1. Some examples of coping with Molecular informatics data legacy data (accuracy)
|
|
|
- Jasper Mason
- 5 years ago
- Views:
Transcription
 2.")
1 Molecular Informatics Tools for Data Analysis and Discovery 1. Some examples of coping with Molecular informatics data legacy data (accuracy) 2. Database searching using a similarity approach fingerprints in 2D and 3D ChemSource 2005 Robert Glen
2 The scale of the data in Molecular Informatics Biological data 3,488,108,873 nucleotide bases over 800 organisms protein x-ray crystal structures 12 million citations in medline 40 main stream databases from EBI Ensemble : 24 million gene predictions Chemical data 60,475,000 chemical substances 3,700,000 chemical reactions 613,000 available reagents Bielstein has 600,000 reaction abstracts >270,000 organic x-ray structures Combichem libraries of Billions of compounds The BIG connection => innovation Patents European patents 150,000,000 pages 150,000 applications/year 400,000 chemical patent hyperstructures ver 100 countries in patent cooperation treaty (PCT) This implies a clear need to make maximum use of this sea of data... Robert C. Glen and S. Aldridge. Developing tools and standards in molecular informatics. Chem. Comm pp
3 Enabling technologies The grid Information and processing as accessible as electricity plug and play The semantic grid Language and knowledge to access the grid Hardware grid Semantic grid
4 Using information from Legacy data 100 years of chemistry in books 20 years of data destruction PDF Computational studies seldom available for analysis and aggregation Need data to be abstracted to a computer readable form Need data to be standardised and checked Need data to be available to robots for processing, checking, analysis and to talk to other robots
5 RSC/UCC markup project Uses CML, natural language processing, knowledge of chemistry An authoring tool A data checker Data abstraction from chemistry papers -> computer readable database Experimental data checker: better information for organic chemists S. E. Adams, J. M. Goodman,, R. J. Kidd, A. D. McNaught, P. Murray-Rust Rust,, F. R. Norton, J. A. Townsend and C. A. Waudby rg. Biomol.. Chem. 2004, 2,, Highlighted in Chemical Science 2004, 1,, C33. Chemical documents: machine understanding and automated information ion extraction J. A. Townsend, S. E. Adams, C. A. Waudby,, V. K. de Souza, J. M. Goodman and P. Murray-Rust rg. Biomol.. Chem. 2004, 2,,
6 Database searching Using a similarity approach Neighborhood Region Active Compound other compounds If the molecular descriptors are valid... the activity of a Compound is shared by most other compounds within its Neighborhood Region i.e. neighbors of a bioactive compound have a higher probability of behaving in a similar bioactive way Molecular similarity: a key technique in molecular informatics. rganic and Biomolecular Chemistry perspective article. R. C. Glen and A. Bender, rg. Biomol. Chem. 2004, 2,

7 The descriptors: Similar to the environment around an ionizable center (atom environments) used previously (Xing, Clark and Glen) E.g. 6-aminoquinoline6 Measured 5.7 predicted 5.4 Start with interesting atom find connections find connections to connections create a tree down to 5 levels bin the atom types for each level create a fingerprint for this atom N2 Level 0 Car--Car Level 1 Car,Car Car,H Level String contains a bin for each required atom type at each level, the number of atom types is accumulated to form the string - 56 bins
8 Method Tabulate many reliable pka measurements Describe the environment around ionizable centers Use partial least squares to create a predictive model Test model with cross validation


9 Using the data 56 bins used to cover all the possibilities Used pls (partial least squares) to create a model pk = pk 0 a c + Σ a i x i + Σ g j y j + Σ q k z... k Used cross validation to validate the model Novel methods for the prediction of pka, logp and logd, Xing L. and Glen R.C.. J. Chem. Inf. Comput. Sci.; 2002; ; 42(4); Recently refined model to improve accuracy
10 pka of acids pka of bases (625) (412) Predicted pk a Predicted pk a R2=0.98 Std.Err.=0.405 N=625 Q 2 =0.92 Measured pk a Measured pk a R 2 =0.99 Std.Err.=0.302 N=412 Q 2 =0.95
11 pka test set Conclusions Series1 4 2 Surprisingly good results - fast measured Predictive for most pk s Useful in biological setting in estimating Pharmacokinetics, active species, metabolism etc. Predicts for all types - sometimes get odd results though, if outside parameter set or the atom types are miss-set set Applying method to other problems e.g. similarity predicted Novel methods for the prediction of pka, logp and logd, Xing L. and Glen R.C.. J. Chem. Inf. Comput. Sci.; 2002; 42(4); Predicting pka by Molecular Tree Structured Fingerprints and PLS. Xing L.,Glen R. C. and Clark, R. D. J. Chem. Inf. Comput. Sci. 2003, 43(3), 870
12 Similarity searching in databases. Andreas Bender 1. Atom centred fingerprints We created a descriptor suitable as a similarity index by looking at all atoms in turn in a molecule and for each atom, generating a depth-3 3 atom environment. No hashing was involved. These are then binned into an integer string - a fingerprint for each atom centre N2 Car--Car Level 0 Level 1 Car,Car Car,H Level 2 etc.
13 2. Information-Gain Based Feature Selection We wish to select the important features. To do this we calculate the entropy of the data as a whole and for each class. This is used to select those features with the highest discrimination, e.g. active or inactive or toxic and non-toxic molecules S = p log 2 p I = S v S S v S v
14 3. Classification The next step is to identify which molecules belong to which class. To do this we use a Naïve Bayesian Classifer using the features (atom environments) we have identified as being important.
15 3. Naïve Bayesian Classifier Include all selected features f i in calculation of P( CL P( CL 1 2 F) F) = P( CL P( CL Ratio > 1: Class membership 1 Ratio < 1: Class membership 2 F: feature vector f i: feature elements 1 2 ) ) i P( P( f f i i CL CL 1 2 ) )
16 MDDR lead discovery MDDR test run: 957 ligands from MDDR 49 5HT3 Receptor antagonists, 40 Angiotensin Converting Enzyme inhibitors (ACE), 111 HMG-Co Co-Reductase inhibitors (HMG), 134 PAF antagonists and 49 Thromboxane A2 antagonists (TXA2) A) Hit rate among ten nearest neighbours for each molecule B) 20-fold Cross Validation, 5 Molecules for query generation
17 MDDR database searches Performance of the Atom Environment Approach, Selecting 20 Features Group of Active Compounds 5HT3 ACE HMG PAF TXA2 verall Expected Hit Rate for Random Selection Hit Rate for this Method Enrichment Factor Fraction Actives Found Percent Database e.g. ACE: We found about 80% of the active molecules among the first 10% of the library
18 Combining data and search performance 100% 90% Mean sample hit rates (in 80% 70% 60% 50% 40% 30% 20% 10% 0% Atom Environm. - 5 Mols Feature Trees Atom Environments ISIS MLSKEYS DAYLIGHT Fingerprints SYBYL Hologram QSAR FLEXSIM-X FLEXSIM-S DCKSIM Briem and Lessel, Perspectives in Drug Discovery and Design 2000, 20, Molecular Similarity Searching using Atom Environments, Information-Based Feature Selection and a Naïve Bayesian Classifier Andreas Bender, Hamse Y. Mussa and Robert C. Glen, University of Cambridge Stephan Reiling, Aventis Pharmaceuticals J. Chem. Inf. Comp. Sci., 2004; 44(1);
19 * Hert J, Willett P, Wilton DJ: Comparison of fingerprint-based methods for virtual screening using multiple bioactive reference structures. J Chem Inf Comput Sci 2004, 44: Comparison using Larger Data Set * 102,000 structures from the MDDR 11 Sets of Active Compounds, ranging in size from 349 to 1246 entries large and diverse data set Performance Measure: Fraction of Active Structures retrieved in Top 5% of sorted library Atom Environments were compared to Unity Fingerprints in Combination with Data Fusion (MAX) and Binary Kernel Discrimination In case of Binary Kernel Discrimination and the Bayes Classifier 10 actives and 100 inactives used for training
20 Comparison of Methods Unity Single AE Single Data-Fusion MAX BKD,10a100i,k=100 AE 10a100i HT3 5HT1A 5HTReup D2 Anta Renin I Ang II Anta Throm I Sub P Anta HIVProt I Cox I Prot Kin C I Average Similarity Searching of Chemical Databases Using Atom Environment Descriptors (MLPRINT 2D): Evaluation of Performance. Bender, A.; Mussa, H. Y.; Glen, R. C.; Reiling, S.J. Chem. Inf. Comput. Sci.,2004; 44(5);
21 Transformation to 3D Two parts: Interaction fingerprint and shape description; here results using only interaction fingerprints are shown, shape description under development Information was merged from multiple molecules by using information-gain feature selection and the Naïve Bayesian Classifier
 Points in Layer 1 Points in Layer 2 Etc.")
22 3D: Environment around a surface point: solvent accessible surface Central Point ( Layer 0 ) Points in Layer 1 Points in Layer 2 Etc.
23 Algorithm Interaction Energies at Surface Points, one Probe at a time EU Binning Scheme Surface Point Environment
24 Algorithm Flow Step Generation of 3D coordinates Calculation of Surface Points Calculation of Interaction Energies Transformation of interaction energies into descriptors Program used Concord msms GRID Perl script Parameters Sphere radius, probe size, triangulation density Probe (and various others) Binning, number of bins, threshold levels
25 Surface Environments comparison with 2D and other methods 100% 90% 80% 70% 60% 50% 40% 30% 20% 10% 0% Average Hit Rate AE - Bayes 5 Mol AE - Tanimoto 1 Mol Feature Trees Surface FP - Bayes AE - Bayes 1 Mol ISIS MLSKEYS Surface FP - Tanimoto DAYLIGHT Fingerprints SYBYL Hologram QSAR FLEXSIM-X FLEXSIM-S DCKSIM
26 Conformational Variance MDDR Dataset (5HT3, ACE, HMG, PAF, TXA2) 10 Randomly selected compounds each 10 Conformations generated by GA search with large window (10 for rigid 5HT3, 100 for ACE, HMG, PAF, TXA2), giving diverse conformations ne force field optimized conformation (Concord-generated) used to find other conformations of the same molecule in whole database of 937 structures, using Tanimoto Coefficient
27 verall findings 64% of conformations found at the top 10 positions -> > 2/3 of compounds identified as being most similar (among list of > 900 structures and structures of same active dataset) >90% of conformations found in Top 5% of sorted database Conclusion: If molecules with the right features are present in the database, they will not be missed (in most cases) because they are represented by a particular conformation
28 Which features are selected for classification? Even if your classifier works, do the selected features make sense? Set of active vs. inactive molecules Information Gain calculated for each feature, those which are much more frequent among actives are suspicious and might constitute the pharmacophore Look at features from ACE, HMG and TXA2

29 Selected Features - HMG Binding Site: HMG + rigid lipophilic ring
30 HMG-15

31 HMG-19
32 ACE Binding Site X X X H X: S1 Zn ++ H S1 H S2 + CH 2 CH 3 NH NH C N H Snake venom peptide analog with putative binding motif to angiotensin used in early compound design (Cushman et al., Biochemistry (1977), 16, ) 5491.) recent crystal structure available

33 Selected Features ACE-31 X X X H X: S1 Zn ++ H S1 H S2 + CH 2 CH 3 NH NH C H N
34 TXA2 Yellow: lipophilic side chains Yamamoto et al., J. Med. Chem (36) 820


35 TXA2-7, and 44

36 Feature Hopping
37 Query 7 8 S H S S N N Ranking position Structure H N N H N P N H N H H H N H N N Ranking position P Structure H H N N S N N H N 9 H N N H N Query (ACE inhibitor) used to screen the database and the highest ranked structures found (out of which all except no. 6,7 and 10 are classified as being ACE inhibitors in the MDDR database). Five of the active structures found (no. 3, 4, 5, 8 and 9) were not found by any of the other seven methods employed. 10 S N N H H H Molecular Surface Point Environments for Virtual Screening and the Elucidation of Binding Patterns (MLPRINT 3D). Bender, A.; Mussa, H. Y.; Gill, G. S.; Glen, R. C. J. Med. Chem. 2004, 47(26),
38 HTS Data Mining and Docking Competition 2005 at McMaster University (ntario) A competition to take 50,000 dihydrofolate reductase inhibitors of known activity (Training Set) and to (blindly) predict the activity of 50,000 new compounds (Test Set) in a high throughput screen. 32 groups took part. We obtained the best results. MLPRINT 2D, was employed for virtual screening of E. coli dihydrofolate reductase (DHFR) inhibitors. Using an original training set of 49,995 compounds, enrichment factors (between one and three) could be achieved on a test library, comprising 50,000 structures We think that these results are poor. Reasons are described below.
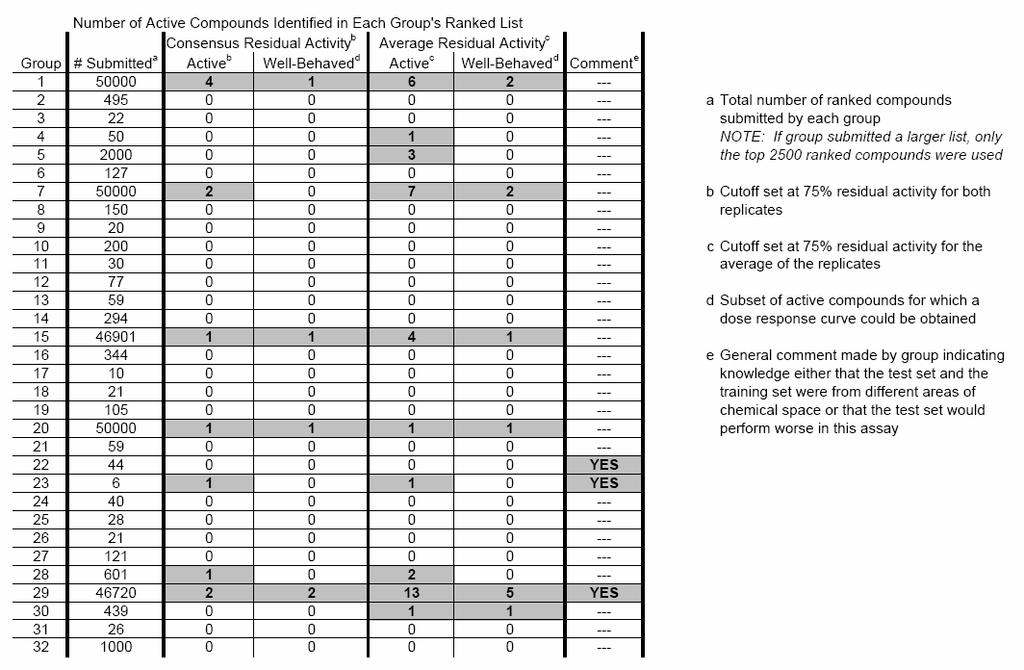
39 Results MolPrint2D
40 Data Set : High-throughput screening of 49,995 compounds was performed by Zolli-Juran et al., identifying 32 hits (defined by less than 75% residual activity in both of two screening runs) comprising several novel scaffolds. bjective: The extraction of the structural knowledge from the compounds and their activities from the first screening ( training set ) and to make predictions about the inhibitory activities of a second set of 50,000 compounds that was to be screened subsequently (42 hits subsequently found in the test set). ur results show ca. 3 fold enrichment in the first 200 compounds ranked. However, this reduced to just over one in the complete set why?
41
42 The Test Set and the Training set contains chemically different structures. Therefore, the method does not always recognise new features in the new set as contributors to activity. We repeated the analysis by randomizing the data and predicting using cross validation.
43 Training and test set were pooled in a second step and randomly split into training and test of equal size again, thus evening out the different chemical characteristics of both libraries. In a ten-fold cross validation study on the new training and test sets, typically 10-fold enrichment could be found in the first 96 positions, 4-fold enrichment in the first 384 positions and 3-fold enrichment in the first 1536 positions, corresponding to 6, 10 and 28 hits (out of a total of 307), respectively.
44 Training and test set were pooled in a second step and randomly split into training and test of equal size again, thus evening out the different chemical characteristics of both libraries. Hit Rates Enrichment Factors First positions Actives < 80% activity; Inactives > 100% activity, 200 Features Ten-fold Random Validation Actives < 85% activity, Inactives > 100% activity, 200 Features (0.7) 10.2 (2.4) 28.0 (3.0) 10.2 (1.2) 4.2 (1.0) 3.0 (0.3) Blind study after randomization note big increase in success In a ten-fold cross validation study on the new training and test sets, typically 10-fold enrichment could be found in the first 96 positions, 4-fold enrichment in the first 384 positions and 3-fold enrichment in the first 1536 positions, corresponding to 6, 10 and 28 hits (out of a total of 307), respectively. Conclusions : n the one hand the work presented here shows that exact-fragment-matching similarity searching methods are not capable of finding completely novel hit structures. Still, they are able to combine knowledge from multiple active structures to give novel combinations of features, as shown previously. n the other hand this work emphasizes the need for an even distribution of chemistry between the training and the test set. Lead hopping, moving from one chemical space to another thus requires analysis based on chemical descriptors (not the structural diagram), which is generally a much more compute intensive calculation.
45 Summary 2D Method: Performs about as well as other 2D methods for single molecule searches, outperforms them by a large margin when combining information from multiple molecules 3D Method: TR invariant, conformationally tolerant; combines high enrichment factors with scaffold hopping discovery of new chemotypes Features shown to correlate with binding patterns Performance (at least in part) due to Bayesian Classifier, which is able to take multiple structures as well as active and inactive information into account Chemically similar training and test sets required for 2D method
46 Acknowledgements Peter Murray-Rust, Jonathan Goodman, Hamse Mussa,, Andreas Bender, Joe Towsend,, Yong Zhang, Simon Tyrrell Unilever, the Royal Society of Chemistry, the Newton Trust, the Department of Trade and Industry, the EPSRC, the BBSRC.
Structure-Activity Modeling - QSAR. Uwe Koch
 Structure-Activity Modeling - QSAR Uwe Koch QSAR Assumption: QSAR attempts to quantify the relationship between activity and molecular strcucture by correlating descriptors with properties Biological activity
Structure-Activity Modeling - QSAR Uwe Koch QSAR Assumption: QSAR attempts to quantify the relationship between activity and molecular strcucture by correlating descriptors with properties Biological activity
Molecular Similarity Searching Using Inference Network
 Molecular Similarity Searching Using Inference Network Ammar Abdo, Naomie Salim* Faculty of Computer Science & Information Systems Universiti Teknologi Malaysia Molecular Similarity Searching Search for
Molecular Similarity Searching Using Inference Network Ammar Abdo, Naomie Salim* Faculty of Computer Science & Information Systems Universiti Teknologi Malaysia Molecular Similarity Searching Search for
An Integrated Approach to in-silico
 An Integrated Approach to in-silico Screening Joseph L. Durant Jr., Douglas. R. Henry, Maurizio Bronzetti, and David. A. Evans MDL Information Systems, Inc. 14600 Catalina St., San Leandro, CA 94577 Goals
An Integrated Approach to in-silico Screening Joseph L. Durant Jr., Douglas. R. Henry, Maurizio Bronzetti, and David. A. Evans MDL Information Systems, Inc. 14600 Catalina St., San Leandro, CA 94577 Goals
Computational chemical biology to address non-traditional drug targets. John Karanicolas
 Computational chemical biology to address non-traditional drug targets John Karanicolas Our computational toolbox Structure-based approaches Ligand-based approaches Detailed MD simulations 2D fingerprints
Computational chemical biology to address non-traditional drug targets John Karanicolas Our computational toolbox Structure-based approaches Ligand-based approaches Detailed MD simulations 2D fingerprints
Machine learning for ligand-based virtual screening and chemogenomics!
 Machine learning for ligand-based virtual screening and chemogenomics! Jean-Philippe Vert Institut Curie - INSERM U900 - Mines ParisTech In silico discovery of molecular probes and drug-like compounds:
Machine learning for ligand-based virtual screening and chemogenomics! Jean-Philippe Vert Institut Curie - INSERM U900 - Mines ParisTech In silico discovery of molecular probes and drug-like compounds:
Universities of Leeds, Sheffield and York
 promoting access to White Rose research papers Universities of Leeds, Sheffield and York http://eprints.whiterose.ac.uk/ This is an author produced version of a paper published in Organic & Biomolecular
promoting access to White Rose research papers Universities of Leeds, Sheffield and York http://eprints.whiterose.ac.uk/ This is an author produced version of a paper published in Organic & Biomolecular
An Enhancement of Bayesian Inference Network for Ligand-Based Virtual Screening using Features Selection
 American Journal of Applied Sciences 8 (4): 368-373, 2011 ISSN 1546-9239 2010 Science Publications An Enhancement of Bayesian Inference Network for Ligand-Based Virtual Screening using Features Selection
American Journal of Applied Sciences 8 (4): 368-373, 2011 ISSN 1546-9239 2010 Science Publications An Enhancement of Bayesian Inference Network for Ligand-Based Virtual Screening using Features Selection
Dr. Sander B. Nabuurs. Computational Drug Discovery group Center for Molecular and Biomolecular Informatics Radboud University Medical Centre
 Dr. Sander B. Nabuurs Computational Drug Discovery group Center for Molecular and Biomolecular Informatics Radboud University Medical Centre The road to new drugs. How to find new hits? High Throughput
Dr. Sander B. Nabuurs Computational Drug Discovery group Center for Molecular and Biomolecular Informatics Radboud University Medical Centre The road to new drugs. How to find new hits? High Throughput
Chemoinformatics and information management. Peter Willett, University of Sheffield, UK
 Chemoinformatics and information management Peter Willett, University of Sheffield, UK verview What is chemoinformatics and why is it necessary Managing structural information Typical facilities in chemoinformatics
Chemoinformatics and information management Peter Willett, University of Sheffield, UK verview What is chemoinformatics and why is it necessary Managing structural information Typical facilities in chemoinformatics
has its own advantages and drawbacks, depending on the questions facing the drug discovery.
 2013 First International Conference on Artificial Intelligence, Modelling & Simulation Comparison of Similarity Coefficients for Chemical Database Retrieval Mukhsin Syuib School of Information Technology
2013 First International Conference on Artificial Intelligence, Modelling & Simulation Comparison of Similarity Coefficients for Chemical Database Retrieval Mukhsin Syuib School of Information Technology
Cheminformatics analysis and learning in a data pipelining environment
 Molecular Diversity (2006) 10: 283 299 DOI: 10.1007/s11030-006-9041-5 c Springer 2006 Review Cheminformatics analysis and learning in a data pipelining environment Moises Hassan 1,, Robert D. Brown 1,
Molecular Diversity (2006) 10: 283 299 DOI: 10.1007/s11030-006-9041-5 c Springer 2006 Review Cheminformatics analysis and learning in a data pipelining environment Moises Hassan 1,, Robert D. Brown 1,
In silico pharmacology for drug discovery
 In silico pharmacology for drug discovery In silico drug design In silico methods can contribute to drug targets identification through application of bionformatics tools. Currently, the application of
In silico pharmacology for drug discovery In silico drug design In silico methods can contribute to drug targets identification through application of bionformatics tools. Currently, the application of
Introduction. OntoChem
 Introduction ntochem Providing drug discovery knowledge & small molecules... Supporting the task of medicinal chemistry Allows selecting best possible small molecule starting point From target to leads
Introduction ntochem Providing drug discovery knowledge & small molecules... Supporting the task of medicinal chemistry Allows selecting best possible small molecule starting point From target to leads
Similarity methods for ligandbased virtual screening
 Similarity methods for ligandbased virtual screening Peter Willett, University of Sheffield Computers in Scientific Discovery 5, 22 nd July 2010 Overview Molecular similarity and its use in virtual screening
Similarity methods for ligandbased virtual screening Peter Willett, University of Sheffield Computers in Scientific Discovery 5, 22 nd July 2010 Overview Molecular similarity and its use in virtual screening
Machine Learning Concepts in Chemoinformatics
 Machine Learning Concepts in Chemoinformatics Martin Vogt B-IT Life Science Informatics Rheinische Friedrich-Wilhelms-Universität Bonn BigChem Winter School 2017 25. October Data Mining in Chemoinformatics
Machine Learning Concepts in Chemoinformatics Martin Vogt B-IT Life Science Informatics Rheinische Friedrich-Wilhelms-Universität Bonn BigChem Winter School 2017 25. October Data Mining in Chemoinformatics
Structural biology and drug design: An overview
 Structural biology and drug design: An overview livier Taboureau Assitant professor Chemoinformatics group-cbs-dtu otab@cbs.dtu.dk Drug discovery Drug and drug design A drug is a key molecule involved
Structural biology and drug design: An overview livier Taboureau Assitant professor Chemoinformatics group-cbs-dtu otab@cbs.dtu.dk Drug discovery Drug and drug design A drug is a key molecule involved
The PhilOEsophy. There are only two fundamental molecular descriptors
 The PhilOEsophy There are only two fundamental molecular descriptors Where can we use shape? Virtual screening More effective than 2D Lead-hopping Shape analogues are not graph analogues Molecular alignment
The PhilOEsophy There are only two fundamental molecular descriptors Where can we use shape? Virtual screening More effective than 2D Lead-hopping Shape analogues are not graph analogues Molecular alignment
Chemogenomic: Approaches to Rational Drug Design. Jonas Skjødt Møller
 Chemogenomic: Approaches to Rational Drug Design Jonas Skjødt Møller Chemogenomic Chemistry Biology Chemical biology Medical chemistry Chemical genetics Chemoinformatics Bioinformatics Chemoproteomics
Chemogenomic: Approaches to Rational Drug Design Jonas Skjødt Møller Chemogenomic Chemistry Biology Chemical biology Medical chemistry Chemical genetics Chemoinformatics Bioinformatics Chemoproteomics
Virtual Libraries and Virtual Screening in Drug Discovery Processes using KNIME
 Virtual Libraries and Virtual Screening in Drug Discovery Processes using KNIME Iván Solt Solutions for Cheminformatics Drug Discovery Strategies for known targets High-Throughput Screening (HTS) Cells
Virtual Libraries and Virtual Screening in Drug Discovery Processes using KNIME Iván Solt Solutions for Cheminformatics Drug Discovery Strategies for known targets High-Throughput Screening (HTS) Cells
A Framework For Genetic-Based Fusion Of Similarity Measures In Chemical Compound Retrieval
 A Framework For Genetic-Based Fusion Of Similarity Measures In Chemical Compound Retrieval Naomie Salim Faculty of Computer Science and Information Systems Universiti Teknologi Malaysia naomie@fsksm.utm.my
A Framework For Genetic-Based Fusion Of Similarity Measures In Chemical Compound Retrieval Naomie Salim Faculty of Computer Science and Information Systems Universiti Teknologi Malaysia naomie@fsksm.utm.my
Using AutoDock for Virtual Screening
 Using AutoDock for Virtual Screening CUHK Croucher ASI Workshop 2011 Stefano Forli, PhD Prof. Arthur J. Olson, Ph.D Molecular Graphics Lab Screening and Virtual Screening The ultimate tool for identifying
Using AutoDock for Virtual Screening CUHK Croucher ASI Workshop 2011 Stefano Forli, PhD Prof. Arthur J. Olson, Ph.D Molecular Graphics Lab Screening and Virtual Screening The ultimate tool for identifying
Author Index Volume
 Perspectives in Drug Discovery and Design, 20: 289, 2000. KLUWER/ESCOM Author Index Volume 20 2000 Bradshaw,J., 1 Knegtel,R.M.A., 191 Rose,P.W., 209 Briem, H., 231 Kostka, T., 245 Kuhn, L.A., 171 Sadowski,
Perspectives in Drug Discovery and Design, 20: 289, 2000. KLUWER/ESCOM Author Index Volume 20 2000 Bradshaw,J., 1 Knegtel,R.M.A., 191 Rose,P.W., 209 Briem, H., 231 Kostka, T., 245 Kuhn, L.A., 171 Sadowski,
Molecular Complexity Effects and Fingerprint-Based Similarity Search Strategies
 Molecular Complexity Effects and Fingerprint-Based Similarity Search Strategies Dissertation zur Erlangung des Doktorgrades (Dr. rer. nat.) der Mathematisch-aturwissenschaftlichen Fakultät der Rheinischen
Molecular Complexity Effects and Fingerprint-Based Similarity Search Strategies Dissertation zur Erlangung des Doktorgrades (Dr. rer. nat.) der Mathematisch-aturwissenschaftlichen Fakultät der Rheinischen
Machine-learning scoring functions for docking
 Machine-learning scoring functions for docking Dr Pedro J Ballester MRC Methodology Research Fellow EMBL-EBI, Cambridge, United Kingdom EBI is an Outstation of the European Molecular Biology Laboratory.
Machine-learning scoring functions for docking Dr Pedro J Ballester MRC Methodology Research Fellow EMBL-EBI, Cambridge, United Kingdom EBI is an Outstation of the European Molecular Biology Laboratory.
De Novo molecular design with Deep Reinforcement Learning
 De Novo molecular design with Deep Reinforcement Learning @olexandr Olexandr Isayev, Ph.D. University of North Carolina at Chapel Hill olexandr@unc.edu http://olexandrisayev.com About me Ph.D. in Chemistry
De Novo molecular design with Deep Reinforcement Learning @olexandr Olexandr Isayev, Ph.D. University of North Carolina at Chapel Hill olexandr@unc.edu http://olexandrisayev.com About me Ph.D. in Chemistry
QSAR Modeling of Human Liver Microsomal Stability Alexey Zakharov
 QSAR Modeling of Human Liver Microsomal Stability Alexey Zakharov CADD Group Chemical Biology Laboratory Frederick National Laboratory for Cancer Research National Cancer Institute, National Institutes
QSAR Modeling of Human Liver Microsomal Stability Alexey Zakharov CADD Group Chemical Biology Laboratory Frederick National Laboratory for Cancer Research National Cancer Institute, National Institutes
A Tiered Screen Protocol for the Discovery of Structurally Diverse HIV Integrase Inhibitors
 A Tiered Screen Protocol for the Discovery of Structurally Diverse HIV Integrase Inhibitors Rajarshi Guha, Debojyoti Dutta, Ting Chen and David J. Wild School of Informatics Indiana University and Dept.
A Tiered Screen Protocol for the Discovery of Structurally Diverse HIV Integrase Inhibitors Rajarshi Guha, Debojyoti Dutta, Ting Chen and David J. Wild School of Informatics Indiana University and Dept.
Evaluation of Molecular Similarity and Molecular Diversity Methods Using Biological Activity Data
 2 Evaluation of Molecular Similarity and Molecular Diversity Methods Using Biological Activity Data Peter Willett Abstract This chapter reviews the techniques available for quantifying the effectiveness
2 Evaluation of Molecular Similarity and Molecular Diversity Methods Using Biological Activity Data Peter Willett Abstract This chapter reviews the techniques available for quantifying the effectiveness
DATA FUSION APPROACHES IN LIGAND-BASED VIRTUAL SCREENING: RECENT DEVELOPMENTS OVERVIEW
 DATA FUSION APPROACHES IN LIGAND-BASED VIRTUAL SCREENING: RECENT DEVELOPMENTS OVERVIEW Mubarak Himmat 1, Naomie Salim 1, Ali Ahmed 1, 2, and Mohammed Mumtaz Al-Dabbagh 1 1 Faculty of Computing, University
DATA FUSION APPROACHES IN LIGAND-BASED VIRTUAL SCREENING: RECENT DEVELOPMENTS OVERVIEW Mubarak Himmat 1, Naomie Salim 1, Ali Ahmed 1, 2, and Mohammed Mumtaz Al-Dabbagh 1 1 Faculty of Computing, University
Data Quality Issues That Can Impact Drug Discovery
 Data Quality Issues That Can Impact Drug Discovery Sean Ekins 1, Joe Olechno 2 Antony J. Williams 3 1 Collaborations in Chemistry, Fuquay Varina, NC. 2 Labcyte Inc, Sunnyvale, CA. 3 Royal Society of Chemistry,
Data Quality Issues That Can Impact Drug Discovery Sean Ekins 1, Joe Olechno 2 Antony J. Williams 3 1 Collaborations in Chemistry, Fuquay Varina, NC. 2 Labcyte Inc, Sunnyvale, CA. 3 Royal Society of Chemistry,
Similarity Search. Uwe Koch
 Similarity Search Uwe Koch Similarity Search The similar property principle: strurally similar molecules tend to have similar properties. However, structure property discontinuities occur frequently. Relevance
Similarity Search Uwe Koch Similarity Search The similar property principle: strurally similar molecules tend to have similar properties. However, structure property discontinuities occur frequently. Relevance
The Schrödinger KNIME extensions
 The Schrödinger KNIME extensions Computational Chemistry and Cheminformatics in a workflow environment Jean-Christophe Mozziconacci Volker Eyrich Topics What are the Schrödinger extensions? Workflow application
The Schrödinger KNIME extensions Computational Chemistry and Cheminformatics in a workflow environment Jean-Christophe Mozziconacci Volker Eyrich Topics What are the Schrödinger extensions? Workflow application
Drug Design 2. Oliver Kohlbacher. Winter 2009/ QSAR Part 4: Selected Chapters
 Drug Design 2 Oliver Kohlbacher Winter 2009/2010 11. QSAR Part 4: Selected Chapters Abt. Simulation biologischer Systeme WSI/ZBIT, Eberhard-Karls-Universität Tübingen Overview GRIND GRid-INDependent Descriptors
Drug Design 2 Oliver Kohlbacher Winter 2009/2010 11. QSAR Part 4: Selected Chapters Abt. Simulation biologischer Systeme WSI/ZBIT, Eberhard-Karls-Universität Tübingen Overview GRIND GRid-INDependent Descriptors
Hit Finding and Optimization Using BLAZE & FORGE
 Hit Finding and Optimization Using BLAZE & FORGE Kevin Cusack,* Maria Argiriadi, Eric Breinlinger, Jeremy Edmunds, Michael Hoemann, Michael Friedman, Sami Osman, Raymond Huntley, Thomas Vargo AbbVie, Immunology
Hit Finding and Optimization Using BLAZE & FORGE Kevin Cusack,* Maria Argiriadi, Eric Breinlinger, Jeremy Edmunds, Michael Hoemann, Michael Friedman, Sami Osman, Raymond Huntley, Thomas Vargo AbbVie, Immunology
Ignasi Belda, PhD CEO. HPC Advisory Council Spain Conference 2015
 Ignasi Belda, PhD CEO HPC Advisory Council Spain Conference 2015 Business lines Molecular Modeling Services We carry out computational chemistry projects using our selfdeveloped and third party technologies
Ignasi Belda, PhD CEO HPC Advisory Council Spain Conference 2015 Business lines Molecular Modeling Services We carry out computational chemistry projects using our selfdeveloped and third party technologies
Navigation in Chemical Space Towards Biological Activity. Peter Ertl Novartis Institutes for BioMedical Research Basel, Switzerland
 Navigation in Chemical Space Towards Biological Activity Peter Ertl Novartis Institutes for BioMedical Research Basel, Switzerland Data Explosion in Chemistry CAS 65 million molecules CCDC 600 000 structures
Navigation in Chemical Space Towards Biological Activity Peter Ertl Novartis Institutes for BioMedical Research Basel, Switzerland Data Explosion in Chemistry CAS 65 million molecules CCDC 600 000 structures
Computational Chemistry in Drug Design. Xavier Fradera Barcelona, 17/4/2007
 Computational Chemistry in Drug Design Xavier Fradera Barcelona, 17/4/2007 verview Introduction and background Drug Design Cycle Computational methods Chemoinformatics Ligand Based Methods Structure Based
Computational Chemistry in Drug Design Xavier Fradera Barcelona, 17/4/2007 verview Introduction and background Drug Design Cycle Computational methods Chemoinformatics Ligand Based Methods Structure Based
Virtual screening in drug discovery
 Virtual screening in drug discovery Pavel Polishchuk Institute of Molecular and Translational Medicine Palacky University pavlo.polishchuk@upol.cz Drug development workflow Vistoli G., et al., Drug Discovery
Virtual screening in drug discovery Pavel Polishchuk Institute of Molecular and Translational Medicine Palacky University pavlo.polishchuk@upol.cz Drug development workflow Vistoli G., et al., Drug Discovery
Universities of Leeds, Sheffield and York
 promoting access to White Rose research papers Universities of Leeds, Sheffield and York http://eprints.whiterose.ac.uk/ This is an author produced version of a paper published in Statistical Analysis
promoting access to White Rose research papers Universities of Leeds, Sheffield and York http://eprints.whiterose.ac.uk/ This is an author produced version of a paper published in Statistical Analysis
Receptor Based Drug Design (1)
") Induced Fit Model For more than 100 years, the behaviour of enzymes had been explained by the "lock-and-key" mechanism developed by pioneering German chemist Emil Fischer. Fischer thought that the chemicals
Induced Fit Model For more than 100 years, the behaviour of enzymes had been explained by the "lock-and-key" mechanism developed by pioneering German chemist Emil Fischer. Fischer thought that the chemicals
Kinome-wide Activity Models from Diverse High-Quality Datasets
 Kinome-wide Activity Models from Diverse High-Quality Datasets Stephan C. Schürer*,1 and Steven M. Muskal 2 1 Department of Molecular and Cellular Pharmacology, Miller School of Medicine and Center for
Kinome-wide Activity Models from Diverse High-Quality Datasets Stephan C. Schürer*,1 and Steven M. Muskal 2 1 Department of Molecular and Cellular Pharmacology, Miller School of Medicine and Center for
EMPIRICAL VS. RATIONAL METHODS OF DISCOVERING NEW DRUGS
 EMPIRICAL VS. RATIONAL METHODS OF DISCOVERING NEW DRUGS PETER GUND Pharmacopeia Inc., CN 5350 Princeton, NJ 08543, USA pgund@pharmacop.com Empirical and theoretical approaches to drug discovery have often
EMPIRICAL VS. RATIONAL METHODS OF DISCOVERING NEW DRUGS PETER GUND Pharmacopeia Inc., CN 5350 Princeton, NJ 08543, USA pgund@pharmacop.com Empirical and theoretical approaches to drug discovery have often
Next Generation Computational Chemistry Tools to Predict Toxicity of CWAs
 Next Generation Computational Chemistry Tools to Predict Toxicity of CWAs William (Bill) Welsh welshwj@umdnj.edu Prospective Funding by DTRA/JSTO-CBD CBIS Conference 1 A State-wide, Regional and National
Next Generation Computational Chemistry Tools to Predict Toxicity of CWAs William (Bill) Welsh welshwj@umdnj.edu Prospective Funding by DTRA/JSTO-CBD CBIS Conference 1 A State-wide, Regional and National
Introduction to Chemoinformatics and Drug Discovery
 Introduction to Chemoinformatics and Drug Discovery Irene Kouskoumvekaki Associate Professor February 15 th, 2013 The Chemical Space There are atoms and space. Everything else is opinion. Democritus (ca.
Introduction to Chemoinformatics and Drug Discovery Irene Kouskoumvekaki Associate Professor February 15 th, 2013 The Chemical Space There are atoms and space. Everything else is opinion. Democritus (ca.
Data Mining in the Chemical Industry. Overview of presentation
 Data Mining in the Chemical Industry Glenn J. Myatt, Ph.D. Partner, Myatt & Johnson, Inc. glenn.myatt@gmail.com verview of presentation verview of the chemical industry Example of the pharmaceutical industry
Data Mining in the Chemical Industry Glenn J. Myatt, Ph.D. Partner, Myatt & Johnson, Inc. glenn.myatt@gmail.com verview of presentation verview of the chemical industry Example of the pharmaceutical industry
Condensed Graph of Reaction: considering a chemical reaction as one single pseudo molecule
 Condensed Graph of Reaction: considering a chemical reaction as one single pseudo molecule Frank Hoonakker 1,3, Nicolas Lachiche 2, Alexandre Varnek 3, and Alain Wagner 3,4 1 Chemoinformatics laboratory,
Condensed Graph of Reaction: considering a chemical reaction as one single pseudo molecule Frank Hoonakker 1,3, Nicolas Lachiche 2, Alexandre Varnek 3, and Alain Wagner 3,4 1 Chemoinformatics laboratory,
This is a repository copy of Chemoinformatics techniques for data mining in files of two-dimensional and three-dimensional chemical molecules.
 This is a repository copy of Chemoinformatics techniques for data mining in files of two-dimensional and three-dimensional chemical molecules. White Rose Research Online URL for this paper: http://eprints.whiterose.ac.uk/8425/
This is a repository copy of Chemoinformatics techniques for data mining in files of two-dimensional and three-dimensional chemical molecules. White Rose Research Online URL for this paper: http://eprints.whiterose.ac.uk/8425/
Clustering Ambiguity: An Overview
 Clustering Ambiguity: An Overview John D. MacCuish Norah E. MacCuish 3 rd Joint Sheffield Conference on Chemoinformatics April 23, 2004 Outline The Problem: Clustering Ambiguity and Chemoinformatics Preliminaries:
Clustering Ambiguity: An Overview John D. MacCuish Norah E. MacCuish 3 rd Joint Sheffield Conference on Chemoinformatics April 23, 2004 Outline The Problem: Clustering Ambiguity and Chemoinformatics Preliminaries:
Bioengineering & Bioinformatics Summer Institute, Dept. Computational Biology, University of Pittsburgh, PGH, PA
 Pharmacophore Model Development for the Identification of Novel Acetylcholinesterase Inhibitors Edwin Kamau Dept Chem & Biochem Kennesa State Uni ersit Kennesa GA 30144 Dept. Chem. & Biochem. Kennesaw
Pharmacophore Model Development for the Identification of Novel Acetylcholinesterase Inhibitors Edwin Kamau Dept Chem & Biochem Kennesa State Uni ersit Kennesa GA 30144 Dept. Chem. & Biochem. Kennesaw
Performing a Pharmacophore Search using CSD-CrossMiner
 Table of Contents Introduction... 2 CSD-CrossMiner Terminology... 2 Overview of CSD-CrossMiner... 3 Searching with a Pharmacophore... 4 Performing a Pharmacophore Search using CSD-CrossMiner Version 2.0
Table of Contents Introduction... 2 CSD-CrossMiner Terminology... 2 Overview of CSD-CrossMiner... 3 Searching with a Pharmacophore... 4 Performing a Pharmacophore Search using CSD-CrossMiner Version 2.0
Biologically Relevant Molecular Comparisons. Mark Mackey
 Biologically Relevant Molecular Comparisons Mark Mackey Agenda > Cresset Technology > Cresset Products > FieldStere > FieldScreen > FieldAlign > FieldTemplater > Cresset and Knime About Cresset > Specialist
Biologically Relevant Molecular Comparisons Mark Mackey Agenda > Cresset Technology > Cresset Products > FieldStere > FieldScreen > FieldAlign > FieldTemplater > Cresset and Knime About Cresset > Specialist
Plan. Day 2: Exercise on MHC molecules.
 Plan Day 1: What is Chemoinformatics and Drug Design? Methods and Algorithms used in Chemoinformatics including SVM. Cross validation and sequence encoding Example and exercise with herg potassium channel:
Plan Day 1: What is Chemoinformatics and Drug Design? Methods and Algorithms used in Chemoinformatics including SVM. Cross validation and sequence encoding Example and exercise with herg potassium channel:
Chemical Space: Modeling Exploration & Understanding
 verview Chemical Space: Modeling Exploration & Understanding Rajarshi Guha School of Informatics Indiana University 16 th August, 2006 utline verview 1 verview 2 3 CDK R utline verview 1 verview 2 3 CDK
verview Chemical Space: Modeling Exploration & Understanding Rajarshi Guha School of Informatics Indiana University 16 th August, 2006 utline verview 1 verview 2 3 CDK R utline verview 1 verview 2 3 CDK
LigandScout. Automated Structure-Based Pharmacophore Model Generation. Gerhard Wolber* and Thierry Langer
 LigandScout Automated Structure-Based Pharmacophore Model Generation Gerhard Wolber* and Thierry Langer * E-Mail: wolber@inteligand.com Pharmacophores from LigandScout Pharmacophores & the Protein Data
LigandScout Automated Structure-Based Pharmacophore Model Generation Gerhard Wolber* and Thierry Langer * E-Mail: wolber@inteligand.com Pharmacophores from LigandScout Pharmacophores & the Protein Data
The Schrödinger KNIME extensions
 The Schrödinger KNIME extensions Computational Chemistry and Cheminformatics in a workflow environment Jean-Christophe Mozziconacci Volker Eyrich KNIME UGM, Berlin, February 2015 The Schrödinger Extensions
The Schrödinger KNIME extensions Computational Chemistry and Cheminformatics in a workflow environment Jean-Christophe Mozziconacci Volker Eyrich KNIME UGM, Berlin, February 2015 The Schrödinger Extensions
Fast similarity searching making the virtual real. Stephen Pickett, GSK
 Fast similarity searching making the virtual real Stephen Pickett, GSK Introduction Introduction to similarity searching Use cases Why is speed so crucial? Why MadFast? Some performance stats Implementation
Fast similarity searching making the virtual real Stephen Pickett, GSK Introduction Introduction to similarity searching Use cases Why is speed so crucial? Why MadFast? Some performance stats Implementation
QSAR/QSPR modeling. Quantitative Structure-Activity Relationships Quantitative Structure-Property-Relationships
 Quantitative Structure-Activity Relationships Quantitative Structure-Property-Relationships QSAR/QSPR modeling Alexandre Varnek Faculté de Chimie, ULP, Strasbourg, FRANCE QSAR/QSPR models Development Validation
Quantitative Structure-Activity Relationships Quantitative Structure-Property-Relationships QSAR/QSPR modeling Alexandre Varnek Faculté de Chimie, ULP, Strasbourg, FRANCE QSAR/QSPR models Development Validation
Overview. Descriptors. Definition. Descriptors. Overview 2D-QSAR. Number Vector Function. Physicochemical property (log P) Atom
 Atom") verview D-QSAR Definition Examples Features counts Topological indices D fingerprints and fragment counts R-group descriptors ow good are D descriptors in practice? Summary Peter Gedeck ovartis Institutes
verview D-QSAR Definition Examples Features counts Topological indices D fingerprints and fragment counts R-group descriptors ow good are D descriptors in practice? Summary Peter Gedeck ovartis Institutes
Creating a Pharmacophore Query from a Reference Molecule & Scaffold Hopping in CSD-CrossMiner
 Table of Contents Creating a Pharmacophore Query from a Reference Molecule & Scaffold Hopping in CSD-CrossMiner Introduction... 2 CSD-CrossMiner Terminology... 2 Overview of CSD-CrossMiner... 3 Features
Table of Contents Creating a Pharmacophore Query from a Reference Molecule & Scaffold Hopping in CSD-CrossMiner Introduction... 2 CSD-CrossMiner Terminology... 2 Overview of CSD-CrossMiner... 3 Features
Virtual affinity fingerprints in drug discovery: The Drug Profile Matching method
 Ágnes Peragovics Virtual affinity fingerprints in drug discovery: The Drug Profile Matching method PhD Theses Supervisor: András Málnási-Csizmadia DSc. Associate Professor Structural Biochemistry Doctoral
Ágnes Peragovics Virtual affinity fingerprints in drug discovery: The Drug Profile Matching method PhD Theses Supervisor: András Málnási-Csizmadia DSc. Associate Professor Structural Biochemistry Doctoral
JCICS Major Research Areas
 JCICS Major Research Areas Chemical Information Text Searching Structure and Substructure Searching Databases Patents George W.A. Milne C571 Lecture Fall 2002 1 JCICS Major Research Areas Chemical Computation
JCICS Major Research Areas Chemical Information Text Searching Structure and Substructure Searching Databases Patents George W.A. Milne C571 Lecture Fall 2002 1 JCICS Major Research Areas Chemical Computation
Progress of Compound Library Design Using In-silico Approach for Collaborative Drug Discovery
 21 th /June/2018@CUGM Progress of Compound Library Design Using In-silico Approach for Collaborative Drug Discovery Kaz Ikeda, Ph.D. Keio University Self Introduction Keio University, Tokyo, Japan (Established
21 th /June/2018@CUGM Progress of Compound Library Design Using In-silico Approach for Collaborative Drug Discovery Kaz Ikeda, Ph.D. Keio University Self Introduction Keio University, Tokyo, Japan (Established
Pose and affinity prediction by ICM in D3R GC3. Max Totrov Molsoft
 Pose and affinity prediction by ICM in D3R GC3 Max Totrov Molsoft Pose prediction method: ICM-dock ICM-dock: - pre-sampling of ligand conformers - multiple trajectory Monte-Carlo with gradient minimization
Pose and affinity prediction by ICM in D3R GC3 Max Totrov Molsoft Pose prediction method: ICM-dock ICM-dock: - pre-sampling of ligand conformers - multiple trajectory Monte-Carlo with gradient minimization
MM-GBSA for Calculating Binding Affinity A rank-ordering study for the lead optimization of Fxa and COX-2 inhibitors
 MM-GBSA for Calculating Binding Affinity A rank-ordering study for the lead optimization of Fxa and COX-2 inhibitors Thomas Steinbrecher Senior Application Scientist Typical Docking Workflow Databases
MM-GBSA for Calculating Binding Affinity A rank-ordering study for the lead optimization of Fxa and COX-2 inhibitors Thomas Steinbrecher Senior Application Scientist Typical Docking Workflow Databases
Retrieving hits through in silico screening and expert assessment M. N. Drwal a,b and R. Griffith a
 Retrieving hits through in silico screening and expert assessment M.. Drwal a,b and R. Griffith a a: School of Medical Sciences/Pharmacology, USW, Sydney, Australia b: Charité Berlin, Germany Abstract:
Retrieving hits through in silico screening and expert assessment M.. Drwal a,b and R. Griffith a a: School of Medical Sciences/Pharmacology, USW, Sydney, Australia b: Charité Berlin, Germany Abstract:
Using Phase for Pharmacophore Modelling. 5th European Life Science Bootcamp March, 2017
 Using Phase for Pharmacophore Modelling 5th European Life Science Bootcamp March, 2017 Phase: Our Pharmacohore generation tool Significant improvements to Phase methods in 2016 New highly interactive interface
Using Phase for Pharmacophore Modelling 5th European Life Science Bootcamp March, 2017 Phase: Our Pharmacohore generation tool Significant improvements to Phase methods in 2016 New highly interactive interface
Generating Small Molecule Conformations from Structural Data
 Generating Small Molecule Conformations from Structural Data Jason Cole cole@ccdc.cam.ac.uk Cambridge Crystallographic Data Centre 1 The Cambridge Crystallographic Data Centre About us A not-for-profit,
Generating Small Molecule Conformations from Structural Data Jason Cole cole@ccdc.cam.ac.uk Cambridge Crystallographic Data Centre 1 The Cambridge Crystallographic Data Centre About us A not-for-profit,
AMRI COMPOUND LIBRARY CONSORTIUM: A NOVEL WAY TO FILL YOUR DRUG PIPELINE
 AMRI COMPOUD LIBRARY COSORTIUM: A OVEL WAY TO FILL YOUR DRUG PIPELIE Muralikrishna Valluri, PhD & Douglas B. Kitchen, PhD Summary The creation of high-quality, innovative small molecule leads is a continual
AMRI COMPOUD LIBRARY COSORTIUM: A OVEL WAY TO FILL YOUR DRUG PIPELIE Muralikrishna Valluri, PhD & Douglas B. Kitchen, PhD Summary The creation of high-quality, innovative small molecule leads is a continual
bcl::cheminfo Suite Enables Machine Learning-Based Drug Discovery Using GPUs Edward W. Lowe, Jr. Nils Woetzel May 17, 2012
 bcl::cheminfo Suite Enables Machine Learning-Based Drug Discovery Using GPUs Edward W. Lowe, Jr. Nils Woetzel May 17, 2012 Outline Machine Learning Cheminformatics Framework QSPR logp QSAR mglur 5 CYP
bcl::cheminfo Suite Enables Machine Learning-Based Drug Discovery Using GPUs Edward W. Lowe, Jr. Nils Woetzel May 17, 2012 Outline Machine Learning Cheminformatics Framework QSPR logp QSAR mglur 5 CYP
Dispensing Processes Profoundly Impact Biological, Computational and Statistical Analyses
 Dispensing Processes Profoundly Impact Biological, Computational and Statistical Analyses Sean Ekins 1, Joe Olechno 2 Antony J. Williams 3 1 Collaborations in Chemistry, Fuquay Varina, NC. 2 Labcyte Inc,
Dispensing Processes Profoundly Impact Biological, Computational and Statistical Analyses Sean Ekins 1, Joe Olechno 2 Antony J. Williams 3 1 Collaborations in Chemistry, Fuquay Varina, NC. 2 Labcyte Inc,
Practical QSAR and Library Design: Advanced tools for research teams
 DS QSAR and Library Design Webinar Practical QSAR and Library Design: Advanced tools for research teams Reservationless-Plus Dial-In Number (US): (866) 519-8942 Reservationless-Plus International Dial-In
DS QSAR and Library Design Webinar Practical QSAR and Library Design: Advanced tools for research teams Reservationless-Plus Dial-In Number (US): (866) 519-8942 Reservationless-Plus International Dial-In
Different conformations of the drugs within the virtual library of FDA approved drugs will be generated.
 Chapter 3 Molecular Modeling 3.1. Introduction In this study pharmacophore models will be created to screen a virtual library of FDA approved drugs for compounds that may inhibit MA-A and MA-B. The virtual
Chapter 3 Molecular Modeling 3.1. Introduction In this study pharmacophore models will be created to screen a virtual library of FDA approved drugs for compounds that may inhibit MA-A and MA-B. The virtual
What is a property-based similarity?
 What is a property-based similarity? Igor V. Tetko (1) GSF - ational Centre for Environment and Health, Institute for Bioinformatics, Ingolstaedter Landstrasse 1, euherberg, 85764, Germany, (2) Institute
What is a property-based similarity? Igor V. Tetko (1) GSF - ational Centre for Environment and Health, Institute for Bioinformatics, Ingolstaedter Landstrasse 1, euherberg, 85764, Germany, (2) Institute
Xia Ning,*, Huzefa Rangwala, and George Karypis
 J. Chem. Inf. Model. XXXX, xxx, 000 A Multi-Assay-Based Structure-Activity Relationship Models: Improving Structure-Activity Relationship Models by Incorporating Activity Information from Related Targets
J. Chem. Inf. Model. XXXX, xxx, 000 A Multi-Assay-Based Structure-Activity Relationship Models: Improving Structure-Activity Relationship Models by Incorporating Activity Information from Related Targets
György M. Keserű H2020 FRAGNET Network Hungarian Academy of Sciences
 Fragment based lead discovery - introduction György M. Keserű H2020 FRAGET etwork Hungarian Academy of Sciences www.fragnet.eu Hit discovery from screening Druglike library Fragment library Large molecules
Fragment based lead discovery - introduction György M. Keserű H2020 FRAGET etwork Hungarian Academy of Sciences www.fragnet.eu Hit discovery from screening Druglike library Fragment library Large molecules
Functional Group Fingerprints CNS Chemistry Wilmington, USA
 Functional Group Fingerprints CS Chemistry Wilmington, USA James R. Arnold Charles L. Lerman William F. Michne James R. Damewood American Chemical Society ational Meeting August, 2004 Philadelphia, PA
Functional Group Fingerprints CS Chemistry Wilmington, USA James R. Arnold Charles L. Lerman William F. Michne James R. Damewood American Chemical Society ational Meeting August, 2004 Philadelphia, PA
est Drive K20 GPUs! Experience The Acceleration Run Computational Chemistry Codes on Tesla K20 GPU today
 est Drive K20 GPUs! Experience The Acceleration Run Computational Chemistry Codes on Tesla K20 GPU today Sign up for FREE GPU Test Drive on remotely hosted clusters www.nvidia.com/gputestd rive Shape Searching
est Drive K20 GPUs! Experience The Acceleration Run Computational Chemistry Codes on Tesla K20 GPU today Sign up for FREE GPU Test Drive on remotely hosted clusters www.nvidia.com/gputestd rive Shape Searching
Virtual Screening: How Are We Doing?
 Virtual Screening: How Are We Doing? Mark E. Snow, James Dunbar, Lakshmi Narasimhan, Jack A. Bikker, Dan Ortwine, Christopher Whitehead, Yiannis Kaznessis, Dave Moreland, Christine Humblet Pfizer Global
Virtual Screening: How Are We Doing? Mark E. Snow, James Dunbar, Lakshmi Narasimhan, Jack A. Bikker, Dan Ortwine, Christopher Whitehead, Yiannis Kaznessis, Dave Moreland, Christine Humblet Pfizer Global
Use of data mining and chemoinformatics in the identification and optimization of high-throughput screening hits for NTDs
 Use of data mining and chemoinformatics in the identification and optimization of high-throughput screening hits for NTDs James Mills; Karl Gibson, Gavin Whitlock, Paul Glossop, Jean-Robert Ioset, Leela
Use of data mining and chemoinformatics in the identification and optimization of high-throughput screening hits for NTDs James Mills; Karl Gibson, Gavin Whitlock, Paul Glossop, Jean-Robert Ioset, Leela
Building innovative drug discovery alliances. Just in KNIME: Successful Process Driven Drug Discovery
 Building innovative drug discovery alliances Just in KIME: Successful Process Driven Drug Discovery Berlin KIME Spring Summit, Feb 2016 Research Informatics @ Evotec Evotec s worldwide operations 2 Pharmaceuticals
Building innovative drug discovery alliances Just in KIME: Successful Process Driven Drug Discovery Berlin KIME Spring Summit, Feb 2016 Research Informatics @ Evotec Evotec s worldwide operations 2 Pharmaceuticals
Chemical Data Retrieval and Management
 Chemical Data Retrieval and Management ChEMBL, ChEBI, and the Chemistry Development Kit Stephan A. Beisken What is EMBL-EBI? Part of the European Molecular Biology Laboratory International, non-profit
Chemical Data Retrieval and Management ChEMBL, ChEBI, and the Chemistry Development Kit Stephan A. Beisken What is EMBL-EBI? Part of the European Molecular Biology Laboratory International, non-profit
A Deep Convolutional Neural Network for Bioactivity Prediction in Structure-based Drug Discovery
 AtomNet A Deep Convolutional Neural Network for Bioactivity Prediction in Structure-based Drug Discovery Izhar Wallach, Michael Dzamba, Abraham Heifets Victor Storchan, Institute for Computational and
AtomNet A Deep Convolutional Neural Network for Bioactivity Prediction in Structure-based Drug Discovery Izhar Wallach, Michael Dzamba, Abraham Heifets Victor Storchan, Institute for Computational and
Plan. Lecture: What is Chemoinformatics and Drug Design? Description of Support Vector Machine (SVM) and its used in Chemoinformatics.
 and its used in Chemoinformatics.") Plan Lecture: What is Chemoinformatics and Drug Design? Description of Support Vector Machine (SVM) and its used in Chemoinformatics. Exercise: Example and exercise with herg potassium channel: Use of
Plan Lecture: What is Chemoinformatics and Drug Design? Description of Support Vector Machine (SVM) and its used in Chemoinformatics. Exercise: Example and exercise with herg potassium channel: Use of
Data Analysis in the Life Sciences - The Fog of Data -
 ALTAA Chair for Bioinformatics & Information Mining Data Analysis in the Life Sciences - The Fog of Data - Michael R. Berthold ALTAA-Chair for Bioinformatics & Information Mining Konstanz University, Germany
ALTAA Chair for Bioinformatics & Information Mining Data Analysis in the Life Sciences - The Fog of Data - Michael R. Berthold ALTAA-Chair for Bioinformatics & Information Mining Konstanz University, Germany
Early Stages of Drug Discovery in the Pharmaceutical Industry
 Early Stages of Drug Discovery in the Pharmaceutical Industry Daniel Seeliger / Jan Kriegl, Discovery Research, Boehringer Ingelheim September 29, 2016 Historical Drug Discovery From Accidential Discovery
Early Stages of Drug Discovery in the Pharmaceutical Industry Daniel Seeliger / Jan Kriegl, Discovery Research, Boehringer Ingelheim September 29, 2016 Historical Drug Discovery From Accidential Discovery
User Guide for LeDock
 User Guide for LeDock Hongtao Zhao, PhD Email: htzhao@lephar.com Website: www.lephar.com Copyright 2017 Hongtao Zhao. All rights reserved. Introduction LeDock is flexible small-molecule docking software,
User Guide for LeDock Hongtao Zhao, PhD Email: htzhao@lephar.com Website: www.lephar.com Copyright 2017 Hongtao Zhao. All rights reserved. Introduction LeDock is flexible small-molecule docking software,
Implementation of novel tools to facilitate fragment-based drug discovery by NMR:
 Implementation of novel tools to facilitate fragment-based drug discovery by NMR: Automated analysis of large sets of ligand-observed NMR binding data and 19 F methods Andreas Lingel Global Discovery Chemistry
Implementation of novel tools to facilitate fragment-based drug discovery by NMR: Automated analysis of large sets of ligand-observed NMR binding data and 19 F methods Andreas Lingel Global Discovery Chemistry
Towards Physics-based Models for ADME/Tox. Tyler Day
 Towards Physics-based Models for ADME/Tox Tyler Day Overview Motivation Application: P450 Site of Metabolism Application: Membrane Permeability Future Directions and Applications Motivation Advantages
Towards Physics-based Models for ADME/Tox Tyler Day Overview Motivation Application: P450 Site of Metabolism Application: Membrane Permeability Future Directions and Applications Motivation Advantages
Ligand Scout Tutorials
 Ligand Scout Tutorials Step : Creating a pharmacophore from a protein-ligand complex. Type ke6 in the upper right area of the screen and press the button Download *+. The protein will be downloaded and
Ligand Scout Tutorials Step : Creating a pharmacophore from a protein-ligand complex. Type ke6 in the upper right area of the screen and press the button Download *+. The protein will be downloaded and
Pooling Experiments for High Throughput Screening in Drug Discovery
 Pooling Experiments for High Throughput Screening in Drug Discovery Jacqueline M. Hughes-Oliver hughesol@stat.ncsu.edu Department of Statistics North Carolina State University Spring Research Conference,
Pooling Experiments for High Throughput Screening in Drug Discovery Jacqueline M. Hughes-Oliver hughesol@stat.ncsu.edu Department of Statistics North Carolina State University Spring Research Conference,
QSAR Modeling of ErbB1 Inhibitors Using Genetic Algorithm-Based Regression
 APPLICATION NOTE QSAR Modeling of ErbB1 Inhibitors Using Genetic Algorithm-Based Regression GAINING EFFICIENCY IN QUANTITATIVE STRUCTURE ACTIVITY RELATIONSHIPS ErbB1 kinase is the cell-surface receptor
APPLICATION NOTE QSAR Modeling of ErbB1 Inhibitors Using Genetic Algorithm-Based Regression GAINING EFFICIENCY IN QUANTITATIVE STRUCTURE ACTIVITY RELATIONSHIPS ErbB1 kinase is the cell-surface receptor
Computational Chemistry in Drug Discovery. Darren Green EBI 2016
 Computational Chemistry in Drug Discovery Darren Green EBI 2016 The Discovery Process gene protein target screen and identify lead Lead optimisation chemical diversity (compound library) test safety& efficacy
Computational Chemistry in Drug Discovery Darren Green EBI 2016 The Discovery Process gene protein target screen and identify lead Lead optimisation chemical diversity (compound library) test safety& efficacy
Research Article. Chemical compound classification based on improved Max-Min kernel
 Available online www.jocpr.com Journal of Chemical and Pharmaceutical Research, 2014, 6(2):368-372 Research Article ISSN : 0975-7384 CODEN(USA) : JCPRC5 Chemical compound classification based on improved
Available online www.jocpr.com Journal of Chemical and Pharmaceutical Research, 2014, 6(2):368-372 Research Article ISSN : 0975-7384 CODEN(USA) : JCPRC5 Chemical compound classification based on improved
Computational Methods and Drug-Likeness. Benjamin Georgi und Philip Groth Pharmakokinetik WS 2003/2004
 Computational Methods and Drug-Likeness Benjamin Georgi und Philip Groth Pharmakokinetik WS 2003/2004 The Problem Drug development in pharmaceutical industry: >8-12 years time ~$800m costs >90% failure
Computational Methods and Drug-Likeness Benjamin Georgi und Philip Groth Pharmakokinetik WS 2003/2004 The Problem Drug development in pharmaceutical industry: >8-12 years time ~$800m costs >90% failure
Prediction and Classif ication of Human G-protein Coupled Receptors Based on Support Vector Machines
 Article Prediction and Classif ication of Human G-protein Coupled Receptors Based on Support Vector Machines Yun-Fei Wang, Huan Chen, and Yan-Hong Zhou* Hubei Bioinformatics and Molecular Imaging Key Laboratory,
Article Prediction and Classif ication of Human G-protein Coupled Receptors Based on Support Vector Machines Yun-Fei Wang, Huan Chen, and Yan-Hong Zhou* Hubei Bioinformatics and Molecular Imaging Key Laboratory,
Chemical library design
 Chemical library design Pavel Polishchuk Institute of Molecular and Translational Medicine Palacky University pavlo.polishchuk@upol.cz Drug development workflow Vistoli G., et al., Drug Discovery Today,
Chemical library design Pavel Polishchuk Institute of Molecular and Translational Medicine Palacky University pavlo.polishchuk@upol.cz Drug development workflow Vistoli G., et al., Drug Discovery Today,
QSAR in Green Chemistry
 QSAR in Green Chemistry Activity Relationship QSAR is the acronym for Quantitative Structure-Activity Relationship Chemistry is based on the premise that similar chemicals will behave similarly The behavior/activity
QSAR in Green Chemistry Activity Relationship QSAR is the acronym for Quantitative Structure-Activity Relationship Chemistry is based on the premise that similar chemicals will behave similarly The behavior/activity
Using Self-Organizing maps to accelerate similarity search
 YOU LOGO Using Self-Organizing maps to accelerate similarity search Fanny Bonachera, Gilles Marcou, Natalia Kireeva, Alexandre Varnek, Dragos Horvath Laboratoire d Infochimie, UM 7177. 1, rue Blaise Pascal,
YOU LOGO Using Self-Organizing maps to accelerate similarity search Fanny Bonachera, Gilles Marcou, Natalia Kireeva, Alexandre Varnek, Dragos Horvath Laboratoire d Infochimie, UM 7177. 1, rue Blaise Pascal,
Interactive Feature Selection with
 Chapter 6 Interactive Feature Selection with TotalBoost g ν We saw in the experimental section that the generalization performance of the corrective and totally corrective boosting algorithms is comparable.
Chapter 6 Interactive Feature Selection with TotalBoost g ν We saw in the experimental section that the generalization performance of the corrective and totally corrective boosting algorithms is comparable.
October 6 University Faculty of pharmacy Computer Aided Drug Design Unit
 October 6 University Faculty of pharmacy Computer Aided Drug Design Unit CADD@O6U.edu.eg CADD Computer-Aided Drug Design Unit The development of new drugs is no longer a process of trial and error or strokes
October 6 University Faculty of pharmacy Computer Aided Drug Design Unit CADD@O6U.edu.eg CADD Computer-Aided Drug Design Unit The development of new drugs is no longer a process of trial and error or strokes