A Robust Grouping Algorithm For Clustering of Similar Protein Folding Units
|
|
|
- Primrose Austin
- 6 years ago
- Views:
Transcription
1 A Robust Grouping Algorithm For Clustering of Similar Protein Folding Units Zhi Li, Nathan E. Brener, S. Sitharama Iyengar*, Guna Seetharaman, and Sumeet Dua Department of Computer Science, Louisiana State University, Baton Rouge, LA 70803, USA S. Ramakumar and K. Manikandan Department of Physics and Bioinformatics Centre, Indian Institute of Science, Bangalore , INDIA Jacob Barhen CESAR Laboratory, Oak Ridge National Laboratory, Oak Ridge, TN 37831, USA * To whom correspondence should be addressed Running Head: Algorithm to Group Similar Protein Folding Units Abstract The properties of a protein depend on its sequence of amino acids and its three-dimensional structure which consists of multiple folds of the peptide chain. If some of the properties depend primarily on the folding structure, then proteins with certain folding units may exhibit properties specific to those units. In that case, a classification of proteins based on folding units would facilitate the selection of proteins with certain desired properties. With this in mind, we propose an efficient clustering algorithm that can be used to classify proteins according to common folding units. Our algorithm has the following steps: Represent the protein structure as a series of conformational angles. Partition the proteins into fragments (folding units) of a specified size. Cluster the fragments into groups. The use of overlapped substrings makes our unique demographic clustering technique not susceptible to noise and outliers. Preliminary implementation of this algorithm indicates that it has the capability to discover secondary structural elements (folding units) in proteins and can be generalized to large protein data banks. The algorithm has been applied to a set of 20 randomly selected proteins from the Protein Data Bank and a set of 12 nonhomologous α/β protein structures from the PDBSELECT solved by X-ray crystallography to better than atomic resolution. The algorithm not only identifies the secondary structural elements such as α-helices and β-strands, but also uncovers different turn types which link extended and helical structures. 1. Introduction A protein is a sequence of amino acids joined by a backbone structure called a peptide chain. In addition to the peptide chain, proteins have a three-dimensional structure which consists of multiple folds of the chain (Orengo, 1994; Murin et al., 1995). The specific properties of the protein depend on both the amino acid sequence and the folding structure. If some of the properties of a protein depend primarily on the folding structure, then proteins with certain folding units may exhibit properties specific to those units. In that case, a classification of proteins based on folding units would facilitate the selection of proteins with certain desired properties. The library of protein fragments (referred to as folded units in our study) derived from the experimentally solved proteins structures is shown to be useful in the process of the ab initio prediction of the 3D structure of proteins from the primary sequence (Kolodny et al., 2002; Haspel et al., 2003; de Brevern and Hazout, 2003). A number of clustering methods have been proposed in the past to identify such representative fragments (Micheletti et al., 2000; Hunter and Subramaniam, 2003) It has also been shown that the protein structures can be represented by a certain length of peptides having a combination of secondary structures and turns (Guruprasad, et al., 2003) and among them a few are shown to be intrinsically stable peptides (Perczel, et al., 2003).
2 2 Currently there is a large quantity of protein structure data available in protein databases (Westbrook et al., 2002), and the amount of data is steadily increasing due to structural genomic projects (Iyengar, 1998). In order to facilitate the search for common folding units in large protein data banks, we propose a new efficient grouping algorithm derived from demographic clustering techniques used in data mining applications (Cabena et al., 1997). Our method has the following computational components: Represent the protein structure as a series of conformational angles (φ,ψ). Partition the proteins into fragments (folding units) containing N pairs of conformational angles. Treat these fragments as points in a 2N-dimensional conformational space. Cluster them into groups according to the distances between them. This algorithm, which is described in detail below, is used to perform case studies on a set of 20 randomly selected proteins from the Protein Data Bank and a set of 12 non-homologous α/β protein structures from the PDBSELECT and the identified clusters are discussed. 2. Data Structure Representation Scheme As mentioned above, a large number of protein 3D structures are now stored in databases, and the number of structure submissions is steadily increasing. We should select an efficient scheme to represent the protein 3D structure in order to facilitate flexible, efficient, and fast searching capabilities for the databases. This will facilitate the discovery of similar structures in proteins. Basically, the protein data banks store the protein s atomic coordinates, as derived from crystallographic studies. Although these coordinates contain the structure information precisely, they are not the best representation for detecting similar folds. A common way of reducing the number of parameters needed to describe the conformation of a protein backbone is to take advantage of the fact that the backbone contains planar units which are connected at C α atoms, with six atoms per planar unit. Two adjacent planar units, (C α,i-1, C i-1, O, N i, H, C αi ) and (C αi, C i, O, N i+1, H, C α,i+1 ), are shown in Figure 1. Each C α atom belongs to two of these planar units. The two adjacent planar units which meet at a C α atom are free to rotate about the C α -N or C α -C bond at the junction. This leads to a wide range of three-dimensional configurations for the protein. Figure 1. The two rotation angles φ and ψ characterize the three-dimensional nature of the protein molecule.
3 There are a number of ways that the protein backbone can be represented (for example, Flocco and Mowbray, 1995; Oldfield and Hubbard, 1994; Smith et al., 1997), including the following: 1. Express the backbone as a series of C α points in 3D space, with 3 coordinates for each point. This is a very precise way to describe the backbone. A large amount of work has been done based on this scheme (Levitt and Chothia, 1976; Russel et al., 1993; Yang and Honig, 2000). But this approach demands too much computation to search for common folding units and hence is not applicable for large databases. 2. Classify the conformations that an amino acid can take into several categories and represent them as symbols (Wintjens, et al., 1998), and implement string alignment to search for similarity between folding units. One of these approaches is to divide the Ramachandran map (Ramachandran and Sasissekharan, 1968) into domains (Rooman et al., 1991); another attempt is to divide the whole conformation space directly into subspaces (Matsuda et al., 1997). Then based on string comparison, we can search for similar folds. These representations greatly decrease the computational tasks by simplifying a 3-D problem to a 1-D problem. But there is a contradiction in this scheme: if the number of subspaces is large, then it is not easy to find similar structures; or, if there are only a few subspaces, the comparison will be too inaccurate. 3. Express the backbone as a series of conformational angles φ and ψ, where φ is the rotation angle of the planar unit about the bond between the C α atom and the nitrogen atom, i.e., the C α -N bond, and ψ is the rotation angle of the planar unit about the bond between the two carbon atoms, i.e., the C α -C bond, as shown in Figure 1. When comparing the similarity of two folding units, we simply compute the difference between each pair of φ angles and each pair of ψ angles on the same position in their respective folding units, and then sum up these differences. In this way, we simplify the 3-D problem to a 2-D one while preserving all of the conformational information. This data representation scheme enables the efficient detection of folding similarities and hence will be used in the present study. An added advantage is that, from the (φ,ψ)s of a cluster of fragments, it is easy to directly identify different secondary structural elements and turn types (Hutchinson and Thornton, 1996) represented by that cluster. The Protein Data Bank (PDB) (Bernstein et al., 1977) and the PDBSELECT (Hobohm and Sander, 1994) are archives of experimentally determined three-dimensional structures of proteins. The archives contain the coordinates of each atom in the proteins. We will extract the atomic coordinates of the backbone atoms and use these to compute the dihedral (conformational) angle pairs (φ,ψ). 3. Grouping Algorithm Once the dihedral angle pairs have been computed, we will use them to search for similar folding units in proteins. The technique we propose to use is based on dividing the protein into fragments of a specified size. For the first study described in this paper, we have selected a fragment length of 8, that is, 8 pairs of dihedral angles. For each protein to be included in the search, we first compute the following series of dihedral angles: { (φ,ψ) 1 (φ,ψ) 2 (φ,ψ) 3 (φ,ψ) 4 (φ,ψ) 5 (φ,ψ) n-1 } where n is the number of amino acids used to obtain the fragments and the range of the dihedral angles is -180 to 180. The peptide chain is then decomposed into a series of overlapping fragments of length 8: Fragment 1: [ (φ,ψ) 1 (φ,ψ) 2 (φ,ψ) 3 (φ,ψ) 4 (φ,ψ) 5 (φ,ψ) 6 (φ,ψ) 7 (φ,ψ) 8 ] Fragment 2: [ (φ,ψ) 2 (φ,ψ) 3 (φ,ψ) 4 (φ,ψ) 5 (φ,ψ) 6 (φ,ψ) 7 (φ,ψ) 8 (φ,ψ) 9 ] Fragment 3: [ (φ,ψ) 3 (φ,ψ) 4 (φ,ψ) 5 (φ,ψ) 6 (φ,ψ) 7 (φ,ψ) 8 (φ,ψ) 9 (φ,ψ) 10 ] Fragment 4: [ (φ,ψ) 4 (φ,ψ) 5 (φ,ψ) 6 (φ,ψ) 7 (φ,ψ) 8 (φ,ψ) 9 (φ,ψ) 10 (φ,ψ) 11 ]. 3
4 Then we apply a grouping algorithm, which is based on the demographic clustering technique of data mining (Cabena et al., 1997). In the following, we treat the fragments as points in a 16-dimensional space. We define the distance between two points A i and A j, DIST (A i, A j ), and the center of group j, C j, as follows: Step 1: DIST (A i, A j ) = ((φ i1 -φ j1 ) 2 + (ψ i1 -ψ j1 ) 2 + (φ i2 -φ j2 ) 2 + (ψ i2 -ψ j2 ) 2 + +(φ i8 -φ j8 ) 2 + (ψ i8 -ψ j8 ) 2 ) ½ for A i [(φ i1, ψ i1 ), (φ i2, ψ i2 ), (φ i8, ψ i8 )] and A j [(φ j1, ψ j1 ), (φ j2, ψ j2 ), (φ j8, ψ j8 )]; ( for every (ψ im -ψ jm ), if ψ im -ψ jm >180, then use 360- ψ im -ψ jm ; repeat the above for (φ im -φ jm ) ). Step 2: Let j be the index that labels the groups. We define the center of group j, C j, as C j = [(φ j1, ψ j1 ), (φ j2, ψ j2 ), (φ j8, ψ j8 )] where φ jm = Σφ im / N j ψ jm = Σψ im / N j ( i = 1, 2,.. N j ; m = 1, 2, 8 ), N j is the number of points in the group, and the sum is over i. Such groups are regarded as folding units in our current work. Algorithm: Input: A set of points in 16-dimensional space and a distance measure R. Output: A set of groups into which the points have been divided, where every point in a group is within the distance R of the group center. Begin: I. Start a stack with all of the points in it. II. Do an operation pop up of a point A 1, create group 1, with center C 1 equal to A 1, set N 1 to 1. III. While ( stack is not empty ) { a. Do an operation pop up of a point A p. b. Compute the distances between A p and each existing group center C j (suppose we have k groups now, then 1 <= j <= k). c. Suppose when j = j min, the distance is a minimum. If DIST ( C jmin, A p ) > R, then create a new group k+1, with center C k+1 equal to A p, set N k+1 to 1. Else 1. Insert A p into group j min, add 1 to N jmin. 2. Compute the new center C jmin of group j min. 3. For i = 1, 2, N jmin { i. Re-compute the distance DIST (A jmin, i, C jmin ) between the point A jmin, i in group j min and the new group center C jmin. ii. If DIST (A jmin, i, C jmin ) > R, push A jmin, i into the stack, subtract 1 from N jmin, go to step 2. } } IV. For each group, re-calculate the distances between the contained points and all of the group centers. If there is any point that has a shorter distance with another group center than with its own group center, move it to the other group where the distance is shorter. If there are no such points, go to END. 4
5 5 V. Re-compute all the group centers. If any point is no longer within distance R of the center of its group, push it into the stack. If there are points in the stack, go back to step III. If there are no points in the stack, go back to step IV. END 4. Case Studies In this section we show how our grouping algorithm can be applied to a set of proteins. The software programs have been implemented in the C language on a high-performance computer system. To test our algorithm, we conducted the following two case studies: Case Study A: 20 Randomly Selected Proteins from the PDB In this study, we randomly selected 20 proteins from the PDB. These proteins have different numbers of amino acids and are from different protein families. Some atoms are not well determined by X-ray crystallography in some proteins; thus, only the amino acids with good resolutions are chosen for computing the fragments. Table 1 shows the 20 proteins that were selected and the number of points (fragments) derived from each one. In our test, we set R (the maximum allowed distance from the center of a group) to 240. Although R seems to be large in this case, it will be significantly decreased if we include more proteins in the study. We obtained a total of 3083 points from these 20 proteins and used our algorithm to group them into 1734 groups. The group center is the average of the coordinates of all the points in the group and thus is usually not an actual fragment from one of the proteins. Therefore, in order to represent each group more reasonably, we choose the fragment that is closest to the group center. Table 2 gives the five largest groups, labeled A, B, C, D, E, and Figure 2 shows the fragments (folding units) that have the minimum distance from the centers of these groups. The table shows that for each group, the fragments are from different proteins, which means that our algorithm is capable of efficiently detecting common folding units in a set of proteins. Case Study B: 12 Non-Homologous α/β Proteins from the PDBSELECT A small set of 12 non-homologous α/β protein structures with sequence identity 25% and refined at resolution of better than 1.0 Å, was selected from the PDBSELECT April 2003 list (Hobohm and Sander, 1994). Resolution cutoff of 1.0 Å or better was chosen to ensure that the parameters used for clustering are to a lesser extent affected by experimental errors. For a residue to be part of a fragment, the torsion angle defining atoms (N, C α and C) of the residue should have the B-factor of less than 60 Å 2 so that the atoms are well defined in the electron density maps. Any missing residues or atoms are considered as a discontinuity in the polypeptide chain. Accordingly, an input set of 3636 fragments has been derived from the selected 12 proteins (Table 3). In this test, we set R (the maximum allowed distance of a fragment from its group center) to 240º. In order to ensure that the deviations are more uniformly distributed along the fragment, the maximum allowed deviation for any main-chain dihedral angle from the corresponding angle in the cluster centroid is taken to be 60º. The deviation can be adjusted to do fine clustering (say 30º) or coarse clustering (say 90º) depending upon the interest of the study. With the R value of 240º and the maximum residue level deviation of 60º, our algorithm grouped the 3636 points into 1858 clusters which include single member clusters. Table 4 gives the five largest clusters, labeled A, B, C, D and E and Figure 3 shows the corresponding fragments (folding units) that have the least distance from the centers of these groups. The top ten clusters identified for varying fragment lengths (6 9) and their secondary structure
6 6 descriptions are given in Table 5. Upon testing on various R values (160, 240 and 320 ) for fragment length 8 (FL8), it is noticed that as expected, the number of clusters decreases as R increases. The nomenclature used to designate the conformation of a given residue is according to Efimov (1991). Focusing on clusters with FL8 for detailed discussion (Table 4 and column FL8 of Table 5), it may be seen that all the clusters represent either the regular secondary structural elements or combination of them. In general, the conformation of helical residues is well defined even though deviations are observed for the N- and C- termini of helices compared to the body of the helix. The deviations are more pronounced at the C-terminal region suggesting fraying of the C-terminus (Soman et al., 1991). The drift of (φ,ψ) from the helical region at the C-terminus presumably maximizes capping interactions. An analysis such as the one reported here would help not only in modeling helical regions but also the helix termini. There are two clusters which correspond to two different kinds of termination of α-helices (clusters 4 and 7 of column FL8 of Table 5). In cluster 4, the residue in the α L conformation is predominantly Glycine and in many cases the residues γα L interlink a β-strand with the preceding α-helix. Focusing now on clusters with fragment length 6 (column FL6 of Table 5), it is seen that here only the β-strand emerges as an independent cluster among the first ten, presumably because β-strands in general are shorter secondary structural elements compared to α-helices. Also the standard deviation in (φ,ψ) associated with β-strands (β 6 ) is approximately 19 whereas the corresponding value for α 6 is 6 suggesting that an α-helix is conformationaly more rigid compared to a β-strand. This may have some implications for the main-chain conformational entropies associated with different types of secondary structural elements in proteins. 5. Conclusion This paper proposes a unique demographic clustering algorithm that can be used to classify proteins according to similar folding units. Such a classification has the potential to facilitate the selection of proteins with specific desired properties. Preliminary implementation of this algorithm indicates that it has the capability to discover secondary structural elements (folding units) in proteins and can be generalized to large protein data banks. This novel clustering technique is likely to be useful in generating different sizes of libraries of protein fragments which may be helpful in homology modeling, ab initio protein structure prediction, building accurate loop conformations and design of peptides with required 3D structures. The algorithm may also be used to find preferred conformers either within a structural class or across structural classes of proteins. 6. References Bernstein, F. C., Koetzle, T. F., Williams, G. J. B., Meyer, E. F., Jr., Brice, M. D., Rodgers, J. R., Kennard, O., Shimanouchi, T. and Tasumi, M. (1977) The Protein Data Bank: A computer-based archival file for macromolecular structures, J. Mol. Biol., 112, ; Cabena, P., Hadjnian, Stadler, Verhees and Zanasi (1997) Discovering Data Mining From Concept to Implementation. Prentice Hall PTR. de Brevern, A. G. and Hazout, S. (2003) Hybrid protein model for optimally defining 3D protein structure fragments, Bioinformatics, 19,
7 7 Efimov, A. V. (1991) Structure of α-α-hairpins with short connections, Protein Engg, 4, Flocco, M. M. and Mowbray, S. L. (1995) C-alpha based torsion angles: A simple tool to analyze protein conformational changes, Protein Sci, 4, Guruprasad, K., Rao, M. J., Adindla, S. and Guruprasad, L. (2003) Combinations of turns in proteins, J. Peptide Res, 62, Iyengar, S. S. (1998) Computer modeling and simulations of complex biological systems. Iyengar, S. S. (editor). CRC Press, Boca Raton, Florida. Haspel, N., Tsai, C. J., Wolfson, H. and Nussinov, R. (2003) Reducing the computational complexity of protein folding via fragment folding and assembly, Protein Sci, 12, Hobohm, U. and Sander, C. (1994) Enlarged representative set of protein structures, Protein Sci, 3, Hunter, C. G. and Subramaniam, S. (2003) Protein fragment clustering and canonical local shapes, Proteins, 50, Hutchinson, E. G. and Thornton, J. M. (1996) PROMOTIF--a program to identify and analyze structural motifs in proteins, Protein Sci, 5, Kolodny, R., Koehi, P., Guibas, L. and Levitt, M. (2002) Small libraries of protein fragments model native protein structures accurately, J. Mol. Biol, 23, Levitt, M. and Chothia, C. (1976) Structural patterns in globular proteins, Nature, 261, Matsuda, H., Taniguchi, F. and Hashimoto, A. (1997) An approach to detection of protein structural motifs using an encoding scheme of backbone conformations, Pac. Symp. Biocomput1997, Micheletti, C., Seno, F. and Maritan, A. (2000) Recurrent oligomers in proteins: An optimal scheme reconciling accurate and concise backbone representations in automated folding and design studies, Proteins, 40, Murzin, A. G., Brenner, S. E., Hubbard, T. and Chothia, C. (1995) SCOP: a structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol, 247, Oldfield, T. J. and Hubbard, R. E. (1994) Analysis of C-alpha geometry in protein structures, Proteins, 18, Orengo, C. (1994) Classification of protein folds, Curr. Opin. Struc. Biol, 4, Perczel, A., Jakli, I. and Csizmadia, I. G. (2003) Intrinsically stable secondary structure elements of proteins: A comprehensive study of folding units of proteins by computation and by analysis of data determined by X-ray crystallography, Chem. Eur. J, 9, Ramachandran, G. N. and Sasisekharan, V. (1968) Conformation of polypeptides and proteins, Advan. Protein Chem, 23,
8 8 Rooman, M. J., Kocher, J. A. and Wodak, S. J. (1991) Prediction of protein backbone conformation based on seven structure assignments, J. Mol. Biol., 221, Russel, R. B. and Barton, G. J. (1993) Multiple protein sequence alignment from tertiary structure comparisons: Assignments of global and residue level confidences, Proteins, 14, Smith, P. E., Blatt, H. D. and Montgomery-Pettit, B. (1997) A simple two-dimensional representation for the common secondary structural elements of polypeptides and proteins, Proteins, 27, Soman, K. V., Karimi, A. and Case, D. A. (1991) Unfolding of an alpha-helix in water, Biopolymers, 31, Westbrook, J., Feng, Z., Jain, S., Bhat, T. N., Thanki, N., Ravichandran, V., Gilliland, G. L., Bluhm, W. F., Weissig, H., Greer, D. S., Bourne, P. E. and Berman, H. M. (2002), The Protein Data Bank: Unifying the archive, Nucleic Acids Research, 30, Wintjens, R, Wodak, S. J. and Rooman, M. (1998) Typical interaction patterns in αβ and βα turn motifs, Protein Engg, 11, Yang, A. S. and Honig, B. (2000) An integrated approach to the analysis and modeling of protein sequences and structures. I. Protein structural alignment and a quantitative measure for protein structural distance, J. Mol. Biol, 301, Acknowledgments This work was supported in part by DOE-ORNL grant no and by an NSF grant. KM thanks CSIR (India) for a fellowship. Facilities at the Bioinformatics center funded by DBT (India) were used and are gratefully acknowledged.
9 9 PDB Name of the Protein Amino Acids Points Entry Selected Derived 1ash HEMOGLOBIN (DOMAIN ONE) bsr RIBONUCLEASE(BOVINE, SEMINAL) (CHAIN A) cca CYTOCHROME C PEROXIDASE cew CYSTATIN clm CALMODULIN (PARAMECIUM TETRAURELIA) crn CRAMBIN ctt CYTIDINE DEAMINASE erb RETINOL BINDING PROTEIN COMPLEX WITH N-ETHYL RETINAMIDE 2 1fut RIBONUCLEASE F hng CD2 (RAT) (CHAIN B) hoe ALPHA-*AMYLASE INHIBITOR HOE-467*A lbu HYDROLASE METALLO (ZN) DD-PEPTIDASE mka BETA-HYDROXYDECANOYL THIOL ESTER DEHYDRASE (CHAIN A) 1mng MANGANESE SUPEROXIDE DISMUTASE (CHAIN A) pkp RIBOSOMAL PROTEIN S udi URACIL-DNA GLYCOSYLASE utg UTEROGLOBIN(OXIDIZED) yal CARICA PAPAYA CHYMOPAPAIN vab MHC CLASS I H-2KB HEAVY CHAIN pti TRYPSIN INHIBITOR Table 1. A short list of proteins that were randomly selected for the demographic clustering of dihedral angles in the peptide chain. For each protein, the table shows the amino acids that were selected and the number of points that were derived.
10 10 Group Name A B C D E φ ψ φ ψ φ ψ φ ψ φ ψ φ ψ φ ψ , φ ψ Points in the Group The Points Sources Points Nearest of mka cew hng udi mka ash: 1 1bsr: 11 1cew: 15 1mka: 16 1mng: 60 1udi: 43 2vab: 56 1bsr: 16 1cew: 13 1hng: 27 1mka: 11 1mng: 1 1udi: 2 2vab: 39 1bsr: 2 1cew: 2 1hng: 11 1mka: 7 1mng: 3 1udi: 3 2vab: 14 1bsr: 6 1cca: 1 1cew: 3 1mka: 4 1mng: 9 1udi: 13 2vab: 4 1bsr: 8 1cew: 4 1hng: 7 1mka: 5 1udi: 5 2vab: 9 Table 2. The top 5 groups detected by our grouping algorithm. For each group, the table gives the coordinates of the group center, the number of points in the group, the point nearest the group center, and the number of points derived from the various proteins.

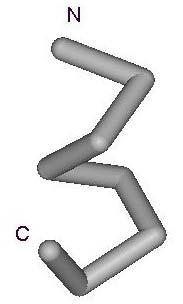
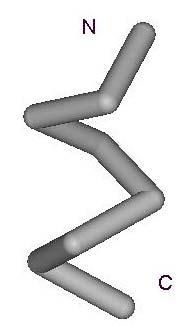
11 11 A B C D E Figure 2. Examples of Folding Units produced by the grouping algorithm. Figures A, B, C, D, E show the conformation of the point nearest the center in groups A, B, C, D, E.
12 Table 3. A list of non-homologous α/β proteins used for the case study and the number of 8 residue fragments derived from each protein. 12 PDB code Name of the protein # fragments 1byi_ Dethiobiotin Synthase 208 1g66A Acetyl xylan esterase II 191 1ga6A Serine-carboxyl proteinase 353 1gci_ Subtilisin 205 1i1wA Endo-1, 4-beta-xylanase 286 1ixh_ Phosphate binding protein 305 1muwA Xylose isomerase 370 1mxtA Cholesterol oxidase 482 1n55A Triosephosphate isomerase 233 1o7jA L-asparaginase 309 1ug6A Beta-glycosidase 410 7a3hA Endoglucanase 284
13 Table 4. The group centers of the top 5 groups detected by our grouping algorithm for a fragment length of 8 (FL8) and the nearest points to the group centers in each group. The root mean square deviations of each position (φ, ψ) and the overall deviation are also given. Group Name A B C D E φ ψ φ ψ φ ψ φ ψ φ ψ φ ψ φ ψ φ ψ Fragments in the Group The Nearest Fragment Description of the fragments 1n55A 110A 117A An α - helix [α 8 ] βi refers to type I β turn 7a3hA 85A 92A An α - helix with type I β turn at the C terminal [α 7 -βi] 1ug6A 63A 70A An α - helix with type I β turn at the C terminal followed by an α L residue [α 6 -βi-α L ] 1o7jA 264A 271A 1i1wA 244A 251A [α 5 -βi-α L -β] [β-α 7 ]
14 14 Table 5. The first 10 ranked clusters identified with the fragment length (FL) varying from 6 to 9 and their corresponding secondary structure combination are listed. Values in parentheses refer to the number of fragments in each cluster. Cluster Number FL 9 FL 8 FL 7 FL 6 1 α 9 (367) α 8 (443) α 7 (522) α 6 (626) 2 α 8 γ (81) α 7 γ (87) α 6 γ (97) α 5 γ (107) 3 α 7 γ α L (45) α 6 γ α L (52) α 5 γ α L (55) β α 5 (59) 4 α 6 γ α L β (39) α 5 γ α L β (39) β α 6 (53) α 4 γ α L (55) 5 β α 8 (35) β α 7 (38) α 4 γ α L β (39) β 6 (50) 6 α 7 γ β (32) α 6 γ β (33) β 6 α (32) β 6 (41) 7 β 2 α 7 (30) α 5 γ β γ (26) α 5 γ β (30) α 3 γ α L β (39) 8 α 5 γ α L β 2 (26) β 2 α 6 (26) α 4 γ β γ (29) α 4 γ β (39) 9 α 5 γ α 3 (25) α 4 γ α 3 (26) β 2 α 5 (28) α γ α L β 3 (34) 10 α 2 γ α 6 (24) α 2 γ α 5 (25) α γ α β 4 (28) β 2 α 4 (33)



15 Figure 3. C α traces of nearest fragments for the first five clusters listed in Table 4. The amino and carboxyl terminal ends of the fragments are denoted as N and C respectively. A B C
16 D E 16
DATE A DAtabase of TIM Barrel Enzymes
 DATE A DAtabase of TIM Barrel Enzymes 2 2.1 Introduction.. 2.2 Objective and salient features of the database 2.2.1 Choice of the dataset.. 2.3 Statistical information on the database.. 2.4 Features....
DATE A DAtabase of TIM Barrel Enzymes 2 2.1 Introduction.. 2.2 Objective and salient features of the database 2.2.1 Choice of the dataset.. 2.3 Statistical information on the database.. 2.4 Features....
Discrete representations of the protein C Xavier F de la Cruz 1, Michael W Mahoney 2 and Byungkook Lee
 Research Paper 223 Discrete representations of the protein C chain Xavier F de la Cruz 1, Michael W Mahoney 2 and Byungkook Lee Background: When a large number of protein conformations are generated and
Research Paper 223 Discrete representations of the protein C chain Xavier F de la Cruz 1, Michael W Mahoney 2 and Byungkook Lee Background: When a large number of protein conformations are generated and
Motif Prediction in Amino Acid Interaction Networks
 Motif Prediction in Amino Acid Interaction Networks Omar GACI and Stefan BALEV Abstract In this paper we represent a protein as a graph where the vertices are amino acids and the edges are interactions
Motif Prediction in Amino Acid Interaction Networks Omar GACI and Stefan BALEV Abstract In this paper we represent a protein as a graph where the vertices are amino acids and the edges are interactions
Protein Structure: Data Bases and Classification Ingo Ruczinski
 Protein Structure: Data Bases and Classification Ingo Ruczinski Department of Biostatistics, Johns Hopkins University Reference Bourne and Weissig Structural Bioinformatics Wiley, 2003 More References
Protein Structure: Data Bases and Classification Ingo Ruczinski Department of Biostatistics, Johns Hopkins University Reference Bourne and Weissig Structural Bioinformatics Wiley, 2003 More References
Supplemental Materials for. Structural Diversity of Protein Segments Follows a Power-law Distribution
 Supplemental Materials for Structural Diversity of Protein Segments Follows a Power-law Distribution Yoshito SAWADA and Shinya HONDA* National Institute of Advanced Industrial Science and Technology (AIST),
Supplemental Materials for Structural Diversity of Protein Segments Follows a Power-law Distribution Yoshito SAWADA and Shinya HONDA* National Institute of Advanced Industrial Science and Technology (AIST),
Number sequence representation of protein structures based on the second derivative of a folded tetrahedron sequence
 Number sequence representation of protein structures based on the second derivative of a folded tetrahedron sequence Naoto Morikawa (nmorika@genocript.com) October 7, 2006. Abstract A protein is a sequence
Number sequence representation of protein structures based on the second derivative of a folded tetrahedron sequence Naoto Morikawa (nmorika@genocript.com) October 7, 2006. Abstract A protein is a sequence
Better Bond Angles in the Protein Data Bank
 Better Bond Angles in the Protein Data Bank C.J. Robinson and D.B. Skillicorn School of Computing Queen s University {robinson,skill}@cs.queensu.ca Abstract The Protein Data Bank (PDB) contains, at least
Better Bond Angles in the Protein Data Bank C.J. Robinson and D.B. Skillicorn School of Computing Queen s University {robinson,skill}@cs.queensu.ca Abstract The Protein Data Bank (PDB) contains, at least
Figure 1. Molecules geometries of 5021 and Each neutral group in CHARMM topology was grouped in dash circle.
 Project I Chemistry 8021, Spring 2005/2/23 This document was turned in by a student as a homework paper. 1. Methods First, the cartesian coordinates of 5021 and 8021 molecules (Fig. 1) are generated, in
Project I Chemistry 8021, Spring 2005/2/23 This document was turned in by a student as a homework paper. 1. Methods First, the cartesian coordinates of 5021 and 8021 molecules (Fig. 1) are generated, in
Introduction to Comparative Protein Modeling. Chapter 4 Part I
 Introduction to Comparative Protein Modeling Chapter 4 Part I 1 Information on Proteins Each modeling study depends on the quality of the known experimental data. Basis of the model Search in the literature
Introduction to Comparative Protein Modeling Chapter 4 Part I 1 Information on Proteins Each modeling study depends on the quality of the known experimental data. Basis of the model Search in the literature
THE TANGO ALGORITHM: SECONDARY STRUCTURE PROPENSITIES, STATISTICAL MECHANICS APPROXIMATION
 THE TANGO ALGORITHM: SECONDARY STRUCTURE PROPENSITIES, STATISTICAL MECHANICS APPROXIMATION AND CALIBRATION Calculation of turn and beta intrinsic propensities. A statistical analysis of a protein structure
THE TANGO ALGORITHM: SECONDARY STRUCTURE PROPENSITIES, STATISTICAL MECHANICS APPROXIMATION AND CALIBRATION Calculation of turn and beta intrinsic propensities. A statistical analysis of a protein structure
Introduction to" Protein Structure
 Introduction to" Protein Structure Function, evolution & experimental methods Thomas Blicher, Center for Biological Sequence Analysis Learning Objectives Outline the basic levels of protein structure.
Introduction to" Protein Structure Function, evolution & experimental methods Thomas Blicher, Center for Biological Sequence Analysis Learning Objectives Outline the basic levels of protein structure.
Supporting Online Material for
 www.sciencemag.org/cgi/content/full/309/5742/1868/dc1 Supporting Online Material for Toward High-Resolution de Novo Structure Prediction for Small Proteins Philip Bradley, Kira M. S. Misura, David Baker*
www.sciencemag.org/cgi/content/full/309/5742/1868/dc1 Supporting Online Material for Toward High-Resolution de Novo Structure Prediction for Small Proteins Philip Bradley, Kira M. S. Misura, David Baker*
Protein Structure. W. M. Grogan, Ph.D. OBJECTIVES
 Protein Structure W. M. Grogan, Ph.D. OBJECTIVES 1. Describe the structure and characteristic properties of typical proteins. 2. List and describe the four levels of structure found in proteins. 3. Relate
Protein Structure W. M. Grogan, Ph.D. OBJECTIVES 1. Describe the structure and characteristic properties of typical proteins. 2. List and describe the four levels of structure found in proteins. 3. Relate
Protein structure similarity based on multi-view images generated from 3D molecular visualization
 Protein structure similarity based on multi-view images generated from 3D molecular visualization Chendra Hadi Suryanto, Shukun Jiang, Kazuhiro Fukui Graduate School of Systems and Information Engineering,
Protein structure similarity based on multi-view images generated from 3D molecular visualization Chendra Hadi Suryanto, Shukun Jiang, Kazuhiro Fukui Graduate School of Systems and Information Engineering,
Giri Narasimhan. CAP 5510: Introduction to Bioinformatics. ECS 254; Phone: x3748
 CAP 5510: Introduction to Bioinformatics Giri Narasimhan ECS 254; Phone: x3748 giri@cis.fiu.edu www.cis.fiu.edu/~giri/teach/bioinfs07.html 2/15/07 CAP5510 1 EM Algorithm Goal: Find θ, Z that maximize Pr
CAP 5510: Introduction to Bioinformatics Giri Narasimhan ECS 254; Phone: x3748 giri@cis.fiu.edu www.cis.fiu.edu/~giri/teach/bioinfs07.html 2/15/07 CAP5510 1 EM Algorithm Goal: Find θ, Z that maximize Pr
Protein Structure Basics
 Protein Structure Basics Presented by Alison Fraser, Christine Lee, Pradhuman Jhala, Corban Rivera Importance of Proteins Muscle structure depends on protein-protein interactions Transport across membranes
Protein Structure Basics Presented by Alison Fraser, Christine Lee, Pradhuman Jhala, Corban Rivera Importance of Proteins Muscle structure depends on protein-protein interactions Transport across membranes
Physiochemical Properties of Residues
 Physiochemical Properties of Residues Various Sources C N Cα R Slide 1 Conformational Propensities Conformational Propensity is the frequency in which a residue adopts a given conformation (in a polypeptide)
Physiochemical Properties of Residues Various Sources C N Cα R Slide 1 Conformational Propensities Conformational Propensity is the frequency in which a residue adopts a given conformation (in a polypeptide)
Procheck output. Bond angles (Procheck) Structure verification and validation Bond lengths (Procheck) Introduction to Bioinformatics.
 Structure verification and validation Bond lengths (Procheck) Introduction to Bioinformatics.") Structure verification and validation Bond lengths (Procheck) Introduction to Bioinformatics Iosif Vaisman Email: ivaisman@gmu.edu ----------------------------------------------------------------- Bond
Structure verification and validation Bond lengths (Procheck) Introduction to Bioinformatics Iosif Vaisman Email: ivaisman@gmu.edu ----------------------------------------------------------------- Bond
Algorithm for Rapid Reconstruction of Protein Backbone from Alpha Carbon Coordinates
 Algorithm for Rapid Reconstruction of Protein Backbone from Alpha Carbon Coordinates MARIUSZ MILIK, 1 *, ANDRZEJ KOLINSKI, 1, 2 and JEFFREY SKOLNICK 1 1 The Scripps Research Institute, Department of Molecular
Algorithm for Rapid Reconstruction of Protein Backbone from Alpha Carbon Coordinates MARIUSZ MILIK, 1 *, ANDRZEJ KOLINSKI, 1, 2 and JEFFREY SKOLNICK 1 1 The Scripps Research Institute, Department of Molecular
A General Model for Amino Acid Interaction Networks
 Author manuscript, published in "N/P" A General Model for Amino Acid Interaction Networks Omar GACI and Stefan BALEV hal-43269, version - Nov 29 Abstract In this paper we introduce the notion of protein
Author manuscript, published in "N/P" A General Model for Amino Acid Interaction Networks Omar GACI and Stefan BALEV hal-43269, version - Nov 29 Abstract In this paper we introduce the notion of protein
Francisco Melo, Damien Devos, Eric Depiereux and Ernest Feytmans
 From: ISMB-97 Proceedings. Copyright 1997, AAAI (www.aaai.org). All rights reserved. ANOLEA: A www Server to Assess Protein Structures Francisco Melo, Damien Devos, Eric Depiereux and Ernest Feytmans Facultés
From: ISMB-97 Proceedings. Copyright 1997, AAAI (www.aaai.org). All rights reserved. ANOLEA: A www Server to Assess Protein Structures Francisco Melo, Damien Devos, Eric Depiereux and Ernest Feytmans Facultés
Protein Structure & Motifs
 & Motifs Biochemistry 201 Molecular Biology January 12, 2000 Doug Brutlag Introduction Proteins are more flexible than nucleic acids in structure because of both the larger number of types of residues
& Motifs Biochemistry 201 Molecular Biology January 12, 2000 Doug Brutlag Introduction Proteins are more flexible than nucleic acids in structure because of both the larger number of types of residues
The typical end scenario for those who try to predict protein
 A method for evaluating the structural quality of protein models by using higher-order pairs scoring Gregory E. Sims and Sung-Hou Kim Berkeley Structural Genomics Center, Lawrence Berkeley National Laboratory,
A method for evaluating the structural quality of protein models by using higher-order pairs scoring Gregory E. Sims and Sung-Hou Kim Berkeley Structural Genomics Center, Lawrence Berkeley National Laboratory,
research papers Creating structure features by data mining the PDB to use as molecular-replacement models 1. Introduction T. J.
 Acta Crystallographica Section D Biological Crystallography ISSN 0907-4449 Creating structure features by data mining the PDB to use as molecular-replacement models T. J. Oldfield Accelrys Inc., Department
Acta Crystallographica Section D Biological Crystallography ISSN 0907-4449 Creating structure features by data mining the PDB to use as molecular-replacement models T. J. Oldfield Accelrys Inc., Department
Introducing Hippy: A visualization tool for understanding the α-helix pair interface
 Introducing Hippy: A visualization tool for understanding the α-helix pair interface Robert Fraser and Janice Glasgow School of Computing, Queen s University, Kingston ON, Canada, K7L3N6 {robert,janice}@cs.queensu.ca
Introducing Hippy: A visualization tool for understanding the α-helix pair interface Robert Fraser and Janice Glasgow School of Computing, Queen s University, Kingston ON, Canada, K7L3N6 {robert,janice}@cs.queensu.ca
Basics of protein structure
 Today: 1. Projects a. Requirements: i. Critical review of one paper ii. At least one computational result b. Noon, Dec. 3 rd written report and oral presentation are due; submit via email to bphys101@fas.harvard.edu
Today: 1. Projects a. Requirements: i. Critical review of one paper ii. At least one computational result b. Noon, Dec. 3 rd written report and oral presentation are due; submit via email to bphys101@fas.harvard.edu
COMP 598 Advanced Computational Biology Methods & Research. Introduction. Jérôme Waldispühl School of Computer Science McGill University
 COMP 598 Advanced Computational Biology Methods & Research Introduction Jérôme Waldispühl School of Computer Science McGill University General informations (1) Office hours: by appointment Office: TR3018
COMP 598 Advanced Computational Biology Methods & Research Introduction Jérôme Waldispühl School of Computer Science McGill University General informations (1) Office hours: by appointment Office: TR3018
Examples of Protein Modeling. Protein Modeling. Primary Structure. Protein Structure Description. Protein Sequence Sources. Importing Sequences to MOE
 Examples of Protein Modeling Protein Modeling Visualization Examination of an experimental structure to gain insight about a research question Dynamics To examine the dynamics of protein structures To
Examples of Protein Modeling Protein Modeling Visualization Examination of an experimental structure to gain insight about a research question Dynamics To examine the dynamics of protein structures To
Design of a Novel Globular Protein Fold with Atomic-Level Accuracy
 Design of a Novel Globular Protein Fold with Atomic-Level Accuracy Brian Kuhlman, Gautam Dantas, Gregory C. Ireton, Gabriele Varani, Barry L. Stoddard, David Baker Presented by Kate Stafford 4 May 05 Protein
Design of a Novel Globular Protein Fold with Atomic-Level Accuracy Brian Kuhlman, Gautam Dantas, Gregory C. Ireton, Gabriele Varani, Barry L. Stoddard, David Baker Presented by Kate Stafford 4 May 05 Protein
Heteropolymer. Mostly in regular secondary structure
 Heteropolymer - + + - Mostly in regular secondary structure 1 2 3 4 C >N trace how you go around the helix C >N C2 >N6 C1 >N5 What s the pattern? Ci>Ni+? 5 6 move around not quite 120 "#$%&'!()*(+2!3/'!4#5'!1/,#64!#6!,6!
Heteropolymer - + + - Mostly in regular secondary structure 1 2 3 4 C >N trace how you go around the helix C >N C2 >N6 C1 >N5 What s the pattern? Ci>Ni+? 5 6 move around not quite 120 "#$%&'!()*(+2!3/'!4#5'!1/,#64!#6!,6!
Secondary and sidechain structures
 Lecture 2 Secondary and sidechain structures James Chou BCMP201 Spring 2008 Images from Petsko & Ringe, Protein Structure and Function. Branden & Tooze, Introduction to Protein Structure. Richardson, J.
Lecture 2 Secondary and sidechain structures James Chou BCMP201 Spring 2008 Images from Petsko & Ringe, Protein Structure and Function. Branden & Tooze, Introduction to Protein Structure. Richardson, J.
THE UNIVERSITY OF MANITOBA. PAPER NO: 409 LOCATION: Fr. Kennedy Gold Gym PAGE NO: 1 of 6 DEPARTMENT & COURSE NO: CHEM 4630 TIME: 3 HOURS
 PAPER NO: 409 LOCATION: Fr. Kennedy Gold Gym PAGE NO: 1 of 6 DEPARTMENT & COURSE NO: CHEM 4630 TIME: 3 HOURS EXAMINATION: Biochemistry of Proteins EXAMINER: J. O'Neil Section 1: You must answer all of
PAPER NO: 409 LOCATION: Fr. Kennedy Gold Gym PAGE NO: 1 of 6 DEPARTMENT & COURSE NO: CHEM 4630 TIME: 3 HOURS EXAMINATION: Biochemistry of Proteins EXAMINER: J. O'Neil Section 1: You must answer all of
CAP 5510 Lecture 3 Protein Structures
 CAP 5510 Lecture 3 Protein Structures Su-Shing Chen Bioinformatics CISE 8/19/2005 Su-Shing Chen, CISE 1 Protein Conformation 8/19/2005 Su-Shing Chen, CISE 2 Protein Conformational Structures Hydrophobicity
CAP 5510 Lecture 3 Protein Structures Su-Shing Chen Bioinformatics CISE 8/19/2005 Su-Shing Chen, CISE 1 Protein Conformation 8/19/2005 Su-Shing Chen, CISE 2 Protein Conformational Structures Hydrophobicity
HOMOLOGY MODELING. The sequence alignment and template structure are then used to produce a structural model of the target.
 HOMOLOGY MODELING Homology modeling, also known as comparative modeling of protein refers to constructing an atomic-resolution model of the "target" protein from its amino acid sequence and an experimental
HOMOLOGY MODELING Homology modeling, also known as comparative modeling of protein refers to constructing an atomic-resolution model of the "target" protein from its amino acid sequence and an experimental
RNA and Protein Structure Prediction
 RNA and Protein Structure Prediction Bioinformatics: Issues and Algorithms CSE 308-408 Spring 2007 Lecture 18-1- Outline Multi-Dimensional Nature of Life RNA Secondary Structure Prediction Protein Structure
RNA and Protein Structure Prediction Bioinformatics: Issues and Algorithms CSE 308-408 Spring 2007 Lecture 18-1- Outline Multi-Dimensional Nature of Life RNA Secondary Structure Prediction Protein Structure
Alpha-helical Topology and Tertiary Structure Prediction of Globular Proteins Scott R. McAllister Christodoulos A. Floudas Princeton University
 Alpha-helical Topology and Tertiary Structure Prediction of Globular Proteins Scott R. McAllister Christodoulos A. Floudas Princeton University Department of Chemical Engineering Program of Applied and
Alpha-helical Topology and Tertiary Structure Prediction of Globular Proteins Scott R. McAllister Christodoulos A. Floudas Princeton University Department of Chemical Engineering Program of Applied and
Conformational Geometry of Peptides and Proteins:
 Conformational Geometry of Peptides and Proteins: Before discussing secondary structure, it is important to appreciate the conformational plasticity of proteins. Each residue in a polypeptide has three
Conformational Geometry of Peptides and Proteins: Before discussing secondary structure, it is important to appreciate the conformational plasticity of proteins. Each residue in a polypeptide has three
Orientational degeneracy in the presence of one alignment tensor.
 Orientational degeneracy in the presence of one alignment tensor. Rotation about the x, y and z axes can be performed in the aligned mode of the program to examine the four degenerate orientations of two
Orientational degeneracy in the presence of one alignment tensor. Rotation about the x, y and z axes can be performed in the aligned mode of the program to examine the four degenerate orientations of two
114 Grundlagen der Bioinformatik, SS 09, D. Huson, July 6, 2009
 114 Grundlagen der Bioinformatik, SS 09, D. Huson, July 6, 2009 9 Protein tertiary structure Sources for this chapter, which are all recommended reading: D.W. Mount. Bioinformatics: Sequences and Genome
114 Grundlagen der Bioinformatik, SS 09, D. Huson, July 6, 2009 9 Protein tertiary structure Sources for this chapter, which are all recommended reading: D.W. Mount. Bioinformatics: Sequences and Genome
ALL LECTURES IN SB Introduction
 1. Introduction 2. Molecular Architecture I 3. Molecular Architecture II 4. Molecular Simulation I 5. Molecular Simulation II 6. Bioinformatics I 7. Bioinformatics II 8. Prediction I 9. Prediction II ALL
1. Introduction 2. Molecular Architecture I 3. Molecular Architecture II 4. Molecular Simulation I 5. Molecular Simulation II 6. Bioinformatics I 7. Bioinformatics II 8. Prediction I 9. Prediction II ALL
Protein Science (1997), 6: Cambridge University Press. Printed in the USA. Copyright 1997 The Protein Society
, 6: Cambridge University Press. Printed in the USA. Copyright 1997 The Protein Society") 1 of 5 1/30/00 8:08 PM Protein Science (1997), 6: 246-248. Cambridge University Press. Printed in the USA. Copyright 1997 The Protein Society FOR THE RECORD LPFC: An Internet library of protein family
1 of 5 1/30/00 8:08 PM Protein Science (1997), 6: 246-248. Cambridge University Press. Printed in the USA. Copyright 1997 The Protein Society FOR THE RECORD LPFC: An Internet library of protein family
Molecular Modeling. Prediction of Protein 3D Structure from Sequence. Vimalkumar Velayudhan. May 21, 2007
 Molecular Modeling Prediction of Protein 3D Structure from Sequence Vimalkumar Velayudhan Jain Institute of Vocational and Advanced Studies May 21, 2007 Vimalkumar Velayudhan Molecular Modeling 1/23 Outline
Molecular Modeling Prediction of Protein 3D Structure from Sequence Vimalkumar Velayudhan Jain Institute of Vocational and Advanced Studies May 21, 2007 Vimalkumar Velayudhan Molecular Modeling 1/23 Outline
STRUCTURAL BIOINFORMATICS. Barry Grant University of Michigan
 STRUCTURAL BIOINFORMATICS Barry Grant University of Michigan www.thegrantlab.org bjgrant@umich.edu Bergen, Norway 28-Sep-2015 Objective: Provide an introduction to the practice of structural bioinformatics,
STRUCTURAL BIOINFORMATICS Barry Grant University of Michigan www.thegrantlab.org bjgrant@umich.edu Bergen, Norway 28-Sep-2015 Objective: Provide an introduction to the practice of structural bioinformatics,
Biomolecules: lecture 10
 Biomolecules: lecture 10 - understanding in detail how protein 3D structures form - realize that protein molecules are not static wire models but instead dynamic, where in principle every atom moves (yet
Biomolecules: lecture 10 - understanding in detail how protein 3D structures form - realize that protein molecules are not static wire models but instead dynamic, where in principle every atom moves (yet
Protein Structure. Hierarchy of Protein Structure. Tertiary structure. independently stable structural unit. includes disulfide bonds
 Protein Structure Hierarchy of Protein Structure 2 3 Structural element Primary structure Secondary structure Super-secondary structure Domain Tertiary structure Quaternary structure Description amino
Protein Structure Hierarchy of Protein Structure 2 3 Structural element Primary structure Secondary structure Super-secondary structure Domain Tertiary structure Quaternary structure Description amino
BCH 4053 Spring 2003 Chapter 6 Lecture Notes
 BCH 4053 Spring 2003 Chapter 6 Lecture Notes 1 CHAPTER 6 Proteins: Secondary, Tertiary, and Quaternary Structure 2 Levels of Protein Structure Primary (sequence) Secondary (ordered structure along peptide
BCH 4053 Spring 2003 Chapter 6 Lecture Notes 1 CHAPTER 6 Proteins: Secondary, Tertiary, and Quaternary Structure 2 Levels of Protein Structure Primary (sequence) Secondary (ordered structure along peptide
From Amino Acids to Proteins - in 4 Easy Steps
 From Amino Acids to Proteins - in 4 Easy Steps Although protein structure appears to be overwhelmingly complex, you can provide your students with a basic understanding of how proteins fold by focusing
From Amino Acids to Proteins - in 4 Easy Steps Although protein structure appears to be overwhelmingly complex, you can provide your students with a basic understanding of how proteins fold by focusing
Secondary Structure. Bioch/BIMS 503 Lecture 2. Structure and Function of Proteins. Further Reading. Φ, Ψ angles alone determine protein structure
 Bioch/BIMS 503 Lecture 2 Structure and Function of Proteins August 28, 2008 Robert Nakamoto rkn3c@virginia.edu 2-0279 Secondary Structure Φ Ψ angles determine protein structure Φ Ψ angles are restricted
Bioch/BIMS 503 Lecture 2 Structure and Function of Proteins August 28, 2008 Robert Nakamoto rkn3c@virginia.edu 2-0279 Secondary Structure Φ Ψ angles determine protein structure Φ Ψ angles are restricted
Protein Structures. Sequences of amino acid residues 20 different amino acids. Quaternary. Primary. Tertiary. Secondary. 10/8/2002 Lecture 12 1
 Protein Structures Sequences of amino acid residues 20 different amino acids Primary Secondary Tertiary Quaternary 10/8/2002 Lecture 12 1 Angles φ and ψ in the polypeptide chain 10/8/2002 Lecture 12 2
Protein Structures Sequences of amino acid residues 20 different amino acids Primary Secondary Tertiary Quaternary 10/8/2002 Lecture 12 1 Angles φ and ψ in the polypeptide chain 10/8/2002 Lecture 12 2
Lecture 10 (10/4/17) Lecture 10 (10/4/17)
 Lecture 10 (10/4/17)") Lecture 10 (10/4/17) Reading: Ch4; 125, 138-141, 141-142 Problems: Ch4 (text); 7, 9, 11 Ch4 (study guide); 1, 2 NEXT Reading: Ch4; 125, 132-136 (structure determination) Ch4; 12-130 (Collagen) Problems:
Lecture 10 (10/4/17) Reading: Ch4; 125, 138-141, 141-142 Problems: Ch4 (text); 7, 9, 11 Ch4 (study guide); 1, 2 NEXT Reading: Ch4; 125, 132-136 (structure determination) Ch4; 12-130 (Collagen) Problems:
CHEM 463: Advanced Inorganic Chemistry Modeling Metalloproteins for Structural Analysis
 CHEM 463: Advanced Inorganic Chemistry Modeling Metalloproteins for Structural Analysis Purpose: The purpose of this laboratory is to introduce some of the basic visualization and modeling tools for viewing
CHEM 463: Advanced Inorganic Chemistry Modeling Metalloproteins for Structural Analysis Purpose: The purpose of this laboratory is to introduce some of the basic visualization and modeling tools for viewing
Bioinformatics. Macromolecular structure
 Bioinformatics Macromolecular structure Contents Determination of protein structure Structure databases Secondary structure elements (SSE) Tertiary structure Structure analysis Structure alignment Domain
Bioinformatics Macromolecular structure Contents Determination of protein structure Structure databases Secondary structure elements (SSE) Tertiary structure Structure analysis Structure alignment Domain
Model Mélange. Physical Models of Peptides and Proteins
 Model Mélange Physical Models of Peptides and Proteins In the Model Mélange activity, you will visit four different stations each featuring a variety of different physical models of peptides or proteins.
Model Mélange Physical Models of Peptides and Proteins In the Model Mélange activity, you will visit four different stations each featuring a variety of different physical models of peptides or proteins.
HMM applications. Applications of HMMs. Gene finding with HMMs. Using the gene finder
 HMM applications Applications of HMMs Gene finding Pairwise alignment (pair HMMs) Characterizing protein families (profile HMMs) Predicting membrane proteins, and membrane protein topology Gene finding
HMM applications Applications of HMMs Gene finding Pairwise alignment (pair HMMs) Characterizing protein families (profile HMMs) Predicting membrane proteins, and membrane protein topology Gene finding
Protein Structures: Experiments and Modeling. Patrice Koehl
 Protein Structures: Experiments and Modeling Patrice Koehl Structural Bioinformatics: Proteins Proteins: Sources of Structure Information Proteins: Homology Modeling Proteins: Ab initio prediction Proteins:
Protein Structures: Experiments and Modeling Patrice Koehl Structural Bioinformatics: Proteins Proteins: Sources of Structure Information Proteins: Homology Modeling Proteins: Ab initio prediction Proteins:
Protein structure analysis. Risto Laakso 10th January 2005
 Protein structure analysis Risto Laakso risto.laakso@hut.fi 10th January 2005 1 1 Summary Various methods of protein structure analysis were examined. Two proteins, 1HLB (Sea cucumber hemoglobin) and 1HLM
Protein structure analysis Risto Laakso risto.laakso@hut.fi 10th January 2005 1 1 Summary Various methods of protein structure analysis were examined. Two proteins, 1HLB (Sea cucumber hemoglobin) and 1HLM
FlexSADRA: Flexible Structural Alignment using a Dimensionality Reduction Approach
 FlexSADRA: Flexible Structural Alignment using a Dimensionality Reduction Approach Shirley Hui and Forbes J. Burkowski University of Waterloo, 200 University Avenue W., Waterloo, Canada ABSTRACT A topic
FlexSADRA: Flexible Structural Alignment using a Dimensionality Reduction Approach Shirley Hui and Forbes J. Burkowski University of Waterloo, 200 University Avenue W., Waterloo, Canada ABSTRACT A topic
Protein Structure. Role of (bio)informatics in drug discovery. Bioinformatics
informatics in drug discovery. Bioinformatics") Bioinformatics Protein Structure Principles & Architecture Marjolein Thunnissen Dep. of Biochemistry & Structural Biology Lund University September 2011 Homology, pattern and 3D structure searches need
Bioinformatics Protein Structure Principles & Architecture Marjolein Thunnissen Dep. of Biochemistry & Structural Biology Lund University September 2011 Homology, pattern and 3D structure searches need
Details of Protein Structure
 Details of Protein Structure Function, evolution & experimental methods Thomas Blicher, Center for Biological Sequence Analysis Anne Mølgaard, Kemisk Institut, Københavns Universitet Learning Objectives
Details of Protein Structure Function, evolution & experimental methods Thomas Blicher, Center for Biological Sequence Analysis Anne Mølgaard, Kemisk Institut, Københavns Universitet Learning Objectives
Identification of Representative Protein Sequence and Secondary Structure Prediction Using SVM Approach
 Identification of Representative Protein Sequence and Secondary Structure Prediction Using SVM Approach Prof. Dr. M. A. Mottalib, Md. Rahat Hossain Department of Computer Science and Information Technology
Identification of Representative Protein Sequence and Secondary Structure Prediction Using SVM Approach Prof. Dr. M. A. Mottalib, Md. Rahat Hossain Department of Computer Science and Information Technology
Get familiar with PDBsum and the PDB Extract atomic coordinates from protein data files Compute bond angles and dihedral angles
 CS483 Assignment #2 Due date: Mar. 1 at the start of class. Protein Geometry Bedbug spit? Just say NO! Purpose of this assignment Get familiar with PDBsum and the PDB Extract atomic coordinates from protein
CS483 Assignment #2 Due date: Mar. 1 at the start of class. Protein Geometry Bedbug spit? Just say NO! Purpose of this assignment Get familiar with PDBsum and the PDB Extract atomic coordinates from protein
F. Piazza Center for Molecular Biophysics and University of Orléans, France. Selected topic in Physical Biology. Lecture 1
 Zhou Pei-Yuan Centre for Applied Mathematics, Tsinghua University November 2013 F. Piazza Center for Molecular Biophysics and University of Orléans, France Selected topic in Physical Biology Lecture 1
Zhou Pei-Yuan Centre for Applied Mathematics, Tsinghua University November 2013 F. Piazza Center for Molecular Biophysics and University of Orléans, France Selected topic in Physical Biology Lecture 1
Protein Secondary Structure Prediction
 Protein Secondary Structure Prediction Doug Brutlag & Scott C. Schmidler Overview Goals and problem definition Existing approaches Classic methods Recent successful approaches Evaluating prediction algorithms
Protein Secondary Structure Prediction Doug Brutlag & Scott C. Schmidler Overview Goals and problem definition Existing approaches Classic methods Recent successful approaches Evaluating prediction algorithms
Homology Modeling. Roberto Lins EPFL - summer semester 2005
 Homology Modeling Roberto Lins EPFL - summer semester 2005 Disclaimer: course material is mainly taken from: P.E. Bourne & H Weissig, Structural Bioinformatics; C.A. Orengo, D.T. Jones & J.M. Thornton,
Homology Modeling Roberto Lins EPFL - summer semester 2005 Disclaimer: course material is mainly taken from: P.E. Bourne & H Weissig, Structural Bioinformatics; C.A. Orengo, D.T. Jones & J.M. Thornton,
Protein structure (and biomolecular structure more generally) CS/CME/BioE/Biophys/BMI 279 Sept. 28 and Oct. 3, 2017 Ron Dror
 CS/CME/BioE/Biophys/BMI 279 Sept. 28 and Oct. 3, 2017 Ron Dror") Protein structure (and biomolecular structure more generally) CS/CME/BioE/Biophys/BMI 279 Sept. 28 and Oct. 3, 2017 Ron Dror Please interrupt if you have questions, and especially if you re confused! Assignment
Protein structure (and biomolecular structure more generally) CS/CME/BioE/Biophys/BMI 279 Sept. 28 and Oct. 3, 2017 Ron Dror Please interrupt if you have questions, and especially if you re confused! Assignment
HIV protease inhibitor. Certain level of function can be found without structure. But a structure is a key to understand the detailed mechanism.
 Proteins are linear polypeptide chains (one or more) Building blocks: 20 types of amino acids. Range from a few 10s-1000s They fold into varying three-dimensional shapes structure medicine Certain level
Proteins are linear polypeptide chains (one or more) Building blocks: 20 types of amino acids. Range from a few 10s-1000s They fold into varying three-dimensional shapes structure medicine Certain level
CS612 - Algorithms in Bioinformatics
 Fall 2017 Databases and Protein Structure Representation October 2, 2017 Molecular Biology as Information Science > 12, 000 genomes sequenced, mostly bacterial (2013) > 5x10 6 unique sequences available
Fall 2017 Databases and Protein Structure Representation October 2, 2017 Molecular Biology as Information Science > 12, 000 genomes sequenced, mostly bacterial (2013) > 5x10 6 unique sequences available
BIRKBECK COLLEGE (University of London)
") BIRKBECK COLLEGE (University of London) SCHOOL OF BIOLOGICAL SCIENCES M.Sc. EXAMINATION FOR INTERNAL STUDENTS ON: Postgraduate Certificate in Principles of Protein Structure MSc Structural Molecular Biology
BIRKBECK COLLEGE (University of London) SCHOOL OF BIOLOGICAL SCIENCES M.Sc. EXAMINATION FOR INTERNAL STUDENTS ON: Postgraduate Certificate in Principles of Protein Structure MSc Structural Molecular Biology
Protein structure prediction. CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror
 Protein structure prediction CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror 1 Outline Why predict protein structure? Can we use (pure) physics-based methods? Knowledge-based methods Two major
Protein structure prediction CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror 1 Outline Why predict protein structure? Can we use (pure) physics-based methods? Knowledge-based methods Two major
Reconstruction of Protein Backbone with the α-carbon Coordinates *
 JOURNAL OF INFORMATION SCIENCE AND ENGINEERING 26, 1107-1119 (2010) Reconstruction of Protein Backbone with the α-carbon Coordinates * JEN-HUI WANG, CHANG-BIAU YANG + AND CHIOU-TING TSENG Department of
JOURNAL OF INFORMATION SCIENCE AND ENGINEERING 26, 1107-1119 (2010) Reconstruction of Protein Backbone with the α-carbon Coordinates * JEN-HUI WANG, CHANG-BIAU YANG + AND CHIOU-TING TSENG Department of
Structural Alignment of Proteins
 Goal Align protein structures Structural Alignment of Proteins 1 2 3 4 5 6 7 8 9 10 11 12 13 14 PHE ASP ILE CYS ARG LEU PRO GLY SER ALA GLU ALA VAL CYS PHE ASN VAL CYS ARG THR PRO --- --- --- GLU ALA ILE
Goal Align protein structures Structural Alignment of Proteins 1 2 3 4 5 6 7 8 9 10 11 12 13 14 PHE ASP ILE CYS ARG LEU PRO GLY SER ALA GLU ALA VAL CYS PHE ASN VAL CYS ARG THR PRO --- --- --- GLU ALA ILE
Packing of Secondary Structures
 7.88 Lecture Notes - 4 7.24/7.88J/5.48J The Protein Folding and Human Disease Professor Gossard Retrieving, Viewing Protein Structures from the Protein Data Base Helix helix packing Packing of Secondary
7.88 Lecture Notes - 4 7.24/7.88J/5.48J The Protein Folding and Human Disease Professor Gossard Retrieving, Viewing Protein Structures from the Protein Data Base Helix helix packing Packing of Secondary
B. β Structure. All contents of this document, unless otherwise noted, are David C. & Jane S. Richardson. All Rights Reserved.
 B. β Structure The other major structural element found in globular proteins is the β sheet. Historically, it was first observed as the β, or extended, form of keratin fibers. An approximate understanding
B. β Structure The other major structural element found in globular proteins is the β sheet. Historically, it was first observed as the β, or extended, form of keratin fibers. An approximate understanding
Protein Dynamics. The space-filling structures of myoglobin and hemoglobin show that there are no pathways for O 2 to reach the heme iron.
 Protein Dynamics The space-filling structures of myoglobin and hemoglobin show that there are no pathways for O 2 to reach the heme iron. Below is myoglobin hydrated with 350 water molecules. Only a small
Protein Dynamics The space-filling structures of myoglobin and hemoglobin show that there are no pathways for O 2 to reach the heme iron. Below is myoglobin hydrated with 350 water molecules. Only a small
Protein Structure and Function. Protein Architecture:
 BCHS 6229 Protein Structure and Function Lecture 2 (October 13, 2011) Protein Architecture: Symmetry relationships and protein structure Primary & Secondary Structure Motifs & Super-secondary Structure
BCHS 6229 Protein Structure and Function Lecture 2 (October 13, 2011) Protein Architecture: Symmetry relationships and protein structure Primary & Secondary Structure Motifs & Super-secondary Structure
D Dobbs ISU - BCB 444/544X 1
 11/7/05 Protein Structure: Classification, Databases, Visualization Announcements BCB 544 Projects - Important Dates: Nov 2 Wed noon - Project proposals due to David/Drena Nov 4 Fri PM - Approvals/responses
11/7/05 Protein Structure: Classification, Databases, Visualization Announcements BCB 544 Projects - Important Dates: Nov 2 Wed noon - Project proposals due to David/Drena Nov 4 Fri PM - Approvals/responses
proteins Comprehensive description of protein structures using protein folding shape code Jiaan Yang* INTRODUCTION
 proteins STRUCTURE O FUNCTION O BIOINFORMATICS Comprehensive description of protein structures using protein folding shape code Jiaan Yang* MicrotechNano, LLC, Indianapolis, Indiana 46234 ABSTRACT Understanding
proteins STRUCTURE O FUNCTION O BIOINFORMATICS Comprehensive description of protein structures using protein folding shape code Jiaan Yang* MicrotechNano, LLC, Indianapolis, Indiana 46234 ABSTRACT Understanding
Ramachandran and his Map
 Ramachandran and his Map C Ramakrishnan Introduction C Ramakrishnan, retired professor from Molecular Biophysics Unit, Indian Institute of Science, Bangalore, has been associated with Professor G N Ramachandran
Ramachandran and his Map C Ramakrishnan Introduction C Ramakrishnan, retired professor from Molecular Biophysics Unit, Indian Institute of Science, Bangalore, has been associated with Professor G N Ramachandran
Ant Colony Approach to Predict Amino Acid Interaction Networks
 Ant Colony Approach to Predict Amino Acid Interaction Networks Omar Gaci, Stefan Balev To cite this version: Omar Gaci, Stefan Balev. Ant Colony Approach to Predict Amino Acid Interaction Networks. IEEE
Ant Colony Approach to Predict Amino Acid Interaction Networks Omar Gaci, Stefan Balev To cite this version: Omar Gaci, Stefan Balev. Ant Colony Approach to Predict Amino Acid Interaction Networks. IEEE
NMR of Nucleic Acids. K.V.R. Chary Workshop on NMR and it s applications in Biological Systems November 26, 2009
 MR of ucleic Acids K.V.R. Chary chary@tifr.res.in Workshop on MR and it s applications in Biological Systems TIFR ovember 26, 2009 ucleic Acids are Polymers of ucleotides Each nucleotide consists of a
MR of ucleic Acids K.V.R. Chary chary@tifr.res.in Workshop on MR and it s applications in Biological Systems TIFR ovember 26, 2009 ucleic Acids are Polymers of ucleotides Each nucleotide consists of a
Lecture 26: Polymers: DNA Packing and Protein folding 26.1 Problem Set 4 due today. Reading for Lectures 22 24: PKT Chapter 8 [ ].
![Lecture 26: Polymers: DNA Packing and Protein folding 26.1 Problem Set 4 due today. Reading for Lectures 22 24: PKT Chapter 8 [ ].](/thumbs/84/90810734.jpg "Lecture 26: Polymers: DNA Packing and Protein folding 26.1 Problem Set 4 due today. Reading for Lectures 22 24: PKT Chapter 8 [ ].") Lecture 26: Polymers: DA Packing and Protein folding 26.1 Problem Set 4 due today. eading for Lectures 22 24: PKT hapter 8 DA Packing for Eukaryotes: The packing problem for the larger eukaryotic genomes
Lecture 26: Polymers: DA Packing and Protein folding 26.1 Problem Set 4 due today. eading for Lectures 22 24: PKT hapter 8 DA Packing for Eukaryotes: The packing problem for the larger eukaryotic genomes
arxiv:q-bio/ v1 [q-bio.mn] 13 Aug 2004
![arxiv:q-bio/ v1 [q-bio.mn] 13 Aug 2004](/thumbs/80/82320686.jpg "arxiv:q-bio/ v1 [q-bio.mn] 13 Aug 2004") Network properties of protein structures Ganesh Bagler and Somdatta Sinha entre for ellular and Molecular Biology, Uppal Road, Hyderabad 57, India (Dated: June 8, 218) arxiv:q-bio/89v1 [q-bio.mn] 13 Aug
Network properties of protein structures Ganesh Bagler and Somdatta Sinha entre for ellular and Molecular Biology, Uppal Road, Hyderabad 57, India (Dated: June 8, 218) arxiv:q-bio/89v1 [q-bio.mn] 13 Aug
Protein Structure Prediction II Lecturer: Serafim Batzoglou Scribe: Samy Hamdouche
 Protein Structure Prediction II Lecturer: Serafim Batzoglou Scribe: Samy Hamdouche The molecular structure of a protein can be broken down hierarchically. The primary structure of a protein is simply its
Protein Structure Prediction II Lecturer: Serafim Batzoglou Scribe: Samy Hamdouche The molecular structure of a protein can be broken down hierarchically. The primary structure of a protein is simply its
Theory and Applications of Residual Dipolar Couplings in Biomolecular NMR
 Theory and Applications of Residual Dipolar Couplings in Biomolecular NMR Residual Dipolar Couplings (RDC s) Relatively new technique ~ 1996 Nico Tjandra, Ad Bax- NIH, Jim Prestegard, UGA Combination of
Theory and Applications of Residual Dipolar Couplings in Biomolecular NMR Residual Dipolar Couplings (RDC s) Relatively new technique ~ 1996 Nico Tjandra, Ad Bax- NIH, Jim Prestegard, UGA Combination of
Protein structure similarities Patrice Koehl
 348 Protein structure similarities Patrice Koehl Comparison of protein structures can reveal distant evolutionary relationships that would not be detected by sequence information alone. This helps to infer
348 Protein structure similarities Patrice Koehl Comparison of protein structures can reveal distant evolutionary relationships that would not be detected by sequence information alone. This helps to infer
BIOCHEMISTRY Course Outline (Fall, 2011)
") BIOCHEMISTRY 402 - Course Outline (Fall, 2011) Number OVERVIEW OF LECTURE TOPICS: of Lectures INSTRUCTOR 1. Structural Components of Proteins G. Brayer (a) Amino Acids and the Polypeptide Chain Backbone...2
BIOCHEMISTRY 402 - Course Outline (Fall, 2011) Number OVERVIEW OF LECTURE TOPICS: of Lectures INSTRUCTOR 1. Structural Components of Proteins G. Brayer (a) Amino Acids and the Polypeptide Chain Backbone...2
CASP3 ab initio protein structure prediction results using a simple distance geometry-based method
 CASP3 ab initio protein structure prediction results using a simple distance geometry-based method Enoch S. Huang 1#, Ram Samudrala 2, and Jay W. Ponder 1* Running title: CASP3 ab initio folding results
CASP3 ab initio protein structure prediction results using a simple distance geometry-based method Enoch S. Huang 1#, Ram Samudrala 2, and Jay W. Ponder 1* Running title: CASP3 ab initio folding results
Optimization of the Sliding Window Size for Protein Structure Prediction
 Optimization of the Sliding Window Size for Protein Structure Prediction Ke Chen* 1, Lukasz Kurgan 1 and Jishou Ruan 2 1 University of Alberta, Department of Electrical and Computer Engineering, Edmonton,
Optimization of the Sliding Window Size for Protein Structure Prediction Ke Chen* 1, Lukasz Kurgan 1 and Jishou Ruan 2 1 University of Alberta, Department of Electrical and Computer Engineering, Edmonton,
Dihedral Angles. Homayoun Valafar. Department of Computer Science and Engineering, USC 02/03/10 CSCE 769
 Dihedral Angles Homayoun Valafar Department of Computer Science and Engineering, USC The precise definition of a dihedral or torsion angle can be found in spatial geometry Angle between to planes Dihedral
Dihedral Angles Homayoun Valafar Department of Computer Science and Engineering, USC The precise definition of a dihedral or torsion angle can be found in spatial geometry Angle between to planes Dihedral
2MHR. Protein structure classification is important because it organizes the protein structure universe that is independent of sequence similarity.
 Protein structure classification is important because it organizes the protein structure universe that is independent of sequence similarity. A global picture of the protein universe will help us to understand
Protein structure classification is important because it organizes the protein structure universe that is independent of sequence similarity. A global picture of the protein universe will help us to understand
Outline. Levels of Protein Structure. Primary (1 ) Structure. Lecture 6:Protein Architecture II: Secondary Structure or From peptides to proteins
 Structure. Lecture 6:Protein Architecture II: Secondary Structure or From peptides to proteins") Lecture 6:Protein Architecture II: Secondary Structure or From peptides to proteins Margaret Daugherty Fall 2004 Outline Four levels of structure are used to describe proteins; Alpha helices and beta sheets
Lecture 6:Protein Architecture II: Secondary Structure or From peptides to proteins Margaret Daugherty Fall 2004 Outline Four levels of structure are used to describe proteins; Alpha helices and beta sheets
As of December 30, 2003, 23,000 solved protein structures
 The protein structure prediction problem could be solved using the current PDB library Yang Zhang and Jeffrey Skolnick* Center of Excellence in Bioinformatics, University at Buffalo, 901 Washington Street,
The protein structure prediction problem could be solved using the current PDB library Yang Zhang and Jeffrey Skolnick* Center of Excellence in Bioinformatics, University at Buffalo, 901 Washington Street,
Homology modeling of Ferredoxin-nitrite reductase from Arabidopsis thaliana
 www.bioinformation.net Hypothesis Volume 6(3) Homology modeling of Ferredoxin-nitrite reductase from Arabidopsis thaliana Karim Kherraz*, Khaled Kherraz, Abdelkrim Kameli Biology department, Ecole Normale
www.bioinformation.net Hypothesis Volume 6(3) Homology modeling of Ferredoxin-nitrite reductase from Arabidopsis thaliana Karim Kherraz*, Khaled Kherraz, Abdelkrim Kameli Biology department, Ecole Normale
Prediction and refinement of NMR structures from sparse experimental data
 Prediction and refinement of NMR structures from sparse experimental data Jeff Skolnick Director Center for the Study of Systems Biology School of Biology Georgia Institute of Technology Overview of talk
Prediction and refinement of NMR structures from sparse experimental data Jeff Skolnick Director Center for the Study of Systems Biology School of Biology Georgia Institute of Technology Overview of talk
Molecular Modelling. part of Bioinformatik von RNA- und Proteinstrukturen. Sonja Prohaska. Leipzig, SS Computational EvoDevo University Leipzig
 part of Bioinformatik von RNA- und Proteinstrukturen Computational EvoDevo University Leipzig Leipzig, SS 2011 Protein Structure levels or organization Primary structure: sequence of amino acids (from
part of Bioinformatik von RNA- und Proteinstrukturen Computational EvoDevo University Leipzig Leipzig, SS 2011 Protein Structure levels or organization Primary structure: sequence of amino acids (from
4 Proteins: Structure, Function, Folding W. H. Freeman and Company
 4 Proteins: Structure, Function, Folding 2013 W. H. Freeman and Company CHAPTER 4 Proteins: Structure, Function, Folding Learning goals: Structure and properties of the peptide bond Structural hierarchy
4 Proteins: Structure, Function, Folding 2013 W. H. Freeman and Company CHAPTER 4 Proteins: Structure, Function, Folding Learning goals: Structure and properties of the peptide bond Structural hierarchy
Principles of Physical Biochemistry
 Principles of Physical Biochemistry Kensal E. van Hold e W. Curtis Johnso n P. Shing Ho Preface x i PART 1 MACROMOLECULAR STRUCTURE AND DYNAMICS 1 1 Biological Macromolecules 2 1.1 General Principles
Principles of Physical Biochemistry Kensal E. van Hold e W. Curtis Johnso n P. Shing Ho Preface x i PART 1 MACROMOLECULAR STRUCTURE AND DYNAMICS 1 1 Biological Macromolecules 2 1.1 General Principles
C--H...O Hydrogen Bonds in /~-sheets
 316 Acta Cryst. (1997). D53, 316-320 C--H...O Hydrogen Bonds in /~-sheets G. FELCY FABIOLA, u S. KRISHNASWAMY, b V. NAGARAJAN" AND VASANTHA PATTABHI ~* ~Department of Crystallography and Biophysics, University
316 Acta Cryst. (1997). D53, 316-320 C--H...O Hydrogen Bonds in /~-sheets G. FELCY FABIOLA, u S. KRISHNASWAMY, b V. NAGARAJAN" AND VASANTHA PATTABHI ~* ~Department of Crystallography and Biophysics, University
Tools for Cryo-EM Map Fitting. Paul Emsley MRC Laboratory of Molecular Biology
 Tools for Cryo-EM Map Fitting Paul Emsley MRC Laboratory of Molecular Biology April 2017 Cryo-EM model-building typically need to move more atoms that one does for crystallography the maps are lower resolution
Tools for Cryo-EM Map Fitting Paul Emsley MRC Laboratory of Molecular Biology April 2017 Cryo-EM model-building typically need to move more atoms that one does for crystallography the maps are lower resolution
Reconstructing Amino Acid Interaction Networks by an Ant Colony Approach
 Author manuscript, published in "Journal of Computational Intelligence in Bioinformatics 2, 2 (2009) 131-146" Reconstructing Amino Acid Interaction Networks by an Ant Colony Approach Omar GACI and Stefan
Author manuscript, published in "Journal of Computational Intelligence in Bioinformatics 2, 2 (2009) 131-146" Reconstructing Amino Acid Interaction Networks by an Ant Colony Approach Omar GACI and Stefan