Why protein structure? The basics of protein Basic measurements for protein structure
|
|
|
- Kathryn McBride
- 5 years ago
- Views:
Transcription
1 Protein structures
2 Protein Structure Why protein structure? The basics of protein Basic measurements for protein structure Levels of protein structure Prediction of protein structure from sequence Finding similarities between protein structures Classification of protein structures
3 Why protein structure? t In the factory of living i cells, proteins are the workers, performing a variety of biological tasks. Each protein has a particular 3-D structure that determines its function. Protein structure is more conserved than protein Protein structure is more conserved than protein sequence, and more closely related to function.
4 Structural information Protein Data Bank: maintained i by the Research Collaboratory of Structural Bioinformatics(RCSB) > protein structures as of April 10 including structures of Protein/Nucleic Acid Complexes, Nucleic Acids, Carbohydrates Most structures are determined by X-ray crystallography. Other methods are NMR and electron microscopy(em). Theoretically predicted structures were removed from PDB a few years ago.




5 PDB Growth Red: Total Blue: Yearly
6 The basics of proteins Proteins are linear heteropolymers: one or more polypeptide chains Building blocks: 20 types of amino acids. Range from a few 10s-1000s1000s Three-dimensional shapes ( fold ) adopted vary enormously.
7 Common structure of Amino Acid
8 Formation of polypeptide chain
9 Basic Measurements for protein structure Bond lengths Bond angles Dihedral (torsion) angles
10
11 Bond Length The distance between bonded atoms is constant Depends on the type of the bond Varies from 1.0 Å(C-H) to 1.5 Å(C-C) BOND LENGTH IS A FUNCTION OF THE POSITIONS OF TWO ATOMS.
12 Bond Length
13 Bond Angles All bond angles are determined d by chemical makeup of the atoms involved, and are constant. Depends on the type of atom, and number of electrons available for bonding. Ranges from 100 to 180 BOND ANGLES IS A FUNCTION OF THE POSITION OF THREE ATOMS.
14 Dihedral Angles These are usually variable Range from in molecules Most famous are φ, ψ, ω and χ DIHEDRAL ANGLES ARE A FUNCTION OF THE POSITION OF FOUR ATOMS.
15
16 Ramachandran plot
17 Levels of protein structure Primary structure Secondary structure Tertiary structure Quaternary structure t
18 Pi Primary structure t This is simply pythe amino acid sequences of polypeptides chains (proteins).


19 Secondary structure Local organization of protein backbone: α-helix, β-strand (groups of β-strands assemble into β-sheet), turn and interconnecting loop. an α-helix various representations and orientations i of a two stranded d β-sheet.
20 The α-helix One of the most closely packed arrangement of residues. Turn: 3.6 residues Pitch: 5.4 Å/turn
21 The β-sheeth t Backbone almost fully extended, loosely packed arrangement of residues.
22 Anti-parallel beta sheet
23 Parallel beta sheet
24





25 β-sheet t (parallel) l) Catechol O-Methyltransferase All strands run in the same direction




26 β-sheet (antiparallel) Urate oxidase All strands run in the opposite direction, more stable
27 Loops and Turns Loops: often contain hydrophilic residue on the surface of proteins Turns: loops with less than 5 residues and often contain G, P
28
29 Tertiary structure Description of the type and location of SSEs is a chain s secondary structure. Three-dimensional i coordinates of the atoms of a chain is its tertiary structure. Quaternary structure: describes the spatial packing of several folded polypeptides p
30 Tertiary structure Packing the secondary structure elements into a compact spatial unit Fold or domain this is the level to which structure prediction is currently possible.
31 Quaternary structure Assembly of homo or heteromeric protein chains. Usually the functional unit of a protein, especially for enzymes
32
33
34
35 Primary and secondary structure are ONEdimensional; Tertiary and quaternary structure are THREE-dimensional. structure usually refers to 3-D structure of protein.
36 PDB Files: the header HEADER OXIDOREDUCTASE(SUPEROXIDE ACCEPTOR) 13-JUL-94 COMPND MANGANESE SUPEROXIDE DISMUTASE (E.C ) COMPLEXED COMPND 2 WITH AZIDE OURCE (THERMUS THERMOPHILUS, HB8) AUTHOR M.S.LAH,M.DIXON,K.A.PATTRIDGE,W.C.STALLINGS,J.A.FEE, AUTHOR 2 M.L.LUDWIG REVDAT 2 15-MAY-95 REVDAT 1 15-OCT-94 JRNL AUTH M.S.LAH,M.DIXON,K.A.PATTRIDGE,W.C.STALLINGS, JRNL AUTH 2 J.A.FEE,M.L.LUDWIG JRNL TITL STRUCTURE-FUNCTION IN E. COLI IRON SUPEROXIDE JRNL TITL 2 DISMUTASE: COMPARISONS WITH THE MANGANESE ENZYME JRNL TITL 3 FROM T. THERMOPHILUS JRNL REF TO BE PUBLISHED REMARK 1 AUTH M.L.LUDWIG,A.L.METZGER,K.A.PATTRIDGE,W.C.STALLINGS REMARK 1 TITL MANGANESE SUPEROXIDE DISMUTASE FROM THERMUS REMARK 1 TITL 2 THERMOPHILUS. A STRUCTURAL MODEL REFINED AT 1.8 REMARK 1 TITL 3 ANGSTROMS RESOLUTION REMARK 1 REF J.MOL.BIOL. V REMARK 1 REFN ASTM JMOBAK UK ISSN REMARK 1 REFERENCE 2 REMARK 1 AUTH W.C.STALLINGS,C.BULL,J.A.FEE,M.S.LAH,M.L.LUDWIG REMARK 1 TITL IRON AND MANGANESE SUPEROXIDE DISMUTASES: REMARK 1 TITL 2 CATALYTIC INFERENCES FROM THE STRUCTURES
37 PDB Files: the coordinates Atom & Residue XYZ Coordinates ATOM 1 N PRO A MNG 192 ATOM 2 CA PRO A MNG 193 ATOM 3 C PRO A MNG 194 ATOM 4 O PRO A MNG 195 ATOM 5 CB PRO A MNG 196 ATOM 6 CG PRO A MNG 197 ATOM 7 CD PRO A MNG 198 ATOM 8 N TYR A MNG 199 ATOM 9 CA TYR A MNG 200 ATOM 10 C TYR A MNG 201 ATOM 11 O TYR A MNG 202 ATOM 12 CB TYR A MNG 203 ATOM 13 CG TYR A MNG 204 ATOM 14 CD1 TYR A MNG 205 ATOM 15 CD2 TYR A MNG 206 ATOM 16 CE1 TYR A MNG 207 ATOM 17 CE2 TYR A MNG 208 ATOM 18 CZ TYR A MNG 209 ATOM 19 OH TYR A MNG 210 ATOM 20 N PRO A MNG 211 ATOM 21 CA PRO A MNG 212

38 Motifs Four helix bundle Helix-loop-helix Coiled coil
39 Secondary structure prediction Given a protein sequence (primary structure) GHWIATRGQLIREAYEDYRHFSSECPFIP Predict its secondary structure content (C=coils H=Alpha Helix E=Beta Strands) CEEEEECHHHHHHHHHHHCCCHHCCCCCC
40 Why Secondary Structure Prediction? Easier problem than 3D structure t prediction (more than 40 years of history). Accurate secondary structure prediction can be an important information for the tertiary structure prediction Improving sequence alignment accuracy Protein function prediction Protein classification Predicting structural change
41 Prediction Methods Statistical i methods Chou-Fasman method, GOR I-IV Nearest neighbors NNSSP, SSPAL Neural network PHD, Psi-Pred, J-Pred Support vector machine
42 Assumptions The entire information for forming secondary structure is contained in the primary sequence. Side groups of residues will determine structure. Examining windows of residues is sufficient to predict structure.
43 Chou-Fasman method Compute parameters a for amino acids Preference to be in alpha helix: P(a) beta sheet: P(b) Turn: P(turn) Frequencies with which the amino acid is in the 1 st, 2 nd, 3 rd, and d4 th position of a turn: f(i), f(i+1), f(i+2), f(i+3). Use a sliding window
44 SSE prediction Alpha-helix prediction Find all regions where 4 of the 6 amino acids in window have P(a) > 100. Extend the region in both directions unless 4 consecutive residues have P(a) < 100. If Σ P(a) > Σ P(b) then the region is predicted to be alpha-helix. Beta-sheet prediction is analogous. Turn prediction Compute P(t) = f(i) + f(i+1) + f(i+2) + f(i+3) for 4 consecutive residues. Predict a turn if P(t) > (check) The average P(turn) > 100 Σ P(turn) > Σ P(a) and Σ P(turn) > Σ P(b)
45 GOR method Use a sliding window of 17 residues Compute the frequencies with which each amino acid occupies the 17 positions in helix, sheet, and turn. Use this to predict the SSE probability of each residue.
46 Performance of SSE prediction A Simple and Fast Secondary Structure Prediction Q3 and SOV are standards Method using Hidden Neural Networks Kuang Lin, Victor A. Simossis, Willam R. Taylor, Jaap Heringa, for computing errors Bioinformatics Advance Access published September 17, 2004


47 Relevance of Protein Structure in the Post-Genome Era structure medicine sequence function
48 Structure-Function Relationship Certain level of function can be found without structure. But a structure is a key to understand the detailed d mechanism. A predicted d structure t is a powerful tool for function inference. Trp repressor as a function switch



49 Structure-Based Drug Design Structure-based rational drug design is a major method for drug discovery. HIV protease inhibitor
50 Experimental techniques for structure determination X-ray Crystallography Nuclear Magnetic Resonance spectroscopy (NMR) Electron Microscopy/Diffraction Free electron lasers?





51 X-ray Crystallography
52 X-ray Crystallography.. From small molecules to viruses Information about the positions of individual atoms Limited it information about dynamics Requires crystals
 Information about distances between pairs of atoms")

53 NMR Limited to molecules up to ~50kDa (good quality up to 30 kda) Information about distances between pairs of atoms A 2-d resonance spectrum with offdiagonal peaks Requires soluble non aggregating Requires soluble, non-aggregating material
54 Protein Folding Problem A protein folds into a unique 3D structure under the physiological condition: determine this structure Lysozyme sequence: KVFGRCELAA AMKRHGLDNY RGYSLGNWVC AAKFESNFNT QATNRNTDGS TDYGILQINS RWWCNDGRTP GSRNLCNIPC SALLSSDITA SVNCAKKIVS DGNGMNAWVA WRNRCKGTDV QAWIRGCRL
55 Levinthal s paradox Consider a 100 residue protein. If each residue can take only 3 positions, there are = possible conformations. If it takes s to convert from 1 structure to another, exhaustive search would take years! Folding must proceed by progressive stabilization of intermediates.
56 Forces driving protein folding It is believed that t hd hydrophobic hbi collapse is a key driving force for protein folding Hydrophobic core Polar surface interacting with solvent Minimum volume (no cavities) Disulfide bond formation stabilizes Hydrogen bonds Polar and electrostatic interactions
57 Effect of a single mutation Hemoglobin is the protein in red blood cells (erythrocytes) y responsible for binding oxygen. The mutation E V in the β chain replaces a charged Glu by a hydrophobic Val on the surface of fhemoglobin The resulting sticky patch causes hemoglobin to agglutinate (stick together) and form fibers which deform the red blood cell and do not carry oxygen efficiently Sickle cell anemia was the first identified molecular disease








58 Sickle Cell Anemia Sequestering hydrophobic residues in the protein core protects proteins from hydrophobic agglutination.
59 Protein Structure Prediction Ab-initio techniques Homology modeling Sequence-sequence comparison Protein threading Sequence-structure comparison
60 Lattice models Simple lattice models (HP-models) Two types of residues: hydrophobic and polar 2-D or 3-D lattice The only force is hydrophobic collapse Score = number of H H contacts
61 Scoring Lattice Models H/P model scoring: count hydrophobic interactions. Score = 5 Sometimes: Penalize for buried polar or surface hydrophobic residues
62 What can we do with lattice models? NP-complete For smaller polypeptides, exhaustive search can be used Looking at the best fold, even in such a simple model, can teach us interesting things about the protein folding process For larger chains, other optimization and search methods must be used Greedy, branch and bound Evolutionary computing, simulated annealing Graph theoretical methods
63 Representing a lattice model Absolute directions UURRDLDRRU Relative directions LFRFRRLLFL Advantage, we can t have UD or RL in absolute Only three directions: LRF What about bumps? LFRRR Give bad score to any configuration that has bumps
64 More realistic models Higher resolution lattices (45 lattice, etc.) Off-lattice models Local moves Optimization/search methods and φ/ψ representations Greedy search Branch and bound EC, Monte Carlo, simulated annealing, etc.
65 Energy functions An energy function to describe the protein bond energy bond angle energy dihedral angel energy van der Waals energy electrostatic energy Minimize the function and obtain the structure. Not practical in general Computationally too expensive Accuracy is poor Empirical force fields Start with a database Look at neighboring residues similar to known protein folds?
66 Difficulties Why is structure prediction and especially ab initio calculations hard? Many degrees of freedom / residue. Computationally too expensive for realistic-sized proteins. Remote non-covalent interactions Nature does not go through all conformations Folding assisted by enzymes & chaperones
67 Protein Structure Prediction Ab-initio techniques Homology modeling Sequence-sequence comparison Protein threading Sequence-structure comparison
68 Homology modeling steps 1. Identify a set of template proteins (with known structures) related to the target t protein. This is based on sequence homology (BLAST, FASTA) with sequence identity of 30% or more. 2. Align the target sequence with the template proteins. This is based on multiple alignment (CLUSTALW). Identify conserved regions. 3. Build a model of the protein backbone, taking the backbone of the template structures (conserved regions) as a model. 4. Model the loops. In regions with gaps, use a loopmodeling procedure to substitute segments of appropriate length. 5. Add sidechains to the model backbone. 6. Evaluate and optimize entire structure.
69 Homology Modeling Servers SWISS-MODEL ESyPred3D
70 Protein Structure Prediction Ab-initio techniques Homology modeling Protein threading Sequence-structure comparison



71 Protein threading Structure is better conserved than sequence Structure can adopt a wide range of mutations. Physical forces favor certain structures. Number of folds is limited. Currently ~700 Total: 1,000 ~10,000 TIM barrel
72 Protein Threading Basic premise The number of unique structural t (domain) folds in nature is fairly small (possibly a few thousand) Statistics from Protein Data Bank (~35,000 structures) 90% of new structures submitted to PDB in the past three years have similar structural folds in PDB
 a query")



73 Concept of Threading o Thread (align or place) a query protein sequence onto a template structure in optimal way o Good alignment gives approximate backbone structure Query sequence MTYKLILNGKTKGETTTEAVDAATAEKVFQYANDNGVDGEWTYTE Template set
74 Threading problem Threading: Given a sequence, and a fold (template), compute the optimal alignment score between the sequence and dthe fold. If we can solve the above problem, then Given a sequence, we can try each known fold, and find the best fold that fits this sequence. Because there are only a few thousands folds, we can find the correct fold for the given sequence. Threading is NP-hard.
75 Components of Threading Template library Use structures from DB classification categories (PDB) Scoring function Single and pairwise energy terms Alignment Consideration of pairwise terms leads to NP-hardness heuristics Confidence assessment Z-score, P-value similar to sequence alignment statistics ti ti Improvements Local threading, multi-structure t t threading








76 Protein Threading structure database Build a template database


77 Protein Threading energy function MTYKLILNGKTKGETTTEAVDAATAEKVFQYANDNGVDGEWTYTE how preferable to put two particular residues nearby: E_p alignment gap penalty: E_g how well a residue fits a structural environment: E_s total energy: E_p + E_s + E_g find a sequence-structure alignment to minimize the energy function




78 Assessing Prediction Reliability MTYKLILNGKTKGETTTEAVDAATAEKVFQYANDNGVDGEWTYTE Score = Score = -720 Score = Score = -900 Which one is the correct structural fold for the target sequence if any? The one with the highest score?

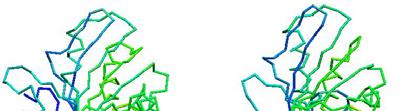





79 Prediction of Protein Structures Examples p a few g good examples p actual actual predicted predicted actual actual predicted predicted



80 Prediction of Protein Structures Not so good example
81 Existing Prediction Programs PROSPECT FUGU THREADER
82
83 CASP/CAFASP CASP: Critical Assessment of Structure Prediction CASP Predictor CAFASP: Critical Assessment of Fully Automated Structure Prediction CAFASP Predictor 1 W t t ti d 1. Won t get tired 2. High-throughput
84 CASP6/CAFASP4 64 targets Resources for predictors No X-ray, NMR machines (of course) CAFASP4 predictors: no manual intervention CASP6 predictors: anything (servers, google, ) Evaluation: CASP6 Assessed by experts+computer p CAFASP4 evaluated by a computer program. Predicted structures are superimposed on the experimental structures. t CASP7 was held last November
85 (a) myoglobin (b) hemoglobin (c) lysozyme (d) transfer RNA (e) antibodies (f) viruses (g) actin (h) the nucleosome (i) myosin (j) ribosome Courtesy of David Goodsell, TSRI
86 Protein structure databases PDB 3D structures SCOP Murzin, Brenner, Hubbard, Chothia Classification Class (mostly alpha, mostly beta, alpha/beta (interspersed), alpha+beta (segregated), multi-domain, membrane) Fold (similar structure) Superfamily (homology, distant sequence similarity) Family (homology and close sequence similarity)
87 The SCOP Database Structural Classification Of Proteins FAMILY: proteins that are >30% similar, or >15% similar and have similar known structure/function SUPERFAMILY: proteins whose families have some sequence and function/structure similarity suggesting a common evolutionary origin COMMON FOLD: superfamilies that have same secondary structures in same arrangement, probably resulting by physics and chemistry CLASS: alpha, beta, alpha beta, alpha+beta, multidomain
88 Protein databases CATH Orengo et al Class (alpha, beta, alpha/beta, few SSEs) Architecture (orientation of SSEs but ignoring connectivity) Topology (orientation and connectivity, based on SSAP = fold of SCOP) Homology (sequence similarity = superfamily of SCOP) S level (high sequence similarity = family of SCOP) SSAP alignment tool (dynamic programming)
89 Protein databases FSSP DALI structure alignment tool (distance matrix) Holm and Sander MMDB VAST structure comparison (hierarchical) Madej, Bryant et al
90 Protein structure comparison Levels of structure description Atom/atom group Residue Fragment Secondary structure t element (SSE) Basis of comparison Geometry/architecture t of coordinates/relative ti positions sequential order of residues along backbone,... physio-chemical properties of residues,
91 How to compare? Key problem: find an optimal correspondence between the arrangements of atoms in two molecular structures (say A and B) in order to align them in 3D Optimality of the alignment is determined using a root mean square measure of the distances between corresponding atoms in the two molecules l Complication: It is not known a priori which atom in molecule B corresponds to a given atom in molecule A (the two molecules may not even have the same number of atoms)
92 Structure Analysis Basic Issues Coordinates for representing 3D structures Cartesian Other (e.g. dihedral angles) Basic operations Translation in 3D space Rotation in 3D space Comparing 3D structures Root mean square distances between points of two molecules are typically used as a measure of how well they are aligned Efficient ways to compute minimal RMSD once correspondences are known (O(n) algorithm) Using eigenvalue analysis of correlation matrix of points Due to the high computational complexity, practical algorithms rely on heuristics
93 Structure Analysis Basic Issues Sequence order dependent approaches Computationally this is easier Interest t in motifs preserving sequence order Sequence order independent approaches More general Active sites may involve non-local AAs Searching with structural information
94 Find the optimal alignment +
95 Optimal Alignment Find the highest number of atoms aligned with the lowest RMSD (Root Mean Squared Deviation) Find a balance between local regions with very Find a balance between local regions with very good alignments and overall alignment
96 Structure Comparison Which atom in structure A corresponds to which atom in structure B? THESESENTENCESALIGN--NICELY S S C THE--SEQUENCE-ALIGNEDNICELY
97 Structural Alignment An optimal superposition of myoglobin and beta-hemoglobin, which are structural neighbors. However, their sequence homology is only 8.5%
98 Structure Comparison Methods to superimpose structures by translation and rotation x 1, y 1, z 1 x 1 + d, y 1, z 1 x 2, y 2, z 2 x 3, y 3, z 3 Translation x 2 + d, y 2, z 2 x 3 + d, y 3, z 3 Rotation
99 Structure Comparison Scoring system to find optimal alignment Answer: Root Mean Square Deviation (RMSD) RMSD n = number of atoms = dd 2 i i d i = distance between 2 corresponding atoms i in 2 structures n
100 Root Mean Square Deviation Root Mean Square Deviation 5 d d d d d ~ 5 ) X (X RMS i 2 BLUE1 RED = =
101 RMSD Unit of RMSD => e.g. Ångstroms - identical structures => RMSD = 0 - similar structures => RMSD is small (1 3 Å) - distant structures => RMSD > 3 Å
102 Pitfalls of RMSD all atoms are treated t equally (e.g. residues on the surface have a higher degree of freedom than those in the core) best alignment does not always mean minimal RMSD significance of RMSD is size dependent
103 Alternative RMSDs armsd = best root-mean-square distance calculated over all aligned alpha-carbon atoms brmsd = the RMSD over the highest scoring residue pairs wrmsd = weighted RMSD Source: W. Taylor(1999), Protein Science, 8:
104 Structural Alignment Methods Distance based methods DALI (Holm and Sander, 1993): Aligning i 2-dimensional i distance matrices STRUCTAL (Subbiah 1993, Gerstein and Levitt 1996): Dynamic programming to minimize the RMSD between two protein backbones. SSAP (Orengo and Taylor, 1990): Double dynamic programming using intra-molecular distance; CE (Shindyalov and Bourne, 1998): Combinatorial Extension of best matching regions Vector based methods VAST (Madej et al., 1995): Graph theory based SSE alignment; 3dSearch (Singh and Brutlag, 1997) and 3D Lookup (Holm and Sander, 1995): Fast SSE index lookup by geometric hashing. TOP (Lu, 2000): SSE vector superpositioning. TOPSCAN (Martin, 2000): Symbolic linear representation of SSE vectors. Both vector and distance based LOCK (Singh and Brutlag, 1997): Hierarchically uses both secondary structures vectors and atomic distances.
105 Basic DP (STRUCTAL) 1. Start with arbitrary alignment of the points in two molecules A and B 2. Superimpose in order to minimize RMSD. 3. Compute a structural alignment (SA) matrix where entry (i,j) (,j) is the score for the structural similarity between the i th point of A and the j th point of B 4. Use DP to compute the next alignment. Gap cost = 0 5. Iterate steps 2--4 until the overall score converges 6. Repeat with a number of initial iti alignments
106 STRUCTAL Given 2S Structures (A&B) B), 2 Basic Comparison Operations 1. Given an alignment optimally SUPERIMPOSE A onto B 2. Find an Alignment between A and B based on their 3D coordinates S ij = M/[1+(d ij /d 0 ) 2 ] M and d 0 are constants
107
108 DALI Method Distance matrix alignment Liisa Holm and Chris Sander, Protein structure comparison by alignment of distance matrices, Journal of Molecular Biology Vol. 233, Liisa Holm and Chris Sander, Mapping the protein universe, Science Vol. 273, Liisa Holm and Chris Sander, Alignment of three-dimensional protein structures: network server for database searching, Methods in Enzymology Vol. 266, 1996.
109 How DALI Works? Based on fact: similar 3D structures have similar intra-molecular distances. Background idea Represent each protein as a 2D matrix storing intramolecular distance. Place one matrix on top of another and slide vertically and horizontally until a common the sub-matrix with the best match is found. Actual implementation Protein A Protein B Break each matrix into small sub-matrices of fixed size. Pair-up similar sub-matrices (one from each protein). Assemble the sub-matrix pairs to get the overall alignment.


110 Structure Representation of DALI 3D shape is described with a distance matrix which stores all intra-molecular l distances between the C α atoms. Distance matrix is independent of coordinate frame. Contains enough information to re-construct the 3D coordinates. Protein A Distance matrix for Protein A Distance matrix for 2drpA and 1bbo d 12 d 13 d 14 d 12 0 d 23 d 24 3 d 13 d 23 0 d 34 4 d 14 d 24 d 34 0




111 Intra-molecular distance for myoglobin
112 DALI Algorithm 1. Decompose distance matrix into elementary contact patterns (sub-matrices of fixed size) Use hexapeptide-hexapeptide contact patterns. 2. Compare contact patterns (pair-wise), and store the matching pairs in pair list. 3. Assemble pairs in the correct order to yield the overall alignment.
113 Assembly of Alignments Non-trivial combinatory yproblem. Assembled in the manner (AB) (A B ), (BC) (B C ),... (i.e., having one overlapping segment with the previous alignment) Available Alignment Methods: Monte Carlo optimization Brach-and-bound Neighbor walk
114 Schematic View of DALI Algorithm 3D (Spatial) 2D (Distance Matrix) 1D (Sequence)
115 Monte Carlo Optimization Used in the earlier versions of DALI. Algorithm Compute a similarity score for the current alignment. Make a random trial change to the current alignment (adding a new pair or deleting an existing pair). Compute the change in the score ( S). If S > 0, the move is always accepted. If S <= 0, the move may be accepted by the probability exp(β * S), where β is a parameter. Once a move is accepted, the change in the alignment becomes permanent. This procedure is iterated until there is no further change in the score, i.e., the system is converged.
116 Branch-and-bound and method Used in the later versions of DALI. Based on Lathrop and Smith s (1996) threading (sequence- structure alignment) algorithm. Solution space consists of all possible placements of residues in protein A relative to the segment of residues of protein B. The algorithm recursively split the solution space that yields the highest upper bound of the similarity score until there is a single alignment trace left.
117 LOCK Uses a hierarchical approach Larger secondary structures such as helixes and strands are represented using vectors and dealt with first Atoms are dealt with afterwards Assumes large secondary structures provide most stability and function to a protein, and are most likely to be preserved during evolution
118 LOCK (Contd.) Key algorithm steps: 1. Represent secondary structures as vectors 2. Obtain initial superposition by computing local alignment of the secondary structure vectors (using dynamic programming) 3. Compute atomic superposition by performing a greedy search to try to minimize root mean square deviation (a RMS distance measure) between pairs of nearest atoms from the two proteins 4. Identify core (well aligned) atoms and try to improve their superposition (possibly at the cost of degrading superposition of non-core atoms) Steps 2, 3, and 4 require iteration at each step
119 Alignment of SSEs Define an orientation-dependent score and an orientationindependent score between SSE vectors. For every pair of query vectors, find all pairs of vectors in database protein that align with a score above a threshold. Two of these vectors must be adjacent. Use orientation independent scores. For each set of four vectors from previous step, find the transformation minimizing rmsd. Apply this transformation to the query. Run dynamic programming using both orientation-dependent ti d t and orientation-independent scores to find the best local alignment. Compute and apply the transformation from the best local alignment. Superpose p in order to minimize rmsd.
120 Atomic superposition Loop find matching gpairs of C α atoms use only those within 3 A find best alignment until rmsd does not change
121 Core identification Loop find the best core (symmetric nns) and align; remove the rest until rmsd does not change
122 VAST Begin with a set of nodes (a,x) where SSEs a and x are of the same type Add an edge between (a,x) and (b,y) if angle and distance between (a,b) is same as between (x,y) Find the maximal clique in this graph; this forms the initial SSE alignment Extend the initial alignment to C α atoms using Gibbs sampling Report statistics on this match
123 Quality of a structure match Statistical theory similar to BLAST Compare the likelihood of a match as compared to a random match Less agreement regarding score matrix z-scores of CE, DALI, and VAST may not be compatible
HIV protease inhibitor. Certain level of function can be found without structure. But a structure is a key to understand the detailed mechanism.
 Proteins are linear polypeptide chains (one or more) Building blocks: 20 types of amino acids. Range from a few 10s-1000s They fold into varying three-dimensional shapes structure medicine Certain level
Proteins are linear polypeptide chains (one or more) Building blocks: 20 types of amino acids. Range from a few 10s-1000s They fold into varying three-dimensional shapes structure medicine Certain level
Protein structure is more conserved than protein sequence, and more closely related to function.
 Why protein structure? The basics of protein Basic measurements for protein structure Levels of protein structure Prediction of protein structure from sequence Finding similarities between protein structures
Why protein structure? The basics of protein Basic measurements for protein structure Levels of protein structure Prediction of protein structure from sequence Finding similarities between protein structures
Basics of protein structure
 Today: 1. Projects a. Requirements: i. Critical review of one paper ii. At least one computational result b. Noon, Dec. 3 rd written report and oral presentation are due; submit via email to bphys101@fas.harvard.edu
Today: 1. Projects a. Requirements: i. Critical review of one paper ii. At least one computational result b. Noon, Dec. 3 rd written report and oral presentation are due; submit via email to bphys101@fas.harvard.edu
Protein Structure: Data Bases and Classification Ingo Ruczinski
 Protein Structure: Data Bases and Classification Ingo Ruczinski Department of Biostatistics, Johns Hopkins University Reference Bourne and Weissig Structural Bioinformatics Wiley, 2003 More References
Protein Structure: Data Bases and Classification Ingo Ruczinski Department of Biostatistics, Johns Hopkins University Reference Bourne and Weissig Structural Bioinformatics Wiley, 2003 More References
CAP 5510 Lecture 3 Protein Structures
 CAP 5510 Lecture 3 Protein Structures Su-Shing Chen Bioinformatics CISE 8/19/2005 Su-Shing Chen, CISE 1 Protein Conformation 8/19/2005 Su-Shing Chen, CISE 2 Protein Conformational Structures Hydrophobicity
CAP 5510 Lecture 3 Protein Structures Su-Shing Chen Bioinformatics CISE 8/19/2005 Su-Shing Chen, CISE 1 Protein Conformation 8/19/2005 Su-Shing Chen, CISE 2 Protein Conformational Structures Hydrophobicity
Protein Structure Prediction II Lecturer: Serafim Batzoglou Scribe: Samy Hamdouche
 Protein Structure Prediction II Lecturer: Serafim Batzoglou Scribe: Samy Hamdouche The molecular structure of a protein can be broken down hierarchically. The primary structure of a protein is simply its
Protein Structure Prediction II Lecturer: Serafim Batzoglou Scribe: Samy Hamdouche The molecular structure of a protein can be broken down hierarchically. The primary structure of a protein is simply its
Bioinformatics. Macromolecular structure
 Bioinformatics Macromolecular structure Contents Determination of protein structure Structure databases Secondary structure elements (SSE) Tertiary structure Structure analysis Structure alignment Domain
Bioinformatics Macromolecular structure Contents Determination of protein structure Structure databases Secondary structure elements (SSE) Tertiary structure Structure analysis Structure alignment Domain
CMPS 6630: Introduction to Computational Biology and Bioinformatics. Structure Comparison
 CMPS 6630: Introduction to Computational Biology and Bioinformatics Structure Comparison Protein Structure Comparison Motivation Understand sequence and structure variability Understand Domain architecture
CMPS 6630: Introduction to Computational Biology and Bioinformatics Structure Comparison Protein Structure Comparison Motivation Understand sequence and structure variability Understand Domain architecture
CMPS 6630: Introduction to Computational Biology and Bioinformatics. Tertiary Structure Prediction
 CMPS 6630: Introduction to Computational Biology and Bioinformatics Tertiary Structure Prediction Tertiary Structure Prediction Why Should Tertiary Structure Prediction Be Possible? Molecules obey the
CMPS 6630: Introduction to Computational Biology and Bioinformatics Tertiary Structure Prediction Tertiary Structure Prediction Why Should Tertiary Structure Prediction Be Possible? Molecules obey the
Introduction to Comparative Protein Modeling. Chapter 4 Part I
 Introduction to Comparative Protein Modeling Chapter 4 Part I 1 Information on Proteins Each modeling study depends on the quality of the known experimental data. Basis of the model Search in the literature
Introduction to Comparative Protein Modeling Chapter 4 Part I 1 Information on Proteins Each modeling study depends on the quality of the known experimental data. Basis of the model Search in the literature
Giri Narasimhan. CAP 5510: Introduction to Bioinformatics. ECS 254; Phone: x3748
 CAP 5510: Introduction to Bioinformatics Giri Narasimhan ECS 254; Phone: x3748 giri@cis.fiu.edu www.cis.fiu.edu/~giri/teach/bioinfs07.html 2/15/07 CAP5510 1 EM Algorithm Goal: Find θ, Z that maximize Pr
CAP 5510: Introduction to Bioinformatics Giri Narasimhan ECS 254; Phone: x3748 giri@cis.fiu.edu www.cis.fiu.edu/~giri/teach/bioinfs07.html 2/15/07 CAP5510 1 EM Algorithm Goal: Find θ, Z that maximize Pr
CMPS 3110: Bioinformatics. Tertiary Structure Prediction
 CMPS 3110: Bioinformatics Tertiary Structure Prediction Tertiary Structure Prediction Why Should Tertiary Structure Prediction Be Possible? Molecules obey the laws of physics! Conformation space is finite
CMPS 3110: Bioinformatics Tertiary Structure Prediction Tertiary Structure Prediction Why Should Tertiary Structure Prediction Be Possible? Molecules obey the laws of physics! Conformation space is finite
ALL LECTURES IN SB Introduction
 1. Introduction 2. Molecular Architecture I 3. Molecular Architecture II 4. Molecular Simulation I 5. Molecular Simulation II 6. Bioinformatics I 7. Bioinformatics II 8. Prediction I 9. Prediction II ALL
1. Introduction 2. Molecular Architecture I 3. Molecular Architecture II 4. Molecular Simulation I 5. Molecular Simulation II 6. Bioinformatics I 7. Bioinformatics II 8. Prediction I 9. Prediction II ALL
Analysis and Prediction of Protein Structure (I)
") Analysis and Prediction of Protein Structure (I) Jianlin Cheng, PhD School of Electrical Engineering and Computer Science University of Central Florida 2006 Free for academic use. Copyright @ Jianlin Cheng
Analysis and Prediction of Protein Structure (I) Jianlin Cheng, PhD School of Electrical Engineering and Computer Science University of Central Florida 2006 Free for academic use. Copyright @ Jianlin Cheng
1. Protein Data Bank (PDB) 1. Protein Data Bank (PDB)
 1. Protein Data Bank (PDB)") Protein structure databases; visualization; and classifications 1. Introduction to Protein Data Bank (PDB) 2. Free graphic software for 3D structure visualization 3. Hierarchical classification of protein
Protein structure databases; visualization; and classifications 1. Introduction to Protein Data Bank (PDB) 2. Free graphic software for 3D structure visualization 3. Hierarchical classification of protein
Structural Alignment of Proteins
 Goal Align protein structures Structural Alignment of Proteins 1 2 3 4 5 6 7 8 9 10 11 12 13 14 PHE ASP ILE CYS ARG LEU PRO GLY SER ALA GLU ALA VAL CYS PHE ASN VAL CYS ARG THR PRO --- --- --- GLU ALA ILE
Goal Align protein structures Structural Alignment of Proteins 1 2 3 4 5 6 7 8 9 10 11 12 13 14 PHE ASP ILE CYS ARG LEU PRO GLY SER ALA GLU ALA VAL CYS PHE ASN VAL CYS ARG THR PRO --- --- --- GLU ALA ILE
Protein Structure Prediction
 Page 1 Protein Structure Prediction Russ B. Altman BMI 214 CS 274 Protein Folding is different from structure prediction --Folding is concerned with the process of taking the 3D shape, usually based on
Page 1 Protein Structure Prediction Russ B. Altman BMI 214 CS 274 Protein Folding is different from structure prediction --Folding is concerned with the process of taking the 3D shape, usually based on
CAP 5510: Introduction to Bioinformatics CGS 5166: Bioinformatics Tools. Giri Narasimhan
 CAP 5510: Introduction to Bioinformatics CGS 5166: Bioinformatics Tools Giri Narasimhan ECS 254; Phone: x3748 giri@cis.fiu.edu www.cis.fiu.edu/~giri/teach/bioinff18.html Proteins and Protein Structure
CAP 5510: Introduction to Bioinformatics CGS 5166: Bioinformatics Tools Giri Narasimhan ECS 254; Phone: x3748 giri@cis.fiu.edu www.cis.fiu.edu/~giri/teach/bioinff18.html Proteins and Protein Structure
Getting To Know Your Protein
 Getting To Know Your Protein Comparative Protein Analysis: Part III. Protein Structure Prediction and Comparison Robert Latek, PhD Sr. Bioinformatics Scientist Whitehead Institute for Biomedical Research
Getting To Know Your Protein Comparative Protein Analysis: Part III. Protein Structure Prediction and Comparison Robert Latek, PhD Sr. Bioinformatics Scientist Whitehead Institute for Biomedical Research
Statistical Machine Learning Methods for Bioinformatics IV. Neural Network & Deep Learning Applications in Bioinformatics
 Statistical Machine Learning Methods for Bioinformatics IV. Neural Network & Deep Learning Applications in Bioinformatics Jianlin Cheng, PhD Department of Computer Science University of Missouri, Columbia
Statistical Machine Learning Methods for Bioinformatics IV. Neural Network & Deep Learning Applications in Bioinformatics Jianlin Cheng, PhD Department of Computer Science University of Missouri, Columbia
Protein Structures. 11/19/2002 Lecture 24 1
 Protein Structures 11/19/2002 Lecture 24 1 All 3 figures are cartoons of an amino acid residue. 11/19/2002 Lecture 24 2 Peptide bonds in chains of residues 11/19/2002 Lecture 24 3 Angles φ and ψ in the
Protein Structures 11/19/2002 Lecture 24 1 All 3 figures are cartoons of an amino acid residue. 11/19/2002 Lecture 24 2 Peptide bonds in chains of residues 11/19/2002 Lecture 24 3 Angles φ and ψ in the
Protein Dynamics. The space-filling structures of myoglobin and hemoglobin show that there are no pathways for O 2 to reach the heme iron.
 Protein Dynamics The space-filling structures of myoglobin and hemoglobin show that there are no pathways for O 2 to reach the heme iron. Below is myoglobin hydrated with 350 water molecules. Only a small
Protein Dynamics The space-filling structures of myoglobin and hemoglobin show that there are no pathways for O 2 to reach the heme iron. Below is myoglobin hydrated with 350 water molecules. Only a small
Protein structure prediction. CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror
 Protein structure prediction CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror 1 Outline Why predict protein structure? Can we use (pure) physics-based methods? Knowledge-based methods Two major
Protein structure prediction CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror 1 Outline Why predict protein structure? Can we use (pure) physics-based methods? Knowledge-based methods Two major
Dihedral Angles. Homayoun Valafar. Department of Computer Science and Engineering, USC 02/03/10 CSCE 769
 Dihedral Angles Homayoun Valafar Department of Computer Science and Engineering, USC The precise definition of a dihedral or torsion angle can be found in spatial geometry Angle between to planes Dihedral
Dihedral Angles Homayoun Valafar Department of Computer Science and Engineering, USC The precise definition of a dihedral or torsion angle can be found in spatial geometry Angle between to planes Dihedral
Protein Secondary Structure Prediction
 Protein Secondary Structure Prediction Doug Brutlag & Scott C. Schmidler Overview Goals and problem definition Existing approaches Classic methods Recent successful approaches Evaluating prediction algorithms
Protein Secondary Structure Prediction Doug Brutlag & Scott C. Schmidler Overview Goals and problem definition Existing approaches Classic methods Recent successful approaches Evaluating prediction algorithms
Physiochemical Properties of Residues
 Physiochemical Properties of Residues Various Sources C N Cα R Slide 1 Conformational Propensities Conformational Propensity is the frequency in which a residue adopts a given conformation (in a polypeptide)
Physiochemical Properties of Residues Various Sources C N Cα R Slide 1 Conformational Propensities Conformational Propensity is the frequency in which a residue adopts a given conformation (in a polypeptide)
Sequence analysis and comparison
 The aim with sequence identification: Sequence analysis and comparison Marjolein Thunnissen Lund September 2012 Is there any known protein sequence that is homologous to mine? Are there any other species
The aim with sequence identification: Sequence analysis and comparison Marjolein Thunnissen Lund September 2012 Is there any known protein sequence that is homologous to mine? Are there any other species
Week 10: Homology Modelling (II) - HHpred
 - HHpred") Week 10: Homology Modelling (II) - HHpred Course: Tools for Structural Biology Fabian Glaser BKU - Technion 1 2 Identify and align related structures by sequence methods is not an easy task All comparative
Week 10: Homology Modelling (II) - HHpred Course: Tools for Structural Biology Fabian Glaser BKU - Technion 1 2 Identify and align related structures by sequence methods is not an easy task All comparative
Procheck output. Bond angles (Procheck) Structure verification and validation Bond lengths (Procheck) Introduction to Bioinformatics.
 Structure verification and validation Bond lengths (Procheck) Introduction to Bioinformatics.") Structure verification and validation Bond lengths (Procheck) Introduction to Bioinformatics Iosif Vaisman Email: ivaisman@gmu.edu ----------------------------------------------------------------- Bond
Structure verification and validation Bond lengths (Procheck) Introduction to Bioinformatics Iosif Vaisman Email: ivaisman@gmu.edu ----------------------------------------------------------------- Bond
Programme Last week s quiz results + Summary Fold recognition Break Exercise: Modelling remote homologues
 Programme 8.00-8.20 Last week s quiz results + Summary 8.20-9.00 Fold recognition 9.00-9.15 Break 9.15-11.20 Exercise: Modelling remote homologues 11.20-11.40 Summary & discussion 11.40-12.00 Quiz 1 Feedback
Programme 8.00-8.20 Last week s quiz results + Summary 8.20-9.00 Fold recognition 9.00-9.15 Break 9.15-11.20 Exercise: Modelling remote homologues 11.20-11.40 Summary & discussion 11.40-12.00 Quiz 1 Feedback
Protein Structure Prediction and Display
 Protein Structure Prediction and Display Goal Take primary structure (sequence) and, using rules derived from known structures, predict the secondary structure that is most likely to be adopted by each
Protein Structure Prediction and Display Goal Take primary structure (sequence) and, using rules derived from known structures, predict the secondary structure that is most likely to be adopted by each
Neural Networks for Protein Structure Prediction Brown, JMB CS 466 Saurabh Sinha
 Neural Networks for Protein Structure Prediction Brown, JMB 1999 CS 466 Saurabh Sinha Outline Goal is to predict secondary structure of a protein from its sequence Artificial Neural Network used for this
Neural Networks for Protein Structure Prediction Brown, JMB 1999 CS 466 Saurabh Sinha Outline Goal is to predict secondary structure of a protein from its sequence Artificial Neural Network used for this
Introduction to" Protein Structure
 Introduction to" Protein Structure Function, evolution & experimental methods Thomas Blicher, Center for Biological Sequence Analysis Learning Objectives Outline the basic levels of protein structure.
Introduction to" Protein Structure Function, evolution & experimental methods Thomas Blicher, Center for Biological Sequence Analysis Learning Objectives Outline the basic levels of protein structure.
Supporting Online Material for
 www.sciencemag.org/cgi/content/full/309/5742/1868/dc1 Supporting Online Material for Toward High-Resolution de Novo Structure Prediction for Small Proteins Philip Bradley, Kira M. S. Misura, David Baker*
www.sciencemag.org/cgi/content/full/309/5742/1868/dc1 Supporting Online Material for Toward High-Resolution de Novo Structure Prediction for Small Proteins Philip Bradley, Kira M. S. Misura, David Baker*
Protein Structure Basics
 Protein Structure Basics Presented by Alison Fraser, Christine Lee, Pradhuman Jhala, Corban Rivera Importance of Proteins Muscle structure depends on protein-protein interactions Transport across membranes
Protein Structure Basics Presented by Alison Fraser, Christine Lee, Pradhuman Jhala, Corban Rivera Importance of Proteins Muscle structure depends on protein-protein interactions Transport across membranes
Details of Protein Structure
 Details of Protein Structure Function, evolution & experimental methods Thomas Blicher, Center for Biological Sequence Analysis Anne Mølgaard, Kemisk Institut, Københavns Universitet Learning Objectives
Details of Protein Structure Function, evolution & experimental methods Thomas Blicher, Center for Biological Sequence Analysis Anne Mølgaard, Kemisk Institut, Københavns Universitet Learning Objectives
Homology Modeling. Roberto Lins EPFL - summer semester 2005
 Homology Modeling Roberto Lins EPFL - summer semester 2005 Disclaimer: course material is mainly taken from: P.E. Bourne & H Weissig, Structural Bioinformatics; C.A. Orengo, D.T. Jones & J.M. Thornton,
Homology Modeling Roberto Lins EPFL - summer semester 2005 Disclaimer: course material is mainly taken from: P.E. Bourne & H Weissig, Structural Bioinformatics; C.A. Orengo, D.T. Jones & J.M. Thornton,
Syllabus of BIOINF 528 (2017 Fall, Bioinformatics Program)
") Syllabus of BIOINF 528 (2017 Fall, Bioinformatics Program) Course Name: Structural Bioinformatics Course Description: Instructor: This course introduces fundamental concepts and methods for structural
Syllabus of BIOINF 528 (2017 Fall, Bioinformatics Program) Course Name: Structural Bioinformatics Course Description: Instructor: This course introduces fundamental concepts and methods for structural
Molecular Modeling lecture 2
 Molecular Modeling 2018 -- lecture 2 Topics 1. Secondary structure 3. Sequence similarity and homology 2. Secondary structure prediction 4. Where do protein structures come from? X-ray crystallography
Molecular Modeling 2018 -- lecture 2 Topics 1. Secondary structure 3. Sequence similarity and homology 2. Secondary structure prediction 4. Where do protein structures come from? X-ray crystallography
2MHR. Protein structure classification is important because it organizes the protein structure universe that is independent of sequence similarity.
 Protein structure classification is important because it organizes the protein structure universe that is independent of sequence similarity. A global picture of the protein universe will help us to understand
Protein structure classification is important because it organizes the protein structure universe that is independent of sequence similarity. A global picture of the protein universe will help us to understand
Building 3D models of proteins
 Building 3D models of proteins Why make a structural model for your protein? The structure can provide clues to the function through structural similarity with other proteins With a structure it is easier
Building 3D models of proteins Why make a structural model for your protein? The structure can provide clues to the function through structural similarity with other proteins With a structure it is easier
Biomolecules: lecture 10
 Biomolecules: lecture 10 - understanding in detail how protein 3D structures form - realize that protein molecules are not static wire models but instead dynamic, where in principle every atom moves (yet
Biomolecules: lecture 10 - understanding in detail how protein 3D structures form - realize that protein molecules are not static wire models but instead dynamic, where in principle every atom moves (yet
Protein structure prediction. CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror
 Protein structure prediction CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror 1 Outline Why predict protein structure? Can we use (pure) physics-based methods? Knowledge-based methods Two major
Protein structure prediction CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror 1 Outline Why predict protein structure? Can we use (pure) physics-based methods? Knowledge-based methods Two major
RNA and Protein Structure Prediction
 RNA and Protein Structure Prediction Bioinformatics: Issues and Algorithms CSE 308-408 Spring 2007 Lecture 18-1- Outline Multi-Dimensional Nature of Life RNA Secondary Structure Prediction Protein Structure
RNA and Protein Structure Prediction Bioinformatics: Issues and Algorithms CSE 308-408 Spring 2007 Lecture 18-1- Outline Multi-Dimensional Nature of Life RNA Secondary Structure Prediction Protein Structure
Can protein model accuracy be. identified? NO! CBS, BioCentrum, Morten Nielsen, DTU
 Can protein model accuracy be identified? Morten Nielsen, CBS, BioCentrum, DTU NO! Identification of Protein-model accuracy Why is it important? What is accuracy RMSD, fraction correct, Protein model correctness/quality
Can protein model accuracy be identified? Morten Nielsen, CBS, BioCentrum, DTU NO! Identification of Protein-model accuracy Why is it important? What is accuracy RMSD, fraction correct, Protein model correctness/quality
THE TANGO ALGORITHM: SECONDARY STRUCTURE PROPENSITIES, STATISTICAL MECHANICS APPROXIMATION
 THE TANGO ALGORITHM: SECONDARY STRUCTURE PROPENSITIES, STATISTICAL MECHANICS APPROXIMATION AND CALIBRATION Calculation of turn and beta intrinsic propensities. A statistical analysis of a protein structure
THE TANGO ALGORITHM: SECONDARY STRUCTURE PROPENSITIES, STATISTICAL MECHANICS APPROXIMATION AND CALIBRATION Calculation of turn and beta intrinsic propensities. A statistical analysis of a protein structure
Protein Structure. W. M. Grogan, Ph.D. OBJECTIVES
 Protein Structure W. M. Grogan, Ph.D. OBJECTIVES 1. Describe the structure and characteristic properties of typical proteins. 2. List and describe the four levels of structure found in proteins. 3. Relate
Protein Structure W. M. Grogan, Ph.D. OBJECTIVES 1. Describe the structure and characteristic properties of typical proteins. 2. List and describe the four levels of structure found in proteins. 3. Relate
Motif Prediction in Amino Acid Interaction Networks
 Motif Prediction in Amino Acid Interaction Networks Omar GACI and Stefan BALEV Abstract In this paper we represent a protein as a graph where the vertices are amino acids and the edges are interactions
Motif Prediction in Amino Acid Interaction Networks Omar GACI and Stefan BALEV Abstract In this paper we represent a protein as a graph where the vertices are amino acids and the edges are interactions
Protein structure analysis. Risto Laakso 10th January 2005
 Protein structure analysis Risto Laakso risto.laakso@hut.fi 10th January 2005 1 1 Summary Various methods of protein structure analysis were examined. Two proteins, 1HLB (Sea cucumber hemoglobin) and 1HLM
Protein structure analysis Risto Laakso risto.laakso@hut.fi 10th January 2005 1 1 Summary Various methods of protein structure analysis were examined. Two proteins, 1HLB (Sea cucumber hemoglobin) and 1HLM
7.91 Amy Keating. Solving structures using X-ray crystallography & NMR spectroscopy
 7.91 Amy Keating Solving structures using X-ray crystallography & NMR spectroscopy How are X-ray crystal structures determined? 1. Grow crystals - structure determination by X-ray crystallography relies
7.91 Amy Keating Solving structures using X-ray crystallography & NMR spectroscopy How are X-ray crystal structures determined? 1. Grow crystals - structure determination by X-ray crystallography relies
COMP 598 Advanced Computational Biology Methods & Research. Introduction. Jérôme Waldispühl School of Computer Science McGill University
 COMP 598 Advanced Computational Biology Methods & Research Introduction Jérôme Waldispühl School of Computer Science McGill University General informations (1) Office hours: by appointment Office: TR3018
COMP 598 Advanced Computational Biology Methods & Research Introduction Jérôme Waldispühl School of Computer Science McGill University General informations (1) Office hours: by appointment Office: TR3018
Protein Structure & Motifs
 & Motifs Biochemistry 201 Molecular Biology January 12, 2000 Doug Brutlag Introduction Proteins are more flexible than nucleic acids in structure because of both the larger number of types of residues
& Motifs Biochemistry 201 Molecular Biology January 12, 2000 Doug Brutlag Introduction Proteins are more flexible than nucleic acids in structure because of both the larger number of types of residues
Protein Structures: Experiments and Modeling. Patrice Koehl
 Protein Structures: Experiments and Modeling Patrice Koehl Structural Bioinformatics: Proteins Proteins: Sources of Structure Information Proteins: Homology Modeling Proteins: Ab initio prediction Proteins:
Protein Structures: Experiments and Modeling Patrice Koehl Structural Bioinformatics: Proteins Proteins: Sources of Structure Information Proteins: Homology Modeling Proteins: Ab initio prediction Proteins:
Heteropolymer. Mostly in regular secondary structure
 Heteropolymer - + + - Mostly in regular secondary structure 1 2 3 4 C >N trace how you go around the helix C >N C2 >N6 C1 >N5 What s the pattern? Ci>Ni+? 5 6 move around not quite 120 "#$%&'!()*(+2!3/'!4#5'!1/,#64!#6!,6!
Heteropolymer - + + - Mostly in regular secondary structure 1 2 3 4 C >N trace how you go around the helix C >N C2 >N6 C1 >N5 What s the pattern? Ci>Ni+? 5 6 move around not quite 120 "#$%&'!()*(+2!3/'!4#5'!1/,#64!#6!,6!
Orientational degeneracy in the presence of one alignment tensor.
 Orientational degeneracy in the presence of one alignment tensor. Rotation about the x, y and z axes can be performed in the aligned mode of the program to examine the four degenerate orientations of two
Orientational degeneracy in the presence of one alignment tensor. Rotation about the x, y and z axes can be performed in the aligned mode of the program to examine the four degenerate orientations of two
114 Grundlagen der Bioinformatik, SS 09, D. Huson, July 6, 2009
 114 Grundlagen der Bioinformatik, SS 09, D. Huson, July 6, 2009 9 Protein tertiary structure Sources for this chapter, which are all recommended reading: D.W. Mount. Bioinformatics: Sequences and Genome
114 Grundlagen der Bioinformatik, SS 09, D. Huson, July 6, 2009 9 Protein tertiary structure Sources for this chapter, which are all recommended reading: D.W. Mount. Bioinformatics: Sequences and Genome
09/06/25. Computergestützte Strukturbiologie (Strukturelle Bioinformatik) Non-uniform distribution of folds. Scheme of protein structure predicition
 Non-uniform distribution of folds. Scheme of protein structure predicition") Sequence identity Structural similarity Computergestützte Strukturbiologie (Strukturelle Bioinformatik) Fold recognition Sommersemester 2009 Peter Güntert Structural similarity X Sequence identity Non-uniform
Sequence identity Structural similarity Computergestützte Strukturbiologie (Strukturelle Bioinformatik) Fold recognition Sommersemester 2009 Peter Güntert Structural similarity X Sequence identity Non-uniform
Number sequence representation of protein structures based on the second derivative of a folded tetrahedron sequence
 Number sequence representation of protein structures based on the second derivative of a folded tetrahedron sequence Naoto Morikawa (nmorika@genocript.com) October 7, 2006. Abstract A protein is a sequence
Number sequence representation of protein structures based on the second derivative of a folded tetrahedron sequence Naoto Morikawa (nmorika@genocript.com) October 7, 2006. Abstract A protein is a sequence
From Amino Acids to Proteins - in 4 Easy Steps
 From Amino Acids to Proteins - in 4 Easy Steps Although protein structure appears to be overwhelmingly complex, you can provide your students with a basic understanding of how proteins fold by focusing
From Amino Acids to Proteins - in 4 Easy Steps Although protein structure appears to be overwhelmingly complex, you can provide your students with a basic understanding of how proteins fold by focusing
Molecular Modeling Lecture 7. Homology modeling insertions/deletions manual realignment
 Molecular Modeling 2018-- Lecture 7 Homology modeling insertions/deletions manual realignment Homology modeling also called comparative modeling Sequences that have similar sequence have similar structure.
Molecular Modeling 2018-- Lecture 7 Homology modeling insertions/deletions manual realignment Homology modeling also called comparative modeling Sequences that have similar sequence have similar structure.
Examples of Protein Modeling. Protein Modeling. Primary Structure. Protein Structure Description. Protein Sequence Sources. Importing Sequences to MOE
 Examples of Protein Modeling Protein Modeling Visualization Examination of an experimental structure to gain insight about a research question Dynamics To examine the dynamics of protein structures To
Examples of Protein Modeling Protein Modeling Visualization Examination of an experimental structure to gain insight about a research question Dynamics To examine the dynamics of protein structures To
Figure 1. Molecules geometries of 5021 and Each neutral group in CHARMM topology was grouped in dash circle.
 Project I Chemistry 8021, Spring 2005/2/23 This document was turned in by a student as a homework paper. 1. Methods First, the cartesian coordinates of 5021 and 8021 molecules (Fig. 1) are generated, in
Project I Chemistry 8021, Spring 2005/2/23 This document was turned in by a student as a homework paper. 1. Methods First, the cartesian coordinates of 5021 and 8021 molecules (Fig. 1) are generated, in
Structure to Function. Molecular Bioinformatics, X3, 2006
 Structure to Function Molecular Bioinformatics, X3, 2006 Structural GeNOMICS Structural Genomics project aims at determination of 3D structures of all proteins: - organize known proteins into families
Structure to Function Molecular Bioinformatics, X3, 2006 Structural GeNOMICS Structural Genomics project aims at determination of 3D structures of all proteins: - organize known proteins into families
Bioinformatics III Structural Bioinformatics and Genome Analysis Part Protein Secondary Structure Prediction. Sepp Hochreiter
 Bioinformatics III Structural Bioinformatics and Genome Analysis Part Protein Secondary Structure Prediction Institute of Bioinformatics Johannes Kepler University, Linz, Austria Chapter 4 Protein Secondary
Bioinformatics III Structural Bioinformatics and Genome Analysis Part Protein Secondary Structure Prediction Institute of Bioinformatics Johannes Kepler University, Linz, Austria Chapter 4 Protein Secondary
Template Free Protein Structure Modeling Jianlin Cheng, PhD
 Template Free Protein Structure Modeling Jianlin Cheng, PhD Associate Professor Computer Science Department Informatics Institute University of Missouri, Columbia 2013 Protein Energy Landscape & Free Sampling
Template Free Protein Structure Modeling Jianlin Cheng, PhD Associate Professor Computer Science Department Informatics Institute University of Missouri, Columbia 2013 Protein Energy Landscape & Free Sampling
Molecular Modeling. Prediction of Protein 3D Structure from Sequence. Vimalkumar Velayudhan. May 21, 2007
 Molecular Modeling Prediction of Protein 3D Structure from Sequence Vimalkumar Velayudhan Jain Institute of Vocational and Advanced Studies May 21, 2007 Vimalkumar Velayudhan Molecular Modeling 1/23 Outline
Molecular Modeling Prediction of Protein 3D Structure from Sequence Vimalkumar Velayudhan Jain Institute of Vocational and Advanced Studies May 21, 2007 Vimalkumar Velayudhan Molecular Modeling 1/23 Outline
Protein structure. Protein structure. Amino acid residue. Cell communication channel. Bioinformatics Methods
 Cell communication channel Bioinformatics Methods Iosif Vaisman Email: ivaisman@gmu.edu SEQUENCE STRUCTURE DNA Sequence Protein Sequence Protein Structure Protein structure ATGAAATTTGGAAACTTCCTTCTCACTTATCAGCCACCT...
Cell communication channel Bioinformatics Methods Iosif Vaisman Email: ivaisman@gmu.edu SEQUENCE STRUCTURE DNA Sequence Protein Sequence Protein Structure Protein structure ATGAAATTTGGAAACTTCCTTCTCACTTATCAGCCACCT...
Protein structure alignments
 Protein structure alignments Proteins that fold in the same way, i.e. have the same fold are often homologs. Structure evolves slower than sequence Sequence is less conserved than structure If BLAST gives
Protein structure alignments Proteins that fold in the same way, i.e. have the same fold are often homologs. Structure evolves slower than sequence Sequence is less conserved than structure If BLAST gives
BIRKBECK COLLEGE (University of London)
") BIRKBECK COLLEGE (University of London) SCHOOL OF BIOLOGICAL SCIENCES M.Sc. EXAMINATION FOR INTERNAL STUDENTS ON: Postgraduate Certificate in Principles of Protein Structure MSc Structural Molecular Biology
BIRKBECK COLLEGE (University of London) SCHOOL OF BIOLOGICAL SCIENCES M.Sc. EXAMINATION FOR INTERNAL STUDENTS ON: Postgraduate Certificate in Principles of Protein Structure MSc Structural Molecular Biology
D Dobbs ISU - BCB 444/544X 1
 11/7/05 Protein Structure: Classification, Databases, Visualization Announcements BCB 544 Projects - Important Dates: Nov 2 Wed noon - Project proposals due to David/Drena Nov 4 Fri PM - Approvals/responses
11/7/05 Protein Structure: Classification, Databases, Visualization Announcements BCB 544 Projects - Important Dates: Nov 2 Wed noon - Project proposals due to David/Drena Nov 4 Fri PM - Approvals/responses
Molecular Modeling lecture 17, Tue, Mar. 19. Rotation Least-squares Superposition Structure-based alignment algorithms
 Molecular Modeling 2019 -- lecture 17, Tue, Mar. 19 Rotation Least-squares Superposition Structure-based alignment algorithms Matrices and vectors Matrix algebra allows you to express multiple equations
Molecular Modeling 2019 -- lecture 17, Tue, Mar. 19 Rotation Least-squares Superposition Structure-based alignment algorithms Matrices and vectors Matrix algebra allows you to express multiple equations
Computational Molecular Modeling
 Computational Molecular Modeling Lecture 1: Structure Models, Properties Chandrajit Bajaj Today s Outline Intro to atoms, bonds, structure, biomolecules, Geometry of Proteins, Nucleic Acids, Ribosomes,
Computational Molecular Modeling Lecture 1: Structure Models, Properties Chandrajit Bajaj Today s Outline Intro to atoms, bonds, structure, biomolecules, Geometry of Proteins, Nucleic Acids, Ribosomes,
Template Free Protein Structure Modeling Jianlin Cheng, PhD
 Template Free Protein Structure Modeling Jianlin Cheng, PhD Professor Department of EECS Informatics Institute University of Missouri, Columbia 2018 Protein Energy Landscape & Free Sampling http://pubs.acs.org/subscribe/archive/mdd/v03/i09/html/willis.html
Template Free Protein Structure Modeling Jianlin Cheng, PhD Professor Department of EECS Informatics Institute University of Missouri, Columbia 2018 Protein Energy Landscape & Free Sampling http://pubs.acs.org/subscribe/archive/mdd/v03/i09/html/willis.html
NMR, X-ray Diffraction, Protein Structure, and RasMol
 NMR, X-ray Diffraction, Protein Structure, and RasMol Introduction So far we have been mostly concerned with the proteins themselves. The techniques (NMR or X-ray diffraction) used to determine a structure
NMR, X-ray Diffraction, Protein Structure, and RasMol Introduction So far we have been mostly concerned with the proteins themselves. The techniques (NMR or X-ray diffraction) used to determine a structure
Homology Modeling (Comparative Structure Modeling) GBCB 5874: Problem Solving in GBCB
 GBCB 5874: Problem Solving in GBCB") Homology Modeling (Comparative Structure Modeling) Aims of Structural Genomics High-throughput 3D structure determination and analysis To determine or predict the 3D structures of all the proteins encoded
Homology Modeling (Comparative Structure Modeling) Aims of Structural Genomics High-throughput 3D structure determination and analysis To determine or predict the 3D structures of all the proteins encoded
Copyright Mark Brandt, Ph.D A third method, cryogenic electron microscopy has seen increasing use over the past few years.
 Structure Determination and Sequence Analysis The vast majority of the experimentally determined three-dimensional protein structures have been solved by one of two methods: X-ray diffraction and Nuclear
Structure Determination and Sequence Analysis The vast majority of the experimentally determined three-dimensional protein structures have been solved by one of two methods: X-ray diffraction and Nuclear
Protein Structure Prediction, Engineering & Design CHEM 430
 Protein Structure Prediction, Engineering & Design CHEM 430 Eero Saarinen The free energy surface of a protein Protein Structure Prediction & Design Full Protein Structure from Sequence - High Alignment
Protein Structure Prediction, Engineering & Design CHEM 430 Eero Saarinen The free energy surface of a protein Protein Structure Prediction & Design Full Protein Structure from Sequence - High Alignment
SCOP. all-β class. all-α class, 3 different folds. T4 endonuclease V. 4-helical cytokines. Globin-like
 SCOP all-β class 4-helical cytokines T4 endonuclease V all-α class, 3 different folds Globin-like TIM-barrel fold α/β class Profilin-like fold α+β class http://scop.mrc-lmb.cam.ac.uk/scop CATH Class, Architecture,
SCOP all-β class 4-helical cytokines T4 endonuclease V all-α class, 3 different folds Globin-like TIM-barrel fold α/β class Profilin-like fold α+β class http://scop.mrc-lmb.cam.ac.uk/scop CATH Class, Architecture,
HMM applications. Applications of HMMs. Gene finding with HMMs. Using the gene finder
 HMM applications Applications of HMMs Gene finding Pairwise alignment (pair HMMs) Characterizing protein families (profile HMMs) Predicting membrane proteins, and membrane protein topology Gene finding
HMM applications Applications of HMMs Gene finding Pairwise alignment (pair HMMs) Characterizing protein families (profile HMMs) Predicting membrane proteins, and membrane protein topology Gene finding
Molecular Modeling lecture 4. Rotation Least-squares Superposition Structure-based alignment algorithms
 Molecular Modeling 2018 -- lecture 4 Rotation Least-squares Superposition Structure-based alignment algorithms Rotation is addition in polar coordinates 2 What happens when you move the mouse to rotate
Molecular Modeling 2018 -- lecture 4 Rotation Least-squares Superposition Structure-based alignment algorithms Rotation is addition in polar coordinates 2 What happens when you move the mouse to rotate
Amino Acid Structures from Klug & Cummings. 10/7/2003 CAP/CGS 5991: Lecture 7 1
 Amino Acid Structures from Klug & Cummings 10/7/2003 CAP/CGS 5991: Lecture 7 1 Amino Acid Structures from Klug & Cummings 10/7/2003 CAP/CGS 5991: Lecture 7 2 Amino Acid Structures from Klug & Cummings
Amino Acid Structures from Klug & Cummings 10/7/2003 CAP/CGS 5991: Lecture 7 1 Amino Acid Structures from Klug & Cummings 10/7/2003 CAP/CGS 5991: Lecture 7 2 Amino Acid Structures from Klug & Cummings
F. Piazza Center for Molecular Biophysics and University of Orléans, France. Selected topic in Physical Biology. Lecture 1
 Zhou Pei-Yuan Centre for Applied Mathematics, Tsinghua University November 2013 F. Piazza Center for Molecular Biophysics and University of Orléans, France Selected topic in Physical Biology Lecture 1
Zhou Pei-Yuan Centre for Applied Mathematics, Tsinghua University November 2013 F. Piazza Center for Molecular Biophysics and University of Orléans, France Selected topic in Physical Biology Lecture 1
Bioinformatics: Secondary Structure Prediction
 Bioinformatics: Secondary Structure Prediction Prof. David Jones d.jones@cs.ucl.ac.uk LMLSTQNPALLKRNIIYWNNVALLWEAGSD The greatest unsolved problem in molecular biology:the Protein Folding Problem? Entries
Bioinformatics: Secondary Structure Prediction Prof. David Jones d.jones@cs.ucl.ac.uk LMLSTQNPALLKRNIIYWNNVALLWEAGSD The greatest unsolved problem in molecular biology:the Protein Folding Problem? Entries
CS612 - Algorithms in Bioinformatics
 Fall 2017 Protein Structure Detection Methods October 30, 2017 Comparative Modeling Comparative modeling is modeling of the unknown based on comparison to what is known In the context of modeling or computing
Fall 2017 Protein Structure Detection Methods October 30, 2017 Comparative Modeling Comparative modeling is modeling of the unknown based on comparison to what is known In the context of modeling or computing
Advanced Certificate in Principles in Protein Structure. You will be given a start time with your exam instructions
 BIRKBECK COLLEGE (University of London) Advanced Certificate in Principles in Protein Structure MSc Structural Molecular Biology Date: Thursday, 1st September 2011 Time: 3 hours You will be given a start
BIRKBECK COLLEGE (University of London) Advanced Certificate in Principles in Protein Structure MSc Structural Molecular Biology Date: Thursday, 1st September 2011 Time: 3 hours You will be given a start
Design of a Novel Globular Protein Fold with Atomic-Level Accuracy
 Design of a Novel Globular Protein Fold with Atomic-Level Accuracy Brian Kuhlman, Gautam Dantas, Gregory C. Ireton, Gabriele Varani, Barry L. Stoddard, David Baker Presented by Kate Stafford 4 May 05 Protein
Design of a Novel Globular Protein Fold with Atomic-Level Accuracy Brian Kuhlman, Gautam Dantas, Gregory C. Ireton, Gabriele Varani, Barry L. Stoddard, David Baker Presented by Kate Stafford 4 May 05 Protein
Protein Structure Determination. Why Bother With Structure? Protein Sequences Far Outnumber Structures. Growth of Structural Data
 Protein Structure Determination Why Bother With Structure? The amino acid sequence of a protein contains interesting information. A protein sequence can be compared to other protein sequences to establish
Protein Structure Determination Why Bother With Structure? The amino acid sequence of a protein contains interesting information. A protein sequence can be compared to other protein sequences to establish
Modeling for 3D structure prediction
 Modeling for 3D structure prediction What is a predicted structure? A structure that is constructed using as the sole source of information data obtained from computer based data-mining. However, mixing
Modeling for 3D structure prediction What is a predicted structure? A structure that is constructed using as the sole source of information data obtained from computer based data-mining. However, mixing
1. What is an ångstrom unit, and why is it used to describe molecular structures?
 1. What is an ångstrom unit, and why is it used to describe molecular structures? The ångstrom unit is a unit of distance suitable for measuring atomic scale objects. 1 ångstrom (Å) = 1 10-10 m. The diameter
1. What is an ångstrom unit, and why is it used to describe molecular structures? The ångstrom unit is a unit of distance suitable for measuring atomic scale objects. 1 ångstrom (Å) = 1 10-10 m. The diameter
A General Model for Amino Acid Interaction Networks
 Author manuscript, published in "N/P" A General Model for Amino Acid Interaction Networks Omar GACI and Stefan BALEV hal-43269, version - Nov 29 Abstract In this paper we introduce the notion of protein
Author manuscript, published in "N/P" A General Model for Amino Acid Interaction Networks Omar GACI and Stefan BALEV hal-43269, version - Nov 29 Abstract In this paper we introduce the notion of protein
Protein Structure Prediction Using Multiple Artificial Neural Network Classifier *
 Protein Structure Prediction Using Multiple Artificial Neural Network Classifier * Hemashree Bordoloi and Kandarpa Kumar Sarma Abstract. Protein secondary structure prediction is the method of extracting
Protein Structure Prediction Using Multiple Artificial Neural Network Classifier * Hemashree Bordoloi and Kandarpa Kumar Sarma Abstract. Protein secondary structure prediction is the method of extracting
Introduction to Computational Structural Biology
 Introduction to Computational Structural Biology Part I 1. Introduction The disciplinary character of Computational Structural Biology The mathematical background required and the topics covered Bibliography
Introduction to Computational Structural Biology Part I 1. Introduction The disciplinary character of Computational Structural Biology The mathematical background required and the topics covered Bibliography
Biophysics 101: Genomics & Computational Biology. Section 8: Protein Structure S T R U C T U R E P R O C E S S. Outline.
 Biophysics 101: Genomics & Computational Biology Section 8: Protein Structure Faisal Reza Nov. 11 th, 2003 B101.pdb from PS5 shown at left with: animated ball and stick model, colored CPK H-bonds on, colored
Biophysics 101: Genomics & Computational Biology Section 8: Protein Structure Faisal Reza Nov. 11 th, 2003 B101.pdb from PS5 shown at left with: animated ball and stick model, colored CPK H-bonds on, colored
Alpha-helical Topology and Tertiary Structure Prediction of Globular Proteins Scott R. McAllister Christodoulos A. Floudas Princeton University
 Alpha-helical Topology and Tertiary Structure Prediction of Globular Proteins Scott R. McAllister Christodoulos A. Floudas Princeton University Department of Chemical Engineering Program of Applied and
Alpha-helical Topology and Tertiary Structure Prediction of Globular Proteins Scott R. McAllister Christodoulos A. Floudas Princeton University Department of Chemical Engineering Program of Applied and
3D Structure. Prediction & Assessment Pt. 2. David Wishart 3-41 Athabasca Hall
 3D Structure Prediction & Assessment Pt. 2 David Wishart 3-41 Athabasca Hall david.wishart@ualberta.ca Objectives Become familiar with methods and algorithms for secondary Structure Prediction Become familiar
3D Structure Prediction & Assessment Pt. 2 David Wishart 3-41 Athabasca Hall david.wishart@ualberta.ca Objectives Become familiar with methods and algorithms for secondary Structure Prediction Become familiar
Study of Mining Protein Structural Properties and its Application
 Study of Mining Protein Structural Properties and its Application A Dissertation Proposal Presented to the Department of Computer Science and Information Engineering College of Electrical Engineering and
Study of Mining Protein Structural Properties and its Application A Dissertation Proposal Presented to the Department of Computer Science and Information Engineering College of Electrical Engineering and
Identification of Representative Protein Sequence and Secondary Structure Prediction Using SVM Approach
 Identification of Representative Protein Sequence and Secondary Structure Prediction Using SVM Approach Prof. Dr. M. A. Mottalib, Md. Rahat Hossain Department of Computer Science and Information Technology
Identification of Representative Protein Sequence and Secondary Structure Prediction Using SVM Approach Prof. Dr. M. A. Mottalib, Md. Rahat Hossain Department of Computer Science and Information Technology
Bioinformatics. Proteins II. - Pattern, Profile, & Structure Database Searching. Robert Latek, Ph.D. Bioinformatics, Biocomputing
 Bioinformatics Proteins II. - Pattern, Profile, & Structure Database Searching Robert Latek, Ph.D. Bioinformatics, Biocomputing WIBR Bioinformatics Course, Whitehead Institute, 2002 1 Proteins I.-III.
Bioinformatics Proteins II. - Pattern, Profile, & Structure Database Searching Robert Latek, Ph.D. Bioinformatics, Biocomputing WIBR Bioinformatics Course, Whitehead Institute, 2002 1 Proteins I.-III.
BCH 4053 Spring 2003 Chapter 6 Lecture Notes
 BCH 4053 Spring 2003 Chapter 6 Lecture Notes 1 CHAPTER 6 Proteins: Secondary, Tertiary, and Quaternary Structure 2 Levels of Protein Structure Primary (sequence) Secondary (ordered structure along peptide
BCH 4053 Spring 2003 Chapter 6 Lecture Notes 1 CHAPTER 6 Proteins: Secondary, Tertiary, and Quaternary Structure 2 Levels of Protein Structure Primary (sequence) Secondary (ordered structure along peptide
Comparing Protein Structures. Why?
 7.91 Amy Keating Comparing Protein Structures Why? detect evolutionary relationships identify recurring motifs detect structure/function relationships predict function assess predicted structures classify
7.91 Amy Keating Comparing Protein Structures Why? detect evolutionary relationships identify recurring motifs detect structure/function relationships predict function assess predicted structures classify