SeqAn and OpenMS Integration Workshop. Temesgen Dadi, Julianus Pfeuffer, Alexander Fillbrunn The Center for Integrative Bioinformatics (CIBI)
|
|
|
- Claude Wilson
- 6 years ago
- Views:
Transcription



1 SeqAn and OpenMS Integration Workshop Temesgen Dadi, Julianus Pfeuffer, Alexander Fillbrunn The Center for Integrative Bioinformatics (CIBI)
2 Mass-spectrometry data analysis in KNIME Julianus Pfeuffer, Alexander Fillbrunn


3 OpenMS OpenMS an open-source C++ framework for computational mass spectrometry Jointly developed at ETH Zürich, FU Berlin, University of Tübingen Open source: BSD 3-clause license Portable: available on Windows, OSX, Linux Vendor-independent: supports all standard formats and vendor-formats through proteowizard OpenMS TOPP tools The OpenMS Proteomics Pipeline tools Building blocks: One application for each analysis step All applications share identical user interfaces Uses PSI standard formats Can be integrated in various workflow systems Galaxy WS-PGRADE/gUSE KNIME Kohlbacher et al., Bioinformatics (2007), 23:e191

4 OpenMS Tools in KNIME Wrapping of OpenMS tools in KNIME via GenericKNIMENodes (GKN) Every tool writes its CommonToolDescription (CTD) via its command line parser GKN generates Java source code for nodes to show up in KNIME Wraps C++ executables and provides file handling nodes

5 Installation of the OpenMS plugin Community-contributions update site (stable & trunk) Bioinformatics & NGS provides > 180 OpenMS TOPP tools as Community nodes SILAC, itraq, TMT, label-free, SWATH, SIP, Search engines: OMSSA, MASCOT, X!TANDEM, MSGFplus, Protein inference: FIDO


6 Data Flow in Shotgun Proteomics Sample HPLC/MS Raw Data 100 GB Sig. Proc. 50 MB Maps Data Reduction Peak Data 1 GB Diff. Quant. Annotated Maps Differentially Expressed Proteins 50 MB Identification 50 kb


7 Quantification Strategies Quantitative Proteomics Relative Quantification Absolute Quantification AQUA SISCAPA Labeled Label-Free In vivo In vitro Spectral Counting MRM Feature-Based 14 N/ 15 N SILAC itraq TMT 16 O/ 18 O After: Lau et al., Proteomics, 2007, 7, 2787

8 Quantitative Data LC-MS Maps Spectra are acquired with rates up to dozens per second Stacking the spectra yields maps Resolution: Up to millions of points per spectrum Tens of thousands of spectra per LC run Huge 2D datasets of up to hundreds of GB per sample MS intensity follows the chromatographic concentration
")
9 LC-MS Data (Map) Quantification (15 nmol/µl, 3x over-expressed, ) 10
10 Label-Free Quantification (LFQ) Label-free quantification is probably the most natural way of quantifying No labeling required, removing further sources of error, no restriction on sample generation, cheap Data on different samples acquired in different measurements higher reproducibility needed Manual analysis difficult Scales very well with the number of samples, basically no limit, no difference in the analysis between 2 or 100 samples
11 LFQ Analysis Strategy 1. Find features in all maps
12 LFQ Analysis Strategy 1. Find features in all maps 2. Align maps
13 LFQ Analysis Strategy 1. Find features in all maps 2. Align maps 3. Link corresponding features
14 LFQ Analysis Strategy 1. Find features in all maps 2. Align maps 3. Link corresponding features 4. Identify features GDAFFGMSCK

15 LFQ Analysis Strategy 1. Find features in all maps 2. Align maps 3. Link corresponding features 4. Identify features 5. Quantify GDAFFGMSCK 1.0 : 1.2 : 0.5
16 Feature-Based Alignment LC-MS maps can contain millions of peaks Retention time of peptides and metabolites can shift between experiments In label-free quantification, maps thus need to be aligned in order to identify corresponding features Alignment can be done on the raw maps (where it is usually called dewarping ) or on already identified features The latter is simpler, as it does not require the alignment of millions of peaks, but just of tens of thousands of features Disadvantage: it replies on an accurate feature finding


17 Feature-Based Alignment ~350,000 peaks ~ 700 features
18 Feature Finding Identify all peaks belonging to one peptide Key idea: Identify suspicious regions (e.g. highest peaks) Fit a model to that region and identify peaks explained by it
19 Feature Finding Extension: collect all data points close to the seed Refinement: remove peaks that are not consistent with the model Fit an optimal model for the reduced set of peaks Iterate this until no further improvement can be achieved
20 m/z Map 1 Multiple Alignment Dewarp k maps onto a comparable coordinate system Choose one map (usually the one with the largest number of features) as reference map (here: map 2 -> T 2 = 1) T 1 Map 2 T 2 Map k rt T k rt Consensus map
21 LFQ with OpenMS in KNIME Identification Feature finding and mapping Map alignment Feature linking Statistical analysis with R Snippets Visualization with KNIME plotting nodes
22 Preprocessing of single maps

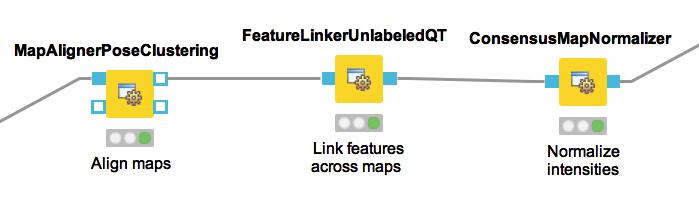
23 Combining information of maps

24 Statistical post-processing and visualization
Overview - MS Proteomics in One Slide. MS masses of peptides. MS/MS fragments of a peptide. Results! Match to sequence database
 Overview - MS Proteomics in One Slide Obtain protein Digest into peptides Acquire spectra in mass spectrometer MS masses of peptides MS/MS fragments of a peptide Results! Match to sequence database 2 But
Overview - MS Proteomics in One Slide Obtain protein Digest into peptides Acquire spectra in mass spectrometer MS masses of peptides MS/MS fragments of a peptide Results! Match to sequence database 2 But
BIOINF 4120 Bioinforma2cs 2 - Structures and Systems -
 BIOINF 4120 Bioinforma2cs 2 - Structures and Systems - Oliver Kohlbacher Summer 2013 16. Quan0ta0ve Proteomics Overview LC- MS- based proteomics - defini0on of maps and features Quan0fica0on approaches
BIOINF 4120 Bioinforma2cs 2 - Structures and Systems - Oliver Kohlbacher Summer 2013 16. Quan0ta0ve Proteomics Overview LC- MS- based proteomics - defini0on of maps and features Quan0fica0on approaches
Isotopic-Labeling and Mass Spectrometry-Based Quantitative Proteomics
 Isotopic-Labeling and Mass Spectrometry-Based Quantitative Proteomics Xiao-jun Li, Ph.D. Current address: Homestead Clinical Day 4 October 19, 2006 Protein Quantification LC-MS/MS Data XLink mzxml file
Isotopic-Labeling and Mass Spectrometry-Based Quantitative Proteomics Xiao-jun Li, Ph.D. Current address: Homestead Clinical Day 4 October 19, 2006 Protein Quantification LC-MS/MS Data XLink mzxml file
Mass Spectrometry and Proteomics - Lecture 5 - Matthias Trost Newcastle University
 Mass Spectrometry and Proteomics - Lecture 5 - Matthias Trost Newcastle University matthias.trost@ncl.ac.uk Previously Proteomics Sample prep 144 Lecture 5 Quantitation techniques Search Algorithms Proteomics
Mass Spectrometry and Proteomics - Lecture 5 - Matthias Trost Newcastle University matthias.trost@ncl.ac.uk Previously Proteomics Sample prep 144 Lecture 5 Quantitation techniques Search Algorithms Proteomics
Protein Quantitation II: Multiple Reaction Monitoring. Kelly Ruggles New York University
 Protein Quantitation II: Multiple Reaction Monitoring Kelly Ruggles kelly@fenyolab.org New York University Traditional Affinity-based proteomics Use antibodies to quantify proteins Western Blot Immunohistochemistry
Protein Quantitation II: Multiple Reaction Monitoring Kelly Ruggles kelly@fenyolab.org New York University Traditional Affinity-based proteomics Use antibodies to quantify proteins Western Blot Immunohistochemistry
Protein Quantitation II: Multiple Reaction Monitoring. Kelly Ruggles New York University
 Protein Quantitation II: Multiple Reaction Monitoring Kelly Ruggles kelly@fenyolab.org New York University Traditional Affinity-based proteomics Use antibodies to quantify proteins Western Blot RPPA Immunohistochemistry
Protein Quantitation II: Multiple Reaction Monitoring Kelly Ruggles kelly@fenyolab.org New York University Traditional Affinity-based proteomics Use antibodies to quantify proteins Western Blot RPPA Immunohistochemistry
Last updated: Copyright
 Last updated: 2012-08-20 Copyright 2004-2012 plabel (v2.4) User s Manual by Bioinformatics Group, Institute of Computing Technology, Chinese Academy of Sciences Tel: 86-10-62601016 Email: zhangkun01@ict.ac.cn,
Last updated: 2012-08-20 Copyright 2004-2012 plabel (v2.4) User s Manual by Bioinformatics Group, Institute of Computing Technology, Chinese Academy of Sciences Tel: 86-10-62601016 Email: zhangkun01@ict.ac.cn,
Chemical Labeling Strategy for Generation of Internal Standards for Targeted Quantitative Proteomics
 Chemical Labeling Strategy for Generation of Internal Standards for Targeted Quantitative Proteomics mtraq Reagents Triplex Christie Hunter, Brian Williamson, Marjorie Minkoff AB SCIEX, USA The utility
Chemical Labeling Strategy for Generation of Internal Standards for Targeted Quantitative Proteomics mtraq Reagents Triplex Christie Hunter, Brian Williamson, Marjorie Minkoff AB SCIEX, USA The utility
Targeted Proteomics Environment
 Targeted Proteomics Environment Quantitative Proteomics with Bruker Q-TOF Instruments and Skyline Brendan MacLean Quantitative Proteomics Spectrum-based Spectral counting Isobaric tags Chromatography-based
Targeted Proteomics Environment Quantitative Proteomics with Bruker Q-TOF Instruments and Skyline Brendan MacLean Quantitative Proteomics Spectrum-based Spectral counting Isobaric tags Chromatography-based
Comprehensive support for quantitation
 Comprehensive support for quantitation One of the major new features in the current release of Mascot is support for quantitation. This is still work in progress. Our goal is to support all of the popular
Comprehensive support for quantitation One of the major new features in the current release of Mascot is support for quantitation. This is still work in progress. Our goal is to support all of the popular
Workflow concept. Data goes through the workflow. A Node contains an operation An edge represents data flow The results are brought together in tables
 PROTEOME DISCOVERER Workflow concept Data goes through the workflow Spectra Peptides Quantitation A Node contains an operation An edge represents data flow The results are brought together in tables Protein
PROTEOME DISCOVERER Workflow concept Data goes through the workflow Spectra Peptides Quantitation A Node contains an operation An edge represents data flow The results are brought together in tables Protein
MassHunter TOF/QTOF Users Meeting
 MassHunter TOF/QTOF Users Meeting 1 Qualitative Analysis Workflows Workflows in Qualitative Analysis allow the user to only see and work with the areas and dialog boxes they need for their specific tasks
MassHunter TOF/QTOF Users Meeting 1 Qualitative Analysis Workflows Workflows in Qualitative Analysis allow the user to only see and work with the areas and dialog boxes they need for their specific tasks
Quan%ta%on with XPRESS. and. ASAPRa%o
 Quan%ta%on with XPRESS and ASAPRa%o 1 Pep%de and Protein Quan%ta%on Raw Mass Spec Data Pep%de Iden%fica%on Pep%de Valida%on Quan%ta%on Protein Assignment Protein List msconvert X!Tandem SpectraST SEQUEST*
Quan%ta%on with XPRESS and ASAPRa%o 1 Pep%de and Protein Quan%ta%on Raw Mass Spec Data Pep%de Iden%fica%on Pep%de Valida%on Quan%ta%on Protein Assignment Protein List msconvert X!Tandem SpectraST SEQUEST*
Agilent MassHunter Profinder: Solving the Challenge of Isotopologue Extraction for Qualitative Flux Analysis
 Agilent MassHunter Profinder: Solving the Challenge of Isotopologue Extraction for Qualitative Flux Analysis Technical Overview Introduction Metabolomics studies measure the relative abundance of metabolites
Agilent MassHunter Profinder: Solving the Challenge of Isotopologue Extraction for Qualitative Flux Analysis Technical Overview Introduction Metabolomics studies measure the relative abundance of metabolites
TUTORIAL EXERCISES WITH ANSWERS
 TUTORIAL EXERCISES WITH ANSWERS Tutorial 1 Settings 1. What is the exact monoisotopic mass difference for peptides carrying a 13 C (and NO additional 15 N) labelled C-terminal lysine residue? a. 6.020129
TUTORIAL EXERCISES WITH ANSWERS Tutorial 1 Settings 1. What is the exact monoisotopic mass difference for peptides carrying a 13 C (and NO additional 15 N) labelled C-terminal lysine residue? a. 6.020129
A Description of the CPTAC Common Data Analysis Pipeline (CDAP)
") A Description of the CPTAC Common Data Analysis Pipeline (CDAP) v. 01/14/2014 Summary The purpose of this document is to describe the software programs and output files of the Common Data Analysis Pipeline
A Description of the CPTAC Common Data Analysis Pipeline (CDAP) v. 01/14/2014 Summary The purpose of this document is to describe the software programs and output files of the Common Data Analysis Pipeline
X!TandemPipeline (Myosine Anabolisée) validating, filtering and grouping MSMS identifications
 validating, filtering and grouping MSMS identifications") X!TandemPipeline 3.3.3 (Myosine Anabolisée) validating, filtering and grouping MSMS identifications Olivier Langella and Benoit Valot langella@moulon.inra.fr; valot@moulon.inra.fr PAPPSO - http://pappso.inra.fr/
X!TandemPipeline 3.3.3 (Myosine Anabolisée) validating, filtering and grouping MSMS identifications Olivier Langella and Benoit Valot langella@moulon.inra.fr; valot@moulon.inra.fr PAPPSO - http://pappso.inra.fr/
profileanalysis Innovation with Integrity Quickly pinpointing and identifying potential biomarkers in Proteomics and Metabolomics research
 profileanalysis Quickly pinpointing and identifying potential biomarkers in Proteomics and Metabolomics research Innovation with Integrity Omics Research Biomarker Discovery Made Easy by ProfileAnalysis
profileanalysis Quickly pinpointing and identifying potential biomarkers in Proteomics and Metabolomics research Innovation with Integrity Omics Research Biomarker Discovery Made Easy by ProfileAnalysis
Designed for Accuracy. Innovation with Integrity. High resolution quantitative proteomics LC-MS
 Designed for Accuracy High resolution quantitative proteomics Innovation with Integrity LC-MS Setting New Standards in Accuracy The development of mass spectrometry based proteomics approaches has dramatically
Designed for Accuracy High resolution quantitative proteomics Innovation with Integrity LC-MS Setting New Standards in Accuracy The development of mass spectrometry based proteomics approaches has dramatically
Proteome-wide label-free quantification with MaxQuant. Jürgen Cox Max Planck Institute of Biochemistry July 2011
 Proteome-wide label-free quantification with MaxQuant Jürgen Cox Max Planck Institute of Biochemistry July 2011 MaxQuant MaxQuant Feature detection Data acquisition Initial Andromeda search Statistics
Proteome-wide label-free quantification with MaxQuant Jürgen Cox Max Planck Institute of Biochemistry July 2011 MaxQuant MaxQuant Feature detection Data acquisition Initial Andromeda search Statistics
Key questions of proteomics. Bioinformatics 2. Proteomics. Foundation of proteomics. What proteins are there? Protein digestion
 s s Key questions of proteomics What proteins are there? Bioinformatics 2 Lecture 2 roteomics How much is there of each of the proteins? - Absolute quantitation - Stoichiometry What (modification/splice)
s s Key questions of proteomics What proteins are there? Bioinformatics 2 Lecture 2 roteomics How much is there of each of the proteins? - Absolute quantitation - Stoichiometry What (modification/splice)
HOWTO, example workflow and data files. (Version )
") HOWTO, example workflow and data files. (Version 20 09 2017) 1 Introduction: SugarQb is a collection of software tools (Nodes) which enable the automated identification of intact glycopeptides from HCD
HOWTO, example workflow and data files. (Version 20 09 2017) 1 Introduction: SugarQb is a collection of software tools (Nodes) which enable the automated identification of intact glycopeptides from HCD
Analysis of Labeled and Non-Labeled Proteomic Data Using Progenesis QI for Proteomics
 Analysis of Labeled and Non-Labeled Proteomic Data Using Progenesis QI for Proteomics Lee Gethings, Gushinder Atwal, Martin Palmer, Chris Hughes, Hans Vissers, and James Langridge Waters Corporation, Wilmslow,
Analysis of Labeled and Non-Labeled Proteomic Data Using Progenesis QI for Proteomics Lee Gethings, Gushinder Atwal, Martin Palmer, Chris Hughes, Hans Vissers, and James Langridge Waters Corporation, Wilmslow,
MassHunter Software Overview
 MassHunter Software Overview 1 Qualitative Analysis Workflows Workflows in Qualitative Analysis allow the user to only see and work with the areas and dialog boxes they need for their specific tasks A
MassHunter Software Overview 1 Qualitative Analysis Workflows Workflows in Qualitative Analysis allow the user to only see and work with the areas and dialog boxes they need for their specific tasks A
Genome wide analysis of protein and mrna half lives reveals dynamic properties of mammalian gene expression
 Genome wide analysis of protein and mrna half lives reveals dynamic properties of mammalian gene expression Matthias Selbach Cell Signaling and Mass Spectrometry Max Delbrück Center for Molecular Medicine
Genome wide analysis of protein and mrna half lives reveals dynamic properties of mammalian gene expression Matthias Selbach Cell Signaling and Mass Spectrometry Max Delbrück Center for Molecular Medicine
Spectronaut Pulsar. User Manual
 Spectronaut Pulsar User Manual 1 General Information... 6 1.1 Computer System Requirements... 6 1.2 Scope of Spectronaut Software... 6 1.3 Spectronaut Pulsar... 6 1.4 Spectronaut Release Features... 7
Spectronaut Pulsar User Manual 1 General Information... 6 1.1 Computer System Requirements... 6 1.2 Scope of Spectronaut Software... 6 1.3 Spectronaut Pulsar... 6 1.4 Spectronaut Release Features... 7
DIA-Umpire: comprehensive computational framework for data independent acquisition proteomics
 DIA-Umpire: comprehensive computational framework for data independent acquisition proteomics Chih-Chiang Tsou 1,2, Dmitry Avtonomov 2, Brett Larsen 3, Monika Tucholska 3, Hyungwon Choi 4 Anne-Claude Gingras
DIA-Umpire: comprehensive computational framework for data independent acquisition proteomics Chih-Chiang Tsou 1,2, Dmitry Avtonomov 2, Brett Larsen 3, Monika Tucholska 3, Hyungwon Choi 4 Anne-Claude Gingras
High-Throughput Protein Quantitation Using Multiple Reaction Monitoring
 High-Throughput Protein Quantitation Using Multiple Reaction Monitoring Application Note Authors Ning Tang, Christine Miller, Joe Roark, Norton Kitagawa and Keith Waddell Agilent Technologies, Inc. Santa
High-Throughput Protein Quantitation Using Multiple Reaction Monitoring Application Note Authors Ning Tang, Christine Miller, Joe Roark, Norton Kitagawa and Keith Waddell Agilent Technologies, Inc. Santa
combined with the computing power of the W4M infrastructure
 An efficient GUI tool for spectra processing from 1D 1H-NMR metabolomics data combined with the computing power of the W4M infrastructure Daniel Jacob Marie Lefebvre Two major metabolomics approaches Metabolic
An efficient GUI tool for spectra processing from 1D 1H-NMR metabolomics data combined with the computing power of the W4M infrastructure Daniel Jacob Marie Lefebvre Two major metabolomics approaches Metabolic
Tutorial 2: Analysis of DIA data in Skyline
 Tutorial 2: Analysis of DIA data in Skyline In this tutorial we will learn how to use Skyline to perform targeted post-acquisition analysis for peptide and inferred protein detection and quantitation using
Tutorial 2: Analysis of DIA data in Skyline In this tutorial we will learn how to use Skyline to perform targeted post-acquisition analysis for peptide and inferred protein detection and quantitation using
PC235: 2008 Lecture 5: Quantitation. Arnold Falick
 PC235: 2008 Lecture 5: Quantitation Arnold Falick falickam@berkeley.edu Summary What you will learn from this lecture: There are many methods to perform quantitation using mass spectrometry (any method
PC235: 2008 Lecture 5: Quantitation Arnold Falick falickam@berkeley.edu Summary What you will learn from this lecture: There are many methods to perform quantitation using mass spectrometry (any method
Automated SWATH Data Analysis Using Targeted Extraction of Ion Chromatograms
 Automated SWATH Data Analysis Using Targeted Extraction of Ion Chromatograms Hannes L. Röst 1,2, Ruedi Aebersold 1,3 and Olga T. Schubert 1,4 1 Institute of Molecular Systems Biology, ETH Zurich, CH-8093
Automated SWATH Data Analysis Using Targeted Extraction of Ion Chromatograms Hannes L. Röst 1,2, Ruedi Aebersold 1,3 and Olga T. Schubert 1,4 1 Institute of Molecular Systems Biology, ETH Zurich, CH-8093
Data pre-processing in liquid chromatography mass spectrometry-based proteomics
 BIOINFORMATICS ORIGINAL PAPER Vol. 21 no. 21 25, pages 454 459 doi:1.193/bioinformatics/bti66 Data and text mining Data pre-processing in liquid chromatography mass spectrometry-based proteomics Xiang
BIOINFORMATICS ORIGINAL PAPER Vol. 21 no. 21 25, pages 454 459 doi:1.193/bioinformatics/bti66 Data and text mining Data pre-processing in liquid chromatography mass spectrometry-based proteomics Xiang
itraq and RNA-Seq analyses provide new insights of Dendrobium officinale seeds (Orchidaceae)
") Page S-1 Supporting Information (SI) itraq and RNA-Seq analyses provide new insights into regulation mechanism of symbiotic germination of Dendrobium officinale seeds (Orchidaceae) Juan Chen 1, Si Si Liu
Page S-1 Supporting Information (SI) itraq and RNA-Seq analyses provide new insights into regulation mechanism of symbiotic germination of Dendrobium officinale seeds (Orchidaceae) Juan Chen 1, Si Si Liu
Extend Your Metabolomics Insight!
 Extend Your Metabolomics Insight! Introducing MassHunter VistaFlux April 2016 Agilent THE Leader in Metabolomics! Fiehn EI Library METLIN MS/MS Library Mass Profiler Professional with Pathway Architect
Extend Your Metabolomics Insight! Introducing MassHunter VistaFlux April 2016 Agilent THE Leader in Metabolomics! Fiehn EI Library METLIN MS/MS Library Mass Profiler Professional with Pathway Architect
Tutorial 1: Setting up your Skyline document
 Tutorial 1: Setting up your Skyline document Caution! For using Skyline the number formats of your computer have to be set to English (United States). Open the Control Panel Clock, Language, and Region
Tutorial 1: Setting up your Skyline document Caution! For using Skyline the number formats of your computer have to be set to English (United States). Open the Control Panel Clock, Language, and Region
Statistical approach to protein quantification
 MCP Papers in Press. Published on November 19, 2013 as Manuscript M112.025445 Statistical approach to protein quantification Authors and affilitations Sarah Gerster a,g, Taejoon Kwon b, Christina Ludwig
MCP Papers in Press. Published on November 19, 2013 as Manuscript M112.025445 Statistical approach to protein quantification Authors and affilitations Sarah Gerster a,g, Taejoon Kwon b, Christina Ludwig
Relative quantification using TMT11plex on a modified Q Exactive HF mass spectrometer
 POSTER NOTE 6558 Relative quantification using TMT11plex on a modified mass spectrometer Authors Tabiwang N. Arrey, 1 Rosa Viner, 2 Ryan D. Bomgarden, 3 Eugen Damoc, 1 Markus Kellmann, 1 Thomas Moehring,
POSTER NOTE 6558 Relative quantification using TMT11plex on a modified mass spectrometer Authors Tabiwang N. Arrey, 1 Rosa Viner, 2 Ryan D. Bomgarden, 3 Eugen Damoc, 1 Markus Kellmann, 1 Thomas Moehring,
Computational Methods for Mass Spectrometry Proteomics
 Computational Methods for Mass Spectrometry Proteomics Eidhammer, Ingvar ISBN-13: 9780470512975 Table of Contents Preface. Acknowledgements. 1 Protein, Proteome, and Proteomics. 1.1 Primary goals for studying
Computational Methods for Mass Spectrometry Proteomics Eidhammer, Ingvar ISBN-13: 9780470512975 Table of Contents Preface. Acknowledgements. 1 Protein, Proteome, and Proteomics. 1.1 Primary goals for studying
The Schrödinger KNIME extensions
 The Schrödinger KNIME extensions Computational Chemistry and Cheminformatics in a workflow environment Jean-Christophe Mozziconacci Volker Eyrich KNIME UGM, Zurich, February 2014 The Schrödinger extensions
The Schrödinger KNIME extensions Computational Chemistry and Cheminformatics in a workflow environment Jean-Christophe Mozziconacci Volker Eyrich KNIME UGM, Zurich, February 2014 The Schrödinger extensions
PeptideProphet: Validation of Peptide Assignments to MS/MS Spectra
 PeptideProphet: Validation of Peptide Assignments to MS/MS Spectra Andrew Keller Day 2 October 17, 2006 Andrew Keller Rosetta Bioinformatics, Seattle Outline Need to validate peptide assignments to MS/MS
PeptideProphet: Validation of Peptide Assignments to MS/MS Spectra Andrew Keller Day 2 October 17, 2006 Andrew Keller Rosetta Bioinformatics, Seattle Outline Need to validate peptide assignments to MS/MS
iprophet: Multi-level integrative analysis of shotgun proteomic data improves peptide and protein identification rates and error estimates
 MCP Papers in Press. Published on August 29, 2011 as Manuscript M111.007690 This is the Pre-Published Version iprophet: Multi-level integrative analysis of shotgun proteomic data improves peptide and protein
MCP Papers in Press. Published on August 29, 2011 as Manuscript M111.007690 This is the Pre-Published Version iprophet: Multi-level integrative analysis of shotgun proteomic data improves peptide and protein
The Source Finder (SoFi)
") 20.12.2016 Wir schaffen Wissen heute für morgen Paul Scherrer Institute F. Canonaco, C. Bozzetti, K. Dällenbach, Y. Sosedova, J. G. Slowik, I. ElHaddad, M. Crippa, U. Baltensperger,A. S. H. Prévôt and
20.12.2016 Wir schaffen Wissen heute für morgen Paul Scherrer Institute F. Canonaco, C. Bozzetti, K. Dällenbach, Y. Sosedova, J. G. Slowik, I. ElHaddad, M. Crippa, U. Baltensperger,A. S. H. Prévôt and
Statistical mass spectrometry-based proteomics
 1 Statistical mass spectrometry-based proteomics Olga Vitek www.stat.purdue.edu Outline What is proteomics? Biological questions and technologies Protein quantification in label-free workflows Joint analysis
1 Statistical mass spectrometry-based proteomics Olga Vitek www.stat.purdue.edu Outline What is proteomics? Biological questions and technologies Protein quantification in label-free workflows Joint analysis
Guide to Peptide Quantitation. Agilent clinical research
 Guide to Peptide Quantitation Agilent clinical research Peptide Quantitation for the Clinical Research Laboratory Peptide quantitation is rapidly growing in clinical research as scientists are translating
Guide to Peptide Quantitation Agilent clinical research Peptide Quantitation for the Clinical Research Laboratory Peptide quantitation is rapidly growing in clinical research as scientists are translating
Developing Algorithms for the Determination of Relative Abundances of Peptides from LC/MS Data
 Developing Algorithms for the Determination of Relative Abundances of Peptides from LC/MS Data RIPS Team Jake Marcus (Project Manager) Anne Eaton Melanie Kanter Aru Ray Faculty Mentors Shawn Cokus Matteo
Developing Algorithms for the Determination of Relative Abundances of Peptides from LC/MS Data RIPS Team Jake Marcus (Project Manager) Anne Eaton Melanie Kanter Aru Ray Faculty Mentors Shawn Cokus Matteo
UCD Conway Institute of Biomolecular & Biomedical Research Graduate Education 2009/2010
 EMERGING PROTEOMIC TECHNOLOGIES - MODULE SCHEDULE & OUTLINE 2010 Course Organiser: Dr. Giuliano Elia Module Co-ordinator: Dr Giuliano Elia Credits: 5 Date & Time Session & Topic Coordinator 14th April
EMERGING PROTEOMIC TECHNOLOGIES - MODULE SCHEDULE & OUTLINE 2010 Course Organiser: Dr. Giuliano Elia Module Co-ordinator: Dr Giuliano Elia Credits: 5 Date & Time Session & Topic Coordinator 14th April
Analyst Software. Peptide and Protein Quantitation Tutorial
 This document is provided to customers who have purchased AB Sciex equipment to use in the operation of such AB Sciex equipment. This document is copyright protected and any reproduction of this document
This document is provided to customers who have purchased AB Sciex equipment to use in the operation of such AB Sciex equipment. This document is copyright protected and any reproduction of this document
Identification of a Set of Conserved Eukaryotic Internal Retention Time Standards for Data-Independent Acquisition Mass Spectrometry
 MCP Papers in Press. Published on July 21, 2015 as Manuscript O114.042267 Identification of a Set of Conserved Eukaryotic Internal Retention Time Standards for Data-Independent Acquisition Mass Spectrometry
MCP Papers in Press. Published on July 21, 2015 as Manuscript O114.042267 Identification of a Set of Conserved Eukaryotic Internal Retention Time Standards for Data-Independent Acquisition Mass Spectrometry
The Schrödinger KNIME extensions
 The Schrödinger KNIME extensions Computational Chemistry and Cheminformatics in a workflow environment Jean-Christophe Mozziconacci Volker Eyrich KNIME UGM, Berlin, February 2015 The Schrödinger Extensions
The Schrödinger KNIME extensions Computational Chemistry and Cheminformatics in a workflow environment Jean-Christophe Mozziconacci Volker Eyrich KNIME UGM, Berlin, February 2015 The Schrödinger Extensions
A Better Scoring Model for De Novo Peptide Sequencing: The Symmetric Difference between Explained and Measured Masses Supplementary Figures
 A Better Scoring Model for De Novo Peptide Sequencing: The Symmetric Difference between Explained and Measured Masses Supplementary Figures Thomas Tschager *, Simon Rösch *, Ludovic Gillet 2 and Peter
A Better Scoring Model for De Novo Peptide Sequencing: The Symmetric Difference between Explained and Measured Masses Supplementary Figures Thomas Tschager *, Simon Rösch *, Ludovic Gillet 2 and Peter
Mass spectrometry-based proteomics has become
 FOCUS: THE ORBITRAP Computational Principles of Determining and Improving Mass Precision and Accuracy for Proteome Measurements in an Orbitrap Jürgen Cox and Matthias Mann Proteomics and Signal Transduction,
FOCUS: THE ORBITRAP Computational Principles of Determining and Improving Mass Precision and Accuracy for Proteome Measurements in an Orbitrap Jürgen Cox and Matthias Mann Proteomics and Signal Transduction,
Quantitation of a target protein in crude samples using targeted peptide quantification by Mass Spectrometry
 Quantitation of a target protein in crude samples using targeted peptide quantification by Mass Spectrometry Jon Hao, Rong Ye, and Mason Tao Poochon Scientific, Frederick, Maryland 21701 Abstract Background:
Quantitation of a target protein in crude samples using targeted peptide quantification by Mass Spectrometry Jon Hao, Rong Ye, and Mason Tao Poochon Scientific, Frederick, Maryland 21701 Abstract Background:
Modeling Mass Spectrometry-Based Protein Analysis
 Chapter 8 Jan Eriksson and David Fenyö Abstract The success of mass spectrometry based proteomics depends on efficient methods for data analysis. These methods require a detailed understanding of the information
Chapter 8 Jan Eriksson and David Fenyö Abstract The success of mass spectrometry based proteomics depends on efficient methods for data analysis. These methods require a detailed understanding of the information
Agilent ESI and APCI sources: for polar to non-polar compounds
 1 Agilent 6400 Series Triple Quadrupole Users Workshop 1 Agilent ESI and APCI sources: for polar to non-polar compounds Nebulizer Pressure Corona current Nebulizer Pressure Vaporizer Vcap Vcap Drying Gas
1 Agilent 6400 Series Triple Quadrupole Users Workshop 1 Agilent ESI and APCI sources: for polar to non-polar compounds Nebulizer Pressure Corona current Nebulizer Pressure Vaporizer Vcap Vcap Drying Gas
Improved Throughput and Reproducibility for Targeted Protein Quantification Using a New High-Performance Triple Quadrupole Mass Spectrometer
 Improved Throughput and Reproducibility for Targeted Protein Quantification Using a New High-Performance Triple Quadrupole Mass Spectrometer Reiko Kiyonami, Mary Blackburn, Andreas FR Hühme: Thermo Fisher
Improved Throughput and Reproducibility for Targeted Protein Quantification Using a New High-Performance Triple Quadrupole Mass Spectrometer Reiko Kiyonami, Mary Blackburn, Andreas FR Hühme: Thermo Fisher
Statistical analysis of isobaric-labeled mass spectrometry data
 Statistical analysis of isobaric-labeled mass spectrometry data Farhad Shakeri July 3, 2018 Core Unit for Bioinformatics Analyses Institute for Genomic Statistics and Bioinformatics University Hospital
Statistical analysis of isobaric-labeled mass spectrometry data Farhad Shakeri July 3, 2018 Core Unit for Bioinformatics Analyses Institute for Genomic Statistics and Bioinformatics University Hospital
Towards the Prediction of Protein Abundance from Tandem Mass Spectrometry Data
 Towards the Prediction of Protein Abundance from Tandem Mass Spectrometry Data Anthony J Bonner Han Liu Abstract This paper addresses a central problem of Proteomics: estimating the amounts of each of
Towards the Prediction of Protein Abundance from Tandem Mass Spectrometry Data Anthony J Bonner Han Liu Abstract This paper addresses a central problem of Proteomics: estimating the amounts of each of
Quantitative Proteomics
 Quantitative Proteomics Quantitation AND Mass Spectrometry Condition A Condition B Identify and quantify differently expressed proteins resulting from a change in the environment (stimulus, disease) Lyse
Quantitative Proteomics Quantitation AND Mass Spectrometry Condition A Condition B Identify and quantify differently expressed proteins resulting from a change in the environment (stimulus, disease) Lyse
Tutorial 1: Library Generation from DDA data
 Tutorial 1: Library Generation from DDA data 1. Introduction Before a targeted, peptide-centric DIA analysis can be performed, a spectral library containing peptide-query parameters needs to be generated.
Tutorial 1: Library Generation from DDA data 1. Introduction Before a targeted, peptide-centric DIA analysis can be performed, a spectral library containing peptide-query parameters needs to be generated.
The Schrödinger KNIME extensions
 The Schrödinger KNIME extensions Computational Chemistry and Cheminformatics in a workflow environment Jean-Christophe Mozziconacci Volker Eyrich Topics What are the Schrödinger extensions? Workflow application
The Schrödinger KNIME extensions Computational Chemistry and Cheminformatics in a workflow environment Jean-Christophe Mozziconacci Volker Eyrich Topics What are the Schrödinger extensions? Workflow application
MS Based Proteomics: Recent Case Studies Using Advanced Instrumentation
 MS Based Proteomics: Recent Case Studies Using Advanced Instrumentation Chris Adams, PH.D. Stanford University Mass Spectrometry http://mass-spec.stanford.edu/ For personal use only. Please do not reuse
MS Based Proteomics: Recent Case Studies Using Advanced Instrumentation Chris Adams, PH.D. Stanford University Mass Spectrometry http://mass-spec.stanford.edu/ For personal use only. Please do not reuse
Protocol. Product Use & Liability. Contact us: InfoLine: Order per fax: www:
 Protocol SpikeTides Set TAA - light SpikeTides Set TAA_L - heavy Peptide Sets for relative quantification of Tumor Associated Antigens (TAAs) in SRM and MRM Assays Contact us: InfoLine: +49-30-6392-7878
Protocol SpikeTides Set TAA - light SpikeTides Set TAA_L - heavy Peptide Sets for relative quantification of Tumor Associated Antigens (TAAs) in SRM and MRM Assays Contact us: InfoLine: +49-30-6392-7878
Compounding insights Thermo Scientific Compound Discoverer Software
 Compounding insights Thermo Scientific Compound Discoverer Software Integrated, complete, toolset solves small-molecule analysis challenges Thermo Scientific Orbitrap mass spectrometers produce information-rich
Compounding insights Thermo Scientific Compound Discoverer Software Integrated, complete, toolset solves small-molecule analysis challenges Thermo Scientific Orbitrap mass spectrometers produce information-rich
MALDI-HDMS E : A Novel Data Independent Acquisition Method for the Enhanced Analysis of 2D-Gel Tryptic Peptide Digests
 -HDMS E : A Novel Data Independent Acquisition Method for the Enhanced Analysis of 2D-Gel Tryptic Peptide Digests Emmanuelle Claude, 1 Mark Towers, 1 and Rachel Craven 2 1 Waters Corporation, Manchester,
-HDMS E : A Novel Data Independent Acquisition Method for the Enhanced Analysis of 2D-Gel Tryptic Peptide Digests Emmanuelle Claude, 1 Mark Towers, 1 and Rachel Craven 2 1 Waters Corporation, Manchester,
ALIGNMENT OF LC-MS DATA USING PEPTIDE FEATURES. A Thesis XINCHENG TANG
 ALIGNMENT OF LC-MS DATA USING PEPTIDE FEATURES A Thesis by XINCHENG TANG Submitted to the Office of Graduate Studies of Texas A&M University in partial fulfillment of the requirements for the degree of
ALIGNMENT OF LC-MS DATA USING PEPTIDE FEATURES A Thesis by XINCHENG TANG Submitted to the Office of Graduate Studies of Texas A&M University in partial fulfillment of the requirements for the degree of
A Software Suite for the Generation and Comparison of Peptide Arrays from Sets. of Data Collected by Liquid Chromatography-Mass Spectrometry
 MCP Papers in Press. Published on July 26, 2005 as Manuscript M500141-MCP200 A Software Suite for the Generation and Comparison of Peptide Arrays from Sets of Data Collected by Liquid Chromatography-Mass
MCP Papers in Press. Published on July 26, 2005 as Manuscript M500141-MCP200 A Software Suite for the Generation and Comparison of Peptide Arrays from Sets of Data Collected by Liquid Chromatography-Mass
The Pitfalls of Peaklist Generation Software Performance on Database Searches
 Proceedings of the 56th ASMS Conference on Mass Spectrometry and Allied Topics, Denver, CO, June 1-5, 2008 The Pitfalls of Peaklist Generation Software Performance on Database Searches Aenoch J. Lynn,
Proceedings of the 56th ASMS Conference on Mass Spectrometry and Allied Topics, Denver, CO, June 1-5, 2008 The Pitfalls of Peaklist Generation Software Performance on Database Searches Aenoch J. Lynn,
for XPS surface analysis
 Thermo Scientific Avantage XPS Software Powerful instrument operation and data processing for XPS surface analysis Avantage Software Atomic Concentration (%) 100 The premier software for surface analysis
Thermo Scientific Avantage XPS Software Powerful instrument operation and data processing for XPS surface analysis Avantage Software Atomic Concentration (%) 100 The premier software for surface analysis
Supplemental Information. Mass Spec Studio for Integrative Structural Biology
 Structure, Volume 22 Supplemental Information Mass Spec Studio for Integrative Structural Biology Martial Rey, Vladimir Sarpe, Kyle M. Burns, Joshua Buse, Charles A.H. Baker, Marc van Dijk, Linda Wordeman,
Structure, Volume 22 Supplemental Information Mass Spec Studio for Integrative Structural Biology Martial Rey, Vladimir Sarpe, Kyle M. Burns, Joshua Buse, Charles A.H. Baker, Marc van Dijk, Linda Wordeman,
SILAC and TMT. IDeA National Resource for Proteomics Workshop for Graduate Students and Post-docs Renny Lan 5/18/2017
 SILAC and TMT IDeA National Resource for Proteomics Workshop for Graduate Students and Post-docs Renny Lan 5/18/2017 UHPLC peak chosen at 26.47 min LC Mass at 571.36 chosen for MS/MS MS/MS MS This is a
SILAC and TMT IDeA National Resource for Proteomics Workshop for Graduate Students and Post-docs Renny Lan 5/18/2017 UHPLC peak chosen at 26.47 min LC Mass at 571.36 chosen for MS/MS MS/MS MS This is a
Targeted protein quantification
 Targeted Quantitative Proteomics Targeted protein quantification with high-resolution, accurate-mass MS Highly selective Very sensitive Complex samples HR/AM A more complete quantitative proteomics picture
Targeted Quantitative Proteomics Targeted protein quantification with high-resolution, accurate-mass MS Highly selective Very sensitive Complex samples HR/AM A more complete quantitative proteomics picture
Automated Quantification of 13C Labeled Peptides
 Automated Quantification of 13C Labeled Peptides amaster sthesis submitted to the faculty of the graduate school of the university of minnesota by Joshua Elliot Goldford in partial fulfillment of the requirements
Automated Quantification of 13C Labeled Peptides amaster sthesis submitted to the faculty of the graduate school of the university of minnesota by Joshua Elliot Goldford in partial fulfillment of the requirements
SRM assay generation and data analysis in Skyline
 in Skyline Preparation 1. Download the example data from www.srmcourse.ch/eupa.html (3 raw files, 1 csv file, 1 sptxt file). 2. The number formats of your computer have to be set to English (United States).
in Skyline Preparation 1. Download the example data from www.srmcourse.ch/eupa.html (3 raw files, 1 csv file, 1 sptxt file). 2. The number formats of your computer have to be set to English (United States).
Overview. Introduction. André Schreiber AB SCIEX Concord, Ontario (Canada)
") Quantitation and Identification of Pharmaceuticals and Personal Care Products (PPCP) in Environmental Samples using Advanced TripleTOF MS/MS Technology André Schreiber AB SCIEX Concord, Ontario (Canada)
Quantitation and Identification of Pharmaceuticals and Personal Care Products (PPCP) in Environmental Samples using Advanced TripleTOF MS/MS Technology André Schreiber AB SCIEX Concord, Ontario (Canada)
BIOINF 4120 Bioinformatics 2 - Structures and Systems - Oliver Kohlbacher Summer Systems Biology Exp. Methods
 BIOINF 4120 Bioinformatics 2 - Structures and Systems - Oliver Kohlbacher Summer 2013 14. Systems Biology Exp. Methods Overview Transcriptomics Basics of microarrays Comparative analysis Interactomics:
BIOINF 4120 Bioinformatics 2 - Structures and Systems - Oliver Kohlbacher Summer 2013 14. Systems Biology Exp. Methods Overview Transcriptomics Basics of microarrays Comparative analysis Interactomics:
An Optimized Interestingness Hotspot Discovery Framework for Large Gridded Spatio-temporal Datasets
 IEEE Big Data 2015 Big Data in Geosciences Workshop An Optimized Interestingness Hotspot Discovery Framework for Large Gridded Spatio-temporal Datasets Fatih Akdag and Christoph F. Eick Department of Computer
IEEE Big Data 2015 Big Data in Geosciences Workshop An Optimized Interestingness Hotspot Discovery Framework for Large Gridded Spatio-temporal Datasets Fatih Akdag and Christoph F. Eick Department of Computer
Agilent MassHunter Quantitative Data Analysis
 Agilent MassHunter Quantitative Data Analysis Presenters: Howard Sanford Stephen Harnos MassHunter Quantitation: Batch and Method Setup Outliers, Data Review, Reporting 1 MassHunter Quantitative Analysis
Agilent MassHunter Quantitative Data Analysis Presenters: Howard Sanford Stephen Harnos MassHunter Quantitation: Batch and Method Setup Outliers, Data Review, Reporting 1 MassHunter Quantitative Analysis
WALKUP LC/MS FOR PHARMACEUTICAL R&D
 Pharmaceutical Workflow Solutions WALKUP LC/MS FOR PHARMACEUTICAL R&D Chemists, Peptide/Protein Chemists, Biologists, and Beyond MASSHUNTER WALKUP A Single User Interface for Robust and Reliable LC/MS
Pharmaceutical Workflow Solutions WALKUP LC/MS FOR PHARMACEUTICAL R&D Chemists, Peptide/Protein Chemists, Biologists, and Beyond MASSHUNTER WALKUP A Single User Interface for Robust and Reliable LC/MS
1. Prepare the MALDI sample plate by spotting an angiotensin standard and the test sample(s).
.") Analysis of a Peptide Sequence from a Proteolytic Digest by MALDI-TOF Post-Source Decay (PSD) and Collision-Induced Dissociation (CID) Standard Operating Procedure Purpose: The following procedure may
Analysis of a Peptide Sequence from a Proteolytic Digest by MALDI-TOF Post-Source Decay (PSD) and Collision-Induced Dissociation (CID) Standard Operating Procedure Purpose: The following procedure may
NPTEL VIDEO COURSE PROTEOMICS PROF. SANJEEVA SRIVASTAVA
 LECTURE-25 Quantitative proteomics: itraq and TMT TRANSCRIPT Welcome to the proteomics course. Today we will talk about quantitative proteomics and discuss about itraq and TMT techniques. The quantitative
LECTURE-25 Quantitative proteomics: itraq and TMT TRANSCRIPT Welcome to the proteomics course. Today we will talk about quantitative proteomics and discuss about itraq and TMT techniques. The quantitative
QTOF-based proteomics and metabolomics for the agro-food chain.
 QTOF-based proteomics and metabolomics for the agro-food chain luigi.lucini@unicatt.it Metabolomics Two scenarios identification of known unknowns and unknown unknowns For known unknowns use spectral or
QTOF-based proteomics and metabolomics for the agro-food chain luigi.lucini@unicatt.it Metabolomics Two scenarios identification of known unknowns and unknown unknowns For known unknowns use spectral or
Making Sense of Differences in LCMS Data: Integrated Tools
 Making Sense of Differences in LCMS Data: Integrated Tools David A. Weil Agilent Technologies MassHunter Overview Page 1 March 2008 How Clean is our Water?... Page 2 Chemical Residue Analysis.... From
Making Sense of Differences in LCMS Data: Integrated Tools David A. Weil Agilent Technologies MassHunter Overview Page 1 March 2008 How Clean is our Water?... Page 2 Chemical Residue Analysis.... From
Computationally Efficient Analysis of Large Array FTIR Data In Chemical Reaction Studies Using Distributed Computing Strategy
 575f Computationally Efficient Analysis of Large Array FTIR Data In Chemical Reaction Studies Using Distributed Computing Strategy Ms Suyun Ong, Dr. Wee Chew, * Dr. Marc Garland Institute of Chemical and
575f Computationally Efficient Analysis of Large Array FTIR Data In Chemical Reaction Studies Using Distributed Computing Strategy Ms Suyun Ong, Dr. Wee Chew, * Dr. Marc Garland Institute of Chemical and
Yifei Bao. Beatrix. Manor Askenazi
 Detection and Correction of Interference in MS1 Quantitation of Peptides Using their Isotope Distributions Yifei Bao Department of Computer Science Stevens Institute of Technology Beatrix Ueberheide Department
Detection and Correction of Interference in MS1 Quantitation of Peptides Using their Isotope Distributions Yifei Bao Department of Computer Science Stevens Institute of Technology Beatrix Ueberheide Department
Mass spectrometry has been used a lot in biology since the late 1950 s. However it really came into play in the late 1980 s once methods were
 Mass spectrometry has been used a lot in biology since the late 1950 s. However it really came into play in the late 1980 s once methods were developed to allow the analysis of large intact (bigger than
Mass spectrometry has been used a lot in biology since the late 1950 s. However it really came into play in the late 1980 s once methods were developed to allow the analysis of large intact (bigger than
Mass spectrometry in proteomics
 I519 Introduction to Bioinformatics, Fall, 2013 Mass spectrometry in proteomics Haixu Tang School of Informatics and Computing Indiana University, Bloomington Modified from: www.bioalgorithms.info Outline
I519 Introduction to Bioinformatics, Fall, 2013 Mass spectrometry in proteomics Haixu Tang School of Informatics and Computing Indiana University, Bloomington Modified from: www.bioalgorithms.info Outline
Workshop: SILAC and Alternative Labeling Strategies in Quantitative Proteomics
 Workshop: SILAC and Alternative Labeling Strategies in Quantitative Proteomics SILAC and Stable Isotope Dimethyl-Labeling Approaches in Quantitative Proteomics Ho-Tak Lau, Hyong-Won Suh, Shao-En Ong UW
Workshop: SILAC and Alternative Labeling Strategies in Quantitative Proteomics SILAC and Stable Isotope Dimethyl-Labeling Approaches in Quantitative Proteomics Ho-Tak Lau, Hyong-Won Suh, Shao-En Ong UW
Multi-residue analysis of pesticides by GC-HRMS
 An Executive Summary Multi-residue analysis of pesticides by GC-HRMS Dr. Hans Mol is senior scientist at RIKILT- Wageningen UR Introduction Regulatory authorities throughout the world set and enforce strict
An Executive Summary Multi-residue analysis of pesticides by GC-HRMS Dr. Hans Mol is senior scientist at RIKILT- Wageningen UR Introduction Regulatory authorities throughout the world set and enforce strict
PeptideProphet: Validation of Peptide Assignments to MS/MS Spectra. Andrew Keller
 PeptideProphet: Validation of Peptide Assignments to MS/MS Spectra Andrew Keller Outline Need to validate peptide assignments to MS/MS spectra Statistical approach to validation Running PeptideProphet
PeptideProphet: Validation of Peptide Assignments to MS/MS Spectra Andrew Keller Outline Need to validate peptide assignments to MS/MS spectra Statistical approach to validation Running PeptideProphet
PRIDE Cluster: building the consensus of proteomics data
 Supplementary Materials PRIDE Cluster: building the consensus of proteomics data Johannes Griss, Joseph Michael Foster, Henning Hermjakob and Juan Antonio Vizcaíno EMBL-European Bioinformatics Institute,
Supplementary Materials PRIDE Cluster: building the consensus of proteomics data Johannes Griss, Joseph Michael Foster, Henning Hermjakob and Juan Antonio Vizcaíno EMBL-European Bioinformatics Institute,
Skyline Small Molecule Targets
 Skyline Small Molecule Targets The Skyline Targeted Proteomics Environment provides informative visual displays of the raw mass spectrometer data you import into your Skyline documents. Originally developed
Skyline Small Molecule Targets The Skyline Targeted Proteomics Environment provides informative visual displays of the raw mass spectrometer data you import into your Skyline documents. Originally developed
Statistical Clustering of Vesicle Patterns Practical Aspects of the Analysis of Large Datasets with R
 Statistical Clustering of Vesicle Patterns Mirko Birbaumer Rmetrics Workshop 3th July 2008 1 / 23 Statistical Clustering of Vesicle Patterns Practical Aspects of the Analysis of Large Datasets with R Mirko
Statistical Clustering of Vesicle Patterns Mirko Birbaumer Rmetrics Workshop 3th July 2008 1 / 23 Statistical Clustering of Vesicle Patterns Practical Aspects of the Analysis of Large Datasets with R Mirko
Protocol. Product Use & Liability. Contact us: InfoLine: Order per fax: www:
 Protocol SpikeTides Sets SpikeTides Sets_L heavy SpikeMix SpikeMix_L heavy Peptide Sets for relative quantification of Proteins in Mass Spectrometry Based Assays Contact us: InfoLine: +49-30-6392-7878
Protocol SpikeTides Sets SpikeTides Sets_L heavy SpikeMix SpikeMix_L heavy Peptide Sets for relative quantification of Proteins in Mass Spectrometry Based Assays Contact us: InfoLine: +49-30-6392-7878
Nature Methods: doi: /nmeth Supplementary Figure 1. Fragment indexing allows efficient spectra similarity comparisons.
 Supplementary Figure 1 Fragment indexing allows efficient spectra similarity comparisons. The cost and efficiency of spectra similarity calculations can be approximated by the number of fragment comparisons
Supplementary Figure 1 Fragment indexing allows efficient spectra similarity comparisons. The cost and efficiency of spectra similarity calculations can be approximated by the number of fragment comparisons
WADA Technical Document TD2003IDCR
 IDENTIFICATION CRITERIA FOR QUALITATIVE ASSAYS INCORPORATING CHROMATOGRAPHY AND MASS SPECTROMETRY The appropriate analytical characteristics must be documented for a particular assay. The Laboratory must
IDENTIFICATION CRITERIA FOR QUALITATIVE ASSAYS INCORPORATING CHROMATOGRAPHY AND MASS SPECTROMETRY The appropriate analytical characteristics must be documented for a particular assay. The Laboratory must
Agilent 6400 Series Triple Quadrupole LC/MS/MS Users Session
 Agilent 6400 Series Triple Quadrupole LC/MS/MS Users Session QQQ Method Development and Optimization MassHunter Quant: Method setup Peak detection optimization Quant troubleshooting David Presser Application
Agilent 6400 Series Triple Quadrupole LC/MS/MS Users Session QQQ Method Development and Optimization MassHunter Quant: Method setup Peak detection optimization Quant troubleshooting David Presser Application
Metabolomics Workflow. Discovery Workflow Guide
 Discovery Workflow Guide Notices Agilent Technologies, Inc. 2011 No part of this manual may be reproduced in any form or by any means (including electronic storage and retrieval or translation into a foreign
Discovery Workflow Guide Notices Agilent Technologies, Inc. 2011 No part of this manual may be reproduced in any form or by any means (including electronic storage and retrieval or translation into a foreign
Agilent MassHunter Quantitative Data Analysis
 Agilent MassHunter Quantitative Data Analysis Presenters: Howard Sanford Stephen Harnos MassHunter Quantitation: Batch Table, Compound Information Setup, Calibration Curve and Globals Settings 1 MassHunter
Agilent MassHunter Quantitative Data Analysis Presenters: Howard Sanford Stephen Harnos MassHunter Quantitation: Batch Table, Compound Information Setup, Calibration Curve and Globals Settings 1 MassHunter
RMassBank: Automatic Recalibration and Processing of Tandem HR-MS Spectra for MassBank
 RMassBank: Automatic Recalibration and Processing of Tandem HR-MS Spectra for MassBank Eawag: Swiss Federal Institute of Aquatic Science and Technology Presenting: Emma Schymanski Coauthors: Michael Stravs,
RMassBank: Automatic Recalibration and Processing of Tandem HR-MS Spectra for MassBank Eawag: Swiss Federal Institute of Aquatic Science and Technology Presenting: Emma Schymanski Coauthors: Michael Stravs,