De Novo molecular design with Deep Reinforcement Learning
|
|
|
- Camron Shaw
- 5 years ago
- Views:
Transcription






1 De Novo molecular design with Deep Reinforcement Olexandr Isayev, Ph.D. University of North Carolina at Chapel Hill
 Minor in CS/ML Worked in Federal research lab on HPC & GPU computing to solve chemical problems Now I am faculty at the University of")
2 About me Ph.D. in Chemistry (computational) Minor in CS/ML Worked in Federal research lab on HPC & GPU computing to solve chemical problems Now I am faculty at the University of North Carolina, Chapel Hill We use ML & AI to solve challenging problems in chemistry olexandr@unc.edu






3 A public-private partnership that supports the discovery of new medicines through open access research


4 The Long and Winding Road to Drug Discovery Data Science approaches useful across the pipeline, but very different techniques aim for success, but if not: fail early, fail cheap

5 internal rate of return (IRR) Source: Endpoints News


6 Drowning in Data but starving for Knowledge


7 The growing appreciation of molecular modeling and informatics 7

8 Behold the rise of the machines
9 Summary of recent AI-based studies on chemical library design Molecular representations Generative models Method of biasing generated compounds Fingerprints SMILES Graphs Autoencoders Generative adversarial models (GANs) Recurrent neural networks (RNNs) Convolutional neural networks (CNNs) None Latent space optimization Fine-tuning on small subset of molecules with the desired property Reinforcement Learning


10 De Novo molecular design with Deep Reinforcement Learning General Approach Application to Molecular design Tm; LogP; pic50; etc Predictive Deep Network Molecules Generative Deep Network Patent pending arxiv:




11 Drug discovery pipeline CHEMICAL STRUCTURES CHEMICAL DESCRIPTORS PREDICTIVE QSAR MODELS PROPERTY/ ACTIVITY CHEMICAL DATABASE QSAR MAGIC VIRTUAL SCREENING HITS (confirmed actives) ~ molecules INACTIVES (confirmed inactives)
12 Design of the ReLeaSE* method Challenges: Generate chemically feasible SMILES Develop SMILESbased QSAR model Employ Predictive ML model to bias library generation *Popova, Mariya, Olexandr Isayev, and Alexander Tropsha. "Deep reinforcement learning for de-novo drug design." arxiv preprint arxiv: (2017).
13 Language of SMILEs
14 Generative model 1.5M molecules from ChEMBL <START>c1ccc(O)cc1<END> c1cc) ( F) cc1<end> c1ccc(o)cc1 NO <START> c1ccc( O) cc1 + loss YES Did the training converge? Softmax loss
15 Reinforcement learning for chemical design Generative model Predictive model FC(F)COc1ccc2c(Nc3ccc(Cl)c(Cl)c3)ncnc2c1 M. Popova, O. Isayev, A. Tropsha. "Deep reinforcement learning for de-novo drug design." arxiv preprint arxiv: (2017).
16 Reinforcement learning for chemical design Generative model Predictive model FC(F)COc1ccc2c(Nc3ccc(Cl)c(Cl)c3)ncnc2c1 M. Popova, O. Isayev, A. Tropsha. "Deep reinforcement learning for de-novo drug design." arxiv preprint arxiv: (2017).
17 Reinforcement learning for chemical design Generative model INACTIVE! Predictive model M. Popova, O. Isayev, A. Tropsha. "Deep reinforcement learning for de-novo drug design." arxiv preprint arxiv: (2017).
18 Reinforcement learning for chemical design Generative model INACTIVE! Predictive model M. Popova, O. Isayev, A. Tropsha. "Deep reinforcement learning for de-novo drug design." arxiv preprint arxiv: (2017).
19 Reinforcement learning for chemical design Generative model INACTIVE! Predictive model M. Popova, O. Isayev, A. Tropsha. "Deep reinforcement learning for de-novo drug design." arxiv preprint arxiv: (2017).


20 Reinforcement learning for chemical design Generative model Predictive model M. Popova, O. Isayev, A. Tropsha. "Deep reinforcement learning for de-novo drug design." arxiv preprint arxiv: (2017).
21 Reinforcement learning for chemical design Generative model Predictive model Fc1ccc2c(Nc3ccc(F)c(F)c3)ncnc2c1 M. Popova, O. Isayev, A. Tropsha. "Deep reinforcement learning for de-novo drug design." arxiv preprint arxiv: (2017).
22 Reinforcement learning for chemical design Generative model Predictive model Fc1ccc2c(Nc3ccc(F)c(F)c3)ncnc2c1 M. Popova, O. Isayev, A. Tropsha. "Deep reinforcement learning for de-novo drug design." arxiv preprint arxiv: (2017).
23 Reinforcement learning for chemical design Generative model ACTIVE! Predictive model M. Popova, O. Isayev, A. Tropsha. "Deep reinforcement learning for de-novo drug design." arxiv preprint arxiv: (2017).
24 Reinforcement learning for chemical design Generative model ACTIVE! Predictive model M. Popova, O. Isayev, A. Tropsha. "Deep reinforcement learning for de-novo drug design." arxiv preprint arxiv: (2017).

25 Reinforcement learning for chemical design Generative model ACTIVE! Predictive model M. Popova, O. Isayev, A. Tropsha. "Deep reinforcement learning for de-novo drug design." arxiv preprint arxiv: (2017).
26 Reinforcement learning for chemical design Generative model Predictive model M. Popova, O. Isayev, A. Tropsha. "Deep reinforcement learning for de-novo drug design." arxiv preprint arxiv: (2017).
27 Technical details Models were trained on Nvidia Titan X and Titan V GPUs Training generative model on ChEMBL took ~ 25 days Training predictive models took ~ 2 hours Biasing generative model with reinforcement learning for one property ~ 1 day Generative model produces 1000s compounds per minute

 Melting")
 M. Popova, O. Isayev, A.")

28 Results: Biasing target properties in the designed libraries Optimized Baseline * JAK2 Inhibition (pic50) Melting temperature (T m ), o C 400 Partition coefficient (logp) M. Popova, O. Isayev, A. Tropsha. "Deep reinforcement learning for de-novo drug design." arxiv preprint arxiv: (2017).

 ZINC37859566 SIMILAR SCAFFOLDS New molecule")
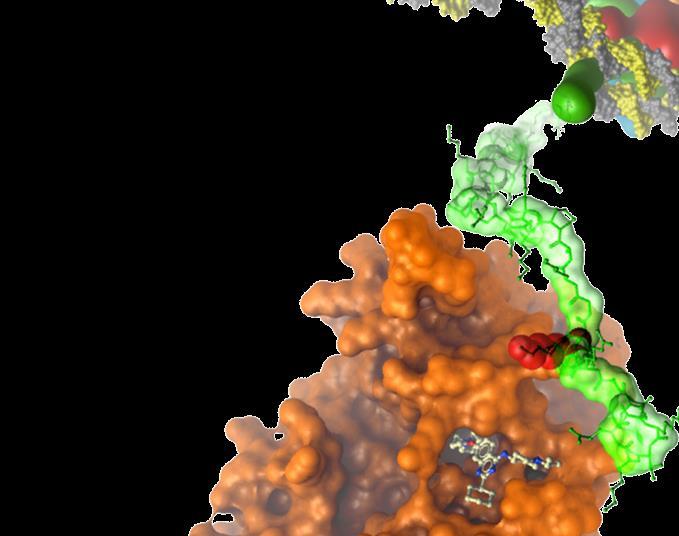
29 JAK2 (Kinase) inhibition Train data distribution Maximized property distribution Minimized property distribution NEW CHEMOTYPE CAS (buffer reagent) ZINC SIMILAR SCAFFOLDS New molecule arxiv:


30 Results: analysis of similarity Distribution of Tanimoto similarity to the nearest neighbor in training dataset for compounds predicted to be active for EGFR by consensus of QSAR models: Similarity= 0.69 Similarity= 0.57 Similarity = Tanimoto similarity 1.0

31 Results: Synthetic accessibility score* of the designed libraries *Ertl, Peter, and Ansgar Schuffenhauer. "Estimation of synthetic accessibility score of drug-like molecules based on molecular complexity and fragment contributions." Journal of cheminformatics 1.1 (2009): 8.


32 Target predictions for generated compounds using SEA* *Keiser MJ, Roth BL, Armbruster BN, Ernsberger P, Irwin JJ, Shoichet BK. Relating protein pharmacology by ligand chemistry. Nat Biotech 25 (2), (2007).


33 Target predictions for generated compounds using SEA* *Keiser MJ, Roth BL, Armbruster BN, Ernsberger P, Irwin JJ, Shoichet BK. Relating protein pharmacology by ligand chemistry. Nat Biotech 25 (2), (2007).


34 Model visualization for JAK2 (projection using t-sne) ZINC pic50 = 8.64 ZINC pic50 = 3.31 ZINC pic50 = 8.23 ZINC pic50 = 3.76 ZINC pic50 = 8.39 pic50 = pic50 = M. Popova, O. Isayev, A. Tropsha. "Deep reinforcement learning for de-novo drug design." arxiv preprint arxiv: (2017).

35 Examples of Stack-RNN cells with interpretable gate activations M. Popova, O. Isayev, A. Tropsha. "Deep reinforcement learning for de-novo drug design." arxiv preprint arxiv: (2017).
36 Summary AI methods coupled with SMILES representation afford biased libraries generation The system naturally embeds reinforcement to produce novel structure with the desired property The system can be tuned to bias libraries towards specific property ranges Next phase is experimental validation of hits by UNC SGC team
Deep Reinforcement Learning for de-novo Drug Design
 Deep Reinforcement Learning for de-novo Drug Design Mariya Popova 1,2,3, Olexandr Isayev 1*, Alexander Tropsha 1* 1 Laboratory for Molecular Modeling, Division of Chemical Biology and Medicinal Chemistry,
Deep Reinforcement Learning for de-novo Drug Design Mariya Popova 1,2,3, Olexandr Isayev 1*, Alexander Tropsha 1* 1 Laboratory for Molecular Modeling, Division of Chemical Biology and Medicinal Chemistry,
In silico generation of novel, drug-like chemical matter using the LSTM deep neural network
 In silico generation of novel, drug-like chemical matter using the LSTM deep neural network Peter Ertl Novartis Institutes for BioMedical Research, Basel, CH September 2018 Neural networks in cheminformatics
In silico generation of novel, drug-like chemical matter using the LSTM deep neural network Peter Ertl Novartis Institutes for BioMedical Research, Basel, CH September 2018 Neural networks in cheminformatics
In silico generation of novel, drug-like chemical matter using the LSTM neural network
 In silico generation of novel, drug-like chemical matter using the LSTM neural network Peter Ertl 1, Richard Lewis 1, Eric Martin 2, Valery Polyakov 2 1 Novartis Institutes for BioMedical Research, Novartis
In silico generation of novel, drug-like chemical matter using the LSTM neural network Peter Ertl 1, Richard Lewis 1, Eric Martin 2, Valery Polyakov 2 1 Novartis Institutes for BioMedical Research, Novartis
Deep Learning: What Makes Neural Networks Great Again?
 6 th Strasbourg Summer School in Chemoinformatics (CS3-2018) Deep Learning: What Makes Neural Networks Great Again? Igor I. Baskin M. V. Lomonosov Moscow State University The First Neural Network (Perceptron)
6 th Strasbourg Summer School in Chemoinformatics (CS3-2018) Deep Learning: What Makes Neural Networks Great Again? Igor I. Baskin M. V. Lomonosov Moscow State University The First Neural Network (Perceptron)
Deep Generative Models for Graph Generation. Jian Tang HEC Montreal CIFAR AI Chair, Mila
 Deep Generative Models for Graph Generation Jian Tang HEC Montreal CIFAR AI Chair, Mila Email: jian.tang@hec.ca Deep Generative Models Goal: model data distribution p(x) explicitly or implicitly, where
Deep Generative Models for Graph Generation Jian Tang HEC Montreal CIFAR AI Chair, Mila Email: jian.tang@hec.ca Deep Generative Models Goal: model data distribution p(x) explicitly or implicitly, where
Virtual screening in drug discovery
 Virtual screening in drug discovery Pavel Polishchuk Institute of Molecular and Translational Medicine Palacky University pavlo.polishchuk@upol.cz Drug development workflow Vistoli G., et al., Drug Discovery
Virtual screening in drug discovery Pavel Polishchuk Institute of Molecular and Translational Medicine Palacky University pavlo.polishchuk@upol.cz Drug development workflow Vistoli G., et al., Drug Discovery
Introduction. OntoChem
 Introduction ntochem Providing drug discovery knowledge & small molecules... Supporting the task of medicinal chemistry Allows selecting best possible small molecule starting point From target to leads
Introduction ntochem Providing drug discovery knowledge & small molecules... Supporting the task of medicinal chemistry Allows selecting best possible small molecule starting point From target to leads
Development of a Structure Generator to Explore Target Areas on Chemical Space
 Development of a Structure Generator to Explore Target Areas on Chemical Space Kimito Funatsu Department of Chemical System Engineering, This materials will be published on Molecular Informatics Drug Development
Development of a Structure Generator to Explore Target Areas on Chemical Space Kimito Funatsu Department of Chemical System Engineering, This materials will be published on Molecular Informatics Drug Development
Machine learning for ligand-based virtual screening and chemogenomics!
 Machine learning for ligand-based virtual screening and chemogenomics! Jean-Philippe Vert Institut Curie - INSERM U900 - Mines ParisTech In silico discovery of molecular probes and drug-like compounds:
Machine learning for ligand-based virtual screening and chemogenomics! Jean-Philippe Vert Institut Curie - INSERM U900 - Mines ParisTech In silico discovery of molecular probes and drug-like compounds:
Molecular Dynamics Graphical Visualization 3-D QSAR Pharmacophore QSAR, COMBINE, Scoring Functions, Homology Modeling,..
 3 Conformational Search Molecular Docking Simulate Annealing Ab Initio QM Molecular Dynamics Graphical Visualization 3-D QSAR Pharmacophore QSAR, COMBINE, Scoring Functions, Homology Modeling,.. Rino Ragno:
3 Conformational Search Molecular Docking Simulate Annealing Ab Initio QM Molecular Dynamics Graphical Visualization 3-D QSAR Pharmacophore QSAR, COMBINE, Scoring Functions, Homology Modeling,.. Rino Ragno:
Structure-Activity Modeling - QSAR. Uwe Koch
 Structure-Activity Modeling - QSAR Uwe Koch QSAR Assumption: QSAR attempts to quantify the relationship between activity and molecular strcucture by correlating descriptors with properties Biological activity
Structure-Activity Modeling - QSAR Uwe Koch QSAR Assumption: QSAR attempts to quantify the relationship between activity and molecular strcucture by correlating descriptors with properties Biological activity
bcl::cheminfo Suite Enables Machine Learning-Based Drug Discovery Using GPUs Edward W. Lowe, Jr. Nils Woetzel May 17, 2012
 bcl::cheminfo Suite Enables Machine Learning-Based Drug Discovery Using GPUs Edward W. Lowe, Jr. Nils Woetzel May 17, 2012 Outline Machine Learning Cheminformatics Framework QSPR logp QSAR mglur 5 CYP
bcl::cheminfo Suite Enables Machine Learning-Based Drug Discovery Using GPUs Edward W. Lowe, Jr. Nils Woetzel May 17, 2012 Outline Machine Learning Cheminformatics Framework QSPR logp QSAR mglur 5 CYP
Computational chemical biology to address non-traditional drug targets. John Karanicolas
 Computational chemical biology to address non-traditional drug targets John Karanicolas Our computational toolbox Structure-based approaches Ligand-based approaches Detailed MD simulations 2D fingerprints
Computational chemical biology to address non-traditional drug targets John Karanicolas Our computational toolbox Structure-based approaches Ligand-based approaches Detailed MD simulations 2D fingerprints
Navigation in Chemical Space Towards Biological Activity. Peter Ertl Novartis Institutes for BioMedical Research Basel, Switzerland
 Navigation in Chemical Space Towards Biological Activity Peter Ertl Novartis Institutes for BioMedical Research Basel, Switzerland Data Explosion in Chemistry CAS 65 million molecules CCDC 600 000 structures
Navigation in Chemical Space Towards Biological Activity Peter Ertl Novartis Institutes for BioMedical Research Basel, Switzerland Data Explosion in Chemistry CAS 65 million molecules CCDC 600 000 structures
Multi objective de novo drug design with conditional graph generative model
 https://doi.org/10.1186/s13321-018-0287-6 RESEARCH ARTICLE Open Access Multi objective de novo drug design with conditional graph generative model Yibo Li, Liangren Zhang * and Zhenming Liu * Abstract
https://doi.org/10.1186/s13321-018-0287-6 RESEARCH ARTICLE Open Access Multi objective de novo drug design with conditional graph generative model Yibo Li, Liangren Zhang * and Zhenming Liu * Abstract
Introduction to Chemoinformatics and Drug Discovery
 Introduction to Chemoinformatics and Drug Discovery Irene Kouskoumvekaki Associate Professor February 15 th, 2013 The Chemical Space There are atoms and space. Everything else is opinion. Democritus (ca.
Introduction to Chemoinformatics and Drug Discovery Irene Kouskoumvekaki Associate Professor February 15 th, 2013 The Chemical Space There are atoms and space. Everything else is opinion. Democritus (ca.
PREDICTION OF HETERODIMERIC PROTEIN COMPLEXES FROM PROTEIN-PROTEIN INTERACTION NETWORKS USING DEEP LEARNING
 PREDICTION OF HETERODIMERIC PROTEIN COMPLEXES FROM PROTEIN-PROTEIN INTERACTION NETWORKS USING DEEP LEARNING Peiying (Colleen) Ruan, PhD, Deep Learning Solution Architect 3/26/2018 Background OUTLINE Method
PREDICTION OF HETERODIMERIC PROTEIN COMPLEXES FROM PROTEIN-PROTEIN INTERACTION NETWORKS USING DEEP LEARNING Peiying (Colleen) Ruan, PhD, Deep Learning Solution Architect 3/26/2018 Background OUTLINE Method
October 6 University Faculty of pharmacy Computer Aided Drug Design Unit
 October 6 University Faculty of pharmacy Computer Aided Drug Design Unit CADD@O6U.edu.eg CADD Computer-Aided Drug Design Unit The development of new drugs is no longer a process of trial and error or strokes
October 6 University Faculty of pharmacy Computer Aided Drug Design Unit CADD@O6U.edu.eg CADD Computer-Aided Drug Design Unit The development of new drugs is no longer a process of trial and error or strokes
Chemical Data Retrieval and Management
 Chemical Data Retrieval and Management ChEMBL, ChEBI, and the Chemistry Development Kit Stephan A. Beisken What is EMBL-EBI? Part of the European Molecular Biology Laboratory International, non-profit
Chemical Data Retrieval and Management ChEMBL, ChEBI, and the Chemistry Development Kit Stephan A. Beisken What is EMBL-EBI? Part of the European Molecular Biology Laboratory International, non-profit
Introducing a Bioinformatics Similarity Search Solution
 Introducing a Bioinformatics Similarity Search Solution 1 Page About the APU 3 The APU as a Driver of Similarity Search 3 Similarity Search in Bioinformatics 3 POC: GSI Joins Forces with the Weizmann Institute
Introducing a Bioinformatics Similarity Search Solution 1 Page About the APU 3 The APU as a Driver of Similarity Search 3 Similarity Search in Bioinformatics 3 POC: GSI Joins Forces with the Weizmann Institute
A Deep Convolutional Neural Network for Bioactivity Prediction in Structure-based Drug Discovery
 AtomNet A Deep Convolutional Neural Network for Bioactivity Prediction in Structure-based Drug Discovery Izhar Wallach, Michael Dzamba, Abraham Heifets Victor Storchan, Institute for Computational and
AtomNet A Deep Convolutional Neural Network for Bioactivity Prediction in Structure-based Drug Discovery Izhar Wallach, Michael Dzamba, Abraham Heifets Victor Storchan, Institute for Computational and
Xia Ning,*, Huzefa Rangwala, and George Karypis
 J. Chem. Inf. Model. XXXX, xxx, 000 A Multi-Assay-Based Structure-Activity Relationship Models: Improving Structure-Activity Relationship Models by Incorporating Activity Information from Related Targets
J. Chem. Inf. Model. XXXX, xxx, 000 A Multi-Assay-Based Structure-Activity Relationship Models: Improving Structure-Activity Relationship Models by Incorporating Activity Information from Related Targets
QSAR Modeling of ErbB1 Inhibitors Using Genetic Algorithm-Based Regression
 APPLICATION NOTE QSAR Modeling of ErbB1 Inhibitors Using Genetic Algorithm-Based Regression GAINING EFFICIENCY IN QUANTITATIVE STRUCTURE ACTIVITY RELATIONSHIPS ErbB1 kinase is the cell-surface receptor
APPLICATION NOTE QSAR Modeling of ErbB1 Inhibitors Using Genetic Algorithm-Based Regression GAINING EFFICIENCY IN QUANTITATIVE STRUCTURE ACTIVITY RELATIONSHIPS ErbB1 kinase is the cell-surface receptor
Machine Learning Concepts in Chemoinformatics
 Machine Learning Concepts in Chemoinformatics Martin Vogt B-IT Life Science Informatics Rheinische Friedrich-Wilhelms-Universität Bonn BigChem Winter School 2017 25. October Data Mining in Chemoinformatics
Machine Learning Concepts in Chemoinformatics Martin Vogt B-IT Life Science Informatics Rheinische Friedrich-Wilhelms-Universität Bonn BigChem Winter School 2017 25. October Data Mining in Chemoinformatics
arxiv: v1 [physics.chem-ph] 6 Apr 2018
![arxiv: v1 [physics.chem-ph] 6 Apr 2018](/thumbs/86/94765438.jpg "arxiv: v1 [physics.chem-ph] 6 Apr 2018") Population-based de novo molecule generation, using grammatical evolution arxiv:1804.02134v1 [physics.chem-ph] 6 Apr 2018 Naruki Yoshikawa a, Kei Terayama a, Teruki Honma b, Kenta Oono c, Koji Tsuda a,d,e,
Population-based de novo molecule generation, using grammatical evolution arxiv:1804.02134v1 [physics.chem-ph] 6 Apr 2018 Naruki Yoshikawa a, Kei Terayama a, Teruki Honma b, Kenta Oono c, Koji Tsuda a,d,e,
Hit Finding and Optimization Using BLAZE & FORGE
 Hit Finding and Optimization Using BLAZE & FORGE Kevin Cusack,* Maria Argiriadi, Eric Breinlinger, Jeremy Edmunds, Michael Hoemann, Michael Friedman, Sami Osman, Raymond Huntley, Thomas Vargo AbbVie, Immunology
Hit Finding and Optimization Using BLAZE & FORGE Kevin Cusack,* Maria Argiriadi, Eric Breinlinger, Jeremy Edmunds, Michael Hoemann, Michael Friedman, Sami Osman, Raymond Huntley, Thomas Vargo AbbVie, Immunology
Development and application of ligand-based computational methods for de-novo drug. design and virtual screening. Alexander Richard Geanes.
 Development and application of ligand-based computational methods for de-novo drug design and virtual screening By Alexander Richard Geanes Thesis Submitted to the Faculty of the Graduate School of Vanderbilt
Development and application of ligand-based computational methods for de-novo drug design and virtual screening By Alexander Richard Geanes Thesis Submitted to the Faculty of the Graduate School of Vanderbilt
Progress of Compound Library Design Using In-silico Approach for Collaborative Drug Discovery
 21 th /June/2018@CUGM Progress of Compound Library Design Using In-silico Approach for Collaborative Drug Discovery Kaz Ikeda, Ph.D. Keio University Self Introduction Keio University, Tokyo, Japan (Established
21 th /June/2018@CUGM Progress of Compound Library Design Using In-silico Approach for Collaborative Drug Discovery Kaz Ikeda, Ph.D. Keio University Self Introduction Keio University, Tokyo, Japan (Established
MSc Drug Design. Module Structure: (15 credits each) Lectures and Tutorials Assessment: 50% coursework, 50% unseen examination.
 Lectures and Tutorials Assessment: 50% coursework, 50% unseen examination.") Module Structure: (15 credits each) Lectures and Assessment: 50% coursework, 50% unseen examination. Module Title Module 1: Bioinformatics and structural biology as applied to drug design MEDC0075 In the
Module Structure: (15 credits each) Lectures and Assessment: 50% coursework, 50% unseen examination. Module Title Module 1: Bioinformatics and structural biology as applied to drug design MEDC0075 In the
Capturing Chemistry. What you see is what you get In the world of mechanism and chemical transformations
 Capturing Chemistry What you see is what you get In the world of mechanism and chemical transformations Dr. Stephan Schürer ead of Intl. Sci. Content Libraria, Inc. sschurer@libraria.com Distribution of
Capturing Chemistry What you see is what you get In the world of mechanism and chemical transformations Dr. Stephan Schürer ead of Intl. Sci. Content Libraria, Inc. sschurer@libraria.com Distribution of
An Integrated Approach to in-silico
 An Integrated Approach to in-silico Screening Joseph L. Durant Jr., Douglas. R. Henry, Maurizio Bronzetti, and David. A. Evans MDL Information Systems, Inc. 14600 Catalina St., San Leandro, CA 94577 Goals
An Integrated Approach to in-silico Screening Joseph L. Durant Jr., Douglas. R. Henry, Maurizio Bronzetti, and David. A. Evans MDL Information Systems, Inc. 14600 Catalina St., San Leandro, CA 94577 Goals
Chemical Space. Space, Diversity, and Synthesis. Jeremy Henle, 4/23/2013
 Chemical Space Space, Diversity, and Synthesis Jeremy Henle, 4/23/2013 Computational Modeling Chemical Space As a diversity construct Outline Quantifying Diversity Diversity Oriented Synthesis Wolf and
Chemical Space Space, Diversity, and Synthesis Jeremy Henle, 4/23/2013 Computational Modeling Chemical Space As a diversity construct Outline Quantifying Diversity Diversity Oriented Synthesis Wolf and
Raed Saeed Khashan. Chapel Hill Approved by: Dr. Alexander Tropsha. Dr. Weifan Zheng. Dr. Alexander Golbraikh. Dr. Wei Wang. Dr.
 DEVELOPMENT AND APPLICATION OF LIGAND-BASED AND STRUCTURE- BASED COMPUTATIONAL DRUG DISCOVERY TOOLS BASED ON FREQUENT SUBGRAPH MINING OF CHEMICAL STRUCTURES Raed Saeed Khashan A dissertation submitted
DEVELOPMENT AND APPLICATION OF LIGAND-BASED AND STRUCTURE- BASED COMPUTATIONAL DRUG DISCOVERY TOOLS BASED ON FREQUENT SUBGRAPH MINING OF CHEMICAL STRUCTURES Raed Saeed Khashan A dissertation submitted
The use of Design of Experiments to develop Efficient Arrays for SAR and Property Exploration
 The use of Design of Experiments to develop Efficient Arrays for SAR and Property Exploration Chris Luscombe, Computational Chemistry GlaxoSmithKline Summary of Talk Traditional approaches SAR Free-Wilson
The use of Design of Experiments to develop Efficient Arrays for SAR and Property Exploration Chris Luscombe, Computational Chemistry GlaxoSmithKline Summary of Talk Traditional approaches SAR Free-Wilson
CHEMINFORMATICS MODELING OF DIVERSE AND DISPARATE BIOLOGICAL DATA AND THE USE OF MODELS TO DISCOVER NOVEL BIOACTIVE MOLECULES
 CHEMINFORMATICS MODELING OF DIVERSE AND DISPARATE BIOLOGICAL DATA AND THE USE OF MODELS TO DISCOVER NOVEL BIOACTIVE MOLECULES Man Luo A dissertation submitted to the faculty of the University of North
CHEMINFORMATICS MODELING OF DIVERSE AND DISPARATE BIOLOGICAL DATA AND THE USE OF MODELS TO DISCOVER NOVEL BIOACTIVE MOLECULES Man Luo A dissertation submitted to the faculty of the University of North
Virtual Libraries and Virtual Screening in Drug Discovery Processes using KNIME
 Virtual Libraries and Virtual Screening in Drug Discovery Processes using KNIME Iván Solt Solutions for Cheminformatics Drug Discovery Strategies for known targets High-Throughput Screening (HTS) Cells
Virtual Libraries and Virtual Screening in Drug Discovery Processes using KNIME Iván Solt Solutions for Cheminformatics Drug Discovery Strategies for known targets High-Throughput Screening (HTS) Cells
Building innovative drug discovery alliances. Just in KNIME: Successful Process Driven Drug Discovery
 Building innovative drug discovery alliances Just in KIME: Successful Process Driven Drug Discovery Berlin KIME Spring Summit, Feb 2016 Research Informatics @ Evotec Evotec s worldwide operations 2 Pharmaceuticals
Building innovative drug discovery alliances Just in KIME: Successful Process Driven Drug Discovery Berlin KIME Spring Summit, Feb 2016 Research Informatics @ Evotec Evotec s worldwide operations 2 Pharmaceuticals
Cheminformatics analysis and learning in a data pipelining environment
 Molecular Diversity (2006) 10: 283 299 DOI: 10.1007/s11030-006-9041-5 c Springer 2006 Review Cheminformatics analysis and learning in a data pipelining environment Moises Hassan 1,, Robert D. Brown 1,
Molecular Diversity (2006) 10: 283 299 DOI: 10.1007/s11030-006-9041-5 c Springer 2006 Review Cheminformatics analysis and learning in a data pipelining environment Moises Hassan 1,, Robert D. Brown 1,
Few-shot learning with KRR
 Few-shot learning with KRR Prudencio Tossou Groupe de Recherche en Apprentissage Automatique Départment d informatique et de génie logiciel Université Laval April 6, 2018 Prudencio Tossou (UL) Few-shot
Few-shot learning with KRR Prudencio Tossou Groupe de Recherche en Apprentissage Automatique Départment d informatique et de génie logiciel Université Laval April 6, 2018 Prudencio Tossou (UL) Few-shot
How IJC is Adding Value to a Molecular Design Business
 How IJC is Adding Value to a Molecular Design Business James Mills Sexis LLP ChemAxon TechTalk Stevenage, ov 2012 james.mills@sexis.co.uk Overview Introduction to Sexis Sexis IJC use cases Data visualisation
How IJC is Adding Value to a Molecular Design Business James Mills Sexis LLP ChemAxon TechTalk Stevenage, ov 2012 james.mills@sexis.co.uk Overview Introduction to Sexis Sexis IJC use cases Data visualisation
Integrated Cheminformatics to Guide Drug Discovery
 Integrated Cheminformatics to Guide Drug Discovery Matthew Segall, Ed Champness, Peter Hunt, Tamsin Mansley CINF Drug Discovery Cheminformatics Approaches August 23 rd 2017 Optibrium, StarDrop, Auto-Modeller,
Integrated Cheminformatics to Guide Drug Discovery Matthew Segall, Ed Champness, Peter Hunt, Tamsin Mansley CINF Drug Discovery Cheminformatics Approaches August 23 rd 2017 Optibrium, StarDrop, Auto-Modeller,
CSC321 Lecture 20: Reversible and Autoregressive Models
 CSC321 Lecture 20: Reversible and Autoregressive Models Roger Grosse Roger Grosse CSC321 Lecture 20: Reversible and Autoregressive Models 1 / 23 Overview Four modern approaches to generative modeling:
CSC321 Lecture 20: Reversible and Autoregressive Models Roger Grosse Roger Grosse CSC321 Lecture 20: Reversible and Autoregressive Models 1 / 23 Overview Four modern approaches to generative modeling:
Using AutoDock for Virtual Screening
 Using AutoDock for Virtual Screening CUHK Croucher ASI Workshop 2011 Stefano Forli, PhD Prof. Arthur J. Olson, Ph.D Molecular Graphics Lab Screening and Virtual Screening The ultimate tool for identifying
Using AutoDock for Virtual Screening CUHK Croucher ASI Workshop 2011 Stefano Forli, PhD Prof. Arthur J. Olson, Ph.D Molecular Graphics Lab Screening and Virtual Screening The ultimate tool for identifying
Chemoinformatics and information management. Peter Willett, University of Sheffield, UK
 Chemoinformatics and information management Peter Willett, University of Sheffield, UK verview What is chemoinformatics and why is it necessary Managing structural information Typical facilities in chemoinformatics
Chemoinformatics and information management Peter Willett, University of Sheffield, UK verview What is chemoinformatics and why is it necessary Managing structural information Typical facilities in chemoinformatics
A Study of Complex Deep Learning Networks on High Performance, Neuromorphic, and Quantum Computers
 A Study of Complex Deep Learning Networks on High Performance, Neuromorphic, and Quantum Computers Thomas E. Potok, Ph.D. Computational Data Analytics Group Oak Ridge National Laboratory ORNL is managed
A Study of Complex Deep Learning Networks on High Performance, Neuromorphic, and Quantum Computers Thomas E. Potok, Ph.D. Computational Data Analytics Group Oak Ridge National Laboratory ORNL is managed
Cross Discipline Analysis made possible with Data Pipelining. J.R. Tozer SciTegic
 Cross Discipline Analysis made possible with Data Pipelining J.R. Tozer SciTegic System Genesis Pipelining tool created to automate data processing in cheminformatics Modular system built with generic
Cross Discipline Analysis made possible with Data Pipelining J.R. Tozer SciTegic System Genesis Pipelining tool created to automate data processing in cheminformatics Modular system built with generic
Cheminformatics Role in Pharmaceutical Industry. Randal Chen Ph.D. Abbott Laboratories Aug. 23, 2004 ACS
 Cheminformatics Role in Pharmaceutical Industry Randal Chen Ph.D. Abbott Laboratories Aug. 23, 2004 ACS Agenda The big picture for pharmaceutical industry Current technological/scientific issues Types
Cheminformatics Role in Pharmaceutical Industry Randal Chen Ph.D. Abbott Laboratories Aug. 23, 2004 ACS Agenda The big picture for pharmaceutical industry Current technological/scientific issues Types
Similarity methods for ligandbased virtual screening
 Similarity methods for ligandbased virtual screening Peter Willett, University of Sheffield Computers in Scientific Discovery 5, 22 nd July 2010 Overview Molecular similarity and its use in virtual screening
Similarity methods for ligandbased virtual screening Peter Willett, University of Sheffield Computers in Scientific Discovery 5, 22 nd July 2010 Overview Molecular similarity and its use in virtual screening
Chemogenomic: Approaches to Rational Drug Design. Jonas Skjødt Møller
 Chemogenomic: Approaches to Rational Drug Design Jonas Skjødt Møller Chemogenomic Chemistry Biology Chemical biology Medical chemistry Chemical genetics Chemoinformatics Bioinformatics Chemoproteomics
Chemogenomic: Approaches to Rational Drug Design Jonas Skjødt Møller Chemogenomic Chemistry Biology Chemical biology Medical chemistry Chemical genetics Chemoinformatics Bioinformatics Chemoproteomics
Pose and affinity prediction by ICM in D3R GC3. Max Totrov Molsoft
 Pose and affinity prediction by ICM in D3R GC3 Max Totrov Molsoft Pose prediction method: ICM-dock ICM-dock: - pre-sampling of ligand conformers - multiple trajectory Monte-Carlo with gradient minimization
Pose and affinity prediction by ICM in D3R GC3 Max Totrov Molsoft Pose prediction method: ICM-dock ICM-dock: - pre-sampling of ligand conformers - multiple trajectory Monte-Carlo with gradient minimization
Supporting Information
 Supporting Information Convolutional Embedding of Attributed Molecular Graphs for Physical Property Prediction Connor W. Coley a, Regina Barzilay b, William H. Green a, Tommi S. Jaakkola b, Klavs F. Jensen
Supporting Information Convolutional Embedding of Attributed Molecular Graphs for Physical Property Prediction Connor W. Coley a, Regina Barzilay b, William H. Green a, Tommi S. Jaakkola b, Klavs F. Jensen
Contents 1 Open-Source Tools, Techniques, and Data in Chemoinformatics
 Contents 1 Open-Source Tools, Techniques, and Data in Chemoinformatics... 1 1.1 Chemoinformatics... 2 1.1.1 Open-Source Tools... 2 1.1.2 Introduction to Programming Languages... 3 1.2 Chemical Structure
Contents 1 Open-Source Tools, Techniques, and Data in Chemoinformatics... 1 1.1 Chemoinformatics... 2 1.1.1 Open-Source Tools... 2 1.1.2 Introduction to Programming Languages... 3 1.2 Chemical Structure
Deep Learning Srihari. Deep Belief Nets. Sargur N. Srihari
 Deep Belief Nets Sargur N. Srihari srihari@cedar.buffalo.edu Topics 1. Boltzmann machines 2. Restricted Boltzmann machines 3. Deep Belief Networks 4. Deep Boltzmann machines 5. Boltzmann machines for continuous
Deep Belief Nets Sargur N. Srihari srihari@cedar.buffalo.edu Topics 1. Boltzmann machines 2. Restricted Boltzmann machines 3. Deep Belief Networks 4. Deep Boltzmann machines 5. Boltzmann machines for continuous
Patent Searching using Bayesian Statistics
 Patent Searching using Bayesian Statistics Willem van Hoorn, Exscientia Ltd Biovia European Forum, London, June 2017 Contents Who are we? Searching molecules in patents What can Pipeline Pilot do for you?
Patent Searching using Bayesian Statistics Willem van Hoorn, Exscientia Ltd Biovia European Forum, London, June 2017 Contents Who are we? Searching molecules in patents What can Pipeline Pilot do for you?
Memory-Augmented Attention Model for Scene Text Recognition
 Memory-Augmented Attention Model for Scene Text Recognition Cong Wang 1,2, Fei Yin 1,2, Cheng-Lin Liu 1,2,3 1 National Laboratory of Pattern Recognition Institute of Automation, Chinese Academy of Sciences
Memory-Augmented Attention Model for Scene Text Recognition Cong Wang 1,2, Fei Yin 1,2, Cheng-Lin Liu 1,2,3 1 National Laboratory of Pattern Recognition Institute of Automation, Chinese Academy of Sciences
Studying the effect of noise on Laplacian-modified Bayesian Analysis and Tanimoto Similarity
 Studying the effect of noise on Laplacian-modified Bayesian nalysis and Tanimoto Similarity David Rogers, Ph.D. SciTegic, Inc. (Division of ccelrys, Inc.) drogers@scitegic.com Description of: nalysis methods
Studying the effect of noise on Laplacian-modified Bayesian nalysis and Tanimoto Similarity David Rogers, Ph.D. SciTegic, Inc. (Division of ccelrys, Inc.) drogers@scitegic.com Description of: nalysis methods
AMRI COMPOUND LIBRARY CONSORTIUM: A NOVEL WAY TO FILL YOUR DRUG PIPELINE
 AMRI COMPOUD LIBRARY COSORTIUM: A OVEL WAY TO FILL YOUR DRUG PIPELIE Muralikrishna Valluri, PhD & Douglas B. Kitchen, PhD Summary The creation of high-quality, innovative small molecule leads is a continual
AMRI COMPOUD LIBRARY COSORTIUM: A OVEL WAY TO FILL YOUR DRUG PIPELIE Muralikrishna Valluri, PhD & Douglas B. Kitchen, PhD Summary The creation of high-quality, innovative small molecule leads is a continual
A Tiered Screen Protocol for the Discovery of Structurally Diverse HIV Integrase Inhibitors
 A Tiered Screen Protocol for the Discovery of Structurally Diverse HIV Integrase Inhibitors Rajarshi Guha, Debojyoti Dutta, Ting Chen and David J. Wild School of Informatics Indiana University and Dept.
A Tiered Screen Protocol for the Discovery of Structurally Diverse HIV Integrase Inhibitors Rajarshi Guha, Debojyoti Dutta, Ting Chen and David J. Wild School of Informatics Indiana University and Dept.
Ignasi Belda, PhD CEO. HPC Advisory Council Spain Conference 2015
 Ignasi Belda, PhD CEO HPC Advisory Council Spain Conference 2015 Business lines Molecular Modeling Services We carry out computational chemistry projects using our selfdeveloped and third party technologies
Ignasi Belda, PhD CEO HPC Advisory Council Spain Conference 2015 Business lines Molecular Modeling Services We carry out computational chemistry projects using our selfdeveloped and third party technologies
The Molecule Cloud - compact visualization of large collections of molecules
 Ertl and Rohde Journal of Cheminformatics 2012, 4:12 METHODOLOGY Open Access The Molecule Cloud - compact visualization of large collections of molecules Peter Ertl * and Bernhard Rohde Abstract Background:
Ertl and Rohde Journal of Cheminformatics 2012, 4:12 METHODOLOGY Open Access The Molecule Cloud - compact visualization of large collections of molecules Peter Ertl * and Bernhard Rohde Abstract Background:
Deep Learning for Gravitational Wave Analysis Results with LIGO Data
 Link to these slides: http://tiny.cc/nips arxiv:1711.03121 Deep Learning for Gravitational Wave Analysis Results with LIGO Data Daniel George & E. A. Huerta NCSA Gravity Group - http://gravity.ncsa.illinois.edu/
Link to these slides: http://tiny.cc/nips arxiv:1711.03121 Deep Learning for Gravitational Wave Analysis Results with LIGO Data Daniel George & E. A. Huerta NCSA Gravity Group - http://gravity.ncsa.illinois.edu/
Artificial Intelligence Methods for Molecular Property Prediction. Evan N. Feinberg Pande Lab
 Artificial Intelligence Methods for Molecular Property Prediction Evan N. Feinberg Pande Lab AI Drug Discovery trends.google.com https://g.co/trends/3y4uf MNIST Performance over Time https://srconstantin.wordpress.com/2017/01/28/performance-trends-in-ai/
Artificial Intelligence Methods for Molecular Property Prediction Evan N. Feinberg Pande Lab AI Drug Discovery trends.google.com https://g.co/trends/3y4uf MNIST Performance over Time https://srconstantin.wordpress.com/2017/01/28/performance-trends-in-ai/
Structure based drug design and LIE models for GPCRs
 Structure based drug design and LIE models for GPCRs Peter Kolb kolb@docking.org Shoichet Lab ACS 237 th National Meeting, March 24, 2009 p.1/26 [Acknowledgements] Brian Shoichet John Irwin Mike Keiser
Structure based drug design and LIE models for GPCRs Peter Kolb kolb@docking.org Shoichet Lab ACS 237 th National Meeting, March 24, 2009 p.1/26 [Acknowledgements] Brian Shoichet John Irwin Mike Keiser
Deep learning / Ian Goodfellow, Yoshua Bengio and Aaron Courville. - Cambridge, MA ; London, Spis treści
 Deep learning / Ian Goodfellow, Yoshua Bengio and Aaron Courville. - Cambridge, MA ; London, 2017 Spis treści Website Acknowledgments Notation xiii xv xix 1 Introduction 1 1.1 Who Should Read This Book?
Deep learning / Ian Goodfellow, Yoshua Bengio and Aaron Courville. - Cambridge, MA ; London, 2017 Spis treści Website Acknowledgments Notation xiii xv xix 1 Introduction 1 1.1 Who Should Read This Book?
DATA FUSION APPROACHES IN LIGAND-BASED VIRTUAL SCREENING: RECENT DEVELOPMENTS OVERVIEW
 DATA FUSION APPROACHES IN LIGAND-BASED VIRTUAL SCREENING: RECENT DEVELOPMENTS OVERVIEW Mubarak Himmat 1, Naomie Salim 1, Ali Ahmed 1, 2, and Mohammed Mumtaz Al-Dabbagh 1 1 Faculty of Computing, University
DATA FUSION APPROACHES IN LIGAND-BASED VIRTUAL SCREENING: RECENT DEVELOPMENTS OVERVIEW Mubarak Himmat 1, Naomie Salim 1, Ali Ahmed 1, 2, and Mohammed Mumtaz Al-Dabbagh 1 1 Faculty of Computing, University
Similarity Search. Uwe Koch
 Similarity Search Uwe Koch Similarity Search The similar property principle: strurally similar molecules tend to have similar properties. However, structure property discontinuities occur frequently. Relevance
Similarity Search Uwe Koch Similarity Search The similar property principle: strurally similar molecules tend to have similar properties. However, structure property discontinuities occur frequently. Relevance
Increasing the Coverage of Medicinal Chemistry-Relevant Space in Commercial Fragments Screening
 pubs.acs.org/jcim Increasing the Coverage of Medicinal Chemistry-Relevant Space in Commercial Fragments Screening N. Yi Mok,, Ruth Brenk,*,,# and Nathan Brown*, Cancer Research UK Cancer Therapeutics Unit,
pubs.acs.org/jcim Increasing the Coverage of Medicinal Chemistry-Relevant Space in Commercial Fragments Screening N. Yi Mok,, Ruth Brenk,*,,# and Nathan Brown*, Cancer Research UK Cancer Therapeutics Unit,
A reliable computational workflow for the selection of optimal screening libraries
 DOI 10.1186/s13321-015-0108-0 RESEARCH ARTICLE Open Access A reliable computational workflow for the selection of optimal screening libraries Yocheved Gilad 1, Katalin Nadassy 2 and Hanoch Senderowitz
DOI 10.1186/s13321-015-0108-0 RESEARCH ARTICLE Open Access A reliable computational workflow for the selection of optimal screening libraries Yocheved Gilad 1, Katalin Nadassy 2 and Hanoch Senderowitz
Juergen Gall. Analyzing Human Behavior in Video Sequences
 Juergen Gall Analyzing Human Behavior in Video Sequences 09. 10. 2 01 7 Juer gen Gall Instit u t e of Com puter Science III Com puter Vision Gr oup 2 Analyzing Human Behavior Analyzing Human Behavior Human
Juergen Gall Analyzing Human Behavior in Video Sequences 09. 10. 2 01 7 Juer gen Gall Instit u t e of Com puter Science III Com puter Vision Gr oup 2 Analyzing Human Behavior Analyzing Human Behavior Human
Introduction to Chemoinformatics
 Introduction to Chemoinformatics Dr. Igor V. Tetko Helmholtz Zentrum München - German Research Center for Environmental Health (GmbH) Institute of Bioinformatics & Systems Biology (HMGU) Kyiv, 10 August
Introduction to Chemoinformatics Dr. Igor V. Tetko Helmholtz Zentrum München - German Research Center for Environmental Health (GmbH) Institute of Bioinformatics & Systems Biology (HMGU) Kyiv, 10 August
Deep Learning Autoencoder Models
 Deep Learning Autoencoder Models Davide Bacciu Dipartimento di Informatica Università di Pisa Intelligent Systems for Pattern Recognition (ISPR) Generative Models Wrap-up Deep Learning Module Lecture Generative
Deep Learning Autoencoder Models Davide Bacciu Dipartimento di Informatica Università di Pisa Intelligent Systems for Pattern Recognition (ISPR) Generative Models Wrap-up Deep Learning Module Lecture Generative
Targeting protein-protein interactions: A hot topic in drug discovery
 Michal Kamenicky; Maria Bräuer; Katrin Volk; Kamil Ödner; Christian Klein; Norbert Müller Targeting protein-protein interactions: A hot topic in drug discovery 104 Biomedizin Innovativ patientinnenfokussierte,
Michal Kamenicky; Maria Bräuer; Katrin Volk; Kamil Ödner; Christian Klein; Norbert Müller Targeting protein-protein interactions: A hot topic in drug discovery 104 Biomedizin Innovativ patientinnenfokussierte,
Topology based deep learning for biomolecular data
 Topology based deep learning for biomolecular data Guowei Wei Departments of Mathematics Michigan State University http://www.math.msu.edu/~wei American Institute of Mathematics July 23-28, 2017 Grant
Topology based deep learning for biomolecular data Guowei Wei Departments of Mathematics Michigan State University http://www.math.msu.edu/~wei American Institute of Mathematics July 23-28, 2017 Grant
Prototype-Based Compound Discovery using. Deep Generative Models
 Prototype-Based Compound Discovery using Deep Generative Models Shahar Harel and Kira Radinsky Department of Computer Science, Technion - Israel Institute of Technology E-mail: sshahar@cs.technion.ac.il;
Prototype-Based Compound Discovery using Deep Generative Models Shahar Harel and Kira Radinsky Department of Computer Science, Technion - Israel Institute of Technology E-mail: sshahar@cs.technion.ac.il;
est Drive K20 GPUs! Experience The Acceleration Run Computational Chemistry Codes on Tesla K20 GPU today
 est Drive K20 GPUs! Experience The Acceleration Run Computational Chemistry Codes on Tesla K20 GPU today Sign up for FREE GPU Test Drive on remotely hosted clusters www.nvidia.com/gputestd rive Shape Searching
est Drive K20 GPUs! Experience The Acceleration Run Computational Chemistry Codes on Tesla K20 GPU today Sign up for FREE GPU Test Drive on remotely hosted clusters www.nvidia.com/gputestd rive Shape Searching
Structural biology and drug design: An overview
 Structural biology and drug design: An overview livier Taboureau Assitant professor Chemoinformatics group-cbs-dtu otab@cbs.dtu.dk Drug discovery Drug and drug design A drug is a key molecule involved
Structural biology and drug design: An overview livier Taboureau Assitant professor Chemoinformatics group-cbs-dtu otab@cbs.dtu.dk Drug discovery Drug and drug design A drug is a key molecule involved
JCICS Major Research Areas
 JCICS Major Research Areas Chemical Information Text Searching Structure and Substructure Searching Databases Patents George W.A. Milne C571 Lecture Fall 2002 1 JCICS Major Research Areas Chemical Computation
JCICS Major Research Areas Chemical Information Text Searching Structure and Substructure Searching Databases Patents George W.A. Milne C571 Lecture Fall 2002 1 JCICS Major Research Areas Chemical Computation
Docking. GBCB 5874: Problem Solving in GBCB
 Docking Benzamidine Docking to Trypsin Relationship to Drug Design Ligand-based design QSAR Pharmacophore modeling Can be done without 3-D structure of protein Receptor/Structure-based design Molecular
Docking Benzamidine Docking to Trypsin Relationship to Drug Design Ligand-based design QSAR Pharmacophore modeling Can be done without 3-D structure of protein Receptor/Structure-based design Molecular
Next Generation Computational Chemistry Tools to Predict Toxicity of CWAs
 Next Generation Computational Chemistry Tools to Predict Toxicity of CWAs William (Bill) Welsh welshwj@umdnj.edu Prospective Funding by DTRA/JSTO-CBD CBIS Conference 1 A State-wide, Regional and National
Next Generation Computational Chemistry Tools to Predict Toxicity of CWAs William (Bill) Welsh welshwj@umdnj.edu Prospective Funding by DTRA/JSTO-CBD CBIS Conference 1 A State-wide, Regional and National
LIBRARY DESIGN FOR COLLABORATIVE DRUG DISCOVERY: EXPANDING DRUGGABLE CHEMOGENOMIC SPACE
 5 th /June/2018@British Embassy in Tokyo LIBRARY DESIGN FOR COLLABORATIVE DRUG DISCOVERY: EXPANDING DRUGGABLE CHEMOGENOMIC SPACE Kazuyoshi Ikeda, Ph.D. Keio University SELF-INTRODUCTION Keio University,
5 th /June/2018@British Embassy in Tokyo LIBRARY DESIGN FOR COLLABORATIVE DRUG DISCOVERY: EXPANDING DRUGGABLE CHEMOGENOMIC SPACE Kazuyoshi Ikeda, Ph.D. Keio University SELF-INTRODUCTION Keio University,
PROVIDING CHEMINFORMATICS SOLUTIONS TO SUPPORT DRUG DISCOVERY DECISIONS
 179 Molecular Informatics: Confronting Complexity, May 13 th - 16 th 2002, Bozen, Italy PROVIDING CHEMINFORMATICS SOLUTIONS TO SUPPORT DRUG DISCOVERY DECISIONS CARLETON R. SAGE, KEVIN R. HOLME, NIANISH
179 Molecular Informatics: Confronting Complexity, May 13 th - 16 th 2002, Bozen, Italy PROVIDING CHEMINFORMATICS SOLUTIONS TO SUPPORT DRUG DISCOVERY DECISIONS CARLETON R. SAGE, KEVIN R. HOLME, NIANISH
Introduction to Machine Learning Midterm Exam
 10-701 Introduction to Machine Learning Midterm Exam Instructors: Eric Xing, Ziv Bar-Joseph 17 November, 2015 There are 11 questions, for a total of 100 points. This exam is open book, open notes, but
10-701 Introduction to Machine Learning Midterm Exam Instructors: Eric Xing, Ziv Bar-Joseph 17 November, 2015 There are 11 questions, for a total of 100 points. This exam is open book, open notes, but
Practical QSAR and Library Design: Advanced tools for research teams
 DS QSAR and Library Design Webinar Practical QSAR and Library Design: Advanced tools for research teams Reservationless-Plus Dial-In Number (US): (866) 519-8942 Reservationless-Plus International Dial-In
DS QSAR and Library Design Webinar Practical QSAR and Library Design: Advanced tools for research teams Reservationless-Plus Dial-In Number (US): (866) 519-8942 Reservationless-Plus International Dial-In
Translating Methods from Pharma to Flavours & Fragrances
 Translating Methods from Pharma to Flavours & Fragrances CINF 27: ACS National Meeting, New Orleans, LA - 18 th March 2018 Peter Hunt, Edmund Champness, Nicholas Foster, Tamsin Mansley & Matthew Segall
Translating Methods from Pharma to Flavours & Fragrances CINF 27: ACS National Meeting, New Orleans, LA - 18 th March 2018 Peter Hunt, Edmund Champness, Nicholas Foster, Tamsin Mansley & Matthew Segall
DivCalc: A Utility for Diversity Analysis and Compound Sampling
 Molecules 2002, 7, 657-661 molecules ISSN 1420-3049 http://www.mdpi.org DivCalc: A Utility for Diversity Analysis and Compound Sampling Rajeev Gangal* SciNova Informatics, 161 Madhumanjiri Apartments,
Molecules 2002, 7, 657-661 molecules ISSN 1420-3049 http://www.mdpi.org DivCalc: A Utility for Diversity Analysis and Compound Sampling Rajeev Gangal* SciNova Informatics, 161 Madhumanjiri Apartments,
Exploring the chemical space of screening results
 Exploring the chemical space of screening results Edmund Champness, Matthew Segall, Chris Leeding, James Chisholm, Iskander Yusof, Nick Foster, Hector Martinez ACS Spring 2013, 7 th April 2013 Optibrium,
Exploring the chemical space of screening results Edmund Champness, Matthew Segall, Chris Leeding, James Chisholm, Iskander Yusof, Nick Foster, Hector Martinez ACS Spring 2013, 7 th April 2013 Optibrium,
Design and Synthesis of the Comprehensive Fragment Library
 YOUR INNOVATIVE CHEMISTRY PARTNER IN DRUG DISCOVERY Design and Synthesis of the Comprehensive Fragment Library A 3D Enabled Library for Medicinal Chemistry Discovery Warren S Wade 1, Kuei-Lin Chang 1,
YOUR INNOVATIVE CHEMISTRY PARTNER IN DRUG DISCOVERY Design and Synthesis of the Comprehensive Fragment Library A 3D Enabled Library for Medicinal Chemistry Discovery Warren S Wade 1, Kuei-Lin Chang 1,
What I Did Last Summer
 What I Did Last Summer LINGOs, GPUs, and Monitoring Vertex Imran Haque Department of Computer Science Pande Lab, Stanford University http://cs.stanford.edu/people/ihaque http://folding.stanford.edu ihaque@cs.stanford.edu
What I Did Last Summer LINGOs, GPUs, and Monitoring Vertex Imran Haque Department of Computer Science Pande Lab, Stanford University http://cs.stanford.edu/people/ihaque http://folding.stanford.edu ihaque@cs.stanford.edu
Deep learning on 3D geometries. Hope Yao Design Informatics Lab Department of Mechanical and Aerospace Engineering
 Deep learning on 3D geometries Hope Yao Design Informatics Lab Department of Mechanical and Aerospace Engineering Overview Background Methods Numerical Result Future improvements Conclusion Background
Deep learning on 3D geometries Hope Yao Design Informatics Lab Department of Mechanical and Aerospace Engineering Overview Background Methods Numerical Result Future improvements Conclusion Background
Tutorials on Library Design E. Lounkine and J. Bajorath (University of Bonn) C. Muller and A. Varnek (University of Strasbourg)
 C. Muller and A. Varnek (University of Strasbourg)") Tutorials on Library Design E. Lounkine and J. Bajorath (University of Bonn) C. Muller and A. Varnek (University of Strasbourg) The purpose of this tutorial is to generate a library of potential inhibitors
Tutorials on Library Design E. Lounkine and J. Bajorath (University of Bonn) C. Muller and A. Varnek (University of Strasbourg) The purpose of this tutorial is to generate a library of potential inhibitors
Tasks ADAS. Self Driving. Non-machine Learning. Traditional MLP. Machine-Learning based method. Supervised CNN. Methods. Deep-Learning based
 UNDERSTANDING CNN ADAS Tasks Self Driving Localizati on Perception Planning/ Control Driver state Vehicle Diagnosis Smart factory Methods Traditional Deep-Learning based Non-machine Learning Machine-Learning
UNDERSTANDING CNN ADAS Tasks Self Driving Localizati on Perception Planning/ Control Driver state Vehicle Diagnosis Smart factory Methods Traditional Deep-Learning based Non-machine Learning Machine-Learning
Unsupervised Learning
 CS 3750 Advanced Machine Learning hkc6@pitt.edu Unsupervised Learning Data: Just data, no labels Goal: Learn some underlying hidden structure of the data P(, ) P( ) Principle Component Analysis (Dimensionality
CS 3750 Advanced Machine Learning hkc6@pitt.edu Unsupervised Learning Data: Just data, no labels Goal: Learn some underlying hidden structure of the data P(, ) P( ) Principle Component Analysis (Dimensionality
Artificial Neural Networks. Introduction to Computational Neuroscience Tambet Matiisen
 Artificial Neural Networks Introduction to Computational Neuroscience Tambet Matiisen 2.04.2018 Artificial neural network NB! Inspired by biology, not based on biology! Applications Automatic speech recognition
Artificial Neural Networks Introduction to Computational Neuroscience Tambet Matiisen 2.04.2018 Artificial neural network NB! Inspired by biology, not based on biology! Applications Automatic speech recognition
CHAPTER 6 QUANTITATIVE STRUCTURE ACTIVITY RELATIONSHIP (QSAR) ANALYSIS
 ANALYSIS") 159 CHAPTER 6 QUANTITATIVE STRUCTURE ACTIVITY RELATIONSHIP (QSAR) ANALYSIS 6.1 INTRODUCTION The purpose of this study is to gain on insight into structural features related the anticancer, antioxidant
159 CHAPTER 6 QUANTITATIVE STRUCTURE ACTIVITY RELATIONSHIP (QSAR) ANALYSIS 6.1 INTRODUCTION The purpose of this study is to gain on insight into structural features related the anticancer, antioxidant
Dr. Sander B. Nabuurs. Computational Drug Discovery group Center for Molecular and Biomolecular Informatics Radboud University Medical Centre
 Dr. Sander B. Nabuurs Computational Drug Discovery group Center for Molecular and Biomolecular Informatics Radboud University Medical Centre The road to new drugs. How to find new hits? High Throughput
Dr. Sander B. Nabuurs Computational Drug Discovery group Center for Molecular and Biomolecular Informatics Radboud University Medical Centre The road to new drugs. How to find new hits? High Throughput
Interactive Feature Selection with
 Chapter 6 Interactive Feature Selection with TotalBoost g ν We saw in the experimental section that the generalization performance of the corrective and totally corrective boosting algorithms is comparable.
Chapter 6 Interactive Feature Selection with TotalBoost g ν We saw in the experimental section that the generalization performance of the corrective and totally corrective boosting algorithms is comparable.
Data Mining. CS57300 Purdue University. March 22, 2018
 Data Mining CS57300 Purdue University March 22, 2018 1 Hypothesis Testing Select 50% users to see headline A Unlimited Clean Energy: Cold Fusion has Arrived Select 50% users to see headline B Wedding War
Data Mining CS57300 Purdue University March 22, 2018 1 Hypothesis Testing Select 50% users to see headline A Unlimited Clean Energy: Cold Fusion has Arrived Select 50% users to see headline B Wedding War
QSAR/QSPR modeling. Quantitative Structure-Activity Relationships Quantitative Structure-Property-Relationships
 Quantitative Structure-Activity Relationships Quantitative Structure-Property-Relationships QSAR/QSPR modeling Alexandre Varnek Faculté de Chimie, ULP, Strasbourg, FRANCE QSAR/QSPR models Development Validation
Quantitative Structure-Activity Relationships Quantitative Structure-Property-Relationships QSAR/QSPR modeling Alexandre Varnek Faculté de Chimie, ULP, Strasbourg, FRANCE QSAR/QSPR models Development Validation
Development of Pharmacophore Model for Indeno[1,2-b]indoles as Human Protein Kinase CK2 Inhibitors and Database Mining
![Development of Pharmacophore Model for Indeno[1,2-b]indoles as Human Protein Kinase CK2 Inhibitors and Database Mining](/thumbs/91/107031261.jpg "Development of Pharmacophore Model for Indeno[1,2-b]indoles as Human Protein Kinase CK2 Inhibitors and Database Mining") Development of Pharmacophore Model for Indeno[1,2-b]indoles as Human Protein Kinase CK2 Inhibitors and Database Mining Samer Haidar 1, Zouhair Bouaziz 2, Christelle Marminon 2, Tiomo Laitinen 3, Anti Poso
Development of Pharmacophore Model for Indeno[1,2-b]indoles as Human Protein Kinase CK2 Inhibitors and Database Mining Samer Haidar 1, Zouhair Bouaziz 2, Christelle Marminon 2, Tiomo Laitinen 3, Anti Poso
BioSolveIT. A Combinatorial Approach for Handling of Protonation and Tautomer Ambiguities in Docking Experiments
 BioSolveIT Biology Problems Solved using Information Technology A Combinatorial Approach for andling of Protonation and Tautomer Ambiguities in Docking Experiments Ingo Dramburg BioSolve IT Gmb An der
BioSolveIT Biology Problems Solved using Information Technology A Combinatorial Approach for andling of Protonation and Tautomer Ambiguities in Docking Experiments Ingo Dramburg BioSolve IT Gmb An der