Molecular Dynamics. Coherency of Molecular Motions. Local Minima & Global Minimum. Theoretical Basis of Molecular Dynamic Calculations
|
|
|
- Drusilla Clarke
- 5 years ago
- Views:
Transcription
1 Molecular Dynamics Molecular Dynamics provide a good approach to determine the preferred conformers and global minimum of a molecule Theoretical Basis of Molecular Dynamic Calculations Molecular dynamics is an extension of the molecular mechanics approach & based on the idea that the atoms of the molecule feel forces and want to move. Each atom is treated as a particle responding to Newton s equations of motion. Integration of these equations with successive time steps lead to the trajectory of the atom over time in the form of a list of positions & velocities This is achieved by the simulations of the dynamic motions of the molecule as it vibrates and undergoes internal rotation Local Minima & Global Minimum Molecular dynamics simulations can be used to obtain low energy conformers. These simulations can run with differing temperatures to obtain different families of conformers. At higher temperatures more conformers are possible, and it becomes feasible to cross energy barriers. Coherency of Molecular Motions The motions of atoms and chemical groups obtained by molecular dynamics simulations reveal subtle underlying molecular machinery. At the beginning of the simulation, motions are frequently interrupted by collisions with neighbouring groups, and each group seems to have an erratic trajectory. However, over longer periods of time, coherent and collective motions start to develop, revealing how some groups can fluctuate somewhat more than others A Typical Molecular Dynamics Run It is possible to generate about 100 conformations and minimise them with molecular mechanics Molecular Mechanics The minimised forms are then used as starting points for dynamics simulations varying time and temperature For each starting point, a number of steps of dynamics at 1 femtosecond intervals are made & the co-ordinates and energy of each point are recorded The conformation representing every picosecond step is saved, and finally, at the end of the study, the atoms trajectory can be displayed as a movie by quickly displaying the sequence of individual frames The co-ordinates and energies of conformers with low energies are saved. 1
2 Molecular Mechanics Molecular Mechanics Molecular Dynamics Molecular Dynamics Conformational Analysis THIS STUDY raison d etre The androgen receptor was docked with both steroidal and non steroidal molecules in the previous exercise It is now interesting to investigate further whether there are different binding modalities between the steroidal & non-steroidal molecules in the androgen receptor active site THIS STUDY raison d etre The ligand docked into the active site of the protein thus re-presents the variable for this molecular dynamics study All other conditions will be kept constant after it is determined that all systems are in equilibrium STUDY VARIABLES STUDY VARIABLES Metribolone High affinity steroid 2

; and a package of molecular simulation programs which includes source code and demos.")
3 STUDY VARIABLES STUDY VARIABLES Metribolone High affinity Steroid Corticosterone Low Affinity Steroid Metribolone High affinity Steroid Corticosterone Low Affinity Steroid Undocked STUDY VARIABLES STUDY VARIABLES Metribolone High affinity Corticosterone Diethylstilboestrol Steroid Low Affinity High Affinity Steroid Undocked Non-Steroid Metribolone Vinclozolin High affinity Corticosterone Diethylstilboestrol Low Affinity Steroid Low Affinity High Affinity Non-Steroid Steroid Undocked Non-Steroid AMBER 8 "Amber" refers to two things: a set of molecular mechanical force fields for the simulation of biomolecules (which are in the public domain, and are used in a variety of simulation programs); and a package of molecular simulation programs which includes source code and demos. The current version of the code is Amber version 8, which is distributed by UCSF subject to a licensing agreement described on the amber website. A good general overview of the Amber codes can be found in: D.A. Pearlman, D.A. Case, J.W. Caldwell, W.R. Ross, T.E. Cheatham, III, S. DeBolt, D. Ferguson, G. Seibel and P. Kollman. AMBER, a computer program for applying molecular mechanics, normal mode analysis, molecular dynamics and free energy calculations to elucidate the structures and energies of molecules. Comp. Phys. Commun. 91, 1-41 (1995). An overview of the Amber protein force fields, and how they were developed, can be found in: J.W. Ponder and D.A. Case. Force fields for protein simulations. Adv. Prot. Chem. 66, (2003). Similar information for nucleic acids is given by T.E. Cheatham, III and M.A. Young. Molecular dynamics simulation of nucleic acids: Successes, limitations and promise. Biopolymers 56, (2001). In this discussion the term amber will refer to the software package which we will be using to run simulations controlled via the Amber force field. The AMBER 8 Suite.. Programs used for this simulation sander: the "main" program used for molecular dynamics simulations, and is also used for replicaexchange, thermodynamic integration, and potential of mean force (PMF) calculations. LEaP: LEaP is an X-windows-based program that provides for basic model building and Amber coordinate and parameter/topology input file creation. It includes a molecular editor which allows for building residues and manipulating molecules. ptraj: This is used to analyze MD trajectories, computing a variety of things, like RMS deviation from a reference structure, hydrogen bonding analysis, time-correlation functions, diffusional behavior, and so on. 3


4 Simulation Plan Create the prmtop and inpcrd files: This is a description of how to generate the initial structure and set up the molecular topology/parameter and coordinate files necessary for performing minimisation or dynamics with sander Minimisation and molecular dynamics in explicit solvent: Setting up and running equilibration simulation and perform basic analysis such as calculating root-mean-squared deviations (RMSd) and plotting various energy terms as a function of time Production simulations for the protein model using TIP3P explicit water. Starting the Simulation. What is the source for the co-ordinates? What representation should be used and what should be simulated? How are prmtop and inpcrd files necessary to parameterise the protein complex & start the simulation built?... Starting Off. The first step in any modelling project is developing the initial model structure Experimentally determined structures are used. These can be found by searching through databases of crystal or NMR structures such as the Protein Data Bank or the Cambridge Structural Database. With nucleic acids, users can also search the Nucleic Acid Database Starting Off. Xleap is the program within the Amber Suite that is used to parameterise the protein ligand complex. The various different residues and their names are defined in library files that xleap loads on startup. The names used for the residues in the pdb files must match those defined in the default xleap library files or in user defined library files. What level of simulation should be attempted? At this point the level of simulation realism to be used must be decided upon: in vacuo models Solvated modelssolvent with implicit or explicit Reading co-ordinates into xleap The protein:ligand complexes were created in Sybyl and pdb files written These were read in to xleap, and where necessary residue nomenclature was changed, and libraries updated to include any unrecognised functional groups When no errors were observed, the protein:ligand complexes were visualised in xleap to ensure that the tertiary structure of the protein had been retained Explicit solvent methods are the most realistic, at the expense of computer intensiveness. This model was adopted for this study 4


.")
5 Solvating the system Explicit waters were added to the system in xleap The protein:ligand complex was placed in an 8A box of water TIP3P water is used to create the box. This is a rigid water molecule in which the two O-H bonds and the H-O-H angle are rigid. Solvating the system- the truncated octahedron Reduces computer intensiveness Less water molecules XLEAP Output Files For each of the 5 systems being studied 2 output files were generated separately from xleap prmtop: The parameter/topology file. This defines the connectivity and parameters for the current model. This information is static, i.e., it does not change during the simulation. The prmtop created also contains the solvated box information inpcrd: The coordinates (and optionally box coordinates and velocities). This is data is not static and changes during the simulations (although the file is unaltered). Running Minimisation and Molecular Dynamics These were run using the Sander module in the Amber8 suite The minimisation and dynamics stages were run on the protein-ligand complexes enclosed in an 8A TIP3P water box Running Minimisation and Molecular Dynamics The minimisation/dynamics process was run in 3 stages as follows: Relaxing the system prior to MD Molecular dynamics at constant temperature Analysing the results Why Minimise? The geometry assigned to the protein:ligand complex may not correspond to the actual minima in the force field being used and may also result in conflicts and overlaps with atoms in other residues It is always wise to minimise the locations of these atoms before commencing molecular dynamics. Failure to successfully minimise these atoms may lead to instabilities when Molecular Dynamics are being run 5

6 Minimisation in Explicit Solvent Running Molecular Dynamics in vacuo, for biomolecular systems, are problematic because these normally exist in a solvated environments, & hence do not accurately represent the system. One solution to this is to use explicit solvent. Using explicit solvent, however, can be expensive computationally This significantly increases the complexity of the simulations increasing both the time required to run the simulations and also the way in which the simulations are set up, and the results visualised Setting up the Minimisation The problems with any bad van der Waal (non bond) and electrostatic interactions in the initial structure, are considerably magnified when simulations are run in explicit solvent. The water molecules have not felt the influence of the solute or charges and moreover there may be gaps between the solvent and solute and solvent and box edges. Such holes can lead to "vacuum" bubbles forming and subsequently an instability in themolecular dynamics simulation. Setting up the Minimisation Thus careful minimisation must be run before slowly heating our system to 300 K. It is also a good idea to allow the water box to relax during a MD equilibration stage prior to running production MD. In this phase it is a good idea, since periodic boundaries will be used, to keep the pressure constant and so allow the volume of the box to change. This approach will allow the water to equilibrate around the solute and come to an equilibrium density. It is essential that this equilibrium phase be monitored in order to be certain that the solvated system has reached equilibrium before production data is obtained from the MD simulation. Setting up the Minimisation In this Molecular Dynamics study, periodic boundary constraints will be applied. After equilibration has been demonstrated and deemed to have been successful, a "production" MD run will be set up. Periodic Boundary Conditions Periodic Boundary Conditions A realistic model of a solution requires a very large number of solvent molecules to be included along with the solute. Simply placing the solute in a box of solvent is not sufficient, however, since while some solvent molecules will be at the boundary between solute and solvent and others will be within the bulk of the solvent a large number will be at the edge of the solvent and the surrounding vacuum. This is obviously not a realistic picture of a bulk fluid. In order to prevent the outer solvent molecules from boiling off into space, and to allow a relatively small number of solvent molecules to reproduce the properties of the bulk, periodic boundary conditions are employed. In this method the particles being simulated are enclosed in a box which is then replicated in all three dimensions to give a periodic array. In this periodic array a particle at position r represents an infinite set of particles at: r + ix + jy+ kz (i,j,k -> -inf, +inf) where x, y and z are the vectors corresponding to the box edges. During the simulation only one of the particles is represented, but the effects are reproduced over all the image particles with each particle not only interacting with the other particles but also with their images in neighbouring boxes. Particles that leave one side of the box re-enter from the opposite side as their image. In this way the total number of particles in the central box remains constant. Upon initial inspection such a method would appear to be very computationally intensive requiring the evaluation of an infinite number of interacting pairs. However, by choosing the non-bonded cutoff distance such that each atom sees only one image of all of the other atoms the complexity is greatly reduced. Hence as long as the box size is more than twice the cut-off distance it is impossible for a particle to interact with any two images of the same particle simultaneously. 6
7 The Cut-Off The choice of cutoff can have a dramatic influence on the results obtained from the MD simulation Fortunately the effect of the cutoff size is not as marked in a solution phase simulation as it is in vacuo The use of the particle mesh Ewald method for treating the long range electrostatics also reduces the effect of the cutoff. However, there is still a trade off between cutoff size and simulation time. Minimisation Stage 1 - Holding the solute fixed The minimisation procedure of the solvated protein:ligand complexes consisted of a two stage approach In the first stage the protein:ligand complex was kept fixed and only the water molecules were minimised. Then in the second stage the entire system was minimised. Positional restraints were placed on each atom in the protein:ligand complex to keep them essentially fixed in the same position. Such restraints work by specifying a reference structure, in this case the starting structure, and then restraining the selected atoms to conform to this structure via the use of a force constant. This can be visualised as basically attaching a spring to each of the solute atoms that connects it to its initial position. Moving the atom from this position results in a force which acts to restore it to the initial position. By varying the magnitude of the force constant the effect can be increased or decreased. Script File for Minimisation Protein:ligand complex min1.in The meaning of each of the terms are as follows: Protein:ligand complex : initial IMIN = 1: Minimisation is turned on (no MD) MAXCYC = 1000: Conduct a total of 1,000 minimisation solvent steps of minimisation. &cntrl NCYC = 500: Initially do 500 steps of steepest descent minimisation followed by imin = 1, 500 steps (MAXCYC - NCYC) steps of conjugate gradient minimisation. maxcyc = 1000, NTB = 1: Use constant volume periodic boundaries CUT = 8: Use a cutoff of 8 ncyc = 500, angstroms. NTR = 1: Use position restraints based on ntb = 1, the GROUP input given in the input file. The GROUP input is the last 5 lines of the input ntr = 1, file: cut = 8 Hold the complex fixed / RES 1 20 END Hold the complex fixed 500 kcal/mol-a**2 is a very large force constant, much larger than is needed Suitablefor minimization, but for dynamics, 10 kcal/mol-a**2 is normal RES END Script File for Minimisation Protein:ligand complex min2.in The meaning of each of the terms are as follows: Protein:ligand complex : second IMIN = 1: Minimisation is turned on (no minimisation whole system MD) MAXCYC = 2500: Conduct a total of &cntrl 2500 steps of minimisation. imin = 1, NCYC = 1000: Initially do 1000 steps of steepest descent minimisation maxcyc = 2500, followed by 1500 steps (MAXCYC - NCYC) steps of conjugate gradient ncyc = 1000, minimisation. ntb = 1, NTB = 1: Use constant volume periodic boundaries CUT = 8: Use a ntr = 0, cutoff of 8 angstroms. cut = 8 NTR = 0: No position restraints This stage of the minimisation thus / minimised the entire system via 2,500 steps of minimisation without the restraints Starting Equilibration Dynamics Now that the system is minimised the next stage in the equilibration protocol is to allow the system to heat up from 0 K to 300 K In order to ensure this happens without any wild fluctuations in the solute a weak restraint will be used, as was used stage 1 of the minimisation, on the solute (protein:ligand) atoms. AMBER 8 supports the new Langevin temperature equilibration scheme (NTT=3) which is significantly better at maintaining and equalising the system temperature. Starting Equilibration Dynamics The ultimate aim is to run production dynamics at constant temperature and pressure since this more closely resembles laboratory conditions However, at the low temperatures, the systems being considered will be at for the first few ps, the calculation of pressure is very inaccurate and so using constant pressure periodic boundaries in this situation can lead to problems Using constant pressure with restraints can also cause problems So initially 20 ps of MD will be run at constant volume. Once our system has equilibrated over approximately 20 ps the restraints will be switched off, a change to a constant pressure simulation will be made before running a further 100 ps of equilibration at 300 K. 7
8 Starting Equilibration Dynamics Starting Equilibration Dynamics Since these simulations are very computationally expensive,it is essential that the computational complexity is reduced as much as possible. One way to do this is to use triangulated water, that is water in which the angle between the hydrogens is kept fixed One such model is the TIP3P water model with which the systems are solvated. When using such a water model it is essential that the hydrogen atom motion of the water also be fixed since failure to do this can lead to very large inaccuracies in the calculation of the densities etc. Since the hydrogen atom motion in the complex is unlikely to effect its large scale dynamics it is possible to fix these hydrogens as well. One method of doing this is to use the SHAKE algorithm in which all bonds involving hydrogen are constrained (NTC=2) This method of removing hydrogen motion has the fortunate effect of removing the highest frequency oscillation in the system, that of the hydrogen vibrations Since it is the highest frequency oscillation that determines the maximum size of the time step, by removing the hydrogen motion the time step may be increased to 2 fs without introducing any instability into the MD simulation This has the effect of allowing the covering of a given amount of phase space in half as much time since only 50,000 steps to cover 100 ps in time as opposed to the 100,000 required with a 1 fs time step Script File for 20 ps Dynamics (weak positional restraints on complex) Protein:ligand complex md1.in Protein:ligand : 20ps MD with res on DNA &cntrl imin = 0, irest = 0, ntx = 1, ntb = 1, cut = 10, ntr = 1, ntc = 2, ntf = 2, tempi = 0.0, temp0 = 300.0, ntt = 3, gamma_ln = 1.0, nstlim = 10000, dt = ntpr = 100, ntwx = 100, ntwr = 1000 / Keep DNA fixed with weak restraints 10.0 RES END END Script File for 20 ps Dynamics (weak positional restraints on complex) IMIN = 0: Minimisation is turned off (MD on) IREST = 0, NTX = 1: This is the first stage of our molecular dynamics. Later these values will change to indicate that a molecular dynamics run from where this run stopped. NTB = 1: Use constant volume periodic boundaries CUT = 8: Use a cutoff of 8 angstroms. NTR = 1: Use position restraints based on the GROUP input given in the input file. In this case the complex will be restrained with a force constant of 10 angstroms. NTC = 2, NTF = 2: SHAKE is be turned on and used to constrain bonds involving hydrogen. TEMPI = 0.0, TEMP0 = 300.0: Simulation with a temperature, of 0 K and will heat up to 300 K. NTT = 3, GAMMA_LN = 1.0: The langevin dynamics control the temperature using a collision frequency of 1.0 ps-1. NSTLIM = 10000, DT = 0.002: A total of 10,000 molecular dynamics steps with a time step of 2 fs per step, possible since SHAKE used, to give a total simulation time of 20 ps. NTPR = 100, NTWX = 100, NTWR = 1000: Write to the output file (NTPR) every 100 steps (200 fs), to the trajectory file (NTWX) every 100 steps and write a restart file (NTWR), in case job crashes, every 1,000 steps. Running MD Equilibration on the whole system Now that the system has been heated at constant volume with weak restraints on the complex, the next stage is to switch to using constant pressure, so that the density of the water can relax At the same time, since the system is at 300 K, it is now possible to safely remove the restraints on the complex This equilibration will be run for 100 ps to give the system plenty of time to relax. Running MD Equilibration on the whole system Protein:ligand complex md2.in Protein:ligand complex :100ps MD &cntrl imin = 0, irest = 1, ntx = 7, ntb = 2, pres0 = 1.0, ntp = 1, taup = 2.0, cut = 8, ntr = 0, ntc = 2, ntf = 2, tempi = 300.0, temp0 = 300.0, ntt = 3, gamma_ln = 1.0, nstlim = 50000, dt = 0.002, ntpr = 100, ntwx = 100, ntwr = 1000 / 8
9 Running MD Equilibration on the whole system IMIN = 0: Minimisation is turned off (run molecular dynamics) IREST = 1, NTX = 7: The MD simulation starts where we left off after the 120 ps of constant volume simulation. IREST tells sander that a simulation will be restarted, so the time is not reset to zero but will start at 20 ps. Previously NTX was set at the default of 1 which meant only the coordinates were read from the restrt file. This time, however, continuation from where finished previously so NTX = 7 which means the coordinates, velocities and box information will be read from a formatted (ASCII) restrt file. NTB = 2, PRES0 = 1.0, NTP = 1, TAUP = 2.0: Use constant pressure periodic boundary with an average pressure of 1 atm (PRES0). Isotropic position scaling should be used to maintain the pressure (NTP=1) and a relaxation time of 2ps should be used (TAUP=2.0). CUT = 8: Use a cut off of 8 angstroms. NTR = 0: Positional restraints no longer used. Running MD Equilibration on the whole system NTC = 2, NTF = 2: SHAKE should be turned on and used to constrain bonds involving hydrogen. TEMPI = 300.0, TEMP0 = 300.0: The system was already heated to 300 K during the first stage of MD so here it will start at 300 K and should be maintained at 300 K. NTT = 3, GAMMA_LN = 1.0: The Langevin dynamics should be used to control the temperature using a collision frequency of 1.0 ps-1. NSTLIM = 50000, DT = 0.002: A total of 50,000 molecular dynamics steps with a time step of 2 fs per step, possible since are now using SHAKE, to give a total simulation time of 100 ps. NTPR = 100, NTWX = 100, NTWR = 1000: Write to the output file (NTPR) every 100 steps (200 fs), to the trajectory file (NTWX) every 100 steps and write a restart file (NTWR), in case job crashes every 1,000 steps. Analysing the results to test the equilibration Now that the system is theoretically equilibrated, it is essential that the success of this equilibrium is verified before moving on to running any production MD simulations through which more information will be gleaned about the complexes. There are a number of system properties that should be monitored to check the quality of the equilibrium. These include: Potential, Kinetic and Total energy Temperature Pressure Volume Density RMSd Analysing the results to test the equilibration These properties may be extracted using a perl script, from the two output files: Output File 1 maps trajectory at constant pressure for 20ps Output File 2 maps trajectory for 100ps at constant temperature This process was carried out for each of the 5 systems under study CHANGE IN ENERGY WITH TIME The black line, which is positive, represents the kinetic energy, The red line is the potential energy (negative) The green line is the total energy. All of the energies increased during the first few ps, corresponding to the heating process from 0 K to 300 K. The kinetic energy then remained constant for the remainder of the simulation implying that the temperature thermostat, which acts on the kinetic energy, was working correctly The potential energy, and consequently the total energy (since total energy = potential energy + kinetic energy), initially increased and then plateaued during the constant volume stage (0 to 20 ps) before decreasing slightly as the system relaxed when the protein restraints were switched off and the system moved to constant pressure conditions (20 to 40 ps). The potential energy then levelled off and remained constant for the remainder of the simulation (40 to 120 ps) indicating that the relaxation was completed and that equilibrium had been attained. The Temperature Profile Here the system temperature started at 0 K and then increased to 300 K over a period of about 5 ps. The temperature then remained more or less constant for the remainder of the simulation indicating the use of Langevin dynamics for temperature regulation was successful. 9
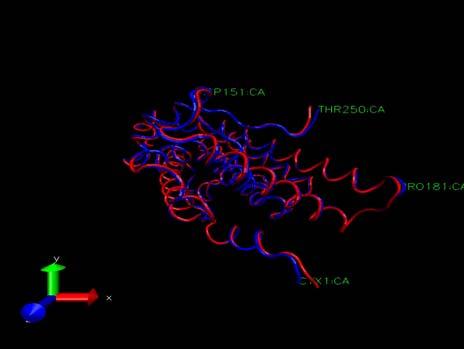
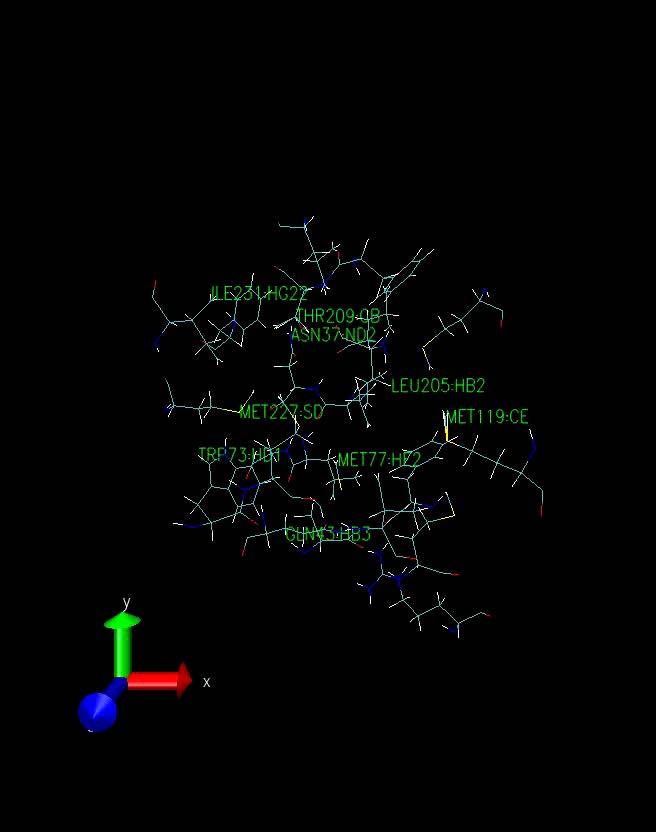
10 The Pressure Profile The Volume Profile For the first 20 ps the pressure is zero. This is to be expected a constant volume simulation was being run in which the pressure was not evaluated. At 20 ps constant pressure conditions were adopted, allowing the volume of the box to change, at which point the pressure dropped sharply becoming negative. The negative pressures correspond to a "force" acting to decrease the box size and the positive pressures to a "force" acting to increase the box size. While the pressure graph seems to show that the pressure fluctuates wildly during the simulation, in actual fact, the mean pressure stabilises as the dynamics proceed which is sufficient to indicate successful equilibration. The volume of the system (in angstroms3) initially decreases as the water box relaxes and reaches an equilibrated density, and thus volume. The smooth transitions in this plot followed by the oscillations about a mean value suggest equilibration has been successful. The Density Profile The system has equilibrated at a density of approximately 1.04 g cm-3 which is reasonable given that the density of pure liquid water at 300 K is approximately 1.00 g cm-3 thus the inference is that the introduction of, in this case the androgen receptor bound to the S-isomer of Vinclozolin and the associated charges has increased the density by around 4 %. The RMSd profile The RMSd of the protein backbone atoms remained low for the first 20 ps, due to the imposed restraints Upon removing the restraints the RMSd increased significantly as the protein relaxed within the solvent After that the RMSd is fairly stable with no wild oscillations Running further equilibration and production MD In the next stage of the simulation a further 1.8 ns of MD were run. Precisely the same conditions were applied as were for the explicit solvent equilibration of the 5 complexes. Namely no restraints on the DNA. Temperature maintained at 300 K using the Langevin thermostat Constant pressure (1 atm) periodic boundaries, SHAKE constraints on the hydrogen atoms and a 2 fs time step. Since 1.8 ns of trajectory were run, output and mdcrd files are written to every 500 fs (250 steps) All of output mdcrd files compressed to save disk space as 1.8 ns of simulation produces very large files Simulation VERY TIME INTENSIVE thus the 1.8 ns broken up into sequential runs i.e. 9 x 200 ps simulations with each successive simulation continuing on from the previous one. Running further equilibration and production MD TIME SPAN (ps) mdrcd OUTPUT FILE Complex1_md2.mdcrd.gz Complex1_md3.mdcrd.gz Complex1_md4.mdcrd.gz Complex1_md5.mdcrd.gz Complex1_md6.mdcrd.gz Complex1_md7.mdcrd.gz Complex1_md8.mdcrd.gz Complex1_md9.mdcrd.gz Complex1_md10.mdcrd.gz 10
11 11
Course in Applied Structural Bioinformatics: Amber Tutorial
 Course in Applied Structural Bioinformatics: Amber Tutorial Lars Skjaerven, Yvan Strahm, Kjell Petersen and Ross Walker September 8, 2010 http://www.bccs.uni.no/ http://www.bioinfo.no/ Contents 1 Introduction
Course in Applied Structural Bioinformatics: Amber Tutorial Lars Skjaerven, Yvan Strahm, Kjell Petersen and Ross Walker September 8, 2010 http://www.bccs.uni.no/ http://www.bioinfo.no/ Contents 1 Introduction
Why Proteins Fold? (Parts of this presentation are based on work of Ashok Kolaskar) CS490B: Introduction to Bioinformatics Mar.
 CS490B: Introduction to Bioinformatics Mar.") Why Proteins Fold? (Parts of this presentation are based on work of Ashok Kolaskar) CS490B: Introduction to Bioinformatics Mar. 25, 2002 Molecular Dynamics: Introduction At physiological conditions, the
Why Proteins Fold? (Parts of this presentation are based on work of Ashok Kolaskar) CS490B: Introduction to Bioinformatics Mar. 25, 2002 Molecular Dynamics: Introduction At physiological conditions, the
Exercise 2: Solvating the Structure Before you continue, follow these steps: Setting up Periodic Boundary Conditions
 Exercise 2: Solvating the Structure HyperChem lets you place a molecular system in a periodic box of water molecules to simulate behavior in aqueous solution, as in a biological system. In this exercise,
Exercise 2: Solvating the Structure HyperChem lets you place a molecular system in a periodic box of water molecules to simulate behavior in aqueous solution, as in a biological system. In this exercise,
Introduction to Molecular Modeling Lab X - Modeling Proteins
 (Rev 12/2/99, Beta Version) Introduction to Molecular Modeling Lab X - Modeling Proteins Table of Contents Introduction...2 Objectives...3 Outline...3 Preparation of the pdb File...4 Using Leap to build
(Rev 12/2/99, Beta Version) Introduction to Molecular Modeling Lab X - Modeling Proteins Table of Contents Introduction...2 Objectives...3 Outline...3 Preparation of the pdb File...4 Using Leap to build
Molecular dynamics simulation of Aquaporin-1. 4 nm
 Molecular dynamics simulation of Aquaporin-1 4 nm Molecular Dynamics Simulations Schrödinger equation i~@ t (r, R) =H (r, R) Born-Oppenheimer approximation H e e(r; R) =E e (R) e(r; R) Nucleic motion described
Molecular dynamics simulation of Aquaporin-1 4 nm Molecular Dynamics Simulations Schrödinger equation i~@ t (r, R) =H (r, R) Born-Oppenheimer approximation H e e(r; R) =E e (R) e(r; R) Nucleic motion described
Medical Research, Medicinal Chemistry, University of Leuven, Leuven, Belgium.
 Supporting Information Towards peptide vaccines against Zika virus: Immunoinformatics combined with molecular dynamics simulations to predict antigenic epitopes of Zika viral proteins Muhammad Usman Mirza
Supporting Information Towards peptide vaccines against Zika virus: Immunoinformatics combined with molecular dynamics simulations to predict antigenic epitopes of Zika viral proteins Muhammad Usman Mirza
An Analysis of Prominent Water Models by Molecular Dynamics Simulations
 Georgia State University ScholarWorks @ Georgia State University Chemistry Theses Department of Chemistry 4-20-2010 An Analysis of Prominent Water Models by Molecular Dynamics Simulations Quentin Ramon
Georgia State University ScholarWorks @ Georgia State University Chemistry Theses Department of Chemistry 4-20-2010 An Analysis of Prominent Water Models by Molecular Dynamics Simulations Quentin Ramon
Molecular Mechanics, Dynamics & Docking
 Molecular Mechanics, Dynamics & Docking Lawrence Hunter, Ph.D. Director, Computational Bioscience Program University of Colorado School of Medicine Larry.Hunter@uchsc.edu http://compbio.uchsc.edu/hunter
Molecular Mechanics, Dynamics & Docking Lawrence Hunter, Ph.D. Director, Computational Bioscience Program University of Colorado School of Medicine Larry.Hunter@uchsc.edu http://compbio.uchsc.edu/hunter
Homology modeling. Dinesh Gupta ICGEB, New Delhi 1/27/2010 5:59 PM
 Homology modeling Dinesh Gupta ICGEB, New Delhi Protein structure prediction Methods: Homology (comparative) modelling Threading Ab-initio Protein Homology modeling Homology modeling is an extrapolation
Homology modeling Dinesh Gupta ICGEB, New Delhi Protein structure prediction Methods: Homology (comparative) modelling Threading Ab-initio Protein Homology modeling Homology modeling is an extrapolation
DISCRETE TUTORIAL. Agustí Emperador. Institute for Research in Biomedicine, Barcelona APPLICATION OF DISCRETE TO FLEXIBLE PROTEIN-PROTEIN DOCKING:
 DISCRETE TUTORIAL Agustí Emperador Institute for Research in Biomedicine, Barcelona APPLICATION OF DISCRETE TO FLEXIBLE PROTEIN-PROTEIN DOCKING: STRUCTURAL REFINEMENT OF DOCKING CONFORMATIONS Emperador
DISCRETE TUTORIAL Agustí Emperador Institute for Research in Biomedicine, Barcelona APPLICATION OF DISCRETE TO FLEXIBLE PROTEIN-PROTEIN DOCKING: STRUCTURAL REFINEMENT OF DOCKING CONFORMATIONS Emperador
Molecular dynamics simulation. CS/CME/BioE/Biophys/BMI 279 Oct. 5 and 10, 2017 Ron Dror
 Molecular dynamics simulation CS/CME/BioE/Biophys/BMI 279 Oct. 5 and 10, 2017 Ron Dror 1 Outline Molecular dynamics (MD): The basic idea Equations of motion Key properties of MD simulations Sample applications
Molecular dynamics simulation CS/CME/BioE/Biophys/BMI 279 Oct. 5 and 10, 2017 Ron Dror 1 Outline Molecular dynamics (MD): The basic idea Equations of motion Key properties of MD simulations Sample applications
Peptide folding in non-aqueous environments investigated with molecular dynamics simulations Soto Becerra, Patricia
 University of Groningen Peptide folding in non-aqueous environments investigated with molecular dynamics simulations Soto Becerra, Patricia IMPORTANT NOTE: You are advised to consult the publisher's version
University of Groningen Peptide folding in non-aqueous environments investigated with molecular dynamics simulations Soto Becerra, Patricia IMPORTANT NOTE: You are advised to consult the publisher's version
The Molecular Dynamics Method
 H-bond energy (kcal/mol) - 4.0 The Molecular Dynamics Method Fibronectin III_1, a mechanical protein that glues cells together in wound healing and in preventing tumor metastasis 0 ATPase, a molecular
H-bond energy (kcal/mol) - 4.0 The Molecular Dynamics Method Fibronectin III_1, a mechanical protein that glues cells together in wound healing and in preventing tumor metastasis 0 ATPase, a molecular
Bioengineering 215. An Introduction to Molecular Dynamics for Biomolecules
 Bioengineering 215 An Introduction to Molecular Dynamics for Biomolecules David Parker May 18, 2007 ntroduction A principal tool to study biological molecules is molecular dynamics simulations (MD). MD
Bioengineering 215 An Introduction to Molecular Dynamics for Biomolecules David Parker May 18, 2007 ntroduction A principal tool to study biological molecules is molecular dynamics simulations (MD). MD
An introduction to Molecular Dynamics. EMBO, June 2016
 An introduction to Molecular Dynamics EMBO, June 2016 What is MD? everything that living things do can be understood in terms of the jiggling and wiggling of atoms. The Feynman Lectures in Physics vol.
An introduction to Molecular Dynamics EMBO, June 2016 What is MD? everything that living things do can be understood in terms of the jiggling and wiggling of atoms. The Feynman Lectures in Physics vol.
Developing Monovalent Ion Parameters for the Optimal Point Charge (OPC) Water Model. John Dood Hope College
 Water Model. John Dood Hope College") Developing Monovalent Ion Parameters for the Optimal Point Charge (OPC) Water Model John Dood Hope College What are MD simulations? Model and predict the structure and dynamics of large macromolecules.
Developing Monovalent Ion Parameters for the Optimal Point Charge (OPC) Water Model John Dood Hope College What are MD simulations? Model and predict the structure and dynamics of large macromolecules.
MARTINI simulation details
 S1 Appendix MARTINI simulation details MARTINI simulation initialization and equilibration In this section, we describe the initialization of simulations from Main Text section Residue-based coarsegrained
S1 Appendix MARTINI simulation details MARTINI simulation initialization and equilibration In this section, we describe the initialization of simulations from Main Text section Residue-based coarsegrained
Applications of Molecular Dynamics
 June 4, 0 Molecular Modeling and Simulation Applications of Molecular Dynamics Agricultural Bioinformatics Research Unit, Graduate School of Agricultural and Life Sciences, The University of Tokyo Tohru
June 4, 0 Molecular Modeling and Simulation Applications of Molecular Dynamics Agricultural Bioinformatics Research Unit, Graduate School of Agricultural and Life Sciences, The University of Tokyo Tohru
Potential Energy (hyper)surface
surface") The Molecular Dynamics Method Thermal motion of a lipid bilayer Water permeation through channels Selective sugar transport Potential Energy (hyper)surface What is Force? Energy U(x) F = " d dx U(x) Conformation
The Molecular Dynamics Method Thermal motion of a lipid bilayer Water permeation through channels Selective sugar transport Potential Energy (hyper)surface What is Force? Energy U(x) F = " d dx U(x) Conformation
Protein Structure Analysis
 BINF 731 Protein Modeling Methods Protein Structure Analysis Iosif Vaisman Ab initio methods: solution of a protein folding problem search in conformational space Energy-based methods: energy minimization
BINF 731 Protein Modeling Methods Protein Structure Analysis Iosif Vaisman Ab initio methods: solution of a protein folding problem search in conformational space Energy-based methods: energy minimization
Routine access to millisecond timescale events with accelerated molecular dynamics
 Routine access to millisecond timescale events with accelerated molecular dynamics Levi C.T. Pierce, Romelia Salomon-Ferrer, Cesar Augusto F. de Oliveira #, J. Andrew McCammon #, Ross C. Walker * SUPPORTING
Routine access to millisecond timescale events with accelerated molecular dynamics Levi C.T. Pierce, Romelia Salomon-Ferrer, Cesar Augusto F. de Oliveira #, J. Andrew McCammon #, Ross C. Walker * SUPPORTING
Structural Bioinformatics (C3210) Molecular Mechanics
 Molecular Mechanics") Structural Bioinformatics (C3210) Molecular Mechanics How to Calculate Energies Calculation of molecular energies is of key importance in protein folding, molecular modelling etc. There are two main computational
Structural Bioinformatics (C3210) Molecular Mechanics How to Calculate Energies Calculation of molecular energies is of key importance in protein folding, molecular modelling etc. There are two main computational
Hands-on : Model Potential Molecular Dynamics
 Hands-on : Model Potential Molecular Dynamics OUTLINE 0. DL_POLY code introduction 0.a Input files 1. THF solvent molecule 1.a Geometry optimization 1.b NVE/NVT dynamics 2. Liquid THF 2.a Equilibration
Hands-on : Model Potential Molecular Dynamics OUTLINE 0. DL_POLY code introduction 0.a Input files 1. THF solvent molecule 1.a Geometry optimization 1.b NVE/NVT dynamics 2. Liquid THF 2.a Equilibration
Computer simulation methods (2) Dr. Vania Calandrini
 Dr. Vania Calandrini") Computer simulation methods (2) Dr. Vania Calandrini in the previous lecture: time average versus ensemble average MC versus MD simulations equipartition theorem (=> computing T) virial theorem (=> computing
Computer simulation methods (2) Dr. Vania Calandrini in the previous lecture: time average versus ensemble average MC versus MD simulations equipartition theorem (=> computing T) virial theorem (=> computing
Protein Dynamics. The space-filling structures of myoglobin and hemoglobin show that there are no pathways for O 2 to reach the heme iron.
 Protein Dynamics The space-filling structures of myoglobin and hemoglobin show that there are no pathways for O 2 to reach the heme iron. Below is myoglobin hydrated with 350 water molecules. Only a small
Protein Dynamics The space-filling structures of myoglobin and hemoglobin show that there are no pathways for O 2 to reach the heme iron. Below is myoglobin hydrated with 350 water molecules. Only a small
Supplementary Information
 Supplementary Information Resveratrol Serves as a Protein-Substrate Interaction Stabilizer in Human SIRT1 Activation Xuben Hou,, David Rooklin, Hao Fang *,,, Yingkai Zhang Department of Medicinal Chemistry
Supplementary Information Resveratrol Serves as a Protein-Substrate Interaction Stabilizer in Human SIRT1 Activation Xuben Hou,, David Rooklin, Hao Fang *,,, Yingkai Zhang Department of Medicinal Chemistry
Example questions for Molecular modelling (Level 4) Dr. Adrian Mulholland
 Dr. Adrian Mulholland") Example questions for Molecular modelling (Level 4) Dr. Adrian Mulholland 1) Question. Two methods which are widely used for the optimization of molecular geometies are the Steepest descents and Newton-Raphson
Example questions for Molecular modelling (Level 4) Dr. Adrian Mulholland 1) Question. Two methods which are widely used for the optimization of molecular geometies are the Steepest descents and Newton-Raphson
Amber 12 Reference Manual
 Amber 12 Reference Manual 2 Amber 12 Reference Manual Principal contributors to the current codes: David A. Case (Rutgers University) Tom Darden (OpenEye) Thomas E. Cheatham III (Utah) Carlos Simmerling
Amber 12 Reference Manual 2 Amber 12 Reference Manual Principal contributors to the current codes: David A. Case (Rutgers University) Tom Darden (OpenEye) Thomas E. Cheatham III (Utah) Carlos Simmerling
How is molecular dynamics being used in life sciences? Davide Branduardi
 How is molecular dynamics being used in life sciences? Davide Branduardi davide.branduardi@schrodinger.com Exploring molecular processes with MD Drug discovery and design Protein-protein interactions Protein-DNA
How is molecular dynamics being used in life sciences? Davide Branduardi davide.branduardi@schrodinger.com Exploring molecular processes with MD Drug discovery and design Protein-protein interactions Protein-DNA
Introduction to Classical Molecular Dynamics. Giovanni Chillemi HPC department, CINECA
 Introduction to Classical Molecular Dynamics Giovanni Chillemi g.chillemi@cineca.it HPC department, CINECA MD ingredients Coordinates Velocities Force field Topology MD Trajectories Input parameters Analysis
Introduction to Classical Molecular Dynamics Giovanni Chillemi g.chillemi@cineca.it HPC department, CINECA MD ingredients Coordinates Velocities Force field Topology MD Trajectories Input parameters Analysis
Free energy simulations
 Free energy simulations Marcus Elstner and Tomáš Kubař January 14, 2013 Motivation a physical quantity that is of most interest in chemistry? free energies Helmholtz F or Gibbs G holy grail of computational
Free energy simulations Marcus Elstner and Tomáš Kubař January 14, 2013 Motivation a physical quantity that is of most interest in chemistry? free energies Helmholtz F or Gibbs G holy grail of computational
Alchemical free energy calculations in OpenMM
 Alchemical free energy calculations in OpenMM Lee-Ping Wang Stanford Department of Chemistry OpenMM Workshop, Stanford University September 7, 2012 Special thanks to: John Chodera, Morgan Lawrenz Outline
Alchemical free energy calculations in OpenMM Lee-Ping Wang Stanford Department of Chemistry OpenMM Workshop, Stanford University September 7, 2012 Special thanks to: John Chodera, Morgan Lawrenz Outline
Computational Modeling of Protein Kinase A and Comparison with Nuclear Magnetic Resonance Data
 Computational Modeling of Protein Kinase A and Comparison with Nuclear Magnetic Resonance Data ABSTRACT Keyword Lei Shi 1 Advisor: Gianluigi Veglia 1,2 Department of Chemistry 1, & Biochemistry, Molecular
Computational Modeling of Protein Kinase A and Comparison with Nuclear Magnetic Resonance Data ABSTRACT Keyword Lei Shi 1 Advisor: Gianluigi Veglia 1,2 Department of Chemistry 1, & Biochemistry, Molecular
A Large-Scale Test of Free-Energy Simulation Estimates of Protein-Ligand Binding Affinities.
 A Large-Scale Test of Free-Energy Simulation Estimates of Protein-Ligand Binding Affinities. Mikulskis, Paulius; Genheden, Samuel; Ryde, Ulf Published in: DOI: 10.1021/ci5004027 2014 Link to publication
A Large-Scale Test of Free-Energy Simulation Estimates of Protein-Ligand Binding Affinities. Mikulskis, Paulius; Genheden, Samuel; Ryde, Ulf Published in: DOI: 10.1021/ci5004027 2014 Link to publication
Molecular Mechanics. I. Quantum mechanical treatment of molecular systems
 Molecular Mechanics I. Quantum mechanical treatment of molecular systems The first principle approach for describing the properties of molecules, including proteins, involves quantum mechanics. For example,
Molecular Mechanics I. Quantum mechanical treatment of molecular systems The first principle approach for describing the properties of molecules, including proteins, involves quantum mechanics. For example,
Supporting Information
 Supporting Information Constant ph molecular dynamics reveals ph-modulated binding of two small-molecule BACE1 inhibitors Christopher R. Ellis 1,, Cheng-Chieh Tsai 1,, Xinjun Hou 2, and Jana Shen 1, 1
Supporting Information Constant ph molecular dynamics reveals ph-modulated binding of two small-molecule BACE1 inhibitors Christopher R. Ellis 1,, Cheng-Chieh Tsai 1,, Xinjun Hou 2, and Jana Shen 1, 1
User Guide for LeDock
 User Guide for LeDock Hongtao Zhao, PhD Email: htzhao@lephar.com Website: www.lephar.com Copyright 2017 Hongtao Zhao. All rights reserved. Introduction LeDock is flexible small-molecule docking software,
User Guide for LeDock Hongtao Zhao, PhD Email: htzhao@lephar.com Website: www.lephar.com Copyright 2017 Hongtao Zhao. All rights reserved. Introduction LeDock is flexible small-molecule docking software,
Proteins are not rigid structures: Protein dynamics, conformational variability, and thermodynamic stability
 Proteins are not rigid structures: Protein dynamics, conformational variability, and thermodynamic stability Dr. Andrew Lee UNC School of Pharmacy (Div. Chemical Biology and Medicinal Chemistry) UNC Med
Proteins are not rigid structures: Protein dynamics, conformational variability, and thermodynamic stability Dr. Andrew Lee UNC School of Pharmacy (Div. Chemical Biology and Medicinal Chemistry) UNC Med
Amber: Building DNAs and Minimizing their Energy Lab 4
 (Rev 9/8/08) Amber: Building DNAs and Minimizing their Energy Lab 4 Table of Contents Objectives...2 Purpose...2 Outline...2 Definitions...3 Setup...3 Building the initial canonical DNA structure...4 Introduction...4
(Rev 9/8/08) Amber: Building DNAs and Minimizing their Energy Lab 4 Table of Contents Objectives...2 Purpose...2 Outline...2 Definitions...3 Setup...3 Building the initial canonical DNA structure...4 Introduction...4
2008 Biowerkzeug Ltd.
 2008 Biowerkzeug Ltd. 1 Contents Summary...3 1 Simulation...4 1.1 Setup...4 1.2 Output...4 2 Settings...5 3 Analysis...9 3.1 Setup...9 3.2 Input options...9 3.3 Descriptions...10 Please note that we cannot
2008 Biowerkzeug Ltd. 1 Contents Summary...3 1 Simulation...4 1.1 Setup...4 1.2 Output...4 2 Settings...5 3 Analysis...9 3.1 Setup...9 3.2 Input options...9 3.3 Descriptions...10 Please note that we cannot
The Molecular Dynamics Method
 The Molecular Dynamics Method Thermal motion of a lipid bilayer Water permeation through channels Selective sugar transport Potential Energy (hyper)surface What is Force? Energy U(x) F = d dx U(x) Conformation
The Molecular Dynamics Method Thermal motion of a lipid bilayer Water permeation through channels Selective sugar transport Potential Energy (hyper)surface What is Force? Energy U(x) F = d dx U(x) Conformation
PROTEIN-PROTEIN DOCKING REFINEMENT USING RESTRAINT MOLECULAR DYNAMICS SIMULATIONS
 TASKQUARTERLYvol.20,No4,2016,pp.353 360 PROTEIN-PROTEIN DOCKING REFINEMENT USING RESTRAINT MOLECULAR DYNAMICS SIMULATIONS MARTIN ZACHARIAS Physics Department T38, Technical University of Munich James-Franck-Str.
TASKQUARTERLYvol.20,No4,2016,pp.353 360 PROTEIN-PROTEIN DOCKING REFINEMENT USING RESTRAINT MOLECULAR DYNAMICS SIMULATIONS MARTIN ZACHARIAS Physics Department T38, Technical University of Munich James-Franck-Str.
Research Article Conformational Search on the Lewis X Structure by Molecular Dynamic: Study of Tri- and Pentasaccharide
 Hindawi Publishing Corporation International Journal of Carbohydrate Chemistry Volume 212, Article ID 725271, 7 pages doi:1.1155/212/725271 Research Article Conformational Search on the Lewis X Structure
Hindawi Publishing Corporation International Journal of Carbohydrate Chemistry Volume 212, Article ID 725271, 7 pages doi:1.1155/212/725271 Research Article Conformational Search on the Lewis X Structure
Don t forget to bring your MD tutorial. Potential Energy (hyper)surface
surface") Don t forget to bring your MD tutorial Lab session starts at 1pm You will have to finish an MD/SMD exercise on α-conotoxin in oxidized and reduced forms Potential Energy (hyper)surface What is Force? Energy
Don t forget to bring your MD tutorial Lab session starts at 1pm You will have to finish an MD/SMD exercise on α-conotoxin in oxidized and reduced forms Potential Energy (hyper)surface What is Force? Energy
Can a continuum solvent model reproduce the free energy landscape of a β-hairpin folding in water?
 Can a continuum solvent model reproduce the free energy landscape of a β-hairpin folding in water? Ruhong Zhou 1 and Bruce J. Berne 2 1 IBM Thomas J. Watson Research Center; and 2 Department of Chemistry,
Can a continuum solvent model reproduce the free energy landscape of a β-hairpin folding in water? Ruhong Zhou 1 and Bruce J. Berne 2 1 IBM Thomas J. Watson Research Center; and 2 Department of Chemistry,
Molecular Dynamics. A very brief introduction
 Molecular Dynamics A very brief introduction Sander Pronk Dept. of Theoretical Physics KTH Royal Institute of Technology & Science For Life Laboratory Stockholm, Sweden Why computer simulations? Two primary
Molecular Dynamics A very brief introduction Sander Pronk Dept. of Theoretical Physics KTH Royal Institute of Technology & Science For Life Laboratory Stockholm, Sweden Why computer simulations? Two primary
Chapter 32. Computations of radial distributions functions, PMFs and diffusion constants
 Chapter 32 Computations of radial distributions functions, PMFs and diffusion constants After studying this chapter, you will be able to perform the 1) Calculations of radial distribution functions between
Chapter 32 Computations of radial distributions functions, PMFs and diffusion constants After studying this chapter, you will be able to perform the 1) Calculations of radial distribution functions between
Gromacs Workshop Spring CSC
 Gromacs Workshop Spring 2007 @ CSC Erik Lindahl Center for Biomembrane Research Stockholm University, Sweden David van der Spoel Dept. Cell & Molecular Biology Uppsala University, Sweden Berk Hess Max-Planck-Institut
Gromacs Workshop Spring 2007 @ CSC Erik Lindahl Center for Biomembrane Research Stockholm University, Sweden David van der Spoel Dept. Cell & Molecular Biology Uppsala University, Sweden Berk Hess Max-Planck-Institut
APBS electrostatics in VMD - Software. APBS! >!Examples! >!Visualization! >! Contents
 Software Search this site Home Announcements An update on mailing lists APBS 1.2.0 released APBS 1.2.1 released APBS 1.3 released New APBS 1.3 Windows Installer PDB2PQR 1.7.1 released PDB2PQR 1.8 released
Software Search this site Home Announcements An update on mailing lists APBS 1.2.0 released APBS 1.2.1 released APBS 1.3 released New APBS 1.3 Windows Installer PDB2PQR 1.7.1 released PDB2PQR 1.8 released
5.1. Hardwares, Softwares and Web server used in Molecular modeling
 5. EXPERIMENTAL The tools, techniques and procedures/methods used for carrying out research work reported in this thesis have been described as follows: 5.1. Hardwares, Softwares and Web server used in
5. EXPERIMENTAL The tools, techniques and procedures/methods used for carrying out research work reported in this thesis have been described as follows: 5.1. Hardwares, Softwares and Web server used in
Computational Analysis of Small Proteins using the WebAmber Interface. By Eddie Gorr Gustavus Adolphus College
 Computational Analysis of Small Proteins using the WebAmber Interface By Eddie Gorr Gustavus Adolphus College Research Goals Upgrade And Revise Web-based Amber interface Use Amber to Explore Interactions
Computational Analysis of Small Proteins using the WebAmber Interface By Eddie Gorr Gustavus Adolphus College Research Goals Upgrade And Revise Web-based Amber interface Use Amber to Explore Interactions
Molecular Dynamics in practice with GROMACS
 Molecular Dynamics in practice with GROMACS GROMACS is one of the wold s fastest software package for molecular dynamics simulations. One can find many helpful materials, manual as well as to download
Molecular Dynamics in practice with GROMACS GROMACS is one of the wold s fastest software package for molecular dynamics simulations. One can find many helpful materials, manual as well as to download
Laboratory 3: Steered molecular dynamics
 Laboratory 3: Steered molecular dynamics Nathan Hammond nhammond@mit.edu Issued: Friday, February 20, 2009 Due: Friday, February 27, 2009 In this lab you will: 1. Do some basic analysis of the molecular
Laboratory 3: Steered molecular dynamics Nathan Hammond nhammond@mit.edu Issued: Friday, February 20, 2009 Due: Friday, February 27, 2009 In this lab you will: 1. Do some basic analysis of the molecular
Hyeyoung Shin a, Tod A. Pascal ab, William A. Goddard III abc*, and Hyungjun Kim a* Korea
 The Scaled Effective Solvent Method for Predicting the Equilibrium Ensemble of Structures with Analysis of Thermodynamic Properties of Amorphous Polyethylene Glycol-Water Mixtures Hyeyoung Shin a, Tod
The Scaled Effective Solvent Method for Predicting the Equilibrium Ensemble of Structures with Analysis of Thermodynamic Properties of Amorphous Polyethylene Glycol-Water Mixtures Hyeyoung Shin a, Tod
Virtual Molecular Dynamics. Indiana University Bloomington, IN Merck and Co., Inc., West Point, PA 19486
 Virtual Molecular Dynamics Yuriy V. Sereda 1, Andrew Abi Mansour, 2 and Peter J. Ortoleva 1 * a Department of Chemistry Indiana University Bloomington, IN 47405 * Contact: ortoleva@indiana.edu 2 Center
Virtual Molecular Dynamics Yuriy V. Sereda 1, Andrew Abi Mansour, 2 and Peter J. Ortoleva 1 * a Department of Chemistry Indiana University Bloomington, IN 47405 * Contact: ortoleva@indiana.edu 2 Center
Molecular Dynamics Simulations. Dr. Noelia Faginas Lago Dipartimento di Chimica,Biologia e Biotecnologie Università di Perugia
 Molecular Dynamics Simulations Dr. Noelia Faginas Lago Dipartimento di Chimica,Biologia e Biotecnologie Università di Perugia 1 An Introduction to Molecular Dynamics Simulations Macroscopic properties
Molecular Dynamics Simulations Dr. Noelia Faginas Lago Dipartimento di Chimica,Biologia e Biotecnologie Università di Perugia 1 An Introduction to Molecular Dynamics Simulations Macroscopic properties
ONETEP PB/SA: Application to G-Quadruplex DNA Stability. Danny Cole
 ONETEP PB/SA: Application to G-Quadruplex DNA Stability Danny Cole Introduction Historical overview of structure and free energy calculation of complex molecules using molecular mechanics and continuum
ONETEP PB/SA: Application to G-Quadruplex DNA Stability Danny Cole Introduction Historical overview of structure and free energy calculation of complex molecules using molecular mechanics and continuum
Tailoring the Properties of Quadruplex Nucleobases for Biological and Nanomaterial Applications
 Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 2014 Supporting Information for: Tailoring the Properties of Quadruplex Nucleobases
Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 2014 Supporting Information for: Tailoring the Properties of Quadruplex Nucleobases
Introduction The gramicidin A (ga) channel forms by head-to-head association of two monomers at their amino termini, one from each bilayer leaflet. Th
 channel forms by head-to-head association of two monomers at their amino termini, one from each bilayer leaflet. Th") Abstract When conductive, gramicidin monomers are linked by six hydrogen bonds. To understand the details of dissociation and how the channel transits from a state with 6H bonds to ones with 4H bonds or
Abstract When conductive, gramicidin monomers are linked by six hydrogen bonds. To understand the details of dissociation and how the channel transits from a state with 6H bonds to ones with 4H bonds or
Molecular dynamics simulations of anti-aggregation effect of ibuprofen. Wenling E. Chang, Takako Takeda, E. Prabhu Raman, and Dmitri Klimov
 Biophysical Journal, Volume 98 Supporting Material Molecular dynamics simulations of anti-aggregation effect of ibuprofen Wenling E. Chang, Takako Takeda, E. Prabhu Raman, and Dmitri Klimov Supplemental
Biophysical Journal, Volume 98 Supporting Material Molecular dynamics simulations of anti-aggregation effect of ibuprofen Wenling E. Chang, Takako Takeda, E. Prabhu Raman, and Dmitri Klimov Supplemental
An Informal AMBER Small Molecule Force Field :
 An Informal AMBER Small Molecule Force Field : parm@frosst Credit Christopher Bayly (1992-2010) initiated, contributed and lead the efforts Daniel McKay (1997-2010) and Jean-François Truchon (2002-2010)
An Informal AMBER Small Molecule Force Field : parm@frosst Credit Christopher Bayly (1992-2010) initiated, contributed and lead the efforts Daniel McKay (1997-2010) and Jean-François Truchon (2002-2010)
Lecture 11: Potential Energy Functions
 Lecture 11: Potential Energy Functions Dr. Ronald M. Levy ronlevy@temple.edu Originally contributed by Lauren Wickstrom (2011) Microscopic/Macroscopic Connection The connection between microscopic interactions
Lecture 11: Potential Energy Functions Dr. Ronald M. Levy ronlevy@temple.edu Originally contributed by Lauren Wickstrom (2011) Microscopic/Macroscopic Connection The connection between microscopic interactions
All-atom Molecular Mechanics. Trent E. Balius AMS 535 / CHE /27/2010
 All-atom Molecular Mechanics Trent E. Balius AMS 535 / CHE 535 09/27/2010 Outline Molecular models Molecular mechanics Force Fields Potential energy function functional form parameters and parameterization
All-atom Molecular Mechanics Trent E. Balius AMS 535 / CHE 535 09/27/2010 Outline Molecular models Molecular mechanics Force Fields Potential energy function functional form parameters and parameterization
XYZ file format Protein Data Bank (pdb) file format Z - matrix
 file format Z - matrix") Chemistry block (exercise 1) In this exercise, students will be introduced how to preform simple quantum chemical calculations. Input files for Gaussian09. Output file structure. Geometry optimization,
Chemistry block (exercise 1) In this exercise, students will be introduced how to preform simple quantum chemical calculations. Input files for Gaussian09. Output file structure. Geometry optimization,
CPMD Tutorial Atosim/RFCT 2009/10
 These exercices were inspired by the CPMD Tutorial of Axel Kohlmeyer http://www.theochem.ruhruni-bochum.de/ axel.kohlmeyer/cpmd-tutor/index.html and by other tutorials. Here is a summary of what we will
These exercices were inspired by the CPMD Tutorial of Axel Kohlmeyer http://www.theochem.ruhruni-bochum.de/ axel.kohlmeyer/cpmd-tutor/index.html and by other tutorials. Here is a summary of what we will
What is Classical Molecular Dynamics?
 What is Classical Molecular Dynamics? Simulation of explicit particles (atoms, ions,... ) Particles interact via relatively simple analytical potential functions Newton s equations of motion are integrated
What is Classical Molecular Dynamics? Simulation of explicit particles (atoms, ions,... ) Particles interact via relatively simple analytical potential functions Newton s equations of motion are integrated
Elbow Flexibility and Ligand-Induced Domain Rearrangements in Antibody Fab NC6.8: Large Effects of a Small Hapten
 614 Biophysical Journal Volume 79 August 2000 614 628 Elbow Flexibility and Ligand-Induced Domain Rearrangements in Antibody Fab NC6.8: Large Effects of a Small Hapten Christoph A. Sotriffer, Bernd M.
614 Biophysical Journal Volume 79 August 2000 614 628 Elbow Flexibility and Ligand-Induced Domain Rearrangements in Antibody Fab NC6.8: Large Effects of a Small Hapten Christoph A. Sotriffer, Bernd M.
Why study protein dynamics?
 Why study protein dynamics? Protein flexibility is crucial for function. One average structure is not enough. Proteins constantly sample configurational space. Transport - binding and moving molecules
Why study protein dynamics? Protein flexibility is crucial for function. One average structure is not enough. Proteins constantly sample configurational space. Transport - binding and moving molecules
Unfolding CspB by means of biased molecular dynamics
 Chapter 4 Unfolding CspB by means of biased molecular dynamics 4.1 Introduction Understanding the mechanism of protein folding has been a major challenge for the last twenty years, as pointed out in the
Chapter 4 Unfolding CspB by means of biased molecular dynamics 4.1 Introduction Understanding the mechanism of protein folding has been a major challenge for the last twenty years, as pointed out in the
Generate topology (Gromacs)
") Running MD code What for...? 1. Minimizzation 2. Molecular Dynamics (classic, brownian, Langevin) 3. Normal Mode Analysis 4. Essential Dynamics and Sampling 5. Free Energy calculations (FEP, Umbrella sampling,
Running MD code What for...? 1. Minimizzation 2. Molecular Dynamics (classic, brownian, Langevin) 3. Normal Mode Analysis 4. Essential Dynamics and Sampling 5. Free Energy calculations (FEP, Umbrella sampling,
freely available at
 freely available at www.gromacs.org Generally 3 to 10 times faster than other Molecular Dynamics programs Very user-friendly: issues clear error messages, no scripting language is required to run the programs,
freely available at www.gromacs.org Generally 3 to 10 times faster than other Molecular Dynamics programs Very user-friendly: issues clear error messages, no scripting language is required to run the programs,
This Answer/Solution Example is taken from a student s actual homework report. I thank him for permission to use it here. JG
 This Answer/Solution Example is taken from a student s actual homework report. I thank him for permission to use it here. JG Chem 8021 Spring 2005 Project II Calculation of Self-Diffusion Coeffecient of
This Answer/Solution Example is taken from a student s actual homework report. I thank him for permission to use it here. JG Chem 8021 Spring 2005 Project II Calculation of Self-Diffusion Coeffecient of
Softwares for Molecular Docking. Lokesh P. Tripathi NCBS 17 December 2007
 Softwares for Molecular Docking Lokesh P. Tripathi NCBS 17 December 2007 Molecular Docking Attempt to predict structures of an intermolecular complex between two or more molecules Receptor-ligand (or drug)
Softwares for Molecular Docking Lokesh P. Tripathi NCBS 17 December 2007 Molecular Docking Attempt to predict structures of an intermolecular complex between two or more molecules Receptor-ligand (or drug)
Conformational Searching using MacroModel and ConfGen. John Shelley Schrödinger Fellow
 Conformational Searching using MacroModel and ConfGen John Shelley Schrödinger Fellow Overview Types of conformational searching applications MacroModel s conformation generation procedure General features
Conformational Searching using MacroModel and ConfGen John Shelley Schrödinger Fellow Overview Types of conformational searching applications MacroModel s conformation generation procedure General features
Introduction to Spark
 1 As you become familiar or continue to explore the Cresset technology and software applications, we encourage you to look through the user manual. This is accessible from the Help menu. However, don t
1 As you become familiar or continue to explore the Cresset technology and software applications, we encourage you to look through the user manual. This is accessible from the Help menu. However, don t
PROTEIN STRUCTURE PREDICTION USING GAS PHASE MOLECULAR DYNAMICS SIMULATION: EOTAXIN-3 CYTOKINE AS A CASE STUDY
 International Conference Mathematical and Computational Biology 2011 International Journal of Modern Physics: Conference Series Vol. 9 (2012) 193 198 World Scientific Publishing Company DOI: 10.1142/S2010194512005259
International Conference Mathematical and Computational Biology 2011 International Journal of Modern Physics: Conference Series Vol. 9 (2012) 193 198 World Scientific Publishing Company DOI: 10.1142/S2010194512005259
Towards fast and accurate binding affinity. prediction with pmemdgti: an efficient. implementation of GPU-accelerated. Thermodynamic Integration
 Towards fast and accurate binding affinity prediction with pmemdgti: an efficient implementation of GPU-accelerated Thermodynamic Integration Tai-Sung Lee,, Yuan Hu, Brad Sherborne, Zhuyan Guo, and Darrin
Towards fast and accurate binding affinity prediction with pmemdgti: an efficient implementation of GPU-accelerated Thermodynamic Integration Tai-Sung Lee,, Yuan Hu, Brad Sherborne, Zhuyan Guo, and Darrin
CS 273 Prof. Serafim Batzoglou Prof. Jean-Claude Latombe Spring Lecture 12 : Energy maintenance (1) Lecturer: Prof. J.C.
 Lecturer: Prof. J.C.") CS 273 Prof. Serafim Batzoglou Prof. Jean-Claude Latombe Spring 2006 Lecture 12 : Energy maintenance (1) Lecturer: Prof. J.C. Latombe Scribe: Neda Nategh How do you update the energy function during the
CS 273 Prof. Serafim Batzoglou Prof. Jean-Claude Latombe Spring 2006 Lecture 12 : Energy maintenance (1) Lecturer: Prof. J.C. Latombe Scribe: Neda Nategh How do you update the energy function during the
Computational Studies of the Photoreceptor Rhodopsin. Scott E. Feller Wabash College
 Computational Studies of the Photoreceptor Rhodopsin Scott E. Feller Wabash College Rhodopsin Photocycle Dark-adapted Rhodopsin hn Isomerize retinal Photorhodopsin ~200 fs Bathorhodopsin Meta-II ms timescale
Computational Studies of the Photoreceptor Rhodopsin Scott E. Feller Wabash College Rhodopsin Photocycle Dark-adapted Rhodopsin hn Isomerize retinal Photorhodopsin ~200 fs Bathorhodopsin Meta-II ms timescale
Formation of water at a Pt(111) surface: A study using reactive force fields (ReaxFF)
 surface: A study using reactive force fields (ReaxFF)") Formation of water at a Pt(111) surface: A study using reactive force fields (ReaxFF) Markus J. Buehler 1, Adri C.T. van Duin 2, Timo Jacob 3, Yunhee Jang 2, Boris Berinov 2, William A. Goddard III 2 1
Formation of water at a Pt(111) surface: A study using reactive force fields (ReaxFF) Markus J. Buehler 1, Adri C.T. van Duin 2, Timo Jacob 3, Yunhee Jang 2, Boris Berinov 2, William A. Goddard III 2 1
Biomolecules are dynamic no single structure is a perfect model
 Molecular Dynamics Simulations of Biomolecules References: A. R. Leach Molecular Modeling Principles and Applications Prentice Hall, 2001. M. P. Allen and D. J. Tildesley "Computer Simulation of Liquids",
Molecular Dynamics Simulations of Biomolecules References: A. R. Leach Molecular Modeling Principles and Applications Prentice Hall, 2001. M. P. Allen and D. J. Tildesley "Computer Simulation of Liquids",
OpenDiscovery: Automated Docking of Ligands to Proteins and Molecular Simulation
 OpenDiscovery: Automated Docking of Ligands to Proteins and Molecular Simulation Gareth Price Computational MiniProject OpenDiscovery Aims + Achievements Produce a high-throughput protocol to screen a
OpenDiscovery: Automated Docking of Ligands to Proteins and Molecular Simulation Gareth Price Computational MiniProject OpenDiscovery Aims + Achievements Produce a high-throughput protocol to screen a
Non-bonded interactions
 speeding up the number-crunching continued Marcus Elstner and Tomáš Kubař December 3, 2013 why care? number of individual pair-wise interactions bonded interactions proportional to N: O(N) non-bonded interactions
speeding up the number-crunching continued Marcus Elstner and Tomáš Kubař December 3, 2013 why care? number of individual pair-wise interactions bonded interactions proportional to N: O(N) non-bonded interactions
A tutorial for molecular dynamics simulations using Amber package
 ISSN 1984-6428 ONLINE www.orbital.ufms.br Vol 4 No. 3 July-September 2012 A tutorial for molecular dynamics simulations using Amber package Marcos Vinícius R. Garcia* a, Wivirkins N. Marciela a, Roberto
ISSN 1984-6428 ONLINE www.orbital.ufms.br Vol 4 No. 3 July-September 2012 A tutorial for molecular dynamics simulations using Amber package Marcos Vinícius R. Garcia* a, Wivirkins N. Marciela a, Roberto
Exploring the energy landscape
 Exploring the energy landscape ChE210D Today's lecture: what are general features of the potential energy surface and how can we locate and characterize minima on it Derivatives of the potential energy
Exploring the energy landscape ChE210D Today's lecture: what are general features of the potential energy surface and how can we locate and characterize minima on it Derivatives of the potential energy
SUPPLEMENTARY MATERIAL. Supplementary material and methods:
 Electronic Supplementary Material (ESI) for Catalysis Science & Technology. This journal is The Royal Society of Chemistry 2015 SUPPLEMENTARY MATERIAL Supplementary material and methods: - Computational
Electronic Supplementary Material (ESI) for Catalysis Science & Technology. This journal is The Royal Society of Chemistry 2015 SUPPLEMENTARY MATERIAL Supplementary material and methods: - Computational
Chemical properties that affect binding of enzyme-inhibiting drugs to enzymes
 Chemical properties that affect binding of enzyme-inhibiting drugs to enzymes Introduction The production of new drugs requires time for development and testing, and can result in large prohibitive costs
Chemical properties that affect binding of enzyme-inhibiting drugs to enzymes Introduction The production of new drugs requires time for development and testing, and can result in large prohibitive costs
Development of Molecular Dynamics Simulation System for Large-Scale Supra-Biomolecules, PABIOS (PArallel BIOmolecular Simulator)
") Development of Molecular Dynamics Simulation System for Large-Scale Supra-Biomolecules, PABIOS (PArallel BIOmolecular Simulator) Group Representative Hisashi Ishida Japan Atomic Energy Research Institute
Development of Molecular Dynamics Simulation System for Large-Scale Supra-Biomolecules, PABIOS (PArallel BIOmolecular Simulator) Group Representative Hisashi Ishida Japan Atomic Energy Research Institute
SAM Teachers Guide Phase Change Overview Learning Objectives Possible Student Pre/Misconceptions
 SAM Teachers Guide Phase Change Overview Students review the atomic arrangements for each state of matter, following trajectories of individual atoms to observe their motion and observing and manipulating
SAM Teachers Guide Phase Change Overview Students review the atomic arrangements for each state of matter, following trajectories of individual atoms to observe their motion and observing and manipulating
Dictionary of ligands
 Dictionary of ligands Some of the web and other resources Small molecules DrugBank: http://www.drugbank.ca/ ZINC: http://zinc.docking.org/index.shtml PRODRUG: http://www.compbio.dundee.ac.uk/web_servers/prodrg_down.html
Dictionary of ligands Some of the web and other resources Small molecules DrugBank: http://www.drugbank.ca/ ZINC: http://zinc.docking.org/index.shtml PRODRUG: http://www.compbio.dundee.ac.uk/web_servers/prodrg_down.html
Comparison of the Efficiency of the LIE and MM/GBSA Methods to Calculate LigandBinding Energies
 Comparison of the Efficiency of the LIE and MM/GBSA Methods to Calculate LigandBinding Energies Genheden, Samuel; Ryde, Ulf Published in: Journal of Chemical Theory and Computation DOI: 10.1021/ct200163c
Comparison of the Efficiency of the LIE and MM/GBSA Methods to Calculate LigandBinding Energies Genheden, Samuel; Ryde, Ulf Published in: Journal of Chemical Theory and Computation DOI: 10.1021/ct200163c
Docking with Water in the Binding Site using GOLD
 Docking with Water in the Binding Site using GOLD Version 2.0 November 2017 GOLD v5.6 Table of Contents Docking with Water in the Binding Site... 2 Case Study... 3 Introduction... 3 Provided Input Files...
Docking with Water in the Binding Site using GOLD Version 2.0 November 2017 GOLD v5.6 Table of Contents Docking with Water in the Binding Site... 2 Case Study... 3 Introduction... 3 Provided Input Files...
The Dominant Interaction Between Peptide and Urea is Electrostatic in Nature: A Molecular Dynamics Simulation Study
 Dror Tobi 1 Ron Elber 1,2 Devarajan Thirumalai 3 1 Department of Biological Chemistry, The Hebrew University, Jerusalem 91904, Israel 2 Department of Computer Science, Cornell University, Ithaca, NY 14853
Dror Tobi 1 Ron Elber 1,2 Devarajan Thirumalai 3 1 Department of Biological Chemistry, The Hebrew University, Jerusalem 91904, Israel 2 Department of Computer Science, Cornell University, Ithaca, NY 14853
Error Analysis of the Poisson P 3 MForce Field Scheme for Particle-Based Simulations of Biological Systems
 Journal of Computational Electronics 4: 179 183, 2005 c 2005 Springer Science + Business Media, Inc. Manufactured in The Netherlands. Error Analysis of the Poisson P 3 MForce Field Scheme for Particle-Based
Journal of Computational Electronics 4: 179 183, 2005 c 2005 Springer Science + Business Media, Inc. Manufactured in The Netherlands. Error Analysis of the Poisson P 3 MForce Field Scheme for Particle-Based
Figure 1: Transition State, Saddle Point, Reaction Pathway
 Computational Chemistry Workshops West Ridge Research Building-UAF Campus 9:00am-4:00pm, Room 009 Electronic Structure - July 19-21, 2016 Molecular Dynamics - July 26-28, 2016 Potential Energy Surfaces
Computational Chemistry Workshops West Ridge Research Building-UAF Campus 9:00am-4:00pm, Room 009 Electronic Structure - July 19-21, 2016 Molecular Dynamics - July 26-28, 2016 Potential Energy Surfaces
A Study of the Thermal Properties of a One. Dimensional Lennard-Jones System
 A Study of the Thermal Properties of a One Dimensional Lennard-Jones System Abstract In this study, the behavior of a one dimensional (1D) Lennard-Jones (LJ) system is simulated. As part of this research,
A Study of the Thermal Properties of a One Dimensional Lennard-Jones System Abstract In this study, the behavior of a one dimensional (1D) Lennard-Jones (LJ) system is simulated. As part of this research,
Preparing a PDB File
 Figure 1: Schematic view of the ligand-binding domain from the vitamin D receptor (PDB file 1IE9). The crystallographic waters are shown as small spheres and the bound ligand is shown as a CPK model. HO
Figure 1: Schematic view of the ligand-binding domain from the vitamin D receptor (PDB file 1IE9). The crystallographic waters are shown as small spheres and the bound ligand is shown as a CPK model. HO
I690/B680 Structural Bioinformatics Spring Protein Structure Determination by NMR Spectroscopy
 I690/B680 Structural Bioinformatics Spring 2006 Protein Structure Determination by NMR Spectroscopy Suggested Reading (1) Van Holde, Johnson, Ho. Principles of Physical Biochemistry, 2 nd Ed., Prentice
I690/B680 Structural Bioinformatics Spring 2006 Protein Structure Determination by NMR Spectroscopy Suggested Reading (1) Van Holde, Johnson, Ho. Principles of Physical Biochemistry, 2 nd Ed., Prentice
Applications of the Spring. Force: molecules
 Applications of the Spring Atoms interact via the coulomb force Force: molecules When atoms are far apart, they are attractive When atoms are too close, they are repulsive Atoms in a molecule display relative
Applications of the Spring Atoms interact via the coulomb force Force: molecules When atoms are far apart, they are attractive When atoms are too close, they are repulsive Atoms in a molecule display relative
IFM Chemistry Computational Chemistry 2010, 7.5 hp LAB2. Computer laboratory exercise 1 (LAB2): Quantum chemical calculations
: Quantum chemical calculations") Computer laboratory exercise 1 (LAB2): Quantum chemical calculations Introduction: The objective of the second computer laboratory exercise is to get acquainted with a program for performing quantum chemical
Computer laboratory exercise 1 (LAB2): Quantum chemical calculations Introduction: The objective of the second computer laboratory exercise is to get acquainted with a program for performing quantum chemical