HOWTO, example workflow and data files. (Version )
|
|
|
- Melinda Joseph
- 6 years ago
- Views:
Transcription
1 HOWTO, example workflow and data files. (Version ) 1
2 Introduction: SugarQb is a collection of software tools (Nodes) which enable the automated identification of intact glycopeptides from HCD MS/MS data sets, using common MS/MS search engines (e.g. MASCOT, SEQUEST HT) in the Proteome Discoverer 1.4 environment. SugarQb is freely available to all researchers. For further information on the algorithm, please refer to the corresponding publication Stadlmann J., Taubenschmid J., et al. Comparative glycoproteomics of stem cells identifies new players in ricin toxicity, Nature (2017). This document is intended to provide you with a quick guide on who to download, install and test SugarQb, analyzing an example data of tryptic glycopeptides derived from human plasma. All relevant.dll files, additional parameter files and a Glycan mass data base are available at: Contact: SugarQb@imba.oeaw.ac.at 2

3 Download and Installation: Download SugarQb for Thermo Scientific Proteome Discoverer 1.4. using the following URL: Save all your files and shutdown Thermo Scientific Proteome Discoverer. Navigate to the folder where you have installed Thermo Scientific Proteome Discoverer (Tip: You can easily find out the path by right clicking the Thermo Scientific Proteome Discoverer desktop icon and open the Properties window. The folder path is written in the field Target.) Copy the.dll files into the Thermo Scientific Proteome Discoverer folder. Unblock the.dll files if required by right clicking each.dll file, opening its properties window and clicking the Unblock button if available. Restart Thermo Scientific Proteome Discoverer, navigate to the licensing page and click on Scan for Missing Features. Subsequently, restart the program once more. 3

4 Example Workflow: Download the test data file LUMOS_SugarQb_Test_humanPlasma_HCDonly.raw from: This data has been generated by analyzing IP HILIC enriched, tryptic glycopeptides derived from a chemically de sialylated human plasma, using HCD on a OrbiTrap Fusion LUMOS instrument. After Installation of the SugarQb Nodes, in Thermo Scientific Proteome Discoverer 1.4., create the following Workflow. Parameter settings of the respective Nodes are detailed below. 4

5 Recommended Settings & Parameters: Spectrum Selector: N.B.: The default settings of the Spectrum Selector Node were modified, to also allow higher mass precursor ions (i.e. up to Da) to be analyzed. 5

6 MS2 Spectrum Processor: This Node provides two MS2 spectrum preprocessing steps: Deisotoping of isotopic clusters and charge deconvolution. For this, spectra are searched for isotopic clusters by determining the distances in m/z values between pairs of peaks. For every cluster detected, only the monoisotopic peaks remain in the spectrum, other peaks are removed. Subsequently, the spectra are deconvolved to charge state 1. Every peak with a charge state greater than 1 will be removed from the spectrum and replaced by a peak at the corresponding singly charged m/z position with the same intensity. Note that the algorithm only works on peaks having charge state information available. For a more detailed description of the algorithm, please refer to: nodes In this exemplary workflow, the following parameter settings are recommended: 6
), and thus allows for a more efficient analysis of glycopeptides. Optimal threshold settings need to be empirically established for each instrument acquisition method.")
7 G Score (optional): The G Score Node filters MS2 spectra based on the occurrence and intensity of various glycan derived oxonium ions (for more details see Stadlmann J., Taubenschmid J., et al. Nature (2017)), and thus allows for a more efficient analysis of glycopeptides. Optimal threshold settings need to be empirically established for each instrument acquisition method. In this example, a G Score threshold of 0.4 was used. N.B. the use of this Node is optional. SugarQb: The SugarQb Node focuses on the identification of the potential [peptide + HexNAc] + fragment ions within MS/MS spectra. For this, the precursor ion masses of a given MS/MS spectrum are iteratively reduced by all masses present in a glycan composition database, minus the mass of one HexNAc residue (i.e amu). This approach generates a set of theoretical [peptide + HexNAc] + fragment ion masses, which are then tried to be matched within the MS/MS spectrum. In cases where an experimental peak matches a theoretical [peptide + HexNAc] + fragment, the concomitant presence of the corresponding potential [peptide] + fragment ion is verified. Only if both peaks are detected, the given spectrum is duplicated with its precursor ion mass set to the mass of the respective potential [peptide + HexNAc] + fragment ion (for more details see Stadlmann J., Taubenschmid J., et al. Nature (2017)). Note, that in this exemplary workflow, charge deconvoluted MS2 spectra (i.e. all fragment ions are expected to be of charge state 1) are analyzed and thus only charge state 1 is allowed. The.txt Glyco Database File used in this example can be downloaded at: The following SugarQb parameter settings are recommended: 7

.")
8 Reporter Ion Filter (Optional): This Node enables the filtering/removal of usually highly abundant, glycan related fragment ions from MS2 spectra. Reporter Ion masses to be completely removed from the MS2 dataset can also be defined in a separate.txt file (i.e. Reporter Ion File Selection). The Reporter Ion File used in this example can be downloaded at: N.B. the use of this Node is optional. 8
9 MS/MS Search Engine Settings & Parameters: For the eventual identification of the glycopeptide amino acid sequences, all MS2 spectra generated by the SugarQb Node (i.e. those with the original and those with the modified precursor ion masses) are searched against a concatenated forward and decoy database of the Uniprot human reference proteome set, considering HexNAc (and its neutral loss of amu) as a variable modification to any asparagine, serine and threonine residue. Here, the use of MASCOT and SEQUEST HT, are exemplified. Of Note, an in house developed MS/MS search engine, MS Amanda, is freely available at: nodes Irrespective of the MS/MS search engine employed, the resulting peptide spectrum matches (PSMs) are then manually filtered. For this, only the best scoring PSMs of each spectrum group (i.e. comprising the MS/MS spectrum with the original precursor ion mass and all its duplicates with the respectively modified precursor ion masses) are kept and filtered to an estimated false discovery rate (FDR) of 1%, employing the standard target decoy approach (Elias, J. E. & Gygi, S. P. Target decoy search strategy for increased confidence in large scale protein identifications by mass spectrometry. Nat Methods 4, (2007)). Currently, PSM filtering is performed, after exporting the search results from Thermo Scientific Proteome Discoverer 1.4. to a.csv file, using a Perl script. These scripts can be downloaded at: N.B. the use of the FDR filtering script is optional. 9

10 MASCOT Recommended MASCOT search parameter settings are listed below. Of note, MASCOT provides additional options to optimize the search engines performance in the identification of glycopeptide amino acid sequences (e.g. handling of the dominant neutral loss of the glycan portion upon HCD fragmentation, or scoring only singly charged fragment ions). Examples of such adjustments are described in the Annex section. 10

11 SEQUEST HT: Recommended SEQUEST HT search parameter settings are listed below. 11

12 ptmrs (Optional): Generally, this tool enables automated and confident localization of modification sites within validated peptide sequences. It calculates individual probability values for each putatively modified site based on the given MS/MS data. ptmrs can also be used to localize N glycosylation sites. For further information on the algorithm of the software, please refer to: nodes The.xml configuration file used in this exemplary workflow can be downloaded at: N.B. the use of this Node is optional. IMP Hyperplex (optional): The IMP Hyperplex has far reaching peptide quantification capabilities. Importantly, this node allows to also extract quantitative data of individual, user defined m/z bins. This information can e.g. be used to manually inspect the agreement between glycan composition and the presence of diagnostic oxonium ions. Ion masses to be considered are defined in a separate.txt configuration file. The configuration file used in this example can be downloaded at: N.B. the use of this Node is optional. 12
, the exported Excel workbook, and a manually filtered result file (.csv) can be downloaded from: www.imba.oeaw.ac.at/sugarqb.")
13 Anticipated Results using the sample data file provided: The Thermo Scientific Proteome Discoverer 1.4. result files (.msf), the exported Excel workbook, and a manually filtered result file (.csv) can be downloaded from: Contact: SugarQb@imba.oeaw.ac.at 13
")

14 Annex Alternative MASCOT Parameters (optional) Instruments Settings: Modification Settings HexNAc(NL) : 14
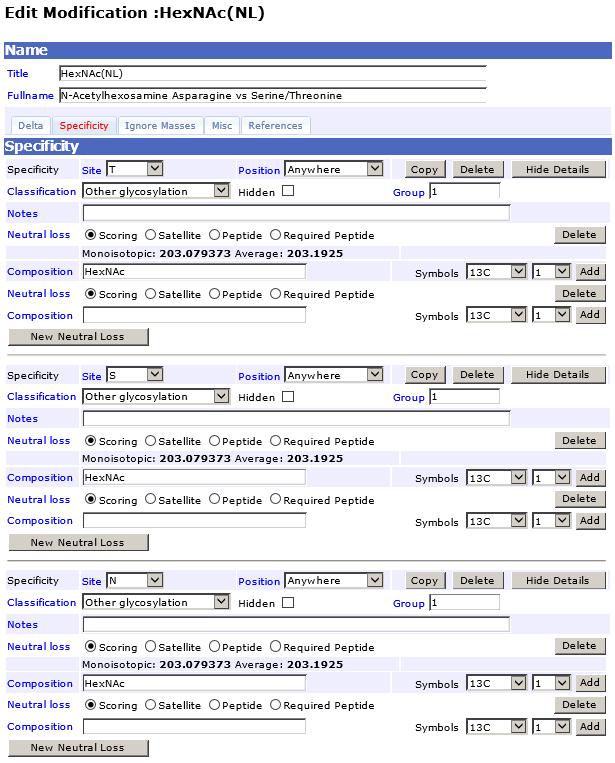
15 15
16 16
Workflow concept. Data goes through the workflow. A Node contains an operation An edge represents data flow The results are brought together in tables
 PROTEOME DISCOVERER Workflow concept Data goes through the workflow Spectra Peptides Quantitation A Node contains an operation An edge represents data flow The results are brought together in tables Protein
PROTEOME DISCOVERER Workflow concept Data goes through the workflow Spectra Peptides Quantitation A Node contains an operation An edge represents data flow The results are brought together in tables Protein
Overview - MS Proteomics in One Slide. MS masses of peptides. MS/MS fragments of a peptide. Results! Match to sequence database
 Overview - MS Proteomics in One Slide Obtain protein Digest into peptides Acquire spectra in mass spectrometer MS masses of peptides MS/MS fragments of a peptide Results! Match to sequence database 2 But
Overview - MS Proteomics in One Slide Obtain protein Digest into peptides Acquire spectra in mass spectrometer MS masses of peptides MS/MS fragments of a peptide Results! Match to sequence database 2 But
Nature Methods: doi: /nmeth Supplementary Figure 1. Fragment indexing allows efficient spectra similarity comparisons.
 Supplementary Figure 1 Fragment indexing allows efficient spectra similarity comparisons. The cost and efficiency of spectra similarity calculations can be approximated by the number of fragment comparisons
Supplementary Figure 1 Fragment indexing allows efficient spectra similarity comparisons. The cost and efficiency of spectra similarity calculations can be approximated by the number of fragment comparisons
SRM assay generation and data analysis in Skyline
 in Skyline Preparation 1. Download the example data from www.srmcourse.ch/eupa.html (3 raw files, 1 csv file, 1 sptxt file). 2. The number formats of your computer have to be set to English (United States).
in Skyline Preparation 1. Download the example data from www.srmcourse.ch/eupa.html (3 raw files, 1 csv file, 1 sptxt file). 2. The number formats of your computer have to be set to English (United States).
Analyst Software. Peptide and Protein Quantitation Tutorial
 This document is provided to customers who have purchased AB Sciex equipment to use in the operation of such AB Sciex equipment. This document is copyright protected and any reproduction of this document
This document is provided to customers who have purchased AB Sciex equipment to use in the operation of such AB Sciex equipment. This document is copyright protected and any reproduction of this document
Spectronaut Pulsar. User Manual
 Spectronaut Pulsar User Manual 1 General Information... 6 1.1 Computer System Requirements... 6 1.2 Scope of Spectronaut Software... 6 1.3 Spectronaut Pulsar... 6 1.4 Spectronaut Release Features... 7
Spectronaut Pulsar User Manual 1 General Information... 6 1.1 Computer System Requirements... 6 1.2 Scope of Spectronaut Software... 6 1.3 Spectronaut Pulsar... 6 1.4 Spectronaut Release Features... 7
Information Dependent Acquisition (IDA) 1
 1") Information Dependent Acquisition (IDA) Information Dependent Acquisition (IDA) enables on the fly acquisition of MS/MS spectra during a chromatographic run. Analyst Software IDA is optimized to generate
Information Dependent Acquisition (IDA) Information Dependent Acquisition (IDA) enables on the fly acquisition of MS/MS spectra during a chromatographic run. Analyst Software IDA is optimized to generate
Self-assembling covalent organic frameworks functionalized. magnetic graphene hydrophilic biocomposite as an ultrasensitive
 Electronic Supplementary Material (ESI) for Nanoscale. This journal is The Royal Society of Chemistry 2017 Electronic Supporting Information for: Self-assembling covalent organic frameworks functionalized
Electronic Supplementary Material (ESI) for Nanoscale. This journal is The Royal Society of Chemistry 2017 Electronic Supporting Information for: Self-assembling covalent organic frameworks functionalized
The Pitfalls of Peaklist Generation Software Performance on Database Searches
 Proceedings of the 56th ASMS Conference on Mass Spectrometry and Allied Topics, Denver, CO, June 1-5, 2008 The Pitfalls of Peaklist Generation Software Performance on Database Searches Aenoch J. Lynn,
Proceedings of the 56th ASMS Conference on Mass Spectrometry and Allied Topics, Denver, CO, June 1-5, 2008 The Pitfalls of Peaklist Generation Software Performance on Database Searches Aenoch J. Lynn,
DIA-Umpire: comprehensive computational framework for data independent acquisition proteomics
 DIA-Umpire: comprehensive computational framework for data independent acquisition proteomics Chih-Chiang Tsou 1,2, Dmitry Avtonomov 2, Brett Larsen 3, Monika Tucholska 3, Hyungwon Choi 4 Anne-Claude Gingras
DIA-Umpire: comprehensive computational framework for data independent acquisition proteomics Chih-Chiang Tsou 1,2, Dmitry Avtonomov 2, Brett Larsen 3, Monika Tucholska 3, Hyungwon Choi 4 Anne-Claude Gingras
TUTORIAL EXERCISES WITH ANSWERS
 TUTORIAL EXERCISES WITH ANSWERS Tutorial 1 Settings 1. What is the exact monoisotopic mass difference for peptides carrying a 13 C (and NO additional 15 N) labelled C-terminal lysine residue? a. 6.020129
TUTORIAL EXERCISES WITH ANSWERS Tutorial 1 Settings 1. What is the exact monoisotopic mass difference for peptides carrying a 13 C (and NO additional 15 N) labelled C-terminal lysine residue? a. 6.020129
Tutorial 1: Setting up your Skyline document
 Tutorial 1: Setting up your Skyline document Caution! For using Skyline the number formats of your computer have to be set to English (United States). Open the Control Panel Clock, Language, and Region
Tutorial 1: Setting up your Skyline document Caution! For using Skyline the number formats of your computer have to be set to English (United States). Open the Control Panel Clock, Language, and Region
Effective Strategies for Improving Peptide Identification with Tandem Mass Spectrometry
 Effective Strategies for Improving Peptide Identification with Tandem Mass Spectrometry by Xi Han A thesis presented to the University of Waterloo in fulfillment of the thesis requirement for the degree
Effective Strategies for Improving Peptide Identification with Tandem Mass Spectrometry by Xi Han A thesis presented to the University of Waterloo in fulfillment of the thesis requirement for the degree
Last updated: Copyright
 Last updated: 2012-08-20 Copyright 2004-2012 plabel (v2.4) User s Manual by Bioinformatics Group, Institute of Computing Technology, Chinese Academy of Sciences Tel: 86-10-62601016 Email: zhangkun01@ict.ac.cn,
Last updated: 2012-08-20 Copyright 2004-2012 plabel (v2.4) User s Manual by Bioinformatics Group, Institute of Computing Technology, Chinese Academy of Sciences Tel: 86-10-62601016 Email: zhangkun01@ict.ac.cn,
All Ions MS/MS: Targeted Screening and Quantitation Using Agilent TOF and Q-TOF LC/MS Systems
 All Ions MS/MS: Targeted Screening and Quantitation Using Agilent TOF and Q-TOF LC/MS Systems Technical Overview Introduction All Ions MS/MS is a technique that is available for Agilent high resolution
All Ions MS/MS: Targeted Screening and Quantitation Using Agilent TOF and Q-TOF LC/MS Systems Technical Overview Introduction All Ions MS/MS is a technique that is available for Agilent high resolution
X!TandemPipeline (Myosine Anabolisée) validating, filtering and grouping MSMS identifications
 validating, filtering and grouping MSMS identifications") X!TandemPipeline 3.3.3 (Myosine Anabolisée) validating, filtering and grouping MSMS identifications Olivier Langella and Benoit Valot langella@moulon.inra.fr; valot@moulon.inra.fr PAPPSO - http://pappso.inra.fr/
X!TandemPipeline 3.3.3 (Myosine Anabolisée) validating, filtering and grouping MSMS identifications Olivier Langella and Benoit Valot langella@moulon.inra.fr; valot@moulon.inra.fr PAPPSO - http://pappso.inra.fr/
Improved 6- Plex TMT Quantification Throughput Using a Linear Ion Trap HCD MS 3 Scan Jane M. Liu, 1,2 * Michael J. Sweredoski, 2 Sonja Hess 2 *
 Improved 6- Plex TMT Quantification Throughput Using a Linear Ion Trap HCD MS 3 Scan Jane M. Liu, 1,2 * Michael J. Sweredoski, 2 Sonja Hess 2 * 1 Department of Chemistry, Pomona College, Claremont, California
Improved 6- Plex TMT Quantification Throughput Using a Linear Ion Trap HCD MS 3 Scan Jane M. Liu, 1,2 * Michael J. Sweredoski, 2 Sonja Hess 2 * 1 Department of Chemistry, Pomona College, Claremont, California
Computational Methods for Mass Spectrometry Proteomics
 Computational Methods for Mass Spectrometry Proteomics Eidhammer, Ingvar ISBN-13: 9780470512975 Table of Contents Preface. Acknowledgements. 1 Protein, Proteome, and Proteomics. 1.1 Primary goals for studying
Computational Methods for Mass Spectrometry Proteomics Eidhammer, Ingvar ISBN-13: 9780470512975 Table of Contents Preface. Acknowledgements. 1 Protein, Proteome, and Proteomics. 1.1 Primary goals for studying
Protein Deconvolution Version 2.0
 Thermo Protein Deconvolution Version 2.0 User Guide XCALI-97414 Revision A August 2012 2012 Thermo Fisher Scientific Inc. All rights reserved. ReSpect is a trademark of Positive Probability Ltd. Xcalibur
Thermo Protein Deconvolution Version 2.0 User Guide XCALI-97414 Revision A August 2012 2012 Thermo Fisher Scientific Inc. All rights reserved. ReSpect is a trademark of Positive Probability Ltd. Xcalibur
Protein Quantitation II: Multiple Reaction Monitoring. Kelly Ruggles New York University
 Protein Quantitation II: Multiple Reaction Monitoring Kelly Ruggles kelly@fenyolab.org New York University Traditional Affinity-based proteomics Use antibodies to quantify proteins Western Blot RPPA Immunohistochemistry
Protein Quantitation II: Multiple Reaction Monitoring Kelly Ruggles kelly@fenyolab.org New York University Traditional Affinity-based proteomics Use antibodies to quantify proteins Western Blot RPPA Immunohistochemistry
PeptideProphet: Validation of Peptide Assignments to MS/MS Spectra. Andrew Keller
 PeptideProphet: Validation of Peptide Assignments to MS/MS Spectra Andrew Keller Outline Need to validate peptide assignments to MS/MS spectra Statistical approach to validation Running PeptideProphet
PeptideProphet: Validation of Peptide Assignments to MS/MS Spectra Andrew Keller Outline Need to validate peptide assignments to MS/MS spectra Statistical approach to validation Running PeptideProphet
MassHunter Software Overview
 MassHunter Software Overview 1 Qualitative Analysis Workflows Workflows in Qualitative Analysis allow the user to only see and work with the areas and dialog boxes they need for their specific tasks A
MassHunter Software Overview 1 Qualitative Analysis Workflows Workflows in Qualitative Analysis allow the user to only see and work with the areas and dialog boxes they need for their specific tasks A
Mass Spectrometry and Proteomics - Lecture 5 - Matthias Trost Newcastle University
 Mass Spectrometry and Proteomics - Lecture 5 - Matthias Trost Newcastle University matthias.trost@ncl.ac.uk Previously Proteomics Sample prep 144 Lecture 5 Quantitation techniques Search Algorithms Proteomics
Mass Spectrometry and Proteomics - Lecture 5 - Matthias Trost Newcastle University matthias.trost@ncl.ac.uk Previously Proteomics Sample prep 144 Lecture 5 Quantitation techniques Search Algorithms Proteomics
High-Field Orbitrap Creating new possibilities
 Thermo Scientific Orbitrap Elite Hybrid Mass Spectrometer High-Field Orbitrap Creating new possibilities Ultrahigh resolution Faster scanning Higher sensitivity Complementary fragmentation The highest
Thermo Scientific Orbitrap Elite Hybrid Mass Spectrometer High-Field Orbitrap Creating new possibilities Ultrahigh resolution Faster scanning Higher sensitivity Complementary fragmentation The highest
Protein Quantitation II: Multiple Reaction Monitoring. Kelly Ruggles New York University
 Protein Quantitation II: Multiple Reaction Monitoring Kelly Ruggles kelly@fenyolab.org New York University Traditional Affinity-based proteomics Use antibodies to quantify proteins Western Blot Immunohistochemistry
Protein Quantitation II: Multiple Reaction Monitoring Kelly Ruggles kelly@fenyolab.org New York University Traditional Affinity-based proteomics Use antibodies to quantify proteins Western Blot Immunohistochemistry
MassHunter TOF/QTOF Users Meeting
 MassHunter TOF/QTOF Users Meeting 1 Qualitative Analysis Workflows Workflows in Qualitative Analysis allow the user to only see and work with the areas and dialog boxes they need for their specific tasks
MassHunter TOF/QTOF Users Meeting 1 Qualitative Analysis Workflows Workflows in Qualitative Analysis allow the user to only see and work with the areas and dialog boxes they need for their specific tasks
Agilent MassHunter Profinder: Solving the Challenge of Isotopologue Extraction for Qualitative Flux Analysis
 Agilent MassHunter Profinder: Solving the Challenge of Isotopologue Extraction for Qualitative Flux Analysis Technical Overview Introduction Metabolomics studies measure the relative abundance of metabolites
Agilent MassHunter Profinder: Solving the Challenge of Isotopologue Extraction for Qualitative Flux Analysis Technical Overview Introduction Metabolomics studies measure the relative abundance of metabolites
SIERRA ANALYTICS, INC. Version Polymerix Software User Manual
 SIERRA ANALYTICS, INC. Version 3.0.0 Polymerix Software User Manual V E R S I O N 3.0.0 M A R C H 2013 Polymerix Software User Manual Copyright 2010 to 2013 Sierra Analytics, Inc. 5815 Stoddard Road, Suite
SIERRA ANALYTICS, INC. Version 3.0.0 Polymerix Software User Manual V E R S I O N 3.0.0 M A R C H 2013 Polymerix Software User Manual Copyright 2010 to 2013 Sierra Analytics, Inc. 5815 Stoddard Road, Suite
Compounding insights Thermo Scientific Compound Discoverer Software
 Compounding insights Thermo Scientific Compound Discoverer Software Integrated, complete, toolset solves small-molecule analysis challenges Thermo Scientific Orbitrap mass spectrometers produce information-rich
Compounding insights Thermo Scientific Compound Discoverer Software Integrated, complete, toolset solves small-molecule analysis challenges Thermo Scientific Orbitrap mass spectrometers produce information-rich
Figure S1. Interaction of PcTS with αsyn. (a) 1 H- 15 N HSQC NMR spectra of 100 µm αsyn in the absence (0:1, black) and increasing equivalent
 1 H- 15 N HSQC NMR spectra of 100 µm αsyn in the absence (0:1, black) and increasing equivalent") Figure S1. Interaction of PcTS with αsyn. (a) 1 H- 15 N HSQC NMR spectra of 100 µm αsyn in the absence (0:1, black) and increasing equivalent concentrations of PcTS (100 µm, blue; 500 µm, green; 1.5 mm,
Figure S1. Interaction of PcTS with αsyn. (a) 1 H- 15 N HSQC NMR spectra of 100 µm αsyn in the absence (0:1, black) and increasing equivalent concentrations of PcTS (100 µm, blue; 500 µm, green; 1.5 mm,
PeptideProphet: Validation of Peptide Assignments to MS/MS Spectra
 PeptideProphet: Validation of Peptide Assignments to MS/MS Spectra Andrew Keller Day 2 October 17, 2006 Andrew Keller Rosetta Bioinformatics, Seattle Outline Need to validate peptide assignments to MS/MS
PeptideProphet: Validation of Peptide Assignments to MS/MS Spectra Andrew Keller Day 2 October 17, 2006 Andrew Keller Rosetta Bioinformatics, Seattle Outline Need to validate peptide assignments to MS/MS
Key Words Q Exactive, Accela, MetQuest, Mass Frontier, Drug Discovery
 Metabolite Stability Screening and Hotspot Metabolite Identification by Combining High-Resolution, Accurate-Mass Nonselective and Selective Fragmentation Tim Stratton, Caroline Ding, Yingying Huang, Dan
Metabolite Stability Screening and Hotspot Metabolite Identification by Combining High-Resolution, Accurate-Mass Nonselective and Selective Fragmentation Tim Stratton, Caroline Ding, Yingying Huang, Dan
Tutorial 1: Library Generation from DDA data
 Tutorial 1: Library Generation from DDA data 1. Introduction Before a targeted, peptide-centric DIA analysis can be performed, a spectral library containing peptide-query parameters needs to be generated.
Tutorial 1: Library Generation from DDA data 1. Introduction Before a targeted, peptide-centric DIA analysis can be performed, a spectral library containing peptide-query parameters needs to be generated.
profileanalysis Innovation with Integrity Quickly pinpointing and identifying potential biomarkers in Proteomics and Metabolomics research
 profileanalysis Quickly pinpointing and identifying potential biomarkers in Proteomics and Metabolomics research Innovation with Integrity Omics Research Biomarker Discovery Made Easy by ProfileAnalysis
profileanalysis Quickly pinpointing and identifying potential biomarkers in Proteomics and Metabolomics research Innovation with Integrity Omics Research Biomarker Discovery Made Easy by ProfileAnalysis
ProMass Deconvolution User Training. Novatia LLC January, 2013
 ProMass Deconvolution User Training Novatia LLC January, 2013 Overview General info about ProMass Features Basics of how ProMass Deconvolution works Example Spectra Manual Deconvolution with ProMass Deconvolution
ProMass Deconvolution User Training Novatia LLC January, 2013 Overview General info about ProMass Features Basics of how ProMass Deconvolution works Example Spectra Manual Deconvolution with ProMass Deconvolution
Comprehensive support for quantitation
 Comprehensive support for quantitation One of the major new features in the current release of Mascot is support for quantitation. This is still work in progress. Our goal is to support all of the popular
Comprehensive support for quantitation One of the major new features in the current release of Mascot is support for quantitation. This is still work in progress. Our goal is to support all of the popular
Relative quantification using TMT11plex on a modified Q Exactive HF mass spectrometer
 POSTER NOTE 6558 Relative quantification using TMT11plex on a modified mass spectrometer Authors Tabiwang N. Arrey, 1 Rosa Viner, 2 Ryan D. Bomgarden, 3 Eugen Damoc, 1 Markus Kellmann, 1 Thomas Moehring,
POSTER NOTE 6558 Relative quantification using TMT11plex on a modified mass spectrometer Authors Tabiwang N. Arrey, 1 Rosa Viner, 2 Ryan D. Bomgarden, 3 Eugen Damoc, 1 Markus Kellmann, 1 Thomas Moehring,
Modeling Mass Spectrometry-Based Protein Analysis
 Chapter 8 Jan Eriksson and David Fenyö Abstract The success of mass spectrometry based proteomics depends on efficient methods for data analysis. These methods require a detailed understanding of the information
Chapter 8 Jan Eriksson and David Fenyö Abstract The success of mass spectrometry based proteomics depends on efficient methods for data analysis. These methods require a detailed understanding of the information
Tutorial 2: Analysis of DIA data in Skyline
 Tutorial 2: Analysis of DIA data in Skyline In this tutorial we will learn how to use Skyline to perform targeted post-acquisition analysis for peptide and inferred protein detection and quantitation using
Tutorial 2: Analysis of DIA data in Skyline In this tutorial we will learn how to use Skyline to perform targeted post-acquisition analysis for peptide and inferred protein detection and quantitation using
Thermo Scientific LTQ Orbitrap Velos Hybrid FT Mass Spectrometer
 IET International Equipment Trading Ltd. www.ietltd.com Proudly serving laboratories worldwide since 1979 CALL +847.913.0777 for Refurbished & Certified Lab Equipment Thermo Scientific LTQ Orbitrap Velos
IET International Equipment Trading Ltd. www.ietltd.com Proudly serving laboratories worldwide since 1979 CALL +847.913.0777 for Refurbished & Certified Lab Equipment Thermo Scientific LTQ Orbitrap Velos
Skyline Small Molecule Targets
 Skyline Small Molecule Targets The Skyline Targeted Proteomics Environment provides informative visual displays of the raw mass spectrometer data you import into your Skyline documents. Originally developed
Skyline Small Molecule Targets The Skyline Targeted Proteomics Environment provides informative visual displays of the raw mass spectrometer data you import into your Skyline documents. Originally developed
Proteomics. November 13, 2007
 Proteomics November 13, 2007 Acknowledgement Slides presented here have been borrowed from presentations by : Dr. Mark A. Knepper (LKEM, NHLBI, NIH) Dr. Nathan Edwards (Center for Bioinformatics and Computational
Proteomics November 13, 2007 Acknowledgement Slides presented here have been borrowed from presentations by : Dr. Mark A. Knepper (LKEM, NHLBI, NIH) Dr. Nathan Edwards (Center for Bioinformatics and Computational
Quantitation of a target protein in crude samples using targeted peptide quantification by Mass Spectrometry
 Quantitation of a target protein in crude samples using targeted peptide quantification by Mass Spectrometry Jon Hao, Rong Ye, and Mason Tao Poochon Scientific, Frederick, Maryland 21701 Abstract Background:
Quantitation of a target protein in crude samples using targeted peptide quantification by Mass Spectrometry Jon Hao, Rong Ye, and Mason Tao Poochon Scientific, Frederick, Maryland 21701 Abstract Background:
A Description of the CPTAC Common Data Analysis Pipeline (CDAP)
") A Description of the CPTAC Common Data Analysis Pipeline (CDAP) v. 01/14/2014 Summary The purpose of this document is to describe the software programs and output files of the Common Data Analysis Pipeline
A Description of the CPTAC Common Data Analysis Pipeline (CDAP) v. 01/14/2014 Summary The purpose of this document is to describe the software programs and output files of the Common Data Analysis Pipeline
Properties of Average Score Distributions of SEQUEST
 Research Properties of Average Score Distributions of SEQUEST THE PROBABILITY RATIO METHOD* S Salvador Martínez-Bartolomé, Pedro Navarro, Fernando Martín-Maroto, Daniel López-Ferrer **, Antonio Ramos-Fernández,
Research Properties of Average Score Distributions of SEQUEST THE PROBABILITY RATIO METHOD* S Salvador Martínez-Bartolomé, Pedro Navarro, Fernando Martín-Maroto, Daniel López-Ferrer **, Antonio Ramos-Fernández,
Protein Identification Using Tandem Mass Spectrometry. Nathan Edwards Informatics Research Applied Biosystems
 Protein Identification Using Tandem Mass Spectrometry Nathan Edwards Informatics Research Applied Biosystems Outline Proteomics context Tandem mass spectrometry Peptide fragmentation Peptide identification
Protein Identification Using Tandem Mass Spectrometry Nathan Edwards Informatics Research Applied Biosystems Outline Proteomics context Tandem mass spectrometry Peptide fragmentation Peptide identification
Spectrum-to-Spectrum Searching Using a. Proteome-wide Spectral Library
 MCP Papers in Press. Published on April 30, 2011 as Manuscript M111.007666 Spectrum-to-Spectrum Searching Using a Proteome-wide Spectral Library Chia-Yu Yen, Stephane Houel, Natalie G. Ahn, and William
MCP Papers in Press. Published on April 30, 2011 as Manuscript M111.007666 Spectrum-to-Spectrum Searching Using a Proteome-wide Spectral Library Chia-Yu Yen, Stephane Houel, Natalie G. Ahn, and William
Site-specific Identification of Lysine Acetylation Stoichiometries in Mammalian Cells
 Supplementary Information Site-specific Identification of Lysine Acetylation Stoichiometries in Mammalian Cells Tong Zhou 1, 2, Ying-hua Chung 1, 2, Jianji Chen 1, Yue Chen 1 1. Department of Biochemistry,
Supplementary Information Site-specific Identification of Lysine Acetylation Stoichiometries in Mammalian Cells Tong Zhou 1, 2, Ying-hua Chung 1, 2, Jianji Chen 1, Yue Chen 1 1. Department of Biochemistry,
Improved Validation of Peptide MS/MS Assignments. Using Spectral Intensity Prediction
 MCP Papers in Press. Published on October 2, 2006 as Manuscript M600320-MCP200 Improved Validation of Peptide MS/MS Assignments Using Spectral Intensity Prediction Shaojun Sun 1, Karen Meyer-Arendt 2,
MCP Papers in Press. Published on October 2, 2006 as Manuscript M600320-MCP200 Improved Validation of Peptide MS/MS Assignments Using Spectral Intensity Prediction Shaojun Sun 1, Karen Meyer-Arendt 2,
De novo Protein Sequencing by Combining Top-Down and Bottom-Up Tandem Mass Spectra. Xiaowen Liu
 De novo Protein Sequencing by Combining Top-Down and Bottom-Up Tandem Mass Spectra Xiaowen Liu Department of BioHealth Informatics, Department of Computer and Information Sciences, Indiana University-Purdue
De novo Protein Sequencing by Combining Top-Down and Bottom-Up Tandem Mass Spectra Xiaowen Liu Department of BioHealth Informatics, Department of Computer and Information Sciences, Indiana University-Purdue
Database Search Strategies for Proteomic Data Sets Generated by Electron Capture Dissociation Mass Spectrometry
 Database Search Strategies for Proteomic Data Sets Generated by Electron Capture Dissociation Mass Spectrometry Steve M. M. Sweet,,# Andrew W. Jones, Debbie L. Cunningham, John K. Heath, Andrew J. Creese,
Database Search Strategies for Proteomic Data Sets Generated by Electron Capture Dissociation Mass Spectrometry Steve M. M. Sweet,,# Andrew W. Jones, Debbie L. Cunningham, John K. Heath, Andrew J. Creese,
Rapid Distinction of Leucine and Isoleucine in Monoclonal Antibodies Using Nanoflow. LCMS n. Discovery Attribute Sciences
 Rapid Distinction of Leucine and Isoleucine in Monoclonal Antibodies Using Nanoflow LCMS n Dhanashri Bagal *, Eddie Kast, Ping Cao Discovery Attribute Sciences Amgen, South San Francisco, California, United
Rapid Distinction of Leucine and Isoleucine in Monoclonal Antibodies Using Nanoflow LCMS n Dhanashri Bagal *, Eddie Kast, Ping Cao Discovery Attribute Sciences Amgen, South San Francisco, California, United
MALDI Calibration. Mass Control List
 MALDI Calibration Thank you for choosing SpheriCal Aqua as your choice of calibrant. You will come to find that it outperforms comparable calibrants available on the market and that it offers you the precision
MALDI Calibration Thank you for choosing SpheriCal Aqua as your choice of calibrant. You will come to find that it outperforms comparable calibrants available on the market and that it offers you the precision
MALDI-HDMS E : A Novel Data Independent Acquisition Method for the Enhanced Analysis of 2D-Gel Tryptic Peptide Digests
 -HDMS E : A Novel Data Independent Acquisition Method for the Enhanced Analysis of 2D-Gel Tryptic Peptide Digests Emmanuelle Claude, 1 Mark Towers, 1 and Rachel Craven 2 1 Waters Corporation, Manchester,
-HDMS E : A Novel Data Independent Acquisition Method for the Enhanced Analysis of 2D-Gel Tryptic Peptide Digests Emmanuelle Claude, 1 Mark Towers, 1 and Rachel Craven 2 1 Waters Corporation, Manchester,
MS-MS Analysis Programs
 MS-MS Analysis Programs Basic Process Genome - Gives AA sequences of proteins Use this to predict spectra Compare data to prediction Determine degree of correctness Make assignment Did we see the protein?
MS-MS Analysis Programs Basic Process Genome - Gives AA sequences of proteins Use this to predict spectra Compare data to prediction Determine degree of correctness Make assignment Did we see the protein?
Supplementary Figure 1
 Supplementary Figure 1 The correlation of n-score cutoff and FDR in both CID-only and CID-ETD fragmentation strategies. A bar diagram of different n-score thresholds applied in the search, plotted against
Supplementary Figure 1 The correlation of n-score cutoff and FDR in both CID-only and CID-ETD fragmentation strategies. A bar diagram of different n-score thresholds applied in the search, plotted against
Mass Spectrometry Based De Novo Peptide Sequencing Error Correction
 Mass Spectrometry Based De Novo Peptide Sequencing Error Correction by Chenyu Yao A thesis presented to the University of Waterloo in fulfillment of the thesis requirement for the degree of Master of Mathematics
Mass Spectrometry Based De Novo Peptide Sequencing Error Correction by Chenyu Yao A thesis presented to the University of Waterloo in fulfillment of the thesis requirement for the degree of Master of Mathematics
High-Throughput Protein Quantitation Using Multiple Reaction Monitoring
 High-Throughput Protein Quantitation Using Multiple Reaction Monitoring Application Note Authors Ning Tang, Christine Miller, Joe Roark, Norton Kitagawa and Keith Waddell Agilent Technologies, Inc. Santa
High-Throughput Protein Quantitation Using Multiple Reaction Monitoring Application Note Authors Ning Tang, Christine Miller, Joe Roark, Norton Kitagawa and Keith Waddell Agilent Technologies, Inc. Santa
Agilent MassHunter Quantitative Data Analysis
 Agilent MassHunter Quantitative Data Analysis Presenters: Howard Sanford Stephen Harnos MassHunter Quantitation: Batch and Method Setup Outliers, Data Review, Reporting 1 MassHunter Quantitative Analysis
Agilent MassHunter Quantitative Data Analysis Presenters: Howard Sanford Stephen Harnos MassHunter Quantitation: Batch and Method Setup Outliers, Data Review, Reporting 1 MassHunter Quantitative Analysis
PRIDE Cluster: building the consensus of proteomics data
 Supplementary Materials PRIDE Cluster: building the consensus of proteomics data Johannes Griss, Joseph Michael Foster, Henning Hermjakob and Juan Antonio Vizcaíno EMBL-European Bioinformatics Institute,
Supplementary Materials PRIDE Cluster: building the consensus of proteomics data Johannes Griss, Joseph Michael Foster, Henning Hermjakob and Juan Antonio Vizcaíno EMBL-European Bioinformatics Institute,
Peptide Targeted Quantification By High Resolution Mass Spectrometry A Paradigm Shift? Zhiqi Hao Thermo Fisher Scientific San Jose, CA
 Peptide Targeted Quantification By High Resolution Mass Spectrometry A Paradigm Shift? Zhiqi Hao Thermo Fisher Scientific San Jose, CA Proteomics is Turning Quantitative Hmmm.. Which ones are my targets?
Peptide Targeted Quantification By High Resolution Mass Spectrometry A Paradigm Shift? Zhiqi Hao Thermo Fisher Scientific San Jose, CA Proteomics is Turning Quantitative Hmmm.. Which ones are my targets?
MS Based Proteomics: Recent Case Studies Using Advanced Instrumentation
 MS Based Proteomics: Recent Case Studies Using Advanced Instrumentation Chris Adams, PH.D. Stanford University Mass Spectrometry http://mass-spec.stanford.edu/ For personal use only. Please do not reuse
MS Based Proteomics: Recent Case Studies Using Advanced Instrumentation Chris Adams, PH.D. Stanford University Mass Spectrometry http://mass-spec.stanford.edu/ For personal use only. Please do not reuse
Learning Score Function Parameters for Improved Spectrum Identification in Tandem Mass Spectrometry Experiments
 pubs.acs.org/jpr Learning Score Function Parameters for Improved Spectrum Identification in Tandem Mass Spectrometry Experiments Marina Spivak, Michael S. Bereman, Michael J. MacCoss, and William Stafford
pubs.acs.org/jpr Learning Score Function Parameters for Improved Spectrum Identification in Tandem Mass Spectrometry Experiments Marina Spivak, Michael S. Bereman, Michael J. MacCoss, and William Stafford
TOMAHAQ Method Construction
 TOMAHAQ Method Construction Triggered by offset mass accurate-mass high-resolution accurate quantitation (TOMAHAQ) can be performed in the standard method editor of the instrument, without modifications
TOMAHAQ Method Construction Triggered by offset mass accurate-mass high-resolution accurate quantitation (TOMAHAQ) can be performed in the standard method editor of the instrument, without modifications
Search for the Gulf of Carpentaria in the remap search bar:
 This tutorial is aimed at getting you started with making maps in Remap (). In this tutorial we are going to develop a simple classification of mangroves in northern Australia. Before getting started with
This tutorial is aimed at getting you started with making maps in Remap (). In this tutorial we are going to develop a simple classification of mangroves in northern Australia. Before getting started with
MSnID Package for Handling MS/MS Identifications
 Vladislav A. Petyuk December 1, 2018 Contents 1 Introduction.............................. 1 2 Starting the project.......................... 3 3 Reading MS/MS data........................ 3 4 Updating
Vladislav A. Petyuk December 1, 2018 Contents 1 Introduction.............................. 1 2 Starting the project.......................... 3 3 Reading MS/MS data........................ 3 4 Updating
Making Sense of Differences in LCMS Data: Integrated Tools
 Making Sense of Differences in LCMS Data: Integrated Tools David A. Weil Agilent Technologies MassHunter Overview Page 1 March 2008 How Clean is our Water?... Page 2 Chemical Residue Analysis.... From
Making Sense of Differences in LCMS Data: Integrated Tools David A. Weil Agilent Technologies MassHunter Overview Page 1 March 2008 How Clean is our Water?... Page 2 Chemical Residue Analysis.... From
Key questions of proteomics. Bioinformatics 2. Proteomics. Foundation of proteomics. What proteins are there? Protein digestion
 s s Key questions of proteomics What proteins are there? Bioinformatics 2 Lecture 2 roteomics How much is there of each of the proteins? - Absolute quantitation - Stoichiometry What (modification/splice)
s s Key questions of proteomics What proteins are there? Bioinformatics 2 Lecture 2 roteomics How much is there of each of the proteins? - Absolute quantitation - Stoichiometry What (modification/splice)
Performing Peptide Bioanalysis Using High Resolution Mass Spectrometry with Target Enhancement MRM Acquisition
 Performing Peptide Bioanalysis Using High Resolution Mass Spectrometry with Target Enhancement MRM Acquisition Yun Wang Alelyunas, Mark D. Wrona, and Nick Tomczyk Waters Corporation, Milford, MA, USA GOAL
Performing Peptide Bioanalysis Using High Resolution Mass Spectrometry with Target Enhancement MRM Acquisition Yun Wang Alelyunas, Mark D. Wrona, and Nick Tomczyk Waters Corporation, Milford, MA, USA GOAL
Quantitation of High Resolution MS Data Using UNIFI: Acquiring and Processing Full Scan or Tof-MRM (Targeted HRMS) Datasets for Quantitative Assays
 Datasets for Quantitative Assays") : Acquiring and Processing Full Scan or Tof-MRM (Targeted HRMS) Datasets for Quantitative Assays Mark Wrona, Jayne Kirk, and Yun Alelyunas Waters Corporation, Milford, MA, USA APPLICATION BENEFITS Ability
: Acquiring and Processing Full Scan or Tof-MRM (Targeted HRMS) Datasets for Quantitative Assays Mark Wrona, Jayne Kirk, and Yun Alelyunas Waters Corporation, Milford, MA, USA APPLICATION BENEFITS Ability
1. Prepare the MALDI sample plate by spotting an angiotensin standard and the test sample(s).
.") Analysis of a Peptide Sequence from a Proteolytic Digest by MALDI-TOF Post-Source Decay (PSD) and Collision-Induced Dissociation (CID) Standard Operating Procedure Purpose: The following procedure may
Analysis of a Peptide Sequence from a Proteolytic Digest by MALDI-TOF Post-Source Decay (PSD) and Collision-Induced Dissociation (CID) Standard Operating Procedure Purpose: The following procedure may
Multi-residue analysis of pesticides by GC-HRMS
 An Executive Summary Multi-residue analysis of pesticides by GC-HRMS Dr. Hans Mol is senior scientist at RIKILT- Wageningen UR Introduction Regulatory authorities throughout the world set and enforce strict
An Executive Summary Multi-residue analysis of pesticides by GC-HRMS Dr. Hans Mol is senior scientist at RIKILT- Wageningen UR Introduction Regulatory authorities throughout the world set and enforce strict
Proteome-wide label-free quantification with MaxQuant. Jürgen Cox Max Planck Institute of Biochemistry July 2011
 Proteome-wide label-free quantification with MaxQuant Jürgen Cox Max Planck Institute of Biochemistry July 2011 MaxQuant MaxQuant Feature detection Data acquisition Initial Andromeda search Statistics
Proteome-wide label-free quantification with MaxQuant Jürgen Cox Max Planck Institute of Biochemistry July 2011 MaxQuant MaxQuant Feature detection Data acquisition Initial Andromeda search Statistics
LECTURE-13. Peptide Mass Fingerprinting HANDOUT. Mass spectrometry is an indispensable tool for qualitative and quantitative analysis of
 LECTURE-13 Peptide Mass Fingerprinting HANDOUT PREAMBLE Mass spectrometry is an indispensable tool for qualitative and quantitative analysis of proteins, drugs and many biological moieties to elucidate
LECTURE-13 Peptide Mass Fingerprinting HANDOUT PREAMBLE Mass spectrometry is an indispensable tool for qualitative and quantitative analysis of proteins, drugs and many biological moieties to elucidate
OECD QSAR Toolbox v.4.1. Step-by-step example for building QSAR model
 OECD QSAR Toolbox v.4.1 Step-by-step example for building QSAR model Background Objectives The exercise Workflow of the exercise Outlook 2 Background This is a step-by-step presentation designed to take
OECD QSAR Toolbox v.4.1 Step-by-step example for building QSAR model Background Objectives The exercise Workflow of the exercise Outlook 2 Background This is a step-by-step presentation designed to take
Agilent All Ions MS/MS
 Agilent All Ions MS/MS Workflow Overview A Determine fragment ions for LC/MS Quant method B Develop final Quant method Develop LC/MS Qualitative Analysis method Process data with Find by Formula Build
Agilent All Ions MS/MS Workflow Overview A Determine fragment ions for LC/MS Quant method B Develop final Quant method Develop LC/MS Qualitative Analysis method Process data with Find by Formula Build
Designed for Accuracy. Innovation with Integrity. High resolution quantitative proteomics LC-MS
 Designed for Accuracy High resolution quantitative proteomics Innovation with Integrity LC-MS Setting New Standards in Accuracy The development of mass spectrometry based proteomics approaches has dramatically
Designed for Accuracy High resolution quantitative proteomics Innovation with Integrity LC-MS Setting New Standards in Accuracy The development of mass spectrometry based proteomics approaches has dramatically
ATLAS of Biochemistry
 ATLAS of Biochemistry USER GUIDE http://lcsb-databases.epfl.ch/atlas/ CONTENT 1 2 3 GET STARTED Create your user account NAVIGATE Curated KEGG reactions ATLAS reactions Pathways Maps USE IT! Fill a gap
ATLAS of Biochemistry USER GUIDE http://lcsb-databases.epfl.ch/atlas/ CONTENT 1 2 3 GET STARTED Create your user account NAVIGATE Curated KEGG reactions ATLAS reactions Pathways Maps USE IT! Fill a gap
NPTEL VIDEO COURSE PROTEOMICS PROF. SANJEEVA SRIVASTAVA
 LECTURE-25 Quantitative proteomics: itraq and TMT TRANSCRIPT Welcome to the proteomics course. Today we will talk about quantitative proteomics and discuss about itraq and TMT techniques. The quantitative
LECTURE-25 Quantitative proteomics: itraq and TMT TRANSCRIPT Welcome to the proteomics course. Today we will talk about quantitative proteomics and discuss about itraq and TMT techniques. The quantitative
Advanced Fragmentation Techniques for BioPharma Characterization
 Advanced Fragmentation Techniques for BioPharma Characterization Global BioPharma Summit The world leader in serving science Different modes of fragmentation to answer different questions or for different
Advanced Fragmentation Techniques for BioPharma Characterization Global BioPharma Summit The world leader in serving science Different modes of fragmentation to answer different questions or for different
Introduction to pepxmltab
 Introduction to pepxmltab Xiaojing Wang October 30, 2018 Contents 1 Introduction 1 2 Convert pepxml to a tabular format 1 3 PSMs Filtering 4 4 Session Information 5 1 Introduction Mass spectrometry (MS)-based
Introduction to pepxmltab Xiaojing Wang October 30, 2018 Contents 1 Introduction 1 2 Convert pepxml to a tabular format 1 3 PSMs Filtering 4 4 Session Information 5 1 Introduction Mass spectrometry (MS)-based
iprophet: Multi-level integrative analysis of shotgun proteomic data improves peptide and protein identification rates and error estimates
 MCP Papers in Press. Published on August 29, 2011 as Manuscript M111.007690 This is the Pre-Published Version iprophet: Multi-level integrative analysis of shotgun proteomic data improves peptide and protein
MCP Papers in Press. Published on August 29, 2011 as Manuscript M111.007690 This is the Pre-Published Version iprophet: Multi-level integrative analysis of shotgun proteomic data improves peptide and protein
Q Exactive TM : A True Qual-Quan HR/AM Mass Spectrometer for Routine Proteomics Applications. Yi Zhang, Ph.D. ThermoFisher Scientific
 Q Exactive TM : A True Qual-Quan HR/AM Mass Spectrometer for Routine Proteomics Applications Yi Zhang, Ph.D. ThermoFisher Scientific Outline Introduction of Q Exactive Performance in Discovery Proteomics
Q Exactive TM : A True Qual-Quan HR/AM Mass Spectrometer for Routine Proteomics Applications Yi Zhang, Ph.D. ThermoFisher Scientific Outline Introduction of Q Exactive Performance in Discovery Proteomics
Isotopic-Labeling and Mass Spectrometry-Based Quantitative Proteomics
 Isotopic-Labeling and Mass Spectrometry-Based Quantitative Proteomics Xiao-jun Li, Ph.D. Current address: Homestead Clinical Day 4 October 19, 2006 Protein Quantification LC-MS/MS Data XLink mzxml file
Isotopic-Labeling and Mass Spectrometry-Based Quantitative Proteomics Xiao-jun Li, Ph.D. Current address: Homestead Clinical Day 4 October 19, 2006 Protein Quantification LC-MS/MS Data XLink mzxml file
EasySDM: A Spatial Data Mining Platform
 EasySDM: A Spatial Data Mining Platform (User Manual) Authors: Amine Abdaoui and Mohamed Ala Al Chikha, Students at the National Computing Engineering School. Algiers. June 2013. 1. Overview EasySDM is
EasySDM: A Spatial Data Mining Platform (User Manual) Authors: Amine Abdaoui and Mohamed Ala Al Chikha, Students at the National Computing Engineering School. Algiers. June 2013. 1. Overview EasySDM is
LogViewer: A Software Tool to Visualize Quality Control Parameters to Optimize Proteomics Experiments using Orbitrap and LTQ-FT Mass Spectrometers
 COMMUNICATION LogViewer: A Software Tool to Visualize Quality Control Parameters to Optimize Proteomics Experiments using Orbitrap and LTQ-FT Mass Spectrometers Michael J. Sweredoski, Geoffrey T. Smith,
COMMUNICATION LogViewer: A Software Tool to Visualize Quality Control Parameters to Optimize Proteomics Experiments using Orbitrap and LTQ-FT Mass Spectrometers Michael J. Sweredoski, Geoffrey T. Smith,
ncounter PlexSet Data Analysis Guidelines
 ncounter PlexSet Data Analysis Guidelines NanoString Technologies, Inc. 530 airview Ave North Seattle, Washington 98109 USA Telephone: 206.378.6266 888.358.6266 E-mail: info@nanostring.com Molecules That
ncounter PlexSet Data Analysis Guidelines NanoString Technologies, Inc. 530 airview Ave North Seattle, Washington 98109 USA Telephone: 206.378.6266 888.358.6266 E-mail: info@nanostring.com Molecules That
MaSS-Simulator: A highly configurable MS/MS simulator for generating test datasets for big data algorithms.
 MaSS-Simulator: A highly configurable MS/MS simulator for generating test datasets for big data algorithms. Muaaz Gul Awan 1 and Fahad Saeed 1 1 Department of Computer Science, Western Michigan University,
MaSS-Simulator: A highly configurable MS/MS simulator for generating test datasets for big data algorithms. Muaaz Gul Awan 1 and Fahad Saeed 1 1 Department of Computer Science, Western Michigan University,
Interpreting raw biological mass spectra using isotopic mass-to-charge ratio and envelope fingerprinting
 Research Article Received: 24 December 2 Revised: 1 February 21 Accepted: March 21 Published online in Wiley Online Library Rapid Commun. Mass Spectrom. 21, 27, 7 77 (wileyonlinelibrary.com) DOI: 1./rcm.55
Research Article Received: 24 December 2 Revised: 1 February 21 Accepted: March 21 Published online in Wiley Online Library Rapid Commun. Mass Spectrom. 21, 27, 7 77 (wileyonlinelibrary.com) DOI: 1./rcm.55
Agilent TOF Screening & Impurity Profiling Julie Cichelli, PhD LC/MS Small Molecule Workshop Dec 6, 2012
 1 Agilent TOF Screening & Impurity Profiling Julie Cichelli, PhD LC/MS Small Molecule Workshop Dec 6, 2012 Review: Technology for Accurate Mass Analysis: TOF LC/MS Mass measurements accurate to several
1 Agilent TOF Screening & Impurity Profiling Julie Cichelli, PhD LC/MS Small Molecule Workshop Dec 6, 2012 Review: Technology for Accurate Mass Analysis: TOF LC/MS Mass measurements accurate to several
Correction of Errors in Tandem Mass Spectrum Extraction Enhances Phosphopeptide Identification
 pubs.acs.org/jpr Correction of Errors in Tandem Mass Spectrum Extraction Enhances Phosphopeptide Identification Piliang Hao,*,,, Yan Ren,, James P. Tam, and Siu Kwan Sze*,, School of Biological Sciences
pubs.acs.org/jpr Correction of Errors in Tandem Mass Spectrum Extraction Enhances Phosphopeptide Identification Piliang Hao,*,,, Yan Ren,, James P. Tam, and Siu Kwan Sze*,, School of Biological Sciences
A graph-based filtering method for top-down mass spectral identification
 Yang and Zhu BMC Genomics 2018, 19(Suppl 7):666 https://doi.org/10.1186/s12864-018-5026-x METHODOLOGY Open Access A graph-based filtering method for top-down mass spectral identification Runmin Yang and
Yang and Zhu BMC Genomics 2018, 19(Suppl 7):666 https://doi.org/10.1186/s12864-018-5026-x METHODOLOGY Open Access A graph-based filtering method for top-down mass spectral identification Runmin Yang and
Exercises for Windows
 Exercises for Windows CAChe User Interface for Windows Select tool Application window Document window (workspace) Style bar Tool palette Select entire molecule Select Similar Group Select Atom tool Rotate
Exercises for Windows CAChe User Interface for Windows Select tool Application window Document window (workspace) Style bar Tool palette Select entire molecule Select Similar Group Select Atom tool Rotate
A Quadrupole-Orbitrap Hybrid Mass Spectrometer Offers Highest Benchtop Performance for In-Depth Analysis of Complex Proteomes
 A Quadrupole-Orbitrap Hybrid Mass Spectrometer Offers Highest Benchtop Performance for In-Depth Analysis of Complex Proteomes Zhiqi Hao 1, Yi Zhang 1, Shannon Eliuk 1, Justin Blethrow 1, Dave Horn 1, Vlad
A Quadrupole-Orbitrap Hybrid Mass Spectrometer Offers Highest Benchtop Performance for In-Depth Analysis of Complex Proteomes Zhiqi Hao 1, Yi Zhang 1, Shannon Eliuk 1, Justin Blethrow 1, Dave Horn 1, Vlad
Proteomics: the first decade and beyond. (2003) Patterson and Aebersold Nat Genet 33 Suppl: from
 Patterson and Aebersold Nat Genet 33 Suppl: from") Advances in mass spectrometry and the generation of large quantities of nucleotide sequence information, combined with computational algorithms that could correlate the two, led to the emergence of proteomics
Advances in mass spectrometry and the generation of large quantities of nucleotide sequence information, combined with computational algorithms that could correlate the two, led to the emergence of proteomics
Analysis of Peptide MS/MS Spectra from Large-Scale Proteomics Experiments Using Spectrum Libraries
 Anal. Chem. 2006, 78, 5678-5684 Analysis of Peptide MS/MS Spectra from Large-Scale Proteomics Experiments Using Spectrum Libraries Barbara E. Frewen, Gennifer E. Merrihew, Christine C. Wu, William Stafford
Anal. Chem. 2006, 78, 5678-5684 Analysis of Peptide MS/MS Spectra from Large-Scale Proteomics Experiments Using Spectrum Libraries Barbara E. Frewen, Gennifer E. Merrihew, Christine C. Wu, William Stafford
Optimization and Use of Peptide Mass Measurement Accuracy in Shotgun Proteomics* S
 Research Optimization and Use of Peptide Mass Measurement Accuracy in Shotgun Proteomics* S Wilhelm Haas, Brendan K. Faherty, Scott A. Gerber, Joshua E. Elias, Sean A. Beausoleil, Corey E. Bakalarski,
Research Optimization and Use of Peptide Mass Measurement Accuracy in Shotgun Proteomics* S Wilhelm Haas, Brendan K. Faherty, Scott A. Gerber, Joshua E. Elias, Sean A. Beausoleil, Corey E. Bakalarski,
CatchmentsUK. User Guide. Wallingford HydroSolutions Ltd. Defining catchments in the UK
 Defining catchments in the UK Wallingford HydroSolutions Ltd Cover photographs (clockwise from top left): istockphoto.com/hazel Proudlove istockphoto.com/antony Spencer istockphoto.com/ann Taylor-Hughes
Defining catchments in the UK Wallingford HydroSolutions Ltd Cover photographs (clockwise from top left): istockphoto.com/hazel Proudlove istockphoto.com/antony Spencer istockphoto.com/ann Taylor-Hughes
Peptide Mass Fingerprinting (PMF) Data Acquisition Using the Voyager DE- PRO Database Search with PMF Data using Ms-Fit
 Data Acquisition Using the Voyager DE- PRO Database Search with PMF Data using Ms-Fit") Peptide Mass Fingerprinting (PMF) Data Acquisition Using the Voyager DE- PRO Introduction page 1 Sample preparation (ZipTip) page 1 Data acquisition using the Voyager DE Pro page 5 Resolution and mass
Peptide Mass Fingerprinting (PMF) Data Acquisition Using the Voyager DE- PRO Introduction page 1 Sample preparation (ZipTip) page 1 Data acquisition using the Voyager DE Pro page 5 Resolution and mass
Introduction to Spark
 1 As you become familiar or continue to explore the Cresset technology and software applications, we encourage you to look through the user manual. This is accessible from the Help menu. However, don t
1 As you become familiar or continue to explore the Cresset technology and software applications, we encourage you to look through the user manual. This is accessible from the Help menu. However, don t