Pipelining Ligands in PHENIX: elbow and REEL
|
|
|
- Christian Fleming
- 5 years ago
- Views:
Transcription

1 Pipelining Ligands in PHENIX: elbow and REEL Nigel W. Moriarty Lawrence Berkeley National Laboratory Physical Biosciences Division
2 Ligands in Crystallography Drug design Biological function studies Generate ligand restraints Fit ligand to density Refine macromolecule and ligand
 Cartesian coordinates file (PDB) phenix.")
3 e-lbow Goals Fast, simple and flexible procedure to include ligands in refinement Reduce the tedium of building 3D ligand models Automate generation of restraints for ligands Comparison of ligand structures with PDB models Chemical input format Reflection Data Protein Information Chemical restraints file (CIF) Cartesian coordinates file (PDB) phenix.refine
4 Ligand pipeline flowchart Chemical input Reflection data Protein model elbow Geometry LigandFit Ligand model phenix.refine Restraints Final model
5 angles elements names atom dihedrals connectivity bond residue name centres chiral orders bond charges formal planes coordinates cartesian Input Formats SMILES PDB xyz Mol2D, Mol3D (v2000, v3000) GAMESS input and output files Sequences PRODRG TXT format Monomer library CIF PDB Ligand
6 The Algorithm
7 The Algorithm Parse SMILES or other input Post process Molecular object Atomic centres Bonds Determine ring and chiral structures Construct a Z-Matrix model of heavy atoms Z-Matrix is an internal coordinate representation Chemically intuitive Provide direct control of bonds, angles and dihedrals

8 Z-Matrix C1 H1 C1 1.1 H2 C1 1.1 H H3 C1 1.1 H H H4 C1 1.1 H H H1 Degrees of freedom = 3n-6 H4 C1 H2 H3
9 The Algorithm Optimise Z-Matrix geometry using simple force field (requires cartesian coordinates) Add hydrogens Optimise just hydrogens using simple force field Optimise using AM1 semiempirical quantum chemical method Check structure and repeat optimisation if necessary Generate geometry restraints

10 Comparison with experiment MSD Chem ligand library contains approximately 6443 ligands SMILES strings a smaller number of experimental cartesian coordinates approximately 3285 comparable ligands Input SMILES string to elbow Compared AM1 geometry to experimental results from the PDB
 Compare to structure deposited in PDB Legend PDB High Low Calculate RMSD using cartesian coordinates, bond, angle & dihedrals")
11 Refinement Tests Use the restraints for ligand obtained from elbow in phenix.refine Refined at resolution 1.2Å or better and 2.5Å (data truncated) Compare to structure deposited in PDB Legend PDB High Low Calculate RMSD using cartesian coordinates, bond, angle & dihedrals e-lbow run time 194 seconds


12 Challenges Disordered or weak density Poor deposited ligand geometry

13 Map Density Correlation Compare the correlation of the model density to the 2mFo-DFc map phenix.refine and elbow provide provide ligand geometries that match the experimental data equally as well as the original libraries used in refinement Results are also good for low resolution, but the comparison is difficult (PDB refinements were at high resolution)

14 Histograms Mean, Sigma xyz: 0.11, 0.07 bond: 0.037, dihedral: 6.0, 4.8 Differences between the original and new geometries are within the restraint standard deviations
15 Flexibility Simple command line interface Scriptable using Python Covalently bound ligands Automatically treats all ligands in a PDB file Close integration with refinement
16 Start COOT with elbow in COOT coot --script $PHENIX/elbow/elbow_in_coot.scm coot --script $PHENIX/elbow/elbow_in_coot.py Build a ligand from SMILES or use the coordinates from an internal molecule Manipulation of ligand geometry to provide a starting geometry AM1 geometry optimisation
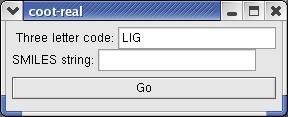


17
18 SMILES Simple run phenix.elbow --smiles c1ccccc1c phenix.elbow --smiles filename.smi phenix.elbow --msd atp PDB file phenix.elbow --file filename.pdb phenix.elbow filename.pdb Atom naming phenix.elbow --smiles c1ccccc1c --template names.pdb phenix.elbow --msd atp --template atp.pdb
19 Output PDB file contains optimised geometry CIF file contains restraints filename.bonding.py is a Python script to change the bonding via --bonding --tripos will output a TRIPOS file --output will change the name of the output files
20 Adjusting geometry phenix.elbow --smiles FC=CF --opt - -view pymol --view pymol run pymol filename.pdb Edit geometry and save as fixed.pdb elbow will read the new geometry as starting geometry for AM1 optimisation
21 Providing geometry Starting geometry for AM1 optimisation phenix.elbow --initial-geometry file.pdb Final geometry to get a CIF file phenix.elbow --final-geometry file.pdb Provide corresponding SMILES for a better CIF file
22 PDB options A PDB file can have many ligands To list all residues in a file phenix.elbow file.pdb --all-residues To run elbow on all unknown ligands phenix.elbow file.pdb --do-all To run elbow on a known ligand phenix.elbow file.pdb --residue ATP
23 PDB options (cont.) To control the auto bond determination -- auto-bond-cutoff=2.5 To control hydrogen addition --add-hydrogens=true To use a large PDB file as template phenix.elbow --smiles O --template large.pdb --residue HOH
24 elbow is Python scripts from elbow.command_line import builder molecule = builder.run(smiles= O, opt=true, quiet=true) print molecule.display()
25 Misc. features --read-only to exit after reading input --pickle to read topology file --pipe to use or open a pipe shell --pymol to use progress in PyMOL if PHENIX installed --quiet & --silent --name sets ligand name --id defaults to LIG
26 elbow & phenix.refine Run phenix.elbow ensuring that the atom naming is correct using a protein-ligand complex PDB file Run phenix.refine using the CIF file Possible to generate CIF link file using the CONECT or LINK record in the proteinligand complex PDB file Add hydrogens to a PDB containing protein model and ligands
27 Other elbow programs phenix.metal_coordinate elbow.join_cif_files, join_pdb_files elbow.join_cif_files combined.cif file1.cif file2.cif elbow.doc
28 elbow Summary e-lbow provides easy to use methods to generate novel ligands and known ligands AM1 provides a computationally efficient method for geometry optimisation of molecules containing any main group elements Provides geometries comparable to existing methods Readily pipelined into automatic refinement using phenix.refine and phenix.ligandfit
29 What is REEL? Restraints Editor elbow Ligands Loads any restraints file Loads any elbow input Restraints Editor Especially Ligands No linking or modifications Restraints Editor Exclusively Ligands Loads a small PDB file Restraints Editor Effectively Ligands REEL will make a real difference A. White, Ph.D., Boehringer-Ingelheim GmbH
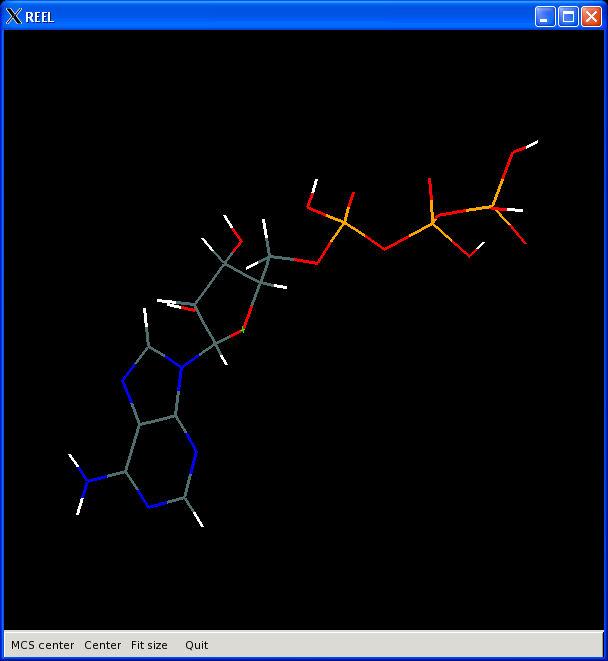
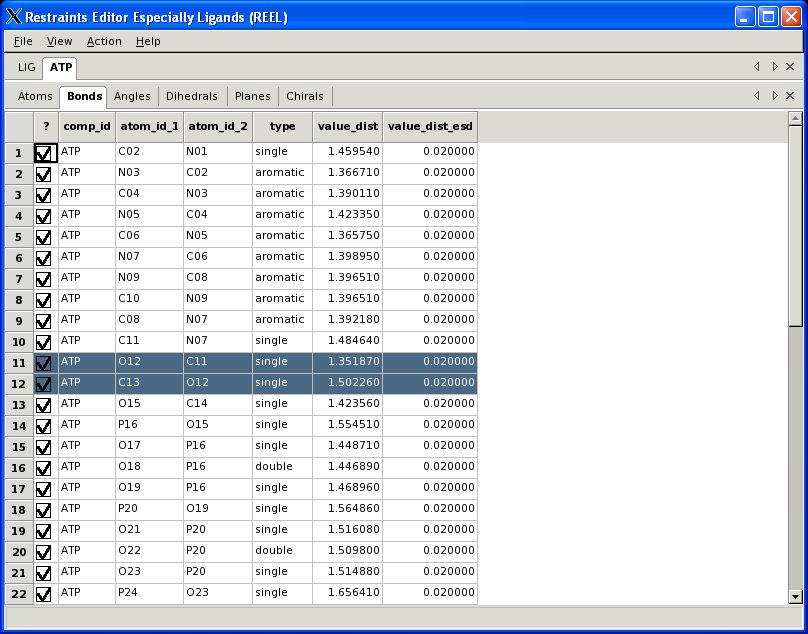
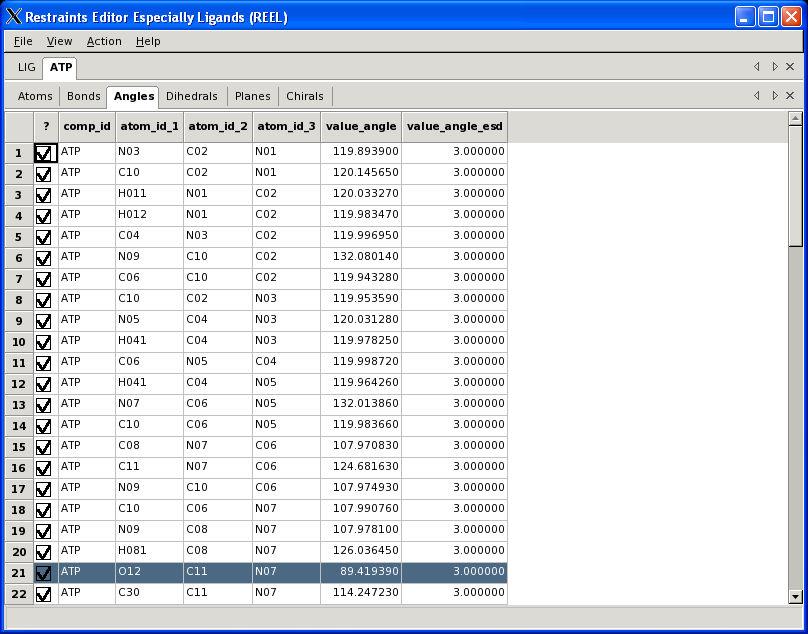
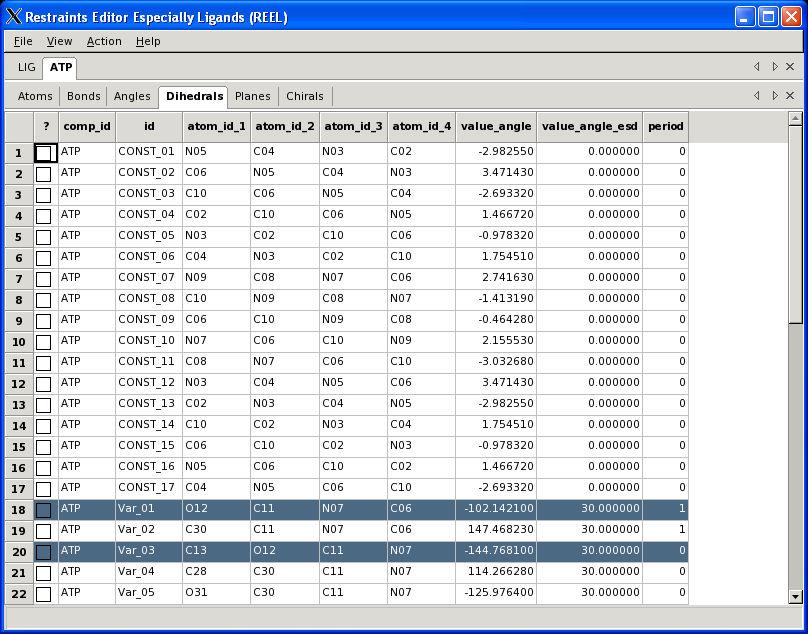
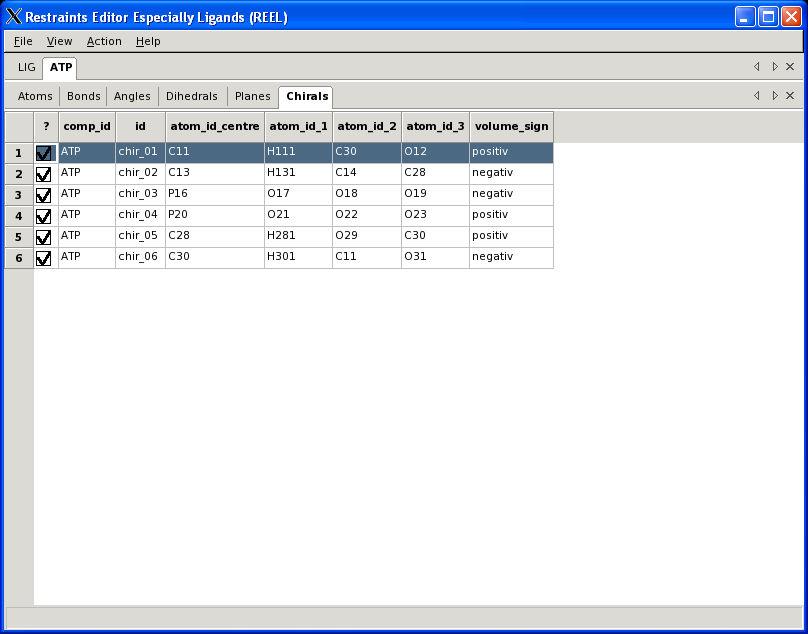
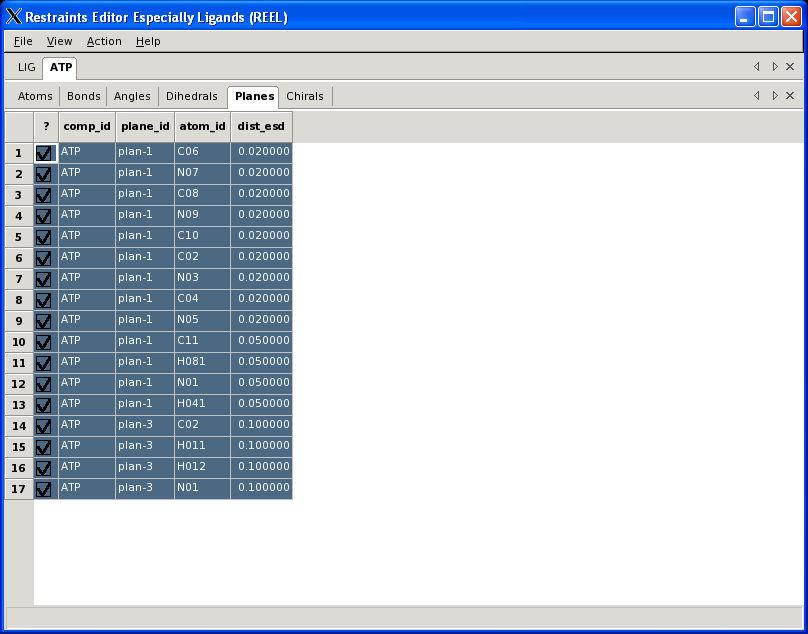
30 Visualisation PHENIX User Meeting March 28th 2008
31 REEL features Generate a geometry for a set of restraints Modify restraints and generate new geometry Fast editing in menu items Highlight atom and restraints are highlighted Multiple ligands simultaneously Highlight restraint and atoms are highlighted Save restraints (CIF) Save geometry (PDB) Run elbow

32 elbow GUI
33 REEL flowchart Chemical input elbow Geometry Restraints REEL Geometry Restraints
34 Demonstration
35 Summary elbow converts many inputs to geometry and restraints REEL allows restraints editing and several elbow features
 Ralf Grosse-Kunstleve (cctbx, phenix.refine, HySS,.) Nick Sauter (iotbx, labelit) Peter Zwart (mmtbx.xtriage, phenix.")
 Alexei Vagin & Garib Murshudov (Monomer Library) Funding: LBNL (DE-AC03-76SF00098) NIH/NIGMS")
36 Acknowledgments Computational Crystallography Initiative Paul Adams Pavel Afonine (phenix.refine) Ralf Grosse-Kunstleve (cctbx, phenix.refine, HySS,.) Nick Sauter (iotbx, labelit) Peter Zwart (mmtbx.xtriage, phenix.refine) Other PHENIX developers Cambridge University, Los Alamos National Laboratory, Texas A&M, Duke University Others CCP4 developers (MTZ library) Alexei Vagin & Garib Murshudov (Monomer Library) Funding: LBNL (DE-AC03-76SF00098) NIH/NIGMS (P01GM063210) NIH/NIGMS (P01GM064692) PHENIX Industrial Consortium
Garib N Murshudov MRC-LMB, Cambridge
 Garib N Murshudov MRC-LMB, Cambridge Contents Introduction AceDRG: two functions Validation of entries in the DB and derived data Generation of new ligand description Jligand for link description Conclusions
Garib N Murshudov MRC-LMB, Cambridge Contents Introduction AceDRG: two functions Validation of entries in the DB and derived data Generation of new ligand description Jligand for link description Conclusions
PHENIX Wizards and Tools
 PHENIX Wizards and Tools Tom Terwilliger Los Alamos National Laboratory terwilliger@lanl.gov l The PHENIX project Computational Crystallography Initiative (LBNL) Paul Adams, Ralf Grosse-Kunstleve, Peter
PHENIX Wizards and Tools Tom Terwilliger Los Alamos National Laboratory terwilliger@lanl.gov l The PHENIX project Computational Crystallography Initiative (LBNL) Paul Adams, Ralf Grosse-Kunstleve, Peter
Dictionary of ligands
 Dictionary of ligands Some of the web and other resources Small molecules DrugBank: http://www.drugbank.ca/ ZINC: http://zinc.docking.org/index.shtml PRODRUG: http://www.compbio.dundee.ac.uk/web_servers/prodrg_down.html
Dictionary of ligands Some of the web and other resources Small molecules DrugBank: http://www.drugbank.ca/ ZINC: http://zinc.docking.org/index.shtml PRODRUG: http://www.compbio.dundee.ac.uk/web_servers/prodrg_down.html
Manipulating Ligands Using Coot. Paul Emsley May 2013
 Manipulating Ligands Using Coot Paul Emsley May 2013 Ligand and Density... Ligand and Density... Ligand and Density... Protein-ligand complex models are often a result of subjective interpretation Scoring
Manipulating Ligands Using Coot Paul Emsley May 2013 Ligand and Density... Ligand and Density... Ligand and Density... Protein-ligand complex models are often a result of subjective interpretation Scoring
Charles Ballard (original GáborBunkóczi) CCP4 Workshop 7 December 2011
 CCP4 Workshop 7 December 2011") Experimental phasing Charles Ballard (original GáborBunkóczi) CCP4 Workshop 7 December 2011 Anomalous diffraction F P protein F A anomalous substructure ano F A " -FA" A F F -* F A F P Phasing Substructure
Experimental phasing Charles Ballard (original GáborBunkóczi) CCP4 Workshop 7 December 2011 Anomalous diffraction F P protein F A anomalous substructure ano F A " -FA" A F F -* F A F P Phasing Substructure
Electronic Supplementary Information (ESI) for Chem. Commun. Unveiling the three- dimensional structure of the green pigment of nitrite- cured meat
 for Chem. Commun. Unveiling the three- dimensional structure of the green pigment of nitrite- cured meat") Electronic Supplementary Information (ESI) for Chem. Commun. Unveiling the three- dimensional structure of the green pigment of nitrite- cured meat Jun Yi* and George B. Richter- Addo* Department of Chemistry
Electronic Supplementary Information (ESI) for Chem. Commun. Unveiling the three- dimensional structure of the green pigment of nitrite- cured meat Jun Yi* and George B. Richter- Addo* Department of Chemistry
Phaser: Experimental phasing
 Phaser: Experimental phasing Using SAD data in Phaser R J Read, Department of Haematology Cambridge Institute for Medical Research Diffraction with anomalous scatterers SAD: single-wavelength anomalous
Phaser: Experimental phasing Using SAD data in Phaser R J Read, Department of Haematology Cambridge Institute for Medical Research Diffraction with anomalous scatterers SAD: single-wavelength anomalous
Likelihood and SAD phasing in Phaser. R J Read, Department of Haematology Cambridge Institute for Medical Research
 Likelihood and SAD phasing in Phaser R J Read, Department of Haematology Cambridge Institute for Medical Research Concept of likelihood Likelihood with dice 4 6 8 10 Roll a seven. Which die?? p(4)=p(6)=0
Likelihood and SAD phasing in Phaser R J Read, Department of Haematology Cambridge Institute for Medical Research Concept of likelihood Likelihood with dice 4 6 8 10 Roll a seven. Which die?? p(4)=p(6)=0
Joana Pereira Lamzin Group EMBL Hamburg, Germany. Small molecules How to identify and build them (with ARP/wARP)
") Joana Pereira Lamzin Group EMBL Hamburg, Germany Small molecules How to identify and build them (with ARP/wARP) The task at hand To find ligand density and build it! Fitting a ligand We have: electron
Joana Pereira Lamzin Group EMBL Hamburg, Germany Small molecules How to identify and build them (with ARP/wARP) The task at hand To find ligand density and build it! Fitting a ligand We have: electron
Lawrence Berkeley National Laboratory Lawrence Berkeley National Laboratory
 Lawrence Berkeley National Laboratory Lawrence Berkeley National Laboratory Title electronic Ligand Builder and Optimisation Workbench (elbow): A tool for ligand coordinate and restraint generation Permalink
Lawrence Berkeley National Laboratory Lawrence Berkeley National Laboratory Title electronic Ligand Builder and Optimisation Workbench (elbow): A tool for ligand coordinate and restraint generation Permalink
The Crystallographic Process
 Phase Improvement Macromolecular Crystallography School Madrid, May 2017 Paul Adams Lawrence Berkeley Laboratory and Department of Bioengineering UC Berkeley The Crystallographic Process Crystallization
Phase Improvement Macromolecular Crystallography School Madrid, May 2017 Paul Adams Lawrence Berkeley Laboratory and Department of Bioengineering UC Berkeley The Crystallographic Process Crystallization
Structure solution from weak anomalous data
 Structure solution from weak anomalous data Phenix Workshop SBGrid-NE-CAT Computing School Harvard Medical School, Boston June 7, 2014 Gábor Bunkóczi, Airlie McCoy, Randy Read (Cambridge University) Nat
Structure solution from weak anomalous data Phenix Workshop SBGrid-NE-CAT Computing School Harvard Medical School, Boston June 7, 2014 Gábor Bunkóczi, Airlie McCoy, Randy Read (Cambridge University) Nat
Performing a Pharmacophore Search using CSD-CrossMiner
 Table of Contents Introduction... 2 CSD-CrossMiner Terminology... 2 Overview of CSD-CrossMiner... 3 Searching with a Pharmacophore... 4 Performing a Pharmacophore Search using CSD-CrossMiner Version 2.0
Table of Contents Introduction... 2 CSD-CrossMiner Terminology... 2 Overview of CSD-CrossMiner... 3 Searching with a Pharmacophore... 4 Performing a Pharmacophore Search using CSD-CrossMiner Version 2.0
This is an author produced version of Privateer: : software for the conformational validation of carbohydrate structures.
 This is an author produced version of Privateer: : software for the conformational validation of carbohydrate structures. White Rose Research Online URL for this paper: http://eprints.whiterose.ac.uk/95794/
This is an author produced version of Privateer: : software for the conformational validation of carbohydrate structures. White Rose Research Online URL for this paper: http://eprints.whiterose.ac.uk/95794/
1.b What are current best practices for selecting an initial target ligand atomic model(s) for structure refinement from X-ray diffraction data?!
 for structure refinement from X-ray diffraction data?!") 1.b What are current best practices for selecting an initial target ligand atomic model(s) for structure refinement from X-ray diffraction data?! Visual analysis: Identification of ligand density from
1.b What are current best practices for selecting an initial target ligand atomic model(s) for structure refinement from X-ray diffraction data?! Visual analysis: Identification of ligand density from
Coot Updates. Paul Emsley Sept 2016
 Coot Updates Paul Emsley Sept 2016 Coot 0.8.4 Released Patterson from intensities Fill-partial-residue uses Backrub-rotamers Sequence dialog is now dynamically updated Outliers Only in Ramachanran Plot
Coot Updates Paul Emsley Sept 2016 Coot 0.8.4 Released Patterson from intensities Fill-partial-residue uses Backrub-rotamers Sequence dialog is now dynamically updated Outliers Only in Ramachanran Plot
Molecular Visualization. Introduction
 Molecular Visualization Jeffry D. Madura Department of Chemistry & Biochemistry Center for Computational Sciences Duquesne University Introduction Assessments of change, dynamics, and cause and effect
Molecular Visualization Jeffry D. Madura Department of Chemistry & Biochemistry Center for Computational Sciences Duquesne University Introduction Assessments of change, dynamics, and cause and effect
Modelling Macromolecules with Coot
 Modelling Macromolecules with Coot Overview Real Space Refinement A Sample of Tools Tools for Cryo-EM Tools for Ligands [Carbohydrates] Paul Emsley MRC Laboratory of Molecular Biology Acknowldegments,
Modelling Macromolecules with Coot Overview Real Space Refinement A Sample of Tools Tools for Cryo-EM Tools for Ligands [Carbohydrates] Paul Emsley MRC Laboratory of Molecular Biology Acknowldegments,
User Guide for LeDock
 User Guide for LeDock Hongtao Zhao, PhD Email: htzhao@lephar.com Website: www.lephar.com Copyright 2017 Hongtao Zhao. All rights reserved. Introduction LeDock is flexible small-molecule docking software,
User Guide for LeDock Hongtao Zhao, PhD Email: htzhao@lephar.com Website: www.lephar.com Copyright 2017 Hongtao Zhao. All rights reserved. Introduction LeDock is flexible small-molecule docking software,
Conformational Searching using MacroModel and ConfGen. John Shelley Schrödinger Fellow
 Conformational Searching using MacroModel and ConfGen John Shelley Schrödinger Fellow Overview Types of conformational searching applications MacroModel s conformation generation procedure General features
Conformational Searching using MacroModel and ConfGen John Shelley Schrödinger Fellow Overview Types of conformational searching applications MacroModel s conformation generation procedure General features
Creating a Pharmacophore Query from a Reference Molecule & Scaffold Hopping in CSD-CrossMiner
 Table of Contents Creating a Pharmacophore Query from a Reference Molecule & Scaffold Hopping in CSD-CrossMiner Introduction... 2 CSD-CrossMiner Terminology... 2 Overview of CSD-CrossMiner... 3 Features
Table of Contents Creating a Pharmacophore Query from a Reference Molecule & Scaffold Hopping in CSD-CrossMiner Introduction... 2 CSD-CrossMiner Terminology... 2 Overview of CSD-CrossMiner... 3 Features
Dock Ligands from a 2D Molecule Sketch
 Dock Ligands from a 2D Molecule Sketch March 31, 2016 Sample to Insight CLC bio, a QIAGEN Company Silkeborgvej 2 Prismet 8000 Aarhus C Denmark Telephone: +45 70 22 32 44 www.clcbio.com support-clcbio@qiagen.com
Dock Ligands from a 2D Molecule Sketch March 31, 2016 Sample to Insight CLC bio, a QIAGEN Company Silkeborgvej 2 Prismet 8000 Aarhus C Denmark Telephone: +45 70 22 32 44 www.clcbio.com support-clcbio@qiagen.com
The Crystallographic Process
 Experimental Phasing Macromolecular Crystallography School Madrid, May 2017 Paul Adams Lawrence Berkeley Laboratory and Department of Bioengineering UC Berkeley The Crystallographic Process Crystallization
Experimental Phasing Macromolecular Crystallography School Madrid, May 2017 Paul Adams Lawrence Berkeley Laboratory and Department of Bioengineering UC Berkeley The Crystallographic Process Crystallization
CSD. Unlock value from crystal structure information in the CSD
 CSD CSD-System Unlock value from crystal structure information in the CSD The Cambridge Structural Database (CSD) is the world s most comprehensive and up-todate knowledge base of crystal structure data,
CSD CSD-System Unlock value from crystal structure information in the CSD The Cambridge Structural Database (CSD) is the world s most comprehensive and up-todate knowledge base of crystal structure data,
Preparing a PDB File
 Figure 1: Schematic view of the ligand-binding domain from the vitamin D receptor (PDB file 1IE9). The crystallographic waters are shown as small spheres and the bound ligand is shown as a CPK model. HO
Figure 1: Schematic view of the ligand-binding domain from the vitamin D receptor (PDB file 1IE9). The crystallographic waters are shown as small spheres and the bound ligand is shown as a CPK model. HO
Docking with Water in the Binding Site using GOLD
 Docking with Water in the Binding Site using GOLD Version 2.0 November 2017 GOLD v5.6 Table of Contents Docking with Water in the Binding Site... 2 Case Study... 3 Introduction... 3 Provided Input Files...
Docking with Water in the Binding Site using GOLD Version 2.0 November 2017 GOLD v5.6 Table of Contents Docking with Water in the Binding Site... 2 Case Study... 3 Introduction... 3 Provided Input Files...
The Schrödinger KNIME extensions
 The Schrödinger KNIME extensions Computational Chemistry and Cheminformatics in a workflow environment Jean-Christophe Mozziconacci Volker Eyrich Topics What are the Schrödinger extensions? Workflow application
The Schrödinger KNIME extensions Computational Chemistry and Cheminformatics in a workflow environment Jean-Christophe Mozziconacci Volker Eyrich Topics What are the Schrödinger extensions? Workflow application
research papers Iterative-build OMIT maps: map improvement by iterative model building and refinement without model bias 1.
 Acta Crystallographica Section D Biological Crystallography ISSN 0907-4449 Iterative-build OMIT maps: map improvement by iterative model building and refinement without model bias Thomas C. Terwilliger,
Acta Crystallographica Section D Biological Crystallography ISSN 0907-4449 Iterative-build OMIT maps: map improvement by iterative model building and refinement without model bias Thomas C. Terwilliger,
Tools for Cryo-EM Map Fitting. Paul Emsley MRC Laboratory of Molecular Biology
 Tools for Cryo-EM Map Fitting Paul Emsley MRC Laboratory of Molecular Biology April 2017 Cryo-EM model-building typically need to move more atoms that one does for crystallography the maps are lower resolution
Tools for Cryo-EM Map Fitting Paul Emsley MRC Laboratory of Molecular Biology April 2017 Cryo-EM model-building typically need to move more atoms that one does for crystallography the maps are lower resolution
Generating Small Molecule Conformations from Structural Data
 Generating Small Molecule Conformations from Structural Data Jason Cole cole@ccdc.cam.ac.uk Cambridge Crystallographic Data Centre 1 The Cambridge Crystallographic Data Centre About us A not-for-profit,
Generating Small Molecule Conformations from Structural Data Jason Cole cole@ccdc.cam.ac.uk Cambridge Crystallographic Data Centre 1 The Cambridge Crystallographic Data Centre About us A not-for-profit,
Introduction to Structure Preparation and Visualization
 Introduction to Structure Preparation and Visualization Created with: Release 2018-4 Prerequisites: Release 2018-2 or higher Access to the internet Categories: Molecular Visualization, Structure-Based
Introduction to Structure Preparation and Visualization Created with: Release 2018-4 Prerequisites: Release 2018-2 or higher Access to the internet Categories: Molecular Visualization, Structure-Based
ICM-Chemist How-To Guide. Version 3.6-1g Last Updated 12/01/2009
 ICM-Chemist How-To Guide Version 3.6-1g Last Updated 12/01/2009 ICM-Chemist HOW TO IMPORT, SKETCH AND EDIT CHEMICALS How to access the ICM Molecular Editor. 1. Click here 2. Start sketching How to sketch
ICM-Chemist How-To Guide Version 3.6-1g Last Updated 12/01/2009 ICM-Chemist HOW TO IMPORT, SKETCH AND EDIT CHEMICALS How to access the ICM Molecular Editor. 1. Click here 2. Start sketching How to sketch
CSD Conformer Generator User Guide
 CSD Conformer Generator User Guide 2018 CSD Release Copyright 2017 Cambridge Crystallographic Data Centre Registered Charity No 800579 Conditions of Use The CSD Conformer Generator is copyright work belonging
CSD Conformer Generator User Guide 2018 CSD Release Copyright 2017 Cambridge Crystallographic Data Centre Registered Charity No 800579 Conditions of Use The CSD Conformer Generator is copyright work belonging
CSD Conformer Generator User Guide
 CSD Conformer Generator User Guide 2017 CSD Release Copyright 2016 Cambridge Crystallographic Data Centre Registered Charity No 800579 Conditions of Use The CSD Conformer Generator is copyright work belonging
CSD Conformer Generator User Guide 2017 CSD Release Copyright 2016 Cambridge Crystallographic Data Centre Registered Charity No 800579 Conditions of Use The CSD Conformer Generator is copyright work belonging
CSD. CSD-Enterprise. Access the CSD and ALL CCDC application software
 CSD CSD-Enterprise Access the CSD and ALL CCDC application software CSD-Enterprise brings it all: access to the Cambridge Structural Database (CSD), the world s comprehensive and up-to-date database of
CSD CSD-Enterprise Access the CSD and ALL CCDC application software CSD-Enterprise brings it all: access to the Cambridge Structural Database (CSD), the world s comprehensive and up-to-date database of
Automated ligand fitting by core-fragment fitting and extension into density
 Acta Crystallographica Section D Biological Crystallography ISSN 0907-4449 Editors: E. N. Baker and Z. Dauter Automated ligand fitting by core-fragment fitting and extension into density Thomas C. Terwilliger,
Acta Crystallographica Section D Biological Crystallography ISSN 0907-4449 Editors: E. N. Baker and Z. Dauter Automated ligand fitting by core-fragment fitting and extension into density Thomas C. Terwilliger,
Tutorial. Getting started. Sample to Insight. March 31, 2016
 Getting started March 31, 2016 Sample to Insight CLC bio, a QIAGEN Company Silkeborgvej 2 Prismet 8000 Aarhus C Denmark Telephone: +45 70 22 32 44 www.clcbio.com support-clcbio@qiagen.com Getting started
Getting started March 31, 2016 Sample to Insight CLC bio, a QIAGEN Company Silkeborgvej 2 Prismet 8000 Aarhus C Denmark Telephone: +45 70 22 32 44 www.clcbio.com support-clcbio@qiagen.com Getting started
Introduction Molecular Structure Script Console External resources Advanced topics. JMol tutorial. Giovanni Morelli.
 Gen 19th, 2017 1 2 Create and edit Display and view Mesurament and labelling Surface and Orbitals 3 4 from Database Protein Enzyme Crystal Structure and Unit Cell 5 Symmetry Animation General information
Gen 19th, 2017 1 2 Create and edit Display and view Mesurament and labelling Surface and Orbitals 3 4 from Database Protein Enzyme Crystal Structure and Unit Cell 5 Symmetry Animation General information
Experimental Phasing with SHELX C/D/E
 WIR SCHAFFEN WISSEN HEUTE FÜR MORGEN Dr. Tim Grüne :: Paul Scherrer Institut :: tim.gruene@psi.ch Experimental Phasing with SHELX C/D/E CCP4 / APS School Chicago 2017 22 nd June 2017 1 - The Phase Problem
WIR SCHAFFEN WISSEN HEUTE FÜR MORGEN Dr. Tim Grüne :: Paul Scherrer Institut :: tim.gruene@psi.ch Experimental Phasing with SHELX C/D/E CCP4 / APS School Chicago 2017 22 nd June 2017 1 - The Phase Problem
electronic reprint PHENIX: a comprehensive Python-based system for macromolecular structure solution
 Acta Crystallographica Section D Biological Crystallography ISSN 0907-4449 Editors: E. N. Baker and Z. Dauter PHENIX: a comprehensive Python-based system for macromolecular structure solution Paul D. Adams,
Acta Crystallographica Section D Biological Crystallography ISSN 0907-4449 Editors: E. N. Baker and Z. Dauter PHENIX: a comprehensive Python-based system for macromolecular structure solution Paul D. Adams,
G L. Achieving high quality protein-ligand X-ray structures for drug design. Oliver Smart Global Phasing Ltd
 Achieving high quality protein-ligand X-ray structures for drug design Oliver Smart Global Phasing Ltd G L Structural Basis of Pharmacology: Deeper Understanding of Drug Discovery through Crystallography
Achieving high quality protein-ligand X-ray structures for drug design Oliver Smart Global Phasing Ltd G L Structural Basis of Pharmacology: Deeper Understanding of Drug Discovery through Crystallography
Molecular Modelling. Computational Chemistry Demystified. RSC Publishing. Interprobe Chemical Services, Lenzie, Kirkintilloch, Glasgow, UK
 Molecular Modelling Computational Chemistry Demystified Peter Bladon Interprobe Chemical Services, Lenzie, Kirkintilloch, Glasgow, UK John E. Gorton Gorton Systems, Glasgow, UK Robert B. Hammond Institute
Molecular Modelling Computational Chemistry Demystified Peter Bladon Interprobe Chemical Services, Lenzie, Kirkintilloch, Glasgow, UK John E. Gorton Gorton Systems, Glasgow, UK Robert B. Hammond Institute
NMR, X-ray Diffraction, Protein Structure, and RasMol
 NMR, X-ray Diffraction, Protein Structure, and RasMol Introduction So far we have been mostly concerned with the proteins themselves. The techniques (NMR or X-ray diffraction) used to determine a structure
NMR, X-ray Diffraction, Protein Structure, and RasMol Introduction So far we have been mostly concerned with the proteins themselves. The techniques (NMR or X-ray diffraction) used to determine a structure
Molecular replacement. New structures from old
 Molecular replacement New structures from old The Phase Problem phase amplitude Phasing by molecular replacement Phases can be calculated from atomic model Rotate and translate related structure Models
Molecular replacement New structures from old The Phase Problem phase amplitude Phasing by molecular replacement Phases can be calculated from atomic model Rotate and translate related structure Models
Flexibility and Constraints in GOLD
 Flexibility and Constraints in GOLD Version 2.1 August 2018 GOLD v5.6.3 Table of Contents Purpose of Docking... 3 GOLD s Evolutionary Algorithm... 4 GOLD and Hermes... 4 Handling Flexibility and Constraints
Flexibility and Constraints in GOLD Version 2.1 August 2018 GOLD v5.6.3 Table of Contents Purpose of Docking... 3 GOLD s Evolutionary Algorithm... 4 GOLD and Hermes... 4 Handling Flexibility and Constraints
Molecular modeling with InsightII
 Molecular modeling with InsightII Yuk Sham Computational Biology/Biochemistry Consultant Phone: (612) 624 7427 (Walter Library) Phone: (612) 624 0783 (VWL) Email: shamy@msi.umn.edu How to run InsightII
Molecular modeling with InsightII Yuk Sham Computational Biology/Biochemistry Consultant Phone: (612) 624 7427 (Walter Library) Phone: (612) 624 0783 (VWL) Email: shamy@msi.umn.edu How to run InsightII
Refine & Validate. In the *.res file, be sure to add the following four commands after the UNIT instruction and before any atoms: ACTA CONF WPDB -2
 Refine & Validate Refinement is simply a way to improve the fit between the measured intensities and the intensities calculated from the model. The peaks in the difference map and the list of worst fitting
Refine & Validate Refinement is simply a way to improve the fit between the measured intensities and the intensities calculated from the model. The peaks in the difference map and the list of worst fitting
Build_model v User Guide
 Build_model v.2.0.1 User Guide MolTech Build_model User Guide 2008-2011 Molecular Technologies Ltd. www.moltech.ru Please send your comments and suggestions to contact@moltech.ru. Table of Contents Input
Build_model v.2.0.1 User Guide MolTech Build_model User Guide 2008-2011 Molecular Technologies Ltd. www.moltech.ru Please send your comments and suggestions to contact@moltech.ru. Table of Contents Input
Examples of Protein Modeling. Protein Modeling. Primary Structure. Protein Structure Description. Protein Sequence Sources. Importing Sequences to MOE
 Examples of Protein Modeling Protein Modeling Visualization Examination of an experimental structure to gain insight about a research question Dynamics To examine the dynamics of protein structures To
Examples of Protein Modeling Protein Modeling Visualization Examination of an experimental structure to gain insight about a research question Dynamics To examine the dynamics of protein structures To
The Schrödinger KNIME extensions
 The Schrödinger KNIME extensions Computational Chemistry and Cheminformatics in a workflow environment Jean-Christophe Mozziconacci Volker Eyrich KNIME UGM, Berlin, February 2015 The Schrödinger Extensions
The Schrödinger KNIME extensions Computational Chemistry and Cheminformatics in a workflow environment Jean-Christophe Mozziconacci Volker Eyrich KNIME UGM, Berlin, February 2015 The Schrödinger Extensions
Scientific Integrity: A crystallographic perspective
 Scientific Integrity: A crystallographic perspective Ian Bruno - Director, Strategic Partnerships The Cambridge Crystallographic Data Centre @ijbruno @ccdc_cambridge Scientific Integrity: Can We Rely on
Scientific Integrity: A crystallographic perspective Ian Bruno - Director, Strategic Partnerships The Cambridge Crystallographic Data Centre @ijbruno @ccdc_cambridge Scientific Integrity: Can We Rely on
Assignment A02: Geometry Definition: File Formats, Redundant Coordinates, PES Scans
 Assignment A02: Geometry Definition: File Formats, Redundant Coordinates, PES Scans In Assignments A00 and A01, you familiarized yourself with GaussView and G09W, you learned the basics about input (GJF)
Assignment A02: Geometry Definition: File Formats, Redundant Coordinates, PES Scans In Assignments A00 and A01, you familiarized yourself with GaussView and G09W, you learned the basics about input (GJF)
tconcoord-gui: Visually Supported Conformational Sampling of Bioactive Molecules
 Software News and Updates tconcoord-gui: Visually Supported Conformational Sampling of Bioactive Molecules DANIEL SEELIGER, BERT L. DE GROOT Computational Biomolecular Dynamics Group, Max-Planck-Institute
Software News and Updates tconcoord-gui: Visually Supported Conformational Sampling of Bioactive Molecules DANIEL SEELIGER, BERT L. DE GROOT Computational Biomolecular Dynamics Group, Max-Planck-Institute
Ultra High Throughput Screening using THINK on the Internet
 Ultra High Throughput Screening using THINK on the Internet Keith Davies Central Chemistry Laboratory, Oxford University Cathy Davies Treweren Consultants, UK Blue Sky Objectives Reduce Development Failures
Ultra High Throughput Screening using THINK on the Internet Keith Davies Central Chemistry Laboratory, Oxford University Cathy Davies Treweren Consultants, UK Blue Sky Objectives Reduce Development Failures
Building small molecules
 Building small molecules Use the Builder (right panel) to build up molecules. Start building clicking a fragment/atom in the builder and it will appear to the workspace. Continue modifying the molecule
Building small molecules Use the Builder (right panel) to build up molecules. Start building clicking a fragment/atom in the builder and it will appear to the workspace. Continue modifying the molecule
Supporting Information
 Supporting Information Structural Basis of the Antiproliferative Activity of Largazole, a Depsipeptide Inhibitor of the Histone Deacetylases Kathryn E. Cole 1, Daniel P. Dowling 1,2, Matthew A. Boone 3,
Supporting Information Structural Basis of the Antiproliferative Activity of Largazole, a Depsipeptide Inhibitor of the Histone Deacetylases Kathryn E. Cole 1, Daniel P. Dowling 1,2, Matthew A. Boone 3,
IFM Chemistry Computational Chemistry 2010, 7.5 hp LAB2. Computer laboratory exercise 1 (LAB2): Quantum chemical calculations
: Quantum chemical calculations") Computer laboratory exercise 1 (LAB2): Quantum chemical calculations Introduction: The objective of the second computer laboratory exercise is to get acquainted with a program for performing quantum chemical
Computer laboratory exercise 1 (LAB2): Quantum chemical calculations Introduction: The objective of the second computer laboratory exercise is to get acquainted with a program for performing quantum chemical
Analyzing Molecular Conformations Using the Cambridge Structural Database. Jason Cole Cambridge Crystallographic Data Centre
 Analyzing Molecular Conformations Using the Cambridge Structural Database Jason Cole Cambridge Crystallographic Data Centre 1 The Cambridge Structural Database (CSD) 905,284* USOPEZ a natural product intermediate,
Analyzing Molecular Conformations Using the Cambridge Structural Database Jason Cole Cambridge Crystallographic Data Centre 1 The Cambridge Structural Database (CSD) 905,284* USOPEZ a natural product intermediate,
Schrodinger ebootcamp #3, Summer EXPLORING METHODS FOR CONFORMER SEARCHING Jas Bhachoo, Senior Applications Scientist
 Schrodinger ebootcamp #3, Summer 2016 EXPLORING METHODS FOR CONFORMER SEARCHING Jas Bhachoo, Senior Applications Scientist Numerous applications Generating conformations MM Agenda http://www.schrodinger.com/macromodel
Schrodinger ebootcamp #3, Summer 2016 EXPLORING METHODS FOR CONFORMER SEARCHING Jas Bhachoo, Senior Applications Scientist Numerous applications Generating conformations MM Agenda http://www.schrodinger.com/macromodel
Version 1.2 October 2017 CSD v5.39
 Mogul Geometry Check Table of Contents Introduction... 2 Example 1. Using Mogul to assess intramolecular geometry... 3 Example 2. Using Mogul to explain activity data... 5 Conclusions... 8 Further Exercises...
Mogul Geometry Check Table of Contents Introduction... 2 Example 1. Using Mogul to assess intramolecular geometry... 3 Example 2. Using Mogul to explain activity data... 5 Conclusions... 8 Further Exercises...
LysinebasedTrypsinActSite. A computer application for modeling Chymotrypsin
 LysinebasedTrypsinActSite A computer application for modeling Chymotrypsin Version.2 May 2006 LysTAS A computer application for modeling chymotrypsin Version.2 May 2006 Table of Contents Page. Introduction
LysinebasedTrypsinActSite A computer application for modeling Chymotrypsin Version.2 May 2006 LysTAS A computer application for modeling chymotrypsin Version.2 May 2006 Table of Contents Page. Introduction
Direct Method. Very few protein diffraction data meet the 2nd condition
 Direct Method Two conditions: -atoms in the structure are equal-weighted -resolution of data are higher than the distance between the atoms in the structure Very few protein diffraction data meet the 2nd
Direct Method Two conditions: -atoms in the structure are equal-weighted -resolution of data are higher than the distance between the atoms in the structure Very few protein diffraction data meet the 2nd
Supplementary Information
 Electronic Supplementary Material (ESI) for ChemComm. This journal is The Royal Society of Chemistry 2015 Supplementary Information Anion clamp allows flexible protein to impose coordination geometry on
Electronic Supplementary Material (ESI) for ChemComm. This journal is The Royal Society of Chemistry 2015 Supplementary Information Anion clamp allows flexible protein to impose coordination geometry on
NMR-Structure determination with the program CNS
 NMR-Structure determination with the program CNS Blockkurs 2013 Exercise 11.10.2013, room Mango? 1 NMR-Structure determination - Overview Amino acid sequence Topology file nef_seq.mtf loop cns_mtf_atom.id
NMR-Structure determination with the program CNS Blockkurs 2013 Exercise 11.10.2013, room Mango? 1 NMR-Structure determination - Overview Amino acid sequence Topology file nef_seq.mtf loop cns_mtf_atom.id
Protein Structure and Visualisation. Introduction to PDB and PyMOL
 Protein Structure and Visualisation Introduction to PDB and PyMOL 1 Feedback Persons http://www.bio-evaluering.dk/ 2 Program 8.00-8.15 Quiz results 8.15-8.50 Introduction to PDB & PyMOL 8.50-9.00 Break
Protein Structure and Visualisation Introduction to PDB and PyMOL 1 Feedback Persons http://www.bio-evaluering.dk/ 2 Program 8.00-8.15 Quiz results 8.15-8.50 Introduction to PDB & PyMOL 8.50-9.00 Break
The Schrödinger KNIME extensions
 The Schrödinger KNIME extensions Computational Chemistry and Cheminformatics in a workflow environment Jean-Christophe Mozziconacci Volker Eyrich KNIME UGM, Zurich, February 2014 The Schrödinger extensions
The Schrödinger KNIME extensions Computational Chemistry and Cheminformatics in a workflow environment Jean-Christophe Mozziconacci Volker Eyrich KNIME UGM, Zurich, February 2014 The Schrödinger extensions
Exercises for Windows
 Exercises for Windows CAChe User Interface for Windows Select tool Application window Document window (workspace) Style bar Tool palette Select entire molecule Select Similar Group Select Atom tool Rotate
Exercises for Windows CAChe User Interface for Windows Select tool Application window Document window (workspace) Style bar Tool palette Select entire molecule Select Similar Group Select Atom tool Rotate
Direct Methods and Many Site Se-Met MAD Problems using BnP. W. Furey
 Direct Methods and Many Site Se-Met MAD Problems using BnP W. Furey Classical Direct Methods Main method for small molecule structure determination Highly automated (almost totally black box ) Solves structures
Direct Methods and Many Site Se-Met MAD Problems using BnP W. Furey Classical Direct Methods Main method for small molecule structure determination Highly automated (almost totally black box ) Solves structures
Supporting Information
 Supporting Information Structural Analysis of the Binding of Type I, I 1/2, and II Inhibitors to Eph Tyrosine Kinases Jing Dong, *1 Hongtao Zhao, 1 Ting Zhou, 1 Dimitrios Spiliotopoulos, 1 Chitra Rajendran,
Supporting Information Structural Analysis of the Binding of Type I, I 1/2, and II Inhibitors to Eph Tyrosine Kinases Jing Dong, *1 Hongtao Zhao, 1 Ting Zhou, 1 Dimitrios Spiliotopoulos, 1 Chitra Rajendran,
Coot, pyrogen & CCP4 SRS. Paul Emsley MRC Laboratory of Molecular Biology, Cambridge, UK June 2015
 Coot, pyrogen & CCP4 SRS Paul Emsley MRC Laboratory of Molecular Biology, Cambridge, UK June 2015 Coot Updates JED-Flip (as it's currently known) added Torsion-spec-dependent rotation of ligand fragment
Coot, pyrogen & CCP4 SRS Paul Emsley MRC Laboratory of Molecular Biology, Cambridge, UK June 2015 Coot Updates JED-Flip (as it's currently known) added Torsion-spec-dependent rotation of ligand fragment
Towards a formal description of models and workflows
 Towards a formal description of models and workflows Heinz A Preisig Process Systems Engineering @ Chemical Engineering NTNU, Trondheim, Norway MoDeNa - FP7 ++ Computational engineering The vision that
Towards a formal description of models and workflows Heinz A Preisig Process Systems Engineering @ Chemical Engineering NTNU, Trondheim, Norway MoDeNa - FP7 ++ Computational engineering The vision that
Q1 current best prac2ce
 Group- A Q1 current best prac2ce Star2ng from some common molecular representa2on with bond orders, configura2on on chiral centers (e.g. ChemDraw, SMILES) NEW! PDB should become resource for refinement
Group- A Q1 current best prac2ce Star2ng from some common molecular representa2on with bond orders, configura2on on chiral centers (e.g. ChemDraw, SMILES) NEW! PDB should become resource for refinement
ATP GTP Problem 2 mm.py
 Problem 1 This problem will give you some experience with the Protein Data Bank (PDB), structure analysis, viewing and assessment and will bring up such issues as evolutionary conservation of function,
Problem 1 This problem will give you some experience with the Protein Data Bank (PDB), structure analysis, viewing and assessment and will bring up such issues as evolutionary conservation of function,
Tutorial I: IQ MOL and Basic DFT and MP2 Calculations 1 / 30
 Tutorial I: IQ MOL and Basic DFT and MP2 Calculations Q-Chem User Workshop, Denver March 21, 2015 1 / 30 2 / 30 Introduction to IQMOL DFT and MP2 Calculations 3 / 30 IQMOL and Q-CHEM IQMOL is an open-source
Tutorial I: IQ MOL and Basic DFT and MP2 Calculations Q-Chem User Workshop, Denver March 21, 2015 1 / 30 2 / 30 Introduction to IQMOL DFT and MP2 Calculations 3 / 30 IQMOL and Q-CHEM IQMOL is an open-source
Identifying Interaction Hot Spots with SuperStar
 Identifying Interaction Hot Spots with SuperStar Version 1.0 November 2017 Table of Contents Identifying Interaction Hot Spots with SuperStar... 2 Case Study... 3 Introduction... 3 Generate SuperStar Maps
Identifying Interaction Hot Spots with SuperStar Version 1.0 November 2017 Table of Contents Identifying Interaction Hot Spots with SuperStar... 2 Case Study... 3 Introduction... 3 Generate SuperStar Maps
Twinning in PDB and REFMAC. Garib N Murshudov Chemistry Department, University of York, UK
 Twinning in PDB and REFMAC Garib N Murshudov Chemistry Department, University of York, UK Contents Crystal peculiarities Problem of twinning Occurrences of twinning in PDB Recognition of potential operators
Twinning in PDB and REFMAC Garib N Murshudov Chemistry Department, University of York, UK Contents Crystal peculiarities Problem of twinning Occurrences of twinning in PDB Recognition of potential operators
Chem 253. Tutorial for Materials Studio
 Chem 253 Tutorial for Materials Studio This tutorial is designed to introduce Materials Studio 7.0, which is a program used for modeling and simulating materials for predicting and rationalizing structure
Chem 253 Tutorial for Materials Studio This tutorial is designed to introduce Materials Studio 7.0, which is a program used for modeling and simulating materials for predicting and rationalizing structure
----- Ver October 24, 2014 Bug about reading MOPAC2012 Ver.14 calculations of 1 atom and 2 atoms molecule was fixed.
 ***** Facio's Release History ***** ----- Ver.18.8.2 ----- October 24, 2014 Bug about reading MOPAC2012 Ver.14 calculations of 1 atom and 2 atoms molecule was fixed. ----- Ver.18.8.1 ----- August 14, 2014
***** Facio's Release History ***** ----- Ver.18.8.2 ----- October 24, 2014 Bug about reading MOPAC2012 Ver.14 calculations of 1 atom and 2 atoms molecule was fixed. ----- Ver.18.8.1 ----- August 14, 2014
The TEN Package. User Guide
 ================================================================= The TEN Package Tools for Elastic Networks User Guide Lars Ackermann, Silke A. Wieninger and G. Matthias Ullmann =================================================================
================================================================= The TEN Package Tools for Elastic Networks User Guide Lars Ackermann, Silke A. Wieninger and G. Matthias Ullmann =================================================================
Ligand Scout Tutorials
 Ligand Scout Tutorials Step : Creating a pharmacophore from a protein-ligand complex. Type ke6 in the upper right area of the screen and press the button Download *+. The protein will be downloaded and
Ligand Scout Tutorials Step : Creating a pharmacophore from a protein-ligand complex. Type ke6 in the upper right area of the screen and press the button Download *+. The protein will be downloaded and
Pose Prediction with GOLD
 Pose Prediction with GOLD Version 3.0 November 2018 GOLD v5.7.0 Table of Contents The Purpose of Docking... 2 GOLD s Evolutionary Algorithm... 3 A Checklist for Docking... 3 GOLD and Hermes... 3 Redocking
Pose Prediction with GOLD Version 3.0 November 2018 GOLD v5.7.0 Table of Contents The Purpose of Docking... 2 GOLD s Evolutionary Algorithm... 3 A Checklist for Docking... 3 GOLD and Hermes... 3 Redocking
Introduction to AutoDock and AutoDock Tools
 Introduction to AutoDock and AutoDock Tools Alexander B. Pacheco User Services Consultant LSU HPC & LONI sys-help@loni.org HPC Training Series Louisiana State University Baton Rouge Mar. 28, 2012 HPC@LSU
Introduction to AutoDock and AutoDock Tools Alexander B. Pacheco User Services Consultant LSU HPC & LONI sys-help@loni.org HPC Training Series Louisiana State University Baton Rouge Mar. 28, 2012 HPC@LSU
April, The energy functions include:
 REDUX A collection of Python scripts for torsion angle Monte Carlo protein molecular simulations and analysis The program is based on unified residue peptide model and is designed for more efficient exploration
REDUX A collection of Python scripts for torsion angle Monte Carlo protein molecular simulations and analysis The program is based on unified residue peptide model and is designed for more efficient exploration
Chapter: 22. Visualization: Making INPUT File and Processing of Output Results
 Chapter: 22 Visualization: Making INPUT File and Processing of Output Results Keywords: visualization, input and output structure, molecular orbital, electron density. In the previous chapters, we have
Chapter: 22 Visualization: Making INPUT File and Processing of Output Results Keywords: visualization, input and output structure, molecular orbital, electron density. In the previous chapters, we have
Lysozyme pka example - Software. APBS! >!Examples! >!pka calculations! >! Lysozyme pka example. Background
 Software Search this site Home Announcements An update on mailing lists APBS 1.2.0 released APBS 1.2.1 released APBS 1.3 released New APBS 1.3 Windows Installer PDB2PQR 1.7.1 released PDB2PQR 1.8 released
Software Search this site Home Announcements An update on mailing lists APBS 1.2.0 released APBS 1.2.1 released APBS 1.3 released New APBS 1.3 Windows Installer PDB2PQR 1.7.1 released PDB2PQR 1.8 released
Application Note. U. Heat of Formation of Ethyl Alcohol and Dimethyl Ether. Introduction
 Application Note U. Introduction The molecular builder (Molecular Builder) is part of the MEDEA standard suite of building tools. This tutorial provides an overview of the Molecular Builder s basic functionality.
Application Note U. Introduction The molecular builder (Molecular Builder) is part of the MEDEA standard suite of building tools. This tutorial provides an overview of the Molecular Builder s basic functionality.
DISCRETE TUTORIAL. Agustí Emperador. Institute for Research in Biomedicine, Barcelona APPLICATION OF DISCRETE TO FLEXIBLE PROTEIN-PROTEIN DOCKING:
 DISCRETE TUTORIAL Agustí Emperador Institute for Research in Biomedicine, Barcelona APPLICATION OF DISCRETE TO FLEXIBLE PROTEIN-PROTEIN DOCKING: STRUCTURAL REFINEMENT OF DOCKING CONFORMATIONS Emperador
DISCRETE TUTORIAL Agustí Emperador Institute for Research in Biomedicine, Barcelona APPLICATION OF DISCRETE TO FLEXIBLE PROTEIN-PROTEIN DOCKING: STRUCTURAL REFINEMENT OF DOCKING CONFORMATIONS Emperador
SUPPLEMENTARY INFORMATION
 DOI: 10.1038/NCHEM.1397 Crystal structures of Λ-[Ru(phen) 2 dppz] 2+ with oligonucleotides containing TA/TA and AT/AT steps show two intercalation modes Hakan Niyazi a, 1 James P. Hall a, Kyra O Sullivan
DOI: 10.1038/NCHEM.1397 Crystal structures of Λ-[Ru(phen) 2 dppz] 2+ with oligonucleotides containing TA/TA and AT/AT steps show two intercalation modes Hakan Niyazi a, 1 James P. Hall a, Kyra O Sullivan
Charge density refinement at ultra high resolution with MoPro software. Christian Jelsch CNRS Université de Lorraine
 Charge density refinement at ultra high resolution with MoPro software Christian Jelsch CNRS Université de Lorraine Laboratoire de Cristallographie & Résonance Magnétique & Modélisations (CRM2) Nancy,
Charge density refinement at ultra high resolution with MoPro software Christian Jelsch CNRS Université de Lorraine Laboratoire de Cristallographie & Résonance Magnétique & Modélisations (CRM2) Nancy,
Integrated Cheminformatics to Guide Drug Discovery
 Integrated Cheminformatics to Guide Drug Discovery Matthew Segall, Ed Champness, Peter Hunt, Tamsin Mansley CINF Drug Discovery Cheminformatics Approaches August 23 rd 2017 Optibrium, StarDrop, Auto-Modeller,
Integrated Cheminformatics to Guide Drug Discovery Matthew Segall, Ed Champness, Peter Hunt, Tamsin Mansley CINF Drug Discovery Cheminformatics Approaches August 23 rd 2017 Optibrium, StarDrop, Auto-Modeller,
ANALYZE. A Program for Cluster Analysis and Characterization of Conformational Ensembles of Polypeptides
 ANALYZE A Program for Cluster Analysis and Characterization of Conformational Ensembles of Polypeptides Calculations of conformational characteristics, such as hydrogen bonds, turn position and types,
ANALYZE A Program for Cluster Analysis and Characterization of Conformational Ensembles of Polypeptides Calculations of conformational characteristics, such as hydrogen bonds, turn position and types,
shelxl: Refinement of Macromolecular Structures from Neutron Data
 ESS Neutron Protein Crystallography 2013 Aarhus, Denmark shelxl: Refinement of Macromolecular Structures from Neutron Data Tim Grüne University of Göttingen Dept. of Structural Chemistry http://shelx.uni-ac.gwdg.de
ESS Neutron Protein Crystallography 2013 Aarhus, Denmark shelxl: Refinement of Macromolecular Structures from Neutron Data Tim Grüne University of Göttingen Dept. of Structural Chemistry http://shelx.uni-ac.gwdg.de
Pymol Practial Guide
 Pymol Practial Guide Pymol is a powerful visualizor very convenient to work with protein molecules. Its interface may seem complex at first, but you will see that with a little practice is simple and powerful
Pymol Practial Guide Pymol is a powerful visualizor very convenient to work with protein molecules. Its interface may seem complex at first, but you will see that with a little practice is simple and powerful
Protein Data Bank Changes Guide. New Changes in Version 3.20 September 15, 2008
 New Records in 3.2 Page 1 Protein Data Bank Changes Guide New Changes in Version 3.20 September 15, 2008 Version 3.20 of the PDB file format introduces a small number of changes and extensions supporting
New Records in 3.2 Page 1 Protein Data Bank Changes Guide New Changes in Version 3.20 September 15, 2008 Version 3.20 of the PDB file format introduces a small number of changes and extensions supporting
PDBe TUTORIAL. PDBePISA (Protein Interfaces, Surfaces and Assemblies)
") PDBe TUTORIAL PDBePISA (Protein Interfaces, Surfaces and Assemblies) http://pdbe.org/pisa/ This tutorial introduces the PDBePISA (PISA for short) service, which is a webbased interactive tool offered by
PDBe TUTORIAL PDBePISA (Protein Interfaces, Surfaces and Assemblies) http://pdbe.org/pisa/ This tutorial introduces the PDBePISA (PISA for short) service, which is a webbased interactive tool offered by
Ensemble refinement of protein crystal structures in PHENIX. Tom Burnley Piet Gros
 Ensemble refinement of protein crystal structures in PHENIX Tom Burnley Piet Gros Incomplete modelling of disorder contributes to R factor gap Only ~5% of residues in the PDB are modelled with more than
Ensemble refinement of protein crystal structures in PHENIX Tom Burnley Piet Gros Incomplete modelling of disorder contributes to R factor gap Only ~5% of residues in the PDB are modelled with more than
ICM-Chemist-Pro How-To Guide. Version 3.6-1h Last Updated 12/29/2009
 ICM-Chemist-Pro How-To Guide Version 3.6-1h Last Updated 12/29/2009 ICM-Chemist-Pro ICM 3D LIGAND EDITOR: SETUP 1. Read in a ligand molecule or PDB file. How to setup the ligand in the ICM 3D Ligand Editor.
ICM-Chemist-Pro How-To Guide Version 3.6-1h Last Updated 12/29/2009 ICM-Chemist-Pro ICM 3D LIGAND EDITOR: SETUP 1. Read in a ligand molecule or PDB file. How to setup the ligand in the ICM 3D Ligand Editor.
The structure of Aquifex aeolicus FtsH in the ADP-bound state reveals a C2-symmetric hexamer
 Volume 71 (2015) Supporting information for article: The structure of Aquifex aeolicus FtsH in the ADP-bound state reveals a C2-symmetric hexamer Marina Vostrukhina, Alexander Popov, Elena Brunstein, Martin
Volume 71 (2015) Supporting information for article: The structure of Aquifex aeolicus FtsH in the ADP-bound state reveals a C2-symmetric hexamer Marina Vostrukhina, Alexander Popov, Elena Brunstein, Martin
Ranking of HIV-protease inhibitors using AutoDock
 Ranking of HIV-protease inhibitors using AutoDock 1. Task Calculate possible binding modes and estimate the binding free energies for 1 3 inhibitors of HIV-protease. You will learn: Some of the theory
Ranking of HIV-protease inhibitors using AutoDock 1. Task Calculate possible binding modes and estimate the binding free energies for 1 3 inhibitors of HIV-protease. You will learn: Some of the theory
BioSolveIT. A Combinatorial Approach for Handling of Protonation and Tautomer Ambiguities in Docking Experiments
 BioSolveIT Biology Problems Solved using Information Technology A Combinatorial Approach for andling of Protonation and Tautomer Ambiguities in Docking Experiments Ingo Dramburg BioSolve IT Gmb An der
BioSolveIT Biology Problems Solved using Information Technology A Combinatorial Approach for andling of Protonation and Tautomer Ambiguities in Docking Experiments Ingo Dramburg BioSolve IT Gmb An der