far we have been mostly concerned with the proteins themselves. The techniques (NMR or X-ray diffraction) used to determine a structure
NMR, X-ray Diffraction, Protein Structure, and RasMol Introduction So far we have been mostly concerned with the proteins themselves. The techniques (NMR or X-ray diffraction) used to determine a structure
Copyright WILEY-VCH Verlag GmbH, D Weinheim, 2000 Angew. Chem Supporting Information For Binding Cesium Ion with Nucleoside Pentamers.
 Copyright WILEY-VCH Verlag GmbH, D-69451 Weinheim, 2000 Angew. Chem. 2000 Supporting Information For Binding Cesium Ion with Nucleoside Pentamers. Templated Self-Assembly of an Isoguanosine Decamer.**
Copyright WILEY-VCH Verlag GmbH, D-69451 Weinheim, 2000 Angew. Chem. 2000 Supporting Information For Binding Cesium Ion with Nucleoside Pentamers. Templated Self-Assembly of an Isoguanosine Decamer.**
shelxl: Refinement of Macromolecular Structures from Neutron Data
 ESS Neutron Protein Crystallography 2013 Aarhus, Denmark shelxl: Refinement of Macromolecular Structures from Neutron Data Tim Grüne University of Göttingen Dept. of Structural Chemistry http://shelx.uni-ac.gwdg.de
ESS Neutron Protein Crystallography 2013 Aarhus, Denmark shelxl: Refinement of Macromolecular Structures from Neutron Data Tim Grüne University of Göttingen Dept. of Structural Chemistry http://shelx.uni-ac.gwdg.de
APPENDIX E. Crystallographic Data for TBA Eu(DO2A)(DPA) Temperature Dependence
(DPA) Temperature Dependence") APPENDIX E Crystallographic Data for TBA Eu(DO2A)(DPA) Temperature Dependence Temperature Designation CCDC Page 100 K MLC18 761599 E2 200 K MLC17 762705 E17 300 K MLC19 763335 E31 E2 CALIFORNIA INSTITUTE
APPENDIX E Crystallographic Data for TBA Eu(DO2A)(DPA) Temperature Dependence Temperature Designation CCDC Page 100 K MLC18 761599 E2 200 K MLC17 762705 E17 300 K MLC19 763335 E31 E2 CALIFORNIA INSTITUTE
How to interpret the BUSTER reciprocal space correlation coefficients plot. Gérard Bricogne
 How to interpret the BUSTER reciprocal space correlation coefficients plot Gérard Bricogne These graphs display the correlation coefficients CC(X,Y) between pairs of quantities X and Y attached to unique
How to interpret the BUSTER reciprocal space correlation coefficients plot Gérard Bricogne These graphs display the correlation coefficients CC(X,Y) between pairs of quantities X and Y attached to unique
Automated identification of functional dynamic contact networks from X-ray crystallography
 1 Automated identification of functional dynamic contact networks from X-ray crystallography Henry van den Bedem, Gira Bhabha, Kun Yang, Peter E. Wright and James S. Fraser Supplementary Figure 1 Supplementary
1 Automated identification of functional dynamic contact networks from X-ray crystallography Henry van den Bedem, Gira Bhabha, Kun Yang, Peter E. Wright and James S. Fraser Supplementary Figure 1 Supplementary
Likelihood and SAD phasing in Phaser. R J Read, Department of Haematology Cambridge Institute for Medical Research
 Likelihood and SAD phasing in Phaser R J Read, Department of Haematology Cambridge Institute for Medical Research Concept of likelihood Likelihood with dice 4 6 8 10 Roll a seven. Which die?? p(4)=p(6)=0
Likelihood and SAD phasing in Phaser R J Read, Department of Haematology Cambridge Institute for Medical Research Concept of likelihood Likelihood with dice 4 6 8 10 Roll a seven. Which die?? p(4)=p(6)=0
Table 1. Crystallographic data collection, phasing and refinement statistics. Native Hg soaked Mn soaked 1 Mn soaked 2
 Table 1. Crystallographic data collection, phasing and refinement statistics Native Hg soaked Mn soaked 1 Mn soaked 2 Data collection Space group P2 1 2 1 2 1 P2 1 2 1 2 1 P2 1 2 1 2 1 P2 1 2 1 2 1 Cell
Table 1. Crystallographic data collection, phasing and refinement statistics Native Hg soaked Mn soaked 1 Mn soaked 2 Data collection Space group P2 1 2 1 2 1 P2 1 2 1 2 1 P2 1 2 1 2 1 P2 1 2 1 2 1 Cell
Full wwpdb/emdatabank EM Map/Model Validation Report i
 Full wwpdb/emdatabank EM Map/Model Validation Report i Sep 25, 2018 07:01 PM EDT PDB ID : 6C0V EMDB ID: : EMD-7325 Title : Molecular structure of human P-glycoprotein in the ATP-bound, outwardfacing conformation
Full wwpdb/emdatabank EM Map/Model Validation Report i Sep 25, 2018 07:01 PM EDT PDB ID : 6C0V EMDB ID: : EMD-7325 Title : Molecular structure of human P-glycoprotein in the ATP-bound, outwardfacing conformation
Twinning. Andrea Thorn
 Twinning Andrea Thorn OVERVIEW Introduction: Definitions, origins of twinning Merohedral twins: Recognition, statistical analysis: H plot, Yeates Padilla plot Example Refinement and R values Reticular
Twinning Andrea Thorn OVERVIEW Introduction: Definitions, origins of twinning Merohedral twins: Recognition, statistical analysis: H plot, Yeates Padilla plot Example Refinement and R values Reticular
PLATON/SQUEEZE. Ton Spek. Bijvoet Center Utrecht University, The Netherlands. PLATON Workshop
 PLATON/SQUEEZE Ton Spek Bijvoet Center Utrecht University, The Netherlands. PLATON Workshop Chicago, 24-July-2010 The Disordered Solvent Problem Molecules of interest often co-crystallize (only) with the
PLATON/SQUEEZE Ton Spek Bijvoet Center Utrecht University, The Netherlands. PLATON Workshop Chicago, 24-July-2010 The Disordered Solvent Problem Molecules of interest often co-crystallize (only) with the
Physics with Neutrons I, WS 2015/2016. Lecture 11, MLZ is a cooperation between:
 Physics with Neutrons I, WS 2015/2016 Lecture 11, 11.1.2016 MLZ is a cooperation between: Organization Exam (after winter term) Registration: via TUM-Online between 16.11.2015 15.1.2015 Email: sebastian.muehlbauer@frm2.tum.de
Physics with Neutrons I, WS 2015/2016 Lecture 11, 11.1.2016 MLZ is a cooperation between: Organization Exam (after winter term) Registration: via TUM-Online between 16.11.2015 15.1.2015 Email: sebastian.muehlbauer@frm2.tum.de
PSD '17 -- Xray Lecture 5, 6. Patterson Space, Molecular Replacement and Heavy Atom Isomorphous Replacement
 PSD '17 -- Xray Lecture 5, 6 Patterson Space, Molecular Replacement and Heavy Atom Isomorphous Replacement The Phase Problem We can t measure the phases! X-ray detectors (film, photomultiplier tubes, CCDs,
PSD '17 -- Xray Lecture 5, 6 Patterson Space, Molecular Replacement and Heavy Atom Isomorphous Replacement The Phase Problem We can t measure the phases! X-ray detectors (film, photomultiplier tubes, CCDs,
Rietveld Structure Refinement of Protein Powder Diffraction Data using GSAS
 Rietveld Structure Refinement of Protein Powder Diffraction Data using GSAS Jon Wright ESRF, Grenoble, France Plan This is a users perspective Cover the protein specific aspects (assuming knowledge of
Rietveld Structure Refinement of Protein Powder Diffraction Data using GSAS Jon Wright ESRF, Grenoble, France Plan This is a users perspective Cover the protein specific aspects (assuming knowledge of
Macromolecular X-ray Crystallography
 Protein Structural Models for CHEM 641 Fall 07 Brian Bahnson Department of Chemistry & Biochemistry University of Delaware Macromolecular X-ray Crystallography Purified Protein X-ray Diffraction Data collection
Protein Structural Models for CHEM 641 Fall 07 Brian Bahnson Department of Chemistry & Biochemistry University of Delaware Macromolecular X-ray Crystallography Purified Protein X-ray Diffraction Data collection
Template Free Protein Structure Modeling Jianlin Cheng, PhD
 Template Free Protein Structure Modeling Jianlin Cheng, PhD Associate Professor Computer Science Department Informatics Institute University of Missouri, Columbia 2013 Protein Energy Landscape & Free Sampling
Template Free Protein Structure Modeling Jianlin Cheng, PhD Associate Professor Computer Science Department Informatics Institute University of Missouri, Columbia 2013 Protein Energy Landscape & Free Sampling
Acta Cryst. (2017). D73, doi: /s
. D73, doi: /s") Acta Cryst. (2017). D73, doi:10.1107/s2059798317010932 Supporting information Volume 73 (2017) Supporting information for article: Designing better diffracting crystals of biotin carboxyl carrier protein
Acta Cryst. (2017). D73, doi:10.1107/s2059798317010932 Supporting information Volume 73 (2017) Supporting information for article: Designing better diffracting crystals of biotin carboxyl carrier protein
Joana Pereira Lamzin Group EMBL Hamburg, Germany. Small molecules How to identify and build them (with ARP/wARP)
") Joana Pereira Lamzin Group EMBL Hamburg, Germany Small molecules How to identify and build them (with ARP/wARP) The task at hand To find ligand density and build it! Fitting a ligand We have: electron
Joana Pereira Lamzin Group EMBL Hamburg, Germany Small molecules How to identify and build them (with ARP/wARP) The task at hand To find ligand density and build it! Fitting a ligand We have: electron
Protein Structure Determination. Part 1 -- X-ray Crystallography
 Protein Structure Determination Part 1 -- X-ray Crystallography Topics covering in this 1/2 course Crystal growth Diffraction theory Symmetry Solving phases using heavy atoms Solving phases using a model
Protein Structure Determination Part 1 -- X-ray Crystallography Topics covering in this 1/2 course Crystal growth Diffraction theory Symmetry Solving phases using heavy atoms Solving phases using a model
disordered, ordered and coherent with the substrate, and ordered but incoherent with the substrate.
 5. Nomenclature of overlayer structures Thus far, we have been discussing an ideal surface, which is in effect the structure of the topmost substrate layer. The surface (selvedge) layers of the solid however
5. Nomenclature of overlayer structures Thus far, we have been discussing an ideal surface, which is in effect the structure of the topmost substrate layer. The surface (selvedge) layers of the solid however
A Primer in X-ray Crystallography for Redox Biologists. Mark Wilson Karolinska Institute June 3 rd, 2014
 A Primer in X-ray Crystallography for Redox Biologists Mark Wilson Karolinska Institute June 3 rd, 2014 X-ray Crystallography Basics Optimistic workflow for crystallography Experiment Schematic Fourier
A Primer in X-ray Crystallography for Redox Biologists Mark Wilson Karolinska Institute June 3 rd, 2014 X-ray Crystallography Basics Optimistic workflow for crystallography Experiment Schematic Fourier
Full wwpdb NMR Structure Validation Report i
 Full wwpdb NMR Structure Validation Report i Feb 17, 2018 06:22 am GMT PDB ID : 141D Title : SOLUTION STRUCTURE OF A CONSERVED DNA SEQUENCE FROM THE HIV-1 GENOME: RESTRAINED MOLECULAR DYNAMICS SIMU- LATION
Full wwpdb NMR Structure Validation Report i Feb 17, 2018 06:22 am GMT PDB ID : 141D Title : SOLUTION STRUCTURE OF A CONSERVED DNA SEQUENCE FROM THE HIV-1 GENOME: RESTRAINED MOLECULAR DYNAMICS SIMU- LATION
Scattering Lecture. February 24, 2014
 Scattering Lecture February 24, 2014 Structure Determination by Scattering Waves of radiation scattered by different objects interfere to give rise to an observable pattern! The wavelength needs to close
Scattering Lecture February 24, 2014 Structure Determination by Scattering Waves of radiation scattered by different objects interfere to give rise to an observable pattern! The wavelength needs to close
 Helpful resources for all X ray lectures Crystallization http://www.hamptonresearch.com under tech support: crystal growth 101 literature Spacegroup tables http://img.chem.ucl.ac.uk/sgp/mainmenu.htm Crystallography
Helpful resources for all X ray lectures Crystallization http://www.hamptonresearch.com under tech support: crystal growth 101 literature Spacegroup tables http://img.chem.ucl.ac.uk/sgp/mainmenu.htm Crystallography
Structure and Dynamics : An Atomic View of Materials
 Structure and Dynamics : An Atomic View of Materials MARTIN T. DOVE Department ofearth Sciences University of Cambridge OXFORD UNIVERSITY PRESS Contents 1 Introduction 1 1.1 Observations 1 1.1.1 Microscopic
Structure and Dynamics : An Atomic View of Materials MARTIN T. DOVE Department ofearth Sciences University of Cambridge OXFORD UNIVERSITY PRESS Contents 1 Introduction 1 1.1 Observations 1 1.1.1 Microscopic
IgE binds asymmetrically to its B cell receptor CD23
 Supplementary Information IgE binds asymmetrically to its B cell receptor CD23 Balvinder Dhaliwal 1*, Marie O. Y. Pang 2, Anthony H. Keeble 2,3, Louisa K. James 2,4, Hannah J. Gould 2, James M. McDonnell
Supplementary Information IgE binds asymmetrically to its B cell receptor CD23 Balvinder Dhaliwal 1*, Marie O. Y. Pang 2, Anthony H. Keeble 2,3, Louisa K. James 2,4, Hannah J. Gould 2, James M. McDonnell
Experimental Phasing with SHELX C/D/E
 WIR SCHAFFEN WISSEN HEUTE FÜR MORGEN Dr. Tim Grüne :: Paul Scherrer Institut :: tim.gruene@psi.ch Experimental Phasing with SHELX C/D/E CCP4 / APS School Chicago 2017 22 nd June 2017 1 - The Phase Problem
WIR SCHAFFEN WISSEN HEUTE FÜR MORGEN Dr. Tim Grüne :: Paul Scherrer Institut :: tim.gruene@psi.ch Experimental Phasing with SHELX C/D/E CCP4 / APS School Chicago 2017 22 nd June 2017 1 - The Phase Problem