PHENIX Wizards and Tools
|
|
|
- Brendan Kelly Butler
- 5 years ago
- Views:
Transcription
1 PHENIX Wizards and Tools Tom Terwilliger Los Alamos National Laboratory l The PHENIX project Computational Crystallography Initiative (LBNL) Paul Adams, Ralf Grosse-Kunstleve, Peter Zwart, Nigel Moriarty, Nicholas Sauter, Pavel Afonine Los Alamos National Lab (LANL) Tom Terwilliger, Li-Wei Hung Cambridge University Randy Read, Airlie McCoy, Gabor Bunkoczi, Rob Oeffner Duke University Jane Richardson, David Richardson, Jeff Headd, Vincent Chen Texas A&M University Tom Ioerger, Jim Sacchettini



2 PHENIX Wizards AutoSol Wizard: Structure solution (MIR/MAD/SAD) with HYSS/Phaser/Solve/Resolve AutoBuild Wizard: Iterative density modification, model- building and refinement with Resolve/phenix.refine/Elbow; model rebuilding in place; touch-up of model; simple OMIT; SA- OMIT; Iterative-build OMIT; OMIT around atoms in a PDB file; protein, RNA, DNA model-building LigandFit Wizard: automated fitting of flexible ligands AutoMR Wizard: Phaser molecular AutoMR Wizard: Phaser molecular replacement followed by automatic rebuilding
3 Determining a SAD structure with PHENIX Solve the structure: phenix.autosol sad.sca 12 se AutoBuild a model and improve phases: phenix.autobuild after_autosol=true Find ligands: phenix.ligandfit sad.sca model=partial.pdb p ligand=atp Refine the model carefully: phenix.refine exptl_fobs_freer_flags.mtz freer flags \ overall_best.pdb #and many more commands



4 Why automate structure determination? Automation makes straightforward cases accessible to a wider group of structural biologists makes difficult cases more feasible for experts can speed up the process can help reduce errors Automation also allows you to try more possibilities estimate uncertainties
 Software carrying out")
 Strategies for structure determination and")
5 Requirements for automation of structure determination of macromolecules by X-ray crystallography y (1) Software carrying out individual steps (2) Seamless connections between steps (3) A way to decide what is good (4) Strategies for structure determination and decisionmaking





6 Why we need good measures of the quality of an electrondensity map: Which h solution is best? Are we on the right track? If map is good: It is easy
7 Why we need good measures of the quality of an electrondensity map: Which h solution is best? Are we on the right track? Contiguous solvent region Flat solvent region If map is good: It is easy Connected density


8 Histogram of electron density values has a positive skew Typical histogram of electron density Low density: Points between atoms and in solvent region High density: Points on top of atoms Number of gr rid points Histogram skewed to the right Electron density
9 Evaluating electron density maps Basis Good map Random map Skew of density (Podjarny, 1977) Highly skewed (very positive at positions of Gaussian histogram atoms, zero elsewhere) Connectivity of regions of high density A few connected regions Many very short can trace entire molecule connected regions (Baker, Krukowski, & Agard, 1993) Correlation of local rms densities (Terwilliger, 1999) Neighboring regions in map have similar rms densities Map has uniform rms density R-factor in 1 st cycle of density modification (Cowtan, 1996) Low R-factor High R-factor
10 Which scoring criteria best reflect the quality of a map? Create real maps Score the maps with each criteria Compare the scores with the actual quality of the maps
11 Creating real maps 247 MAD, SAD, MIR datasets with final model available (PHENIX library and JCSG publicly-available data) Run AutoSol Wizard on each dataset. Calculate maps for each solution considered (opposing hands, additional sites, including various derivatives for MIR)




 Model")

12 Score maps based on each criteria Calculate map correlation coefficient (CC) to model map (no density modification, shift origin if necessary) Model map 1VQB, 2.6 Å, SG C2 SOLVE MAD map CC=0.62 Inverse-hand map CC=0.55
13 Skew of electron density 0.8 density Skew of electron A A A CC to model map
")
14 Correlation of local RMS density (Solvent next to solvent, protein next to protein)
15 Using scoring criteria to estimate the quality of a map Skew depends on CC Estimate CC from skew kew of electron de ensity CC to model ma ap CC= S Skew= CC to model map Skew of electron density
16 How accurate are estimates of map quality? Bayesian estimates of CC using Skew Actual quality CC to mod del map Predicted CC to model map Estimated quality
17 Estimated map quality in practice Evaluating solutions to a 2-wavelength MAD experiment (JCSG Tm3681, 1VPM, SeMet 1.6 Å data) Data for HYSS Sites Estimated CC ± 2SD Actual CC Peak ± Peak (inverse hand) ± F A F A (inverse) ± 0.73 ± ± Sites from diff Fourier ± What to do next: Follow up on all the solutions that MIGHT be the best (within 2 SD of the top)
 A distribution of densities like those of model")
18 Statistical density modification (RESOLVE) Principle: phase probability information from probability of the map and from experiment: P( )= P map probability ( ) P experiment ( ) Phases that lead to a believable map are more probable than those that do not A believable map is a map that has a relatively flat solvent region NCS (if appropriate) A distribution of densities like those of model proteins Method: calculate how map probability varies with electron density deduce how map probability varies with phase combine with experimental phase information
19 Map probability phasing: Getting a new probability distribution for each phase given estimates of all others 1. Identify expected features of map (flat far from center) 2. Calculate map with current estimates of all structure factors except one (k) 3. Test all possible phases for structure factor k (for each phase, calculate new map including k) 4. Probability of phase estimated from agreement of map with expectations 5. Phase probability of reflection k from map is independent of starting phase probability because reflection k is omitted from the map A function that is (relatively) flat far from the origin Function calculated from estimates of all structure factors but one (k) Test each possible phase of structure factor k. P( ) is high for phase that leads to flat region

) found at")

20 A map-probability function allowing different weighting of information from different parts of the map Log-probability of the map is sum over all points in map of local log-probability A map with a flat (blank) solvent region is a likely map Local log-probability is believability bilit of the value of electron density ( (x)) found at this point If the point is in the PROTEIN region, most values of electron density ( (x)) are believable If the point is in the SOLVENT region, only values of electron density near zero are believable
21 Rapid building of models for regions containing regular secondary-structure Helices: Identification: rods of density at low resolution Strands: Identification: structure as nearly-parallel pairs of tubes Any protein chains (trace_chain): Identification: C positions consistent with density and geometry of protein chains RNA/DNA: Identification: match of density to averaged A or B-form template

22 Model -helix; 3 Å map

23 Model -helix; 7 Å map

24 Find points along tubes of density in 7 Å map

25 Trace along tubes of density in 7 Å map
26 Trace main-chain with ideal helix, allowing curvature 2 Å radius, 5.4 Å /turn ideal helix

27 Identify direction and C position from overlap with 4 Å radius helices offset +/- 1 Å from main-chain 4 Å radius, 5.4 Å /turn ideal helix offset +1 Å along x 4 Å radius, 5.4 Å /turn ideal helix offset -1 Å along x

28 Choose best-fitting helices; link together if necessary

29 Comparison with model helix
30 A real case: 1T5S SAD map (3.1 Å)
31 A real case: 1T5S SAD map (7 Å)
32 Finding helices in 1T5S SAD map (7 Å)
33 Finding helices in 1T5S SAD map (3.1 Å)
34 Helices from 1T5S SAD map compared with 1T5S (3.1 Å)
35 PHENIX AutoSol Wizard Scale and analyze X-ray data (SAD/MAD/MIR/Multiple datasets) HYSS heavy-atom search on each dataset t Initial phasing (2.5 Å) Score and rank solutions (Which hand? Which dataset gave best solution?) Final phasing and density modification Build secondary-structure-only model
36 AutoSol fully automatic tests with structure library (MAD datasets, HYSS search, SOLVE/RESOLVE phases)
37 AutoSol fully automatic tests with structure library (MAD datasets, HYSS search, Phaser phases)
38 AutoSol fully automatic tests with structure library (MIR datasets, HYSS search, SOLVE/RESOLVE phases)
39 RESOLVE model-building at moderate resolution FFT-based identification of helices and strands Extension with tripeptide libraries Probabilistic sequence alignment Automatic molecular assembly
40 Initial model-building strand fragments
41 Chain extension (result: many overlapping fragments)
42 Main-chain as a series of fragments (choosing the best fragment at each location)
43 Side-chain template matching to identify sequence alignment to map (IF5A data) Relative probability for each amino acid at each position (Correct amino acids in bold) # G A S V I L M C F Y K R W H E D Q N P T
44 Addition of side-chains to fixed main-chain positions
45 Iterative density modification, model-building and refinement with the PHENIX AutoBuild Wizard Fp, Fp, phases, HL HL coefficients Density modify (with (with NCS, NCS, density density histograms, histograms, solvent solvent flattening, flattening, fragment fragment ID, ID, local local pattern pattern ID) ID) Build and and score models Refine with with phenix.refine Density modify including model information Evaluate final final model
46 Generating one very good model with the PHENIX AutoBuild Wizard Near-final model Rebuild model in overlapping 6-residue segments Combine best parts of each model 5 rebuilt models Final model
47 AutoBuild tests with structure library Fully automated iterative model-building, final R/Rfree AutoBuild R and Fr reer valu ues (one e-good-m model) R Rfree Resolution (Å)
48 AutoBuild tests with structure library Final Rfree with one-good-model vs standard AutoBuild FreeR d-model One-goo AutoBuild Free R
49 What can you do with automated procedures for structure solution and model-building? If a task is modular and automated you can run it many times checking different space groups, datasets to use checking if your model is biasing your map checking if you always get the same model







50 Iterative-Build OMIT procedure 2mFo-DFc omit map After building outside OMIT region 10 cycles 1HP7 molecular replacement with 1AS4 R/Rfree after initial refinement: 0.41/0.48
51 Multiple-model representation of uncertainties 20 models built iltfor 1CQP, no waters, Dmin=2.6 26A R= ; Rfree= The variation among models is a lower bound on their uncertainty
52 What else can you do with automated procedures for structure solution and model-building? If a task is modular and automated you can run it focusing on different parts of the structure build the RNA and then the protein build the helices in a low resolution map... use cross-crystal averaging in density modification build a protein model and then add ligands
53 Building RNA Group II intron at 3.5 Å. Data courtesy of J. Doudna
54 Finding helices Ca 2+ ATPase SAD map at 3.1 Å. Data courtesy of P. Nissen

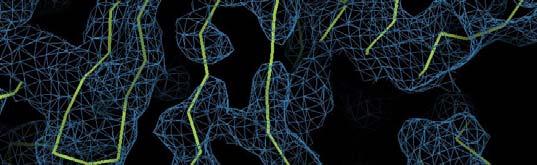









55 Statistical density modification with cross-crystal averaging Cell receptor at 3.5/3.7 Å. Data courtesy of J. Zhu Crystal 1 (4 copies) Crystal 2 (2 copies) RESOLVE density modification PHENIX Multi-crystal averaging




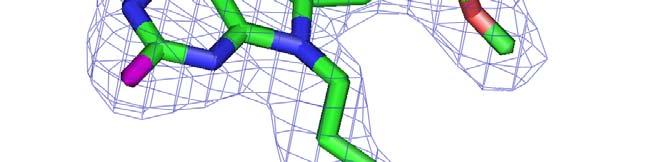
56 Automated fitting of flexible ligands FAD (0.95 Å, 1N1P) 8-(2,5-DIMETHOXY-BENZYL)-2- FLUORO-9-PENT-9H-PURIN-6-YLAMINE (2.2 Å 1UYI)



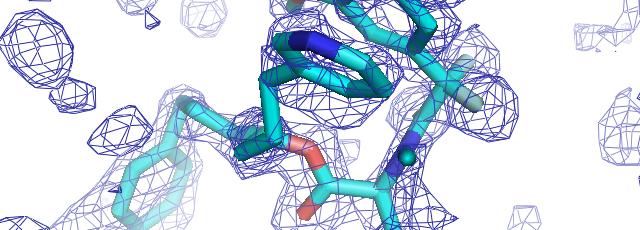
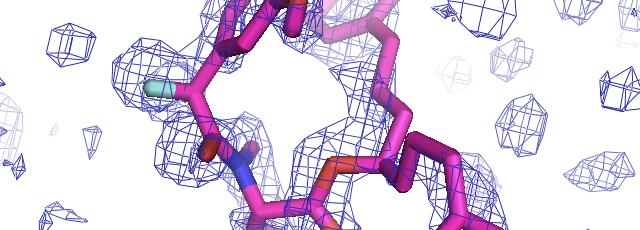

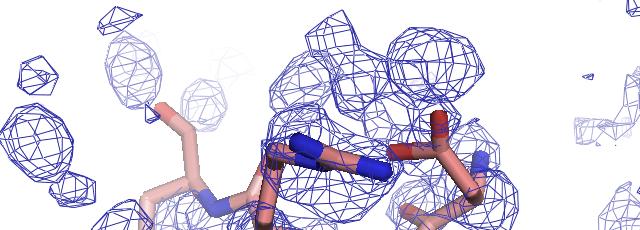


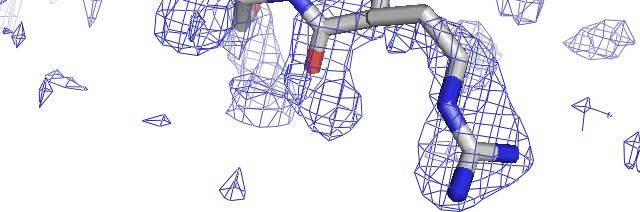
57 phenix.find_all_ligands 1J4R (3 molecules of FKB12) FKB12 Peptide decoy Site 1 Site 2 Site 3
58 The future: many hard problems remain in macromolecular crystallography y Automatically identifying and building all ligands, metals, waters Building multiple conformers Building poorly-defined regions Building complexes of protein and nucleic acid Representation of uncertainties in models Choosing optimal data (multiple crystals, multiple soaks) to use Automatic analysis of radiation damage Optimal structure solution in the presence of twinning and many more
59 PHENIX AT ARGONNE CCP4 WORKSHOP Paul Adams Tom Terwilliger org phenix.doc for help phenix.autosol, phenix.autobuild phenix.refine
 Tom Terwilliger, Li-Wei")

60 The PHENIX project Computational Crystallography Initiative (LBNL) Paul Adams, Ralf Grosse-Kunstleve, Peter Zwart, Nigel Moriarty, Nicholas Sauter, Pavel Afonine Los Alamos National Lab (LANL) Tom Terwilliger, Li-Wei Hung Cambridge University Randy Read, Airlie McCoy, Gabor Bunkoczi, Rob Oeffner Duke University Jane Richardson, David Richardson, Jeff Headd, Vincent Chen Texas A&M University Tom Ioerger, Jim Sacchettini
Structure solution from weak anomalous data
 Structure solution from weak anomalous data Phenix Workshop SBGrid-NE-CAT Computing School Harvard Medical School, Boston June 7, 2014 Gábor Bunkóczi, Airlie McCoy, Randy Read (Cambridge University) Nat
Structure solution from weak anomalous data Phenix Workshop SBGrid-NE-CAT Computing School Harvard Medical School, Boston June 7, 2014 Gábor Bunkóczi, Airlie McCoy, Randy Read (Cambridge University) Nat
Charles Ballard (original GáborBunkóczi) CCP4 Workshop 7 December 2011
 CCP4 Workshop 7 December 2011") Experimental phasing Charles Ballard (original GáborBunkóczi) CCP4 Workshop 7 December 2011 Anomalous diffraction F P protein F A anomalous substructure ano F A " -FA" A F F -* F A F P Phasing Substructure
Experimental phasing Charles Ballard (original GáborBunkóczi) CCP4 Workshop 7 December 2011 Anomalous diffraction F P protein F A anomalous substructure ano F A " -FA" A F F -* F A F P Phasing Substructure
Phaser: Experimental phasing
 Phaser: Experimental phasing Using SAD data in Phaser R J Read, Department of Haematology Cambridge Institute for Medical Research Diffraction with anomalous scatterers SAD: single-wavelength anomalous
Phaser: Experimental phasing Using SAD data in Phaser R J Read, Department of Haematology Cambridge Institute for Medical Research Diffraction with anomalous scatterers SAD: single-wavelength anomalous
The Crystallographic Process
 Experimental Phasing Macromolecular Crystallography School Madrid, May 2017 Paul Adams Lawrence Berkeley Laboratory and Department of Bioengineering UC Berkeley The Crystallographic Process Crystallization
Experimental Phasing Macromolecular Crystallography School Madrid, May 2017 Paul Adams Lawrence Berkeley Laboratory and Department of Bioengineering UC Berkeley The Crystallographic Process Crystallization
SOLVE and RESOLVE: automated structure solution, density modification and model building
 Journal of Synchrotron Radiation ISSN 0909-0495 SOLVE and RESOLVE: automated structure solution, density modification and model building Thomas Terwilliger Copyright International Union of Crystallography
Journal of Synchrotron Radiation ISSN 0909-0495 SOLVE and RESOLVE: automated structure solution, density modification and model building Thomas Terwilliger Copyright International Union of Crystallography
Pipelining Ligands in PHENIX: elbow and REEL
 Pipelining Ligands in PHENIX: elbow and REEL Nigel W. Moriarty Lawrence Berkeley National Laboratory Physical Biosciences Division Ligands in Crystallography Drug design Biological function studies Generate
Pipelining Ligands in PHENIX: elbow and REEL Nigel W. Moriarty Lawrence Berkeley National Laboratory Physical Biosciences Division Ligands in Crystallography Drug design Biological function studies Generate
research papers Iterative-build OMIT maps: map improvement by iterative model building and refinement without model bias 1.
 Acta Crystallographica Section D Biological Crystallography ISSN 0907-4449 Iterative-build OMIT maps: map improvement by iterative model building and refinement without model bias Thomas C. Terwilliger,
Acta Crystallographica Section D Biological Crystallography ISSN 0907-4449 Iterative-build OMIT maps: map improvement by iterative model building and refinement without model bias Thomas C. Terwilliger,
The Crystallographic Process
 Phase Improvement Macromolecular Crystallography School Madrid, May 2017 Paul Adams Lawrence Berkeley Laboratory and Department of Bioengineering UC Berkeley The Crystallographic Process Crystallization
Phase Improvement Macromolecular Crystallography School Madrid, May 2017 Paul Adams Lawrence Berkeley Laboratory and Department of Bioengineering UC Berkeley The Crystallographic Process Crystallization
Likelihood and SAD phasing in Phaser. R J Read, Department of Haematology Cambridge Institute for Medical Research
 Likelihood and SAD phasing in Phaser R J Read, Department of Haematology Cambridge Institute for Medical Research Concept of likelihood Likelihood with dice 4 6 8 10 Roll a seven. Which die?? p(4)=p(6)=0
Likelihood and SAD phasing in Phaser R J Read, Department of Haematology Cambridge Institute for Medical Research Concept of likelihood Likelihood with dice 4 6 8 10 Roll a seven. Which die?? p(4)=p(6)=0
Electronic Supplementary Information (ESI) for Chem. Commun. Unveiling the three- dimensional structure of the green pigment of nitrite- cured meat
 for Chem. Commun. Unveiling the three- dimensional structure of the green pigment of nitrite- cured meat") Electronic Supplementary Information (ESI) for Chem. Commun. Unveiling the three- dimensional structure of the green pigment of nitrite- cured meat Jun Yi* and George B. Richter- Addo* Department of Chemistry
Electronic Supplementary Information (ESI) for Chem. Commun. Unveiling the three- dimensional structure of the green pigment of nitrite- cured meat Jun Yi* and George B. Richter- Addo* Department of Chemistry
Molecular replacement. New structures from old
 Molecular replacement New structures from old The Phase Problem phase amplitude Phasing by molecular replacement Phases can be calculated from atomic model Rotate and translate related structure Models
Molecular replacement New structures from old The Phase Problem phase amplitude Phasing by molecular replacement Phases can be calculated from atomic model Rotate and translate related structure Models
Automated ligand fitting by core-fragment fitting and extension into density
 Acta Crystallographica Section D Biological Crystallography ISSN 0907-4449 Editors: E. N. Baker and Z. Dauter Automated ligand fitting by core-fragment fitting and extension into density Thomas C. Terwilliger,
Acta Crystallographica Section D Biological Crystallography ISSN 0907-4449 Editors: E. N. Baker and Z. Dauter Automated ligand fitting by core-fragment fitting and extension into density Thomas C. Terwilliger,
Pathogenic C9ORF72 Antisense Repeat RNA Forms a Double Helix with Tandem C:C Mismatches
 Supporting Information Pathogenic C9ORF72 Antisense Repeat RNA Forms a Double Helix with Tandem C:C Mismatches David W. Dodd, Diana R. Tomchick, David R. Corey, and Keith T. Gagnon METHODS S1 RNA synthesis.
Supporting Information Pathogenic C9ORF72 Antisense Repeat RNA Forms a Double Helix with Tandem C:C Mismatches David W. Dodd, Diana R. Tomchick, David R. Corey, and Keith T. Gagnon METHODS S1 RNA synthesis.
CCP4 Diamond 2014 SHELXC/D/E. Andrea Thorn
 CCP4 Diamond 2014 SHELXC/D/E Andrea Thorn SHELXC/D/E workflow SHELXC: α calculation, file preparation SHELXD: Marker atom search = substructure search SHELXE: density modification Maps and coordinate files
CCP4 Diamond 2014 SHELXC/D/E Andrea Thorn SHELXC/D/E workflow SHELXC: α calculation, file preparation SHELXD: Marker atom search = substructure search SHELXE: density modification Maps and coordinate files
electronic reprint PHENIX: a comprehensive Python-based system for macromolecular structure solution
 Acta Crystallographica Section D Biological Crystallography ISSN 0907-4449 Editors: E. N. Baker and Z. Dauter PHENIX: a comprehensive Python-based system for macromolecular structure solution Paul D. Adams,
Acta Crystallographica Section D Biological Crystallography ISSN 0907-4449 Editors: E. N. Baker and Z. Dauter PHENIX: a comprehensive Python-based system for macromolecular structure solution Paul D. Adams,
SHELXC/D/E. Andrea Thorn
 SHELXC/D/E Andrea Thorn What is experimental phasing? Experimental phasing is what you do if MR doesn t work. What is experimental phasing? Experimental phasing methods depend on intensity differences.
SHELXC/D/E Andrea Thorn What is experimental phasing? Experimental phasing is what you do if MR doesn t work. What is experimental phasing? Experimental phasing methods depend on intensity differences.
Molecular Replacement. Airlie McCoy
 Molecular Replacement Airlie McCoy Molecular Replacement Find orienta5on and posi5on where model overlies the target structure Borrow the phases Then it becomes a refinement problem the phases change known
Molecular Replacement Airlie McCoy Molecular Replacement Find orienta5on and posi5on where model overlies the target structure Borrow the phases Then it becomes a refinement problem the phases change known
research papers 1. Introduction Thomas C. Terwilliger a * and Joel Berendzen b
 Acta Crystallographica Section D Biological Crystallography ISSN 0907-4449 Discrimination of solvent from protein regions in native Fouriers as a means of evaluating heavy-atom solutions in the MIR and
Acta Crystallographica Section D Biological Crystallography ISSN 0907-4449 Discrimination of solvent from protein regions in native Fouriers as a means of evaluating heavy-atom solutions in the MIR and
Supporting Information
 Supporting Information Structural Analysis of the Binding of Type I, I 1/2, and II Inhibitors to Eph Tyrosine Kinases Jing Dong, *1 Hongtao Zhao, 1 Ting Zhou, 1 Dimitrios Spiliotopoulos, 1 Chitra Rajendran,
Supporting Information Structural Analysis of the Binding of Type I, I 1/2, and II Inhibitors to Eph Tyrosine Kinases Jing Dong, *1 Hongtao Zhao, 1 Ting Zhou, 1 Dimitrios Spiliotopoulos, 1 Chitra Rajendran,
Protein Data Bank Changes Guide. New Changes in Version 3.20 September 15, 2008
 New Records in 3.2 Page 1 Protein Data Bank Changes Guide New Changes in Version 3.20 September 15, 2008 Version 3.20 of the PDB file format introduces a small number of changes and extensions supporting
New Records in 3.2 Page 1 Protein Data Bank Changes Guide New Changes in Version 3.20 September 15, 2008 Version 3.20 of the PDB file format introduces a small number of changes and extensions supporting
Web-based Auto-Rickshaw for validation of the X-ray experiment at the synchrotron beamline
 Web-based Auto-Rickshaw for validation of the X-ray experiment at the synchrotron beamline Auto-Rickshaw http://www.embl-hamburg.de/auto-rickshaw A platform for automated crystal structure determination
Web-based Auto-Rickshaw for validation of the X-ray experiment at the synchrotron beamline Auto-Rickshaw http://www.embl-hamburg.de/auto-rickshaw A platform for automated crystal structure determination
Supplementary Information
 Electronic Supplementary Material (ESI) for ChemComm. This journal is The Royal Society of Chemistry 2015 Supplementary Information Anion clamp allows flexible protein to impose coordination geometry on
Electronic Supplementary Material (ESI) for ChemComm. This journal is The Royal Society of Chemistry 2015 Supplementary Information Anion clamp allows flexible protein to impose coordination geometry on
Resolution Dependence of an Ab Initio Phasing Method in Protein X-ray Crystallography
 crystals Article Resolution Dependence of an Ab Initio Phasing Method in Protein X-ray Crystallography Mengchao Jiang, Hongxing He ID, Yunpeng Cheng and Wu-Pei Su * Department of Physics and Texas Center
crystals Article Resolution Dependence of an Ab Initio Phasing Method in Protein X-ray Crystallography Mengchao Jiang, Hongxing He ID, Yunpeng Cheng and Wu-Pei Su * Department of Physics and Texas Center
MR model selection, preparation and assessing the solution
 Ronan Keegan CCP4 Group MR model selection, preparation and assessing the solution DLS-CCP4 Data Collection and Structure Solution Workshop 2018 Overview Introduction Step-by-step guide to performing Molecular
Ronan Keegan CCP4 Group MR model selection, preparation and assessing the solution DLS-CCP4 Data Collection and Structure Solution Workshop 2018 Overview Introduction Step-by-step guide to performing Molecular
Joana Pereira Lamzin Group EMBL Hamburg, Germany. Small molecules How to identify and build them (with ARP/wARP)
") Joana Pereira Lamzin Group EMBL Hamburg, Germany Small molecules How to identify and build them (with ARP/wARP) The task at hand To find ligand density and build it! Fitting a ligand We have: electron
Joana Pereira Lamzin Group EMBL Hamburg, Germany Small molecules How to identify and build them (with ARP/wARP) The task at hand To find ligand density and build it! Fitting a ligand We have: electron
Tools for Cryo-EM Map Fitting. Paul Emsley MRC Laboratory of Molecular Biology
 Tools for Cryo-EM Map Fitting Paul Emsley MRC Laboratory of Molecular Biology April 2017 Cryo-EM model-building typically need to move more atoms that one does for crystallography the maps are lower resolution
Tools for Cryo-EM Map Fitting Paul Emsley MRC Laboratory of Molecular Biology April 2017 Cryo-EM model-building typically need to move more atoms that one does for crystallography the maps are lower resolution
Molecular Replacement (Alexei Vagin s lecture)
") Molecular Replacement (Alexei Vagin s lecture) Contents What is Molecular Replacement Functions in Molecular Replacement Weighting scheme Information from data and model Some special techniques of Molecular
Molecular Replacement (Alexei Vagin s lecture) Contents What is Molecular Replacement Functions in Molecular Replacement Weighting scheme Information from data and model Some special techniques of Molecular
Direct Method. Very few protein diffraction data meet the 2nd condition
 Direct Method Two conditions: -atoms in the structure are equal-weighted -resolution of data are higher than the distance between the atoms in the structure Very few protein diffraction data meet the 2nd
Direct Method Two conditions: -atoms in the structure are equal-weighted -resolution of data are higher than the distance between the atoms in the structure Very few protein diffraction data meet the 2nd
Direct Methods and Many Site Se-Met MAD Problems using BnP. W. Furey
 Direct Methods and Many Site Se-Met MAD Problems using BnP W. Furey Classical Direct Methods Main method for small molecule structure determination Highly automated (almost totally black box ) Solves structures
Direct Methods and Many Site Se-Met MAD Problems using BnP W. Furey Classical Direct Methods Main method for small molecule structure determination Highly automated (almost totally black box ) Solves structures
GC376 (compound 28). Compound 23 (GC373) (0.50 g, 1.24 mmol), sodium bisulfite (0.119 g,
. Compound 23 (GC373) (0.50 g, 1.24 mmol), sodium bisulfite (0.119 g,") Supplemental Material Synthesis of GC376 GC376 (compound 28). Compound 23 (GC373) (0.50 g, 1.24 mmol), sodium bisulfite (0.119 g, 1.12 mmol), ethyl acetate (2 ml), ethanol (1 ml) and water (0.40 ml) were
Supplemental Material Synthesis of GC376 GC376 (compound 28). Compound 23 (GC373) (0.50 g, 1.24 mmol), sodium bisulfite (0.119 g, 1.12 mmol), ethyl acetate (2 ml), ethanol (1 ml) and water (0.40 ml) were
From x-ray crystallography to electron microscopy and back -- how best to exploit the continuum of structure-determination methods now available
 From x-ray crystallography to electron microscopy and back -- how best to exploit the continuum of structure-determination methods now available Scripps EM course, November 14, 2007 What aspects of contemporary
From x-ray crystallography to electron microscopy and back -- how best to exploit the continuum of structure-determination methods now available Scripps EM course, November 14, 2007 What aspects of contemporary
research papers A general method for phasing novel complex RNA crystal structures without heavy-atom derivatives
 Acta Crystallographica Section D Biological Crystallography ISSN 0907-4449 A general method for phasing novel complex RNA crystal structures without heavy-atom derivatives Michael P. Robertson and William
Acta Crystallographica Section D Biological Crystallography ISSN 0907-4449 A general method for phasing novel complex RNA crystal structures without heavy-atom derivatives Michael P. Robertson and William
Experimental phasing in Crank2
 Experimental phasing in Crank2 Pavol Skubak and Navraj Pannu Biophysical Structural Chemistry, Leiden University, The Netherlands http://www.bfsc.leidenuniv.nl/software/crank/ X-ray structure solution
Experimental phasing in Crank2 Pavol Skubak and Navraj Pannu Biophysical Structural Chemistry, Leiden University, The Netherlands http://www.bfsc.leidenuniv.nl/software/crank/ X-ray structure solution
Ensemble refinement of protein crystal structures in PHENIX. Tom Burnley Piet Gros
 Ensemble refinement of protein crystal structures in PHENIX Tom Burnley Piet Gros Incomplete modelling of disorder contributes to R factor gap Only ~5% of residues in the PDB are modelled with more than
Ensemble refinement of protein crystal structures in PHENIX Tom Burnley Piet Gros Incomplete modelling of disorder contributes to R factor gap Only ~5% of residues in the PDB are modelled with more than
Full wwpdb X-ray Structure Validation Report i
 Full wwpdb X-ray Structure Validation Report i Mar 8, 2018 10:24 pm GMT PDB ID : 1A30 Title : HIV-1 PROTEASE COMPLEXED WITH A TRIPEPTIDE INHIBITOR Authors : Louis, J.M.; Dyda, F.; Nashed, N.T.; Kimmel,
Full wwpdb X-ray Structure Validation Report i Mar 8, 2018 10:24 pm GMT PDB ID : 1A30 Title : HIV-1 PROTEASE COMPLEXED WITH A TRIPEPTIDE INHIBITOR Authors : Louis, J.M.; Dyda, F.; Nashed, N.T.; Kimmel,
Crystal lattice Real Space. Reflections Reciprocal Space. I. Solving Phases II. Model Building for CHEM 645. Purified Protein. Build model.
 I. Solving Phases II. Model Building for CHEM 645 Purified Protein Solve Phase Build model and refine Crystal lattice Real Space Reflections Reciprocal Space ρ (x, y, z) pronounced rho F hkl 2 I F (h,
I. Solving Phases II. Model Building for CHEM 645 Purified Protein Solve Phase Build model and refine Crystal lattice Real Space Reflections Reciprocal Space ρ (x, y, z) pronounced rho F hkl 2 I F (h,
Supporting Information
 Supporting Information Structural Basis of the Antiproliferative Activity of Largazole, a Depsipeptide Inhibitor of the Histone Deacetylases Kathryn E. Cole 1, Daniel P. Dowling 1,2, Matthew A. Boone 3,
Supporting Information Structural Basis of the Antiproliferative Activity of Largazole, a Depsipeptide Inhibitor of the Histone Deacetylases Kathryn E. Cole 1, Daniel P. Dowling 1,2, Matthew A. Boone 3,
The SHELX approach to the experimental phasing of macromolecules. George M. Sheldrick, Göttingen University
 The SHELX approach to the experimental phasing of macromolecules IUCr 2011 Madrid George M. Sheldrick, Göttingen University http://shelx.uni-ac.gwdg.de/shelx/ Experimental phasing of macromolecules Except
The SHELX approach to the experimental phasing of macromolecules IUCr 2011 Madrid George M. Sheldrick, Göttingen University http://shelx.uni-ac.gwdg.de/shelx/ Experimental phasing of macromolecules Except
Protein Structure Prediction, Engineering & Design CHEM 430
 Protein Structure Prediction, Engineering & Design CHEM 430 Eero Saarinen The free energy surface of a protein Protein Structure Prediction & Design Full Protein Structure from Sequence - High Alignment
Protein Structure Prediction, Engineering & Design CHEM 430 Eero Saarinen The free energy surface of a protein Protein Structure Prediction & Design Full Protein Structure from Sequence - High Alignment
Full wwpdb X-ray Structure Validation Report i
 Full wwpdb X-ray Structure Validation Report i Mar 10, 2018 01:44 am GMT PDB ID : 1MWP Title : N-TERMINAL DOMAIN OF THE AMYLOID PRECURSOR PROTEIN Authors : Rossjohn, J.; Cappai, R.; Feil, S.C.; Henry,
Full wwpdb X-ray Structure Validation Report i Mar 10, 2018 01:44 am GMT PDB ID : 1MWP Title : N-TERMINAL DOMAIN OF THE AMYLOID PRECURSOR PROTEIN Authors : Rossjohn, J.; Cappai, R.; Feil, S.C.; Henry,
Protein Crystallography
 Protein Crystallography Part II Tim Grüne Dept. of Structural Chemistry Prof. G. Sheldrick University of Göttingen http://shelx.uni-ac.gwdg.de tg@shelx.uni-ac.gwdg.de Overview The Reciprocal Lattice The
Protein Crystallography Part II Tim Grüne Dept. of Structural Chemistry Prof. G. Sheldrick University of Göttingen http://shelx.uni-ac.gwdg.de tg@shelx.uni-ac.gwdg.de Overview The Reciprocal Lattice The
Statistical density modification with non-crystallographic symmetry
 Acta Crystallographica Section D Biological Crystallography ISSN 0907-4449 Statistical density modification with non-crystallographic symmetry Thomas C. Terwilliger Copyright International Union of Crystallography
Acta Crystallographica Section D Biological Crystallography ISSN 0907-4449 Statistical density modification with non-crystallographic symmetry Thomas C. Terwilliger Copyright International Union of Crystallography
Protein crystallography. Garry Taylor
 Protein crystallography Garry Taylor X-ray Crystallography - the Basics Grow crystals Collect X-ray data Determine phases Calculate ρ-map Interpret map Refine coordinates Do the biology. Nitrogen at -180
Protein crystallography Garry Taylor X-ray Crystallography - the Basics Grow crystals Collect X-ray data Determine phases Calculate ρ-map Interpret map Refine coordinates Do the biology. Nitrogen at -180
Full wwpdb X-ray Structure Validation Report i
 Full wwpdb X-ray Structure Validation Report i Mar 8, 2018 06:13 pm GMT PDB ID : 5G5C Title : Structure of the Pyrococcus furiosus Esterase Pf2001 with space group C2221 Authors : Varejao, N.; Reverter,
Full wwpdb X-ray Structure Validation Report i Mar 8, 2018 06:13 pm GMT PDB ID : 5G5C Title : Structure of the Pyrococcus furiosus Esterase Pf2001 with space group C2221 Authors : Varejao, N.; Reverter,
Automated Protein Model Building with ARP/wARP
 Joana Pereira Lamzin Group EMBL Hamburg, Germany Automated Protein Model Building with ARP/wARP (and nucleic acids too) Electron density map: the ultimate result of a crystallography experiment How would
Joana Pereira Lamzin Group EMBL Hamburg, Germany Automated Protein Model Building with ARP/wARP (and nucleic acids too) Electron density map: the ultimate result of a crystallography experiment How would
Electron Density at various resolutions, and fitting a model as accurately as possible.
 Section 9, Electron Density Maps 900 Electron Density at various resolutions, and fitting a model as accurately as possible. ρ xyz = (Vol) -1 h k l m hkl F hkl e iφ hkl e-i2π( hx + ky + lz ) Amplitude
Section 9, Electron Density Maps 900 Electron Density at various resolutions, and fitting a model as accurately as possible. ρ xyz = (Vol) -1 h k l m hkl F hkl e iφ hkl e-i2π( hx + ky + lz ) Amplitude
Computational aspects of high-throughput crystallographic macromolecular structure determination
 Cop[Book Title], Edited by [Editor s Name]. ISBN 0-471-XXXXX-X Copyright 2007 Wiley[Imprint], Inc. Chapter 4 Computational aspects of high-throughput crystallographic macromolecular structure determination
Cop[Book Title], Edited by [Editor s Name]. ISBN 0-471-XXXXX-X Copyright 2007 Wiley[Imprint], Inc. Chapter 4 Computational aspects of high-throughput crystallographic macromolecular structure determination
Experimental phasing, Pattersons and SHELX Andrea Thorn
 Experimental phasing, Pattersons and SHELX Andrea Thorn What is experimental phasing? Experimental phasing is what you do if MR doesn t work. What is experimental phasing? Experimental phasing methods
Experimental phasing, Pattersons and SHELX Andrea Thorn What is experimental phasing? Experimental phasing is what you do if MR doesn t work. What is experimental phasing? Experimental phasing methods
Neural Networks for Protein Structure Prediction Brown, JMB CS 466 Saurabh Sinha
 Neural Networks for Protein Structure Prediction Brown, JMB 1999 CS 466 Saurabh Sinha Outline Goal is to predict secondary structure of a protein from its sequence Artificial Neural Network used for this
Neural Networks for Protein Structure Prediction Brown, JMB 1999 CS 466 Saurabh Sinha Outline Goal is to predict secondary structure of a protein from its sequence Artificial Neural Network used for this
Full wwpdb X-ray Structure Validation Report i
 Full wwpdb X-ray Structure Validation Report i Mar 14, 2018 02:00 pm GMT PDB ID : 3RRQ Title : Crystal structure of the extracellular domain of human PD-1 Authors : Lazar-Molnar, E.; Ramagopal, U.A.; Nathenson,
Full wwpdb X-ray Structure Validation Report i Mar 14, 2018 02:00 pm GMT PDB ID : 3RRQ Title : Crystal structure of the extracellular domain of human PD-1 Authors : Lazar-Molnar, E.; Ramagopal, U.A.; Nathenson,
The structure of Aquifex aeolicus FtsH in the ADP-bound state reveals a C2-symmetric hexamer
 Volume 71 (2015) Supporting information for article: The structure of Aquifex aeolicus FtsH in the ADP-bound state reveals a C2-symmetric hexamer Marina Vostrukhina, Alexander Popov, Elena Brunstein, Martin
Volume 71 (2015) Supporting information for article: The structure of Aquifex aeolicus FtsH in the ADP-bound state reveals a C2-symmetric hexamer Marina Vostrukhina, Alexander Popov, Elena Brunstein, Martin
ACORN - a flexible and efficient ab initio procedure to solve a protein structure when atomic resolution data is available
 ACORN - a flexible and efficient ab initio procedure to solve a protein structure when atomic resolution data is available Yao Jia-xing Department of Chemistry, University of York, Heslington, York, YO10
ACORN - a flexible and efficient ab initio procedure to solve a protein structure when atomic resolution data is available Yao Jia-xing Department of Chemistry, University of York, Heslington, York, YO10
Principles of Physical Biochemistry
 Principles of Physical Biochemistry Kensal E. van Hold e W. Curtis Johnso n P. Shing Ho Preface x i PART 1 MACROMOLECULAR STRUCTURE AND DYNAMICS 1 1 Biological Macromolecules 2 1.1 General Principles
Principles of Physical Biochemistry Kensal E. van Hold e W. Curtis Johnso n P. Shing Ho Preface x i PART 1 MACROMOLECULAR STRUCTURE AND DYNAMICS 1 1 Biological Macromolecules 2 1.1 General Principles
Full wwpdb X-ray Structure Validation Report i
 Full wwpdb X-ray Structure Validation Report i Jan 28, 2019 11:10 AM EST PDB ID : 6A5H Title : The structure of [4+2] and [6+4] cyclase in the biosynthetic pathway of unidentified natural product Authors
Full wwpdb X-ray Structure Validation Report i Jan 28, 2019 11:10 AM EST PDB ID : 6A5H Title : The structure of [4+2] and [6+4] cyclase in the biosynthetic pathway of unidentified natural product Authors
Giri Narasimhan. CAP 5510: Introduction to Bioinformatics. ECS 254; Phone: x3748
 CAP 5510: Introduction to Bioinformatics Giri Narasimhan ECS 254; Phone: x3748 giri@cis.fiu.edu www.cis.fiu.edu/~giri/teach/bioinfs07.html 2/15/07 CAP5510 1 EM Algorithm Goal: Find θ, Z that maximize Pr
CAP 5510: Introduction to Bioinformatics Giri Narasimhan ECS 254; Phone: x3748 giri@cis.fiu.edu www.cis.fiu.edu/~giri/teach/bioinfs07.html 2/15/07 CAP5510 1 EM Algorithm Goal: Find θ, Z that maximize Pr
Anomalous dispersion
 Selenomethionine MAD Selenomethionine is the amino acid methionine with the Sulfur replaced by a Selenium. Selenium is a heavy atom that also has the propery of "anomalous scatter" at some wavelengths,
Selenomethionine MAD Selenomethionine is the amino acid methionine with the Sulfur replaced by a Selenium. Selenium is a heavy atom that also has the propery of "anomalous scatter" at some wavelengths,
Full wwpdb X-ray Structure Validation Report i
 Full wwpdb X-ray Structure Validation Report i Jan 14, 2019 11:10 AM EST PDB ID : 6GYW Title : Crystal structure of DacA from Staphylococcus aureus Authors : Tosi, T.; Freemont, P.S.; Grundling, A. Deposited
Full wwpdb X-ray Structure Validation Report i Jan 14, 2019 11:10 AM EST PDB ID : 6GYW Title : Crystal structure of DacA from Staphylococcus aureus Authors : Tosi, T.; Freemont, P.S.; Grundling, A. Deposited
HTCondor and macromolecular structure validation
 HTCondor and macromolecular structure validation Vincent Chen John Markley/Eldon Ulrich, NMRFAM/BMRB, UW@Madison David & Jane Richardson, Duke University Macromolecules David S. Goodsell 1999 Two questions
HTCondor and macromolecular structure validation Vincent Chen John Markley/Eldon Ulrich, NMRFAM/BMRB, UW@Madison David & Jane Richardson, Duke University Macromolecules David S. Goodsell 1999 Two questions
The structure of a nucleolytic ribozyme that employs a catalytic metal ion. Yijin Liu, Timothy J. Wilson and David M.J. Lilley
 SUPPLEMENTARY INFORMATION The structure of a nucleolytic ribozyme that employs a catalytic metal ion Yijin Liu, Timothy J. Wilson and David M.J. Lilley Cancer Research UK Nucleic Acid Structure Research
SUPPLEMENTARY INFORMATION The structure of a nucleolytic ribozyme that employs a catalytic metal ion Yijin Liu, Timothy J. Wilson and David M.J. Lilley Cancer Research UK Nucleic Acid Structure Research
Nature Structural & Molecular Biology: doi: /nsmb Supplementary Figure 1
 Supplementary Figure 1 Crystallization. a, Crystallization constructs of the ET B receptor are shown, with all of the modifications to the human wild-type the ET B receptor indicated. Residues interacting
Supplementary Figure 1 Crystallization. a, Crystallization constructs of the ET B receptor are shown, with all of the modifications to the human wild-type the ET B receptor indicated. Residues interacting
Full wwpdb X-ray Structure Validation Report i
 Full wwpdb X-ray Structure Validation Report i Jan 17, 2019 09:42 AM EST PDB ID : 6D3Z Title : Protease SFTI complex Authors : Law, R.H.P.; Wu, G. Deposited on : 2018-04-17 Resolution : 2.00 Å(reported)
Full wwpdb X-ray Structure Validation Report i Jan 17, 2019 09:42 AM EST PDB ID : 6D3Z Title : Protease SFTI complex Authors : Law, R.H.P.; Wu, G. Deposited on : 2018-04-17 Resolution : 2.00 Å(reported)
Orientational degeneracy in the presence of one alignment tensor.
 Orientational degeneracy in the presence of one alignment tensor. Rotation about the x, y and z axes can be performed in the aligned mode of the program to examine the four degenerate orientations of two
Orientational degeneracy in the presence of one alignment tensor. Rotation about the x, y and z axes can be performed in the aligned mode of the program to examine the four degenerate orientations of two
Introduction to Comparative Protein Modeling. Chapter 4 Part I
 Introduction to Comparative Protein Modeling Chapter 4 Part I 1 Information on Proteins Each modeling study depends on the quality of the known experimental data. Basis of the model Search in the literature
Introduction to Comparative Protein Modeling Chapter 4 Part I 1 Information on Proteins Each modeling study depends on the quality of the known experimental data. Basis of the model Search in the literature
Axel T. Brunger, Debanu Das, Ashley M. Deacon, Joanna Grant, Thomas C. Terwilliger, Randy J. Read, Paul D. Adams, Michael Levitt and Gunnar F.
 Acta Crystallographica Section D Biological Crystallography ISSN 0907-4449 Editors: E. N. Baker and Z. Dauter Application of DEN refinement and automated model building to a difficult case of molecular-replacement
Acta Crystallographica Section D Biological Crystallography ISSN 0907-4449 Editors: E. N. Baker and Z. Dauter Application of DEN refinement and automated model building to a difficult case of molecular-replacement
Garib N Murshudov MRC-LMB, Cambridge
 Garib N Murshudov MRC-LMB, Cambridge Contents Introduction AceDRG: two functions Validation of entries in the DB and derived data Generation of new ligand description Jligand for link description Conclusions
Garib N Murshudov MRC-LMB, Cambridge Contents Introduction AceDRG: two functions Validation of entries in the DB and derived data Generation of new ligand description Jligand for link description Conclusions
Full wwpdb X-ray Structure Validation Report i
 Full wwpdb X-ray Structure Validation Report i Mar 13, 2018 04:03 pm GMT PDB ID : 5NMJ Title : Chicken GRIFIN (crystallisation ph: 6.5) Authors : Ruiz, F.M.; Romero, A. Deposited on : 2017-04-06 Resolution
Full wwpdb X-ray Structure Validation Report i Mar 13, 2018 04:03 pm GMT PDB ID : 5NMJ Title : Chicken GRIFIN (crystallisation ph: 6.5) Authors : Ruiz, F.M.; Romero, A. Deposited on : 2017-04-06 Resolution
Week 10: Homology Modelling (II) - HHpred
 - HHpred") Week 10: Homology Modelling (II) - HHpred Course: Tools for Structural Biology Fabian Glaser BKU - Technion 1 2 Identify and align related structures by sequence methods is not an easy task All comparative
Week 10: Homology Modelling (II) - HHpred Course: Tools for Structural Biology Fabian Glaser BKU - Technion 1 2 Identify and align related structures by sequence methods is not an easy task All comparative
Prediction and refinement of NMR structures from sparse experimental data
 Prediction and refinement of NMR structures from sparse experimental data Jeff Skolnick Director Center for the Study of Systems Biology School of Biology Georgia Institute of Technology Overview of talk
Prediction and refinement of NMR structures from sparse experimental data Jeff Skolnick Director Center for the Study of Systems Biology School of Biology Georgia Institute of Technology Overview of talk
X-ray Crystallography
 2009/11/25 [ 1 ] X-ray Crystallography Andrew Torda, wintersemester 2009 / 2010 X-ray numerically most important more than 4/5 structures Goal a set of x, y, z coordinates different properties to NMR History
2009/11/25 [ 1 ] X-ray Crystallography Andrew Torda, wintersemester 2009 / 2010 X-ray numerically most important more than 4/5 structures Goal a set of x, y, z coordinates different properties to NMR History
Assignment 2 Atomic-Level Molecular Modeling
 Assignment 2 Atomic-Level Molecular Modeling CS/BIOE/CME/BIOPHYS/BIOMEDIN 279 Due: November 3, 2016 at 3:00 PM The goal of this assignment is to understand the biological and computational aspects of macromolecular
Assignment 2 Atomic-Level Molecular Modeling CS/BIOE/CME/BIOPHYS/BIOMEDIN 279 Due: November 3, 2016 at 3:00 PM The goal of this assignment is to understand the biological and computational aspects of macromolecular
Examples of Protein Modeling. Protein Modeling. Primary Structure. Protein Structure Description. Protein Sequence Sources. Importing Sequences to MOE
 Examples of Protein Modeling Protein Modeling Visualization Examination of an experimental structure to gain insight about a research question Dynamics To examine the dynamics of protein structures To
Examples of Protein Modeling Protein Modeling Visualization Examination of an experimental structure to gain insight about a research question Dynamics To examine the dynamics of protein structures To
Rietveld Structure Refinement of Protein Powder Diffraction Data using GSAS
 Rietveld Structure Refinement of Protein Powder Diffraction Data using GSAS Jon Wright ESRF, Grenoble, France Plan This is a users perspective Cover the protein specific aspects (assuming knowledge of
Rietveld Structure Refinement of Protein Powder Diffraction Data using GSAS Jon Wright ESRF, Grenoble, France Plan This is a users perspective Cover the protein specific aspects (assuming knowledge of
SCOP. all-β class. all-α class, 3 different folds. T4 endonuclease V. 4-helical cytokines. Globin-like
 SCOP all-β class 4-helical cytokines T4 endonuclease V all-α class, 3 different folds Globin-like TIM-barrel fold α/β class Profilin-like fold α+β class http://scop.mrc-lmb.cam.ac.uk/scop CATH Class, Architecture,
SCOP all-β class 4-helical cytokines T4 endonuclease V all-α class, 3 different folds Globin-like TIM-barrel fold α/β class Profilin-like fold α+β class http://scop.mrc-lmb.cam.ac.uk/scop CATH Class, Architecture,
NMR, X-ray Diffraction, Protein Structure, and RasMol
 NMR, X-ray Diffraction, Protein Structure, and RasMol Introduction So far we have been mostly concerned with the proteins themselves. The techniques (NMR or X-ray diffraction) used to determine a structure
NMR, X-ray Diffraction, Protein Structure, and RasMol Introduction So far we have been mostly concerned with the proteins themselves. The techniques (NMR or X-ray diffraction) used to determine a structure
Exploiting Protein Conformational Change to Optimize Adenosine-Derived Inhibitors of HSP70
 SUPPRTIG IFRMATI Exploiting Protein Conformational Change to ptimize Adenosine-Derived Inhibitors of HSP70 Matthew D. Cheeseman, 1 Isaac M. Westwood, 1,2 livier Barbeau, 1 Martin Rowlands, 1 Sarah Dobson,
SUPPRTIG IFRMATI Exploiting Protein Conformational Change to ptimize Adenosine-Derived Inhibitors of HSP70 Matthew D. Cheeseman, 1 Isaac M. Westwood, 1,2 livier Barbeau, 1 Martin Rowlands, 1 Sarah Dobson,
The structure of a nucleolytic ribozyme that employs a catalytic metal ion Liu, Yijin; Wilson, Timothy; Lilley, David
 University of Dundee The structure of a nucleolytic ribozyme that employs a catalytic metal ion Liu, Yijin; Wilson, Timothy; Lilley, David Published in: Nature Chemical Biology DOI: 10.1038/nchembio.2333
University of Dundee The structure of a nucleolytic ribozyme that employs a catalytic metal ion Liu, Yijin; Wilson, Timothy; Lilley, David Published in: Nature Chemical Biology DOI: 10.1038/nchembio.2333
TLS and all that. Ethan A Merritt. CCP4 Summer School 2011 (Argonne, IL) Abstract
 Abstract") TLS and all that Ethan A Merritt CCP4 Summer School 2011 (Argonne, IL) Abstract We can never know the position of every atom in a crystal structure perfectly. Each atom has an associated positional uncertainty.
TLS and all that Ethan A Merritt CCP4 Summer School 2011 (Argonne, IL) Abstract We can never know the position of every atom in a crystal structure perfectly. Each atom has an associated positional uncertainty.
Pymol Practial Guide
 Pymol Practial Guide Pymol is a powerful visualizor very convenient to work with protein molecules. Its interface may seem complex at first, but you will see that with a little practice is simple and powerful
Pymol Practial Guide Pymol is a powerful visualizor very convenient to work with protein molecules. Its interface may seem complex at first, but you will see that with a little practice is simple and powerful
Template Free Protein Structure Modeling Jianlin Cheng, PhD
 Template Free Protein Structure Modeling Jianlin Cheng, PhD Associate Professor Computer Science Department Informatics Institute University of Missouri, Columbia 2013 Protein Energy Landscape & Free Sampling
Template Free Protein Structure Modeling Jianlin Cheng, PhD Associate Professor Computer Science Department Informatics Institute University of Missouri, Columbia 2013 Protein Energy Landscape & Free Sampling
Design of a Novel Globular Protein Fold with Atomic-Level Accuracy
 Design of a Novel Globular Protein Fold with Atomic-Level Accuracy Brian Kuhlman, Gautam Dantas, Gregory C. Ireton, Gabriele Varani, Barry L. Stoddard, David Baker Presented by Kate Stafford 4 May 05 Protein
Design of a Novel Globular Protein Fold with Atomic-Level Accuracy Brian Kuhlman, Gautam Dantas, Gregory C. Ireton, Gabriele Varani, Barry L. Stoddard, David Baker Presented by Kate Stafford 4 May 05 Protein
PAN-modular Structure of Parasite Sarcocystis muris Microneme Protein SML-2 at 1.95 Å Resolution and the Complex with 1-Thio-β-D-Galactose
 Supplementary Material to the paper: PAN-modular Structure of Parasite Sarcocystis muris Microneme Protein SML-2 at 1.95 Å Resolution and the Complex with 1-Thio-β-D-Galactose Jürgen J. Müller, a Manfred
Supplementary Material to the paper: PAN-modular Structure of Parasite Sarcocystis muris Microneme Protein SML-2 at 1.95 Å Resolution and the Complex with 1-Thio-β-D-Galactose Jürgen J. Müller, a Manfred
Full wwpdb X-ray Structure Validation Report i
 Full wwpdb X-ray Structure Validation Report i Mar 8, 2018 08:34 pm GMT PDB ID : 1RUT Title : Complex of LMO4 LIM domains 1 and 2 with the ldb1 LID domain Authors : Deane, J.E.; Ryan, D.P.; Maher, M.J.;
Full wwpdb X-ray Structure Validation Report i Mar 8, 2018 08:34 pm GMT PDB ID : 1RUT Title : Complex of LMO4 LIM domains 1 and 2 with the ldb1 LID domain Authors : Deane, J.E.; Ryan, D.P.; Maher, M.J.;
Experimental Phasing with SHELX C/D/E
 WIR SCHAFFEN WISSEN HEUTE FÜR MORGEN Dr. Tim Grüne :: Paul Scherrer Institut :: tim.gruene@psi.ch Experimental Phasing with SHELX C/D/E CCP4 / APS School Chicago 2017 22 nd June 2017 1 - The Phase Problem
WIR SCHAFFEN WISSEN HEUTE FÜR MORGEN Dr. Tim Grüne :: Paul Scherrer Institut :: tim.gruene@psi.ch Experimental Phasing with SHELX C/D/E CCP4 / APS School Chicago 2017 22 nd June 2017 1 - The Phase Problem
wwpdb X-ray Structure Validation Summary Report
 wwpdb X-ray Structure Validation Summary Report io Jan 31, 2016 06:45 PM GMT PDB ID : 1CBS Title : CRYSTAL STRUCTURE OF CELLULAR RETINOIC-ACID-BINDING PROTEINS I AND II IN COMPLEX WITH ALL-TRANS-RETINOIC
wwpdb X-ray Structure Validation Summary Report io Jan 31, 2016 06:45 PM GMT PDB ID : 1CBS Title : CRYSTAL STRUCTURE OF CELLULAR RETINOIC-ACID-BINDING PROTEINS I AND II IN COMPLEX WITH ALL-TRANS-RETINOIC
Supporting Information. Full and Partial Agonism of a Designed Enzyme Switch
 Supporting Information Full and Partial Agonism of a Designed Enzyme Switch S. Jimmy Budiardjo 1, Timothy J. Licknack 2, Michael B. Cory 2, Dora Kapros 2, Anuradha Roy 3, Scott Lovell 4, Justin Douglas
Supporting Information Full and Partial Agonism of a Designed Enzyme Switch S. Jimmy Budiardjo 1, Timothy J. Licknack 2, Michael B. Cory 2, Dora Kapros 2, Anuradha Roy 3, Scott Lovell 4, Justin Douglas
Direct-method SAD phasing with partial-structure iteration: towards automation
 Acta Crystallographica Section D Biological Crystallography ISSN 0907-4449 Editors: E. N. Baker and Z. Dauter Direct-method SAD phasing with partial-structure iteration: towards automation J. W. Wang,
Acta Crystallographica Section D Biological Crystallography ISSN 0907-4449 Editors: E. N. Baker and Z. Dauter Direct-method SAD phasing with partial-structure iteration: towards automation J. W. Wang,
1. Protein Data Bank (PDB) 1. Protein Data Bank (PDB)
 1. Protein Data Bank (PDB)") Protein structure databases; visualization; and classifications 1. Introduction to Protein Data Bank (PDB) 2. Free graphic software for 3D structure visualization 3. Hierarchical classification of protein
Protein structure databases; visualization; and classifications 1. Introduction to Protein Data Bank (PDB) 2. Free graphic software for 3D structure visualization 3. Hierarchical classification of protein
Macromolecular Crystallography Part II
 Molecular Biology Course 2009 Macromolecular Crystallography Part II Tim Grüne University of Göttingen Dept. of Structural Chemistry November 2009 http://shelx.uni-ac.gwdg.de tg@shelx.uni-ac.gwdg.de From
Molecular Biology Course 2009 Macromolecular Crystallography Part II Tim Grüne University of Göttingen Dept. of Structural Chemistry November 2009 http://shelx.uni-ac.gwdg.de tg@shelx.uni-ac.gwdg.de From
Analysis and Prediction of Protein Structure (I)
") Analysis and Prediction of Protein Structure (I) Jianlin Cheng, PhD School of Electrical Engineering and Computer Science University of Central Florida 2006 Free for academic use. Copyright @ Jianlin Cheng
Analysis and Prediction of Protein Structure (I) Jianlin Cheng, PhD School of Electrical Engineering and Computer Science University of Central Florida 2006 Free for academic use. Copyright @ Jianlin Cheng
Better Bond Angles in the Protein Data Bank
 Better Bond Angles in the Protein Data Bank C.J. Robinson and D.B. Skillicorn School of Computing Queen s University {robinson,skill}@cs.queensu.ca Abstract The Protein Data Bank (PDB) contains, at least
Better Bond Angles in the Protein Data Bank C.J. Robinson and D.B. Skillicorn School of Computing Queen s University {robinson,skill}@cs.queensu.ca Abstract The Protein Data Bank (PDB) contains, at least
Structural Mechanism for the Fidelity Modulation of DNA Polymerase λ. 128 Academia Road Sec. 2, Nankang, Taipei, 115, Taiwan
 SUPPORTING INFORMATION Structural Mechanism for the Fidelity Modulation of DNA Polymerase λ Mu-Sen Liu, 1,3 Hsin-Yue Tsai, 1,# Xiao-Xia Liu, 1,# Meng-Chiao Ho, 1,3 Wen-Jin Wu, 1,* and Ming-Daw Tsai 1,2,3,*
SUPPORTING INFORMATION Structural Mechanism for the Fidelity Modulation of DNA Polymerase λ Mu-Sen Liu, 1,3 Hsin-Yue Tsai, 1,# Xiao-Xia Liu, 1,# Meng-Chiao Ho, 1,3 Wen-Jin Wu, 1,* and Ming-Daw Tsai 1,2,3,*
Acta Crystallographica Section F
 Supporting information Acta Crystallographica Section F Volume 70 (2014) Supporting information for article: Chemical conversion of cisplatin and carboplatin with histidine in a model protein crystallised
Supporting information Acta Crystallographica Section F Volume 70 (2014) Supporting information for article: Chemical conversion of cisplatin and carboplatin with histidine in a model protein crystallised
Protein structure prediction. CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror
 Protein structure prediction CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror 1 Outline Why predict protein structure? Can we use (pure) physics-based methods? Knowledge-based methods Two major
Protein structure prediction CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror 1 Outline Why predict protein structure? Can we use (pure) physics-based methods? Knowledge-based methods Two major
research papers HKL-3000: the integration of data reduction and structure solution from diffraction images to an initial model in minutes
 Acta Crystallographica Section D Biological Crystallography ISSN 0907-4449 HKL-3000: the integration of data reduction and structure solution from diffraction images to an initial model in minutes Wladek
Acta Crystallographica Section D Biological Crystallography ISSN 0907-4449 HKL-3000: the integration of data reduction and structure solution from diffraction images to an initial model in minutes Wladek
Automated identification of functional dynamic contact networks from X-ray crystallography
 1 Automated identification of functional dynamic contact networks from X-ray crystallography Henry van den Bedem, Gira Bhabha, Kun Yang, Peter E. Wright and James S. Fraser Supplementary Figure 1 Supplementary
1 Automated identification of functional dynamic contact networks from X-ray crystallography Henry van den Bedem, Gira Bhabha, Kun Yang, Peter E. Wright and James S. Fraser Supplementary Figure 1 Supplementary
2013 Eric Pitman Summer Workshop in Computational Science....an introduction to R, statistics, programming, and getting to know datasets
 2013 Eric Pitman Summer Workshop in Computational Science...an introduction to R, statistics, programming, and getting to know datasets Introducing the Workshop Project Here's what we'll cover: The story
2013 Eric Pitman Summer Workshop in Computational Science...an introduction to R, statistics, programming, and getting to know datasets Introducing the Workshop Project Here's what we'll cover: The story
Protein Structure Prediction
 Page 1 Protein Structure Prediction Russ B. Altman BMI 214 CS 274 Protein Folding is different from structure prediction --Folding is concerned with the process of taking the 3D shape, usually based on
Page 1 Protein Structure Prediction Russ B. Altman BMI 214 CS 274 Protein Folding is different from structure prediction --Folding is concerned with the process of taking the 3D shape, usually based on
Molecular Biology Course 2006 Protein Crystallography Part II
 Molecular Biology Course 2006 Protein Crystallography Part II Tim Grüne University of Göttingen Dept. of Structural Chemistry December 2006 http://shelx.uni-ac.gwdg.de tg@shelx.uni-ac.gwdg.de Overview
Molecular Biology Course 2006 Protein Crystallography Part II Tim Grüne University of Göttingen Dept. of Structural Chemistry December 2006 http://shelx.uni-ac.gwdg.de tg@shelx.uni-ac.gwdg.de Overview
Bio nformatics. Lecture 23. Saad Mneimneh
 Bio nformatics Lecture 23 Protein folding The goal is to determine the three-dimensional structure of a protein based on its amino acid sequence Assumption: amino acid sequence completely and uniquely
Bio nformatics Lecture 23 Protein folding The goal is to determine the three-dimensional structure of a protein based on its amino acid sequence Assumption: amino acid sequence completely and uniquely
CRYSTALLOGRAPHY AND STORYTELLING WITH DATA. President, Association of Women in Science, Bethesda Chapter STEM Consultant
 CRYSTALLOGRAPHY AND STORYTELLING WITH DATA President, Association of Women in Science, Bethesda Chapter STEM Consultant MY STORY Passion for Science BS Biology Major MS Biotechnology & Project in Bioinformatics
CRYSTALLOGRAPHY AND STORYTELLING WITH DATA President, Association of Women in Science, Bethesda Chapter STEM Consultant MY STORY Passion for Science BS Biology Major MS Biotechnology & Project in Bioinformatics