11/18/2010. Pairwise sequence. Copyright notice. November 22, Announcements. Outline: pairwise alignment. Learning objectives
|
|
|
- Daniella Scott
- 6 years ago
- Views:
Transcription
. Copyright 2009 by John Wiley & Sons, Inc.")

1 Copyright notice November 22, 2010 Pairwise sequence alignment Jonathan Pevsner, Ph.D. Bioinformatics Johns Hopkins School Med. Many of the images in this powerpoint presentation are from Bioinformatics and Functional Genomics by Jonathan Pevsner (ISBN ). Copyright 2009 by John Wiley & Sons, Inc. These images and materials may not be used without permission from the publisher. We welcome instructors to use these powerpoints for educational purposes, but please acknowledge the source. The book has a homepage at including hyperlinks to the book chapters. nnouncements The moodle quiz from lecture 1 is due one week later by today at noon. fter then the quiz closes and won t be available to you. The quiz from today s lecture ( opens at 10:30 am) is due in one week later at noon. Because of the Thanksgiving break, I m extending the deadline a day to Tuesday November 30 (5:00 pm). Outline: pairwise alignment Overview and examples Definitions: homologs, paralogs, orthologs ssigning g scores to aligned amino acids: Dayhoff s PM matrices lignment algorithms: Needleman-Wunsch, Smith-Waterman Learning objectives Pairwise alignments in the 1950s Define homologs, paralogs, orthologs Perform pairwise alignments (NCBI BLST) Understand how scores are assigned to aligned amino acids using Dayhoff s PM matrices xplain how the Needleman-Wunsch algorithm performs global pairwise alignments -corticotropin (sheep) Corticotropin (pig) Oxytocin Vasopressin ala gly glu asp asp glu asp gly ala glu asp glu CYINCPLG CYFNCPG Page 46 1

 It is used in the analysis of genomes Page 47 Pairwise alignment: protein sequences can be more informative than DN protein is more informative (20 vs 4")


2 globins: myoglobin arly example of sequence alignment: globins (1961) Pairwise sequence alignment is the most fundamental operation of bioinformatics It is used to decide if two proteins (or genes) are related structurally or functionally It is used to identify domains or motifs that are shared between proteins H.C. Watson and J.C. Kendrew, Comparison Between the mino-cid Sequences of Sperm Whale Myoglobin and of Human Hæmoglobin. Nature 190: , It is the basis of BLST searching (next week) It is used in the analysis of genomes Page 47 Pairwise alignment: protein sequences can be more informative than DN protein is more informative (20 vs 4 characters); many amino acids share related biophysical properties codons are degenerate: changes in the third position often do not alter the amino acid that is specified protein sequences offer a longer look-back time DN sequences can be translated into protein, and then used in pairwise alignments Page 54 Pairwise alignment: protein sequences can be more informative than DN Many times, DN alignments are appropriate --to confirm the identity of a cdn --to study noncoding regions of DN --to study DN polymorphisms --example: Neanderthal vs modern human DN uery: 181 catcaactacaactccaaagacacccttacacccactaggatatcaacaaacctacccac 240 Sbjct: 189 catcaactgcaaccccaaagccacccct-cacccactaggatatcaacaaacctacccac 247 Outline: pairwise alignment Overview and examples Definitions: homologs, paralogs, orthologs ssigning g scores to aligned amino acids: Dayhoff s PM matrices lignment algorithms: Needleman-Wunsch, Smith-Waterman 2
 for the purpose of assessing the degree of similarity and the possibility of homology.")
 2HHB myoglobin (NP_005359) 2MM1 Orthologs Homologous sequences in different species that arose")
 family in various organisms.")
3 Definition: pairwise alignment Definition: homology Pairwise alignment The process of lining up two sequences to achieve maximal levels of identity (and conservation, in the case of amino acid sequences) for the purpose of assessing the degree of similarity and the possibility of homology. Homology Similarity attributed to descent from a common ancestor. Page 53 Page 49 Definitions: two types of homology Beta globin (NP_000509) 2HHB myoglobin (NP_005359) 2MM1 Orthologs Homologous sequences in different species that arose from a common ancestral gene during speciation; may or may not be responsible for a similar il function. Paralogs Homologous sequences within a single species that arose by gene duplication. Page 49 Page 49 Orthologs: members of a gene (protein) family in various organisms. This tree shows globin orthologs. Paralogs: members of a gene (protein) family within a species. This tree shows human globin paralogs. You can view these sequences at (document 3.1) Page 51 Page 52 3






 and click BLST.")
4 Orthologs and paralogs are often viewed in a single tree General approach to pairwise alignment Choose two sequences Select an algorithm that generates a score llow gaps (insertions, deletions) Score reflects degree of similarity il it lignments can be global or local stimate probability that the alignment occurred by chance Source: NCBI Calculation of an alignment score Find BLST from the home page of NCBI and select protein BLST Source: Page 52 Choose align two or more sequences nter the two sequences (as accession numbers or in the fasta format) and click BLST. Optionally select lgorithm parameters and note the matrix option. Page 52 4

5 Pairwise alignment result of human beta globin and myoglobin Myoglobin efseq Information about this alignment: score, expect value, identities, positives, gaps Pairwise alignment result of human beta globin and myoglobin: the score is a sum of match, mismatch, gap creation, and gap extension scores uery = HBB Subject = MB Middle row displays identities; + sign for similar matches Page 53 Page 53 Pairwise alignment result of human beta globin and myoglobin: the score is a sum of match, mismatch, gap creation, and gap extension scores Definitions: homology Homology Similarity attributed to descent from a common ancestor. V matching V earns +4 T matching L earns -1 These scores come from a scoring matrix! Page 53 Page 50 Definitions: identity, similarity, conservation Identity The extent to which two (nucleotide or amino acid) sequences are invariant. Similarity The extent to which nucleotide or protein sequences are related. It is based upon identity plus conservation. Definition: pairwise alignment Pairwise alignment The process of lining up two sequences to achieve maximal levels of identity (and conservation, for amino acid sequences) for the purpose of assessing the degree of similarity and the possibility of homology. Conservation Changes at a specific position of an amino acid or (less commonly, DN) sequence that preserve the physico-chemical properties of the original residue. Page 51 Page 53 5
6 Mind the gaps Gaps First gap position scores -11 Second gap position scores -1 Gap creation tends to have a large negative score; Gap extension involves a small penalty Page 55 Positions at which a letter is paired with a null are called gaps. Gap scores are typically negative. Since a single mutational event may cause the insertion i or deletion of more than one residue, the presence of a gap is ascribed more significance than the length of the gap. Thus there are separate penalties for gap creation and gap extension. In BLST, it is rarely necessary to change gap values from the default. Pairwise alignment of retinol-binding protein and -lactoglobulin: xample of an alignment with internal, terminal gaps 1 MKWVWLLLLWDCVSSFVKNFDKFSGTWYMKKDPG 50 BP..... :..: : 1...MKCLLLLLTCGLIVT..TMKGLDIKVGTWYSLMSD. 44 lactoglobulin 51 LFLDNIVFSVDTGMSTKGV.LLNNWD..VCDMVGTFTDT 97 BP : ::.. :. 45 ISLLDSPLV.YVLKPTPGDLILLKWNGCKKIIKTK 93 lactoglobulin 98 DPKFKMKYWGVSFLKGNDDHWIVDTDYDTYV...YSC 136 BP. : IPVFKIDLNNKVL...VLDTDYKKYLLFCMNSPSLC 135 lactoglobulin 137 LLNLDGTCDSYSFVFSDPNGLPPKIV.LCLYLIV 185 BP. :. 136 CLVTPVDDLKFDKLKLPMHILSFNPTLCHI lactoglobulin Pairwise alignment of retinol-binding protein from human (top) and rainbow trout (O. mykiss): xample of an alignment with few gaps 1.MKWVWLLLL.WDCVSSFVKNFDKFSGTWYMKKDP 48 ::.... : :. :.. 1 MLICVLCLTCW...DCVSNIVMNFDSYTGWYVKKDP GLFLDNIVFSVDTGMSTKGVLLNNWDVCDMVGTFTDTD 98 : :... : :.. 48 VGLFLLDNVVFSVDSGKMTTHGVIILNNWMCNMFGTFDTPD PKFKMKYWGVSFLKGNDDHWIVDTDYDTYVYSCLLNLDGTCDS 148 : : :: :.. 98 PKFKMYWGSYLTGNDDHWVIDTDYDNYIHYSCVDLDGTCLDG YSFVFSDPNGLPPKIVLCLYLIVHNGYCDGSNLL 199 : :.. :. : : : 148 YSFIFSHPTGLPDKIVTDKKKICFLGKYVGHTGFCSS Pairwise sequence alignment allows us to look back billions of years ago (BY) Multiple sequence alignment of glyceraldehyde 3-phosphate dehydrogenases: example of extremely high conservation Origin of life arliest fossils Origin of eukaryotes ukaryote/ archaea Fungi/animal Plant/animal insects fly GKKVIISP SD.PM..F VCGVNLDYK PDMKVVSNS CTTNCLPL human GKVIISP SD.PM..F VMGVNHKYD NSLKIISNS CTTNCLPL plant GKKVIISP SD.PM..F VVGVNHTY PNMDIVSNS CTTNCLPL bacterium GKKVVMTGP SKDNTPM..F VKGNFDKY. GDIVSNS CTTNCLPL yeast GKKVVITP SS.TPM..F VMGVNKYT SDLKIVSNS CTTNCLPL archaeon GDKVLISP PKGDPVKL VYGVNHDYD G.DVVSNS CTTNSITPV When you do a pairwise alignment of homologous human and plant proteins, you are studying sequences that last shared a common ancestor 1.5 billion years ago! Page 56 fly KVINDNFIV GLMTTVHT TTKTVDGP SGKLWDGG NIIPST human KVIHDNFGIV GLMTTVHI TTKTVDGP SGKLWDGG LNIIPST plant KVVHFGIL GLMTTVHT TTKTVDGP SMKDWGGG SNIIPSST bacterium KVINDNFGII GLMTTVHT TTKTVDGP SHKDWGGG SNIIPSST yeast KVINDFGI GLMTTVHSL TTKTVDGP SHKDWGGT SGNIIPSST archaeon KVLDFGIN GLTTVHY TGSNLMDGP NGKP. NIIPTST fly GKVGKVI PLNGKLTGM FVPTPNVS VVDLTVLGK GSYDIKK human GKVGKVI PLNGKLTGM FVPTNVS VVDLTCLK PKYDDIKKV plant GKVGKVL PLNGKLTGM FVPTSNVS VVDLTCLK GSYDVK bacterium GKVGKVL PLNGKLTGM FVPTPNVS VVDLTVLK TYIK yeast GKVGKVL PLGKLTGM FVPTVDVS VVDLTVKLNK TTYDIKKV archaeon GTVL PLGKLDGM IVPVPNGS ITFVVDLDD DVTSDVN Page 57 6
7 Outline: pairwise alignment Overview and examples Definitions: homologs, paralogs, orthologs ssigning g scores to aligned amino acids: Dayhoff s PM matrices lignment algorithms: Needleman-Wunsch, Smith-Waterman lys found at 58% of arg sites mile Zuckerkandl and Linus Pauling (1965) considered substitution frequencies in 18 globins (myoglobins and hemoglobins from human to lamprey). Black: identity Gray: very conservative substitutions (>40% occurrence) White: fairly conservative substitutions (>21% occurrence) ed: no substitutions observed Page N D C Where we re heading: to a PM250 log odds scoring matrix that assigns scores and is forgiving of mismatches (such as +17 for W to W G or -5 for W to T) H I L K M F P S T W Y V N D C G H I L K M F P S T W Y V Page 93 Page N D C G H I and to a whole series of scoring matrices such as PM10 that are strict and do not tolerate mismatches (such as +13 for W to W or -19 for W to T) L K M F P S T W Y V N D C G H I L K M F P S T W Y V Page 69 Dayhoff s 34 protein superfamilies Protein PMs per 100 million years Ig kappa chain 37 Kappa casein 33 luteinizing hormone b 30 lactalbumin 27 complement component 3 27 epidermal growth factor 26 proopiomelanocortin 21 pancreatic ribonuclease 21 haptoglobin alpha 20 serum albumin 19 phospholipase 2, group IB 19 prolactin 17 carbonic anhydrase C 16 Hemoglobin 12 Hemoglobin 12 Page 59 7
8 Dayhoff s 34 protein superfamilies Dayhoff s 34 protein superfamilies Protein PMs per 100 million years Protein PMs per 100 million years Ig kappa chain 37 Kappa casein 33 luteinizing hormone b 30 lactalbumin 27 complement component 3 27 epidermal growth factor 26 proopiomelanocortin 21 pancreatic ribonuclease 21 haptoglobin human (NP_005203) alpha versus mouse (NP_031812) 20 serum albumin 19 phospholipase 2, group IB 19 prolactin 17 carbonic anhydrase C 16 Hemoglobin 12 Hemoglobin 12 apolipoprotein -II 10 lysozyme 9.8 gastrin 9.8 myoglobin 8.9 nerve growth factor 8.5 myelin basic protein 7.4 thyroid stimulating hormone b 7.4 parathyroid hormone 7.3 parvalbumin 7.0 trypsin 5.9 insulin 4.4 calcitonin 4.3 arginine vasopressin 3.6 adenylate kinase Page 59 Dayhoff s 34 protein superfamilies Pairwise alignment of human (NP_005203) versus mouse (NP_031812) ubiquitin Protein PMs per 100 million years triosephosphate isomerase vasoactive intestinal peptide 2.6 glyceraldehyde phosph. dehydrogease 2.2 cytochrome c collagen 1.7 troponin C, skeletal muscle 1.5 alpha crystallin B chain 1.5 glucagon 1.2 glutamate dehydrogenase 0.9 histone H2B, member 0.9 ubiquitin 0 Page 59 Dayhoff s approach to assigning scores for any two aligned amino acid residues Dayhoff et al. defined the score of two aligned residues i,j as 10 times the log of how likely it is to observe these two residues (based on the empirical observation of how often they are aligned in nature) divided by the background probability of finding these amino acids by chance. This provides a score for each pair of residues. Page 58 Dayhoff s numbers of accepted point mutations : what amino acid substitutions occur in proteins? la rg N sn D sp C Cys Gln Glu G Gly N D C G H Dayhoff (1978) p.346. Page 61 8
9 Multiple sequence alignment of glyceraldehyde 3-phosphate dehydrogenases: columns of residues may have high or low conservation fly GKKVIISP SD.PM..F VCGVNLDYK PDMKVVSNS CTTNCLPL human GKVIISP SD.PM..F VMGVNHKYD NSLKIISNS CTTNCLPL plant GKKVIISP SD.PM..F VVGVNHTY PNMDIVSNS CTTNCLPL bacterium GKKVVMTGP SKDNTPM..F VKGNFDKY. GDIVSNS CTTNCLPL yeast GKKVVITP SS.TPM..F VMGVNKYT SDLKIVSNS CTTNCLPL archaeon GDKVLISP PKGDPVKL VYGVNHDYD G.DVVSNS CTTNSITPV fly KVINDNFIV GLMTTVHT TTKTVDGP SGKLWDGG NIIPST human KVIHDNFGIV GLMTTVHI TTKTVDGP SGKLWDGG LNIIPST plant KVVHFGIL GLMTTVHT TTKTVDGP SMKDWGGG SNIIPSST bacterium KVINDNFGII GLMTTVHT TTKTVDGP SHKDWGGG SNIIPSST yeast KVINDFGI GLMTTVHSL TTKTVDGP SHKDWGGT SGNIIPSST archaeon KVLDFGIN GLTTVHY TGSNLMDGP NGKP. NIIPTST fly GKVGKVI PLNGKLTGM FVPTPNVS VVDLTVLGK GSYDIKK human GKVGKVI PLNGKLTGM FVPTNVS VVDLTCLK PKYDDIKKV plant GKVGKVL PLNGKLTGM FVPTSNVS VVDLTCLK GSYDVK bacterium GKVGKVL PLNGKLTGM FVPTPNVS VVDLTVLK TYIK yeast GKVGKVL PLGKLTGM FVPTVDVS VVDLTVKLNK TTYDIKKV archaeon GTVL PLGKLDGM IVPVPNGS ITFVVDLDD DVTSDVN The relative mutability of amino acids sn 134 His 66 Ser 120 rg 65 sp 106 Lys 56 Glu 102 Pro 56 la 100 Gly 49 Thr 97 Tyr 41 Ile 96 Phe 41 Met 94 Leu 40 Gln 93 Cys 20 Val 74 Trp 18 Page 57 Page 63 Normalized frequencies of amino acids Gly 8.9% rg 4.1% la 8.7% sn 4.0% Leu 8.5% Phe 4.0% Lys 8.1% Gln 3.8% Ser 7.0% Ile 3.7% Val 6.5% His 3.4% Thr 5.8% Cys 3.3% Pro 5.1% Tyr 3.0% Glu 5.0% Met 1.5% sp 4.7% Trp 1.0% blue=6 codons; red=1 codon These frequencies f i sum to 1 Page 63 Dayhoff s numbers of accepted point mutations : what amino acid substitutions occur in proteins? la rg N sn D sp C Cys Gln Glu G Gly N D C G H Page 61 Dayhoff s PM1 mutation probability matrix la rg N sn D sp N D Original amino acid C Cys Gln Glu G H Gly His C G H I Page 66 N D Dayhoff s PM1 mutation probability matrix la rg N sn D sp C Cys Gln Glu G Gly H His C G H I ach element of the matrix shows the probability that an original amino acid (top) will be replaced by another amino acid (side) 9
10 Substitution Matrix substitution matrix contains values proportional to the probability that amino acid i mutates into amino acid j for all pairs of amino acids. Substitution matrices are constructed by assembling a large and diverse sample of verified pairwise alignments (or multiple sequence alignments) of amino acids. Substitution matrices should reflect the true probabilities of mutations occurring through a period of evolution. The two major types of substitution matrices are PM and BLOSUM. PM matrices: Point-accepted mutations PM matrices are based on global alignments of closely related proteins. The PM1 is the matrix calculated from comparisons of sequences with no more than 1% divergence. t an evolutionary interval of PM1, one change has occurred over a length of 100 amino acids. Other PM matrices are extrapolated from PM1. For PM250, 250 changes have occurred for two proteins over a length of 100 amino acids. ll the PM data come from closely related proteins (>85% amino acid identity). Page 63 Dayhoff s PM1 mutation probability matrix la rg N sn D sp N D C Cys Gln Glu G H Gly His C G H I Page 66 Dayhoff s PM0 mutation probability matrix: the rules for extremely slowly evolving proteins PM0 la rg N sn D sp C Cys Gln Glu 100% 0% 0% 0% 0% 0% 0% 0% 100% 0% 0% 0% 0% 0% N 0% 0% 100% 0% 0% 0% 0% D 0% 0% 0% 100% 0% 0% 0% C 0% 0% 0% 0% 100% 0% 0% 0% 0% 0% 0% 0% 100% 0% 0% 0% 0% 0% 0% 0% 100% G 0% 0% 0% 0% 0% 0% 0% Top: original amino acid Side: replacement amino acid Page 68 Dayhoff s PM2000 mutation probability matrix: the rules for very distantly related proteins PM la rg N sn D sp C Cys Gln Glu G Gly 8.7% 8.7% 8.7% 8.7% 8.7% 8.7% 8.7% 8.7% 4.1% 4.1% 4.1% 4.1% 4.1% 4.1% 4.1% 4.1% N 4.0% 4.0% 4.0% 4.0% 4.0% 4.0% 4.0% 4.0% D 4.7% 4.7% 4.7% 4.7% 4.7% 4.7% 4.7% 4.7% C 3.3% 3.3% 3.3% 3.3% 3.3% 3.3% 3.3% 3.3% 3.8% 3.8% 3.8% 3.8% 3.8% 3.8% 3.8% 3.8% 5.0% 5.0% 5.0% 5.0% 5.0% 5.0% 5.0% 5.0% G 8.9% 8.9% 8.9% 8.9% 8.9% 8.9% 8.9% 8.9% Top: original amino acid Side: replacement amino acid Page 68 PM250 mutation probability matrix N D C G H I L K M F P S T W Y V N D C G H I L K M F P S T W Y V Top: original amino acid Side: replacement amino acid Page 68 10
11 2-2 6 N D C G H I L PM250 log odds scoring matrix K M F P S T W Y V N D C G H I L K M F P S T W Y V Why do we go from a mutation probability matrix to a log odds matrix? We want a scoring matrix so that when we do a pairwise alignment (or a BLST search) we know what score to assign to two aligned amino acid residues. Logarithms are easier to use for a scoring system. They allow us to sum the scores of aligned residues (rather than having to multiply them). Page 69 Page 69 How do we go from a mutation probability matrix to a log odds matrix? What do the numbers mean in a log odds matrix? The cells in a log odds matrix consist of an odds ratio : the probability that an alignment is authentic the probability that the alignment was random The score S for an alignment of residues a,b is given by: S(a,b) = 10 log 10 (M ab /p b ) s an example, for tryptophan, score of +2 indicates that the amino acid replacement occurs 1.6 times as frequently as expected by chance. score of 0 is neutral. score of 10 indicates that the correspondence of two amino acids in an alignment that accurately represents homology (evolutionary descent) is one tenth as frequent as the chance alignment of these amino acids. S(trp,trp) = 10 log 10 (0.55/0.010) = 17.4 Page 69 Page 58 two nearly identical proteins two distantly related proteins More conserved at versus mouse globin Less conserved at versus bacterial globin page 72 11
12 BLOSUM Matrices BLOSUM Matrices BLOSUM matrices are based on local alignments. BLOSUM stands for blocks substitution matrix. BLOSUM62 is a matrix calculated from comparisons of sequences with no less than 62% divergence. identity Percent amino acid Page 70 BLOSUM62 BLOSUM Matrices BLOSUM Matrices identity Percent amino acid BLOSUM80 BLOSUM BLOSUM30 ll BLOSUM matrices are based on observed alignments; they are not extrapolated from comparisons of closely related proteins. The BLOCKS database contains thousands of groups of multiple sequence alignments. BLOSUM62 is the default matrix in BLST 2.0. Though it is tailored for comparisons of moderately distant proteins, it performs well in detecting closer relationships. search for distant relatives may be more sensitive with a different matrix. Page N D C G H I L K M F P S T W Y V N D C G H I L K M F P S T W Y V Blosum62 scoring matrix Page 73 PM matrices: Point-accepted mutations PM matrices are based on global alignments of closely related proteins. The PM1 is the matrix calculated from comparisons of sequences with no more than 1% divergence. t an evolutionary interval of PM1, one change has occurred over a length of 100 amino acids. Other PM matrices are extrapolated from PM1. For PM250, 250 changes have occurred for two proteins over a length of 100 amino acids. ll the PM data come from closely related proteins (>85% amino acid identity). Page 74 12
13 Two randomly diverging protein sequences change in a negatively exponential fashion t PM1, two proteins are 99% identical t PM10.7, there are 10 differences per 100 residues t PM80, there are 50 differences per 100 residues t PM250, there are 80 differences per 100 residues Percent t identity entity twilight zone Percent id twilight zone volutionary distance in PMs Differences per 100 residues Page 74 Page 75 PM: ccepted point mutation Outline: pairwise alignment Two proteins with 50% identity may have 80 changes per 100 residues. (Why? Because any residue can be subject to back mutations.) Proteins with 20% to 25% identity are in the twilight zone and may be statistically significantly related. PM or accepted point mutation refers to the hits or matches between two sequences (Dayhoff & ck, 1968) Overview and examples Definitions: homologs, paralogs, orthologs ssigning g scores to aligned amino acids: Dayhoff s PM matrices lignment algorithms: Needleman-Wunsch, Smith-Waterman Page 75 Two kinds of sequence alignment: global and local We will first consider the global alignment algorithm of Needleman and Wunsch (1970). We will then explore the local alignment algorithm of Smith and Waterman (1981). Finally, we will consider BLST, a heuristic version of Smith-Waterman. We will cover BLST in detail on Monday. Global alignment with the algorithm of Needleman and Wunsch (1970) Two sequences can be compared in a matrix along x- and y-axes. If they are identical, a path along a diagonal can be drawn Find the optimal subpaths, and add them up to achieve the best score. This involves --adding gaps when needed --allowing for conservative substitutions --choosing a scoring system (simple or complicated) Page 76 N-W is guaranteed to find optimal alignment(s) Page 76 13


![up a matrix [2] score the matrix](/docs-images/76/74095409/images/14-3.jpg "[3] identify the optimal")
![alignment(s) 2 1 [1] identity](/docs-images/76/74095409/images/14-5.jpg "(stay along a diagonal) [2]")

 [4] gap in the other sequence")
 Page 76 Page 77 Start")

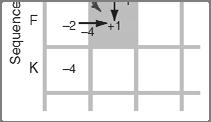
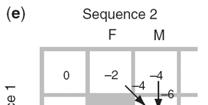
14 Three steps to global alignment with the Needleman-Wunsch algorithm Four possible outcomes in aligning two sequences [1] set up a matrix [2] score the matrix [3] identify the optimal alignment(s) 2 1 [1] identity (stay along a diagonal) [2] mismatch (stay along a diagonal) [3] gap in one sequence (move vertically!) [4] gap in the other sequence (move horizontally!) Page 76 Page 77 Start Needleman-Wunsch with an identity matrix Page 77 Page 77 Start Needleman-Wunsch with an identity matrix Fill in the matrix using dynamic programming Page 77 Page 78 14



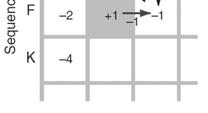
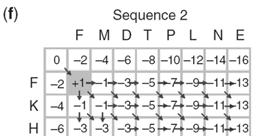
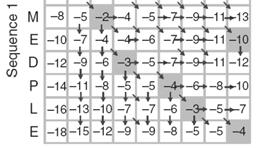
15 Fill in the matrix using dynamic programming Fill in the matrix using dynamic programming Page 78 Page 78 Fill in the matrix using dynamic programming Fill in the matrix using dynamic programming Page 78 Page 78 Fill in the matrix using dynamic programming Fill in the matrix using dynamic programming Page 78 Page 78 15



 alpha globin (NP_000549) Page 81 Global alignment versus local alignment")
.")
16 Traceback to find the optimal (best) pairwise alignment Needleman-Wunsch: dynamic programming N-W is guaranteed to find optimal alignments, although the algorithm does not search all possible alignments. It is an example of a dynamic programming algorithm: an optimal path (alignment) is identified by incrementally extending optimal subpaths. Thus, a series of decisions is made at each step of the alignment to find the pair of residues with the best score. Page 79 Page 80 Try using needle to implement a Needleman- Wunsch global alignment algorithm to find the optimum alignment (including gaps): ueries: beta globin (NP_000509) alpha globin (NP_000549) Page 81 Global alignment versus local alignment Global alignment (Needleman-Wunsch) extends from one end of each sequence to the other. Local alignment finds optimally matching regions within two sequences ( subsequences ). Local alignment is almost always used for database searches such as BLST. It is useful to find domains (or limited regions of homology) within sequences. Smith and Waterman (1981) solved the problem of performing optimal local sequence alignment. Other methods (BLST, FST) are faster but less thorough. Page 82 16
 15% identity No values in the scoring matrix can be negative!")
![S > 0 The score in each cell is the maximum of four values: [1] s(i-1, j-1) + the new score at [i,j] (a match or](/docs-images/76/74095409/images/17-3.jpg "mismatch) [2] s(i,j-1) gap penalty [3] s(i-1,j) gap penalty [4] zero 30% identity NP_824492, NP_337032 page 81 Page 82")


.")
17 Global alignment (top) includes matches ignored by local alignment (bottom) How the Smith-Waterman algorithm works Set up a matrix between two proteins (size m+1, n+1) 15% identity No values in the scoring matrix can be negative! S > 0 The score in each cell is the maximum of four values: [1] s(i-1, j-1) + the new score at [i,j] (a match or mismatch) [2] s(i,j-1) gap penalty [3] s(i-1,j) gap penalty [4] zero 30% identity NP_824492, NP_ page 81 Page 82 Smith-Waterman algorithm allows the alignment of subsets of sequences Try using SSCH to perform a rigorous Smith- Waterman local alignment: Page 83 ueries: beta globin (NP_000509) alpha globin (NP_000549) apid, heuristic versions of Smith-Waterman: FST and BLST Smith-Waterman is very rigorous and it is guaranteed to find an optimal alignment. But Smith-Waterman is slow. It requires computer space and time proportional to the product of the two sequences being aligned (or the product of a query against an entire database). Gotoh (1982) and Myers and Miller (1988) improved the algorithms so both global and local alignment require less time and space. FST and BLST provide rapid alternatives to S-W. Page 84 17
.")
.")
 with itself. The output includes a dot plot.")


18 Statistical significance of pairwise alignment Pairwise alignments with dot plots: graphical displays of relatedness with NCBI s BLST We will discuss the statistical significance of alignment scores in the next lecture (BLST). basic question is how to determine whether a particular alignment score is likely l to have occurred by chance. ccording to the null hypothesis, two aligned sequences are not homologous (evolutionarily related). Can we reject the null hypothesis at a particular significance level alpha? [1] Compare human cytoglobin (NP_599030, length 190 amino acids) with itself. The output includes a dot plot. The data points showing amino acid identities appear as a diagonal line. [2] Compare cytoglobin with a globin from the snail Biomphalaria glabrata (accession CJ44466, length 2,148 amino acids. See lots of repeated regions! Page 85 Pairwise alignments with dot plots: cytoglobin versus itself yields a straight line Pairwise alignments with dot plots: cytoglobin versus itself (but with 15 amino acids deleted from one copy) Page 85 Page 85 Pairwise alignments with dot plots: cytoglobin versus a snail globin Next in the course... Take the quiz (on pairwise alignment), due in a week (because of the Thanksgiving break, it s due TUSDY at 5 pm). Next Monday: BLST Page 85 18
Biochemistry 324 Bioinformatics. Pairwise sequence alignment
 Biochemistry 324 Bioinformatics Pairwise sequence alignment How do we compare genes/proteins? When we have sequenced a genome, we try and identify the function of unknown genes by finding a similar gene
Biochemistry 324 Bioinformatics Pairwise sequence alignment How do we compare genes/proteins? When we have sequenced a genome, we try and identify the function of unknown genes by finding a similar gene
Sequence Alignments. Dynamic programming approaches, scoring, and significance. Lucy Skrabanek ICB, WMC January 31, 2013
 Sequence Alignments Dynamic programming approaches, scoring, and significance Lucy Skrabanek ICB, WMC January 31, 213 Sequence alignment Compare two (or more) sequences to: Find regions of conservation
Sequence Alignments Dynamic programming approaches, scoring, and significance Lucy Skrabanek ICB, WMC January 31, 213 Sequence alignment Compare two (or more) sequences to: Find regions of conservation
Algorithms in Bioinformatics FOUR Pairwise Sequence Alignment. Pairwise Sequence Alignment. Convention: DNA Sequences 5. Sequence Alignment
 Algorithms in Bioinformatics FOUR Sami Khuri Department of Computer Science San José State University Pairwise Sequence Alignment Homology Similarity Global string alignment Local string alignment Dot
Algorithms in Bioinformatics FOUR Sami Khuri Department of Computer Science San José State University Pairwise Sequence Alignment Homology Similarity Global string alignment Local string alignment Dot
EECS 730 Introduction to Bioinformatics Sequence Alignment. Luke Huan Electrical Engineering and Computer Science
 EECS 730 Introduction to Bioinformatics Sequence Alignment Luke Huan Electrical Engineering and Computer Science http://people.eecs.ku.edu/~jhuan/ Administrative Term-project In a group of 2 students Think
EECS 730 Introduction to Bioinformatics Sequence Alignment Luke Huan Electrical Engineering and Computer Science http://people.eecs.ku.edu/~jhuan/ Administrative Term-project In a group of 2 students Think
Alignment & BLAST. By: Hadi Mozafari KUMS
 Alignment & BLAST By: Hadi Mozafari KUMS SIMILARITY - ALIGNMENT Comparison of primary DNA or protein sequences to other primary or secondary sequences Expecting that the function of the similar sequence
Alignment & BLAST By: Hadi Mozafari KUMS SIMILARITY - ALIGNMENT Comparison of primary DNA or protein sequences to other primary or secondary sequences Expecting that the function of the similar sequence
3. SEQUENCE ANALYSIS BIOINFORMATICS COURSE MTAT
 3. SEQUENCE ANALYSIS BIOINFORMATICS COURSE MTAT.03.239 25.09.2012 SEQUENCE ANALYSIS IS IMPORTANT FOR... Prediction of function Gene finding the process of identifying the regions of genomic DNA that encode
3. SEQUENCE ANALYSIS BIOINFORMATICS COURSE MTAT.03.239 25.09.2012 SEQUENCE ANALYSIS IS IMPORTANT FOR... Prediction of function Gene finding the process of identifying the regions of genomic DNA that encode
THEORY. Based on sequence Length According to the length of sequence being compared it is of following two types
 Exp 11- THEORY Sequence Alignment is a process of aligning two sequences to achieve maximum levels of identity between them. This help to derive functional, structural and evolutionary relationships between
Exp 11- THEORY Sequence Alignment is a process of aligning two sequences to achieve maximum levels of identity between them. This help to derive functional, structural and evolutionary relationships between
Pairwise & Multiple sequence alignments
 Pairwise & Multiple sequence alignments Urmila Kulkarni-Kale Bioinformatics Centre 411 007 urmila@bioinfo.ernet.in Basis for Sequence comparison Theory of evolution: gene sequences have evolved/derived
Pairwise & Multiple sequence alignments Urmila Kulkarni-Kale Bioinformatics Centre 411 007 urmila@bioinfo.ernet.in Basis for Sequence comparison Theory of evolution: gene sequences have evolved/derived
CISC 889 Bioinformatics (Spring 2004) Sequence pairwise alignment (I)
 Sequence pairwise alignment (I)") CISC 889 Bioinformatics (Spring 2004) Sequence pairwise alignment (I) Contents Alignment algorithms Needleman-Wunsch (global alignment) Smith-Waterman (local alignment) Heuristic algorithms FASTA BLAST
CISC 889 Bioinformatics (Spring 2004) Sequence pairwise alignment (I) Contents Alignment algorithms Needleman-Wunsch (global alignment) Smith-Waterman (local alignment) Heuristic algorithms FASTA BLAST
CONCEPT OF SEQUENCE COMPARISON. Natapol Pornputtapong 18 January 2018
 CONCEPT OF SEQUENCE COMPARISON Natapol Pornputtapong 18 January 2018 SEQUENCE ANALYSIS - A ROSETTA STONE OF LIFE Sequence analysis is the process of subjecting a DNA, RNA or peptide sequence to any of
CONCEPT OF SEQUENCE COMPARISON Natapol Pornputtapong 18 January 2018 SEQUENCE ANALYSIS - A ROSETTA STONE OF LIFE Sequence analysis is the process of subjecting a DNA, RNA or peptide sequence to any of
Sequence analysis and comparison
 The aim with sequence identification: Sequence analysis and comparison Marjolein Thunnissen Lund September 2012 Is there any known protein sequence that is homologous to mine? Are there any other species
The aim with sequence identification: Sequence analysis and comparison Marjolein Thunnissen Lund September 2012 Is there any known protein sequence that is homologous to mine? Are there any other species
Similarity or Identity? When are molecules similar?
 Similarity or Identity? When are molecules similar? Mapping Identity A -> A T -> T G -> G C -> C or Leu -> Leu Pro -> Pro Arg -> Arg Phe -> Phe etc If we map similarity using identity, how similar are
Similarity or Identity? When are molecules similar? Mapping Identity A -> A T -> T G -> G C -> C or Leu -> Leu Pro -> Pro Arg -> Arg Phe -> Phe etc If we map similarity using identity, how similar are
Massachusetts Institute of Technology Computational Evolutionary Biology, Fall, 2005 Notes for November 7: Molecular evolution
 Massachusetts Institute of Technology 6.877 Computational Evolutionary Biology, Fall, 2005 Notes for November 7: Molecular evolution 1. Rates of amino acid replacement The initial motivation for the neutral
Massachusetts Institute of Technology 6.877 Computational Evolutionary Biology, Fall, 2005 Notes for November 7: Molecular evolution 1. Rates of amino acid replacement The initial motivation for the neutral
Sequence Alignment (chapter 6)
") Sequence lignment (chapter 6) he biological problem lobal alignment Local alignment Multiple alignment Introduction to bioinformatics, utumn 6 Background: comparative genomics Basic question in biology:
Sequence lignment (chapter 6) he biological problem lobal alignment Local alignment Multiple alignment Introduction to bioinformatics, utumn 6 Background: comparative genomics Basic question in biology:
In-Depth Assessment of Local Sequence Alignment
 2012 International Conference on Environment Science and Engieering IPCBEE vol.3 2(2012) (2012)IACSIT Press, Singapoore In-Depth Assessment of Local Sequence Alignment Atoosa Ghahremani and Mahmood A.
2012 International Conference on Environment Science and Engieering IPCBEE vol.3 2(2012) (2012)IACSIT Press, Singapoore In-Depth Assessment of Local Sequence Alignment Atoosa Ghahremani and Mahmood A.
Module: Sequence Alignment Theory and Applications Session: Introduction to Searching and Sequence Alignment
 Module: Sequence Alignment Theory and Applications Session: Introduction to Searching and Sequence Alignment Introduction to Bioinformatics online course : IBT Jonathan Kayondo Learning Objectives Understand
Module: Sequence Alignment Theory and Applications Session: Introduction to Searching and Sequence Alignment Introduction to Bioinformatics online course : IBT Jonathan Kayondo Learning Objectives Understand
Practical considerations of working with sequencing data
 Practical considerations of working with sequencing data File Types Fastq ->aligner -> reference(genome) coordinates Coordinate files SAM/BAM most complete, contains all of the info in fastq and more!
Practical considerations of working with sequencing data File Types Fastq ->aligner -> reference(genome) coordinates Coordinate files SAM/BAM most complete, contains all of the info in fastq and more!
CSE 549: Computational Biology. Substitution Matrices
 CSE 9: Computational Biology Substitution Matrices How should we score alignments So far, we ve looked at arbitrary schemes for scoring mutations. How can we assign scores in a more meaningful way? Are
CSE 9: Computational Biology Substitution Matrices How should we score alignments So far, we ve looked at arbitrary schemes for scoring mutations. How can we assign scores in a more meaningful way? Are
Bioinformatics Exercises
 Bioinformatics Exercises AP Biology Teachers Workshop Susan Cates, Ph.D. Evolution of Species Phylogenetic Trees show the relatedness of organisms Common Ancestor (Root of the tree) 1 Rooted vs. Unrooted
Bioinformatics Exercises AP Biology Teachers Workshop Susan Cates, Ph.D. Evolution of Species Phylogenetic Trees show the relatedness of organisms Common Ancestor (Root of the tree) 1 Rooted vs. Unrooted
Algorithms in Bioinformatics
 Algorithms in Bioinformatics Sami Khuri Department of omputer Science San José State University San José, alifornia, USA khuri@cs.sjsu.edu www.cs.sjsu.edu/faculty/khuri Pairwise Sequence Alignment Homology
Algorithms in Bioinformatics Sami Khuri Department of omputer Science San José State University San José, alifornia, USA khuri@cs.sjsu.edu www.cs.sjsu.edu/faculty/khuri Pairwise Sequence Alignment Homology
Sequence Alignment: A General Overview. COMP Fall 2010 Luay Nakhleh, Rice University
 Sequence Alignment: A General Overview COMP 571 - Fall 2010 Luay Nakhleh, Rice University Life through Evolution All living organisms are related to each other through evolution This means: any pair of
Sequence Alignment: A General Overview COMP 571 - Fall 2010 Luay Nakhleh, Rice University Life through Evolution All living organisms are related to each other through evolution This means: any pair of
Sara C. Madeira. Universidade da Beira Interior. (Thanks to Ana Teresa Freitas, IST for useful resources on this subject)
") Bioinformática Sequence Alignment Pairwise Sequence Alignment Universidade da Beira Interior (Thanks to Ana Teresa Freitas, IST for useful resources on this subject) 1 16/3/29 & 23/3/29 27/4/29 Outline
Bioinformática Sequence Alignment Pairwise Sequence Alignment Universidade da Beira Interior (Thanks to Ana Teresa Freitas, IST for useful resources on this subject) 1 16/3/29 & 23/3/29 27/4/29 Outline
Quantifying sequence similarity
 Quantifying sequence similarity Bas E. Dutilh Systems Biology: Bioinformatic Data Analysis Utrecht University, February 16 th 2016 After this lecture, you can define homology, similarity, and identity
Quantifying sequence similarity Bas E. Dutilh Systems Biology: Bioinformatic Data Analysis Utrecht University, February 16 th 2016 After this lecture, you can define homology, similarity, and identity
Pairwise sequence alignments
 Pairwise sequence alignments Volker Flegel VI, October 2003 Page 1 Outline Introduction Definitions Biological context of pairwise alignments Computing of pairwise alignments Some programs VI, October
Pairwise sequence alignments Volker Flegel VI, October 2003 Page 1 Outline Introduction Definitions Biological context of pairwise alignments Computing of pairwise alignments Some programs VI, October
Pairwise sequence alignments. Vassilios Ioannidis (From Volker Flegel )
") Pairwise sequence alignments Vassilios Ioannidis (From Volker Flegel ) Outline Introduction Definitions Biological context of pairwise alignments Computing of pairwise alignments Some programs Importance
Pairwise sequence alignments Vassilios Ioannidis (From Volker Flegel ) Outline Introduction Definitions Biological context of pairwise alignments Computing of pairwise alignments Some programs Importance
Introduction to Bioinformatics
 Introduction to Bioinformatics Lecture : p he biological problem p lobal alignment p Local alignment p Multiple alignment 6 Background: comparative genomics p Basic question in biology: what properties
Introduction to Bioinformatics Lecture : p he biological problem p lobal alignment p Local alignment p Multiple alignment 6 Background: comparative genomics p Basic question in biology: what properties
Bioinformatics (GLOBEX, Summer 2015) Pairwise sequence alignment
 Pairwise sequence alignment") Bioinformatics (GLOBEX, Summer 2015) Pairwise sequence alignment Substitution score matrices, PAM, BLOSUM Needleman-Wunsch algorithm (Global) Smith-Waterman algorithm (Local) BLAST (local, heuristic) E-value
Bioinformatics (GLOBEX, Summer 2015) Pairwise sequence alignment Substitution score matrices, PAM, BLOSUM Needleman-Wunsch algorithm (Global) Smith-Waterman algorithm (Local) BLAST (local, heuristic) E-value
SEQUENCE ALIGNMENT BACKGROUND: BIOINFORMATICS. Prokaryotes and Eukaryotes. DNA and RNA
 SEQUENCE ALIGNMENT BACKGROUND: BIOINFORMATICS 1 Prokaryotes and Eukaryotes 2 DNA and RNA 3 4 Double helix structure Codons Codons are triplets of bases from the RNA sequence. Each triplet defines an amino-acid.
SEQUENCE ALIGNMENT BACKGROUND: BIOINFORMATICS 1 Prokaryotes and Eukaryotes 2 DNA and RNA 3 4 Double helix structure Codons Codons are triplets of bases from the RNA sequence. Each triplet defines an amino-acid.
Bio 1B Lecture Outline (please print and bring along) Fall, 2007
 Fall, 2007") Bio 1B Lecture Outline (please print and bring along) Fall, 2007 B.D. Mishler, Dept. of Integrative Biology 2-6810, bmishler@berkeley.edu Evolution lecture #5 -- Molecular genetics and molecular evolution
Bio 1B Lecture Outline (please print and bring along) Fall, 2007 B.D. Mishler, Dept. of Integrative Biology 2-6810, bmishler@berkeley.edu Evolution lecture #5 -- Molecular genetics and molecular evolution
Chapter 2: Sequence Alignment
 Chapter 2: Sequence lignment 2.2 Scoring Matrices Prof. Yechiam Yemini (YY) Computer Science Department Columbia University COMS4761-2007 Overview PM: scoring based on evolutionary statistics Elementary
Chapter 2: Sequence lignment 2.2 Scoring Matrices Prof. Yechiam Yemini (YY) Computer Science Department Columbia University COMS4761-2007 Overview PM: scoring based on evolutionary statistics Elementary
Computational Biology
 Computational Biology Lecture 6 31 October 2004 1 Overview Scoring matrices (Thanks to Shannon McWeeney) BLAST algorithm Start sequence alignment 2 1 What is a homologous sequence? A homologous sequence,
Computational Biology Lecture 6 31 October 2004 1 Overview Scoring matrices (Thanks to Shannon McWeeney) BLAST algorithm Start sequence alignment 2 1 What is a homologous sequence? A homologous sequence,
Background: comparative genomics. Sequence similarity. Homologs. Similarity vs homology (2) Similarity vs homology. Sequence Alignment (chapter 6)
 Similarity vs homology. Sequence Alignment (chapter 6)") Sequence lignment (chapter ) he biological problem lobal alignment Local alignment Multiple alignment Background: comparative genomics Basic question in biology: what properties are shared among organisms?
Sequence lignment (chapter ) he biological problem lobal alignment Local alignment Multiple alignment Background: comparative genomics Basic question in biology: what properties are shared among organisms?
Sequence analysis and Genomics
 Sequence analysis and Genomics October 12 th November 23 rd 2 PM 5 PM Prof. Peter Stadler Dr. Katja Nowick Katja: group leader TFome and Transcriptome Evolution Bioinformatics group Paul-Flechsig-Institute
Sequence analysis and Genomics October 12 th November 23 rd 2 PM 5 PM Prof. Peter Stadler Dr. Katja Nowick Katja: group leader TFome and Transcriptome Evolution Bioinformatics group Paul-Flechsig-Institute
M.O. Dayhoff, R.M. Schwartz, and B. C, Orcutt
 A Model of volutionary Change in Proteins M.O. Dayhoff, R.M. Schwartz, and B. C, Orcutt n the eight years since we last examined the amino acid exchanges seen in closely related proteins,' the information
A Model of volutionary Change in Proteins M.O. Dayhoff, R.M. Schwartz, and B. C, Orcutt n the eight years since we last examined the amino acid exchanges seen in closely related proteins,' the information
An Introduction to Sequence Similarity ( Homology ) Searching
 Searching") An Introduction to Sequence Similarity ( Homology ) Searching Gary D. Stormo 1 UNIT 3.1 1 Washington University, School of Medicine, St. Louis, Missouri ABSTRACT Homologous sequences usually have the same,
An Introduction to Sequence Similarity ( Homology ) Searching Gary D. Stormo 1 UNIT 3.1 1 Washington University, School of Medicine, St. Louis, Missouri ABSTRACT Homologous sequences usually have the same,
Large-Scale Genomic Surveys
 Bioinformatics Subtopics Fold Recognition Secondary Structure Prediction Docking & Drug Design Protein Geometry Protein Flexibility Homology Modeling Sequence Alignment Structure Classification Gene Prediction
Bioinformatics Subtopics Fold Recognition Secondary Structure Prediction Docking & Drug Design Protein Geometry Protein Flexibility Homology Modeling Sequence Alignment Structure Classification Gene Prediction
Exploring Evolution & Bioinformatics
 Chapter 6 Exploring Evolution & Bioinformatics Jane Goodall The human sequence (red) differs from the chimpanzee sequence (blue) in only one amino acid in a protein chain of 153 residues for myoglobin
Chapter 6 Exploring Evolution & Bioinformatics Jane Goodall The human sequence (red) differs from the chimpanzee sequence (blue) in only one amino acid in a protein chain of 153 residues for myoglobin
Sequence Alignment Techniques and Their Uses
 Sequence Alignment Techniques and Their Uses Sarah Fiorentino Since rapid sequencing technology and whole genomes sequencing, the amount of sequence information has grown exponentially. With all of this
Sequence Alignment Techniques and Their Uses Sarah Fiorentino Since rapid sequencing technology and whole genomes sequencing, the amount of sequence information has grown exponentially. With all of this
Single alignment: Substitution Matrix. 16 march 2017
 Single alignment: Substitution Matrix 16 march 2017 BLOSUM Matrix BLOSUM Matrix [2] (Blocks Amino Acid Substitution Matrices ) It is based on the amino acids substitutions observed in ~2000 conserved block
Single alignment: Substitution Matrix 16 march 2017 BLOSUM Matrix BLOSUM Matrix [2] (Blocks Amino Acid Substitution Matrices ) It is based on the amino acids substitutions observed in ~2000 conserved block
Introduction to Comparative Protein Modeling. Chapter 4 Part I
 Introduction to Comparative Protein Modeling Chapter 4 Part I 1 Information on Proteins Each modeling study depends on the quality of the known experimental data. Basis of the model Search in the literature
Introduction to Comparative Protein Modeling Chapter 4 Part I 1 Information on Proteins Each modeling study depends on the quality of the known experimental data. Basis of the model Search in the literature
METHODS FOR DETERMINING PHYLOGENY. In Chapter 11, we discovered that classifying organisms into groups was, and still is, a difficult task.
 Chapter 12 (Strikberger) Molecular Phylogenies and Evolution METHODS FOR DETERMINING PHYLOGENY In Chapter 11, we discovered that classifying organisms into groups was, and still is, a difficult task. Modern
Chapter 12 (Strikberger) Molecular Phylogenies and Evolution METHODS FOR DETERMINING PHYLOGENY In Chapter 11, we discovered that classifying organisms into groups was, and still is, a difficult task. Modern
8 Grundlagen der Bioinformatik, SoSe 11, D. Huson, April 18, 2011
 8 Grundlagen der Bioinformatik, SoSe 11, D. Huson, April 18, 2011 2 Pairwise alignment We will discuss: 1. Strings 2. Dot matrix method for comparing sequences 3. Edit distance and alignment 4. The number
8 Grundlagen der Bioinformatik, SoSe 11, D. Huson, April 18, 2011 2 Pairwise alignment We will discuss: 1. Strings 2. Dot matrix method for comparing sequences 3. Edit distance and alignment 4. The number
Motivating the need for optimal sequence alignments...
 1 Motivating the need for optimal sequence alignments... 2 3 Note that this actually combines two objectives of optimal sequence alignments: (i) use the score of the alignment o infer homology; (ii) use
1 Motivating the need for optimal sequence alignments... 2 3 Note that this actually combines two objectives of optimal sequence alignments: (i) use the score of the alignment o infer homology; (ii) use
5. MULTIPLE SEQUENCE ALIGNMENT BIOINFORMATICS COURSE MTAT
 5. MULTIPLE SEQUENCE ALIGNMENT BIOINFORMATICS COURSE MTAT.03.239 03.10.2012 ALIGNMENT Alignment is the task of locating equivalent regions of two or more sequences to maximize their similarity. Homology:
5. MULTIPLE SEQUENCE ALIGNMENT BIOINFORMATICS COURSE MTAT.03.239 03.10.2012 ALIGNMENT Alignment is the task of locating equivalent regions of two or more sequences to maximize their similarity. Homology:
8 Grundlagen der Bioinformatik, SS 09, D. Huson, April 28, 2009
 8 Grundlagen der Bioinformatik, SS 09, D. Huson, April 28, 2009 2 Pairwise alignment We will discuss: 1. Strings 2. Dot matrix method for comparing sequences 3. Edit distance and alignment 4. The number
8 Grundlagen der Bioinformatik, SS 09, D. Huson, April 28, 2009 2 Pairwise alignment We will discuss: 1. Strings 2. Dot matrix method for comparing sequences 3. Edit distance and alignment 4. The number
Sequence Analysis 17: lecture 5. Substitution matrices Multiple sequence alignment
 Sequence Analysis 17: lecture 5 Substitution matrices Multiple sequence alignment Substitution matrices Used to score aligned positions, usually of amino acids. Expressed as the log-likelihood ratio of
Sequence Analysis 17: lecture 5 Substitution matrices Multiple sequence alignment Substitution matrices Used to score aligned positions, usually of amino acids. Expressed as the log-likelihood ratio of
Bioinformatics. Scoring Matrices. David Gilbert Bioinformatics Research Centre
 Bioinformatics Scoring Matrices David Gilbert Bioinformatics Research Centre www.brc.dcs.gla.ac.uk Department of Computing Science, University of Glasgow Learning Objectives To explain the requirement
Bioinformatics Scoring Matrices David Gilbert Bioinformatics Research Centre www.brc.dcs.gla.ac.uk Department of Computing Science, University of Glasgow Learning Objectives To explain the requirement
7.36/7.91 recitation CB Lecture #4
 7.36/7.91 recitation 2-19-2014 CB Lecture #4 1 Announcements / Reminders Homework: - PS#1 due Feb. 20th at noon. - Late policy: ½ credit if received within 24 hrs of due date, otherwise no credit - Answer
7.36/7.91 recitation 2-19-2014 CB Lecture #4 1 Announcements / Reminders Homework: - PS#1 due Feb. 20th at noon. - Late policy: ½ credit if received within 24 hrs of due date, otherwise no credit - Answer
Properties of amino acids in proteins
 Properties of amino acids in proteins one of the primary roles of DNA (but not the only one!) is to code for proteins A typical bacterium builds thousands types of proteins, all from ~20 amino acids repeated
Properties of amino acids in proteins one of the primary roles of DNA (but not the only one!) is to code for proteins A typical bacterium builds thousands types of proteins, all from ~20 amino acids repeated
C E N T R. Introduction to bioinformatics 2007 E B I O I N F O R M A T I C S V U F O R I N T. Lecture 5 G R A T I V. Pair-wise Sequence Alignment
 C E N T R E F O R I N T E G R A T I V E B I O I N F O R M A T I C S V U Introduction to bioinformatics 2007 Lecture 5 Pair-wise Sequence Alignment Bioinformatics Nothing in Biology makes sense except in
C E N T R E F O R I N T E G R A T I V E B I O I N F O R M A T I C S V U Introduction to bioinformatics 2007 Lecture 5 Pair-wise Sequence Alignment Bioinformatics Nothing in Biology makes sense except in
BIO 285/CSCI 285/MATH 285 Bioinformatics Programming Lecture 8 Pairwise Sequence Alignment 2 And Python Function Instructor: Lei Qian Fisk University
 BIO 285/CSCI 285/MATH 285 Bioinformatics Programming Lecture 8 Pairwise Sequence Alignment 2 And Python Function Instructor: Lei Qian Fisk University Measures of Sequence Similarity Alignment with dot
BIO 285/CSCI 285/MATH 285 Bioinformatics Programming Lecture 8 Pairwise Sequence Alignment 2 And Python Function Instructor: Lei Qian Fisk University Measures of Sequence Similarity Alignment with dot
Administration. ndrew Torda April /04/2008 [ 1 ]
![Administration. ndrew Torda April /04/2008 [ 1 ]](/thumbs/73/68891262.jpg "Administration. ndrew Torda April /04/2008 [ 1 ]") ndrew Torda April 2008 Administration 22/04/2008 [ 1 ] Sprache? zu verhandeln (Englisch, Hochdeutsch, Bayerisch) Selection of topics Proteins / DNA / RNA Two halves to course week 1-7 Prof Torda (larger
ndrew Torda April 2008 Administration 22/04/2008 [ 1 ] Sprache? zu verhandeln (Englisch, Hochdeutsch, Bayerisch) Selection of topics Proteins / DNA / RNA Two halves to course week 1-7 Prof Torda (larger
Pairwise Sequence Alignment
 Introduction to Bioinformatics Pairwise Sequence Alignment Prof. Dr. Nizamettin AYDIN naydin@yildiz.edu.tr Outline Introduction to sequence alignment pair wise sequence alignment The Dot Matrix Scoring
Introduction to Bioinformatics Pairwise Sequence Alignment Prof. Dr. Nizamettin AYDIN naydin@yildiz.edu.tr Outline Introduction to sequence alignment pair wise sequence alignment The Dot Matrix Scoring
Introduction to sequence alignment. Local alignment the Smith-Waterman algorithm
 Lecture 2, 12/3/2003: Introduction to sequence alignment The Needleman-Wunsch algorithm for global sequence alignment: description and properties Local alignment the Smith-Waterman algorithm 1 Computational
Lecture 2, 12/3/2003: Introduction to sequence alignment The Needleman-Wunsch algorithm for global sequence alignment: description and properties Local alignment the Smith-Waterman algorithm 1 Computational
Genomes and Their Evolution
 Chapter 21 Genomes and Their Evolution PowerPoint Lecture Presentations for Biology Eighth Edition Neil Campbell and Jane Reece Lectures by Chris Romero, updated by Erin Barley with contributions from
Chapter 21 Genomes and Their Evolution PowerPoint Lecture Presentations for Biology Eighth Edition Neil Campbell and Jane Reece Lectures by Chris Romero, updated by Erin Barley with contributions from
Homology Modeling. Roberto Lins EPFL - summer semester 2005
 Homology Modeling Roberto Lins EPFL - summer semester 2005 Disclaimer: course material is mainly taken from: P.E. Bourne & H Weissig, Structural Bioinformatics; C.A. Orengo, D.T. Jones & J.M. Thornton,
Homology Modeling Roberto Lins EPFL - summer semester 2005 Disclaimer: course material is mainly taken from: P.E. Bourne & H Weissig, Structural Bioinformatics; C.A. Orengo, D.T. Jones & J.M. Thornton,
Introduction to Bioinformatics
 Introduction to Bioinformatics Jianlin Cheng, PhD Department of Computer Science Informatics Institute 2011 Topics Introduction Biological Sequence Alignment and Database Search Analysis of gene expression
Introduction to Bioinformatics Jianlin Cheng, PhD Department of Computer Science Informatics Institute 2011 Topics Introduction Biological Sequence Alignment and Database Search Analysis of gene expression
First generation sequencing and pairwise alignment (High-tech, not high throughput) Analysis of Biological Sequences
 Analysis of Biological Sequences") First generation sequencing and pairwise alignment (High-tech, not high throughput) Analysis of Biological Sequences 140.638 where do sequences come from? DNA is not hard to extract (getting DNA from a
First generation sequencing and pairwise alignment (High-tech, not high throughput) Analysis of Biological Sequences 140.638 where do sequences come from? DNA is not hard to extract (getting DNA from a
Chapter 5. Proteomics and the analysis of protein sequence Ⅱ
 Proteomics Chapter 5. Proteomics and the analysis of protein sequence Ⅱ 1 Pairwise similarity searching (1) Figure 5.5: manual alignment One of the amino acids in the top sequence has no equivalent and
Proteomics Chapter 5. Proteomics and the analysis of protein sequence Ⅱ 1 Pairwise similarity searching (1) Figure 5.5: manual alignment One of the amino acids in the top sequence has no equivalent and
BIOINFORMATICS: An Introduction
 BIOINFORMATICS: An Introduction What is Bioinformatics? The term was first coined in 1988 by Dr. Hwa Lim The original definition was : a collective term for data compilation, organisation, analysis and
BIOINFORMATICS: An Introduction What is Bioinformatics? The term was first coined in 1988 by Dr. Hwa Lim The original definition was : a collective term for data compilation, organisation, analysis and
 Tree of Life iological Sequence nalysis Chapter http://tolweb.org/tree/ Phylogenetic Prediction ll organisms on Earth have a common ancestor. ll species are related. The relationship is called a phylogeny
Tree of Life iological Sequence nalysis Chapter http://tolweb.org/tree/ Phylogenetic Prediction ll organisms on Earth have a common ancestor. ll species are related. The relationship is called a phylogeny
InDel 3-5. InDel 8-9. InDel 3-5. InDel 8-9. InDel InDel 8-9
 Lecture 5 Alignment I. Introduction. For sequence data, the process of generating an alignment establishes positional homologies; that is, alignment provides the identification of homologous phylogenetic
Lecture 5 Alignment I. Introduction. For sequence data, the process of generating an alignment establishes positional homologies; that is, alignment provides the identification of homologous phylogenetic
Bioinformatics for Computer Scientists (Part 2 Sequence Alignment) Sepp Hochreiter
 Sepp Hochreiter") Bioinformatics for Computer Scientists (Part 2 Sequence Alignment) Institute of Bioinformatics Johannes Kepler University, Linz, Austria Sequence Alignment 2. Sequence Alignment Sequence Alignment 2.1
Bioinformatics for Computer Scientists (Part 2 Sequence Alignment) Institute of Bioinformatics Johannes Kepler University, Linz, Austria Sequence Alignment 2. Sequence Alignment Sequence Alignment 2.1
Bioinformatics and BLAST
 Bioinformatics and BLAST Overview Recap of last time Similarity discussion Algorithms: Needleman-Wunsch Smith-Waterman BLAST Implementation issues and current research Recap from Last Time Genome consists
Bioinformatics and BLAST Overview Recap of last time Similarity discussion Algorithms: Needleman-Wunsch Smith-Waterman BLAST Implementation issues and current research Recap from Last Time Genome consists
Homology Modeling (Comparative Structure Modeling) GBCB 5874: Problem Solving in GBCB
 GBCB 5874: Problem Solving in GBCB") Homology Modeling (Comparative Structure Modeling) Aims of Structural Genomics High-throughput 3D structure determination and analysis To determine or predict the 3D structures of all the proteins encoded
Homology Modeling (Comparative Structure Modeling) Aims of Structural Genomics High-throughput 3D structure determination and analysis To determine or predict the 3D structures of all the proteins encoded
Lecture 4: Evolutionary Models and Substitution Matrices (PAM and BLOSUM)
") Bioinformatics II Probability and Statistics Universität Zürich and ETH Zürich Spring Semester 2009 Lecture 4: Evolutionary Models and Substitution Matrices (PAM and BLOSUM) Dr Fraser Daly adapted from
Bioinformatics II Probability and Statistics Universität Zürich and ETH Zürich Spring Semester 2009 Lecture 4: Evolutionary Models and Substitution Matrices (PAM and BLOSUM) Dr Fraser Daly adapted from
Genomics and bioinformatics summary. Finding genes -- computer searches
 Genomics and bioinformatics summary 1. Gene finding: computer searches, cdnas, ESTs, 2. Microarrays 3. Use BLAST to find homologous sequences 4. Multiple sequence alignments (MSAs) 5. Trees quantify sequence
Genomics and bioinformatics summary 1. Gene finding: computer searches, cdnas, ESTs, 2. Microarrays 3. Use BLAST to find homologous sequences 4. Multiple sequence alignments (MSAs) 5. Trees quantify sequence
Sequence Alignment: Scoring Schemes. COMP 571 Luay Nakhleh, Rice University
 Sequence Alignment: Scoring Schemes COMP 571 Luay Nakhleh, Rice University Scoring Schemes Recall that an alignment score is aimed at providing a scale to measure the degree of similarity (or difference)
Sequence Alignment: Scoring Schemes COMP 571 Luay Nakhleh, Rice University Scoring Schemes Recall that an alignment score is aimed at providing a scale to measure the degree of similarity (or difference)
Pairwise sequence alignment
 Department of Evolutionary Biology Example Alignment between very similar human alpha- and beta globins: GSAQVKGHGKKVADALTNAVAHVDDMPNALSALSDLHAHKL G+ +VK+HGKKV A+++++AH+D++ +++++LS+LH KL GNPKVKAHGKKVLGAFSDGLAHLDNLKGTFATLSELHCDKL
Department of Evolutionary Biology Example Alignment between very similar human alpha- and beta globins: GSAQVKGHGKKVADALTNAVAHVDDMPNALSALSDLHAHKL G+ +VK+HGKKV A+++++AH+D++ +++++LS+LH KL GNPKVKAHGKKVLGAFSDGLAHLDNLKGTFATLSELHCDKL
... and searches for related sequences probably make up the vast bulk of bioinformatics activities.
 1 2 ... and searches for related sequences probably make up the vast bulk of bioinformatics activities. 3 The terms homology and similarity are often confused and used incorrectly. Homology is a quality.
1 2 ... and searches for related sequences probably make up the vast bulk of bioinformatics activities. 3 The terms homology and similarity are often confused and used incorrectly. Homology is a quality.
Tools and Algorithms in Bioinformatics
 Tools and Algorithms in Bioinformatics GCBA815, Fall 2013 Week3: Blast Algorithm, theory and practice Babu Guda, Ph.D. Department of Genetics, Cell Biology & Anatomy Bioinformatics and Systems Biology
Tools and Algorithms in Bioinformatics GCBA815, Fall 2013 Week3: Blast Algorithm, theory and practice Babu Guda, Ph.D. Department of Genetics, Cell Biology & Anatomy Bioinformatics and Systems Biology
Lecture 1, 31/10/2001: Introduction to sequence alignment. The Needleman-Wunsch algorithm for global sequence alignment: description and properties
 Lecture 1, 31/10/2001: Introduction to sequence alignment The Needleman-Wunsch algorithm for global sequence alignment: description and properties 1 Computational sequence-analysis The major goal of computational
Lecture 1, 31/10/2001: Introduction to sequence alignment The Needleman-Wunsch algorithm for global sequence alignment: description and properties 1 Computational sequence-analysis The major goal of computational
BLAST Database Searching. BME 110: CompBio Tools Todd Lowe April 8, 2010
 BLAST Database Searching BME 110: CompBio Tools Todd Lowe April 8, 2010 Admin Reading: Read chapter 7, and the NCBI Blast Guide and tutorial http://www.ncbi.nlm.nih.gov/blast/why.shtml Read Chapter 8 for
BLAST Database Searching BME 110: CompBio Tools Todd Lowe April 8, 2010 Admin Reading: Read chapter 7, and the NCBI Blast Guide and tutorial http://www.ncbi.nlm.nih.gov/blast/why.shtml Read Chapter 8 for
Tools and Algorithms in Bioinformatics
 Tools and Algorithms in Bioinformatics GCBA815, Fall 2015 Week-4 BLAST Algorithm Continued Multiple Sequence Alignment Babu Guda, Ph.D. Department of Genetics, Cell Biology & Anatomy Bioinformatics and
Tools and Algorithms in Bioinformatics GCBA815, Fall 2015 Week-4 BLAST Algorithm Continued Multiple Sequence Alignment Babu Guda, Ph.D. Department of Genetics, Cell Biology & Anatomy Bioinformatics and
Proteins: Characteristics and Properties of Amino Acids
 SBI4U:Biochemistry Macromolecules Eachaminoacidhasatleastoneamineandoneacidfunctionalgroupasthe nameimplies.thedifferentpropertiesresultfromvariationsinthestructuresof differentrgroups.thergroupisoftenreferredtoastheaminoacidsidechain.
SBI4U:Biochemistry Macromolecules Eachaminoacidhasatleastoneamineandoneacidfunctionalgroupasthe nameimplies.thedifferentpropertiesresultfromvariationsinthestructuresof differentrgroups.thergroupisoftenreferredtoastheaminoacidsidechain.
Sequence Database Search Techniques I: Blast and PatternHunter tools
 Sequence Database Search Techniques I: Blast and PatternHunter tools Zhang Louxin National University of Singapore Outline. Database search 2. BLAST (and filtration technique) 3. PatternHunter (empowered
Sequence Database Search Techniques I: Blast and PatternHunter tools Zhang Louxin National University of Singapore Outline. Database search 2. BLAST (and filtration technique) 3. PatternHunter (empowered
Global alignments - review
 Global alignments - review Take two sequences: X[j] and Y[j] M[i-1, j-1] ± 1 M[i, j] = max M[i, j-1] 2 M[i-1, j] 2 The best alignment for X[1 i] and Y[1 j] is called M[i, j] X[j] Initiation: M[,]= pply
Global alignments - review Take two sequences: X[j] and Y[j] M[i-1, j-1] ± 1 M[i, j] = max M[i, j-1] 2 M[i-1, j] 2 The best alignment for X[1 i] and Y[1 j] is called M[i, j] X[j] Initiation: M[,]= pply
USING BLAST TO IDENTIFY PROTEINS THAT ARE EVOLUTIONARILY RELATED ACROSS SPECIES
 USING BLAST TO IDENTIFY PROTEINS THAT ARE EVOLUTIONARILY RELATED ACROSS SPECIES HOW CAN BIOINFORMATICS BE USED AS A TOOL TO DETERMINE EVOLUTIONARY RELATIONSHPS AND TO BETTER UNDERSTAND PROTEIN HERITAGE?
USING BLAST TO IDENTIFY PROTEINS THAT ARE EVOLUTIONARILY RELATED ACROSS SPECIES HOW CAN BIOINFORMATICS BE USED AS A TOOL TO DETERMINE EVOLUTIONARY RELATIONSHPS AND TO BETTER UNDERSTAND PROTEIN HERITAGE?
NMR Assignments using NMRView II: Sequential Assignments
 NMR Assignments using NMRView II: Sequential Assignments DO THE FOLLOWING, IF YOU HAVE NOT ALREADY DONE SO: For Mac OS X, you should have a subdirectory nmrview. At UGA this is /Users/bcmb8190/nmrview.
NMR Assignments using NMRView II: Sequential Assignments DO THE FOLLOWING, IF YOU HAVE NOT ALREADY DONE SO: For Mac OS X, you should have a subdirectory nmrview. At UGA this is /Users/bcmb8190/nmrview.
Supplementary Figure 3 a. Structural comparison between the two determined structures for the IL 23:MA12 complex. The overall RMSD between the two
 Supplementary Figure 1. Biopanningg and clone enrichment of Alphabody binders against human IL 23. Positive clones in i phage ELISA with optical density (OD) 3 times higher than background are shown for
Supplementary Figure 1. Biopanningg and clone enrichment of Alphabody binders against human IL 23. Positive clones in i phage ELISA with optical density (OD) 3 times higher than background are shown for
Lecture 10 (10/4/17) Lecture 10 (10/4/17)
 Lecture 10 (10/4/17)") Lecture 10 (10/4/17) Reading: Ch4; 125, 138-141, 141-142 Problems: Ch4 (text); 7, 9, 11 Ch4 (study guide); 1, 2 NEXT Reading: Ch4; 125, 132-136 (structure determination) Ch4; 12-130 (Collagen) Problems:
Lecture 10 (10/4/17) Reading: Ch4; 125, 138-141, 141-142 Problems: Ch4 (text); 7, 9, 11 Ch4 (study guide); 1, 2 NEXT Reading: Ch4; 125, 132-136 (structure determination) Ch4; 12-130 (Collagen) Problems:
Translation. A ribosome, mrna, and trna.
 Translation The basic processes of translation are conserved among prokaryotes and eukaryotes. Prokaryotic Translation A ribosome, mrna, and trna. In the initiation of translation in prokaryotes, the Shine-Dalgarno
Translation The basic processes of translation are conserved among prokaryotes and eukaryotes. Prokaryotic Translation A ribosome, mrna, and trna. In the initiation of translation in prokaryotes, the Shine-Dalgarno
20 Grundlagen der Bioinformatik, SS 08, D. Huson, May 27, Global and local alignment of two sequences using dynamic programming
 20 Grundlagen der Bioinformatik, SS 08, D. Huson, May 27, 2008 4 Pairwise alignment We will discuss: 1. Strings 2. Dot matrix method for comparing sequences 3. Edit distance 4. Global and local alignment
20 Grundlagen der Bioinformatik, SS 08, D. Huson, May 27, 2008 4 Pairwise alignment We will discuss: 1. Strings 2. Dot matrix method for comparing sequences 3. Edit distance 4. Global and local alignment
Sequence comparison: Score matrices
 Sequence comparison: Score matrices http://facultywashingtonedu/jht/gs559_2013/ Genome 559: Introduction to Statistical and omputational Genomics Prof James H Thomas FYI - informal inductive proof of best
Sequence comparison: Score matrices http://facultywashingtonedu/jht/gs559_2013/ Genome 559: Introduction to Statistical and omputational Genomics Prof James H Thomas FYI - informal inductive proof of best
Introduction to protein alignments
 Introduction to protein alignments Comparative Analysis of Proteins Experimental evidence from one or more proteins can be used to infer function of related protein(s). Gene A Gene X Protein A compare
Introduction to protein alignments Comparative Analysis of Proteins Experimental evidence from one or more proteins can be used to infer function of related protein(s). Gene A Gene X Protein A compare
Scoring Matrices. Shifra Ben-Dor Irit Orr
 Scoring Matrices Shifra Ben-Dor Irit Orr Scoring matrices Sequence alignment and database searching programs compare sequences to each other as a series of characters. All algorithms (programs) for comparison
Scoring Matrices Shifra Ben-Dor Irit Orr Scoring matrices Sequence alignment and database searching programs compare sequences to each other as a series of characters. All algorithms (programs) for comparison
PAM-1 Matrix 10,000. From: Ala Arg Asn Asp Cys Gln Glu To:
 119-1 atrix 10,000 rom: la rg sn sp ys ln lu o: la 9867 2 9 10 3 8 17 rg 1 9913 1 0 1 10 0 sn 4 1 9822 36 0 4 6 sp 6 0 42 9859 0 6 53 ys 1 1 0 0 9973 0 0 ln 3 9 4 5 0 9876 27 lu 10 0 7 56 0 35 9865 120
119-1 atrix 10,000 rom: la rg sn sp ys ln lu o: la 9867 2 9 10 3 8 17 rg 1 9913 1 0 1 10 0 sn 4 1 9822 36 0 4 6 sp 6 0 42 9859 0 6 53 ys 1 1 0 0 9973 0 0 ln 3 9 4 5 0 9876 27 lu 10 0 7 56 0 35 9865 120
Alignment Strategies for Large Scale Genome Alignments
 Alignment Strategies for Large Scale Genome Alignments CSHL Computational Genomics 9 November 2003 Algorithms for Biological Sequence Comparison algorithm value scoring gap time calculated matrix penalty
Alignment Strategies for Large Scale Genome Alignments CSHL Computational Genomics 9 November 2003 Algorithms for Biological Sequence Comparison algorithm value scoring gap time calculated matrix penalty
Comparative genomics: Overview & Tools + MUMmer algorithm
 Comparative genomics: Overview & Tools + MUMmer algorithm Urmila Kulkarni-Kale Bioinformatics Centre University of Pune, Pune 411 007. urmila@bioinfo.ernet.in Genome sequence: Fact file 1995: The first
Comparative genomics: Overview & Tools + MUMmer algorithm Urmila Kulkarni-Kale Bioinformatics Centre University of Pune, Pune 411 007. urmila@bioinfo.ernet.in Genome sequence: Fact file 1995: The first
Sequence comparison: Score matrices. Genome 559: Introduction to Statistical and Computational Genomics Prof. James H. Thomas
 Sequence comparison: Score matrices Genome 559: Introduction to Statistical and omputational Genomics Prof James H Thomas FYI - informal inductive proof of best alignment path onsider the last step in
Sequence comparison: Score matrices Genome 559: Introduction to Statistical and omputational Genomics Prof James H Thomas FYI - informal inductive proof of best alignment path onsider the last step in
Sequence alignment methods. Pairwise alignment. The universe of biological sequence analysis
 he universe of biological sequence analysis Word/pattern recognition- Identification of restriction enzyme cleavage sites Sequence alignment methods PstI he universe of biological sequence analysis - prediction
he universe of biological sequence analysis Word/pattern recognition- Identification of restriction enzyme cleavage sites Sequence alignment methods PstI he universe of biological sequence analysis - prediction
Pairwise Alignment. Guan-Shieng Huang. Dept. of CSIE, NCNU. Pairwise Alignment p.1/55
 Pairwise Alignment Guan-Shieng Huang shieng@ncnu.edu.tw Dept. of CSIE, NCNU Pairwise Alignment p.1/55 Approach 1. Problem definition 2. Computational method (algorithms) 3. Complexity and performance Pairwise
Pairwise Alignment Guan-Shieng Huang shieng@ncnu.edu.tw Dept. of CSIE, NCNU Pairwise Alignment p.1/55 Approach 1. Problem definition 2. Computational method (algorithms) 3. Complexity and performance Pairwise
Lecture 2, 5/12/2001: Local alignment the Smith-Waterman algorithm. Alignment scoring schemes and theory: substitution matrices and gap models
 Lecture 2, 5/12/2001: Local alignment the Smith-Waterman algorithm Alignment scoring schemes and theory: substitution matrices and gap models 1 Local sequence alignments Local sequence alignments are necessary
Lecture 2, 5/12/2001: Local alignment the Smith-Waterman algorithm Alignment scoring schemes and theory: substitution matrices and gap models 1 Local sequence alignments Local sequence alignments are necessary
Bioinformatics for Biologists
 Bioinformatics for Biologists Sequence Analysis: Part I. Pairwise alignment and database searching Fran Lewitter, Ph.D. Head, Biocomputing Whitehead Institute Bioinformatics Definitions The use of computational
Bioinformatics for Biologists Sequence Analysis: Part I. Pairwise alignment and database searching Fran Lewitter, Ph.D. Head, Biocomputing Whitehead Institute Bioinformatics Definitions The use of computational
Week 10: Homology Modelling (II) - HHpred
 - HHpred") Week 10: Homology Modelling (II) - HHpred Course: Tools for Structural Biology Fabian Glaser BKU - Technion 1 2 Identify and align related structures by sequence methods is not an easy task All comparative
Week 10: Homology Modelling (II) - HHpred Course: Tools for Structural Biology Fabian Glaser BKU - Technion 1 2 Identify and align related structures by sequence methods is not an easy task All comparative
Secondary Structure. Bioch/BIMS 503 Lecture 2. Structure and Function of Proteins. Further Reading. Φ, Ψ angles alone determine protein structure
 Bioch/BIMS 503 Lecture 2 Structure and Function of Proteins August 28, 2008 Robert Nakamoto rkn3c@virginia.edu 2-0279 Secondary Structure Φ Ψ angles determine protein structure Φ Ψ angles are restricted
Bioch/BIMS 503 Lecture 2 Structure and Function of Proteins August 28, 2008 Robert Nakamoto rkn3c@virginia.edu 2-0279 Secondary Structure Φ Ψ angles determine protein structure Φ Ψ angles are restricted
Sequence comparison: Score matrices. Genome 559: Introduction to Statistical and Computational Genomics Prof. James H. Thomas
 Sequence comparison: Score matrices Genome 559: Introduction to Statistical and omputational Genomics Prof James H Thomas Informal inductive proof of best alignment path onsider the last step in the best
Sequence comparison: Score matrices Genome 559: Introduction to Statistical and omputational Genomics Prof James H Thomas Informal inductive proof of best alignment path onsider the last step in the best
Amino Acids and Peptides
 Amino Acids Amino Acids and Peptides Amino acid a compound that contains both an amino group and a carboxyl group α-amino acid an amino acid in which the amino group is on the carbon adjacent to the carboxyl
Amino Acids Amino Acids and Peptides Amino acid a compound that contains both an amino group and a carboxyl group α-amino acid an amino acid in which the amino group is on the carbon adjacent to the carboxyl
Copyright 2000 N. AYDIN. All rights reserved. 1
 Introduction to Bioinformatics Prof. Dr. Nizamettin AYDIN naydin@yildiz.edu.tr Multiple Sequence Alignment Outline Multiple sequence alignment introduction to msa methods of msa progressive global alignment
Introduction to Bioinformatics Prof. Dr. Nizamettin AYDIN naydin@yildiz.edu.tr Multiple Sequence Alignment Outline Multiple sequence alignment introduction to msa methods of msa progressive global alignment
Early History up to Schedule. Proteins DNA & RNA Schwann and Schleiden Cell Theory Charles Darwin publishes Origin of Species
 Schedule Bioinformatics and Computational Biology: History and Biological Background (JH) 0.0 he Parsimony criterion GKN.0 Stochastic Models of Sequence Evolution GKN 7.0 he Likelihood criterion GKN 0.0
Schedule Bioinformatics and Computational Biology: History and Biological Background (JH) 0.0 he Parsimony criterion GKN.0 Stochastic Models of Sequence Evolution GKN 7.0 he Likelihood criterion GKN 0.0