Sequence Alignment: Scoring Schemes. COMP 571 Luay Nakhleh, Rice University
|
|
|
- Toby McDaniel
- 6 years ago
- Views:
Transcription
1 Sequence Alignment: Scoring Schemes COMP 571 Luay Nakhleh, Rice University
2 Scoring Schemes Recall that an alignment score is aimed at providing a scale to measure the degree of similarity (or difference) between two sequences and thus make it possible to quickly distinguish among the many subtly different alignments that can be generated for any two sequences Scoring schemes contain two separate elements: the first assigns a value to a pair of aligned residues the second assigns penalties to gaps
3 Deriving a Substitution Matrix The alignment score attempts to measure the likelihood of a common evolutionary ancestor To achieve this mathematically, we consider the alignment of two residues from two sequences under two competing models: a random model, R, and a match (nonrandom, evolutionary) model, M
4 The Random Model (R) All sequences are assumed to be random selections from a given pool of residues, with every position in the sequence totally independent of every other Thus for a protein sequence, if the proportion of amino acid type a in the pool is pa, this fraction will be reproduced in the amino acid composition of the protein In this model, the probability of residue a being aligned with residue b is simply papb
5 The Match Model (M) Sequences are related, due to an evolutionary process, and there is a high correlation between aligned residues The probability of occurrence of particular residues thus depends not on the pool of available residues, but on the residue at the equivalent position in the sequence of the common ancestor In this model, the probability of residue a being aligned with residue b is qa,b, where the actual values of qa,b depend on the properties of the evolutionary process
6 The Odds Ratio So, we have P(a,b R)=papb and P(a,b M)=qa,b These two models can be compared by taking the odds ratio qa,b/papb If this ratio is greater than 1, the match model is more likely to have produced the alignment of these residues
7 The Odds Ratio The odds ratio for the entire alignment is taken as the product of the odds ratios for the different positions u ( qa,b p a p b where u ranges over all positions in the alignment ) u
8 The Log-odds Ratio It is frequently more practical to deal with sums rather than products, especially when small numbers are involved This can be achieved by taking logarithms of the odds ratio to give the log-odds ratio. This ratio can be summed over all positions of the alignment to give S, the score of the alignment: S = u log ( qa,b p a p b ) u = u (s a,b ) u where sa,b is the substitution matrix element associated with the alignment of residue types a and b
9 The Log-odds Ratio A positive value of sa,b means that the probability of those two residues being aligned is greater in the match model than in the random model The converse is true for negative sa,b values S is a measure of the relative likelihood of the whole alignment arising due to the match model as compared with the random model However, a positive S is not a sufficient test of the alignment s significance (more on the significance of scores later)
10 PAM Scoring Matrices It is strongly argued that the scoring matrices are best developed based on experimental data, thus reflecting the kind of relationships occurring in nature The first scoring matrices developed from known data were the PAM matrices Point accepted mutations matrix, derived by Dayhoff et al. Dayhoff et al. estimated the substitution probabilities by using known mutational histories (mutation here means substitution) 34 protein superfamilies were used, divided into 71 groups of near homologous sequences (>85% identity to reduce the number of superimposed mutations) and a phylogenetic tree was constructed for each group (including the inference of the most likely ancestral sequences at each internal node)
11 PAM Scoring Matrices Then, the accepted point mutations on each edge were estimated A mutation is accepted if it is accepted by the species This usually means that the new amino acid must have the same effect (must function in a similar way) as the old one, which usually requires strong physio-chemical similarity, dependent on how critical the position of the amino acid is
12 PAM Scoring Matrices Let τ be a time interval of evolution, measured in numbers of mutations per residue Dayhoff s procedure used the following steps: 1. Divide the set of sequences into groups of similar sequences, and make a multiple alignment of each group 2. Construct phylogenetic trees for each group, and estimate the mutations on the edges 3. Define an evolutionary model to explain the evolution 4. Construct substitution matrices (the substitution matrix for an evolutionary interval τ given for each pair (a,b) of residues an estimate for the probability of a to mutate to b in a time interval τ) 5. Construct scoring matrices from the substitution matrices
13 The Evolutionary Model The evolutionary model used has the following assumption: the probability of a mutation in one position of a sequence is only dependent on which amino acid is in that position It is independent of position and neighbor residues, and independent of previous mutations in the position The biological clock is also assumed, which means that the rate of mutations is constant over time Hence, the time of evolution can be measured by the number of mutations observed in a certain number of residues This is measured in point accepted mutations (PAMs), and 1 PAM means one accepted mutation per 100 residues
14 Calculating the Substitution Matrix The substitution matrix is calculated by observing the number of accepted mutations in the constructed phylogenetic trees (1572 in the first experiment)
15 Calculating the Substitution Matrix The task is then to calculate a value for the relation between the amino acids a and b in terms of mutations This is done by first estimating the probability that a will be replaced by b in a certain evolutionary time τ, and denote this by M τ ab τ is measured in PAMs, and first we look at τ=1 (M 1 ab) When τ=1, the time specification is often omitted, and the probability denoted by Mab Note that Mab need not be equal to Mba Mab depends on (1) the probability that a mutates and (2) the probability that a mutates to b given that a mutates
16 Calculating the Substitution Matrix The procedure can be described as follows 1. Find all accepted mutations in the data. From this calculate fab (the number of mutations from a to b or b to a), fa (the total number of mutations that involve a), and f (the sum of fa for all a) 2. Calculate the frequency pa for all a (this is the relative occurrence of amino acid a in the data) 3. Calculate the relative mutability ma, which is a measure of the probability that a will mutate in the evolutionary time of interest. ma depends on fa (ma should increase with increasing fa) and pa (ma should decrease with increasing pa). Hence, ma can be defined as ma=k fa / pa, where K is a constant (for the value of K, see the next slide) 4. For determining Mab we can now use the facts that (a) the probability that a mutates (in time 1 PAM) is ma, and (b) the probability that a mutates to b, given that a mutates, is fab/fa. Therefore, for a b, Mab = ma fab/fa for a=b, Maa = 1 - ma
17 The Constant K The probability that an arbitrary mutation contains a is fa/(f/2) The probability that it is from a is (since fab=fba) is 1/2 (fa/(f/2))= fa/f Among 100 residues there are 100pa occurrences of a, hence the probability for any one of these to mutate is m a = 1 100p a f a f = 1 100f As a check, we can find expected number of mutations per 100 residues (100p a )m a = a a 100p a f a f100p a = 1 f f a p a f a = f f =1 a
18
19 Matrices for General Evolutionary Times Due to the independence properties of the model (Markov model), M z, for an arbitrary evolutionary time z, can be computed as M raised to the power z (matrix M multiplied by itself z times)
20
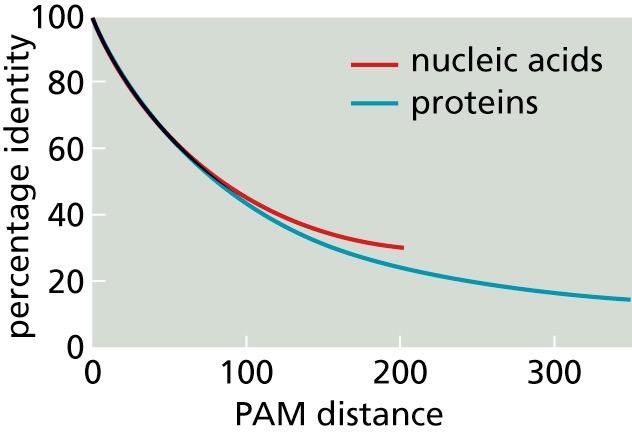
21 Substitution Matrices These matrices tell how many mutations have been accepted, but not the percentage of residues that have mutated: some may have mutated more than once, others not at all Suppose two sequences q and d have evolutionary distance τ (τ mutations per 100 residues have occurred in the transition from the ancestral sequence, say q, to the derived one, d) With 100 ( 1 c p c M τ cc ) we find how many residues on average are different per 100 residues
22
23 Obtaining a Scoring Matrix So far we have obtained a substitution matrix, but not a scoring matrix Using the log-odds ratio, we need to divide the probability under the match model (given by the substitution matrix) by the probability under the random model S ab = log M ab p b
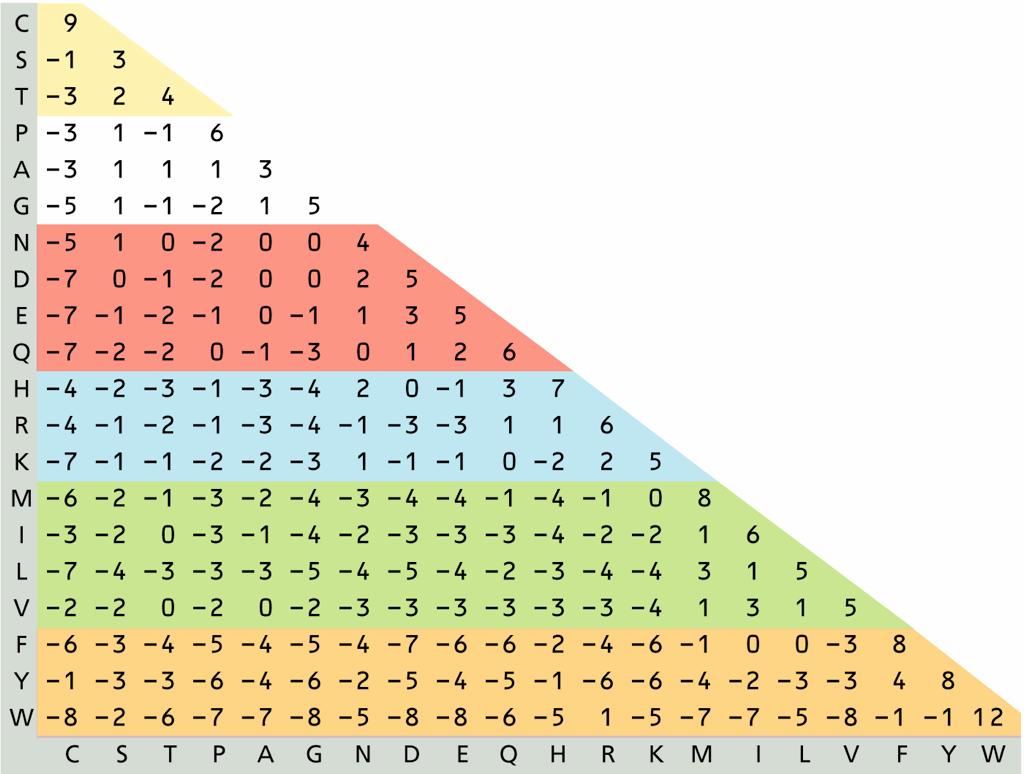
24 PAM120
25 BLOSUM Scoring Matrices In the Dayhoff model, the scoring values are derived from protein sequences with at least 85% identity Alignments are, however, most often performed on sequences of less similarity, and the scoring matrices for use in these cases are calculated from the 1 PAM matrix Henikoff and Henikoff (1992) have therefore developed scoring matrices based on known alignments of more diverse sequences
26 BLOSUM Scoring Matrices They take a group of related proteins and produce a set of blocks representing this group, where a block is defined as an ungapped region of aligned amino acids An example of two blocks is K I F I M K N L F K T R K I F K T K K L F E S R K I F K G R G D E V K G D S K K G D P K A G D A E R G D A A K
27 BLOSUM Scoring Matrices The Henikoffs used over 2000 blocks in order to derive their scoring matrices For each column in each block they counted the number of occurrences of each pair of amino acids, when all pairs of segments were used Then the frequency distribution of all 210 different pairs of amino acids were found A block of length w from an alignment of m sequences makes (wm(m-1))/2 pairs of amino acids
28 BLOSUM Scoring Matrices We define hab as the number of occurrences of the amino acid pair (ab) (note that hab=hba) T as the total number of pairs in the alignment T = c h ce, e c c,e M where is interpreted as a total ordering over the amino acids f ab=hab/t (the frequency of observed pairs)
29 BLOSUM Scoring Matrices Example K I F I M K N L F K T R K I F K T K K L F E S R K I F K G R hkr=6 hkk=1 hrr=3 G D E V K G D S K K G D P K A G D A E R G D A A K For the two blocks: hkr=9; there are 110 pairs Hence: fkr=9/110
30 Log-odds Matrix For each pair (ab), the expected probability that they are aligned by chance, eab, must be calculated Then fab>eab, the observed frequency is higher than expected by chance, which indicates a biological relation between the amino acids a and b fab<eab, the observed frequency is less than expected by chance, which indicates a biological aversion between the amino acids a and b fab=eab, which indicates biological neutrality between the amino acids a and b
31 Log-odds Matrix To calculate the expected number of occurrences of the amino acid pairs, assume that the observed frequencies are equal to the frequencies in the actual population From this the expected probability that a specific amino acid a is in a pair can be calculated: the number of residues in the considered data is 2T amino acid a occurs 2haa+ e ahae times amino acid a occurs with a frequency of p a = 2h aa + e a h ae 2T = f aa + e a f ae 2 Suppose now that all pairs are separated, and that new pairs are drawn according to the observed frequencies; we get eaa=papa and eab=2papb (a b) In order to obtain the log-odds matrix we need to calculate the ratio between the observed and the expected frequencies for each amino acid pair This is simply fab/eab, and working with the logarithm of the odds, we take R ab = log 2 f ab e ab
32 Developing Scoring Matrices for Different Evolutionary Distances When comparing two sequences q and d with an evolutionary distance X, one should use segment pairs corresponding to this distance for constructing an appropriate scoring matrix For developing a matrix for an X% identity (if we take identity to reflect evolutionary distance), similar blocks with X or higher percentage identity are grouped into one group, and treated as one segment
33 Developing Scoring Matrices for Different Evolutionary Distances The procedure for developing a BLOSUM X matrix 1. Collect a set of multiple alignments 2. Find the blocks 3. Group the segments with an X% identity 4. Count the occurrences of all pairs of amino acids 5. Develop the matrix, as explained before BLOSUM-62 is often used as the standard for ungapped alignments For gapped alignments, BLOSUM-50 is more often used
34 BLOSUM-62
35 Comparing BLOSUM and PAM Matrices The basis for constructing the two sets of matrices is different BLOSUM matrices with a low percentage correspond to PAM matrices for large evolutionary distances By use of relative entropy, it can be found that PAM250 corresponds to BLOSUM-45 and PAM160 corresponds to BLOSUM-62, and PAM120 corresponds to BLOSUM-80
36 Comparing BLOSUM and PAM Matrices When comparing sequences it is always a question of which PAM or BLOSUM matrix to use, especially when the evolutionary distance between the sequences is unknown Different studies have concluded that for the PAM matrices it is generally best to try PAM40, PAM120, and PAM250 When used for local alignments, lower PAM matrices find short local alignments, but higher PAM matrices find longer but weaker local alignments
37 Comparing BLOSUM and PAM Matrices Often a quick alignment is done first (using, for example, the identity scoring matrix), the evolutionary distance estimated, and the corresponding scoring matrix used However, several different matrices should be used, and the alignment that is judged to be evolutionarily the most accurate should be chosen
38 Scoring Matrices for Nucleotide Sequences The same techniques as those just described can be applied to nucleotides, although often simple scoring schemes such as +5 for a match and -4 for a mismatch are used
39 Gap Penalty Models A scoring scheme is required for insertions and deletions in alignments, as they are common evolutionary events The simplest method is to assign a gap penalty g on aligning any residue with a gap; that is, g=-e for a positive number E If the gap is ngap residues long, then this linear gap penalty is defined as g(ngap) = -ngape
40 Gap Penalty Models The observed preference for fewer and longer gaps can be modeled by using a higher penalty to initiate a gap (the gap opening penalty, or GOP, designated I) and then a lower penalty to extend an existing gap (the gap extension penalty, or GEP, designated E) This leads to the affine gap penalty forumla g(ngap)=-i-(ngap-1)e Typical ranges of the parameters for protein alignment are 7-15 for I and for E
41 Gap Penalty Models Very high gap penalty results in gaps only at beginning and end, and 10% sequence identity Very low gap penalty results in many more gaps, and 18% sequence identity
Bioinformatics. Scoring Matrices. David Gilbert Bioinformatics Research Centre
 Bioinformatics Scoring Matrices David Gilbert Bioinformatics Research Centre www.brc.dcs.gla.ac.uk Department of Computing Science, University of Glasgow Learning Objectives To explain the requirement
Bioinformatics Scoring Matrices David Gilbert Bioinformatics Research Centre www.brc.dcs.gla.ac.uk Department of Computing Science, University of Glasgow Learning Objectives To explain the requirement
Lecture 4: Evolutionary Models and Substitution Matrices (PAM and BLOSUM)
") Bioinformatics II Probability and Statistics Universität Zürich and ETH Zürich Spring Semester 2009 Lecture 4: Evolutionary Models and Substitution Matrices (PAM and BLOSUM) Dr Fraser Daly adapted from
Bioinformatics II Probability and Statistics Universität Zürich and ETH Zürich Spring Semester 2009 Lecture 4: Evolutionary Models and Substitution Matrices (PAM and BLOSUM) Dr Fraser Daly adapted from
Sequence Alignment: A General Overview. COMP Fall 2010 Luay Nakhleh, Rice University
 Sequence Alignment: A General Overview COMP 571 - Fall 2010 Luay Nakhleh, Rice University Life through Evolution All living organisms are related to each other through evolution This means: any pair of
Sequence Alignment: A General Overview COMP 571 - Fall 2010 Luay Nakhleh, Rice University Life through Evolution All living organisms are related to each other through evolution This means: any pair of
Algorithms in Bioinformatics FOUR Pairwise Sequence Alignment. Pairwise Sequence Alignment. Convention: DNA Sequences 5. Sequence Alignment
 Algorithms in Bioinformatics FOUR Sami Khuri Department of Computer Science San José State University Pairwise Sequence Alignment Homology Similarity Global string alignment Local string alignment Dot
Algorithms in Bioinformatics FOUR Sami Khuri Department of Computer Science San José State University Pairwise Sequence Alignment Homology Similarity Global string alignment Local string alignment Dot
Scoring Matrices. Shifra Ben-Dor Irit Orr
 Scoring Matrices Shifra Ben-Dor Irit Orr Scoring matrices Sequence alignment and database searching programs compare sequences to each other as a series of characters. All algorithms (programs) for comparison
Scoring Matrices Shifra Ben-Dor Irit Orr Scoring matrices Sequence alignment and database searching programs compare sequences to each other as a series of characters. All algorithms (programs) for comparison
Lecture 4: Evolutionary models and substitution matrices (PAM and BLOSUM).
.") 1 Bioinformatics: In-depth PROBABILITY & STATISTICS Spring Semester 2011 University of Zürich and ETH Zürich Lecture 4: Evolutionary models and substitution matrices (PAM and BLOSUM). Dr. Stefanie Muff
1 Bioinformatics: In-depth PROBABILITY & STATISTICS Spring Semester 2011 University of Zürich and ETH Zürich Lecture 4: Evolutionary models and substitution matrices (PAM and BLOSUM). Dr. Stefanie Muff
Sara C. Madeira. Universidade da Beira Interior. (Thanks to Ana Teresa Freitas, IST for useful resources on this subject)
") Bioinformática Sequence Alignment Pairwise Sequence Alignment Universidade da Beira Interior (Thanks to Ana Teresa Freitas, IST for useful resources on this subject) 1 16/3/29 & 23/3/29 27/4/29 Outline
Bioinformática Sequence Alignment Pairwise Sequence Alignment Universidade da Beira Interior (Thanks to Ana Teresa Freitas, IST for useful resources on this subject) 1 16/3/29 & 23/3/29 27/4/29 Outline
Quantifying sequence similarity
 Quantifying sequence similarity Bas E. Dutilh Systems Biology: Bioinformatic Data Analysis Utrecht University, February 16 th 2016 After this lecture, you can define homology, similarity, and identity
Quantifying sequence similarity Bas E. Dutilh Systems Biology: Bioinformatic Data Analysis Utrecht University, February 16 th 2016 After this lecture, you can define homology, similarity, and identity
Biochemistry 324 Bioinformatics. Pairwise sequence alignment
 Biochemistry 324 Bioinformatics Pairwise sequence alignment How do we compare genes/proteins? When we have sequenced a genome, we try and identify the function of unknown genes by finding a similar gene
Biochemistry 324 Bioinformatics Pairwise sequence alignment How do we compare genes/proteins? When we have sequenced a genome, we try and identify the function of unknown genes by finding a similar gene
Sequence Analysis 17: lecture 5. Substitution matrices Multiple sequence alignment
 Sequence Analysis 17: lecture 5 Substitution matrices Multiple sequence alignment Substitution matrices Used to score aligned positions, usually of amino acids. Expressed as the log-likelihood ratio of
Sequence Analysis 17: lecture 5 Substitution matrices Multiple sequence alignment Substitution matrices Used to score aligned positions, usually of amino acids. Expressed as the log-likelihood ratio of
Lecture 2, 5/12/2001: Local alignment the Smith-Waterman algorithm. Alignment scoring schemes and theory: substitution matrices and gap models
 Lecture 2, 5/12/2001: Local alignment the Smith-Waterman algorithm Alignment scoring schemes and theory: substitution matrices and gap models 1 Local sequence alignments Local sequence alignments are necessary
Lecture 2, 5/12/2001: Local alignment the Smith-Waterman algorithm Alignment scoring schemes and theory: substitution matrices and gap models 1 Local sequence alignments Local sequence alignments are necessary
Practical considerations of working with sequencing data
 Practical considerations of working with sequencing data File Types Fastq ->aligner -> reference(genome) coordinates Coordinate files SAM/BAM most complete, contains all of the info in fastq and more!
Practical considerations of working with sequencing data File Types Fastq ->aligner -> reference(genome) coordinates Coordinate files SAM/BAM most complete, contains all of the info in fastq and more!
CSE 549: Computational Biology. Substitution Matrices
 CSE 9: Computational Biology Substitution Matrices How should we score alignments So far, we ve looked at arbitrary schemes for scoring mutations. How can we assign scores in a more meaningful way? Are
CSE 9: Computational Biology Substitution Matrices How should we score alignments So far, we ve looked at arbitrary schemes for scoring mutations. How can we assign scores in a more meaningful way? Are
Single alignment: Substitution Matrix. 16 march 2017
 Single alignment: Substitution Matrix 16 march 2017 BLOSUM Matrix BLOSUM Matrix [2] (Blocks Amino Acid Substitution Matrices ) It is based on the amino acids substitutions observed in ~2000 conserved block
Single alignment: Substitution Matrix 16 march 2017 BLOSUM Matrix BLOSUM Matrix [2] (Blocks Amino Acid Substitution Matrices ) It is based on the amino acids substitutions observed in ~2000 conserved block
THEORY. Based on sequence Length According to the length of sequence being compared it is of following two types
 Exp 11- THEORY Sequence Alignment is a process of aligning two sequences to achieve maximum levels of identity between them. This help to derive functional, structural and evolutionary relationships between
Exp 11- THEORY Sequence Alignment is a process of aligning two sequences to achieve maximum levels of identity between them. This help to derive functional, structural and evolutionary relationships between
Sequence analysis and comparison
 The aim with sequence identification: Sequence analysis and comparison Marjolein Thunnissen Lund September 2012 Is there any known protein sequence that is homologous to mine? Are there any other species
The aim with sequence identification: Sequence analysis and comparison Marjolein Thunnissen Lund September 2012 Is there any known protein sequence that is homologous to mine? Are there any other species
Computational Biology
 Computational Biology Lecture 6 31 October 2004 1 Overview Scoring matrices (Thanks to Shannon McWeeney) BLAST algorithm Start sequence alignment 2 1 What is a homologous sequence? A homologous sequence,
Computational Biology Lecture 6 31 October 2004 1 Overview Scoring matrices (Thanks to Shannon McWeeney) BLAST algorithm Start sequence alignment 2 1 What is a homologous sequence? A homologous sequence,
Module: Sequence Alignment Theory and Applications Session: Introduction to Searching and Sequence Alignment
 Module: Sequence Alignment Theory and Applications Session: Introduction to Searching and Sequence Alignment Introduction to Bioinformatics online course : IBT Jonathan Kayondo Learning Objectives Understand
Module: Sequence Alignment Theory and Applications Session: Introduction to Searching and Sequence Alignment Introduction to Bioinformatics online course : IBT Jonathan Kayondo Learning Objectives Understand
Advanced topics in bioinformatics
 Feinberg Graduate School of the Weizmann Institute of Science Advanced topics in bioinformatics Shmuel Pietrokovski & Eitan Rubin Spring 2003 Course WWW site: http://bioinformatics.weizmann.ac.il/courses/atib
Feinberg Graduate School of the Weizmann Institute of Science Advanced topics in bioinformatics Shmuel Pietrokovski & Eitan Rubin Spring 2003 Course WWW site: http://bioinformatics.weizmann.ac.il/courses/atib
3. SEQUENCE ANALYSIS BIOINFORMATICS COURSE MTAT
 3. SEQUENCE ANALYSIS BIOINFORMATICS COURSE MTAT.03.239 25.09.2012 SEQUENCE ANALYSIS IS IMPORTANT FOR... Prediction of function Gene finding the process of identifying the regions of genomic DNA that encode
3. SEQUENCE ANALYSIS BIOINFORMATICS COURSE MTAT.03.239 25.09.2012 SEQUENCE ANALYSIS IS IMPORTANT FOR... Prediction of function Gene finding the process of identifying the regions of genomic DNA that encode
First generation sequencing and pairwise alignment (High-tech, not high throughput) Analysis of Biological Sequences
 Analysis of Biological Sequences") First generation sequencing and pairwise alignment (High-tech, not high throughput) Analysis of Biological Sequences 140.638 where do sequences come from? DNA is not hard to extract (getting DNA from a
First generation sequencing and pairwise alignment (High-tech, not high throughput) Analysis of Biological Sequences 140.638 where do sequences come from? DNA is not hard to extract (getting DNA from a
Pairwise sequence alignment
 Department of Evolutionary Biology Example Alignment between very similar human alpha- and beta globins: GSAQVKGHGKKVADALTNAVAHVDDMPNALSALSDLHAHKL G+ +VK+HGKKV A+++++AH+D++ +++++LS+LH KL GNPKVKAHGKKVLGAFSDGLAHLDNLKGTFATLSELHCDKL
Department of Evolutionary Biology Example Alignment between very similar human alpha- and beta globins: GSAQVKGHGKKVADALTNAVAHVDDMPNALSALSDLHAHKL G+ +VK+HGKKV A+++++AH+D++ +++++LS+LH KL GNPKVKAHGKKVLGAFSDGLAHLDNLKGTFATLSELHCDKL
Sequence Alignments. Dynamic programming approaches, scoring, and significance. Lucy Skrabanek ICB, WMC January 31, 2013
 Sequence Alignments Dynamic programming approaches, scoring, and significance Lucy Skrabanek ICB, WMC January 31, 213 Sequence alignment Compare two (or more) sequences to: Find regions of conservation
Sequence Alignments Dynamic programming approaches, scoring, and significance Lucy Skrabanek ICB, WMC January 31, 213 Sequence alignment Compare two (or more) sequences to: Find regions of conservation
Scoring Matrices. Shifra Ben Dor Irit Orr
 Scoring Matrices Shifra Ben Dor Irit Orr Scoring matrices Sequence alignment and database searching programs compare sequences to each other as a series of characters. All algorithms (programs) for comparison
Scoring Matrices Shifra Ben Dor Irit Orr Scoring matrices Sequence alignment and database searching programs compare sequences to each other as a series of characters. All algorithms (programs) for comparison
BINF 730. DNA Sequence Alignment Why?
 BINF 730 Lecture 2 Seuence Alignment DNA Seuence Alignment Why? Recognition sites might be common restriction enzyme start seuence stop seuence other regulatory seuences Homology evolutionary common progenitor
BINF 730 Lecture 2 Seuence Alignment DNA Seuence Alignment Why? Recognition sites might be common restriction enzyme start seuence stop seuence other regulatory seuences Homology evolutionary common progenitor
Substitution matrices
 Introduction to Bioinformatics Substitution matrices Jacques van Helden Jacques.van-Helden@univ-amu.fr Université d Aix-Marseille, France Lab. Technological Advances for Genomics and Clinics (TAGC, INSERM
Introduction to Bioinformatics Substitution matrices Jacques van Helden Jacques.van-Helden@univ-amu.fr Université d Aix-Marseille, France Lab. Technological Advances for Genomics and Clinics (TAGC, INSERM
Local Alignment Statistics
 Local Alignment Statistics Stephen Altschul National Center for Biotechnology Information National Library of Medicine National Institutes of Health Bethesda, MD Central Issues in Biological Sequence Comparison
Local Alignment Statistics Stephen Altschul National Center for Biotechnology Information National Library of Medicine National Institutes of Health Bethesda, MD Central Issues in Biological Sequence Comparison
Pairwise sequence alignments
 Pairwise sequence alignments Volker Flegel VI, October 2003 Page 1 Outline Introduction Definitions Biological context of pairwise alignments Computing of pairwise alignments Some programs VI, October
Pairwise sequence alignments Volker Flegel VI, October 2003 Page 1 Outline Introduction Definitions Biological context of pairwise alignments Computing of pairwise alignments Some programs VI, October
Sequence comparison: Score matrices. Genome 559: Introduction to Statistical and Computational Genomics Prof. James H. Thomas
 Sequence comparison: Score matrices Genome 559: Introduction to Statistical and omputational Genomics Prof James H Thomas Informal inductive proof of best alignment path onsider the last step in the best
Sequence comparison: Score matrices Genome 559: Introduction to Statistical and omputational Genomics Prof James H Thomas Informal inductive proof of best alignment path onsider the last step in the best
Algorithms in Bioinformatics
 Algorithms in Bioinformatics Sami Khuri Department of omputer Science San José State University San José, alifornia, USA khuri@cs.sjsu.edu www.cs.sjsu.edu/faculty/khuri Pairwise Sequence Alignment Homology
Algorithms in Bioinformatics Sami Khuri Department of omputer Science San José State University San José, alifornia, USA khuri@cs.sjsu.edu www.cs.sjsu.edu/faculty/khuri Pairwise Sequence Alignment Homology
Week 10: Homology Modelling (II) - HHpred
 - HHpred") Week 10: Homology Modelling (II) - HHpred Course: Tools for Structural Biology Fabian Glaser BKU - Technion 1 2 Identify and align related structures by sequence methods is not an easy task All comparative
Week 10: Homology Modelling (II) - HHpred Course: Tools for Structural Biology Fabian Glaser BKU - Technion 1 2 Identify and align related structures by sequence methods is not an easy task All comparative
Sequence comparison: Score matrices
 Sequence comparison: Score matrices http://facultywashingtonedu/jht/gs559_2013/ Genome 559: Introduction to Statistical and omputational Genomics Prof James H Thomas FYI - informal inductive proof of best
Sequence comparison: Score matrices http://facultywashingtonedu/jht/gs559_2013/ Genome 559: Introduction to Statistical and omputational Genomics Prof James H Thomas FYI - informal inductive proof of best
Pairwise Alignment. Guan-Shieng Huang. Dept. of CSIE, NCNU. Pairwise Alignment p.1/55
 Pairwise Alignment Guan-Shieng Huang shieng@ncnu.edu.tw Dept. of CSIE, NCNU Pairwise Alignment p.1/55 Approach 1. Problem definition 2. Computational method (algorithms) 3. Complexity and performance Pairwise
Pairwise Alignment Guan-Shieng Huang shieng@ncnu.edu.tw Dept. of CSIE, NCNU Pairwise Alignment p.1/55 Approach 1. Problem definition 2. Computational method (algorithms) 3. Complexity and performance Pairwise
Sequence comparison: Score matrices. Genome 559: Introduction to Statistical and Computational Genomics Prof. James H. Thomas
 Sequence comparison: Score matrices Genome 559: Introduction to Statistical and omputational Genomics Prof James H Thomas FYI - informal inductive proof of best alignment path onsider the last step in
Sequence comparison: Score matrices Genome 559: Introduction to Statistical and omputational Genomics Prof James H Thomas FYI - informal inductive proof of best alignment path onsider the last step in
BIO 285/CSCI 285/MATH 285 Bioinformatics Programming Lecture 8 Pairwise Sequence Alignment 2 And Python Function Instructor: Lei Qian Fisk University
 BIO 285/CSCI 285/MATH 285 Bioinformatics Programming Lecture 8 Pairwise Sequence Alignment 2 And Python Function Instructor: Lei Qian Fisk University Measures of Sequence Similarity Alignment with dot
BIO 285/CSCI 285/MATH 285 Bioinformatics Programming Lecture 8 Pairwise Sequence Alignment 2 And Python Function Instructor: Lei Qian Fisk University Measures of Sequence Similarity Alignment with dot
Bioinformatics (GLOBEX, Summer 2015) Pairwise sequence alignment
 Pairwise sequence alignment") Bioinformatics (GLOBEX, Summer 2015) Pairwise sequence alignment Substitution score matrices, PAM, BLOSUM Needleman-Wunsch algorithm (Global) Smith-Waterman algorithm (Local) BLAST (local, heuristic) E-value
Bioinformatics (GLOBEX, Summer 2015) Pairwise sequence alignment Substitution score matrices, PAM, BLOSUM Needleman-Wunsch algorithm (Global) Smith-Waterman algorithm (Local) BLAST (local, heuristic) E-value
Pairwise sequence alignments. Vassilios Ioannidis (From Volker Flegel )
") Pairwise sequence alignments Vassilios Ioannidis (From Volker Flegel ) Outline Introduction Definitions Biological context of pairwise alignments Computing of pairwise alignments Some programs Importance
Pairwise sequence alignments Vassilios Ioannidis (From Volker Flegel ) Outline Introduction Definitions Biological context of pairwise alignments Computing of pairwise alignments Some programs Importance
Copyright 2000 N. AYDIN. All rights reserved. 1
 Introduction to Bioinformatics Prof. Dr. Nizamettin AYDIN naydin@yildiz.edu.tr Multiple Sequence Alignment Outline Multiple sequence alignment introduction to msa methods of msa progressive global alignment
Introduction to Bioinformatics Prof. Dr. Nizamettin AYDIN naydin@yildiz.edu.tr Multiple Sequence Alignment Outline Multiple sequence alignment introduction to msa methods of msa progressive global alignment
Chapter 2: Sequence Alignment
 Chapter 2: Sequence lignment 2.2 Scoring Matrices Prof. Yechiam Yemini (YY) Computer Science Department Columbia University COMS4761-2007 Overview PM: scoring based on evolutionary statistics Elementary
Chapter 2: Sequence lignment 2.2 Scoring Matrices Prof. Yechiam Yemini (YY) Computer Science Department Columbia University COMS4761-2007 Overview PM: scoring based on evolutionary statistics Elementary
Large-Scale Genomic Surveys
 Bioinformatics Subtopics Fold Recognition Secondary Structure Prediction Docking & Drug Design Protein Geometry Protein Flexibility Homology Modeling Sequence Alignment Structure Classification Gene Prediction
Bioinformatics Subtopics Fold Recognition Secondary Structure Prediction Docking & Drug Design Protein Geometry Protein Flexibility Homology Modeling Sequence Alignment Structure Classification Gene Prediction
Moreover, the circular logic
 Moreover, the circular logic How do we know what is the right distance without a good alignment? And how do we construct a good alignment without knowing what substitutions were made previously? ATGCGT--GCAAGT
Moreover, the circular logic How do we know what is the right distance without a good alignment? And how do we construct a good alignment without knowing what substitutions were made previously? ATGCGT--GCAAGT
Basic Local Alignment Search Tool
 Basic Local Alignment Search Tool Alignments used to uncover homologies between sequences combined with phylogenetic studies o can determine orthologous and paralogous relationships Local Alignment uses
Basic Local Alignment Search Tool Alignments used to uncover homologies between sequences combined with phylogenetic studies o can determine orthologous and paralogous relationships Local Alignment uses
Sequence Database Search Techniques I: Blast and PatternHunter tools
 Sequence Database Search Techniques I: Blast and PatternHunter tools Zhang Louxin National University of Singapore Outline. Database search 2. BLAST (and filtration technique) 3. PatternHunter (empowered
Sequence Database Search Techniques I: Blast and PatternHunter tools Zhang Louxin National University of Singapore Outline. Database search 2. BLAST (and filtration technique) 3. PatternHunter (empowered
CONCEPT OF SEQUENCE COMPARISON. Natapol Pornputtapong 18 January 2018
 CONCEPT OF SEQUENCE COMPARISON Natapol Pornputtapong 18 January 2018 SEQUENCE ANALYSIS - A ROSETTA STONE OF LIFE Sequence analysis is the process of subjecting a DNA, RNA or peptide sequence to any of
CONCEPT OF SEQUENCE COMPARISON Natapol Pornputtapong 18 January 2018 SEQUENCE ANALYSIS - A ROSETTA STONE OF LIFE Sequence analysis is the process of subjecting a DNA, RNA or peptide sequence to any of
Tools and Algorithms in Bioinformatics
 Tools and Algorithms in Bioinformatics GCBA815, Fall 2013 Week3: Blast Algorithm, theory and practice Babu Guda, Ph.D. Department of Genetics, Cell Biology & Anatomy Bioinformatics and Systems Biology
Tools and Algorithms in Bioinformatics GCBA815, Fall 2013 Week3: Blast Algorithm, theory and practice Babu Guda, Ph.D. Department of Genetics, Cell Biology & Anatomy Bioinformatics and Systems Biology
Pairwise Sequence Alignment
 Introduction to Bioinformatics Pairwise Sequence Alignment Prof. Dr. Nizamettin AYDIN naydin@yildiz.edu.tr Outline Introduction to sequence alignment pair wise sequence alignment The Dot Matrix Scoring
Introduction to Bioinformatics Pairwise Sequence Alignment Prof. Dr. Nizamettin AYDIN naydin@yildiz.edu.tr Outline Introduction to sequence alignment pair wise sequence alignment The Dot Matrix Scoring
Pairwise & Multiple sequence alignments
 Pairwise & Multiple sequence alignments Urmila Kulkarni-Kale Bioinformatics Centre 411 007 urmila@bioinfo.ernet.in Basis for Sequence comparison Theory of evolution: gene sequences have evolved/derived
Pairwise & Multiple sequence alignments Urmila Kulkarni-Kale Bioinformatics Centre 411 007 urmila@bioinfo.ernet.in Basis for Sequence comparison Theory of evolution: gene sequences have evolved/derived
In-Depth Assessment of Local Sequence Alignment
 2012 International Conference on Environment Science and Engieering IPCBEE vol.3 2(2012) (2012)IACSIT Press, Singapoore In-Depth Assessment of Local Sequence Alignment Atoosa Ghahremani and Mahmood A.
2012 International Conference on Environment Science and Engieering IPCBEE vol.3 2(2012) (2012)IACSIT Press, Singapoore In-Depth Assessment of Local Sequence Alignment Atoosa Ghahremani and Mahmood A.
Phylogenetic inference
 Phylogenetic inference Bas E. Dutilh Systems Biology: Bioinformatic Data Analysis Utrecht University, March 7 th 016 After this lecture, you can discuss (dis-) advantages of different information types
Phylogenetic inference Bas E. Dutilh Systems Biology: Bioinformatic Data Analysis Utrecht University, March 7 th 016 After this lecture, you can discuss (dis-) advantages of different information types
An Introduction to Sequence Similarity ( Homology ) Searching
 Searching") An Introduction to Sequence Similarity ( Homology ) Searching Gary D. Stormo 1 UNIT 3.1 1 Washington University, School of Medicine, St. Louis, Missouri ABSTRACT Homologous sequences usually have the same,
An Introduction to Sequence Similarity ( Homology ) Searching Gary D. Stormo 1 UNIT 3.1 1 Washington University, School of Medicine, St. Louis, Missouri ABSTRACT Homologous sequences usually have the same,
Sequence Bioinformatics. Multiple Sequence Alignment Waqas Nasir
 Sequence Bioinformatics Multiple Sequence Alignment Waqas Nasir 2010-11-12 Multiple Sequence Alignment One amino acid plays coy; a pair of homologous sequences whisper; many aligned sequences shout out
Sequence Bioinformatics Multiple Sequence Alignment Waqas Nasir 2010-11-12 Multiple Sequence Alignment One amino acid plays coy; a pair of homologous sequences whisper; many aligned sequences shout out
Phylogenetics: Bayesian Phylogenetic Analysis. COMP Spring 2015 Luay Nakhleh, Rice University
 Phylogenetics: Bayesian Phylogenetic Analysis COMP 571 - Spring 2015 Luay Nakhleh, Rice University Bayes Rule P(X = x Y = y) = P(X = x, Y = y) P(Y = y) = P(X = x)p(y = y X = x) P x P(X = x 0 )P(Y = y X
Phylogenetics: Bayesian Phylogenetic Analysis COMP 571 - Spring 2015 Luay Nakhleh, Rice University Bayes Rule P(X = x Y = y) = P(X = x, Y = y) P(Y = y) = P(X = x)p(y = y X = x) P x P(X = x 0 )P(Y = y X
Lecture 5,6 Local sequence alignment
 Lecture 5,6 Local sequence alignment Chapter 6 in Jones and Pevzner Fall 2018 September 4,6, 2018 Evolution as a tool for biological insight Nothing in biology makes sense except in the light of evolution
Lecture 5,6 Local sequence alignment Chapter 6 in Jones and Pevzner Fall 2018 September 4,6, 2018 Evolution as a tool for biological insight Nothing in biology makes sense except in the light of evolution
Homology Modeling (Comparative Structure Modeling) GBCB 5874: Problem Solving in GBCB
 GBCB 5874: Problem Solving in GBCB") Homology Modeling (Comparative Structure Modeling) Aims of Structural Genomics High-throughput 3D structure determination and analysis To determine or predict the 3D structures of all the proteins encoded
Homology Modeling (Comparative Structure Modeling) Aims of Structural Genomics High-throughput 3D structure determination and analysis To determine or predict the 3D structures of all the proteins encoded
BLAST: Target frequencies and information content Dannie Durand
 Computational Genomics and Molecular Biology, Fall 2016 1 BLAST: Target frequencies and information content Dannie Durand BLAST has two components: a fast heuristic for searching for similar sequences
Computational Genomics and Molecular Biology, Fall 2016 1 BLAST: Target frequencies and information content Dannie Durand BLAST has two components: a fast heuristic for searching for similar sequences
Dr. Amira A. AL-Hosary
 Phylogenetic analysis Amira A. AL-Hosary PhD of infectious diseases Department of Animal Medicine (Infectious Diseases) Faculty of Veterinary Medicine Assiut University-Egypt Phylogenetic Basics: Biological
Phylogenetic analysis Amira A. AL-Hosary PhD of infectious diseases Department of Animal Medicine (Infectious Diseases) Faculty of Veterinary Medicine Assiut University-Egypt Phylogenetic Basics: Biological
Evolutionary Models. Evolutionary Models
 Edit Operators In standard pairwise alignment, what are the allowed edit operators that transform one sequence into the other? Describe how each of these edit operations are represented on a sequence alignment
Edit Operators In standard pairwise alignment, what are the allowed edit operators that transform one sequence into the other? Describe how each of these edit operations are represented on a sequence alignment
Bioinformatics for Computer Scientists (Part 2 Sequence Alignment) Sepp Hochreiter
 Sepp Hochreiter") Bioinformatics for Computer Scientists (Part 2 Sequence Alignment) Institute of Bioinformatics Johannes Kepler University, Linz, Austria Sequence Alignment 2. Sequence Alignment Sequence Alignment 2.1
Bioinformatics for Computer Scientists (Part 2 Sequence Alignment) Institute of Bioinformatics Johannes Kepler University, Linz, Austria Sequence Alignment 2. Sequence Alignment Sequence Alignment 2.1
Amira A. AL-Hosary PhD of infectious diseases Department of Animal Medicine (Infectious Diseases) Faculty of Veterinary Medicine Assiut
 Faculty of Veterinary Medicine Assiut") Amira A. AL-Hosary PhD of infectious diseases Department of Animal Medicine (Infectious Diseases) Faculty of Veterinary Medicine Assiut University-Egypt Phylogenetic analysis Phylogenetic Basics: Biological
Amira A. AL-Hosary PhD of infectious diseases Department of Animal Medicine (Infectious Diseases) Faculty of Veterinary Medicine Assiut University-Egypt Phylogenetic analysis Phylogenetic Basics: Biological
Page 1. Evolutionary Trees. Why build evolutionary tree? Outline
 Page Evolutionary Trees Russ. ltman MI S 7 Outline. Why build evolutionary trees?. istance-based vs. character-based methods. istance-based: Ultrametric Trees dditive Trees. haracter-based: Perfect phylogeny
Page Evolutionary Trees Russ. ltman MI S 7 Outline. Why build evolutionary trees?. istance-based vs. character-based methods. istance-based: Ultrametric Trees dditive Trees. haracter-based: Perfect phylogeny
CHAPTERS 24-25: Evidence for Evolution and Phylogeny
 CHAPTERS 24-25: Evidence for Evolution and Phylogeny 1. For each of the following, indicate how it is used as evidence of evolution by natural selection or shown as an evolutionary trend: a. Paleontology
CHAPTERS 24-25: Evidence for Evolution and Phylogeny 1. For each of the following, indicate how it is used as evidence of evolution by natural selection or shown as an evolutionary trend: a. Paleontology
进化树构建方法的概率方法 第 4 章 : 进化树构建的概率方法 问题介绍. 部分 lid 修改自 i i f l 的 ih l i
 第 4 章 : 进化树构建的概率方法 问题介绍 进化树构建方法的概率方法 部分 lid 修改自 i i f l 的 ih l i 部分 Slides 修改自 University of Basel 的 Michael Springmann 课程 CS302 Seminar Life Science Informatics 的讲义 Phylogenetic Tree branch internal node
第 4 章 : 进化树构建的概率方法 问题介绍 进化树构建方法的概率方法 部分 lid 修改自 i i f l 的 ih l i 部分 Slides 修改自 University of Basel 的 Michael Springmann 课程 CS302 Seminar Life Science Informatics 的讲义 Phylogenetic Tree branch internal node
Homology Modeling. Roberto Lins EPFL - summer semester 2005
 Homology Modeling Roberto Lins EPFL - summer semester 2005 Disclaimer: course material is mainly taken from: P.E. Bourne & H Weissig, Structural Bioinformatics; C.A. Orengo, D.T. Jones & J.M. Thornton,
Homology Modeling Roberto Lins EPFL - summer semester 2005 Disclaimer: course material is mainly taken from: P.E. Bourne & H Weissig, Structural Bioinformatics; C.A. Orengo, D.T. Jones & J.M. Thornton,
Sequence analysis and Genomics
 Sequence analysis and Genomics October 12 th November 23 rd 2 PM 5 PM Prof. Peter Stadler Dr. Katja Nowick Katja: group leader TFome and Transcriptome Evolution Bioinformatics group Paul-Flechsig-Institute
Sequence analysis and Genomics October 12 th November 23 rd 2 PM 5 PM Prof. Peter Stadler Dr. Katja Nowick Katja: group leader TFome and Transcriptome Evolution Bioinformatics group Paul-Flechsig-Institute
Lecture Notes: Markov chains
 Computational Genomics and Molecular Biology, Fall 5 Lecture Notes: Markov chains Dannie Durand At the beginning of the semester, we introduced two simple scoring functions for pairwise alignments: a similarity
Computational Genomics and Molecular Biology, Fall 5 Lecture Notes: Markov chains Dannie Durand At the beginning of the semester, we introduced two simple scoring functions for pairwise alignments: a similarity
20 Grundlagen der Bioinformatik, SS 08, D. Huson, May 27, Global and local alignment of two sequences using dynamic programming
 20 Grundlagen der Bioinformatik, SS 08, D. Huson, May 27, 2008 4 Pairwise alignment We will discuss: 1. Strings 2. Dot matrix method for comparing sequences 3. Edit distance 4. Global and local alignment
20 Grundlagen der Bioinformatik, SS 08, D. Huson, May 27, 2008 4 Pairwise alignment We will discuss: 1. Strings 2. Dot matrix method for comparing sequences 3. Edit distance 4. Global and local alignment
... and searches for related sequences probably make up the vast bulk of bioinformatics activities.
 1 2 ... and searches for related sequences probably make up the vast bulk of bioinformatics activities. 3 The terms homology and similarity are often confused and used incorrectly. Homology is a quality.
1 2 ... and searches for related sequences probably make up the vast bulk of bioinformatics activities. 3 The terms homology and similarity are often confused and used incorrectly. Homology is a quality.
"Nothing in biology makes sense except in the light of evolution Theodosius Dobzhansky
 MOLECULAR PHYLOGENY "Nothing in biology makes sense except in the light of evolution Theodosius Dobzhansky EVOLUTION - theory that groups of organisms change over time so that descendeants differ structurally
MOLECULAR PHYLOGENY "Nothing in biology makes sense except in the light of evolution Theodosius Dobzhansky EVOLUTION - theory that groups of organisms change over time so that descendeants differ structurally
Substitution Matrices
 C E N T R F O R I N T E G R A T I V E B I O I N F O R M A T I C S V U E [1] Substitution matrices Sequence analysis 2006 Substitution Matrices Introduction to bioinformatics 2007 Lecture 8 C E N T R F
C E N T R F O R I N T E G R A T I V E B I O I N F O R M A T I C S V U E [1] Substitution matrices Sequence analysis 2006 Substitution Matrices Introduction to bioinformatics 2007 Lecture 8 C E N T R F
Tools and Algorithms in Bioinformatics
 Tools and Algorithms in Bioinformatics GCBA815, Fall 2015 Week-4 BLAST Algorithm Continued Multiple Sequence Alignment Babu Guda, Ph.D. Department of Genetics, Cell Biology & Anatomy Bioinformatics and
Tools and Algorithms in Bioinformatics GCBA815, Fall 2015 Week-4 BLAST Algorithm Continued Multiple Sequence Alignment Babu Guda, Ph.D. Department of Genetics, Cell Biology & Anatomy Bioinformatics and
5. MULTIPLE SEQUENCE ALIGNMENT BIOINFORMATICS COURSE MTAT
 5. MULTIPLE SEQUENCE ALIGNMENT BIOINFORMATICS COURSE MTAT.03.239 03.10.2012 ALIGNMENT Alignment is the task of locating equivalent regions of two or more sequences to maximize their similarity. Homology:
5. MULTIPLE SEQUENCE ALIGNMENT BIOINFORMATICS COURSE MTAT.03.239 03.10.2012 ALIGNMENT Alignment is the task of locating equivalent regions of two or more sequences to maximize their similarity. Homology:
COPIA: A New Software for Finding Consensus Patterns. Chengzhi Liang. A thesis. presented to the University ofwaterloo. in fulfilment of the
 COPIA: A New Software for Finding Consensus Patterns in Unaligned Protein Sequences by Chengzhi Liang A thesis presented to the University ofwaterloo in fulfilment of the thesis requirement for the degree
COPIA: A New Software for Finding Consensus Patterns in Unaligned Protein Sequences by Chengzhi Liang A thesis presented to the University ofwaterloo in fulfilment of the thesis requirement for the degree
Sequence Analysis, '18 -- lecture 9. Families and superfamilies. Sequence weights. Profiles. Logos. Building a representative model for a gene.
 Sequence Analysis, '18 -- lecture 9 Families and superfamilies. Sequence weights. Profiles. Logos. Building a representative model for a gene. How can I represent thousands of homolog sequences in a compact
Sequence Analysis, '18 -- lecture 9 Families and superfamilies. Sequence weights. Profiles. Logos. Building a representative model for a gene. How can I represent thousands of homolog sequences in a compact
An Introduction to Bioinformatics Algorithms Hidden Markov Models
 Hidden Markov Models Outline 1. CG-Islands 2. The Fair Bet Casino 3. Hidden Markov Model 4. Decoding Algorithm 5. Forward-Backward Algorithm 6. Profile HMMs 7. HMM Parameter Estimation 8. Viterbi Training
Hidden Markov Models Outline 1. CG-Islands 2. The Fair Bet Casino 3. Hidden Markov Model 4. Decoding Algorithm 5. Forward-Backward Algorithm 6. Profile HMMs 7. HMM Parameter Estimation 8. Viterbi Training
Similarity or Identity? When are molecules similar?
 Similarity or Identity? When are molecules similar? Mapping Identity A -> A T -> T G -> G C -> C or Leu -> Leu Pro -> Pro Arg -> Arg Phe -> Phe etc If we map similarity using identity, how similar are
Similarity or Identity? When are molecules similar? Mapping Identity A -> A T -> T G -> G C -> C or Leu -> Leu Pro -> Pro Arg -> Arg Phe -> Phe etc If we map similarity using identity, how similar are
CISC 889 Bioinformatics (Spring 2004) Sequence pairwise alignment (I)
 Sequence pairwise alignment (I)") CISC 889 Bioinformatics (Spring 2004) Sequence pairwise alignment (I) Contents Alignment algorithms Needleman-Wunsch (global alignment) Smith-Waterman (local alignment) Heuristic algorithms FASTA BLAST
CISC 889 Bioinformatics (Spring 2004) Sequence pairwise alignment (I) Contents Alignment algorithms Needleman-Wunsch (global alignment) Smith-Waterman (local alignment) Heuristic algorithms FASTA BLAST
Phylogenetics: Parsimony
 1 Phylogenetics: Parsimony COMP 571 Luay Nakhleh, Rice University he Problem 2 Input: Multiple alignment of a set S of sequences Output: ree leaf-labeled with S Assumptions Characters are mutually independent
1 Phylogenetics: Parsimony COMP 571 Luay Nakhleh, Rice University he Problem 2 Input: Multiple alignment of a set S of sequences Output: ree leaf-labeled with S Assumptions Characters are mutually independent
Overview Multiple Sequence Alignment
 Overview Multiple Sequence Alignment Inge Jonassen Bioinformatics group Dept. of Informatics, UoB Inge.Jonassen@ii.uib.no Definition/examples Use of alignments The alignment problem scoring alignments
Overview Multiple Sequence Alignment Inge Jonassen Bioinformatics group Dept. of Informatics, UoB Inge.Jonassen@ii.uib.no Definition/examples Use of alignments The alignment problem scoring alignments
Multiple Sequence Alignment, Gunnar Klau, December 9, 2005, 17:
 Multiple Sequence Alignment, Gunnar Klau, December 9, 2005, 17:50 5001 5 Multiple Sequence Alignment The first part of this exposition is based on the following sources, which are recommended reading:
Multiple Sequence Alignment, Gunnar Klau, December 9, 2005, 17:50 5001 5 Multiple Sequence Alignment The first part of this exposition is based on the following sources, which are recommended reading:
C E N T R. Introduction to bioinformatics 2007 E B I O I N F O R M A T I C S V U F O R I N T. Lecture 5 G R A T I V. Pair-wise Sequence Alignment
 C E N T R E F O R I N T E G R A T I V E B I O I N F O R M A T I C S V U Introduction to bioinformatics 2007 Lecture 5 Pair-wise Sequence Alignment Bioinformatics Nothing in Biology makes sense except in
C E N T R E F O R I N T E G R A T I V E B I O I N F O R M A T I C S V U Introduction to bioinformatics 2007 Lecture 5 Pair-wise Sequence Alignment Bioinformatics Nothing in Biology makes sense except in
Effects of Gap Open and Gap Extension Penalties
 Brigham Young University BYU ScholarsArchive All Faculty Publications 200-10-01 Effects of Gap Open and Gap Extension Penalties Hyrum Carroll hyrumcarroll@gmail.com Mark J. Clement clement@cs.byu.edu See
Brigham Young University BYU ScholarsArchive All Faculty Publications 200-10-01 Effects of Gap Open and Gap Extension Penalties Hyrum Carroll hyrumcarroll@gmail.com Mark J. Clement clement@cs.byu.edu See
Motivating the need for optimal sequence alignments...
 1 Motivating the need for optimal sequence alignments... 2 3 Note that this actually combines two objectives of optimal sequence alignments: (i) use the score of the alignment o infer homology; (ii) use
1 Motivating the need for optimal sequence alignments... 2 3 Note that this actually combines two objectives of optimal sequence alignments: (i) use the score of the alignment o infer homology; (ii) use
Exercise 5. Sequence Profiles & BLAST
 Exercise 5 Sequence Profiles & BLAST 1 Substitution Matrix (BLOSUM62) Likelihood to substitute one amino acid with another Figure taken from https://en.wikipedia.org/wiki/blosum 2 Substitution Matrix (BLOSUM62)
Exercise 5 Sequence Profiles & BLAST 1 Substitution Matrix (BLOSUM62) Likelihood to substitute one amino acid with another Figure taken from https://en.wikipedia.org/wiki/blosum 2 Substitution Matrix (BLOSUM62)
Introduction to Bioinformatics
 Introduction to Bioinformatics Jianlin Cheng, PhD Department of Computer Science Informatics Institute 2011 Topics Introduction Biological Sequence Alignment and Database Search Analysis of gene expression
Introduction to Bioinformatics Jianlin Cheng, PhD Department of Computer Science Informatics Institute 2011 Topics Introduction Biological Sequence Alignment and Database Search Analysis of gene expression
Pairwise alignment. 2.1 Introduction GSAQVKGHGKKVADALTNAVAHVDDMPNALSALSD----LHAHKL
 2 Pairwise alignment 2.1 Introduction The most basic sequence analysis task is to ask if two sequences are related. This is usually done by first aligning the sequences (or parts of them) and then deciding
2 Pairwise alignment 2.1 Introduction The most basic sequence analysis task is to ask if two sequences are related. This is usually done by first aligning the sequences (or parts of them) and then deciding
M.O. Dayhoff, R.M. Schwartz, and B. C, Orcutt
 A Model of volutionary Change in Proteins M.O. Dayhoff, R.M. Schwartz, and B. C, Orcutt n the eight years since we last examined the amino acid exchanges seen in closely related proteins,' the information
A Model of volutionary Change in Proteins M.O. Dayhoff, R.M. Schwartz, and B. C, Orcutt n the eight years since we last examined the amino acid exchanges seen in closely related proteins,' the information
Local Alignment: Smith-Waterman algorithm
 Local Alignment: Smith-Waterman algorithm Example: a shared common domain of two protein sequences; extended sections of genomic DNA sequence. Sensitive to detect similarity in highly diverged sequences.
Local Alignment: Smith-Waterman algorithm Example: a shared common domain of two protein sequences; extended sections of genomic DNA sequence. Sensitive to detect similarity in highly diverged sequences.
Phylogenetics: Building Phylogenetic Trees
 1 Phylogenetics: Building Phylogenetic Trees COMP 571 Luay Nakhleh, Rice University 2 Four Questions Need to be Answered What data should we use? Which method should we use? Which evolutionary model should
1 Phylogenetics: Building Phylogenetic Trees COMP 571 Luay Nakhleh, Rice University 2 Four Questions Need to be Answered What data should we use? Which method should we use? Which evolutionary model should
Chapter 5. Proteomics and the analysis of protein sequence Ⅱ
 Proteomics Chapter 5. Proteomics and the analysis of protein sequence Ⅱ 1 Pairwise similarity searching (1) Figure 5.5: manual alignment One of the amino acids in the top sequence has no equivalent and
Proteomics Chapter 5. Proteomics and the analysis of protein sequence Ⅱ 1 Pairwise similarity searching (1) Figure 5.5: manual alignment One of the amino acids in the top sequence has no equivalent and
Today s Lecture: HMMs
 Today s Lecture: HMMs Definitions Examples Probability calculations WDAG Dynamic programming algorithms: Forward Viterbi Parameter estimation Viterbi training 1 Hidden Markov Models Probability models
Today s Lecture: HMMs Definitions Examples Probability calculations WDAG Dynamic programming algorithms: Forward Viterbi Parameter estimation Viterbi training 1 Hidden Markov Models Probability models
A New Similarity Measure among Protein Sequences
 A New Similarity Measure among Protein Sequences Kuen-Pin Wu, Hsin-Nan Lin, Ting-Yi Sung and Wen-Lian Hsu * Institute of Information Science Academia Sinica, Taipei 115, Taiwan Abstract Protein sequence
A New Similarity Measure among Protein Sequences Kuen-Pin Wu, Hsin-Nan Lin, Ting-Yi Sung and Wen-Lian Hsu * Institute of Information Science Academia Sinica, Taipei 115, Taiwan Abstract Protein sequence
Chapter 7: Rapid alignment methods: FASTA and BLAST
 Chapter 7: Rapid alignment methods: FASTA and BLAST The biological problem Search strategies FASTA BLAST Introduction to bioinformatics, Autumn 2007 117 BLAST: Basic Local Alignment Search Tool BLAST (Altschul
Chapter 7: Rapid alignment methods: FASTA and BLAST The biological problem Search strategies FASTA BLAST Introduction to bioinformatics, Autumn 2007 117 BLAST: Basic Local Alignment Search Tool BLAST (Altschul
Phylogenetics: Building Phylogenetic Trees. COMP Fall 2010 Luay Nakhleh, Rice University
 Phylogenetics: Building Phylogenetic Trees COMP 571 - Fall 2010 Luay Nakhleh, Rice University Four Questions Need to be Answered What data should we use? Which method should we use? Which evolutionary
Phylogenetics: Building Phylogenetic Trees COMP 571 - Fall 2010 Luay Nakhleh, Rice University Four Questions Need to be Answered What data should we use? Which method should we use? Which evolutionary
EECS730: Introduction to Bioinformatics
 EECS730: Introduction to Bioinformatics Lecture 07: profile Hidden Markov Model http://bibiserv.techfak.uni-bielefeld.de/sadr2/databasesearch/hmmer/profilehmm.gif Slides adapted from Dr. Shaojie Zhang
EECS730: Introduction to Bioinformatics Lecture 07: profile Hidden Markov Model http://bibiserv.techfak.uni-bielefeld.de/sadr2/databasesearch/hmmer/profilehmm.gif Slides adapted from Dr. Shaojie Zhang
Sequence Alignment Techniques and Their Uses
 Sequence Alignment Techniques and Their Uses Sarah Fiorentino Since rapid sequencing technology and whole genomes sequencing, the amount of sequence information has grown exponentially. With all of this
Sequence Alignment Techniques and Their Uses Sarah Fiorentino Since rapid sequencing technology and whole genomes sequencing, the amount of sequence information has grown exponentially. With all of this
Hidden Markov Models
 Hidden Markov Models Outline 1. CG-Islands 2. The Fair Bet Casino 3. Hidden Markov Model 4. Decoding Algorithm 5. Forward-Backward Algorithm 6. Profile HMMs 7. HMM Parameter Estimation 8. Viterbi Training
Hidden Markov Models Outline 1. CG-Islands 2. The Fair Bet Casino 3. Hidden Markov Model 4. Decoding Algorithm 5. Forward-Backward Algorithm 6. Profile HMMs 7. HMM Parameter Estimation 8. Viterbi Training
Statistical Distributions of Optimal Global Alignment Scores of Random Protein Sequences
 BMC Bioinformatics This Provisional PDF corresponds to the article as it appeared upon acceptance. The fully-formatted PDF version will become available shortly after the date of publication, from the
BMC Bioinformatics This Provisional PDF corresponds to the article as it appeared upon acceptance. The fully-formatted PDF version will become available shortly after the date of publication, from the
Alignment principles and homology searching using (PSI-)BLAST. Jaap Heringa Centre for Integrative Bioinformatics VU (IBIVU)
BLAST. Jaap Heringa Centre for Integrative Bioinformatics VU (IBIVU)") Alignment principles and homology searching using (PSI-)BLAST Jaap Heringa Centre for Integrative Bioinformatics VU (IBIVU) http://ibivu.cs.vu.nl Bioinformatics Nothing in Biology makes sense except in
Alignment principles and homology searching using (PSI-)BLAST Jaap Heringa Centre for Integrative Bioinformatics VU (IBIVU) http://ibivu.cs.vu.nl Bioinformatics Nothing in Biology makes sense except in
EVOLUTIONARY DISTANCES
 EVOLUTIONARY DISTANCES FROM STRINGS TO TREES Luca Bortolussi 1 1 Dipartimento di Matematica ed Informatica Università degli studi di Trieste luca@dmi.units.it Trieste, 14 th November 2007 OUTLINE 1 STRINGS:
EVOLUTIONARY DISTANCES FROM STRINGS TO TREES Luca Bortolussi 1 1 Dipartimento di Matematica ed Informatica Università degli studi di Trieste luca@dmi.units.it Trieste, 14 th November 2007 OUTLINE 1 STRINGS:
Lecture 4. Models of DNA and protein change. Likelihood methods
 Lecture 4. Models of DNA and protein change. Likelihood methods Joe Felsenstein Department of Genome Sciences and Department of Biology Lecture 4. Models of DNA and protein change. Likelihood methods p.1/36
Lecture 4. Models of DNA and protein change. Likelihood methods Joe Felsenstein Department of Genome Sciences and Department of Biology Lecture 4. Models of DNA and protein change. Likelihood methods p.1/36