Exploring Evolution & Bioinformatics
|
|
|
- Grant Phillips
- 5 years ago
- Views:
Transcription
 differs")


1 Chapter 6 Exploring Evolution & Bioinformatics Jane Goodall The human sequence (red) differs from the chimpanzee sequence (blue) in only one amino acid in a protein chain of 153 residues for myoglobin
2 Like members of a human family, members of molecular families often have features in common. Such family resemblance is most easily detected by comparing threedimensional structure, the aspect of a molecule most closely linked to function. Consider as an example ribonuclease from cows, which was introduced in our consideration of protein folding (Section 2.6). Comparing structures reveals that the three-dimensional structure of this protein and that of a human ribonuclease are quite similar (Figure 6.1).

3 Although the degree of overlap between these two structures is not unexpected, given their nearly identical biological functions, similarities revealed by other such comparisons are sometimes surprising. For example, angiogenin, a protein that stimulates the growth of new blood vessels, also turns out to be structurally similar to ribonuclease so similar that it is clear that both angiogenin and ribonuclease are clearly members of the same protein family Angiogenin and ribonuclease must have had a common ancestor at some earlier stage of evolution.
4 Three-dimensional structures have been determined for only a small proportion of the total number of proteins. In contrast, gene sequences and the corresponding amino acid sequences are available for a great number of proteins, largely owing to the tremendous power of DNA cloning and sequencing techniques including applications to complete genome sequencing. Evolutionary relationships also are manifest in amino acid sequences. For example, 35% of the amino acids in corresponding positions are identical in the sequences of bovine ribonuclease and angiogenin. Is this level sufficiently high to ensure an evolutionary relationship? In this chapter, we shall examine the methods that are used to compare amino acid sequences and to deduce such evolutionary relationships. Sequence-comparison methods have become one of the most powerful tools in modem biochemistry.
5 By examining the footprints present in modern protein sequences, the biochemist can become a molecular archeologist able to learn about events in the evolutionary past. Sequence comparisons can often reveal pathways of evolutionary descent and estimated dates of specific evolutionary landmarks. Molecular evolution can also be studied experimentally. In some cases, DNA from fossils can be amplified by PCR methods and sequenced, giving a direct view into the past. In addition, investigators can observe molecular evolution taking place in the laboratory, through experiments based on nucleic acid replication. The results of such studies are revealing more about how evolution proceeds.
6 6.1 Homologs Are Descended from a Common Ancestor The most fundamental relationship between two entities is homology; two molecules are said to be homologous if they have been derived from a common ancestor. Homologous molecules, or homologs, can be divided into two classes. Paralogs are homologs that are present within one species. Paralogs often differ in their detailed biochemical functions. Orthologs are homologs that are present within different species and have very similar or identical functions. How can we tell whether two human proteins are paralogs or whether a yeast protein is the ortholog of a human protein?
7 6.2 Statistical Analysis of Sequence Alignments Can Detect Homology Both nucleic acid and protein sequences can be compared to detect homology. Because nucleic acids are composed of fewer building blocks than proteins (4 bases versus 20 amino acids), the likelihood of random agreement between two DNA or RNA sequences is significantly greater than that for protein sequences. For this reason, detection of homology between protein sequences is typically far more effective.
8 Let us consider a class of proteins called the globins. Myoglobin is a protein that binds oxygen in muscle, whereas hemoglobin is the oxygen-carrying protein in blood (Chapter 7). Both proteins cradle a heme group, an iron-containing organic molecule that binds the oxygen. Each human hemoglobin molecule is composed of four heme-containing polypeptide chains, two identical a chains and two identical β-chains. Here, we consider only the α-chain. To examine the similarity between the amino acid sequence of the human α-chain and that of human myoglobin (Figure 6.4), we apply a method, referred to as a sequence alignment, in which the two sequences are systematically aligned with respect to each other to identify regions of significant overlap.
9 How can we tell where to align the two sequences? In the course of evolution, the sequences of two proteins that have an ancestor in common will have diverged in a variety of ways. Insertions and deletions may have occurred at the ends of the proteins or within the functional domains themselves. Individual amino acids may have been mutated to other residues of varying degrees of similarity To understand how the methods of sequence alignment take these potential into account,

10 let us first consider the simplest approach, where we slide one sequence past the other, one amino acid at a time, and count the number of matched residues, or sequence identities. For α-hemoglobin and myoglobin, the best alignment reveals 23 sequence identities, spread throughout the central parts of the sequences. However, we see that another alignment, featuring 22 identities, is nearly as good. This alignment is shifted by six residues and yields identities that are concentrated toward the aminoterminal end of the sequences.

11 By introducing a gap into one of the sequences, the identities found in both alignments will be represented 38 identities : 38 x (+10) = gap : 1 x (-25) = 25 Score: 355 Insertion of gaps allows the alignment method to compensate for the insertions or deletions of nucleotides that may have taken place in the gene for one molecule but not the other in the course of evolution. These methods use scoring systems to compare different alignments, including penalties for gaps to prevent the insertion of an unreasonable number of them. Next, we must determine the significance of this score and level of identity
12 The Statistical Significance of Alignments Can Be Estimated by Shuffling The similarities in sequence in Figure 6.6 appear striking, yet there remains the possibility that a grouping of sequence identities has occurred by chance alone. Hence, we can assess the significance of our alignment by shuffling, or randomly rearranging, one of the sequences, repeat the sequence alignment, and determine a new alignment score.
13 When this procedure is applied to the sequences of myoglobin and a-hemoglobin the authentic alignment clearly stands out (Figure 6.8). Its score is far above the mean for the alignment scores based on shuffled sequences. The probability that such a deviation occurred by chance alone are approximately 1 in Thus, we can comfortably conclude that the two sequences are genuinely similar; the simplest explanation for this similarity is that these sequences are homologous that is, that the two molecules have descended by divergence from a common ancestor.
14 Distant Evolutionary Relationships Can Be Detected Through the Use of Substitution Matrices No credit is given for any pairing that is not an identity. Not all substitutions are equivalent. Amino acid changes classified as structurally conservative or nonconservative. A conservative substitution replace one amino acid with another that is similar in size and chemical properties. Conservative substitutions may have only minor effects on protein structure and often can be tolerated without compromising protein function. In contrast, in a nonconservative substitution, an amino acid is replaced by one that is structurally dissimilar. Conservative and single-nucleotide substitutions are likely to be more common than are substitutions with more radical effects. How can we account for the type of substitution when comparing sequences?

15 Blosum-62 substitution matrix Blosum: Blocks of amino acid substitution matrix

16 Notice that scores are not the same for each residue, owing to the less frequently occurring amino acids such as cysteine (C) and tryptophan (W) will align by chance less often than the more common residues align. Furthermore, structurally conservative substitutions such as lysine (K) for arginine (R) and isoleucine (I) for valine (V) have relatively high scores, whereas nonconservative substitutions such as lysine for tryptophan result in negative scores. In addition, the introduction of a single residue gap lowers the alignment score by 12 points and the extension of an existing gap costs 2 points per residue.
 and relatively few are strongly disfavored types (Figure 6.11).")
17 With the use of this scoring system, the alignment shown in Figure 6.6 receives a score of 115. In many regions, most substitutions are conservative (defined as those substitutions with scores greater than 0) and relatively few are strongly disfavored types (Figure 6.11). Yellow: conservative substitution Orange: identity This scoring system detects homology between less obviously related sequences with greater sensitivity than would a comparison of identities only.

18 Consider, for example, the protein leghemoglobin, an oxygen-binding protein found in the roots of some plants. Scoring based on identities by chance alone are 1 in 20. In contrast, users of the substitution matrix the odds of the alignment occurring by chance approximately 1 in 300.

19 Thus, an analysis performed by using the substitution matrix reaches a much firmer conclusion about the evolutionary relationship between these proteins Experience with sequence analysis has led to sequence identities greater than 25% are probably homologous. less than 15% identical is unlikely to indicate statistically significant similarity. between 15 and 25% identical, further analysis is necessary to determine the statistical significance of the alignment. It must be emphasized that the lack of a statistically significant degree of sequence similarity does not rule out homology.
20 Databases Can Be Searched to Identify Homologous Sequences When the sequence of a protein is first determined, comparing it with all previously characterized sequences can be a source of tremendous insight into its evolutionary relatives and, hence, its structure and function. The sequence-alignment methods just described are used to compare an individual sequence with all members of a database of known sequences. Database searches are most often accomplished by using resources available on the Internet at the National Center for Biotechnology Information ( The procedure used is referred to as a BLAST (Basic Local Alignment Search Tool) search. An amino acid sequence is typed or pasted into the Web browser, and a search is performed, most often against a non-redundant database of all known sequences. At the end of 2009, this database included more than 10 million sequences. A BLAST search yields a list of sequence alignments, each accompanied by an estimate giving the likelihood that the alignment occurred by chance (Figure 6.14).

21
22 In 1995, investigators reported the first complete sequence of the genome of a free-living organism, the bacterium Haemophilus infuenzae. With the sequences available, they performed a BLAST search with each deduced protein sequence. Of 1,743 identified protein-coding regions, also called open reading frames, 1,007 (58%) could be linked to some protein of known function that had been previously characterized in another organism. An additional 347 open reading frames could be linked to sequences in the database for which no function had yet been assigned ("hypothetical proteins"). The remaining 389 sequences did not match any sequence present in the database at that time. Thus, investigators were able to identify likely functions for more than half the proteins within this organism solely by sequence comparisons. Sequence-comparison methods have become one of the most powerful tools in modem biochemistry.
23 6.3 Examination of Three-Dimensional Structure Enhances Our Understanding of Evolutionary Relationships Sequence comparison is a powerful tool for extending our knowledge of protein function and kinship. However, biomolecules generally function as intricate three-dimensional structures rather than as linear polymers. Mutations occur at the level of sequence, but the effects of the mutations are at the level of function, and function is directly related to tertiary structure. Consequently, to gain a deeper understanding of evolutionary relationships between proteins, we must examine three-dimensional structures, especially in conjunction with sequence information. The techniques of structural determination are presented in Chapter 3.
.")
24 Tertiary Structure Is More Conserved Than Primary Structure Tertiary structures of the globins are extremely similar even though the similarity between human myoglobin and lupine leghemoglobin is just barely detectable at the sequence level and that between human α hemoglobin and lupine leghemoglobin is not statistically significant (15.6% identity). This structural similarity firmly establishes that the framework that binds the heme group and facilitates the reversible binding of oxygen has been conserved over a long evolutionary period.
, which assists protein folding inside cells.")
25 In a growing number of other cases, however, a comparison of three-dimensional structures has revealed striking similarities between proteins that were not expected to be related. A case in point is the protein actin, a major component of the cytoskeleton, and heat shock protein 70 (Hsp-70), which assists protein folding inside cells. These two proteins were found to be noticeably similar in structure despite only 15.6% sequence identity. On the basis of their three-dimensional structures, actin and Hsp-70 are paralogs.
26 Knowledge of Three-Dimensional Structures Can Aid in the Evaluation of Sequence Alignments The sequence-comparison methods described thus far treat all positions within a sequence equally. However, we know from examining families of homologous proteins for which at least one three-dimensional structure is known that regions and residues critical to protein function are more strongly conserved than are other residues. For example, each type of globin contains a bound heme group with an iron atom at its center. A histidine residue that interacts directly with this iron (residue 64 in human myoglobin) is conserved in all globins. After we have identified key residues or highly conserved sequences within a family of proteins, we can sometimes identify other family members even when the overall level of sequence similarity is below statistical significance. Thus it may be useful to generate a sequence template a map of conserved residues that are structurally and functionally important and are characteristic of particular families of proteins

27 Repeated Motifs Can Be Detected by Aligning Sequences with Themselves More than 10% of all proteins contain sets of two or more domains that are similar to one another. Sequence search methods can often detect internally repeated sequences that have been characterized in other proteins. Their presence can be detected by attempting to align a given sequence with itself. For the TATA-box-binding protein, a key protein in controlling gene transcription such an alignment is highly significant: 30% of the amino acids are identical

28 The determination of the three-dimensional structure of the TATA-boxbinding protein confirmed the presence of repeated structures; the protein is formed of two nearly identical domains. The evidence is convincing that the gene encoding this protein evolved by duplication of a gene encoding a single domain.
29 Convergent Evolution Illustrates Common Solutions to Biochemical Challenges Thus far, we have been exploring proteins derived from common ancestors that is, through divergent evolution. Other cases have been found of proteins that are structurally similar in important ways but are not descended from a common ancestor. How might two unrelated proteins come to resemble each other structurally? Two proteins evolving independently may have converged on a similar structure to perform a similar biochemical activity. Perhaps that structure was an especially effective solution to a biochemical problem that organisms face. The process by which very different evolutionary pathways lead to the same solution is called convergent evolution.
30 One example of convergent evolution is found among the serine proteases. These enzymes, to be considered in more detail in Chapter 9, cleave peptide bonds by hydrolysis. Figure 6.18 shows the structure of the active sites that is, the sites on the proteins at which the hydrolysis reaction takes place for two such enzymes, chymotrypsin and subtilisin. These active-site structures are remarkably similar. In each case, a serine residue, a histidine residue, and an aspartic acid residue are positioned in space in nearly identical arrangements. However, the key serine, histidine, and aspartic acid residues do not occupy similar positions or even appear in the same order within the two sequences.

31 As we will see, this is the case because chymotrypsin and subtilisin use the same mechanistic solution to the problem of peptide hydrolysis. At first glance, this similarity might suggest that these proteins are homologous. However, striking differences in the overall structures of these proteins make an evolutionary relationship extremely unlikely (Figure 6.19). Whereas chymotrypsin consists almost entirely of β-sheets, subtilisin contains extensive α-helical structure. It is extremely unlikely that two proteins evolving from a common ancestor could have retained similar active-site structures while other aspects of the structure changed so dramatically.
32 Comparison of RNA Sequences Can Be a Source of Insight into RNA Secondary Structures Homologous RNA sequences can be compared in a manner similar to that already described, Such comparisons can be a source of important insights into evolutionary relationships; in addition, they provide clues to the three dimensional structure of the RNA itself. In a family of sequences that form similar base-paired structures, base sequences may vary, but base-pairing ability is conserved. Consider, for example, a region from a large RNA molecule present in the ribosomes of all organisms reveals that the bases in positions 9 and 22 retain the ability to form a Watson- Crick base pair even though the identities of the bases in these positions vary.
33 6.4 Evolutionary Trees Can Be Constructed on the Basis of Sequence Information The observation that homology is often manifested as sequence similarity suggests an evolutionary tree

34 Such comparisons reveal only the relative divergence times for example, that myoglobin diverged from hemoglobin twice as long ago as the α-chain diverged from the β-chain. How can we estimate the approximate dates of gene duplications and other evolutionary events? Evolutionary trees can be calibrated by comparing the deduced branch points with divergence times determined from the fossil record. For example, the duplication leading to the two chains of hemoglobin appears to have occurred 350 million years ago. This estimate is supported by the observation that jawless fish such as the lamprey, which diverged from bony fish approximately 400 million years ago, contain hemoglobin built from a single type of subunit (Figure 6. 22).
35 6.5 Modern Techniques Make the Experimental Exploration of Evolution Possible Two techniques of biochemistry have made it possible to examine the course of evolution more directly and not simply by inference. The polymerase chain reaction allows the direct examination of ancient DNA sequences, releasing us, at least in some cases, from the constraints of being able to examine existing genomes from living organisms only. Molecular evolution may be investigated through the use of combinatorial chemistry, the process of producing large populations of molecules en masse and selecting for a biochemical property. This exciting process provides a glimpse into the types of molecules that may have existed very early in evolution.
36 Ancient DNA Can Sometimes Be Amplified and Sequenced The tremendous chemical stability of DNA makes the molecule well suited to its role as the storage site of genetic information. So stable is the molecule that samples of DNA have survived for many thousands of years under appropriate conditions. With the development of PCR methods, such ancient DNA can sometimes be amplified and sequenced. This approach has been applied to mitochondrial DNA from a Neanderthal fossil estimated at 38,000 years of age excavated from Vindija Cave, Croatia, in Remarkably, investigators have completely sequenced the mitochondrial genome from this specimen. Comparison of the Neanderthal mitochondrial sequence with those from Homo sapiens individuals revealed between 201 and 234 substitutions, considerably fewer than the approximately 1,500 differences between human beings and chimpanzees over the same regions.
37 Further analysis suggested that the common ancestor of modern human beings and Neanderthals lived approximately 660,000 years ago. An evolutionary tree constructed by using these data and others revealed that the Neanderthal was not an intermediate between chimpanzees and human beings but, instead, was an evolutionary "dead end" that became extinct (Figure 6.23 ). A few earlier studies claimed to determine the sequences of far more ancient DNA such as that found in insects trapped in amber, but these studies appear to have been flawed. The source of these sequences turned out to be contaminating modern DNA. Successful sequencing of ancient DNA requires sufficient DNA for reliable amplification and the rigorous exclusion of all sources of contamination.



38 7 May 2010 Neanderthal Genome 2006 Nov

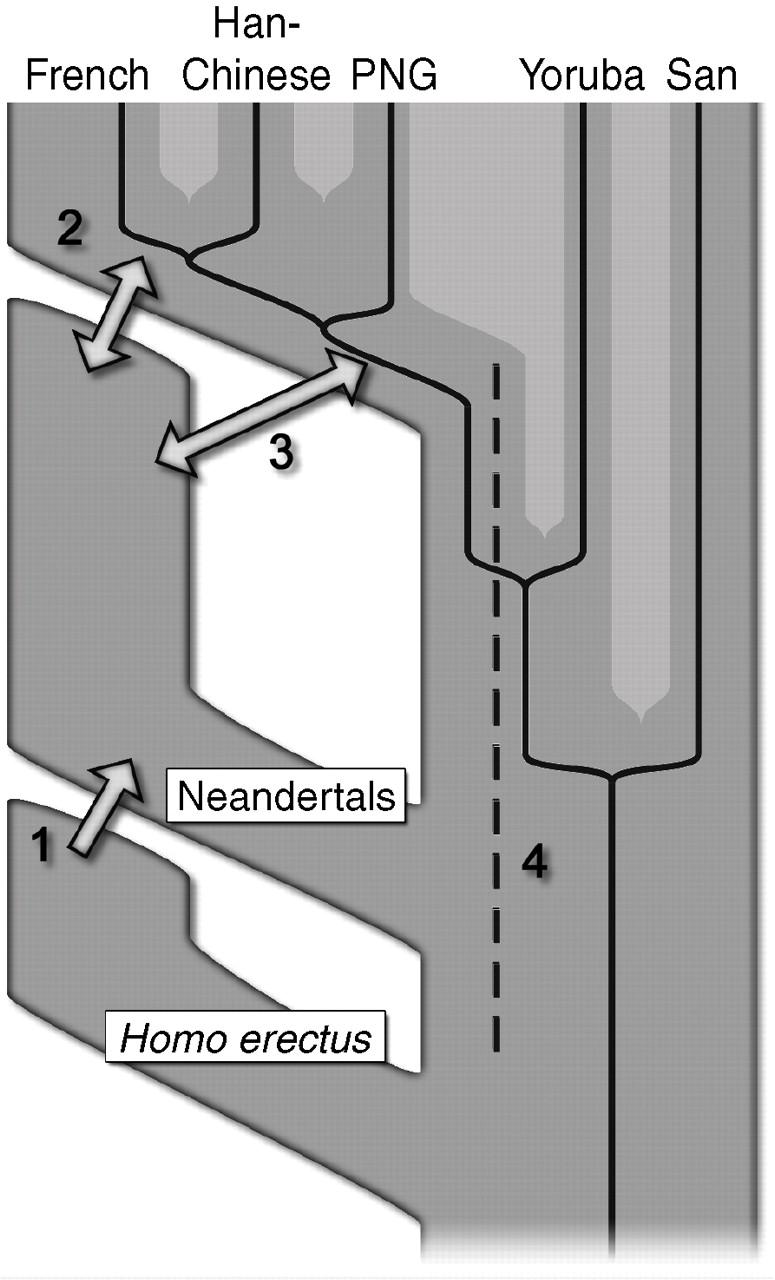

39 7 May 2010
40 Molecular Evolution Can Be Examined Experimentally Evolution requires three processes: (1) the generation of a diverse population (2) the selection of members based on some criterion of fitness (3) reproduction to enrich the population in these more-fit members. Nucleic acid molecules are capable of undergoing all three processes in vitro under appropriate conditions. The results of such studies enable us to glimpse how evolutionary processes might have generated catalytic activities and specific binding abilities important biochemical functions in all living systems.
41 A diverse population of nucleic acid molecules can be synthesized in the laboratory by the process of combinatorial chemistry When an initial population has been generated, it is subjected to a selection process that isolates specific molecules with desired binding or reactivity properties. Finally, molecules that have survived the selection process are replicated through the use of PCR; Errors that occur naturally in the course of the replication process introduce additional variation into the population in each generation.
42 Conserved 2 nd structure The new population was subjected to additional rounds of selection for ATP-binding activity. After eight generations, members of the selected population were characterized by sequencing. Seventeen different sequences were obtained, 16 of which could form the structure shown in Figure Each of these molecules bound ATP with high affinity, as indicated by dissociation constants less than 50 µm. The collection of molecules that were bound well by the ATP affinity column were allowed to replicate by reverse transcription into DNA, amplification by PCR, and transcription back into RNA. The somewhat error-prone replication processes introduced additional mutations into the population in each cycle.

43 The folded structure of the ATP-binding region from one of these RNAs was determined by nuclear magnetic resonance methods (Figure 6.26). As expected, this 40-nucleotide molecule is composed of two Watson- Crick base-paired helical regions separated by an 11-nucleotide loop. This loop folds back on itself in an intricate way to form a deep pocket into which the adenine ring can fit. Thus, a structure had evolved that was capable of a specific interaction.
METHODS FOR DETERMINING PHYLOGENY. In Chapter 11, we discovered that classifying organisms into groups was, and still is, a difficult task.
 Chapter 12 (Strikberger) Molecular Phylogenies and Evolution METHODS FOR DETERMINING PHYLOGENY In Chapter 11, we discovered that classifying organisms into groups was, and still is, a difficult task. Modern
Chapter 12 (Strikberger) Molecular Phylogenies and Evolution METHODS FOR DETERMINING PHYLOGENY In Chapter 11, we discovered that classifying organisms into groups was, and still is, a difficult task. Modern
Algorithms in Bioinformatics FOUR Pairwise Sequence Alignment. Pairwise Sequence Alignment. Convention: DNA Sequences 5. Sequence Alignment
 Algorithms in Bioinformatics FOUR Sami Khuri Department of Computer Science San José State University Pairwise Sequence Alignment Homology Similarity Global string alignment Local string alignment Dot
Algorithms in Bioinformatics FOUR Sami Khuri Department of Computer Science San José State University Pairwise Sequence Alignment Homology Similarity Global string alignment Local string alignment Dot
Chapter 5. Proteomics and the analysis of protein sequence Ⅱ
 Proteomics Chapter 5. Proteomics and the analysis of protein sequence Ⅱ 1 Pairwise similarity searching (1) Figure 5.5: manual alignment One of the amino acids in the top sequence has no equivalent and
Proteomics Chapter 5. Proteomics and the analysis of protein sequence Ⅱ 1 Pairwise similarity searching (1) Figure 5.5: manual alignment One of the amino acids in the top sequence has no equivalent and
BIOINFORMATICS: An Introduction
 BIOINFORMATICS: An Introduction What is Bioinformatics? The term was first coined in 1988 by Dr. Hwa Lim The original definition was : a collective term for data compilation, organisation, analysis and
BIOINFORMATICS: An Introduction What is Bioinformatics? The term was first coined in 1988 by Dr. Hwa Lim The original definition was : a collective term for data compilation, organisation, analysis and
Lecture 14 - Cells. Astronomy Winter Lecture 14 Cells: The Building Blocks of Life
 Lecture 14 Cells: The Building Blocks of Life Astronomy 141 Winter 2012 This lecture describes Cells, the basic structural units of all life on Earth. Basic components of cells: carbohydrates, lipids,
Lecture 14 Cells: The Building Blocks of Life Astronomy 141 Winter 2012 This lecture describes Cells, the basic structural units of all life on Earth. Basic components of cells: carbohydrates, lipids,
Properties of amino acids in proteins
 Properties of amino acids in proteins one of the primary roles of DNA (but not the only one!) is to code for proteins A typical bacterium builds thousands types of proteins, all from ~20 amino acids repeated
Properties of amino acids in proteins one of the primary roles of DNA (but not the only one!) is to code for proteins A typical bacterium builds thousands types of proteins, all from ~20 amino acids repeated
Module: Sequence Alignment Theory and Applications Session: Introduction to Searching and Sequence Alignment
 Module: Sequence Alignment Theory and Applications Session: Introduction to Searching and Sequence Alignment Introduction to Bioinformatics online course : IBT Jonathan Kayondo Learning Objectives Understand
Module: Sequence Alignment Theory and Applications Session: Introduction to Searching and Sequence Alignment Introduction to Bioinformatics online course : IBT Jonathan Kayondo Learning Objectives Understand
Sequence Alignment: A General Overview. COMP Fall 2010 Luay Nakhleh, Rice University
 Sequence Alignment: A General Overview COMP 571 - Fall 2010 Luay Nakhleh, Rice University Life through Evolution All living organisms are related to each other through evolution This means: any pair of
Sequence Alignment: A General Overview COMP 571 - Fall 2010 Luay Nakhleh, Rice University Life through Evolution All living organisms are related to each other through evolution This means: any pair of
Quantifying sequence similarity
 Quantifying sequence similarity Bas E. Dutilh Systems Biology: Bioinformatic Data Analysis Utrecht University, February 16 th 2016 After this lecture, you can define homology, similarity, and identity
Quantifying sequence similarity Bas E. Dutilh Systems Biology: Bioinformatic Data Analysis Utrecht University, February 16 th 2016 After this lecture, you can define homology, similarity, and identity
Protein Architecture V: Evolution, Function & Classification. Lecture 9: Amino acid use units. Caveat: collagen is a. Margaret A. Daugherty.
 Lecture 9: Protein Architecture V: Evolution, Function & Classification Margaret A. Daugherty Fall 2004 Amino acid use *Proteins don t use aa s equally; eg, most proteins not repeating units. Caveat: collagen
Lecture 9: Protein Architecture V: Evolution, Function & Classification Margaret A. Daugherty Fall 2004 Amino acid use *Proteins don t use aa s equally; eg, most proteins not repeating units. Caveat: collagen
Homology Modeling (Comparative Structure Modeling) GBCB 5874: Problem Solving in GBCB
 GBCB 5874: Problem Solving in GBCB") Homology Modeling (Comparative Structure Modeling) Aims of Structural Genomics High-throughput 3D structure determination and analysis To determine or predict the 3D structures of all the proteins encoded
Homology Modeling (Comparative Structure Modeling) Aims of Structural Genomics High-throughput 3D structure determination and analysis To determine or predict the 3D structures of all the proteins encoded
Biochemistry 324 Bioinformatics. Pairwise sequence alignment
 Biochemistry 324 Bioinformatics Pairwise sequence alignment How do we compare genes/proteins? When we have sequenced a genome, we try and identify the function of unknown genes by finding a similar gene
Biochemistry 324 Bioinformatics Pairwise sequence alignment How do we compare genes/proteins? When we have sequenced a genome, we try and identify the function of unknown genes by finding a similar gene
Sequence analysis and comparison
 The aim with sequence identification: Sequence analysis and comparison Marjolein Thunnissen Lund September 2012 Is there any known protein sequence that is homologous to mine? Are there any other species
The aim with sequence identification: Sequence analysis and comparison Marjolein Thunnissen Lund September 2012 Is there any known protein sequence that is homologous to mine? Are there any other species
Bio 1B Lecture Outline (please print and bring along) Fall, 2007
 Fall, 2007") Bio 1B Lecture Outline (please print and bring along) Fall, 2007 B.D. Mishler, Dept. of Integrative Biology 2-6810, bmishler@berkeley.edu Evolution lecture #5 -- Molecular genetics and molecular evolution
Bio 1B Lecture Outline (please print and bring along) Fall, 2007 B.D. Mishler, Dept. of Integrative Biology 2-6810, bmishler@berkeley.edu Evolution lecture #5 -- Molecular genetics and molecular evolution
Computational approaches for functional genomics
 Computational approaches for functional genomics Kalin Vetsigian October 31, 2001 The rapidly increasing number of completely sequenced genomes have stimulated the development of new methods for finding
Computational approaches for functional genomics Kalin Vetsigian October 31, 2001 The rapidly increasing number of completely sequenced genomes have stimulated the development of new methods for finding
Enzyme Catalysis & Biotechnology
 L28-1 Enzyme Catalysis & Biotechnology Bovine Pancreatic RNase A Biochemistry, Life, and all that L28-2 A brief word about biochemistry traditionally, chemical engineers used organic and inorganic chemistry
L28-1 Enzyme Catalysis & Biotechnology Bovine Pancreatic RNase A Biochemistry, Life, and all that L28-2 A brief word about biochemistry traditionally, chemical engineers used organic and inorganic chemistry
Evidence for Evolution
 Evidence for Evolution 1. 2. 3. 4. 5. Paleontology Comparative Anatomy Embryology Comparative Biochemistry Geographical Distribution How old is everything? The History of Earth as a Clock Station 1: Paleontology
Evidence for Evolution 1. 2. 3. 4. 5. Paleontology Comparative Anatomy Embryology Comparative Biochemistry Geographical Distribution How old is everything? The History of Earth as a Clock Station 1: Paleontology
Berg Tymoczko Stryer Biochemistry Sixth Edition Chapter 1:
 Berg Tymoczko Stryer Biochemistry Sixth Edition Chapter 1: Biochemistry: An Evolving Science Tips on note taking... Remember copies of my lectures are available on my webpage If you forget to print them
Berg Tymoczko Stryer Biochemistry Sixth Edition Chapter 1: Biochemistry: An Evolving Science Tips on note taking... Remember copies of my lectures are available on my webpage If you forget to print them
Motivating the need for optimal sequence alignments...
 1 Motivating the need for optimal sequence alignments... 2 3 Note that this actually combines two objectives of optimal sequence alignments: (i) use the score of the alignment o infer homology; (ii) use
1 Motivating the need for optimal sequence alignments... 2 3 Note that this actually combines two objectives of optimal sequence alignments: (i) use the score of the alignment o infer homology; (ii) use
Bioinformatics Exercises
 Bioinformatics Exercises AP Biology Teachers Workshop Susan Cates, Ph.D. Evolution of Species Phylogenetic Trees show the relatedness of organisms Common Ancestor (Root of the tree) 1 Rooted vs. Unrooted
Bioinformatics Exercises AP Biology Teachers Workshop Susan Cates, Ph.D. Evolution of Species Phylogenetic Trees show the relatedness of organisms Common Ancestor (Root of the tree) 1 Rooted vs. Unrooted
C3020 Molecular Evolution. Exercises #3: Phylogenetics
 C3020 Molecular Evolution Exercises #3: Phylogenetics Consider the following sequences for five taxa 1-5 and the known outgroup O, which has the ancestral states (note that sequence 3 has changed from
C3020 Molecular Evolution Exercises #3: Phylogenetics Consider the following sequences for five taxa 1-5 and the known outgroup O, which has the ancestral states (note that sequence 3 has changed from
Bioinformatics. Scoring Matrices. David Gilbert Bioinformatics Research Centre
 Bioinformatics Scoring Matrices David Gilbert Bioinformatics Research Centre www.brc.dcs.gla.ac.uk Department of Computing Science, University of Glasgow Learning Objectives To explain the requirement
Bioinformatics Scoring Matrices David Gilbert Bioinformatics Research Centre www.brc.dcs.gla.ac.uk Department of Computing Science, University of Glasgow Learning Objectives To explain the requirement
Practical considerations of working with sequencing data
 Practical considerations of working with sequencing data File Types Fastq ->aligner -> reference(genome) coordinates Coordinate files SAM/BAM most complete, contains all of the info in fastq and more!
Practical considerations of working with sequencing data File Types Fastq ->aligner -> reference(genome) coordinates Coordinate files SAM/BAM most complete, contains all of the info in fastq and more!
Protein Structure Analysis and Verification. Course S Basics for Biosystems of the Cell exercise work. Maija Nevala, BIO, 67485U 16.1.
 Protein Structure Analysis and Verification Course S-114.2500 Basics for Biosystems of the Cell exercise work Maija Nevala, BIO, 67485U 16.1.2008 1. Preface When faced with an unknown protein, scientists
Protein Structure Analysis and Verification Course S-114.2500 Basics for Biosystems of the Cell exercise work Maija Nevala, BIO, 67485U 16.1.2008 1. Preface When faced with an unknown protein, scientists
"Nothing in biology makes sense except in the light of evolution Theodosius Dobzhansky
 MOLECULAR PHYLOGENY "Nothing in biology makes sense except in the light of evolution Theodosius Dobzhansky EVOLUTION - theory that groups of organisms change over time so that descendeants differ structurally
MOLECULAR PHYLOGENY "Nothing in biology makes sense except in the light of evolution Theodosius Dobzhansky EVOLUTION - theory that groups of organisms change over time so that descendeants differ structurally
(Lys), resulting in translation of a polypeptide without the Lys amino acid. resulting in translation of a polypeptide without the Lys amino acid.
, resulting in translation of a polypeptide without the Lys amino acid. resulting in translation of a polypeptide without the Lys amino acid.") 1. A change that makes a polypeptide defective has been discovered in its amino acid sequence. The normal and defective amino acid sequences are shown below. Researchers are attempting to reproduce the
1. A change that makes a polypeptide defective has been discovered in its amino acid sequence. The normal and defective amino acid sequences are shown below. Researchers are attempting to reproduce the
THEORY. Based on sequence Length According to the length of sequence being compared it is of following two types
 Exp 11- THEORY Sequence Alignment is a process of aligning two sequences to achieve maximum levels of identity between them. This help to derive functional, structural and evolutionary relationships between
Exp 11- THEORY Sequence Alignment is a process of aligning two sequences to achieve maximum levels of identity between them. This help to derive functional, structural and evolutionary relationships between
CONCEPT OF SEQUENCE COMPARISON. Natapol Pornputtapong 18 January 2018
 CONCEPT OF SEQUENCE COMPARISON Natapol Pornputtapong 18 January 2018 SEQUENCE ANALYSIS - A ROSETTA STONE OF LIFE Sequence analysis is the process of subjecting a DNA, RNA or peptide sequence to any of
CONCEPT OF SEQUENCE COMPARISON Natapol Pornputtapong 18 January 2018 SEQUENCE ANALYSIS - A ROSETTA STONE OF LIFE Sequence analysis is the process of subjecting a DNA, RNA or peptide sequence to any of
Genomics and bioinformatics summary. Finding genes -- computer searches
 Genomics and bioinformatics summary 1. Gene finding: computer searches, cdnas, ESTs, 2. Microarrays 3. Use BLAST to find homologous sequences 4. Multiple sequence alignments (MSAs) 5. Trees quantify sequence
Genomics and bioinformatics summary 1. Gene finding: computer searches, cdnas, ESTs, 2. Microarrays 3. Use BLAST to find homologous sequences 4. Multiple sequence alignments (MSAs) 5. Trees quantify sequence
Lecture 11 Friday, October 21, 2011
 Lecture 11 Friday, October 21, 2011 Phylogenetic tree (phylogeny) Darwin and classification: In the Origin, Darwin said that descent from a common ancestral species could explain why the Linnaean system
Lecture 11 Friday, October 21, 2011 Phylogenetic tree (phylogeny) Darwin and classification: In the Origin, Darwin said that descent from a common ancestral species could explain why the Linnaean system
Molecular evolution - Part 1. Pawan Dhar BII
 Molecular evolution - Part 1 Pawan Dhar BII Theodosius Dobzhansky Nothing in biology makes sense except in the light of evolution Age of life on earth: 3.85 billion years Formation of planet: 4.5 billion
Molecular evolution - Part 1 Pawan Dhar BII Theodosius Dobzhansky Nothing in biology makes sense except in the light of evolution Age of life on earth: 3.85 billion years Formation of planet: 4.5 billion
Chapter 7: Covalent Structure of Proteins. Voet & Voet: Pages ,
 Chapter 7: Covalent Structure of Proteins Voet & Voet: Pages 163-164, 185-194 Slide 1 Structure & Function Function is best understood in terms of structure Four levels of structure that apply to proteins
Chapter 7: Covalent Structure of Proteins Voet & Voet: Pages 163-164, 185-194 Slide 1 Structure & Function Function is best understood in terms of structure Four levels of structure that apply to proteins
Sequence comparison: Score matrices. Genome 559: Introduction to Statistical and Computational Genomics Prof. James H. Thomas
 Sequence comparison: Score matrices Genome 559: Introduction to Statistical and omputational Genomics Prof James H Thomas Informal inductive proof of best alignment path onsider the last step in the best
Sequence comparison: Score matrices Genome 559: Introduction to Statistical and omputational Genomics Prof James H Thomas Informal inductive proof of best alignment path onsider the last step in the best
Graph Alignment and Biological Networks
 Graph Alignment and Biological Networks Johannes Berg http://www.uni-koeln.de/ berg Institute for Theoretical Physics University of Cologne Germany p.1/12 Networks in molecular biology New large-scale
Graph Alignment and Biological Networks Johannes Berg http://www.uni-koeln.de/ berg Institute for Theoretical Physics University of Cologne Germany p.1/12 Networks in molecular biology New large-scale
Homology and Information Gathering and Domain Annotation for Proteins
 Homology and Information Gathering and Domain Annotation for Proteins Outline Homology Information Gathering for Proteins Domain Annotation for Proteins Examples and exercises The concept of homology The
Homology and Information Gathering and Domain Annotation for Proteins Outline Homology Information Gathering for Proteins Domain Annotation for Proteins Examples and exercises The concept of homology The
Pairwise & Multiple sequence alignments
 Pairwise & Multiple sequence alignments Urmila Kulkarni-Kale Bioinformatics Centre 411 007 urmila@bioinfo.ernet.in Basis for Sequence comparison Theory of evolution: gene sequences have evolved/derived
Pairwise & Multiple sequence alignments Urmila Kulkarni-Kale Bioinformatics Centre 411 007 urmila@bioinfo.ernet.in Basis for Sequence comparison Theory of evolution: gene sequences have evolved/derived
Genomes and Their Evolution
 Chapter 21 Genomes and Their Evolution PowerPoint Lecture Presentations for Biology Eighth Edition Neil Campbell and Jane Reece Lectures by Chris Romero, updated by Erin Barley with contributions from
Chapter 21 Genomes and Their Evolution PowerPoint Lecture Presentations for Biology Eighth Edition Neil Campbell and Jane Reece Lectures by Chris Romero, updated by Erin Barley with contributions from
Introduction to" Protein Structure
 Introduction to" Protein Structure Function, evolution & experimental methods Thomas Blicher, Center for Biological Sequence Analysis Learning Objectives Outline the basic levels of protein structure.
Introduction to" Protein Structure Function, evolution & experimental methods Thomas Blicher, Center for Biological Sequence Analysis Learning Objectives Outline the basic levels of protein structure.
Statistical Machine Learning Methods for Biomedical Informatics II. Hidden Markov Model for Biological Sequences
 Statistical Machine Learning Methods for Biomedical Informatics II. Hidden Markov Model for Biological Sequences Jianlin Cheng, PhD William and Nancy Thompson Missouri Distinguished Professor Department
Statistical Machine Learning Methods for Biomedical Informatics II. Hidden Markov Model for Biological Sequences Jianlin Cheng, PhD William and Nancy Thompson Missouri Distinguished Professor Department
Lecture 4: Evolutionary Models and Substitution Matrices (PAM and BLOSUM)
") Bioinformatics II Probability and Statistics Universität Zürich and ETH Zürich Spring Semester 2009 Lecture 4: Evolutionary Models and Substitution Matrices (PAM and BLOSUM) Dr Fraser Daly adapted from
Bioinformatics II Probability and Statistics Universität Zürich and ETH Zürich Spring Semester 2009 Lecture 4: Evolutionary Models and Substitution Matrices (PAM and BLOSUM) Dr Fraser Daly adapted from
UoN, CAS, DBSC BIOL102 lecture notes by: Dr. Mustafa A. Mansi. The Phylogenetic Systematics (Phylogeny and Systematics)
") - Phylogeny? - Systematics? The Phylogenetic Systematics (Phylogeny and Systematics) - Phylogenetic systematics? Connection between phylogeny and classification. - Phylogenetic systematics informs the
- Phylogeny? - Systematics? The Phylogenetic Systematics (Phylogeny and Systematics) - Phylogenetic systematics? Connection between phylogeny and classification. - Phylogenetic systematics informs the
Newly made RNA is called primary transcript and is modified in three ways before leaving the nucleus:
 m Eukaryotic mrna processing Newly made RNA is called primary transcript and is modified in three ways before leaving the nucleus: Cap structure a modified guanine base is added to the 5 end. Poly-A tail
m Eukaryotic mrna processing Newly made RNA is called primary transcript and is modified in three ways before leaving the nucleus: Cap structure a modified guanine base is added to the 5 end. Poly-A tail
Homology Modeling. Roberto Lins EPFL - summer semester 2005
 Homology Modeling Roberto Lins EPFL - summer semester 2005 Disclaimer: course material is mainly taken from: P.E. Bourne & H Weissig, Structural Bioinformatics; C.A. Orengo, D.T. Jones & J.M. Thornton,
Homology Modeling Roberto Lins EPFL - summer semester 2005 Disclaimer: course material is mainly taken from: P.E. Bourne & H Weissig, Structural Bioinformatics; C.A. Orengo, D.T. Jones & J.M. Thornton,
Sequence comparison: Score matrices
 Sequence comparison: Score matrices http://facultywashingtonedu/jht/gs559_2013/ Genome 559: Introduction to Statistical and omputational Genomics Prof James H Thomas FYI - informal inductive proof of best
Sequence comparison: Score matrices http://facultywashingtonedu/jht/gs559_2013/ Genome 559: Introduction to Statistical and omputational Genomics Prof James H Thomas FYI - informal inductive proof of best
Computational Biology: Basics & Interesting Problems
 Computational Biology: Basics & Interesting Problems Summary Sources of information Biological concepts: structure & terminology Sequencing Gene finding Protein structure prediction Sources of information
Computational Biology: Basics & Interesting Problems Summary Sources of information Biological concepts: structure & terminology Sequencing Gene finding Protein structure prediction Sources of information
CHAPTERS 24-25: Evidence for Evolution and Phylogeny
 CHAPTERS 24-25: Evidence for Evolution and Phylogeny 1. For each of the following, indicate how it is used as evidence of evolution by natural selection or shown as an evolutionary trend: a. Paleontology
CHAPTERS 24-25: Evidence for Evolution and Phylogeny 1. For each of the following, indicate how it is used as evidence of evolution by natural selection or shown as an evolutionary trend: a. Paleontology
Introduction to Comparative Protein Modeling. Chapter 4 Part I
 Introduction to Comparative Protein Modeling Chapter 4 Part I 1 Information on Proteins Each modeling study depends on the quality of the known experimental data. Basis of the model Search in the literature
Introduction to Comparative Protein Modeling Chapter 4 Part I 1 Information on Proteins Each modeling study depends on the quality of the known experimental data. Basis of the model Search in the literature
SCIENTIFIC EVIDENCE TO SUPPORT THE THEORY OF EVOLUTION. Using Anatomy, Embryology, Biochemistry, and Paleontology
 SCIENTIFIC EVIDENCE TO SUPPORT THE THEORY OF EVOLUTION Using Anatomy, Embryology, Biochemistry, and Paleontology Scientific Fields Different fields of science have contributed evidence for the theory of
SCIENTIFIC EVIDENCE TO SUPPORT THE THEORY OF EVOLUTION Using Anatomy, Embryology, Biochemistry, and Paleontology Scientific Fields Different fields of science have contributed evidence for the theory of
Introduction to Bioinformatics
 Introduction to Bioinformatics Lecture : p he biological problem p lobal alignment p Local alignment p Multiple alignment 6 Background: comparative genomics p Basic question in biology: what properties
Introduction to Bioinformatics Lecture : p he biological problem p lobal alignment p Local alignment p Multiple alignment 6 Background: comparative genomics p Basic question in biology: what properties
From gene to protein. Premedical biology
 From gene to protein Premedical biology Central dogma of Biology, Molecular Biology, Genetics transcription replication reverse transcription translation DNA RNA Protein RNA chemically similar to DNA,
From gene to protein Premedical biology Central dogma of Biology, Molecular Biology, Genetics transcription replication reverse transcription translation DNA RNA Protein RNA chemically similar to DNA,
8/23/2014. Phylogeny and the Tree of Life
 Phylogeny and the Tree of Life Chapter 26 Objectives Explain the following characteristics of the Linnaean system of classification: a. binomial nomenclature b. hierarchical classification List the major
Phylogeny and the Tree of Life Chapter 26 Objectives Explain the following characteristics of the Linnaean system of classification: a. binomial nomenclature b. hierarchical classification List the major
Copyright Mark Brandt, Ph.D A third method, cryogenic electron microscopy has seen increasing use over the past few years.
 Structure Determination and Sequence Analysis The vast majority of the experimentally determined three-dimensional protein structures have been solved by one of two methods: X-ray diffraction and Nuclear
Structure Determination and Sequence Analysis The vast majority of the experimentally determined three-dimensional protein structures have been solved by one of two methods: X-ray diffraction and Nuclear
BLAST. Varieties of BLAST
 BLAST Basic Local Alignment Search Tool (1990) Altschul, Gish, Miller, Myers, & Lipman Uses short-cuts or heuristics to improve search speed Like speed-reading, does not examine every nucleotide of database
BLAST Basic Local Alignment Search Tool (1990) Altschul, Gish, Miller, Myers, & Lipman Uses short-cuts or heuristics to improve search speed Like speed-reading, does not examine every nucleotide of database
Protein Structure. W. M. Grogan, Ph.D. OBJECTIVES
 Protein Structure W. M. Grogan, Ph.D. OBJECTIVES 1. Describe the structure and characteristic properties of typical proteins. 2. List and describe the four levels of structure found in proteins. 3. Relate
Protein Structure W. M. Grogan, Ph.D. OBJECTIVES 1. Describe the structure and characteristic properties of typical proteins. 2. List and describe the four levels of structure found in proteins. 3. Relate
1. The Fossil Record 2. Biogeography 3. Comparative Anatomy 4. Comparative Embryology 5. Molecular Biology
 What Darwin Observed. copy 1. The Fossil Record 2. Biogeography 3. Comparative Anatomy 4. Comparative Embryology 5. Molecular Biology Activity in groups copy Provides a chronological record of organisms
What Darwin Observed. copy 1. The Fossil Record 2. Biogeography 3. Comparative Anatomy 4. Comparative Embryology 5. Molecular Biology Activity in groups copy Provides a chronological record of organisms
Ohio Tutorials are designed specifically for the Ohio Learning Standards to prepare students for the Ohio State Tests and end-ofcourse
 Tutorial Outline Ohio Tutorials are designed specifically for the Ohio Learning Standards to prepare students for the Ohio State Tests and end-ofcourse exams. Biology Tutorials offer targeted instruction,
Tutorial Outline Ohio Tutorials are designed specifically for the Ohio Learning Standards to prepare students for the Ohio State Tests and end-ofcourse exams. Biology Tutorials offer targeted instruction,
Chapter 26: Phylogeny and the Tree of Life Phylogenies Show Evolutionary Relationships
 Chapter 26: Phylogeny and the Tree of Life You Must Know The taxonomic categories and how they indicate relatedness. How systematics is used to develop phylogenetic trees. How to construct a phylogenetic
Chapter 26: Phylogeny and the Tree of Life You Must Know The taxonomic categories and how they indicate relatedness. How systematics is used to develop phylogenetic trees. How to construct a phylogenetic
Warm-Up- Review Natural Selection and Reproduction for quiz today!!!! Notes on Evidence of Evolution Work on Vocabulary and Lab
 Date: Agenda Warm-Up- Review Natural Selection and Reproduction for quiz today!!!! Notes on Evidence of Evolution Work on Vocabulary and Lab Ask questions based on 5.1 and 5.2 Quiz on 5.1 and 5.2 How
Date: Agenda Warm-Up- Review Natural Selection and Reproduction for quiz today!!!! Notes on Evidence of Evolution Work on Vocabulary and Lab Ask questions based on 5.1 and 5.2 Quiz on 5.1 and 5.2 How
Sequence comparison: Score matrices. Genome 559: Introduction to Statistical and Computational Genomics Prof. James H. Thomas
 Sequence comparison: Score matrices Genome 559: Introduction to Statistical and omputational Genomics Prof James H Thomas FYI - informal inductive proof of best alignment path onsider the last step in
Sequence comparison: Score matrices Genome 559: Introduction to Statistical and omputational Genomics Prof James H Thomas FYI - informal inductive proof of best alignment path onsider the last step in
Chapter 17. From Gene to Protein. Biology Kevin Dees
 Chapter 17 From Gene to Protein DNA The information molecule Sequences of bases is a code DNA organized in to chromosomes Chromosomes are organized into genes What do the genes actually say??? Reflecting
Chapter 17 From Gene to Protein DNA The information molecule Sequences of bases is a code DNA organized in to chromosomes Chromosomes are organized into genes What do the genes actually say??? Reflecting
Biol478/ August
 Biol478/595 29 August # Day Inst. Topic Hwk Reading August 1 M 25 MG Introduction 2 W 27 MG Sequences and Evolution Handouts 3 F 29 MG Sequences and Evolution September M 1 Labor Day 4 W 3 MG Database
Biol478/595 29 August # Day Inst. Topic Hwk Reading August 1 M 25 MG Introduction 2 W 27 MG Sequences and Evolution Handouts 3 F 29 MG Sequences and Evolution September M 1 Labor Day 4 W 3 MG Database
Unit 1: Chemistry - Guided Notes
 Scientific Method Notes: Unit 1: Chemistry - Guided Notes 1 Common Elements in Biology: Atoms are made up of: 1. 2. 3. In order to be stable, an atom of an element needs a full valence shell of electrons.
Scientific Method Notes: Unit 1: Chemistry - Guided Notes 1 Common Elements in Biology: Atoms are made up of: 1. 2. 3. In order to be stable, an atom of an element needs a full valence shell of electrons.
Bioinformatics Chapter 1. Introduction
 Bioinformatics Chapter 1. Introduction Outline! Biological Data in Digital Symbol Sequences! Genomes Diversity, Size, and Structure! Proteins and Proteomes! On the Information Content of Biological Sequences!
Bioinformatics Chapter 1. Introduction Outline! Biological Data in Digital Symbol Sequences! Genomes Diversity, Size, and Structure! Proteins and Proteomes! On the Information Content of Biological Sequences!
Amira A. AL-Hosary PhD of infectious diseases Department of Animal Medicine (Infectious Diseases) Faculty of Veterinary Medicine Assiut
 Faculty of Veterinary Medicine Assiut") Amira A. AL-Hosary PhD of infectious diseases Department of Animal Medicine (Infectious Diseases) Faculty of Veterinary Medicine Assiut University-Egypt Phylogenetic analysis Phylogenetic Basics: Biological
Amira A. AL-Hosary PhD of infectious diseases Department of Animal Medicine (Infectious Diseases) Faculty of Veterinary Medicine Assiut University-Egypt Phylogenetic analysis Phylogenetic Basics: Biological
Chapters 25 and 26. Searching for Homology. Phylogeny
 Chapters 25 and 26 The Origin of Life as we know it. Phylogeny traces evolutionary history of taxa Systematics- analyzes relationships (modern and past) of organisms Figure 25.1 A gallery of fossils The
Chapters 25 and 26 The Origin of Life as we know it. Phylogeny traces evolutionary history of taxa Systematics- analyzes relationships (modern and past) of organisms Figure 25.1 A gallery of fossils The
BLAST Database Searching. BME 110: CompBio Tools Todd Lowe April 8, 2010
 BLAST Database Searching BME 110: CompBio Tools Todd Lowe April 8, 2010 Admin Reading: Read chapter 7, and the NCBI Blast Guide and tutorial http://www.ncbi.nlm.nih.gov/blast/why.shtml Read Chapter 8 for
BLAST Database Searching BME 110: CompBio Tools Todd Lowe April 8, 2010 Admin Reading: Read chapter 7, and the NCBI Blast Guide and tutorial http://www.ncbi.nlm.nih.gov/blast/why.shtml Read Chapter 8 for
Organic Chemistry Option II: Chemical Biology
 Organic Chemistry Option II: Chemical Biology Recommended books: Dr Stuart Conway Department of Chemistry, Chemistry Research Laboratory, University of Oxford email: stuart.conway@chem.ox.ac.uk Teaching
Organic Chemistry Option II: Chemical Biology Recommended books: Dr Stuart Conway Department of Chemistry, Chemistry Research Laboratory, University of Oxford email: stuart.conway@chem.ox.ac.uk Teaching
Evidence of Evolution
 Evidence of Evolution Biology Name Date Block Background Much evidence has been found to indicate that living things have evolved or changed gradually during their natural history. The study of fossils
Evidence of Evolution Biology Name Date Block Background Much evidence has been found to indicate that living things have evolved or changed gradually during their natural history. The study of fossils
3. SEQUENCE ANALYSIS BIOINFORMATICS COURSE MTAT
 3. SEQUENCE ANALYSIS BIOINFORMATICS COURSE MTAT.03.239 25.09.2012 SEQUENCE ANALYSIS IS IMPORTANT FOR... Prediction of function Gene finding the process of identifying the regions of genomic DNA that encode
3. SEQUENCE ANALYSIS BIOINFORMATICS COURSE MTAT.03.239 25.09.2012 SEQUENCE ANALYSIS IS IMPORTANT FOR... Prediction of function Gene finding the process of identifying the regions of genomic DNA that encode
Outline. Evolution: Speciation and More Evidence. Key Concepts: Evolution is a FACT. 1. Key concepts 2. Speciation 3. More evidence 4.
 Evolution: Speciation and More Evidence Evolution is a FACT 1. Key concepts 2. Speciation 3. More evidence 4. Conclusions Outline Key Concepts: A species consist of one or more populations of individuals
Evolution: Speciation and More Evidence Evolution is a FACT 1. Key concepts 2. Speciation 3. More evidence 4. Conclusions Outline Key Concepts: A species consist of one or more populations of individuals
Chapter 1. DNA is made from the building blocks adenine, guanine, cytosine, and. Answer: d
 Chapter 1 1. Matching Questions DNA is made from the building blocks adenine, guanine, cytosine, and. Answer: d 2. Matching Questions : Unbranched polymer that, when folded into its three-dimensional shape,
Chapter 1 1. Matching Questions DNA is made from the building blocks adenine, guanine, cytosine, and. Answer: d 2. Matching Questions : Unbranched polymer that, when folded into its three-dimensional shape,
Statistical Machine Learning Methods for Bioinformatics II. Hidden Markov Model for Biological Sequences
 Statistical Machine Learning Methods for Bioinformatics II. Hidden Markov Model for Biological Sequences Jianlin Cheng, PhD Department of Computer Science University of Missouri 2008 Free for Academic
Statistical Machine Learning Methods for Bioinformatics II. Hidden Markov Model for Biological Sequences Jianlin Cheng, PhD Department of Computer Science University of Missouri 2008 Free for Academic
Theory of Evolution. Chapter 15
 Theory of Evolution Chapter 15 The History of Evolutionary Thought Evolution The development of new types of organisms from preexisting types of organisms over time. Also could be described as a heritable
Theory of Evolution Chapter 15 The History of Evolutionary Thought Evolution The development of new types of organisms from preexisting types of organisms over time. Also could be described as a heritable
Biology. Slide 1 of 36. End Show. Copyright Pearson Prentice Hall
 Biology 1 of 36 2 of 36 Formation of Earth Formation of Earth Hypotheses about Earth s early history are based on a relatively small amount of evidence. Gaps and uncertainties make it likely that scientific
Biology 1 of 36 2 of 36 Formation of Earth Formation of Earth Hypotheses about Earth s early history are based on a relatively small amount of evidence. Gaps and uncertainties make it likely that scientific
Advanced Certificate in Principles in Protein Structure. You will be given a start time with your exam instructions
 BIRKBECK COLLEGE (University of London) Advanced Certificate in Principles in Protein Structure MSc Structural Molecular Biology Date: Thursday, 1st September 2011 Time: 3 hours You will be given a start
BIRKBECK COLLEGE (University of London) Advanced Certificate in Principles in Protein Structure MSc Structural Molecular Biology Date: Thursday, 1st September 2011 Time: 3 hours You will be given a start
Chem Lecture 3 Hemoglobin & Myoglobin
 Chem 452 - Lecture 3 Hemoglobin & Myoglobin 111003 Hemoglobin (Hb) and Myoglobin (Mb) function as oxygen transport and storage molecules in higher organisms. There functions have been long studied and,
Chem 452 - Lecture 3 Hemoglobin & Myoglobin 111003 Hemoglobin (Hb) and Myoglobin (Mb) function as oxygen transport and storage molecules in higher organisms. There functions have been long studied and,
4. The Michaelis-Menten combined rate constant Km, is defined for the following kinetic mechanism as k 1 k 2 E + S ES E + P k -1
 Fall 2000 CH 595C Exam 1 Answer Key Multiple Choice 1. One of the reasons that enzymes are such efficient catalysts is that a) the energy level of the enzyme-transition state complex is much higher than
Fall 2000 CH 595C Exam 1 Answer Key Multiple Choice 1. One of the reasons that enzymes are such efficient catalysts is that a) the energy level of the enzyme-transition state complex is much higher than
Basic Local Alignment Search Tool
 Basic Local Alignment Search Tool Alignments used to uncover homologies between sequences combined with phylogenetic studies o can determine orthologous and paralogous relationships Local Alignment uses
Basic Local Alignment Search Tool Alignments used to uncover homologies between sequences combined with phylogenetic studies o can determine orthologous and paralogous relationships Local Alignment uses
2 Genome evolution: gene fusion versus gene fission
 2 Genome evolution: gene fusion versus gene fission Berend Snel, Peer Bork and Martijn A. Huynen Trends in Genetics 16 (2000) 9-11 13 Chapter 2 Introduction With the advent of complete genome sequencing,
2 Genome evolution: gene fusion versus gene fission Berend Snel, Peer Bork and Martijn A. Huynen Trends in Genetics 16 (2000) 9-11 13 Chapter 2 Introduction With the advent of complete genome sequencing,
Christian Sigrist. November 14 Protein Bioinformatics: Sequence-Structure-Function 2018 Basel
 Christian Sigrist General Definition on Conserved Regions Conserved regions in proteins can be classified into 5 different groups: Domains: specific combination of secondary structures organized into a
Christian Sigrist General Definition on Conserved Regions Conserved regions in proteins can be classified into 5 different groups: Domains: specific combination of secondary structures organized into a
Ch 3: Chemistry of Life. Chemistry Water Macromolecules Enzymes
 Ch 3: Chemistry of Life Chemistry Water Macromolecules Enzymes Chemistry Atom = smallest unit of matter that cannot be broken down by chemical means Element = substances that have similar properties and
Ch 3: Chemistry of Life Chemistry Water Macromolecules Enzymes Chemistry Atom = smallest unit of matter that cannot be broken down by chemical means Element = substances that have similar properties and
An Introduction to Sequence Similarity ( Homology ) Searching
 Searching") An Introduction to Sequence Similarity ( Homology ) Searching Gary D. Stormo 1 UNIT 3.1 1 Washington University, School of Medicine, St. Louis, Missouri ABSTRACT Homologous sequences usually have the same,
An Introduction to Sequence Similarity ( Homology ) Searching Gary D. Stormo 1 UNIT 3.1 1 Washington University, School of Medicine, St. Louis, Missouri ABSTRACT Homologous sequences usually have the same,
Dr. Amira A. AL-Hosary
 Phylogenetic analysis Amira A. AL-Hosary PhD of infectious diseases Department of Animal Medicine (Infectious Diseases) Faculty of Veterinary Medicine Assiut University-Egypt Phylogenetic Basics: Biological
Phylogenetic analysis Amira A. AL-Hosary PhD of infectious diseases Department of Animal Medicine (Infectious Diseases) Faculty of Veterinary Medicine Assiut University-Egypt Phylogenetic Basics: Biological
Structures and Functions of Living Organisms (LS1)
") EALR 4: Big Idea: Core Content: Life Science Structures and Functions of Living Organisms (LS1) Processes Within Cells In prior grades students learned that all living systems are composed of cells which
EALR 4: Big Idea: Core Content: Life Science Structures and Functions of Living Organisms (LS1) Processes Within Cells In prior grades students learned that all living systems are composed of cells which
Algorithms in Bioinformatics
 Algorithms in Bioinformatics Sami Khuri Department of omputer Science San José State University San José, alifornia, USA khuri@cs.sjsu.edu www.cs.sjsu.edu/faculty/khuri Pairwise Sequence Alignment Homology
Algorithms in Bioinformatics Sami Khuri Department of omputer Science San José State University San José, alifornia, USA khuri@cs.sjsu.edu www.cs.sjsu.edu/faculty/khuri Pairwise Sequence Alignment Homology
Tutorial 4 Substitution matrices and PSI-BLAST
 Tutorial 4 Substitution matrices and PSI-BLAST 1 Agenda Substitution Matrices PAM - Point Accepted Mutations BLOSUM - Blocks Substitution Matrix PSI-BLAST Cool story of the day: Why should we care about
Tutorial 4 Substitution matrices and PSI-BLAST 1 Agenda Substitution Matrices PAM - Point Accepted Mutations BLOSUM - Blocks Substitution Matrix PSI-BLAST Cool story of the day: Why should we care about
Local Alignment Statistics
 Local Alignment Statistics Stephen Altschul National Center for Biotechnology Information National Library of Medicine National Institutes of Health Bethesda, MD Central Issues in Biological Sequence Comparison
Local Alignment Statistics Stephen Altschul National Center for Biotechnology Information National Library of Medicine National Institutes of Health Bethesda, MD Central Issues in Biological Sequence Comparison
Computational Biology
 Computational Biology Lecture 6 31 October 2004 1 Overview Scoring matrices (Thanks to Shannon McWeeney) BLAST algorithm Start sequence alignment 2 1 What is a homologous sequence? A homologous sequence,
Computational Biology Lecture 6 31 October 2004 1 Overview Scoring matrices (Thanks to Shannon McWeeney) BLAST algorithm Start sequence alignment 2 1 What is a homologous sequence? A homologous sequence,
Sequence Alignment (chapter 6)
") Sequence lignment (chapter 6) he biological problem lobal alignment Local alignment Multiple alignment Introduction to bioinformatics, utumn 6 Background: comparative genomics Basic question in biology:
Sequence lignment (chapter 6) he biological problem lobal alignment Local alignment Multiple alignment Introduction to bioinformatics, utumn 6 Background: comparative genomics Basic question in biology:
BIOINFORMATICS LAB AP BIOLOGY
 BIOINFORMATICS LAB AP BIOLOGY Bioinformatics is the science of collecting and analyzing complex biological data. Bioinformatics combines computer science, statistics and biology to allow scientists to
BIOINFORMATICS LAB AP BIOLOGY Bioinformatics is the science of collecting and analyzing complex biological data. Bioinformatics combines computer science, statistics and biology to allow scientists to
Piecing It Together. 1) The envelope contains puzzle pieces for 5 vertebrate embryos in 3 different stages of
 The envelope contains puzzle pieces for 5 vertebrate embryos in 3 different stages of") Piecing It Together 1) The envelope contains puzzle pieces for 5 vertebrate embryos in 3 different stages of development. Lay out the pieces so that you have matched up each animal name card with its 3
Piecing It Together 1) The envelope contains puzzle pieces for 5 vertebrate embryos in 3 different stages of development. Lay out the pieces so that you have matched up each animal name card with its 3
1. In most cases, genes code for and it is that
 Name Chapter 10 Reading Guide From DNA to Protein: Gene Expression Concept 10.1 Genetics Shows That Genes Code for Proteins 1. In most cases, genes code for and it is that determine. 2. Describe what Garrod
Name Chapter 10 Reading Guide From DNA to Protein: Gene Expression Concept 10.1 Genetics Shows That Genes Code for Proteins 1. In most cases, genes code for and it is that determine. 2. Describe what Garrod
98 Washington State K-12 Science Learning Standards Version 1.2
 EALR 4: Big Idea: Core Content: Life Science Structures and Functions of Living Organisms (LS1) Processes Within Cells In prior grades students learned that all living systems are composed of cells which
EALR 4: Big Idea: Core Content: Life Science Structures and Functions of Living Organisms (LS1) Processes Within Cells In prior grades students learned that all living systems are composed of cells which
Bioinformatics. Dept. of Computational Biology & Bioinformatics
 Bioinformatics Dept. of Computational Biology & Bioinformatics 3 Bioinformatics - play with sequences & structures Dept. of Computational Biology & Bioinformatics 4 ORGANIZATION OF LIFE ROLE OF BIOINFORMATICS
Bioinformatics Dept. of Computational Biology & Bioinformatics 3 Bioinformatics - play with sequences & structures Dept. of Computational Biology & Bioinformatics 4 ORGANIZATION OF LIFE ROLE OF BIOINFORMATICS
Biomolecules. Energetics in biology. Biomolecules inside the cell
 Biomolecules Energetics in biology Biomolecules inside the cell Energetics in biology The production of energy, its storage, and its use are central to the economy of the cell. Energy may be defined as
Biomolecules Energetics in biology Biomolecules inside the cell Energetics in biology The production of energy, its storage, and its use are central to the economy of the cell. Energy may be defined as
Phylogenetic inference
 Phylogenetic inference Bas E. Dutilh Systems Biology: Bioinformatic Data Analysis Utrecht University, March 7 th 016 After this lecture, you can discuss (dis-) advantages of different information types
Phylogenetic inference Bas E. Dutilh Systems Biology: Bioinformatic Data Analysis Utrecht University, March 7 th 016 After this lecture, you can discuss (dis-) advantages of different information types
MATHEMATICAL MODELS - Vol. III - Mathematical Modeling and the Human Genome - Hilary S. Booth MATHEMATICAL MODELING AND THE HUMAN GENOME
 MATHEMATICAL MODELING AND THE HUMAN GENOME Hilary S. Booth Australian National University, Australia Keywords: Human genome, DNA, bioinformatics, sequence analysis, evolution. Contents 1. Introduction:
MATHEMATICAL MODELING AND THE HUMAN GENOME Hilary S. Booth Australian National University, Australia Keywords: Human genome, DNA, bioinformatics, sequence analysis, evolution. Contents 1. Introduction:
Phylogeny & Systematics
 Phylogeny & Systematics Phylogeny & Systematics An unexpected family tree. What are the evolutionary relationships among a human, a mushroom, and a tulip? Molecular systematics has revealed that despite
Phylogeny & Systematics Phylogeny & Systematics An unexpected family tree. What are the evolutionary relationships among a human, a mushroom, and a tulip? Molecular systematics has revealed that despite
A. Incorrect! In the binomial naming convention the Kingdom is not part of the name.
 Microbiology Problem Drill 08: Classification of Microorganisms No. 1 of 10 1. In the binomial system of naming which term is always written in lowercase? (A) Kingdom (B) Domain (C) Genus (D) Specific
Microbiology Problem Drill 08: Classification of Microorganisms No. 1 of 10 1. In the binomial system of naming which term is always written in lowercase? (A) Kingdom (B) Domain (C) Genus (D) Specific
B. Phylogeny and Systematics:
 Tracing Phylogeny A. Fossils: Some fossils form as is weathered and eroded from the land and carried by rivers to seas and where the particles settle to the bottom. Deposits pile up and the older sediments
Tracing Phylogeny A. Fossils: Some fossils form as is weathered and eroded from the land and carried by rivers to seas and where the particles settle to the bottom. Deposits pile up and the older sediments