Workflow concept. Data goes through the workflow. A Node contains an operation An edge represents data flow The results are brought together in tables
|
|
|
- Reynard Adams
- 6 years ago
- Views:
Transcription
1 PROTEOME DISCOVERER
2 Workflow concept Data goes through the workflow Spectra Peptides Quantitation A Node contains an operation An edge represents data flow The results are brought together in tables Protein (group) table Peptide (group) table
3 Two aspects of the analysis Identification MS1 + MS2 Quantification MS1
4 Identification

5 Decision tree based MS
6 How to analyze? Decision tree is implemented in acquisition software Data analysis is slightly different for each fragmentation / detector method Rebuild the decision tree in the workflow!
7 Overview of the steps 1. Select the file(s) to use 2. Select only the MS2 spectra 3. Choose the different fragmentation methods 4. Create a specific workflow for each fragmentation type 1. Spectrum filtering 2. Database search 5. PSM Validation, PTM scoring
8 Example dataset 1 Human cancer cell-line with 3 treatments Fractionated with SCX Decision tree: HCD / ETD-FT / ETD
9 1. Select files to use
10 2. Extract the ms2 spectra

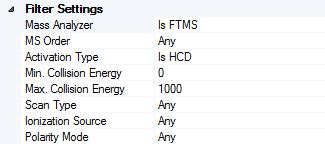
11 3. Select spectrum origins

12 Side note:

13 For ETD: precursor removal
14 For all: size reduction
15 Identifications: Mascot
16 MASCOT search engine

17 Proteomics Mass Spectrometry Trypsin Digest 17
18 Single Stage MS MS 18
19 Tandem Mass Spectrometry (MS/MS) Precursor selection 19
20 Tandem Mass Spectrometry (MS/MS) Precursor selection + collision induced dissociation (CID) MS/MS 20
21 How do search engines work? Input Spectrums Sequence database (proteins) Search parameters Match spectrum to possible peptides Score



22 Precursor m/z Charge spectrum Precursor mass Mass range Range of possible peptide masses
23 A peptide database We now know a range where the peptide mass has to be in Which peptides confirm to this criterium? Taken from protein database Modifications taken into account Miscleavages taken into account




24 Protein database Fixed modifications Variable modifications Digest, modify Allowed missed cleavages Peptide database with modifications indexed by mass
25 Matching the spectra Now we re able to match the spectrum For that, theoretical spectra are composed This is done with the information from the peptide database


26 Peptide database With modifications Indexed by mass Range of possible peptide masses Theoretical spectra Compare and score MS/MSspectrum Ranked list of peptides
27 How to build a theoretical spectrum Information is needed about Amino acid masses Modification masses Methods of fragmentation
28 Peptide Fragmentation Peptide: S-G-F-L-E-E-D-E-L-K MW ion ion MW 88 b 1 S GFLEEDELK y b 2 SG FLEEDELK y b 3 SGF LEEDELK y b 4 SGFL EEDELK y b 5 SGFLE EDELK y b 6 SGFLEE DELK y b 7 SGFLEED ELK y b 8 SGFLEEDE LK y b 9 SGFLEEDEL K y


29 Identifications: Mascot
30 PSM Thresholds
31 PSM thresholds Several options: Target/decoy databases Percolator
32 False Discovery Rate This is a number indicating how many of your hits are false If the FDR is 0.01 (1%), we expect 1 out of 100 peptide hits to be a false identification The number is estimated by the number of hits in the decoy database search result: FDR = decoy hits / target hits
33 Decoy hits Too much noise Mixed spectrum Overall low signal MS2 peaks do match with a peptide
34 Two ways of FDR calculation Concatenated database One search A spectrum matches either one or the other ( Competitive ) Separate decoy database (in Mascot) Two searches A spectrum may match both decoy or target seq ( non-competitive ) FDR = decoy hits / total hits forward reverse d forward reverse d
35 Limitation: one dimension Mascot score Mass delta (ppm) Mascot score
36 Introducing Percolator Percolator (Lukas Käll, McCoss Lab 2007) Machine learning Multiple features
37 What is a support vector machine Classification algorithm Best hyperplane between two groups
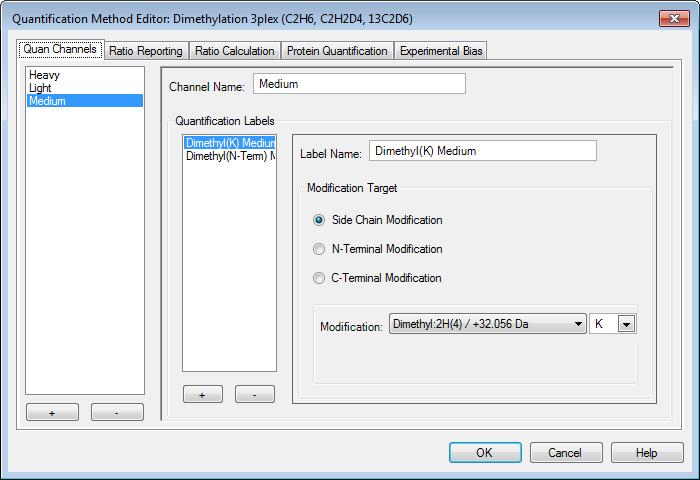
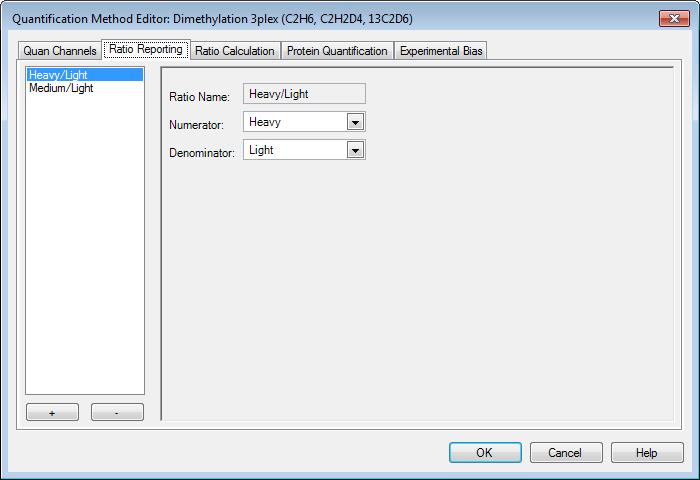
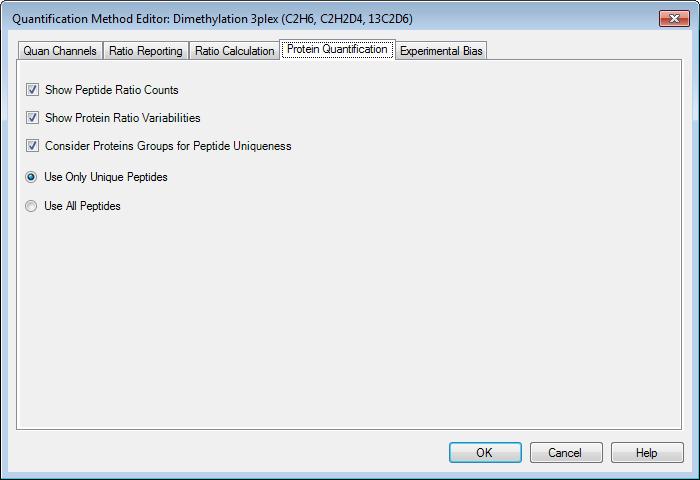
38 What is a support vector machine Classification algorithm Best hyperplane between two groups

39 Soft edges for SVMs
40 Using several features Score Mass delta (ppm) Delta score Number of matched ions
41 Higher dimensions
42 PTM scoring PhosphoRS node Parallel to percolator node PhosphoRS version 2 names change PhosphoRS score Mao will explain in the afternoon
43 Quantification
 Node to extract the")
44 Labeled Quantification Peptide modifications Extracted Ion Chromatrogram (XIC) Node to extract the intensities
45 How to interpret the XICs The XICs need to be grouped Isotopes Known modification (e.g. dimethylation, SILAC) The node needs details Quantification method


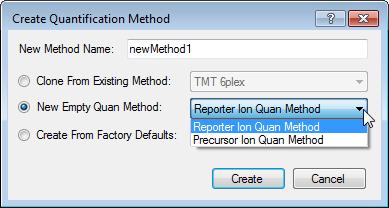
46 Manage quant methods

47
48 PD Output
49 Understanding PD output PD run log Node result columns Protein output Peptide output NB: the modified peptide column comes from the Mascot node, whilst the phosphors site localizations are from that node Modified peptide does not represent best localizations
50 Peptides tab List Peptides Grouping (right click) Grouped Ungrouped (PTMs) Grouping parameters are in the Result Filters tab Based on sequence Based on sequence and mass (i.e. modifications) PD will filter the PTMs based on the settings in the Result Filters tab

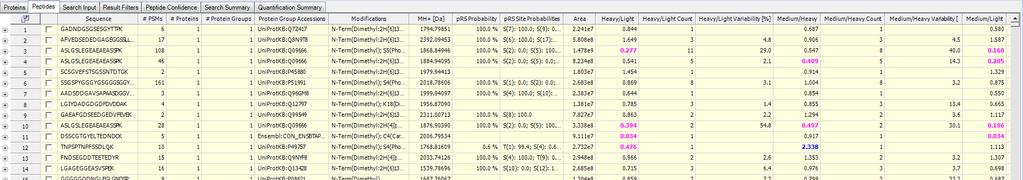
51 Peptide output < 0.5 >2
52 Protein tab List proteins Grouping (right click) Grouped Ungrouped Grouping based on shared peptides
53 What s Grouped? Grouping allows for the calculation of standard deviations Show the highest scores
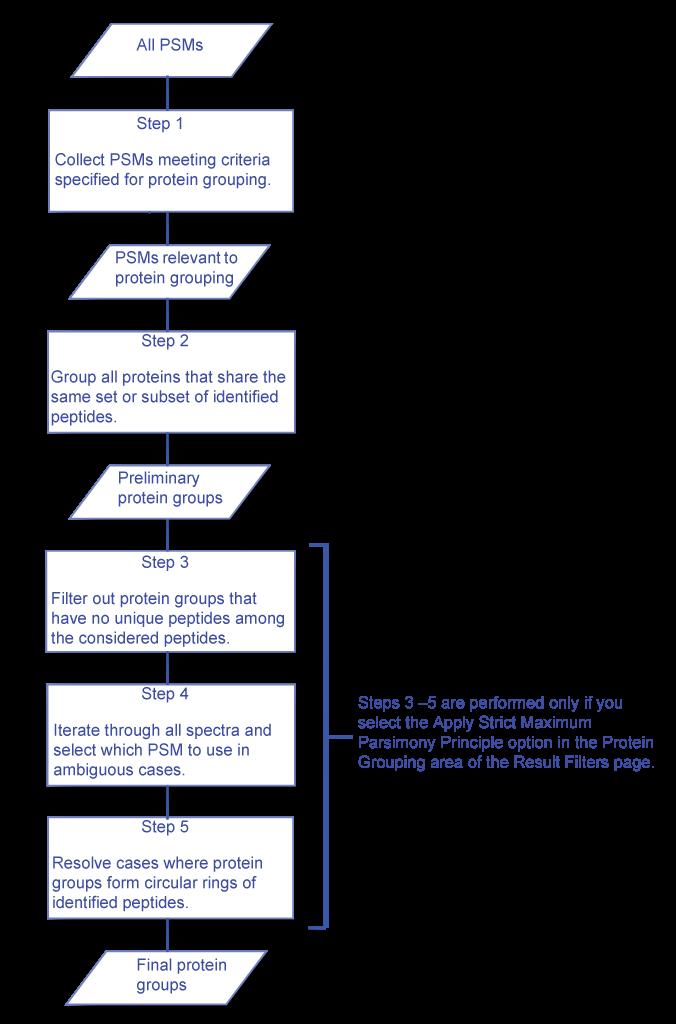
54
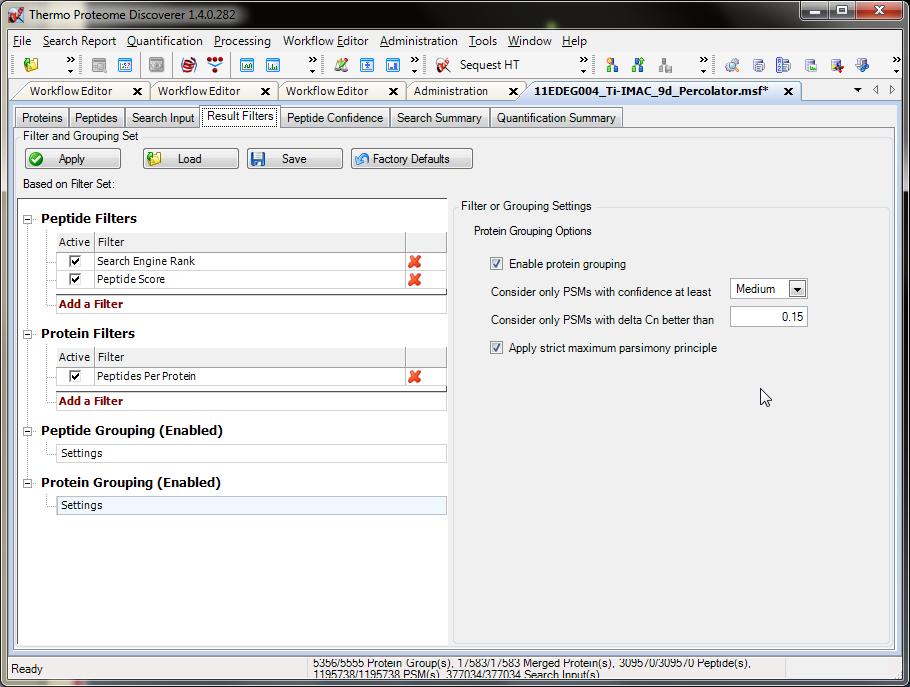
55 Filters
56 Example dataset 2 Yeast time course experiment Labeled with TMT 6 -plex Fractionated with SCX

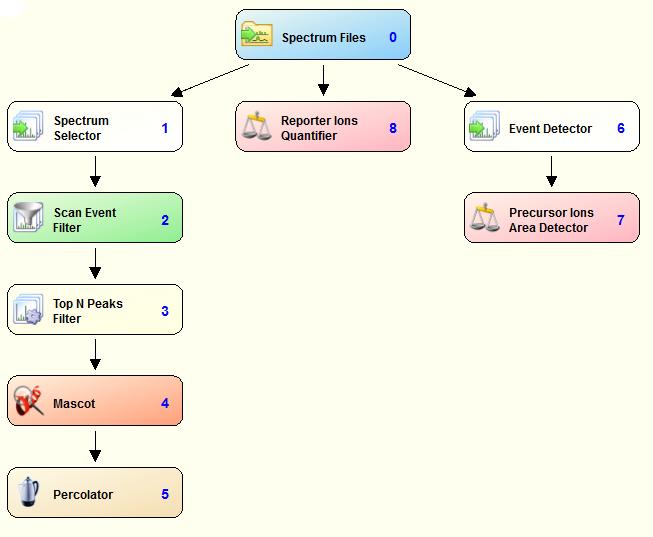
57 build this!
58 Alternative TMT analysis Isobar package Nice Statistical analysis Nice output Freely available Disadvantages Written in R Somewhat hard to use Ask Bas
Protein Identification Using Tandem Mass Spectrometry. Nathan Edwards Informatics Research Applied Biosystems
 Protein Identification Using Tandem Mass Spectrometry Nathan Edwards Informatics Research Applied Biosystems Outline Proteomics context Tandem mass spectrometry Peptide fragmentation Peptide identification
Protein Identification Using Tandem Mass Spectrometry Nathan Edwards Informatics Research Applied Biosystems Outline Proteomics context Tandem mass spectrometry Peptide fragmentation Peptide identification
Proteomics. November 13, 2007
 Proteomics November 13, 2007 Acknowledgement Slides presented here have been borrowed from presentations by : Dr. Mark A. Knepper (LKEM, NHLBI, NIH) Dr. Nathan Edwards (Center for Bioinformatics and Computational
Proteomics November 13, 2007 Acknowledgement Slides presented here have been borrowed from presentations by : Dr. Mark A. Knepper (LKEM, NHLBI, NIH) Dr. Nathan Edwards (Center for Bioinformatics and Computational
HOWTO, example workflow and data files. (Version )
") HOWTO, example workflow and data files. (Version 20 09 2017) 1 Introduction: SugarQb is a collection of software tools (Nodes) which enable the automated identification of intact glycopeptides from HCD
HOWTO, example workflow and data files. (Version 20 09 2017) 1 Introduction: SugarQb is a collection of software tools (Nodes) which enable the automated identification of intact glycopeptides from HCD
Overview - MS Proteomics in One Slide. MS masses of peptides. MS/MS fragments of a peptide. Results! Match to sequence database
 Overview - MS Proteomics in One Slide Obtain protein Digest into peptides Acquire spectra in mass spectrometer MS masses of peptides MS/MS fragments of a peptide Results! Match to sequence database 2 But
Overview - MS Proteomics in One Slide Obtain protein Digest into peptides Acquire spectra in mass spectrometer MS masses of peptides MS/MS fragments of a peptide Results! Match to sequence database 2 But
Mass Spectrometry and Proteomics - Lecture 5 - Matthias Trost Newcastle University
 Mass Spectrometry and Proteomics - Lecture 5 - Matthias Trost Newcastle University matthias.trost@ncl.ac.uk Previously Proteomics Sample prep 144 Lecture 5 Quantitation techniques Search Algorithms Proteomics
Mass Spectrometry and Proteomics - Lecture 5 - Matthias Trost Newcastle University matthias.trost@ncl.ac.uk Previously Proteomics Sample prep 144 Lecture 5 Quantitation techniques Search Algorithms Proteomics
Computational Methods for Mass Spectrometry Proteomics
 Computational Methods for Mass Spectrometry Proteomics Eidhammer, Ingvar ISBN-13: 9780470512975 Table of Contents Preface. Acknowledgements. 1 Protein, Proteome, and Proteomics. 1.1 Primary goals for studying
Computational Methods for Mass Spectrometry Proteomics Eidhammer, Ingvar ISBN-13: 9780470512975 Table of Contents Preface. Acknowledgements. 1 Protein, Proteome, and Proteomics. 1.1 Primary goals for studying
Last updated: Copyright
 Last updated: 2012-08-20 Copyright 2004-2012 plabel (v2.4) User s Manual by Bioinformatics Group, Institute of Computing Technology, Chinese Academy of Sciences Tel: 86-10-62601016 Email: zhangkun01@ict.ac.cn,
Last updated: 2012-08-20 Copyright 2004-2012 plabel (v2.4) User s Manual by Bioinformatics Group, Institute of Computing Technology, Chinese Academy of Sciences Tel: 86-10-62601016 Email: zhangkun01@ict.ac.cn,
TOMAHAQ Method Construction
 TOMAHAQ Method Construction Triggered by offset mass accurate-mass high-resolution accurate quantitation (TOMAHAQ) can be performed in the standard method editor of the instrument, without modifications
TOMAHAQ Method Construction Triggered by offset mass accurate-mass high-resolution accurate quantitation (TOMAHAQ) can be performed in the standard method editor of the instrument, without modifications
Improved 6- Plex TMT Quantification Throughput Using a Linear Ion Trap HCD MS 3 Scan Jane M. Liu, 1,2 * Michael J. Sweredoski, 2 Sonja Hess 2 *
 Improved 6- Plex TMT Quantification Throughput Using a Linear Ion Trap HCD MS 3 Scan Jane M. Liu, 1,2 * Michael J. Sweredoski, 2 Sonja Hess 2 * 1 Department of Chemistry, Pomona College, Claremont, California
Improved 6- Plex TMT Quantification Throughput Using a Linear Ion Trap HCD MS 3 Scan Jane M. Liu, 1,2 * Michael J. Sweredoski, 2 Sonja Hess 2 * 1 Department of Chemistry, Pomona College, Claremont, California
Key questions of proteomics. Bioinformatics 2. Proteomics. Foundation of proteomics. What proteins are there? Protein digestion
 s s Key questions of proteomics What proteins are there? Bioinformatics 2 Lecture 2 roteomics How much is there of each of the proteins? - Absolute quantitation - Stoichiometry What (modification/splice)
s s Key questions of proteomics What proteins are there? Bioinformatics 2 Lecture 2 roteomics How much is there of each of the proteins? - Absolute quantitation - Stoichiometry What (modification/splice)
Thermo Scientific LTQ Orbitrap Velos Hybrid FT Mass Spectrometer
 IET International Equipment Trading Ltd. www.ietltd.com Proudly serving laboratories worldwide since 1979 CALL +847.913.0777 for Refurbished & Certified Lab Equipment Thermo Scientific LTQ Orbitrap Velos
IET International Equipment Trading Ltd. www.ietltd.com Proudly serving laboratories worldwide since 1979 CALL +847.913.0777 for Refurbished & Certified Lab Equipment Thermo Scientific LTQ Orbitrap Velos
A TMT-labeled Spectral Library for Peptide Sequencing
 A TMT-labeled Spectral Library for Peptide Sequencing by Jianqiao Shen A thesis presented to the University of Waterloo in fulfillment of the thesis requirement for the degree of Master of Mathematics
A TMT-labeled Spectral Library for Peptide Sequencing by Jianqiao Shen A thesis presented to the University of Waterloo in fulfillment of the thesis requirement for the degree of Master of Mathematics
MassHunter Software Overview
 MassHunter Software Overview 1 Qualitative Analysis Workflows Workflows in Qualitative Analysis allow the user to only see and work with the areas and dialog boxes they need for their specific tasks A
MassHunter Software Overview 1 Qualitative Analysis Workflows Workflows in Qualitative Analysis allow the user to only see and work with the areas and dialog boxes they need for their specific tasks A
Relative quantification using TMT11plex on a modified Q Exactive HF mass spectrometer
 POSTER NOTE 6558 Relative quantification using TMT11plex on a modified mass spectrometer Authors Tabiwang N. Arrey, 1 Rosa Viner, 2 Ryan D. Bomgarden, 3 Eugen Damoc, 1 Markus Kellmann, 1 Thomas Moehring,
POSTER NOTE 6558 Relative quantification using TMT11plex on a modified mass spectrometer Authors Tabiwang N. Arrey, 1 Rosa Viner, 2 Ryan D. Bomgarden, 3 Eugen Damoc, 1 Markus Kellmann, 1 Thomas Moehring,
PeptideProphet: Validation of Peptide Assignments to MS/MS Spectra. Andrew Keller
 PeptideProphet: Validation of Peptide Assignments to MS/MS Spectra Andrew Keller Outline Need to validate peptide assignments to MS/MS spectra Statistical approach to validation Running PeptideProphet
PeptideProphet: Validation of Peptide Assignments to MS/MS Spectra Andrew Keller Outline Need to validate peptide assignments to MS/MS spectra Statistical approach to validation Running PeptideProphet
Proteome-wide label-free quantification with MaxQuant. Jürgen Cox Max Planck Institute of Biochemistry July 2011
 Proteome-wide label-free quantification with MaxQuant Jürgen Cox Max Planck Institute of Biochemistry July 2011 MaxQuant MaxQuant Feature detection Data acquisition Initial Andromeda search Statistics
Proteome-wide label-free quantification with MaxQuant Jürgen Cox Max Planck Institute of Biochemistry July 2011 MaxQuant MaxQuant Feature detection Data acquisition Initial Andromeda search Statistics
NPTEL VIDEO COURSE PROTEOMICS PROF. SANJEEVA SRIVASTAVA
 LECTURE-25 Quantitative proteomics: itraq and TMT TRANSCRIPT Welcome to the proteomics course. Today we will talk about quantitative proteomics and discuss about itraq and TMT techniques. The quantitative
LECTURE-25 Quantitative proteomics: itraq and TMT TRANSCRIPT Welcome to the proteomics course. Today we will talk about quantitative proteomics and discuss about itraq and TMT techniques. The quantitative
MaSS-Simulator: A highly configurable MS/MS simulator for generating test datasets for big data algorithms.
 MaSS-Simulator: A highly configurable MS/MS simulator for generating test datasets for big data algorithms. Muaaz Gul Awan 1 and Fahad Saeed 1 1 Department of Computer Science, Western Michigan University,
MaSS-Simulator: A highly configurable MS/MS simulator for generating test datasets for big data algorithms. Muaaz Gul Awan 1 and Fahad Saeed 1 1 Department of Computer Science, Western Michigan University,
Supplementary Figure 1
 Supplementary Figure 1 The correlation of n-score cutoff and FDR in both CID-only and CID-ETD fragmentation strategies. A bar diagram of different n-score thresholds applied in the search, plotted against
Supplementary Figure 1 The correlation of n-score cutoff and FDR in both CID-only and CID-ETD fragmentation strategies. A bar diagram of different n-score thresholds applied in the search, plotted against
Comprehensive support for quantitation
 Comprehensive support for quantitation One of the major new features in the current release of Mascot is support for quantitation. This is still work in progress. Our goal is to support all of the popular
Comprehensive support for quantitation One of the major new features in the current release of Mascot is support for quantitation. This is still work in progress. Our goal is to support all of the popular
X!TandemPipeline (Myosine Anabolisée) validating, filtering and grouping MSMS identifications
 validating, filtering and grouping MSMS identifications") X!TandemPipeline 3.3.3 (Myosine Anabolisée) validating, filtering and grouping MSMS identifications Olivier Langella and Benoit Valot langella@moulon.inra.fr; valot@moulon.inra.fr PAPPSO - http://pappso.inra.fr/
X!TandemPipeline 3.3.3 (Myosine Anabolisée) validating, filtering and grouping MSMS identifications Olivier Langella and Benoit Valot langella@moulon.inra.fr; valot@moulon.inra.fr PAPPSO - http://pappso.inra.fr/
Protein Quantitation II: Multiple Reaction Monitoring. Kelly Ruggles New York University
 Protein Quantitation II: Multiple Reaction Monitoring Kelly Ruggles kelly@fenyolab.org New York University Traditional Affinity-based proteomics Use antibodies to quantify proteins Western Blot RPPA Immunohistochemistry
Protein Quantitation II: Multiple Reaction Monitoring Kelly Ruggles kelly@fenyolab.org New York University Traditional Affinity-based proteomics Use antibodies to quantify proteins Western Blot RPPA Immunohistochemistry
SPECTRA LIBRARY ASSISTED DE NOVO PEPTIDE SEQUENCING FOR HCD AND ETD SPECTRA PAIRS
 SPECTRA LIBRARY ASSISTED DE NOVO PEPTIDE SEQUENCING FOR HCD AND ETD SPECTRA PAIRS 1 Yan Yan Department of Computer Science University of Western Ontario, Canada OUTLINE Background Tandem mass spectrometry
SPECTRA LIBRARY ASSISTED DE NOVO PEPTIDE SEQUENCING FOR HCD AND ETD SPECTRA PAIRS 1 Yan Yan Department of Computer Science University of Western Ontario, Canada OUTLINE Background Tandem mass spectrometry
Increasing the Multiplexing of Protein Quantitation from 6- to 10-Plex with Reporter Ion Isotopologues
 Increasing the Multiplexing of Protein Quantitation from 6- to 1-Plex with Reporter Ion Isotopologues Rosa Viner, 1 Ryan Bomgarden, 2 Michael Blank, 1 John Rogers 2 1 Thermo Fisher Scientific, San Jose,
Increasing the Multiplexing of Protein Quantitation from 6- to 1-Plex with Reporter Ion Isotopologues Rosa Viner, 1 Ryan Bomgarden, 2 Michael Blank, 1 John Rogers 2 1 Thermo Fisher Scientific, San Jose,
Effective Strategies for Improving Peptide Identification with Tandem Mass Spectrometry
 Effective Strategies for Improving Peptide Identification with Tandem Mass Spectrometry by Xi Han A thesis presented to the University of Waterloo in fulfillment of the thesis requirement for the degree
Effective Strategies for Improving Peptide Identification with Tandem Mass Spectrometry by Xi Han A thesis presented to the University of Waterloo in fulfillment of the thesis requirement for the degree
Protein Quantitation II: Multiple Reaction Monitoring. Kelly Ruggles New York University
 Protein Quantitation II: Multiple Reaction Monitoring Kelly Ruggles kelly@fenyolab.org New York University Traditional Affinity-based proteomics Use antibodies to quantify proteins Western Blot Immunohistochemistry
Protein Quantitation II: Multiple Reaction Monitoring Kelly Ruggles kelly@fenyolab.org New York University Traditional Affinity-based proteomics Use antibodies to quantify proteins Western Blot Immunohistochemistry
MS-based proteomics to investigate proteins and their modifications
 MS-based proteomics to investigate proteins and their modifications Francis Impens VIB Proteomics Core October th 217 Overview Mass spectrometry-based proteomics: general workflow Identification of protein
MS-based proteomics to investigate proteins and their modifications Francis Impens VIB Proteomics Core October th 217 Overview Mass spectrometry-based proteomics: general workflow Identification of protein
Learning Score Function Parameters for Improved Spectrum Identification in Tandem Mass Spectrometry Experiments
 pubs.acs.org/jpr Learning Score Function Parameters for Improved Spectrum Identification in Tandem Mass Spectrometry Experiments Marina Spivak, Michael S. Bereman, Michael J. MacCoss, and William Stafford
pubs.acs.org/jpr Learning Score Function Parameters for Improved Spectrum Identification in Tandem Mass Spectrometry Experiments Marina Spivak, Michael S. Bereman, Michael J. MacCoss, and William Stafford
Spectronaut Pulsar. User Manual
 Spectronaut Pulsar User Manual 1 General Information... 6 1.1 Computer System Requirements... 6 1.2 Scope of Spectronaut Software... 6 1.3 Spectronaut Pulsar... 6 1.4 Spectronaut Release Features... 7
Spectronaut Pulsar User Manual 1 General Information... 6 1.1 Computer System Requirements... 6 1.2 Scope of Spectronaut Software... 6 1.3 Spectronaut Pulsar... 6 1.4 Spectronaut Release Features... 7
Self-assembling covalent organic frameworks functionalized. magnetic graphene hydrophilic biocomposite as an ultrasensitive
 Electronic Supplementary Material (ESI) for Nanoscale. This journal is The Royal Society of Chemistry 2017 Electronic Supporting Information for: Self-assembling covalent organic frameworks functionalized
Electronic Supplementary Material (ESI) for Nanoscale. This journal is The Royal Society of Chemistry 2017 Electronic Supporting Information for: Self-assembling covalent organic frameworks functionalized
High-Field Orbitrap Creating new possibilities
 Thermo Scientific Orbitrap Elite Hybrid Mass Spectrometer High-Field Orbitrap Creating new possibilities Ultrahigh resolution Faster scanning Higher sensitivity Complementary fragmentation The highest
Thermo Scientific Orbitrap Elite Hybrid Mass Spectrometer High-Field Orbitrap Creating new possibilities Ultrahigh resolution Faster scanning Higher sensitivity Complementary fragmentation The highest
Tandem MS = MS / MS. ESI-MS give information on the mass of a molecule but none on the structure
 Tandem MS = MS / MS ESI-MS give information on the mass of a molecule but none on the structure In tandem MS (MSMS) (pseudo-)molecular ions are selected in MS1 and fragmented by collision with gas. collision
Tandem MS = MS / MS ESI-MS give information on the mass of a molecule but none on the structure In tandem MS (MSMS) (pseudo-)molecular ions are selected in MS1 and fragmented by collision with gas. collision
A Description of the CPTAC Common Data Analysis Pipeline (CDAP)
") A Description of the CPTAC Common Data Analysis Pipeline (CDAP) v. 01/14/2014 Summary The purpose of this document is to describe the software programs and output files of the Common Data Analysis Pipeline
A Description of the CPTAC Common Data Analysis Pipeline (CDAP) v. 01/14/2014 Summary The purpose of this document is to describe the software programs and output files of the Common Data Analysis Pipeline
iprophet: Multi-level integrative analysis of shotgun proteomic data improves peptide and protein identification rates and error estimates
 MCP Papers in Press. Published on August 29, 2011 as Manuscript M111.007690 This is the Pre-Published Version iprophet: Multi-level integrative analysis of shotgun proteomic data improves peptide and protein
MCP Papers in Press. Published on August 29, 2011 as Manuscript M111.007690 This is the Pre-Published Version iprophet: Multi-level integrative analysis of shotgun proteomic data improves peptide and protein
Nature Methods: doi: /nmeth Supplementary Figure 1. Fragment indexing allows efficient spectra similarity comparisons.
 Supplementary Figure 1 Fragment indexing allows efficient spectra similarity comparisons. The cost and efficiency of spectra similarity calculations can be approximated by the number of fragment comparisons
Supplementary Figure 1 Fragment indexing allows efficient spectra similarity comparisons. The cost and efficiency of spectra similarity calculations can be approximated by the number of fragment comparisons
PeptideProphet: Validation of Peptide Assignments to MS/MS Spectra
 PeptideProphet: Validation of Peptide Assignments to MS/MS Spectra Andrew Keller Day 2 October 17, 2006 Andrew Keller Rosetta Bioinformatics, Seattle Outline Need to validate peptide assignments to MS/MS
PeptideProphet: Validation of Peptide Assignments to MS/MS Spectra Andrew Keller Day 2 October 17, 2006 Andrew Keller Rosetta Bioinformatics, Seattle Outline Need to validate peptide assignments to MS/MS
MS-MS Analysis Programs
 MS-MS Analysis Programs Basic Process Genome - Gives AA sequences of proteins Use this to predict spectra Compare data to prediction Determine degree of correctness Make assignment Did we see the protein?
MS-MS Analysis Programs Basic Process Genome - Gives AA sequences of proteins Use this to predict spectra Compare data to prediction Determine degree of correctness Make assignment Did we see the protein?
Isotopic-Labeling and Mass Spectrometry-Based Quantitative Proteomics
 Isotopic-Labeling and Mass Spectrometry-Based Quantitative Proteomics Xiao-jun Li, Ph.D. Current address: Homestead Clinical Day 4 October 19, 2006 Protein Quantification LC-MS/MS Data XLink mzxml file
Isotopic-Labeling and Mass Spectrometry-Based Quantitative Proteomics Xiao-jun Li, Ph.D. Current address: Homestead Clinical Day 4 October 19, 2006 Protein Quantification LC-MS/MS Data XLink mzxml file
Spectrum-to-Spectrum Searching Using a. Proteome-wide Spectral Library
 MCP Papers in Press. Published on April 30, 2011 as Manuscript M111.007666 Spectrum-to-Spectrum Searching Using a Proteome-wide Spectral Library Chia-Yu Yen, Stephane Houel, Natalie G. Ahn, and William
MCP Papers in Press. Published on April 30, 2011 as Manuscript M111.007666 Spectrum-to-Spectrum Searching Using a Proteome-wide Spectral Library Chia-Yu Yen, Stephane Houel, Natalie G. Ahn, and William
Properties of Average Score Distributions of SEQUEST
 Research Properties of Average Score Distributions of SEQUEST THE PROBABILITY RATIO METHOD* S Salvador Martínez-Bartolomé, Pedro Navarro, Fernando Martín-Maroto, Daniel López-Ferrer **, Antonio Ramos-Fernández,
Research Properties of Average Score Distributions of SEQUEST THE PROBABILITY RATIO METHOD* S Salvador Martínez-Bartolomé, Pedro Navarro, Fernando Martín-Maroto, Daniel López-Ferrer **, Antonio Ramos-Fernández,
SRM assay generation and data analysis in Skyline
 in Skyline Preparation 1. Download the example data from www.srmcourse.ch/eupa.html (3 raw files, 1 csv file, 1 sptxt file). 2. The number formats of your computer have to be set to English (United States).
in Skyline Preparation 1. Download the example data from www.srmcourse.ch/eupa.html (3 raw files, 1 csv file, 1 sptxt file). 2. The number formats of your computer have to be set to English (United States).
Tutorial 2: Analysis of DIA data in Skyline
 Tutorial 2: Analysis of DIA data in Skyline In this tutorial we will learn how to use Skyline to perform targeted post-acquisition analysis for peptide and inferred protein detection and quantitation using
Tutorial 2: Analysis of DIA data in Skyline In this tutorial we will learn how to use Skyline to perform targeted post-acquisition analysis for peptide and inferred protein detection and quantitation using
Qualitative Proteomics (how to obtain high-confidence high-throughput protein identification!)
") Qualitative Proteomics (how to obtain high-confidence high-throughput protein identification!) James A. Mobley, Ph.D. Director of Research in Urology Associate Director of Mass Spectrometry (contact: mobleyja@uab.edu)
Qualitative Proteomics (how to obtain high-confidence high-throughput protein identification!) James A. Mobley, Ph.D. Director of Research in Urology Associate Director of Mass Spectrometry (contact: mobleyja@uab.edu)
Mass Spectrometry Based De Novo Peptide Sequencing Error Correction
 Mass Spectrometry Based De Novo Peptide Sequencing Error Correction by Chenyu Yao A thesis presented to the University of Waterloo in fulfillment of the thesis requirement for the degree of Master of Mathematics
Mass Spectrometry Based De Novo Peptide Sequencing Error Correction by Chenyu Yao A thesis presented to the University of Waterloo in fulfillment of the thesis requirement for the degree of Master of Mathematics
Methods for proteome analysis of obesity (Adipose tissue)
") Methods for proteome analysis of obesity (Adipose tissue) I. Sample preparation and liquid chromatography-tandem mass spectrometric analysis Instruments, softwares, and materials AB SCIEX Triple TOF 5600
Methods for proteome analysis of obesity (Adipose tissue) I. Sample preparation and liquid chromatography-tandem mass spectrometric analysis Instruments, softwares, and materials AB SCIEX Triple TOF 5600
Purdue-UAB Botanicals Center for Age- Related Disease
 Purdue-UAB Botanicals Center for Age- Related Disease MALDI-TOF Mass Spectrometry Fingerprinting Technique Landon Wilson MALDI-TOF mass spectrometry is an advanced technique for rapid protein identification
Purdue-UAB Botanicals Center for Age- Related Disease MALDI-TOF Mass Spectrometry Fingerprinting Technique Landon Wilson MALDI-TOF mass spectrometry is an advanced technique for rapid protein identification
Tutorial 1: Setting up your Skyline document
 Tutorial 1: Setting up your Skyline document Caution! For using Skyline the number formats of your computer have to be set to English (United States). Open the Control Panel Clock, Language, and Region
Tutorial 1: Setting up your Skyline document Caution! For using Skyline the number formats of your computer have to be set to English (United States). Open the Control Panel Clock, Language, and Region
De novo peptide sequencing methods for tandem mass. spectra
 De novo peptide sequencing methods for tandem mass spectra A Thesis Submitted to the College of Graduate Studies and Research in Partial Fulfillment of the Requirements for the degree of Doctor of Philosophy
De novo peptide sequencing methods for tandem mass spectra A Thesis Submitted to the College of Graduate Studies and Research in Partial Fulfillment of the Requirements for the degree of Doctor of Philosophy
Quantitation of a target protein in crude samples using targeted peptide quantification by Mass Spectrometry
 Quantitation of a target protein in crude samples using targeted peptide quantification by Mass Spectrometry Jon Hao, Rong Ye, and Mason Tao Poochon Scientific, Frederick, Maryland 21701 Abstract Background:
Quantitation of a target protein in crude samples using targeted peptide quantification by Mass Spectrometry Jon Hao, Rong Ye, and Mason Tao Poochon Scientific, Frederick, Maryland 21701 Abstract Background:
Peptide Targeted Quantification By High Resolution Mass Spectrometry A Paradigm Shift? Zhiqi Hao Thermo Fisher Scientific San Jose, CA
 Peptide Targeted Quantification By High Resolution Mass Spectrometry A Paradigm Shift? Zhiqi Hao Thermo Fisher Scientific San Jose, CA Proteomics is Turning Quantitative Hmmm.. Which ones are my targets?
Peptide Targeted Quantification By High Resolution Mass Spectrometry A Paradigm Shift? Zhiqi Hao Thermo Fisher Scientific San Jose, CA Proteomics is Turning Quantitative Hmmm.. Which ones are my targets?
Figure S1. Interaction of PcTS with αsyn. (a) 1 H- 15 N HSQC NMR spectra of 100 µm αsyn in the absence (0:1, black) and increasing equivalent
 1 H- 15 N HSQC NMR spectra of 100 µm αsyn in the absence (0:1, black) and increasing equivalent") Figure S1. Interaction of PcTS with αsyn. (a) 1 H- 15 N HSQC NMR spectra of 100 µm αsyn in the absence (0:1, black) and increasing equivalent concentrations of PcTS (100 µm, blue; 500 µm, green; 1.5 mm,
Figure S1. Interaction of PcTS with αsyn. (a) 1 H- 15 N HSQC NMR spectra of 100 µm αsyn in the absence (0:1, black) and increasing equivalent concentrations of PcTS (100 µm, blue; 500 µm, green; 1.5 mm,
LECTURE-13. Peptide Mass Fingerprinting HANDOUT. Mass spectrometry is an indispensable tool for qualitative and quantitative analysis of
 LECTURE-13 Peptide Mass Fingerprinting HANDOUT PREAMBLE Mass spectrometry is an indispensable tool for qualitative and quantitative analysis of proteins, drugs and many biological moieties to elucidate
LECTURE-13 Peptide Mass Fingerprinting HANDOUT PREAMBLE Mass spectrometry is an indispensable tool for qualitative and quantitative analysis of proteins, drugs and many biological moieties to elucidate
On Optimizing the Non-metric Similarity Search in Tandem Mass Spectra by Clustering
 On Optimizing the Non-metric Similarity Search in Tandem Mass Spectra by Clustering Jiří Novák, David Hoksza, Jakub Lokoč, and Tomáš Skopal Siret Research Group, Faculty of Mathematics and Physics, Charles
On Optimizing the Non-metric Similarity Search in Tandem Mass Spectra by Clustering Jiří Novák, David Hoksza, Jakub Lokoč, and Tomáš Skopal Siret Research Group, Faculty of Mathematics and Physics, Charles
DIA-Umpire: comprehensive computational framework for data independent acquisition proteomics
 DIA-Umpire: comprehensive computational framework for data independent acquisition proteomics Chih-Chiang Tsou 1,2, Dmitry Avtonomov 2, Brett Larsen 3, Monika Tucholska 3, Hyungwon Choi 4 Anne-Claude Gingras
DIA-Umpire: comprehensive computational framework for data independent acquisition proteomics Chih-Chiang Tsou 1,2, Dmitry Avtonomov 2, Brett Larsen 3, Monika Tucholska 3, Hyungwon Choi 4 Anne-Claude Gingras
Proteomics: the first decade and beyond. (2003) Patterson and Aebersold Nat Genet 33 Suppl: from
 Patterson and Aebersold Nat Genet 33 Suppl: from") Advances in mass spectrometry and the generation of large quantities of nucleotide sequence information, combined with computational algorithms that could correlate the two, led to the emergence of proteomics
Advances in mass spectrometry and the generation of large quantities of nucleotide sequence information, combined with computational algorithms that could correlate the two, led to the emergence of proteomics
Mass spectrometry has been used a lot in biology since the late 1950 s. However it really came into play in the late 1980 s once methods were
 Mass spectrometry has been used a lot in biology since the late 1950 s. However it really came into play in the late 1980 s once methods were developed to allow the analysis of large intact (bigger than
Mass spectrometry has been used a lot in biology since the late 1950 s. However it really came into play in the late 1980 s once methods were developed to allow the analysis of large intact (bigger than
TUTORIAL EXERCISES WITH ANSWERS
 TUTORIAL EXERCISES WITH ANSWERS Tutorial 1 Settings 1. What is the exact monoisotopic mass difference for peptides carrying a 13 C (and NO additional 15 N) labelled C-terminal lysine residue? a. 6.020129
TUTORIAL EXERCISES WITH ANSWERS Tutorial 1 Settings 1. What is the exact monoisotopic mass difference for peptides carrying a 13 C (and NO additional 15 N) labelled C-terminal lysine residue? a. 6.020129
Quantitative Proteomics
 Quantitative Proteomics Quantitation AND Mass Spectrometry Condition A Condition B Identify and quantify differently expressed proteins resulting from a change in the environment (stimulus, disease) Lyse
Quantitative Proteomics Quantitation AND Mass Spectrometry Condition A Condition B Identify and quantify differently expressed proteins resulting from a change in the environment (stimulus, disease) Lyse
Tandem mass spectra were extracted from the Xcalibur data system format. (.RAW) and charge state assignment was performed using in house software
 and charge state assignment was performed using in house software") Supplementary Methods Software Interpretation of Tandem mass spectra Tandem mass spectra were extracted from the Xcalibur data system format (.RAW) and charge state assignment was performed using in house
Supplementary Methods Software Interpretation of Tandem mass spectra Tandem mass spectra were extracted from the Xcalibur data system format (.RAW) and charge state assignment was performed using in house
MassHunter TOF/QTOF Users Meeting
 MassHunter TOF/QTOF Users Meeting 1 Qualitative Analysis Workflows Workflows in Qualitative Analysis allow the user to only see and work with the areas and dialog boxes they need for their specific tasks
MassHunter TOF/QTOF Users Meeting 1 Qualitative Analysis Workflows Workflows in Qualitative Analysis allow the user to only see and work with the areas and dialog boxes they need for their specific tasks
ASCQ_ME: a new engine for peptide mass fingerprint directly from mass spectrum without mass list extraction
 ASCQ_ME: a new engine for peptide mass fingerprint directly from mass spectrum without mass list extraction Jean-Charles BOISSON1, Laetitia JOURDAN1, El-Ghazali TALBI1, Cécile CREN-OLIVE2 et Christian
ASCQ_ME: a new engine for peptide mass fingerprint directly from mass spectrum without mass list extraction Jean-Charles BOISSON1, Laetitia JOURDAN1, El-Ghazali TALBI1, Cécile CREN-OLIVE2 et Christian
Protein analysis using mass spectrometry
 Protein analysis using mass spectrometry Michael Stadlmeier 2017/12/18 Literature http://www.carellgroup.de/teaching/master 3 What is Proteomics? The proteome is: the entire set of proteins in a given
Protein analysis using mass spectrometry Michael Stadlmeier 2017/12/18 Literature http://www.carellgroup.de/teaching/master 3 What is Proteomics? The proteome is: the entire set of proteins in a given
1. Prepare the MALDI sample plate by spotting an angiotensin standard and the test sample(s).
.") Analysis of a Peptide Sequence from a Proteolytic Digest by MALDI-TOF Post-Source Decay (PSD) and Collision-Induced Dissociation (CID) Standard Operating Procedure Purpose: The following procedure may
Analysis of a Peptide Sequence from a Proteolytic Digest by MALDI-TOF Post-Source Decay (PSD) and Collision-Induced Dissociation (CID) Standard Operating Procedure Purpose: The following procedure may
MSnID Package for Handling MS/MS Identifications
 Vladislav A. Petyuk December 1, 2018 Contents 1 Introduction.............................. 1 2 Starting the project.......................... 3 3 Reading MS/MS data........................ 3 4 Updating
Vladislav A. Petyuk December 1, 2018 Contents 1 Introduction.............................. 1 2 Starting the project.......................... 3 3 Reading MS/MS data........................ 3 4 Updating
Atomic masses. Atomic masses of elements. Atomic masses of isotopes. Nominal and exact atomic masses. Example: CO, N 2 ja C 2 H 4
 High-Resolution Mass spectrometry (HR-MS, HRAM-MS) (FT mass spectrometry) MS that enables identifying elemental compositions (empirical formulas) from accurate m/z data 9.05.2017 1 Atomic masses (atomic
High-Resolution Mass spectrometry (HR-MS, HRAM-MS) (FT mass spectrometry) MS that enables identifying elemental compositions (empirical formulas) from accurate m/z data 9.05.2017 1 Atomic masses (atomic
Quantitation of TMT-Labeled Peptides Using Higher-Energy Collisional Dissociation on the Velos Pro Ion Trap Mass Spectrometer
 Application Note: 520 Quantitation of TMT-Labeled Peptides Using Higher-Energy Collisional Dissociation on the Velos Pro Ion Trap Mass Spectrometer Roger G. Biringer, Julie A. Horner, Rosa Viner, Andreas
Application Note: 520 Quantitation of TMT-Labeled Peptides Using Higher-Energy Collisional Dissociation on the Velos Pro Ion Trap Mass Spectrometer Roger G. Biringer, Julie A. Horner, Rosa Viner, Andreas
Relative Quantitation of TMT-Labeled Proteomes Focus on Sensitivity and Precision
 Relative Quantitation of TMT-Labeled Proteomes Focus on Sensitivity and Precision R. Viner 1, M. Scigelova 2, M. Zeller 2, M. Oppermann 2, T. Moehring 2 and V. Zabrouskov 1 1 Thermo Fisher Scientific,
Relative Quantitation of TMT-Labeled Proteomes Focus on Sensitivity and Precision R. Viner 1, M. Scigelova 2, M. Zeller 2, M. Oppermann 2, T. Moehring 2 and V. Zabrouskov 1 1 Thermo Fisher Scientific,
Modeling Mass Spectrometry-Based Protein Analysis
 Chapter 8 Jan Eriksson and David Fenyö Abstract The success of mass spectrometry based proteomics depends on efficient methods for data analysis. These methods require a detailed understanding of the information
Chapter 8 Jan Eriksson and David Fenyö Abstract The success of mass spectrometry based proteomics depends on efficient methods for data analysis. These methods require a detailed understanding of the information
SILAC and TMT. IDeA National Resource for Proteomics Workshop for Graduate Students and Post-docs Renny Lan 5/18/2017
 SILAC and TMT IDeA National Resource for Proteomics Workshop for Graduate Students and Post-docs Renny Lan 5/18/2017 UHPLC peak chosen at 26.47 min LC Mass at 571.36 chosen for MS/MS MS/MS MS This is a
SILAC and TMT IDeA National Resource for Proteomics Workshop for Graduate Students and Post-docs Renny Lan 5/18/2017 UHPLC peak chosen at 26.47 min LC Mass at 571.36 chosen for MS/MS MS/MS MS This is a
De novo Protein Sequencing by Combining Top-Down and Bottom-Up Tandem Mass Spectra. Xiaowen Liu
 De novo Protein Sequencing by Combining Top-Down and Bottom-Up Tandem Mass Spectra Xiaowen Liu Department of BioHealth Informatics, Department of Computer and Information Sciences, Indiana University-Purdue
De novo Protein Sequencing by Combining Top-Down and Bottom-Up Tandem Mass Spectra Xiaowen Liu Department of BioHealth Informatics, Department of Computer and Information Sciences, Indiana University-Purdue
All Ions MS/MS: Targeted Screening and Quantitation Using Agilent TOF and Q-TOF LC/MS Systems
 All Ions MS/MS: Targeted Screening and Quantitation Using Agilent TOF and Q-TOF LC/MS Systems Technical Overview Introduction All Ions MS/MS is a technique that is available for Agilent high resolution
All Ions MS/MS: Targeted Screening and Quantitation Using Agilent TOF and Q-TOF LC/MS Systems Technical Overview Introduction All Ions MS/MS is a technique that is available for Agilent high resolution
PRIDE Cluster: building the consensus of proteomics data
 Supplementary Materials PRIDE Cluster: building the consensus of proteomics data Johannes Griss, Joseph Michael Foster, Henning Hermjakob and Juan Antonio Vizcaíno EMBL-European Bioinformatics Institute,
Supplementary Materials PRIDE Cluster: building the consensus of proteomics data Johannes Griss, Joseph Michael Foster, Henning Hermjakob and Juan Antonio Vizcaíno EMBL-European Bioinformatics Institute,
QuasiNovo: Algorithms for De Novo Peptide Sequencing
 University of South Carolina Scholar Commons Theses and Dissertations 2013 QuasiNovo: Algorithms for De Novo Peptide Sequencing James Paul Cleveland University of South Carolina Follow this and additional
University of South Carolina Scholar Commons Theses and Dissertations 2013 QuasiNovo: Algorithms for De Novo Peptide Sequencing James Paul Cleveland University of South Carolina Follow this and additional
Q Exactive TM : A True Qual-Quan HR/AM Mass Spectrometer for Routine Proteomics Applications. Yi Zhang, Ph.D. ThermoFisher Scientific
 Q Exactive TM : A True Qual-Quan HR/AM Mass Spectrometer for Routine Proteomics Applications Yi Zhang, Ph.D. ThermoFisher Scientific Outline Introduction of Q Exactive Performance in Discovery Proteomics
Q Exactive TM : A True Qual-Quan HR/AM Mass Spectrometer for Routine Proteomics Applications Yi Zhang, Ph.D. ThermoFisher Scientific Outline Introduction of Q Exactive Performance in Discovery Proteomics
for the Novice Mass Spectrometry (^>, John Greaves and John Roboz yc**' CRC Press J Taylor & Francis Group Boca Raton London New York
 Mass Spectrometry for the Novice John Greaves and John Roboz (^>, yc**' CRC Press J Taylor & Francis Group Boca Raton London New York CRC Press is an imprint of the Taylor & Francis Croup, an informa business
Mass Spectrometry for the Novice John Greaves and John Roboz (^>, yc**' CRC Press J Taylor & Francis Group Boca Raton London New York CRC Press is an imprint of the Taylor & Francis Croup, an informa business
The Pitfalls of Peaklist Generation Software Performance on Database Searches
 Proceedings of the 56th ASMS Conference on Mass Spectrometry and Allied Topics, Denver, CO, June 1-5, 2008 The Pitfalls of Peaklist Generation Software Performance on Database Searches Aenoch J. Lynn,
Proceedings of the 56th ASMS Conference on Mass Spectrometry and Allied Topics, Denver, CO, June 1-5, 2008 The Pitfalls of Peaklist Generation Software Performance on Database Searches Aenoch J. Lynn,
Tandem Mass Spectrometry: Generating function, alignment and assembly
 Tandem Mass Spectrometry: Generating function, alignment and assembly With slides from Sangtae Kim and from Jones & Pevzner 2004 Determining reliability of identifications Can we use Target/Decoy to estimate
Tandem Mass Spectrometry: Generating function, alignment and assembly With slides from Sangtae Kim and from Jones & Pevzner 2004 Determining reliability of identifications Can we use Target/Decoy to estimate
BST 226 Statistical Methods for Bioinformatics David M. Rocke. January 22, 2014 BST 226 Statistical Methods for Bioinformatics 1
 BST 226 Statistical Methods for Bioinformatics David M. Rocke January 22, 2014 BST 226 Statistical Methods for Bioinformatics 1 Mass Spectrometry Mass spectrometry (mass spec, MS) comprises a set of instrumental
BST 226 Statistical Methods for Bioinformatics David M. Rocke January 22, 2014 BST 226 Statistical Methods for Bioinformatics 1 Mass Spectrometry Mass spectrometry (mass spec, MS) comprises a set of instrumental
Database Search Strategies for Proteomic Data Sets Generated by Electron Capture Dissociation Mass Spectrometry
 Database Search Strategies for Proteomic Data Sets Generated by Electron Capture Dissociation Mass Spectrometry Steve M. M. Sweet,,# Andrew W. Jones, Debbie L. Cunningham, John K. Heath, Andrew J. Creese,
Database Search Strategies for Proteomic Data Sets Generated by Electron Capture Dissociation Mass Spectrometry Steve M. M. Sweet,,# Andrew W. Jones, Debbie L. Cunningham, John K. Heath, Andrew J. Creese,
itraq and RNA-Seq analyses provide new insights of Dendrobium officinale seeds (Orchidaceae)
") Page S-1 Supporting Information (SI) itraq and RNA-Seq analyses provide new insights into regulation mechanism of symbiotic germination of Dendrobium officinale seeds (Orchidaceae) Juan Chen 1, Si Si Liu
Page S-1 Supporting Information (SI) itraq and RNA-Seq analyses provide new insights into regulation mechanism of symbiotic germination of Dendrobium officinale seeds (Orchidaceae) Juan Chen 1, Si Si Liu
Statistical analysis of isobaric-labeled mass spectrometry data
 Statistical analysis of isobaric-labeled mass spectrometry data Farhad Shakeri July 3, 2018 Core Unit for Bioinformatics Analyses Institute for Genomic Statistics and Bioinformatics University Hospital
Statistical analysis of isobaric-labeled mass spectrometry data Farhad Shakeri July 3, 2018 Core Unit for Bioinformatics Analyses Institute for Genomic Statistics and Bioinformatics University Hospital
MS Based Proteomics: Recent Case Studies Using Advanced Instrumentation
 MS Based Proteomics: Recent Case Studies Using Advanced Instrumentation Chris Adams, PH.D. Stanford University Mass Spectrometry http://mass-spec.stanford.edu/ For personal use only. Please do not reuse
MS Based Proteomics: Recent Case Studies Using Advanced Instrumentation Chris Adams, PH.D. Stanford University Mass Spectrometry http://mass-spec.stanford.edu/ For personal use only. Please do not reuse
Tutorial 1: Library Generation from DDA data
 Tutorial 1: Library Generation from DDA data 1. Introduction Before a targeted, peptide-centric DIA analysis can be performed, a spectral library containing peptide-query parameters needs to be generated.
Tutorial 1: Library Generation from DDA data 1. Introduction Before a targeted, peptide-centric DIA analysis can be performed, a spectral library containing peptide-query parameters needs to be generated.
Agilent All Ions MS/MS
 Agilent All Ions MS/MS Workflow Overview A Determine fragment ions for LC/MS Quant method B Develop final Quant method Develop LC/MS Qualitative Analysis method Process data with Find by Formula Build
Agilent All Ions MS/MS Workflow Overview A Determine fragment ions for LC/MS Quant method B Develop final Quant method Develop LC/MS Qualitative Analysis method Process data with Find by Formula Build
A new algorithm for the evaluation of shotgun peptide sequencing in proteomics: support vector machine classification of peptide MS/MS spectra
 A new algorithm for the evaluation of shotgun peptide sequencing in proteomics: support vector machine classification of peptide MS/MS spectra and SEQUEST scores D.C. Anderson*, Weiqun Li, Donald G. Payan,
A new algorithm for the evaluation of shotgun peptide sequencing in proteomics: support vector machine classification of peptide MS/MS spectra and SEQUEST scores D.C. Anderson*, Weiqun Li, Donald G. Payan,
Introduction to pepxmltab
 Introduction to pepxmltab Xiaojing Wang October 30, 2018 Contents 1 Introduction 1 2 Convert pepxml to a tabular format 1 3 PSMs Filtering 4 4 Session Information 5 1 Introduction Mass spectrometry (MS)-based
Introduction to pepxmltab Xiaojing Wang October 30, 2018 Contents 1 Introduction 1 2 Convert pepxml to a tabular format 1 3 PSMs Filtering 4 4 Session Information 5 1 Introduction Mass spectrometry (MS)-based
A graph-based filtering method for top-down mass spectral identification
 Yang and Zhu BMC Genomics 2018, 19(Suppl 7):666 https://doi.org/10.1186/s12864-018-5026-x METHODOLOGY Open Access A graph-based filtering method for top-down mass spectral identification Runmin Yang and
Yang and Zhu BMC Genomics 2018, 19(Suppl 7):666 https://doi.org/10.1186/s12864-018-5026-x METHODOLOGY Open Access A graph-based filtering method for top-down mass spectral identification Runmin Yang and
Performing Peptide Bioanalysis Using High Resolution Mass Spectrometry with Target Enhancement MRM Acquisition
 Performing Peptide Bioanalysis Using High Resolution Mass Spectrometry with Target Enhancement MRM Acquisition Yun Wang Alelyunas, Mark D. Wrona, and Nick Tomczyk Waters Corporation, Milford, MA, USA GOAL
Performing Peptide Bioanalysis Using High Resolution Mass Spectrometry with Target Enhancement MRM Acquisition Yun Wang Alelyunas, Mark D. Wrona, and Nick Tomczyk Waters Corporation, Milford, MA, USA GOAL
An Unsupervised, Model-Free, Machine-Learning Combiner for Peptide Identifications from Tandem Mass Spectra
 Clin Proteom (2009) 5:23 36 DOI 0.007/s204-009-9024-5 An Unsupervised, Model-Free, Machine-Learning Combiner for Peptide Identifications from Tandem Mass Spectra Nathan Edwards Xue Wu Chau-Wen Tseng Published
Clin Proteom (2009) 5:23 36 DOI 0.007/s204-009-9024-5 An Unsupervised, Model-Free, Machine-Learning Combiner for Peptide Identifications from Tandem Mass Spectra Nathan Edwards Xue Wu Chau-Wen Tseng Published
Integrated enzyme reactor and high resolving chromatography in sub-chip dimensions for. sensitive protein mass spectrometry
 Integrated enzyme reactor and high resolving chromatography in sub-chip dimensions for sensitive protein mass spectrometry Hanne Kolsrud Hustoft 1,*, Ole Kristian Brandtzaeg 1, Magnus Rogeberg 1,3, Dorna
Integrated enzyme reactor and high resolving chromatography in sub-chip dimensions for sensitive protein mass spectrometry Hanne Kolsrud Hustoft 1,*, Ole Kristian Brandtzaeg 1, Magnus Rogeberg 1,3, Dorna
WADA Technical Document TD2015IDCR
 MINIMUM CRITERIA FOR CHROMATOGRAPHIC-MASS SPECTROMETRIC CONFIRMATION OF THE IDENTITY OF ANALYTES FOR DOPING CONTROL PURPOSES. The ability of a method to identify an analyte is a function of the entire
MINIMUM CRITERIA FOR CHROMATOGRAPHIC-MASS SPECTROMETRIC CONFIRMATION OF THE IDENTITY OF ANALYTES FOR DOPING CONTROL PURPOSES. The ability of a method to identify an analyte is a function of the entire
Andromeda: A Peptide Search Engine Integrated into the MaxQuant Environment
 pubs.acs.org/jpr Andromeda: A Peptide Search Engine Integrated into the MaxQuant Environment J urgen Cox,*, Nadin Neuhauser, Annette Michalski, Richard A. Scheltema, Jesper V. Olsen, and Matthias Mann*,,
pubs.acs.org/jpr Andromeda: A Peptide Search Engine Integrated into the MaxQuant Environment J urgen Cox,*, Nadin Neuhauser, Annette Michalski, Richard A. Scheltema, Jesper V. Olsen, and Matthias Mann*,,
MSc Chemistry Analytical Sciences. Advances in Data Dependent and Data Independent Acquisition for data analysis in proteomic research
 MSc Chemistry Analytical Sciences Literature Thesis Advances in Data Dependent and Data Independent Acquisition for data analysis in proteomic research by Florian L. R. Lucas 11198877 September 2016 12
MSc Chemistry Analytical Sciences Literature Thesis Advances in Data Dependent and Data Independent Acquisition for data analysis in proteomic research by Florian L. R. Lucas 11198877 September 2016 12
Protein identification problem from a Bayesian pointofview
 Statistics and Its Interface Volume 5 (2012 21 37 Protein identification problem from a Bayesian pointofview YongFugaLi,RandyJ.Arnold,PredragRadivojac and Haixu Tang We present a generic Bayesian framework
Statistics and Its Interface Volume 5 (2012 21 37 Protein identification problem from a Bayesian pointofview YongFugaLi,RandyJ.Arnold,PredragRadivojac and Haixu Tang We present a generic Bayesian framework
STATISTICAL METHODS FOR THE ANALYSIS OF MASS SPECTROMETRY- BASED PROTEOMICS DATA. A Dissertation XUAN WANG
 STATISTICAL METHODS FOR THE ANALYSIS OF MASS SPECTROMETRY- BASED PROTEOMICS DATA A Dissertation by XUAN WANG Submitted to the Office of Graduate Studies of Texas A&M University in partial fulfillment of
STATISTICAL METHODS FOR THE ANALYSIS OF MASS SPECTROMETRY- BASED PROTEOMICS DATA A Dissertation by XUAN WANG Submitted to the Office of Graduate Studies of Texas A&M University in partial fulfillment of
Isobaric Labeling-Based Relative Quantification in Shotgun Proteomics
 This is an open access article published under an ACS AuthorChoice License, which permits copying and redistribution of the article or any adaptations for non-commercial purposes. pubs.acs.org/jpr Isobaric
This is an open access article published under an ACS AuthorChoice License, which permits copying and redistribution of the article or any adaptations for non-commercial purposes. pubs.acs.org/jpr Isobaric
Improved Validation of Peptide MS/MS Assignments. Using Spectral Intensity Prediction
 MCP Papers in Press. Published on October 2, 2006 as Manuscript M600320-MCP200 Improved Validation of Peptide MS/MS Assignments Using Spectral Intensity Prediction Shaojun Sun 1, Karen Meyer-Arendt 2,
MCP Papers in Press. Published on October 2, 2006 as Manuscript M600320-MCP200 Improved Validation of Peptide MS/MS Assignments Using Spectral Intensity Prediction Shaojun Sun 1, Karen Meyer-Arendt 2,
Designed for Accuracy. Innovation with Integrity. High resolution quantitative proteomics LC-MS
 Designed for Accuracy High resolution quantitative proteomics Innovation with Integrity LC-MS Setting New Standards in Accuracy The development of mass spectrometry based proteomics approaches has dramatically
Designed for Accuracy High resolution quantitative proteomics Innovation with Integrity LC-MS Setting New Standards in Accuracy The development of mass spectrometry based proteomics approaches has dramatically
MASS ANALYSER. Mass analysers - separate the ions according to their mass-to-charge ratio. sample. Vacuum pumps
 ION ANALYZERS MASS ANALYSER sample Vacuum pumps Mass analysers - separate the ions according to their mass-to-charge ratio MASS ANALYSER Separate the ions according to their mass-to-charge ratio in space
ION ANALYZERS MASS ANALYSER sample Vacuum pumps Mass analysers - separate the ions according to their mass-to-charge ratio MASS ANALYSER Separate the ions according to their mass-to-charge ratio in space
High-Throughput Protein Quantitation Using Multiple Reaction Monitoring
 High-Throughput Protein Quantitation Using Multiple Reaction Monitoring Application Note Authors Ning Tang, Christine Miller, Joe Roark, Norton Kitagawa and Keith Waddell Agilent Technologies, Inc. Santa
High-Throughput Protein Quantitation Using Multiple Reaction Monitoring Application Note Authors Ning Tang, Christine Miller, Joe Roark, Norton Kitagawa and Keith Waddell Agilent Technologies, Inc. Santa