Quan%ta%on with XPRESS. and. ASAPRa%o
|
|
|
- Adam Harmon
- 5 years ago
- Views:
Transcription

1 Quan%ta%on with XPRESS and ASAPRa%o 1

2 Pep%de and Protein Quan%ta%on Raw Mass Spec Data Pep%de Iden%fica%on Pep%de Valida%on Quan%ta%on Protein Assignment Protein List msconvert X!Tandem SpectraST SEQUEST* Mascot* Pep%deProphet iprophet PTMProphet ASAPRa%o XPRESS Libra ProteinProphet SBEAMS PIPE2 mzml pepxml protxml
3 Lecture Outline Principles of quan%ta%ve proteomics using LC- ESI- MS/MS Pep%de and Protein Quan%ta%on with XPRESS Running XPRESS Looking at results Pep%de and Protein Quan%ta%on with ASAPRa%o Running ASAPRaBo Looking at results Exercises 3


4 Summary of LC- ESI- MS/MS Protein mixtures are digested into pep%des Pep%des are concentrated and frac%onated by separa%on technologies such as SCX, IEF, RP, etc. While elu%ng from RP column, pep%des are ionized by ESI and analyzed by MS/MS Pep%des are iden%fied from CID spectra Pep%des are usually quan%fied from MS signatures Except in the case of itraq Denatured protein complex Peptides RP chromatographic separation of peptides Identify proteins in complex Db search Mass Spec 4
5 Complica%ons Shotgun MS detects pep%des not proteins MulBple pepbdes per protein MulBple proteins per pepbde Strong Ca%on- Exchange Chromatograph Fair but not great separabon power Same pepbde separated into several fracbons 5
6 Reversed- Phase Chromatography Reproducible: but a few erra%c data points may exist Tapered frit! - 2 to 4 kv! UV detector HPLC Peek microcross! Analytical Column (100 µm i.d.)! Grounded! waste close Precolumn! Peptide eluting profiling 6-way Divert Valve time 6
7 Electrospray Ioniza%on Mul%ple charge states: from +1 to +4 M + z H + = M(H + ) z m/z = (M+z*H)/z LC +1 ESI + HV - time
8 ESI- Tandem Mass Spectrometry identified peptides quantify peptides MS MS/MS (CID) identify peptides 8
9 Pep%de Iden%fica%on Match CID (MS/MS) spectra with database SEQUEST, MASCOT, X!Tandem, Mul%ple IDs for the same pep%de different isotopes: light and heavy different charge states: +1, +2, +3 repeabng IDs: same isotope and same charge state!!!!!! y4 y5 y6 y7 y8 y9 y10 y11 y12 y13 y14 y15 y16 y17 D I N N T I Q S L T A D A b2 b3 b4 b5 b6 b7 b8 b9 b10 b11 b12 b13 b14 A D A T L S Q I T N N I D b
10 Single Ion Chromatogram 2D view: m/z, intensity 3D view: m/z, intensity, time MS scans intensity intensity m/z Single Ion Current (SIC) Trace intensity time (scan #) m/z= scan # 10
11 Pep%de Quan%ta%on Area under SIC is propor%onal to pep%de abundance PROBLEM Ioniza%on efficiency of each pep%de is different Depends on the pepbde molecular properbes (e.g. number of basic residues) ONE SOLUTION Samples labeled with different stable isotopes Chemically idenbcal PepBdes are idenbfied before quanbficabon DisBnguishable by MS in mass shi\ PepBde abundance rabo measured by rabo of SIC areas 11

12 Different Labeling Methods Metabolic labeling 13 C, 15 N, SILAC Chemical reac%on ICAT, cleavable ICAT itraq Enzyme reac%on 18 O 12
13 Summary of Quan%ta%ve LC- MS/MS Approach Samples are isotopically labeled Simultaneously iden%fy & quan%fy thousands of proteins in complex samples PepBde ion must be idenbfied in MS 2 spectrum to be quanbfied Accuracy: ±10-30% Dynamic range: ~100 fold TPP provides 2 op%ons: Xpress and ASAPRa%o 13
14 Protein Iden%fica%on and Quan%fica%on Hierarchy Structure protein VNG0679G peptide GCPTAELRFDDMR LGDKGCPTAELR LC peak haloicat2_33 (scan 1274) haloicat2_32 (scan 1306) haloicat2_33 (scan 1024) CID heavy, +2 heavy, +2 light, +2 light, +3 heavy, +3 Peptide IDs & Ratios Protein IDs & Ratios 14
15 Lecture Outline Principles of quanbtabve proteomics using LC- ESI- MS/ MS Pep%de and Protein Quan%ta%on with XPRESS Running XPRESS Looking at results PepBde and Protein QuanBtaBon with ASAPRaBo Running ASAPRaBo Looking at results Exercises 15

16 XPRESS Publica%on Han DK, Eng J, Zhou H, and Aebersold R. (2001) Nature Biotechnology 19:
17 XPRESS Pep%de Ra%o Calculated from SIC of charge state in which pep%de was iden%fied Smoothing done with a Buherworth low- pass filter No background es%ma%on Works with different labeling methods ICAT, SILAC, etc 17
18 XPRESS Protein Ra%o Calculated as the Geometric Mean of the cons%tuent pep%de ra%os Uncertainty is also calculated 18

19 Running XPRESS: Petunia Interface 19
20 Running XPRESS: Command- line Use the X flag for xinteract xpressoptions [will run XPRESS analysis with any specified options that follow the 'X']: -m<num> change XPRESS mass tolerance (default=1.0) -l<str> change labeled residues (default='c') -n<str>,<num> change XPRESS residue mass difference for <str> to <num> (default=9.0) -b heavy labeled peptide elutes before light labeled -F<num> partner fix elution peak area as +-<num> scans (<num> optional, default=5) from peak apex -L for ratio, set/fix light to 1, vary heavy -H for ratio, set/fix heavy to 1, vary light -M for metabolic labeling; ignore all other parameters, assume IDs are normal and quantify w/corresponding 15N heavy pair -N for metabolic labeling; ignore all other parameters, assume IDs are 15N heavy and quantify corresponding 14N light pair 20
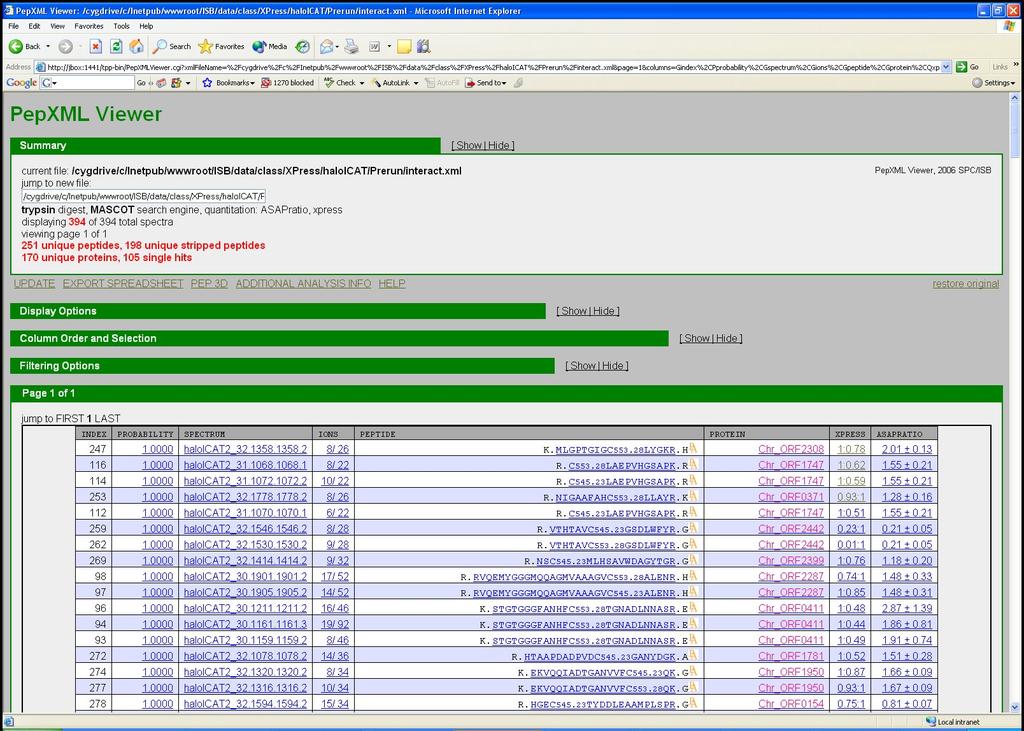
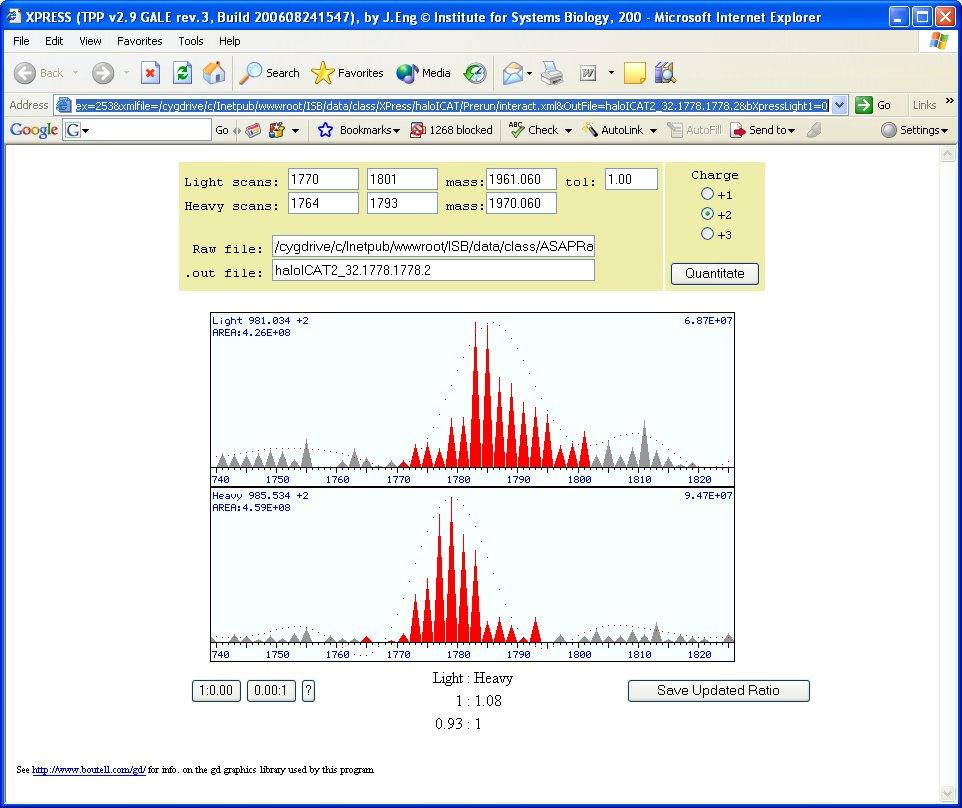
21 XPRESS Pep%deProphet Results 21
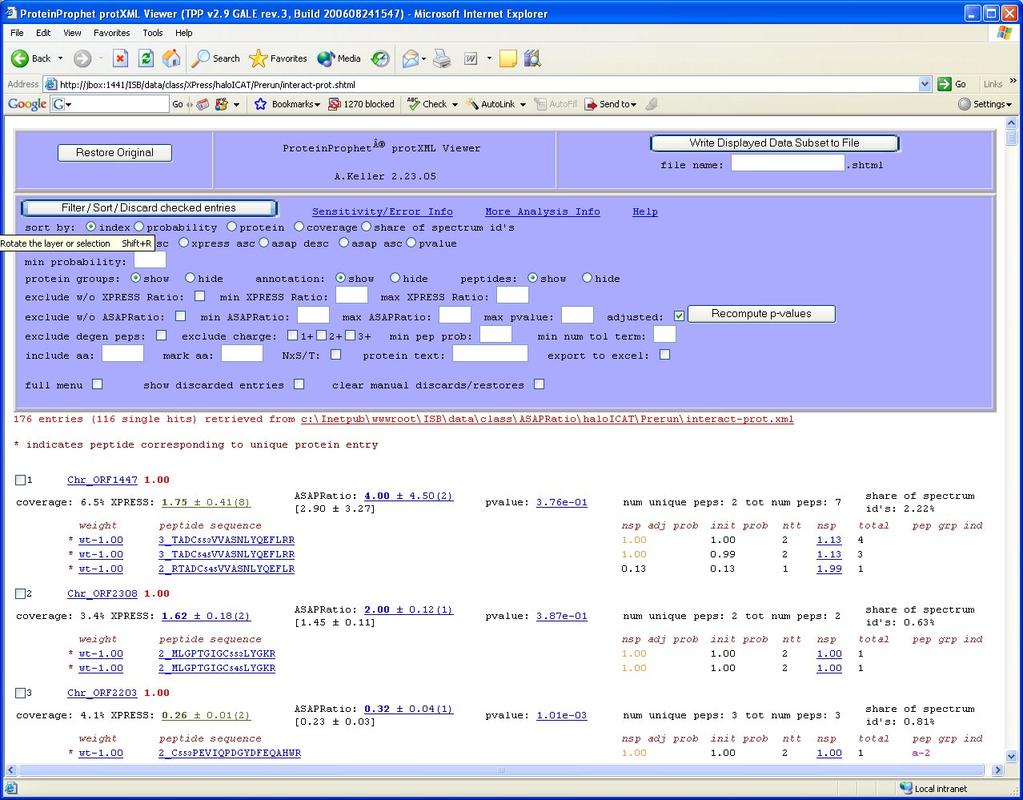

22 XPRESS ProteinProphet Results 22
23 Lecture Outline Principles of quanbtabve proteomics using LC- ESI- MS/MS PepBde and Protein QuanBtaBon with XPRESS Running XPRESS Looking at results Pep%de and Protein Quan%ta%on with ASAPRa%o Running ASAPRa%o Looking at results Exercises 23
24 Defini%ons protein Protein Ratio VNG0679G Unique peptide ratio peptide GCPTAELRFDDMR LGDKGCPTAELR LC peak haloicat2_33 (scan 1274) haloicat2_32 (scan 1306) haloicat2_33 (scan 1024) CID heavy, +2 heavy, +2 light, +2 light, +3 heavy, +3 Peptide (CID) ratio 24
25 ASAPRa%o Methodology Reconstruc%on of single- ion chromatograms Evalua%on of pep%de abundance ra%os Evalua%on of unique pep%de abundance ra%os Evalua%on of protein abundance ra%os Sample- dependent ra%o normaliza%on Large- scale protein profiling Anal. Chem.; 2003; 75(23) pp
26 Reconstruc%on of Single- Ion Chromatogram Assume pep%de iden%fica%on correct Raw chromatogram Summarize MS intensibes within a m/z window and trace the sum in Bme Smooth chromatogram Savitzsky- Golay smooth filter Subtract background and calculate area Es%mate elu%on %me of isotopic partner 26
27 Example on Single- Ion Chromatogram Red: raw Blue: fitting Green: area Pink: background T-bar: CID 27
28 Pep%de Charge Distribu%on Out of 1857 peptides
29 Single-Ion Chromatogram of +2 Ion 29
30 Single-Ion Chromatogram of +3 Ion 30
31 Single-Ion Chromatogram of +4 Ion 31
32 Evalua%on of Pep%de Abundance Ra%o Evaluate a pep%de ra%o with error from each available charge state Use Dixon s test to iden%fy any outliers Weight charge states by chromatogram areas Use sta%s%cal methods to calculate pep%de ra%o and error 32
33 Example on Peptide Ratio mean +- SD (CV%) CV = SD/mean SD: Std.Dev, CV: Coeff. Of Variation 33
34 Evalua%on of Unique Pep%de Abundance Ra%o protein VNG0679G Unique peptide ratio peptide GCPTAELRFDDMR LGDKGCPTAELR LC peak Step 2 haloicat2_33 (scan 1274) haloicat2_32 (scan 1306) haloicat2_33 (scan 1024) CID heavy, +2 heavy, +2 light, +2 light, +3 heavy, +3 Step 1 Peptide ratio 34
35 Evalua%on of Unique Pep%de Ra%o Group abundance ra%os of same pep%de and same RP elu%on peak together isotopic forms, charge states, repeats Most of them same If not: - Weight data points by their largest chromatogram areas - Calculate mean and standard deviabon - Use Dixon s test for outliers Step 1 35
36 Evalua%on of Unique Pep%de Ra%o Step 2 Group abundance ra%os of same pep%de but different RP elu%on peaks together SCX fracbons, RP elubon Bmes Weight data points by their largest chromatogram areas Calculate mean and standard devia%on Use Dixon s test for outliers 36

37 Example of Unique Peptide Ratio Step 2 Step 1 Step 1 37
38 Evalua%on of Protein Abundance Ra%o Collect all unique pep%de ra%os of same protein together Use Dixon s test on outliers misidenbficabon, modificabon, etc. Weight data points by error Use sta%s%cal methods to calculate mean and standard devia%on 38
 1.97 2.")
39 Example on Protein Ratio 0.54 (outlier)
40 Sample- Dependent Ra%o Normaliza%on To Correct Systematic Error Due to Sample Handling
41 Sample- Dependent Ra%o Normaliza%on Condi%on: Background Proteins Dominant Fit log10(unique pep%de ra%o) with normal distribu%on (Fig. 5, ASAPRa%o paper) Normalize protein ra%os by peak ra%o 41
42 Large- Scale Protein Profiling Evaluate p value for each protein p value: probability of a protein belonging to background group P value depends on: Specify significance level (by user) 42
43 ASAPRatio Main Features Able to handle various labeling methods (except itraq) Es%mate error on pep%de and protein ra%os Calculate pep%de ra%os from mul%ple charge states Not just from charge state in which the CID was matched Chromatogram signal background subtrac%on to increase the dynamic range Calculate protein ra%os based on pep%des that were assigned to proteins by ProteinProphet Evaluate p- value for protein profiling Detect outliers: Dixon s test Easy to use user interface for manual valida%on of ra%os 43

44 How to Use TPP for Data Analysis in Quan%ta%ve Proteomics Start TPP Click on Analyze Peptides Select the xml files that you want to analyze Same as when running PeptideProphet 44

45 How to Use TPP for Data Analysis in Quan%ta%ve Proteomics Select RUN XPRESS Select RUN ASAPRatio 45
46 How to Use TPP for Data Analysis in Quan%ta%ve Proteomics interact.prot.xml 46
47 How to Interpret ASAPRa%o Results 47
48 How to Interpret ASAPRatio Results Protein ratio and its standard deviation Protein p-value for differential expression Number of unique peptides Normalized protein ratio and its standard deviation 48
49 How to Interpret ASAPRatio Results Interface for protein ratio 49
50 How to Interpret ASAPRatio Results Protein profiling based on their ratios Normalized ratio: r* = r/r 0 P-value: significance in differential expression; how far is the data from r 0 50
51 How to Interpret ASAPRatio Results Individual peptides 51
52 How to Interpret ASAPRatio Results Details on individual peptides 52
53 How to Interpret ASAPRatio Results Interface for peptide ratio 53
54 How to Interpret ASAPRatio Results 54

55 How to Interpret ASAPRatio Results 55
56 How to Interpret ASAPRatio Results 56
57 How to Interpret ASAPRatio Results Changes can be made Click Evaluate Ratio for new results Notice new interim ratio If you like the changes, click on Interim Ratio under Set Accepted Ratio to for record 57
58 How to Interpret ASAPRatio Results Changes can be made Click Evaluate Ratio for new results Notice new interim ratio If you like the changes, click on Interim Ratio under Set Accepted Ratio to for record 58

59 How to Interpret ASAPRa%o Results Sort by p values first and verify potentially interesting data Identify and verify troublesome unique peptide ratios 59

60 How to Interpret ASAPRa%o Results For peptides of same experiment, verify one peptide ratio and reject others Pay attention to unusual data: large error, 1:0, 0:1, or unknown 60
61 Lecture Outline Principles of quanbtabve proteomics using LC- ESI- MS/ MS PepBde and Protein QuanBtaBon with XPRESS Running XPRESS Looking at results PepBde and Protein QuanBtaBon with ASAPRaBo Running ASAPRaBo Looking at results Exercises 61
Isotopic-Labeling and Mass Spectrometry-Based Quantitative Proteomics
 Isotopic-Labeling and Mass Spectrometry-Based Quantitative Proteomics Xiao-jun Li, Ph.D. Current address: Homestead Clinical Day 4 October 19, 2006 Protein Quantification LC-MS/MS Data XLink mzxml file
Isotopic-Labeling and Mass Spectrometry-Based Quantitative Proteomics Xiao-jun Li, Ph.D. Current address: Homestead Clinical Day 4 October 19, 2006 Protein Quantification LC-MS/MS Data XLink mzxml file
BIOINF 4120 Bioinforma2cs 2 - Structures and Systems -
 BIOINF 4120 Bioinforma2cs 2 - Structures and Systems - Oliver Kohlbacher Summer 2013 16. Quan0ta0ve Proteomics Overview LC- MS- based proteomics - defini0on of maps and features Quan0fica0on approaches
BIOINF 4120 Bioinforma2cs 2 - Structures and Systems - Oliver Kohlbacher Summer 2013 16. Quan0ta0ve Proteomics Overview LC- MS- based proteomics - defini0on of maps and features Quan0fica0on approaches
PeptideProphet: Validation of Peptide Assignments to MS/MS Spectra. Andrew Keller
 PeptideProphet: Validation of Peptide Assignments to MS/MS Spectra Andrew Keller Outline Need to validate peptide assignments to MS/MS spectra Statistical approach to validation Running PeptideProphet
PeptideProphet: Validation of Peptide Assignments to MS/MS Spectra Andrew Keller Outline Need to validate peptide assignments to MS/MS spectra Statistical approach to validation Running PeptideProphet
Overview - MS Proteomics in One Slide. MS masses of peptides. MS/MS fragments of a peptide. Results! Match to sequence database
 Overview - MS Proteomics in One Slide Obtain protein Digest into peptides Acquire spectra in mass spectrometer MS masses of peptides MS/MS fragments of a peptide Results! Match to sequence database 2 But
Overview - MS Proteomics in One Slide Obtain protein Digest into peptides Acquire spectra in mass spectrometer MS masses of peptides MS/MS fragments of a peptide Results! Match to sequence database 2 But
PeptideProphet: Validation of Peptide Assignments to MS/MS Spectra
 PeptideProphet: Validation of Peptide Assignments to MS/MS Spectra Andrew Keller Day 2 October 17, 2006 Andrew Keller Rosetta Bioinformatics, Seattle Outline Need to validate peptide assignments to MS/MS
PeptideProphet: Validation of Peptide Assignments to MS/MS Spectra Andrew Keller Day 2 October 17, 2006 Andrew Keller Rosetta Bioinformatics, Seattle Outline Need to validate peptide assignments to MS/MS
Comprehensive support for quantitation
 Comprehensive support for quantitation One of the major new features in the current release of Mascot is support for quantitation. This is still work in progress. Our goal is to support all of the popular
Comprehensive support for quantitation One of the major new features in the current release of Mascot is support for quantitation. This is still work in progress. Our goal is to support all of the popular
Tandem mass spectra were extracted from the Xcalibur data system format. (.RAW) and charge state assignment was performed using in house software
 and charge state assignment was performed using in house software") Supplementary Methods Software Interpretation of Tandem mass spectra Tandem mass spectra were extracted from the Xcalibur data system format (.RAW) and charge state assignment was performed using in house
Supplementary Methods Software Interpretation of Tandem mass spectra Tandem mass spectra were extracted from the Xcalibur data system format (.RAW) and charge state assignment was performed using in house
Introduction into Selected Reaction Monitoring (SRM) Christina Ludwig
 Christina Ludwig") Introduction into Selected Reaction Monitoring (SRM) Christina Ludwig EuPA Bioinformatics course 28.11.2013 Overview A) What is selected reac/on monitoring, how does it work and why is it useful? B) How
Introduction into Selected Reaction Monitoring (SRM) Christina Ludwig EuPA Bioinformatics course 28.11.2013 Overview A) What is selected reac/on monitoring, how does it work and why is it useful? B) How
Quantitation of a target protein in crude samples using targeted peptide quantification by Mass Spectrometry
 Quantitation of a target protein in crude samples using targeted peptide quantification by Mass Spectrometry Jon Hao, Rong Ye, and Mason Tao Poochon Scientific, Frederick, Maryland 21701 Abstract Background:
Quantitation of a target protein in crude samples using targeted peptide quantification by Mass Spectrometry Jon Hao, Rong Ye, and Mason Tao Poochon Scientific, Frederick, Maryland 21701 Abstract Background:
Computational Methods for Mass Spectrometry Proteomics
 Computational Methods for Mass Spectrometry Proteomics Eidhammer, Ingvar ISBN-13: 9780470512975 Table of Contents Preface. Acknowledgements. 1 Protein, Proteome, and Proteomics. 1.1 Primary goals for studying
Computational Methods for Mass Spectrometry Proteomics Eidhammer, Ingvar ISBN-13: 9780470512975 Table of Contents Preface. Acknowledgements. 1 Protein, Proteome, and Proteomics. 1.1 Primary goals for studying
MS-based proteomics to investigate proteins and their modifications
 MS-based proteomics to investigate proteins and their modifications Francis Impens VIB Proteomics Core October th 217 Overview Mass spectrometry-based proteomics: general workflow Identification of protein
MS-based proteomics to investigate proteins and their modifications Francis Impens VIB Proteomics Core October th 217 Overview Mass spectrometry-based proteomics: general workflow Identification of protein
SeqAn and OpenMS Integration Workshop. Temesgen Dadi, Julianus Pfeuffer, Alexander Fillbrunn The Center for Integrative Bioinformatics (CIBI)
") SeqAn and OpenMS Integration Workshop Temesgen Dadi, Julianus Pfeuffer, Alexander Fillbrunn The Center for Integrative Bioinformatics (CIBI) Mass-spectrometry data analysis in KNIME Julianus Pfeuffer,
SeqAn and OpenMS Integration Workshop Temesgen Dadi, Julianus Pfeuffer, Alexander Fillbrunn The Center for Integrative Bioinformatics (CIBI) Mass-spectrometry data analysis in KNIME Julianus Pfeuffer,
Mass Spectrometry and Proteomics - Lecture 5 - Matthias Trost Newcastle University
 Mass Spectrometry and Proteomics - Lecture 5 - Matthias Trost Newcastle University matthias.trost@ncl.ac.uk Previously Proteomics Sample prep 144 Lecture 5 Quantitation techniques Search Algorithms Proteomics
Mass Spectrometry and Proteomics - Lecture 5 - Matthias Trost Newcastle University matthias.trost@ncl.ac.uk Previously Proteomics Sample prep 144 Lecture 5 Quantitation techniques Search Algorithms Proteomics
Improved 6- Plex TMT Quantification Throughput Using a Linear Ion Trap HCD MS 3 Scan Jane M. Liu, 1,2 * Michael J. Sweredoski, 2 Sonja Hess 2 *
 Improved 6- Plex TMT Quantification Throughput Using a Linear Ion Trap HCD MS 3 Scan Jane M. Liu, 1,2 * Michael J. Sweredoski, 2 Sonja Hess 2 * 1 Department of Chemistry, Pomona College, Claremont, California
Improved 6- Plex TMT Quantification Throughput Using a Linear Ion Trap HCD MS 3 Scan Jane M. Liu, 1,2 * Michael J. Sweredoski, 2 Sonja Hess 2 * 1 Department of Chemistry, Pomona College, Claremont, California
A Software Suite for the Generation and Comparison of Peptide Arrays from Sets. of Data Collected by Liquid Chromatography-Mass Spectrometry
 MCP Papers in Press. Published on July 26, 2005 as Manuscript M500141-MCP200 A Software Suite for the Generation and Comparison of Peptide Arrays from Sets of Data Collected by Liquid Chromatography-Mass
MCP Papers in Press. Published on July 26, 2005 as Manuscript M500141-MCP200 A Software Suite for the Generation and Comparison of Peptide Arrays from Sets of Data Collected by Liquid Chromatography-Mass
Quantitative Proteomics
 Quantitative Proteomics Quantitation AND Mass Spectrometry Condition A Condition B Identify and quantify differently expressed proteins resulting from a change in the environment (stimulus, disease) Lyse
Quantitative Proteomics Quantitation AND Mass Spectrometry Condition A Condition B Identify and quantify differently expressed proteins resulting from a change in the environment (stimulus, disease) Lyse
NPTEL VIDEO COURSE PROTEOMICS PROF. SANJEEVA SRIVASTAVA
 LECTURE-25 Quantitative proteomics: itraq and TMT TRANSCRIPT Welcome to the proteomics course. Today we will talk about quantitative proteomics and discuss about itraq and TMT techniques. The quantitative
LECTURE-25 Quantitative proteomics: itraq and TMT TRANSCRIPT Welcome to the proteomics course. Today we will talk about quantitative proteomics and discuss about itraq and TMT techniques. The quantitative
MassHunter TOF/QTOF Users Meeting
 MassHunter TOF/QTOF Users Meeting 1 Qualitative Analysis Workflows Workflows in Qualitative Analysis allow the user to only see and work with the areas and dialog boxes they need for their specific tasks
MassHunter TOF/QTOF Users Meeting 1 Qualitative Analysis Workflows Workflows in Qualitative Analysis allow the user to only see and work with the areas and dialog boxes they need for their specific tasks
Key questions of proteomics. Bioinformatics 2. Proteomics. Foundation of proteomics. What proteins are there? Protein digestion
 s s Key questions of proteomics What proteins are there? Bioinformatics 2 Lecture 2 roteomics How much is there of each of the proteins? - Absolute quantitation - Stoichiometry What (modification/splice)
s s Key questions of proteomics What proteins are there? Bioinformatics 2 Lecture 2 roteomics How much is there of each of the proteins? - Absolute quantitation - Stoichiometry What (modification/splice)
Mass spectrometry has been used a lot in biology since the late 1950 s. However it really came into play in the late 1980 s once methods were
 Mass spectrometry has been used a lot in biology since the late 1950 s. However it really came into play in the late 1980 s once methods were developed to allow the analysis of large intact (bigger than
Mass spectrometry has been used a lot in biology since the late 1950 s. However it really came into play in the late 1980 s once methods were developed to allow the analysis of large intact (bigger than
Monday (March 28)- Mass spectrometry Tuesday (March 29)- Experiment 6: Separa>ons ICP-MS calcula>ons
- Mass spectrometry Tuesday (March 29)- Experiment 6: Separa>ons ICP-MS calcula>ons") LOOKING AHEAD Monday (March 28)- Mass spectrometry Tuesday (March 29)- Experiment 6: Separa>ons ICP-MS calcula>ons Thursday (March 31)- Poster crea>on? Hayes on travel Monday (April 4)- Chromatographic
LOOKING AHEAD Monday (March 28)- Mass spectrometry Tuesday (March 29)- Experiment 6: Separa>ons ICP-MS calcula>ons Thursday (March 31)- Poster crea>on? Hayes on travel Monday (April 4)- Chromatographic
MassHunter Software Overview
 MassHunter Software Overview 1 Qualitative Analysis Workflows Workflows in Qualitative Analysis allow the user to only see and work with the areas and dialog boxes they need for their specific tasks A
MassHunter Software Overview 1 Qualitative Analysis Workflows Workflows in Qualitative Analysis allow the user to only see and work with the areas and dialog boxes they need for their specific tasks A
Protein Quantitation II: Multiple Reaction Monitoring. Kelly Ruggles New York University
 Protein Quantitation II: Multiple Reaction Monitoring Kelly Ruggles kelly@fenyolab.org New York University Traditional Affinity-based proteomics Use antibodies to quantify proteins Western Blot RPPA Immunohistochemistry
Protein Quantitation II: Multiple Reaction Monitoring Kelly Ruggles kelly@fenyolab.org New York University Traditional Affinity-based proteomics Use antibodies to quantify proteins Western Blot RPPA Immunohistochemistry
Designed for Accuracy. Innovation with Integrity. High resolution quantitative proteomics LC-MS
 Designed for Accuracy High resolution quantitative proteomics Innovation with Integrity LC-MS Setting New Standards in Accuracy The development of mass spectrometry based proteomics approaches has dramatically
Designed for Accuracy High resolution quantitative proteomics Innovation with Integrity LC-MS Setting New Standards in Accuracy The development of mass spectrometry based proteomics approaches has dramatically
Methods for proteome analysis of obesity (Adipose tissue)
") Methods for proteome analysis of obesity (Adipose tissue) I. Sample preparation and liquid chromatography-tandem mass spectrometric analysis Instruments, softwares, and materials AB SCIEX Triple TOF 5600
Methods for proteome analysis of obesity (Adipose tissue) I. Sample preparation and liquid chromatography-tandem mass spectrometric analysis Instruments, softwares, and materials AB SCIEX Triple TOF 5600
iprophet: Multi-level integrative analysis of shotgun proteomic data improves peptide and protein identification rates and error estimates
 MCP Papers in Press. Published on August 29, 2011 as Manuscript M111.007690 This is the Pre-Published Version iprophet: Multi-level integrative analysis of shotgun proteomic data improves peptide and protein
MCP Papers in Press. Published on August 29, 2011 as Manuscript M111.007690 This is the Pre-Published Version iprophet: Multi-level integrative analysis of shotgun proteomic data improves peptide and protein
Tutorial 1: Setting up your Skyline document
 Tutorial 1: Setting up your Skyline document Caution! For using Skyline the number formats of your computer have to be set to English (United States). Open the Control Panel Clock, Language, and Region
Tutorial 1: Setting up your Skyline document Caution! For using Skyline the number formats of your computer have to be set to English (United States). Open the Control Panel Clock, Language, and Region
High-Throughput Protein Quantitation Using Multiple Reaction Monitoring
 High-Throughput Protein Quantitation Using Multiple Reaction Monitoring Application Note Authors Ning Tang, Christine Miller, Joe Roark, Norton Kitagawa and Keith Waddell Agilent Technologies, Inc. Santa
High-Throughput Protein Quantitation Using Multiple Reaction Monitoring Application Note Authors Ning Tang, Christine Miller, Joe Roark, Norton Kitagawa and Keith Waddell Agilent Technologies, Inc. Santa
PC235: 2008 Lecture 5: Quantitation. Arnold Falick
 PC235: 2008 Lecture 5: Quantitation Arnold Falick falickam@berkeley.edu Summary What you will learn from this lecture: There are many methods to perform quantitation using mass spectrometry (any method
PC235: 2008 Lecture 5: Quantitation Arnold Falick falickam@berkeley.edu Summary What you will learn from this lecture: There are many methods to perform quantitation using mass spectrometry (any method
All Ions MS/MS: Targeted Screening and Quantitation Using Agilent TOF and Q-TOF LC/MS Systems
 All Ions MS/MS: Targeted Screening and Quantitation Using Agilent TOF and Q-TOF LC/MS Systems Technical Overview Introduction All Ions MS/MS is a technique that is available for Agilent high resolution
All Ions MS/MS: Targeted Screening and Quantitation Using Agilent TOF and Q-TOF LC/MS Systems Technical Overview Introduction All Ions MS/MS is a technique that is available for Agilent high resolution
DIA-Umpire: comprehensive computational framework for data independent acquisition proteomics
 DIA-Umpire: comprehensive computational framework for data independent acquisition proteomics Chih-Chiang Tsou 1,2, Dmitry Avtonomov 2, Brett Larsen 3, Monika Tucholska 3, Hyungwon Choi 4 Anne-Claude Gingras
DIA-Umpire: comprehensive computational framework for data independent acquisition proteomics Chih-Chiang Tsou 1,2, Dmitry Avtonomov 2, Brett Larsen 3, Monika Tucholska 3, Hyungwon Choi 4 Anne-Claude Gingras
WADA Technical Document TD2015IDCR
 MINIMUM CRITERIA FOR CHROMATOGRAPHIC-MASS SPECTROMETRIC CONFIRMATION OF THE IDENTITY OF ANALYTES FOR DOPING CONTROL PURPOSES. The ability of a method to identify an analyte is a function of the entire
MINIMUM CRITERIA FOR CHROMATOGRAPHIC-MASS SPECTROMETRIC CONFIRMATION OF THE IDENTITY OF ANALYTES FOR DOPING CONTROL PURPOSES. The ability of a method to identify an analyte is a function of the entire
Performing Peptide Bioanalysis Using High Resolution Mass Spectrometry with Target Enhancement MRM Acquisition
 Performing Peptide Bioanalysis Using High Resolution Mass Spectrometry with Target Enhancement MRM Acquisition Yun Wang Alelyunas, Mark D. Wrona, and Nick Tomczyk Waters Corporation, Milford, MA, USA GOAL
Performing Peptide Bioanalysis Using High Resolution Mass Spectrometry with Target Enhancement MRM Acquisition Yun Wang Alelyunas, Mark D. Wrona, and Nick Tomczyk Waters Corporation, Milford, MA, USA GOAL
Workflow concept. Data goes through the workflow. A Node contains an operation An edge represents data flow The results are brought together in tables
 PROTEOME DISCOVERER Workflow concept Data goes through the workflow Spectra Peptides Quantitation A Node contains an operation An edge represents data flow The results are brought together in tables Protein
PROTEOME DISCOVERER Workflow concept Data goes through the workflow Spectra Peptides Quantitation A Node contains an operation An edge represents data flow The results are brought together in tables Protein
Bruker Daltonics. EASY-nLC. Tailored HPLC for nano-lc-ms Proteomics. Nano-HPLC. think forward
 Bruker Daltonics EASY-nLC Tailored HPLC for nano-lc-ms Proteomics think forward Nano-HPLC World-Class Performance with a Small Footprint Bruker Daltonics presents a nano-lc system, perfectly integrated
Bruker Daltonics EASY-nLC Tailored HPLC for nano-lc-ms Proteomics think forward Nano-HPLC World-Class Performance with a Small Footprint Bruker Daltonics presents a nano-lc system, perfectly integrated
Bioanalytical Chem: 4590: LC-MSMS of analgesics LC-MS Experiment Liquid Chromatography Mass Spectrometry (LC/MS)
") Liquid Chromatography Mass Spectrometry (LC/MS) Prelab Questions: Questions to be answered before doing the experiment. The answers are due at the beginning of each experiment without exception (the questions
Liquid Chromatography Mass Spectrometry (LC/MS) Prelab Questions: Questions to be answered before doing the experiment. The answers are due at the beginning of each experiment without exception (the questions
Targeted Proteomics Environment
 Targeted Proteomics Environment Quantitative Proteomics with Bruker Q-TOF Instruments and Skyline Brendan MacLean Quantitative Proteomics Spectrum-based Spectral counting Isobaric tags Chromatography-based
Targeted Proteomics Environment Quantitative Proteomics with Bruker Q-TOF Instruments and Skyline Brendan MacLean Quantitative Proteomics Spectrum-based Spectral counting Isobaric tags Chromatography-based
SRM assay generation and data analysis in Skyline
 in Skyline Preparation 1. Download the example data from www.srmcourse.ch/eupa.html (3 raw files, 1 csv file, 1 sptxt file). 2. The number formats of your computer have to be set to English (United States).
in Skyline Preparation 1. Download the example data from www.srmcourse.ch/eupa.html (3 raw files, 1 csv file, 1 sptxt file). 2. The number formats of your computer have to be set to English (United States).
UCD Conway Institute of Biomolecular & Biomedical Research Graduate Education 2009/2010
 EMERGING PROTEOMIC TECHNOLOGIES - MODULE SCHEDULE & OUTLINE 2010 Course Organiser: Dr. Giuliano Elia Module Co-ordinator: Dr Giuliano Elia Credits: 5 Date & Time Session & Topic Coordinator 14th April
EMERGING PROTEOMIC TECHNOLOGIES - MODULE SCHEDULE & OUTLINE 2010 Course Organiser: Dr. Giuliano Elia Module Co-ordinator: Dr Giuliano Elia Credits: 5 Date & Time Session & Topic Coordinator 14th April
1. Prepare the MALDI sample plate by spotting an angiotensin standard and the test sample(s).
.") Analysis of a Peptide Sequence from a Proteolytic Digest by MALDI-TOF Post-Source Decay (PSD) and Collision-Induced Dissociation (CID) Standard Operating Procedure Purpose: The following procedure may
Analysis of a Peptide Sequence from a Proteolytic Digest by MALDI-TOF Post-Source Decay (PSD) and Collision-Induced Dissociation (CID) Standard Operating Procedure Purpose: The following procedure may
Protein Quantitation II: Multiple Reaction Monitoring. Kelly Ruggles New York University
 Protein Quantitation II: Multiple Reaction Monitoring Kelly Ruggles kelly@fenyolab.org New York University Traditional Affinity-based proteomics Use antibodies to quantify proteins Western Blot Immunohistochemistry
Protein Quantitation II: Multiple Reaction Monitoring Kelly Ruggles kelly@fenyolab.org New York University Traditional Affinity-based proteomics Use antibodies to quantify proteins Western Blot Immunohistochemistry
Introduction to Proteomics & Bottom-up Proteomics
 Used for MS Short Course at Tsinghua by R. Graham Cooks, Hao Chen, Zheng Ouyang, Andy Tao, Yu Xia and Lingjun Li Introduction to Proteomics & Bottom-up Proteomics W. Andy Tao Purdue University watao@purdue.edu
Used for MS Short Course at Tsinghua by R. Graham Cooks, Hao Chen, Zheng Ouyang, Andy Tao, Yu Xia and Lingjun Li Introduction to Proteomics & Bottom-up Proteomics W. Andy Tao Purdue University watao@purdue.edu
X!TandemPipeline (Myosine Anabolisée) validating, filtering and grouping MSMS identifications
 validating, filtering and grouping MSMS identifications") X!TandemPipeline 3.3.3 (Myosine Anabolisée) validating, filtering and grouping MSMS identifications Olivier Langella and Benoit Valot langella@moulon.inra.fr; valot@moulon.inra.fr PAPPSO - http://pappso.inra.fr/
X!TandemPipeline 3.3.3 (Myosine Anabolisée) validating, filtering and grouping MSMS identifications Olivier Langella and Benoit Valot langella@moulon.inra.fr; valot@moulon.inra.fr PAPPSO - http://pappso.inra.fr/
Chemistry Instrumental Analysis Lecture 37. Chem 4631
 Chemistry 4631 Instrumental Analysis Lecture 37 Most analytes separated by HPLC are thermally stable and non-volatile (liquids) (unlike in GC) so not ionized easily by EI or CI techniques. MS must be at
Chemistry 4631 Instrumental Analysis Lecture 37 Most analytes separated by HPLC are thermally stable and non-volatile (liquids) (unlike in GC) so not ionized easily by EI or CI techniques. MS must be at
Biological Mass Spectrometry
 Biochemistry 412 Biological Mass Spectrometry February 13 th, 2007 Proteomics The study of the complete complement of proteins found in an organism Degrees of Freedom for Protein Variability Covalent Modifications
Biochemistry 412 Biological Mass Spectrometry February 13 th, 2007 Proteomics The study of the complete complement of proteins found in an organism Degrees of Freedom for Protein Variability Covalent Modifications
PosterREPRINT AN INTERACTIVE PHYSICOCHEMICAL PROPERTY PROFILING SOFTWARE FOR EARLY CANDIDATE ANALYSIS IN DRUG DISCOVERY INTRODUCTION
 INTRODUCTION Full characterization of the physicochemical properties of new chemical entities (NCE s) typically takes place in pharmaceutical development laboratories. However, in an effort to minimize
INTRODUCTION Full characterization of the physicochemical properties of new chemical entities (NCE s) typically takes place in pharmaceutical development laboratories. However, in an effort to minimize
Isotope Dilution Mass Spectrometry
 Isotope Dilution Mass Spectrometry J. Ignacio Garcia Alonso and Pablo Rodriguez-Gonzalez Faculty of Chemistry, University of Oviedo, Oviedo, Spain E-mail: jiga@uniovi.es, rodriguezpablo@uniovi.es RSC Publishing
Isotope Dilution Mass Spectrometry J. Ignacio Garcia Alonso and Pablo Rodriguez-Gonzalez Faculty of Chemistry, University of Oviedo, Oviedo, Spain E-mail: jiga@uniovi.es, rodriguezpablo@uniovi.es RSC Publishing
Mass spectrometry-based proteomics has become
 FOCUS: THE ORBITRAP Computational Principles of Determining and Improving Mass Precision and Accuracy for Proteome Measurements in an Orbitrap Jürgen Cox and Matthias Mann Proteomics and Signal Transduction,
FOCUS: THE ORBITRAP Computational Principles of Determining and Improving Mass Precision and Accuracy for Proteome Measurements in an Orbitrap Jürgen Cox and Matthias Mann Proteomics and Signal Transduction,
6 x 5 Ways to Ensure Your LC-MS/MS is Healthy
 6 x 5 Ways to Ensure Your LC-MS/MS is Healthy (Also known as - Tracking Performance with the 6 x 5 LC-MS/MS Peptide Reference Mixture) Mike Rosenblatt, Ph.D. Group Leader Mass Spec Reagents 215. We monitor
6 x 5 Ways to Ensure Your LC-MS/MS is Healthy (Also known as - Tracking Performance with the 6 x 5 LC-MS/MS Peptide Reference Mixture) Mike Rosenblatt, Ph.D. Group Leader Mass Spec Reagents 215. We monitor
WADA Technical Document TD2003IDCR
 IDENTIFICATION CRITERIA FOR QUALITATIVE ASSAYS INCORPORATING CHROMATOGRAPHY AND MASS SPECTROMETRY The appropriate analytical characteristics must be documented for a particular assay. The Laboratory must
IDENTIFICATION CRITERIA FOR QUALITATIVE ASSAYS INCORPORATING CHROMATOGRAPHY AND MASS SPECTROMETRY The appropriate analytical characteristics must be documented for a particular assay. The Laboratory must
Mixture Mode for Peptide Mass Fingerprinting ASMS 2003
 Mixture Mode for Peptide Mass Fingerprinting ASMS 2003 1 Mixture Mode: New in Mascot 1.9 All peptide mass fingerprint searches now test for the possibility that the sample is a mixture of proteins. Mascot
Mixture Mode for Peptide Mass Fingerprinting ASMS 2003 1 Mixture Mode: New in Mascot 1.9 All peptide mass fingerprint searches now test for the possibility that the sample is a mixture of proteins. Mascot
Tandem MS = MS / MS. ESI-MS give information on the mass of a molecule but none on the structure
 Tandem MS = MS / MS ESI-MS give information on the mass of a molecule but none on the structure In tandem MS (MSMS) (pseudo-)molecular ions are selected in MS1 and fragmented by collision with gas. collision
Tandem MS = MS / MS ESI-MS give information on the mass of a molecule but none on the structure In tandem MS (MSMS) (pseudo-)molecular ions are selected in MS1 and fragmented by collision with gas. collision
Quantitation of High Resolution MS Data Using UNIFI: Acquiring and Processing Full Scan or Tof-MRM (Targeted HRMS) Datasets for Quantitative Assays
 Datasets for Quantitative Assays") : Acquiring and Processing Full Scan or Tof-MRM (Targeted HRMS) Datasets for Quantitative Assays Mark Wrona, Jayne Kirk, and Yun Alelyunas Waters Corporation, Milford, MA, USA APPLICATION BENEFITS Ability
: Acquiring and Processing Full Scan or Tof-MRM (Targeted HRMS) Datasets for Quantitative Assays Mark Wrona, Jayne Kirk, and Yun Alelyunas Waters Corporation, Milford, MA, USA APPLICATION BENEFITS Ability
TANDEM MASS SPECTROSCOPY
 TANDEM MASS SPECTROSCOPY 1 MASS SPECTROMETER TYPES OF MASS SPECTROMETER PRINCIPLE TANDEM MASS SPECTROMETER INSTRUMENTATION QUADRAPOLE MASS ANALYZER TRIPLE QUADRAPOLE MASS ANALYZER TIME OF FLIGHT MASS ANALYSER
TANDEM MASS SPECTROSCOPY 1 MASS SPECTROMETER TYPES OF MASS SPECTROMETER PRINCIPLE TANDEM MASS SPECTROMETER INSTRUMENTATION QUADRAPOLE MASS ANALYZER TRIPLE QUADRAPOLE MASS ANALYZER TIME OF FLIGHT MASS ANALYSER
Tutorial 2: Analysis of DIA data in Skyline
 Tutorial 2: Analysis of DIA data in Skyline In this tutorial we will learn how to use Skyline to perform targeted post-acquisition analysis for peptide and inferred protein detection and quantitation using
Tutorial 2: Analysis of DIA data in Skyline In this tutorial we will learn how to use Skyline to perform targeted post-acquisition analysis for peptide and inferred protein detection and quantitation using
Towards the Prediction of Protein Abundance from Tandem Mass Spectrometry Data
 Towards the Prediction of Protein Abundance from Tandem Mass Spectrometry Data Anthony J Bonner Han Liu Abstract This paper addresses a central problem of Proteomics: estimating the amounts of each of
Towards the Prediction of Protein Abundance from Tandem Mass Spectrometry Data Anthony J Bonner Han Liu Abstract This paper addresses a central problem of Proteomics: estimating the amounts of each of
From mass to compound iden3ty
 Mass Spectrometry Analy&cal tool measuring molecular weight of molecules in chemistry, biology and pharmacology Only picomolar concentra&ons required Within 5 ppm for small organic molecules For a 40 kda
Mass Spectrometry Analy&cal tool measuring molecular weight of molecules in chemistry, biology and pharmacology Only picomolar concentra&ons required Within 5 ppm for small organic molecules For a 40 kda
Tutorial 1: Library Generation from DDA data
 Tutorial 1: Library Generation from DDA data 1. Introduction Before a targeted, peptide-centric DIA analysis can be performed, a spectral library containing peptide-query parameters needs to be generated.
Tutorial 1: Library Generation from DDA data 1. Introduction Before a targeted, peptide-centric DIA analysis can be performed, a spectral library containing peptide-query parameters needs to be generated.
Data pre-processing in liquid chromatography mass spectrometry-based proteomics
 BIOINFORMATICS ORIGINAL PAPER Vol. 21 no. 21 25, pages 454 459 doi:1.193/bioinformatics/bti66 Data and text mining Data pre-processing in liquid chromatography mass spectrometry-based proteomics Xiang
BIOINFORMATICS ORIGINAL PAPER Vol. 21 no. 21 25, pages 454 459 doi:1.193/bioinformatics/bti66 Data and text mining Data pre-processing in liquid chromatography mass spectrometry-based proteomics Xiang
CRL MASS SPECTROMETRY FACILITY USER MANUAL LCT CLASSIC A & B
 Mass Spectrometry Instrument Training Guide Page 1 of 18 24/10/2012 CRL MASS SPECTROMETRY FACILITY USER MANUAL LCT CLASSIC A & B 1st Floor Mass Spec Lab: 00.097 This is a guide to using the LCT classic
Mass Spectrometry Instrument Training Guide Page 1 of 18 24/10/2012 CRL MASS SPECTROMETRY FACILITY USER MANUAL LCT CLASSIC A & B 1st Floor Mass Spec Lab: 00.097 This is a guide to using the LCT classic
Developing Algorithms for the Determination of Relative Abundances of Peptides from LC/MS Data
 Developing Algorithms for the Determination of Relative Abundances of Peptides from LC/MS Data RIPS Team Jake Marcus (Project Manager) Anne Eaton Melanie Kanter Aru Ray Faculty Mentors Shawn Cokus Matteo
Developing Algorithms for the Determination of Relative Abundances of Peptides from LC/MS Data RIPS Team Jake Marcus (Project Manager) Anne Eaton Melanie Kanter Aru Ray Faculty Mentors Shawn Cokus Matteo
Protein Deconvolution Version 2.0
 Thermo Protein Deconvolution Version 2.0 User Guide XCALI-97414 Revision A August 2012 2012 Thermo Fisher Scientific Inc. All rights reserved. ReSpect is a trademark of Positive Probability Ltd. Xcalibur
Thermo Protein Deconvolution Version 2.0 User Guide XCALI-97414 Revision A August 2012 2012 Thermo Fisher Scientific Inc. All rights reserved. ReSpect is a trademark of Positive Probability Ltd. Xcalibur
Protein analysis using mass spectrometry
 Protein analysis using mass spectrometry Michael Stadlmeier 2017/12/18 Literature http://www.carellgroup.de/teaching/master 3 What is Proteomics? The proteome is: the entire set of proteins in a given
Protein analysis using mass spectrometry Michael Stadlmeier 2017/12/18 Literature http://www.carellgroup.de/teaching/master 3 What is Proteomics? The proteome is: the entire set of proteins in a given
Qualitative Proteomics (how to obtain high-confidence high-throughput protein identification!)
") Qualitative Proteomics (how to obtain high-confidence high-throughput protein identification!) James A. Mobley, Ph.D. Director of Research in Urology Associate Director of Mass Spectrometry (contact: mobleyja@uab.edu)
Qualitative Proteomics (how to obtain high-confidence high-throughput protein identification!) James A. Mobley, Ph.D. Director of Research in Urology Associate Director of Mass Spectrometry (contact: mobleyja@uab.edu)
BIOINF 4120 Bioinformatics 2 - Structures and Systems - Oliver Kohlbacher Summer Systems Biology Exp. Methods
 BIOINF 4120 Bioinformatics 2 - Structures and Systems - Oliver Kohlbacher Summer 2013 14. Systems Biology Exp. Methods Overview Transcriptomics Basics of microarrays Comparative analysis Interactomics:
BIOINF 4120 Bioinformatics 2 - Structures and Systems - Oliver Kohlbacher Summer 2013 14. Systems Biology Exp. Methods Overview Transcriptomics Basics of microarrays Comparative analysis Interactomics:
An extended siderophore suite from Synechococcus sp. PCC 7002 revealed by LC- ICPMS-ESIMS
 Electronic Supplementary Material (ESI) for Metallomics. This journal is The Royal Society of Chemistry 2015 An extended siderophore suite from Synechococcus sp. PCC 7002 revealed by LC- ICPMS-ESIMS Rene
Electronic Supplementary Material (ESI) for Metallomics. This journal is The Royal Society of Chemistry 2015 An extended siderophore suite from Synechococcus sp. PCC 7002 revealed by LC- ICPMS-ESIMS Rene
Multi-residue analysis of pesticides by GC-HRMS
 An Executive Summary Multi-residue analysis of pesticides by GC-HRMS Dr. Hans Mol is senior scientist at RIKILT- Wageningen UR Introduction Regulatory authorities throughout the world set and enforce strict
An Executive Summary Multi-residue analysis of pesticides by GC-HRMS Dr. Hans Mol is senior scientist at RIKILT- Wageningen UR Introduction Regulatory authorities throughout the world set and enforce strict
Analysis of Illegal Dyes in Food Matrices using Automated Online Sample Preparation with LC/MS
 Application Note: 56 Analysis of Illegal Dyes in Food Matrices using Automated Online Sample Preparation with LC/MS Yang Shi, Catherine Lafontaine, Matthew Berube, John Fink, François Espourteille Thermo
Application Note: 56 Analysis of Illegal Dyes in Food Matrices using Automated Online Sample Preparation with LC/MS Yang Shi, Catherine Lafontaine, Matthew Berube, John Fink, François Espourteille Thermo
HOWTO, example workflow and data files. (Version )
") HOWTO, example workflow and data files. (Version 20 09 2017) 1 Introduction: SugarQb is a collection of software tools (Nodes) which enable the automated identification of intact glycopeptides from HCD
HOWTO, example workflow and data files. (Version 20 09 2017) 1 Introduction: SugarQb is a collection of software tools (Nodes) which enable the automated identification of intact glycopeptides from HCD
Rapid and Accurate Forensics Analysis using High Resolution All Ions MS/MS
 Rapid and Accurate Forensics Analysis using High Resolution All Ions MS/MS Application Note Forensic Toxicology Authors Martin Josefsson, and Markus Roman National Board of Forensic Medicine Linköping,
Rapid and Accurate Forensics Analysis using High Resolution All Ions MS/MS Application Note Forensic Toxicology Authors Martin Josefsson, and Markus Roman National Board of Forensic Medicine Linköping,
Proteomics: the first decade and beyond. (2003) Patterson and Aebersold Nat Genet 33 Suppl: from
 Patterson and Aebersold Nat Genet 33 Suppl: from") Advances in mass spectrometry and the generation of large quantities of nucleotide sequence information, combined with computational algorithms that could correlate the two, led to the emergence of proteomics
Advances in mass spectrometry and the generation of large quantities of nucleotide sequence information, combined with computational algorithms that could correlate the two, led to the emergence of proteomics
Advances in quantitative proteomics using stable isotope tags
 Advances in quantitative proteomics using stable isotope tags Mark R. Flory, Timothy J. Griffin, Daniel Martin and Ruedi Aebersold A great deal of current biological and clinical research is directed at
Advances in quantitative proteomics using stable isotope tags Mark R. Flory, Timothy J. Griffin, Daniel Martin and Ruedi Aebersold A great deal of current biological and clinical research is directed at
SEAMLESS INTEGRATION OF MASS DETECTION INTO THE UV CHROMATOGRAPHIC WORKFLOW
 SEAMLESS INTEGRATION OF MASS DETECTION INTO THE UV CHROMATOGRAPHIC WORKFLOW Paula Hong, John Van Antwerp, and Patricia McConville Waters Corporation, Milford, MA, USA Historically UV detection has been
SEAMLESS INTEGRATION OF MASS DETECTION INTO THE UV CHROMATOGRAPHIC WORKFLOW Paula Hong, John Van Antwerp, and Patricia McConville Waters Corporation, Milford, MA, USA Historically UV detection has been
Guide to Peptide Quantitation. Agilent clinical research
 Guide to Peptide Quantitation Agilent clinical research Peptide Quantitation for the Clinical Research Laboratory Peptide quantitation is rapidly growing in clinical research as scientists are translating
Guide to Peptide Quantitation Agilent clinical research Peptide Quantitation for the Clinical Research Laboratory Peptide quantitation is rapidly growing in clinical research as scientists are translating
An Effective Workflow for Impurity Analysis Incorporating High Quality HRAM LCMS & MSMS with Intelligent Automated Data Mining
 An Effective Workflow for Impurity Analysis Incorporating High Quality HRAM LCMS & MSMS with Intelligent Automated Data Mining Dave Weil, Ph.D. and Jim Lau, Ph.D. Typical Method Conditions: 1260 UHPLC
An Effective Workflow for Impurity Analysis Incorporating High Quality HRAM LCMS & MSMS with Intelligent Automated Data Mining Dave Weil, Ph.D. and Jim Lau, Ph.D. Typical Method Conditions: 1260 UHPLC
Mass Spectrometry. Hyphenated Techniques GC-MS LC-MS and MS-MS
 Mass Spectrometry Hyphenated Techniques GC-MS LC-MS and MS-MS Reasons for Using Chromatography with MS Mixture analysis by MS alone is difficult Fragmentation from ionization (EI or CI) Fragments from
Mass Spectrometry Hyphenated Techniques GC-MS LC-MS and MS-MS Reasons for Using Chromatography with MS Mixture analysis by MS alone is difficult Fragmentation from ionization (EI or CI) Fragments from
Translational Biomarker Core
 Translational Biomarker Core Instrumentation Thermo Scientific TSQ Quantum Triple Quadrupole Mass Spectrometers. There are two TSQ Quantum Ultra AM instruments available in the TBC. The TSQ Quantum Ultra
Translational Biomarker Core Instrumentation Thermo Scientific TSQ Quantum Triple Quadrupole Mass Spectrometers. There are two TSQ Quantum Ultra AM instruments available in the TBC. The TSQ Quantum Ultra
Live Webinar : How to be more Successful with your ACQUITY QDa Detector?
 Live Webinar : How to be more Successful with your ACQUITY QDa Detector? Q&A Transcript ---------------- Q - How do you generate multiple charges reproductively? A - If you use the same settings on the
Live Webinar : How to be more Successful with your ACQUITY QDa Detector? Q&A Transcript ---------------- Q - How do you generate multiple charges reproductively? A - If you use the same settings on the
Proteome-wide label-free quantification with MaxQuant. Jürgen Cox Max Planck Institute of Biochemistry July 2011
 Proteome-wide label-free quantification with MaxQuant Jürgen Cox Max Planck Institute of Biochemistry July 2011 MaxQuant MaxQuant Feature detection Data acquisition Initial Andromeda search Statistics
Proteome-wide label-free quantification with MaxQuant Jürgen Cox Max Planck Institute of Biochemistry July 2011 MaxQuant MaxQuant Feature detection Data acquisition Initial Andromeda search Statistics
Applications of Mass Spectrometry for Biotherapeutic Characterization
 Applications of Mass Spectrometry for Biotherapeutic Characterization Case Studies of Disulfide Characterization and Separation free Modes of Analysis Steven L. Cockrill Amgen Colorado Analytical Sciences
Applications of Mass Spectrometry for Biotherapeutic Characterization Case Studies of Disulfide Characterization and Separation free Modes of Analysis Steven L. Cockrill Amgen Colorado Analytical Sciences
Effective Strategies for Improving Peptide Identification with Tandem Mass Spectrometry
 Effective Strategies for Improving Peptide Identification with Tandem Mass Spectrometry by Xi Han A thesis presented to the University of Waterloo in fulfillment of the thesis requirement for the degree
Effective Strategies for Improving Peptide Identification with Tandem Mass Spectrometry by Xi Han A thesis presented to the University of Waterloo in fulfillment of the thesis requirement for the degree
A Description of the CPTAC Common Data Analysis Pipeline (CDAP)
") A Description of the CPTAC Common Data Analysis Pipeline (CDAP) v. 01/14/2014 Summary The purpose of this document is to describe the software programs and output files of the Common Data Analysis Pipeline
A Description of the CPTAC Common Data Analysis Pipeline (CDAP) v. 01/14/2014 Summary The purpose of this document is to describe the software programs and output files of the Common Data Analysis Pipeline
Agilent MassHunter Profinder: Solving the Challenge of Isotopologue Extraction for Qualitative Flux Analysis
 Agilent MassHunter Profinder: Solving the Challenge of Isotopologue Extraction for Qualitative Flux Analysis Technical Overview Introduction Metabolomics studies measure the relative abundance of metabolites
Agilent MassHunter Profinder: Solving the Challenge of Isotopologue Extraction for Qualitative Flux Analysis Technical Overview Introduction Metabolomics studies measure the relative abundance of metabolites
TUTORIAL EXERCISES WITH ANSWERS
 TUTORIAL EXERCISES WITH ANSWERS Tutorial 1 Settings 1. What is the exact monoisotopic mass difference for peptides carrying a 13 C (and NO additional 15 N) labelled C-terminal lysine residue? a. 6.020129
TUTORIAL EXERCISES WITH ANSWERS Tutorial 1 Settings 1. What is the exact monoisotopic mass difference for peptides carrying a 13 C (and NO additional 15 N) labelled C-terminal lysine residue? a. 6.020129
ALIGNMENT OF LC-MS DATA USING PEPTIDE FEATURES. A Thesis XINCHENG TANG
 ALIGNMENT OF LC-MS DATA USING PEPTIDE FEATURES A Thesis by XINCHENG TANG Submitted to the Office of Graduate Studies of Texas A&M University in partial fulfillment of the requirements for the degree of
ALIGNMENT OF LC-MS DATA USING PEPTIDE FEATURES A Thesis by XINCHENG TANG Submitted to the Office of Graduate Studies of Texas A&M University in partial fulfillment of the requirements for the degree of
The Power of LC MALDI: Identification of Proteins by LC MALDI MS/MS Using the Applied Biosystems 4700 Proteomics Analyzer with TOF/TOF Optics
 APPLICATION NOTE TOF MS The Power of LC MALDI: Identification of Proteins by LC MALDI MS/MS Using the Applied Biosystems 4700 Proteomics Analyzer with TOF/TOF Optics Purpose The Applied Biosystems 4700
APPLICATION NOTE TOF MS The Power of LC MALDI: Identification of Proteins by LC MALDI MS/MS Using the Applied Biosystems 4700 Proteomics Analyzer with TOF/TOF Optics Purpose The Applied Biosystems 4700
Nature Methods: doi: /nmeth Supplementary Figure 1. Fragment indexing allows efficient spectra similarity comparisons.
 Supplementary Figure 1 Fragment indexing allows efficient spectra similarity comparisons. The cost and efficiency of spectra similarity calculations can be approximated by the number of fragment comparisons
Supplementary Figure 1 Fragment indexing allows efficient spectra similarity comparisons. The cost and efficiency of spectra similarity calculations can be approximated by the number of fragment comparisons
Welcome! Course 7: Concepts for LC-MS
 Welcome! Mass Spectrometry meets Cheminformatics Tobias Kind and Julie Leary UC Davis Course 7: Concepts for LC-MS Class website: CHE 241 - Spring 28 - CRN 16583 Slides: http://fiehnlab.ucdavis.edu/staff/kind/teaching/
Welcome! Mass Spectrometry meets Cheminformatics Tobias Kind and Julie Leary UC Davis Course 7: Concepts for LC-MS Class website: CHE 241 - Spring 28 - CRN 16583 Slides: http://fiehnlab.ucdavis.edu/staff/kind/teaching/
Analysis of a Verapamil Microsomal Incubation using Metabolite ID and Mass Frontier TM
 Application Note: 320 Analysis of a Verapamil Microsomal Incubation using Metabolite ID and Mass Frontier TM Key Words Metabolism Study Structure Elucidation Metabolite ID Mass Frontier Chromatography
Application Note: 320 Analysis of a Verapamil Microsomal Incubation using Metabolite ID and Mass Frontier TM Key Words Metabolism Study Structure Elucidation Metabolite ID Mass Frontier Chromatography
Aplicació de la proteòmica a la cerca de Biomarcadors proteics Barcelona, 08 de Juny 2010
 Aplicació de la proteòmica a la cerca de Biomarcadors proteics Barcelona, 8 de Juny 21 Eliandre de Oliveira Plataforma de Proteòmica Parc Científic de Barcelona Protein Chemistry Proteomics Hypothesis-free
Aplicació de la proteòmica a la cerca de Biomarcadors proteics Barcelona, 8 de Juny 21 Eliandre de Oliveira Plataforma de Proteòmica Parc Científic de Barcelona Protein Chemistry Proteomics Hypothesis-free
SILAC and TMT. IDeA National Resource for Proteomics Workshop for Graduate Students and Post-docs Renny Lan 5/18/2017
 SILAC and TMT IDeA National Resource for Proteomics Workshop for Graduate Students and Post-docs Renny Lan 5/18/2017 UHPLC peak chosen at 26.47 min LC Mass at 571.36 chosen for MS/MS MS/MS MS This is a
SILAC and TMT IDeA National Resource for Proteomics Workshop for Graduate Students and Post-docs Renny Lan 5/18/2017 UHPLC peak chosen at 26.47 min LC Mass at 571.36 chosen for MS/MS MS/MS MS This is a
Robust Estimation of Peptide Abundance Ratios and Rigorous Scoring of Their Variability and Bias in Quantitative Shotgun Proteomics
 Anal. Chem. 2006, 78, 7110-7120 Robust Estimation of Peptide Abundance Ratios and Rigorous Scoring of Their Variability and Bias in Quantitative Shotgun Proteomics Chongle Pan,,,, Guruprasad Kora,, David
Anal. Chem. 2006, 78, 7110-7120 Robust Estimation of Peptide Abundance Ratios and Rigorous Scoring of Their Variability and Bias in Quantitative Shotgun Proteomics Chongle Pan,,,, Guruprasad Kora,, David
I. Proteomics by Mass Spectrometry 1. What is an internal standard and what does it accomplish analytically?
 Name I. Proteomics by Mass Spectrometry 1. What is an internal standard and what does it accomplish analytically? Internal standards are standards added intentionally to all samples, standards and blanks.
Name I. Proteomics by Mass Spectrometry 1. What is an internal standard and what does it accomplish analytically? Internal standards are standards added intentionally to all samples, standards and blanks.
Overview. Introduction. André Schreiber AB SCIEX Concord, Ontario (Canada)
") Quantitation and Identification of Pharmaceuticals and Personal Care Products (PPCP) in Environmental Samples using Advanced TripleTOF MS/MS Technology André Schreiber AB SCIEX Concord, Ontario (Canada)
Quantitation and Identification of Pharmaceuticals and Personal Care Products (PPCP) in Environmental Samples using Advanced TripleTOF MS/MS Technology André Schreiber AB SCIEX Concord, Ontario (Canada)
Peptide Isolation Using the Prep 150 LC System
 Jo-Ann M. Jablonski and Andrew J. Aubin Waters Corporation, Milford, MA, USA APPLICATION BENEFITS The Prep 150 LC System, an affordable, highly reliable system for preparative chromatography, is suitable
Jo-Ann M. Jablonski and Andrew J. Aubin Waters Corporation, Milford, MA, USA APPLICATION BENEFITS The Prep 150 LC System, an affordable, highly reliable system for preparative chromatography, is suitable
Indiana University, Fall 2014 P309 Intermediate Physics Lab. Lecture 1: Experimental UncertainBes
 Indiana University, Fall 2014 P309 Intermediate Physics Lab Lecture 1: Experimental UncertainBes Reading: Bevington & Robinson, Chapters 1-3 Handouts from hmp://physics.indiana.edu/~courses/p309/f14/ Experimental
Indiana University, Fall 2014 P309 Intermediate Physics Lab Lecture 1: Experimental UncertainBes Reading: Bevington & Robinson, Chapters 1-3 Handouts from hmp://physics.indiana.edu/~courses/p309/f14/ Experimental
Feature File 1. Feature Detection. Similarity Comparison (Precursor and product ions) Spectra Generation. Decoy spectra.
 Spectra Generation. Decoy spectra.") Supplementary Figure 1. The workflow of Group-DIA. DIA Data Data File 1 Data File 2 Data File 3 Feature Detection Feature File 1 Feature File 2 Feature File 3 Retention Time Alignment Intensity Extraction
Supplementary Figure 1. The workflow of Group-DIA. DIA Data Data File 1 Data File 2 Data File 3 Feature Detection Feature File 1 Feature File 2 Feature File 3 Retention Time Alignment Intensity Extraction
Analyst Software. Peptide and Protein Quantitation Tutorial
 This document is provided to customers who have purchased AB Sciex equipment to use in the operation of such AB Sciex equipment. This document is copyright protected and any reproduction of this document
This document is provided to customers who have purchased AB Sciex equipment to use in the operation of such AB Sciex equipment. This document is copyright protected and any reproduction of this document
Identification of proteins by enzyme digestion, mass
 Method for Screening Peptide Fragment Ion Mass Spectra Prior to Database Searching Roger E. Moore, Mary K. Young, and Terry D. Lee Beckman Research Institute of the City of Hope, Duarte, California, USA
Method for Screening Peptide Fragment Ion Mass Spectra Prior to Database Searching Roger E. Moore, Mary K. Young, and Terry D. Lee Beckman Research Institute of the City of Hope, Duarte, California, USA
GUIDELINES ON THE USE OF MASS SPECTROMETRY (MS) FOR IDENTIFICATION, CONFIRMATION AND QUANTITATIVE DETERMINATION OF RESIDUES CAC/GL
 FOR IDENTIFICATION, CONFIRMATION AND QUANTITATIVE DETERMINATION OF RESIDUES CAC/GL") CAC/GL 56-2005 Page 1 of 6 GUIDELINES ON THE USE OF MASS SPECTROMETRY (MS) FOR IDENTIFICATION, CONFIRMATION AND QUANTITATIVE DETERMINATION OF RESIDUES CONFIRMATORY TESTS CAC/GL 56-2005 When analyses are
CAC/GL 56-2005 Page 1 of 6 GUIDELINES ON THE USE OF MASS SPECTROMETRY (MS) FOR IDENTIFICATION, CONFIRMATION AND QUANTITATIVE DETERMINATION OF RESIDUES CONFIRMATORY TESTS CAC/GL 56-2005 When analyses are