Woods Hole brief primer on Multiple Sequence Alignment presented by Mark Holder
|
|
|
- Stephany Jennings
- 6 years ago
- Views:
Transcription
1 Woods Hole brief primer on Multiple Sequence Alignment presented by Mark Holder Many forms of sequence alignment are used in bioinformatics: Structural Alignment Local alignments Global, evolutionary alignment Inputs: unaligned sequences thought to be homologous over their full length we often ignore events like transpositions or inversions
2 The goal of MSA is to introduce gaps such that: residues in the same column are homologous all descended from a residue in the common ancestor, and all descendants of a residue are put in the same column.
3 G C A C T A A G T G T A C G T A C T A C G T A G T A C - T A C G T A - G T A - G T A G T A G T slide by Derrick Zwickl
4 C A G T A C G T A G T A C G T A C G T A G T A C A C A - A C G T G T G T G T A G T C A C G T A G T slide by Derrick Zwickl
5 Main points Accuracy of alignment is important. Pairwise alignment is tractable. Most MSA programs use progressive alignment: a series of pairwise operations. these algorithms are not guaranteed to return the optimal solution. the criteria used are not ideal from an evolutionary standpoint.
6 Main points (continued) Simultaneous inference of MSA and tree is the most appropriate choice, but is computationally demanding. See: Poisson Indel Process (Bouchard-Côté and Jordan, 2013), Bali-Phy, Handel, AliFritz, and POY software Many people cull ambiguously aligned regions.
7 human chimp gorilla orang KRSV KRV KSV KPRV How we align these sequences affects tree estimation. human KRSV human KRSV human KRSV chimp KR-V chimp K-RV chimp KR-V gorilla KS-V gorilla K-SV gorilla K-SV orang KPRV orang KPRV orang KPRV
8 How we align affects estimation of tests about processes of molecular evolution. Redelings (2014) studied jointly estimating alignments and ω. ω is the ratio of the rate of a non-synonymous substitution type to the rate of a synonymous substitution type (more from Joe Bielawski). A model with ω > 1 allows for positive, diversifying selection.
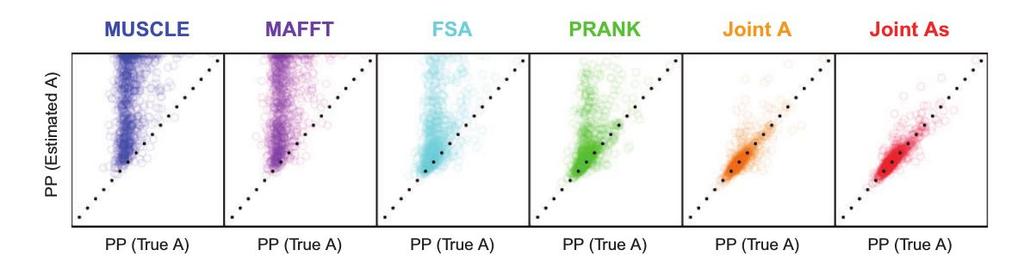
9
10 Expressing homology detection as a bioinformatics challenge The problem is recast as: reward matches (+ scores) penalize rare substitutions ( scores), penalize gaps ( scores), try to find an alignment that maximizes the total score
11 Pairwise alignment Gap penalties and a substitution matrix imply a score for any alignment. Pairwise alignment involves finding the alignment that maximizes this score. substitution matrices assign positive values to matches or substitutions between similar residues (for example Leucine Isoleucine). infrequent types of substitutions receive negative scores indels are rare, so gaps are heavily penalized (negative scores).
12 BLOSUM 62 Substitution matrix A R N D C Q E G H I L K M F P S T W Y V A 4 R -1 5 N D C Q E G H I L K M F P S T W Y V A R N D C Q E G H I L K M F P S T W Y V
13 Scoring an alignment with the BLOSUM 62 matrix Pongo V D E V G G E L G R L F V V P T Q Gorilla V E V A G D L G R L L I V Y P S R Score The score for the alignment is D ij = k d (k) ij If i indicates Pongo and j indicates Gorilla. (k) is just an index for the column. D ij = 12
14 Scoring an alignment with gaps If we were to use a gap penalty of -8: Pongo V D E V G G E L G R L - F V V P T Q Gorilla V - E V A G D L G R L L I V Y P S R Score By introducing gaps we have improved the score: D ij = 40
15 Gap Penalties Penalizing gaps more heavily than substitutions avoids alignments like this: Pongo Gorilla VDEVGGE-LGRLFVVPTQ VDEVGG-DLGRLFVVPTQ
16 Affine gap penalties are often used to accommodate multi-site indels: where: GP is the gap penalty. GP = GO + (l)ge GO is the gap-opening penalty GE is the gap-extension penalty l is the length of the gap
17 Finding an optimal alignment Gorilla V E V A G D L G R L L I V Y P S R V Pongo D E V G G E L G R L F V V P T Q
18 pongo V D E V G G E L G R L F V V P T Q gorilla V E V A G D L G R L L I V Y P S R score Gorilla V E V A G D L G R L L I V Y P S R V Pongo D E V G G E L G R L F V V P T Q
19 Pongo V D E V G G E L G R L - F V V P T Q Gorilla V - E V A G D L G R L L I V Y P S R Score Gorilla V E V A G D L G R L L I V Y P S R V Pongo D E V G G E L G R L F V V P T Q
20 Needleman-Wunsch algorithm (paraphrased) Work from the top left (beginning of both sequences) For each cell store the highest score possible for that cell and a back pointer to tell point to the previous step in the best path When you reach the lower right corner, you know the optimal score and the back pointers tell you the alignment. The highest score calculation at each cell only depends on the cell s 3 possible previous neighbors. If one sequence is length M 1, and the other is length M 2, then Needleman-Wunsch only takes O(M 1 M 2 ) calculations. But there are a much larger number of possible alignments (# alignments grows exponentially).
21 V E V A G D V D E V G G 0
22 Using a gap penalty of -5 V E V A G D V D E V G G
23 V D E V G G V -5 4 E -10 V A G D
24 V D E V G G V E V -15 A G D
25 V D E V G G V E V A -20 G D
26 Pairwise alignment is a beautiful topic in bioinformatics Clever programming tricks let us find the bestscoring pairwise alignment quickly The additive scoring system: can incorporate biological knowledge (via empirically-based substitution matrices) can almost be justified in terms of powerful statistical methodology (maximum likelihood).
27 Pairwise alignment costs Paul Lewis will explain likelihood tomorrow, Additive costs can be justified as approximations to the log of likelihoods if: we can identify the events that must have occurred in generate the data, and we can assign (relative) probabilities based on whether these events are rare or common.
28 Pongo V D E V G G E L G R L - F V V P T Q Gorilla V - E V A G D L G R L L I V Y P S R Score Pongo Gorilla
29 Pongo V D E V G G E L G R L - F V V P T Q Gorilla V - E V A G D L G R L L I V Y P S R Score Pongo Gorilla V V P(pos. 1) = P(V V )
30 Pongo V D E V G G E L G R L - F V V P T Q Gorilla V - E V A G D L G R L L I V Y P S R Score Pongo Gorilla V V D - P(pos. 1 2) = P(V V ) P(D )
31 Pongo V D E V G G E L G R L - F V V P T Q Gorilla V - E V A G D L G R L L I V Y P S R Score Pongo Gorilla V V D - E E P(pos. 1 3) = P(V V ) P(D ) P(E E)
32 Pongo V D E V G G E L G R L - F V V P T Q Gorilla V - E V A G D L G R L L I V Y P S R Score Pongo Gorilla V V D - E E ln P(pos. 1 3) = ln P(V V ) + ln P(D ) + ln P(E E)
33 side note on probability models We usually project from: models of instantaneous rates of events the microscopic scale (sensu Rivas & Eddy) to: probabilities of descendant sequences the macroscopic scale Rivas and Eddy [preprint] have recently gone backwards
34 Rivas and Eddy L1 vs TKF91

35 Fig 1.3 from a great review by Lunter et al. 2005
36 Rivas and Eddy models Their models are natural to implement with dynamic programming linear reversible LR is a single-residue indel model similar to TKF91, Affine Insert Fragment AIF is an affine indel model similar to TKF92
37 Pairwise alignment summary The sum of the substitution and gap cost can serve as a proxy for the log-likelihood under a reasonable model (the LR or AIF model of Rivas and Eddy, preprint). Dynamic programming can let us find the alignment that has the highest likelihood.
38 from (Rausch and Reinert, 2011)
39 Multiple sequence alignment is an ugly topic in bioinformatics Clever programming tricks help, but we still have to rely on heuristics approaches that provide good solutions, but are not guaranteed to find the best solution. The additive scoring system suffers from the fact that we do not observe ancestral sequences.
40 U is the set of unaligned sequences T is the genealogy tree that describes the ancestry of the sequences H is an indel history (specification of where all inserstions and deletions in the history of U occur on the tree T ). A is an alignment of the sequences U We might want: P(T U) or P(A U) or P(T, A U). BaliPhy approximates these quantities, but it is tough to do for large datasets. Ĥ which maximizes P(H U, T ). ProtPal (Westesson et al., 2012) approximates this (but we have to know T )
41 Sum of Pairs scoring Most MSA programs optimize a fairly strange score: SP = w ij d(a i, A j ) i j where A i is the alignment pruned down to just sequence i, and w ij is a weight for the comparison between sequences i and j. d(a i, A j ) can be: a measure of the distance from A i to A j or a measure of the consistency of the alignment A with a pairwise alignment of i to j. We usually cannot guarantee that we have found the alignment that optimizes the sum of pairs score.
42 Aligning more than two sequences B D A C E
43 Progressive alignment An approximate method for producing multiple sequence alignments using a guide tree. Perform pairwise alignments to produce a distance matrix Produce a guide tree from the distances Use the guide tree to specify the ordering used for aligning sequences, closest to furthest. Feng and Doolittle 1987 and Higgins and Sharp, 1988
44 A PEEKSAVTALWGKVNVDEVGG B GEEKAAVLALWDKVNEEEVGG C PADKTNVKAAWGKVGAHAGEYGA D AADKTNVKAAWSKVGGHAGEYGA E EHEWQLVLHVWAKVEADVAGHGQ pairwise alignment A - B.17 - C D E tree inference A PEEKSAVTALWGKVNVDEVGG B GEEKAAVLALWDKVNEEEVGG C PADKTNVKAAWGKVGAHAGEYGA + D AADKTNVKAAWSKVGGHAGEYGA E EHEWQLVLHVWAKVEADVAGHGQ B D A C E alignment stage A PEEKSAVTALWGKVN--VDEVGG B GEEKAAVLALWDKVN--EEEVGG C PADKTNVKAAWGKVGAHAGEYGA D AADKTNVKAAWSKVGGHAGEYGA E EHEWQLVLHVWAKVEADVAGHGQ
45 Alignment stage of progressive alignments Sequences of clades become grouped as the algorithm descends the tree. Alignment at each step involves Sequence-Sequence, Sequence-Group, or Group-Group
46 Aligning multiple sequences B D A C E Seq-Seq Seq-Seq Seq-Group 0.09 Group-Group
47 Group-to-Group alignment V E V A G D L G R L L I Y P S R A V E D E V G G L G M R L F V P T Q L D D E V - G A G V R L F V P T Q V E I A G D L - - L L L Y P T R V V E V A G E L - - L L L Y P T K I
48 Group-to-group alignments Adding a gap to a group means that every member of that group gets a gap at that position. from Edgar (2004)
49 Group-to-group alignment Usually the scores for each edge in the Needleman-Wunsch graph are calculated using a sum of pairs scoring system. Many tools 1 uses weights assigned to each sequence in a group to down-weight closely related sequences so that they are not overrepresented - this is a weighted sum-of-pair scoring system. 1 e.g. Clustal and MAFFT
50 Greedy choices leading to failure to find the best alignment Consider the scoring scheme: match = 0 mismatch = -3 gap = -7 Guide Tree: Sp1 Sp2 Sp3 Sequences: Sp2 GCCGTAG Sp1 GACCGTG Sp3 GACCGTAG
51 Greedy choices leading to failure to find the best alignment match = 0 mismatch = -3 gap = -7 ungapped1vs2 Sp1 G A C C G T G Sp2 G C C G T A G Score Total= -12 would be preferred over gapped1vs2: Sp1 G A C C G T - G Sp2 G - C C G T A G Score Total= -14
52 Adding a Sp3 to ungapped1vs2: Sp1 G - A C C G T G Sp2 G - C C G T A G Sp3 G A C C G T A G This implies 1 indel, and 4 substitutions. Score = -19 If we had been able to use gapped1vs2 then we could have: Sp1 G A C C G T - G Sp2 G - C C G T A G Sp3 G A C C G T A G score = -14 = sort of...
53 Score = -19 if we count events (but sum of pairs score would differ)
54 Score = -14 if we count events (but sum of pairs score would differ)
55 Polishing (aka iterative alignment can correct some errors caused by greedy heuristics) 1. break the alignment into 2 groups of sequences (often by breaking an edge in the merge tree). 2. realign those 2 groups to each other 3. keep the realignment if it improves the score Opal also uses random 3-group polishing.
56 Weighted sum of pairs scoring system Assigns a score for a group-to-group by averaging (or summing) the scores all of the implied pairwise alignments. Frequently w ij = w i w j Group 1 Group 2 Seq weight AA taxon A 0.3 V taxon C 0.24 A taxon E 0.19 I Seq weight AA taxon B 0.15 V taxon D 0.25 M D G1,G2 = i j w iw j d ij n i n j
57 Group 1 Group 2 Seq weight AA taxon A 0.3 V taxon C 0.24 A taxon E 0.19 I Seq weight AA taxon B 0.15 V taxon D 0.25 M D G1,G2 = i j w iw j d ij n i n j = 1 6 [d(v, V )w Aw B + d(v, M)w A w D + d(a, V )w C w B d(a, M)w C w D + d(i, V )w E w B + d(i, M)w E w D ] = 1 ( ) =
58 Sum-of-pairs is an odd scoring system G A A A A L
59 Imperfect scoring system. Consider one position in a group-to-group alignment: (A,A,G) (A,A,L) (A,A,G) (A,A,L) The sum-of-pairs score for aligning would be: 4 9 (A A) (A L) (G A) + 1 (G L) 9
60 But in the context of the tree we might be pretty certain of an A A event G A A A A L A A Note: weighted sum-of-pairs would help reflect the effect of ancestry better (but still not perfectly; sum-of-pairs techniques are simply not very sophisticated forms of ancestral sequence reconstruction).
61 Consistency based alignment In our sum of pair scoring, we could just check how often each pair of sequences (one from G h and one from G v ) display the same alignment that they did in pairwise alignment. T-Coffee (Notredame et al., 2000) introduced the idea of performing group-to-group alignments during progressive alignment using this sense of consistency.
62 Indirect consistency arguments In the pairwise alignments, if h 10 g 12 and p 17 g 12 then h 10 should align with p 17
63 Probabilistic measures of consistency The simplest assessment of the consistency of an MSA to pairwise alignments uses just the optimal pairwise alignment of each pair. Opal (Wheeler and Kececioglu, 2007) uses some suboptimal alignments. Do et al. (2005) made an important advance by proposing the use of the probability that two residues are aligned during consistency-based alignment (ProbCons). The same sort of dynamic programming traversal of the alignment grid can give us a probability that 2 residues are aligned.
64 Sequence annealing AMAP (Schwartz and Pachter, 2007) and FSA (Bradley et al., 2009) use a sequence annealing approach: start with trivial alignments (all residues opposite gaps), anchor regions by aligning long matches, merge columns as long as the score keeps improving. Fig. 3 of (Bradley et al., 2009)
65 Progressive alignment Uses a guide tree to change the MSA problem into a series of pairwise alignment problems; May not return the alignment with the best weighted sumof-pairs scores. Early alignment decisions get locked in Most aligners try to polish the alignment, but we cannot guarantee that we have found the optimal alignment; Reconstruction of ancestral sequences is usually done in a quick-and-dirty, implicit fashion or is not done at all;
66 PRANK Löytynoja and Goldman (2005) showed most progressive alignment techniques were particularly prone to compression because of poor ancestral reconstruction:

67 PRANK Flagging inserted residues allows PRANK to effectively skip over these positions in the ancestor, producing more phylogenetically-sensible alignments:
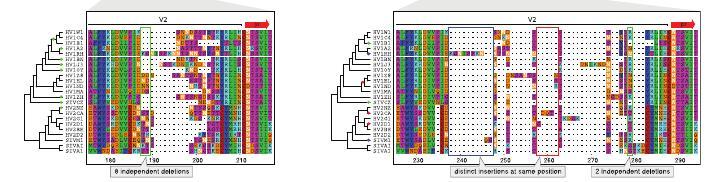
68

69 ProtPal is similar to PRANK, but is retains a set of inferred ancestral seqs. Fig. 5 of Westesson et al. (2012)
70 Impact of the guide tree Using a guide tree can bias subsequent tree inference toward the guide tree. This can also cause inflated support. Ironically, this effect may be more of a problem for a more evolutionarily-sensible aligner such as PRANK or ProtPal!
71 Opinion Opinion Dealing with alignment ambiguity TRENDS in Eco (a) X Y X Z Y Z (a) X (b) (b) Outgroup Taxon A Taxon B Taxon C Taxon D Taxon E (d) Outgroup T A G A Outgroup G C A C T T C A G A G C Outgroup A C T C A T G A G A Taxon A T A G A Taxon G C A AC T T C A G A G C Taxon A C T AC A T G A G A TRENDS in Ecology Taxon & Evolution B T A Vol.16 G T No.12 Taxon G A A Decem BG C T C ber A G 2001 T G A Taxon A G C BC A T G A G T Taxon C T A G T Taxon G A A CG C T C A G T G A Taxon A G C C A T G A G T Taxon D T A G Taxon A G C D T C A G A G Taxon C DC A T G A G A Taxon E T A G Taxon A G C E T C A G A G Taxon C EC A T G A G A data sets because strong phylogenet X Y Z X Y X Z Y Z (c) (c) (d) required to generate incongruence; A B latter criteria might lead to choosing containg the least phylogenetic info T A G A G C Outgroup A C T C A T G A G A Outgroup G C A C T T C A G A G C A C T C A G T A G A G C Taxon A C T A C A T GA G A Taxon G C A AC T T C A G A G C A C T C A G T A G T G A Taxon A G C B C A T G A G T Taxon G A A BG C T C A G T G A A G C C A G T A G T G A Taxon A G C C A T GA G T Taxon G A A CG C T C A G T G A A G C C A G T A G A G C Taxon D C A T GA G - Taxon - Elision - A DG C T C A G A G C C A G T A G A G C Taxon E C A T GA G - Taxon - - A EG C T C A G A G C C A G A B C from M. S. Y. Lee, TREE, 2001 DE In the elision method, a range of pla D alignments is generated as detailed instead of being analysed separately (e) X (e) Y X Z X Y Y Z combined ( concatenated ) into 0 a 1 sin 2 1,1 4, 6, 8, 9
72 682 Opinion Dealing with alignment ambiguity - deletion TRENDS in Eco (a) X Y Z (b) X Outgroup T A G A G C A C T C A G Taxon A T A G A G C A C T C A G Taxon B T A G T G A A G C C A G Taxon C T A G T G A A G C C A G Taxon D T A G A G C C A G Taxon E T A G A G C C A G Outgroup Taxon A Taxon B Taxon C Taxon D Taxon E T A G A T A G A T A G T T A G T T A G A T A G A X Z (c) Outgroup T A G Outgroup C A G Taxon A T A G Taxon C A GA Taxon B T A G Taxon C A G B Taxon C T A G Taxon C A GC Taxon D T A G Taxon C A GD Taxon E T A G Taxon C A GE X Y X Z Y Z (d) T A Outgroup G A G C A T C A T G C A G C A C T C A G T A Taxon G A G AC A T C A T G C A G C A C T C A G T A Taxon G T G BA A T G A C G C T A G A A G C C A G T A Taxon G T G CA A T G A C G C T A G A A G C C A G T A Taxon G - - D- A T G A C G C? A G?? C A G T A Taxon G - - E- A T G A C G C? A G?? C A G A B DE D from M. S. Y. Lee, TREE, 2001 (e) X Y Z X Y
73 Filtering GUIDANCE score (Penn et al., 2010) which reflects sensitivity of the alignment to poorly supported parts of the guide tree; and Head-or-Tails algorithm (Landan and Graur, 2007) which compares the alignment of the sequences to the alignment that you would get from reversed forms of the sequences. SOAP (Löytynoja and Milinkovitch, 2001) looks for parts of the alignment that are sensitive to alignment parameters trimal (Capella-Gutiérrez et al., 2009) uses column properties and consistency Other tools, such as Gblocks, examine properties of the columns in the matrix.
74 Simultaneous tree inference and alignment Ideally we would jointly estimate with uncertainty Allows for application of statistical models to improve inference and assessments of reliability Just now becoming feasible: BAliPhy (Redelings and Suchard, 2005) See also: POY (Wheeler, Gladstein, Laet, 2002), Handel (Holmes and Bruno, 2001), and BEAST(Lunter et al., 2005; Drummond and Rambaut, 2007). SATé (Liu et al., 2009) and PASTA (Mirarab et al., 2014) are iterative (back and forth between tree and alignment estimation)
75 SATé repeats the following steps until termination A B C D Decompose based on input tree A C B D Align subproblems A C B D Estimate ML tree on merged alignment ABCD Merge subproblems
76 SATé simulation results
77 Conclusions Evolutionary multiple sequence alignment is still a very active area of research. We are hampered by: lacking a good criterion to optimize (when T is unknown). being forced to use rough heuristics to optimize the sum of pairs scores. Filtering throws away information, but may be helpful Most phylogenetic inference tools ignore information from the indel process (but see Rivas et al. (2008); Rivas and Eddy (2013) and talk to DZ)
78 Recommendations See if BaliPhy can run on your data! If not, try FSA Look at your alignments Be aware that the standard aligners only consider substitutions and indels. If your data shows inversions, duplications,... there can be serious artifacts. if you have protein-coding sequences, use amino acid or codon-aware aligners if you have sequences from an RNA with important secondary structure, check out Infernal

79 Fig. 2 from Katoh & Standley 2013
80 References Bouchard-Côté, A. and Jordan, M. I. (2013). Evolutionary inference via the poisson indel process. Proceedings of the National Academy of Sciences, 110(4): Bradley, R. K., Roberts, A., Smoot, M., Juvekar, S., Do, J., Dewey, C., Holmes, I., and Pachter, L. (2009). Fast Statistical Alignment. PLoS Computational Biology, 5(5). Capella-Gutiérrez, S., Sillla-Martínex, J. M., and Gabaldón, T. (2009). trimal: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. Do, C. B., Mahabhashyam, M. S., Brudno, M., and Batzoglou, S. (2005). Probcons: Probabilistic consistency-based multiple sequence alignment. Genome research, 15(2):
81 Drummond, A. J. and Rambaut, A. (2007). Beast: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol., 7:214. Edgar, R. (2004). MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics, 5:113. Holmes, I. and Bruno, W. J. (2001). Evolutionary HMMs: a Bayesian approach to multiple alignment. Bioinformatics (Oxford, England), 17(9): Landan, G. and Graur, D. (2007). Heads or tails: a simple reliability check for multiple sequence alignments. Molecular Biology and Evolution, 24(6): Liu, K., Raghavan, S., Nelesen, S., Linder, C., and Warnow, T. (2009). Rapid and accurate large-scale coestimation of sequence alignments and phylogenetic trees. Science, 324(5934): Löytynoja, A. and Goldman, N. (2005). An algorithm for progressive multiple alignment of sequences with insertions. Proceedings of the National
82 Academy of Sciences of the United States of America, 102(30): Löytynoja, A. and Milinkovitch, M. C. (2001). Soap, cleaning multiple alignments from unstable blocks. Bioinformatics, 17(6): Lunter, G., Miklós, I., Drummond, A., Jensen, J. L., and Hein, J. (2005). Bayesian coestimation of phylogeny and sequence alignment. BMC Bioinformatics, 6:83. Mirarab, S., Nguyen, N., and Warnow, T. (2014). Pasta: ultra-large multiple sequence alignment. In Research in Computational Molecular Biology, pages Springer. Notredame, C., Higgins, D. G., and Heringa, J. (2000). T-coffee: a novel method for fast and accurate multiple sequence alignment. Journal of Molecular Biology, 302(1): Penn, O., Privman, E., Landan, G., Graur, D., and Pupko, T. (2010). An alignment confidence score capturing robustness to guide tree uncertainty. Molecular Biology and Evolution, 27(8):
83 Rausch, T. and Reinert, K. (2011). Problem Solving Handbook in Computational Biology and Bioinformatics, chapter Practical Multiple Sequence Alignment, pages Springer. Redelings, B. (2014). Erasing errors due to alignment ambiguity when estimating positive selection. Molecular Biology and Evolution. Redelings, B. and Suchard, M. (2005). Joint bayesian estimation of alignment and phylogeny. Systematic Biology, 54(3): Rivas, E. and Eddy, S. (2013). A probabilistic evolutionary model compatible with standard affine gap cost sequence alignment. In Review. Rivas, E., Eddy, S. R., and Haussler, D. (2008). Probabilistic phylogenetic inference with insertions and deletions. PLoS Computational Biology, 4(9):e Schwartz, A. S. and Pachter, L. (2007). Multiple alignment by sequence annealing. Bioinformatics, 23(2):e24 e29.
84 Westesson, O., Lunter, G., Paten, B., and Holmes, I. (2012). Accurate reconstruction of insertion-deletion histories by statistical phylogenetics. PLoS ONE, 7(4):e Wheeler, T. J. and Kececioglu, J. D. (2007). Multiple alignment by aligning alignments. Bioinformatics, 23(13):i559 i568.
85 Some of the tricks clustal uses to produce better alignments Chooses substitution matrix (PAM or BLOSUM series for amino acids) based on sequence similarity AA residues in a neighborhood affect gap opening penalty (easier to have gaps in hydrophilic loops) Gap penalties are raised if a column has no gaps, but there are gaps nearby. Low scoring alignments may be postponed until a later stage.
86 Terminal gaps Using normal gap costs causes problems if one sequence is missing the starting or ending residues: instead of : Pongo Gorilla Pongo Gorilla VDEFKLIVEGELGRLFVVPTQ VD GELGRLFVVPTQ VDEFKLIVEGELGRLFVVPTQ VDGELGRLFVVPTQ (Methods that utilize local alignment information are more appropriate in these cases).
87 Using free terminal gaps to avoid this problem, but you have to watch out for: Pongo Gorilla VDEPFRFKLTNRGTSHIILVAPR VDEPNRGTSHIILVAPR
Thanks to Paul Lewis, Jeff Thorne, and Joe Felsenstein for the use of slides
 hanks to Paul Lewis, Jeff horne, and Joe Felsenstein for the use of slides Hennigian logic reconstructs the tree if we know polarity of characters and there is no homoplasy UPM infers a tree from a distance
hanks to Paul Lewis, Jeff horne, and Joe Felsenstein for the use of slides Hennigian logic reconstructs the tree if we know polarity of characters and there is no homoplasy UPM infers a tree from a distance
InDel 3-5. InDel 8-9. InDel 3-5. InDel 8-9. InDel InDel 8-9
 Lecture 5 Alignment I. Introduction. For sequence data, the process of generating an alignment establishes positional homologies; that is, alignment provides the identification of homologous phylogenetic
Lecture 5 Alignment I. Introduction. For sequence data, the process of generating an alignment establishes positional homologies; that is, alignment provides the identification of homologous phylogenetic
The statistical and informatics challenges posed by ascertainment biases in phylogenetic data collection
 The statistical and informatics challenges posed by ascertainment biases in phylogenetic data collection Mark T. Holder and Jordan M. Koch Department of Ecology and Evolutionary Biology, University of
The statistical and informatics challenges posed by ascertainment biases in phylogenetic data collection Mark T. Holder and Jordan M. Koch Department of Ecology and Evolutionary Biology, University of
Algorithms in Bioinformatics FOUR Pairwise Sequence Alignment. Pairwise Sequence Alignment. Convention: DNA Sequences 5. Sequence Alignment
 Algorithms in Bioinformatics FOUR Sami Khuri Department of Computer Science San José State University Pairwise Sequence Alignment Homology Similarity Global string alignment Local string alignment Dot
Algorithms in Bioinformatics FOUR Sami Khuri Department of Computer Science San José State University Pairwise Sequence Alignment Homology Similarity Global string alignment Local string alignment Dot
Using Ensembles of Hidden Markov Models for Grand Challenges in Bioinformatics
 Using Ensembles of Hidden Markov Models for Grand Challenges in Bioinformatics Tandy Warnow Founder Professor of Engineering The University of Illinois at Urbana-Champaign http://tandy.cs.illinois.edu
Using Ensembles of Hidden Markov Models for Grand Challenges in Bioinformatics Tandy Warnow Founder Professor of Engineering The University of Illinois at Urbana-Champaign http://tandy.cs.illinois.edu
Mul$ple Sequence Alignment Methods. Tandy Warnow Departments of Bioengineering and Computer Science h?p://tandy.cs.illinois.edu
 Mul$ple Sequence Alignment Methods Tandy Warnow Departments of Bioengineering and Computer Science h?p://tandy.cs.illinois.edu Species Tree Orangutan Gorilla Chimpanzee Human From the Tree of the Life
Mul$ple Sequence Alignment Methods Tandy Warnow Departments of Bioengineering and Computer Science h?p://tandy.cs.illinois.edu Species Tree Orangutan Gorilla Chimpanzee Human From the Tree of the Life
CONCEPT OF SEQUENCE COMPARISON. Natapol Pornputtapong 18 January 2018
 CONCEPT OF SEQUENCE COMPARISON Natapol Pornputtapong 18 January 2018 SEQUENCE ANALYSIS - A ROSETTA STONE OF LIFE Sequence analysis is the process of subjecting a DNA, RNA or peptide sequence to any of
CONCEPT OF SEQUENCE COMPARISON Natapol Pornputtapong 18 January 2018 SEQUENCE ANALYSIS - A ROSETTA STONE OF LIFE Sequence analysis is the process of subjecting a DNA, RNA or peptide sequence to any of
Sara C. Madeira. Universidade da Beira Interior. (Thanks to Ana Teresa Freitas, IST for useful resources on this subject)
") Bioinformática Sequence Alignment Pairwise Sequence Alignment Universidade da Beira Interior (Thanks to Ana Teresa Freitas, IST for useful resources on this subject) 1 16/3/29 & 23/3/29 27/4/29 Outline
Bioinformática Sequence Alignment Pairwise Sequence Alignment Universidade da Beira Interior (Thanks to Ana Teresa Freitas, IST for useful resources on this subject) 1 16/3/29 & 23/3/29 27/4/29 Outline
CS 581 Algorithmic Computational Genomics. Tandy Warnow University of Illinois at Urbana-Champaign
 CS 581 Algorithmic Computational Genomics Tandy Warnow University of Illinois at Urbana-Champaign Today Explain the course Introduce some of the research in this area Describe some open problems Talk about
CS 581 Algorithmic Computational Genomics Tandy Warnow University of Illinois at Urbana-Champaign Today Explain the course Introduce some of the research in this area Describe some open problems Talk about
THEORY. Based on sequence Length According to the length of sequence being compared it is of following two types
 Exp 11- THEORY Sequence Alignment is a process of aligning two sequences to achieve maximum levels of identity between them. This help to derive functional, structural and evolutionary relationships between
Exp 11- THEORY Sequence Alignment is a process of aligning two sequences to achieve maximum levels of identity between them. This help to derive functional, structural and evolutionary relationships between
Sequence Bioinformatics. Multiple Sequence Alignment Waqas Nasir
 Sequence Bioinformatics Multiple Sequence Alignment Waqas Nasir 2010-11-12 Multiple Sequence Alignment One amino acid plays coy; a pair of homologous sequences whisper; many aligned sequences shout out
Sequence Bioinformatics Multiple Sequence Alignment Waqas Nasir 2010-11-12 Multiple Sequence Alignment One amino acid plays coy; a pair of homologous sequences whisper; many aligned sequences shout out
Moreover, the circular logic
 Moreover, the circular logic How do we know what is the right distance without a good alignment? And how do we construct a good alignment without knowing what substitutions were made previously? ATGCGT--GCAAGT
Moreover, the circular logic How do we know what is the right distance without a good alignment? And how do we construct a good alignment without knowing what substitutions were made previously? ATGCGT--GCAAGT
An Introduction to Sequence Similarity ( Homology ) Searching
 Searching") An Introduction to Sequence Similarity ( Homology ) Searching Gary D. Stormo 1 UNIT 3.1 1 Washington University, School of Medicine, St. Louis, Missouri ABSTRACT Homologous sequences usually have the same,
An Introduction to Sequence Similarity ( Homology ) Searching Gary D. Stormo 1 UNIT 3.1 1 Washington University, School of Medicine, St. Louis, Missouri ABSTRACT Homologous sequences usually have the same,
3. SEQUENCE ANALYSIS BIOINFORMATICS COURSE MTAT
 3. SEQUENCE ANALYSIS BIOINFORMATICS COURSE MTAT.03.239 25.09.2012 SEQUENCE ANALYSIS IS IMPORTANT FOR... Prediction of function Gene finding the process of identifying the regions of genomic DNA that encode
3. SEQUENCE ANALYSIS BIOINFORMATICS COURSE MTAT.03.239 25.09.2012 SEQUENCE ANALYSIS IS IMPORTANT FOR... Prediction of function Gene finding the process of identifying the regions of genomic DNA that encode
5. MULTIPLE SEQUENCE ALIGNMENT BIOINFORMATICS COURSE MTAT
 5. MULTIPLE SEQUENCE ALIGNMENT BIOINFORMATICS COURSE MTAT.03.239 03.10.2012 ALIGNMENT Alignment is the task of locating equivalent regions of two or more sequences to maximize their similarity. Homology:
5. MULTIPLE SEQUENCE ALIGNMENT BIOINFORMATICS COURSE MTAT.03.239 03.10.2012 ALIGNMENT Alignment is the task of locating equivalent regions of two or more sequences to maximize their similarity. Homology:
Phylogenetic inference
 Phylogenetic inference Bas E. Dutilh Systems Biology: Bioinformatic Data Analysis Utrecht University, March 7 th 016 After this lecture, you can discuss (dis-) advantages of different information types
Phylogenetic inference Bas E. Dutilh Systems Biology: Bioinformatic Data Analysis Utrecht University, March 7 th 016 After this lecture, you can discuss (dis-) advantages of different information types
Erasing Errors Due to Alignment Ambiguity When Estimating Positive Selection
 Article (Methods) Erasing Errors Due to Alignment Ambiguity When Estimating Positive Selection Authors Benjamin Redelings 1,2 1 Biology Department, Duke University 2 National Evolutionary Synthesis Center
Article (Methods) Erasing Errors Due to Alignment Ambiguity When Estimating Positive Selection Authors Benjamin Redelings 1,2 1 Biology Department, Duke University 2 National Evolutionary Synthesis Center
Tools and Algorithms in Bioinformatics
 Tools and Algorithms in Bioinformatics GCBA815, Fall 2015 Week-4 BLAST Algorithm Continued Multiple Sequence Alignment Babu Guda, Ph.D. Department of Genetics, Cell Biology & Anatomy Bioinformatics and
Tools and Algorithms in Bioinformatics GCBA815, Fall 2015 Week-4 BLAST Algorithm Continued Multiple Sequence Alignment Babu Guda, Ph.D. Department of Genetics, Cell Biology & Anatomy Bioinformatics and
Introduction to Phylogenetics Workshop on Molecular Evolution 2018 Marine Biological Lab, Woods Hole, MA. USA. Mark T. Holder University of Kansas
 Introduction to Phylogenetics Workshop on Molecular Evolution 2018 Marine Biological Lab, Woods Hole, MA. USA Mark T. Holder University of Kansas Outline 1. phylogenetics is crucial for comparative biology
Introduction to Phylogenetics Workshop on Molecular Evolution 2018 Marine Biological Lab, Woods Hole, MA. USA Mark T. Holder University of Kansas Outline 1. phylogenetics is crucial for comparative biology
Effects of Gap Open and Gap Extension Penalties
 Brigham Young University BYU ScholarsArchive All Faculty Publications 200-10-01 Effects of Gap Open and Gap Extension Penalties Hyrum Carroll hyrumcarroll@gmail.com Mark J. Clement clement@cs.byu.edu See
Brigham Young University BYU ScholarsArchive All Faculty Publications 200-10-01 Effects of Gap Open and Gap Extension Penalties Hyrum Carroll hyrumcarroll@gmail.com Mark J. Clement clement@cs.byu.edu See
SEPP and TIPP for metagenomic analysis. Tandy Warnow Department of Computer Science University of Texas
 SEPP and TIPP for metagenomic analysis Tandy Warnow Department of Computer Science University of Texas Phylogeny (evolutionary tree) Orangutan From the Tree of the Life Website, University of Arizona Gorilla
SEPP and TIPP for metagenomic analysis Tandy Warnow Department of Computer Science University of Texas Phylogeny (evolutionary tree) Orangutan From the Tree of the Life Website, University of Arizona Gorilla
Quantifying sequence similarity
 Quantifying sequence similarity Bas E. Dutilh Systems Biology: Bioinformatic Data Analysis Utrecht University, February 16 th 2016 After this lecture, you can define homology, similarity, and identity
Quantifying sequence similarity Bas E. Dutilh Systems Biology: Bioinformatic Data Analysis Utrecht University, February 16 th 2016 After this lecture, you can define homology, similarity, and identity
Probalign: Multiple sequence alignment using partition function posterior probabilities
 Sequence Analysis Probalign: Multiple sequence alignment using partition function posterior probabilities Usman Roshan 1* and Dennis R. Livesay 2 1 Department of Computer Science, New Jersey Institute
Sequence Analysis Probalign: Multiple sequence alignment using partition function posterior probabilities Usman Roshan 1* and Dennis R. Livesay 2 1 Department of Computer Science, New Jersey Institute
CS 581 Algorithmic Computational Genomics. Tandy Warnow University of Illinois at Urbana-Champaign
 CS 581 Algorithmic Computational Genomics Tandy Warnow University of Illinois at Urbana-Champaign Course Staff Professor Tandy Warnow Office hours Tuesdays after class (2-3 PM) in Siebel 3235 Email address:
CS 581 Algorithmic Computational Genomics Tandy Warnow University of Illinois at Urbana-Champaign Course Staff Professor Tandy Warnow Office hours Tuesdays after class (2-3 PM) in Siebel 3235 Email address:
Practical considerations of working with sequencing data
 Practical considerations of working with sequencing data File Types Fastq ->aligner -> reference(genome) coordinates Coordinate files SAM/BAM most complete, contains all of the info in fastq and more!
Practical considerations of working with sequencing data File Types Fastq ->aligner -> reference(genome) coordinates Coordinate files SAM/BAM most complete, contains all of the info in fastq and more!
Sequence Alignment: Scoring Schemes. COMP 571 Luay Nakhleh, Rice University
 Sequence Alignment: Scoring Schemes COMP 571 Luay Nakhleh, Rice University Scoring Schemes Recall that an alignment score is aimed at providing a scale to measure the degree of similarity (or difference)
Sequence Alignment: Scoring Schemes COMP 571 Luay Nakhleh, Rice University Scoring Schemes Recall that an alignment score is aimed at providing a scale to measure the degree of similarity (or difference)
SEPP and TIPP for metagenomic analysis. Tandy Warnow Department of Computer Science University of Texas
 SEPP and TIPP for metagenomic analysis Tandy Warnow Department of Computer Science University of Texas Metagenomics: Venter et al., Exploring the Sargasso Sea: Scientists Discover One Million New Genes
SEPP and TIPP for metagenomic analysis Tandy Warnow Department of Computer Science University of Texas Metagenomics: Venter et al., Exploring the Sargasso Sea: Scientists Discover One Million New Genes
Multiple sequence alignment
 Multiple sequence alignment Multiple sequence alignment: today s goals to define what a multiple sequence alignment is and how it is generated; to describe profile HMMs to introduce databases of multiple
Multiple sequence alignment Multiple sequence alignment: today s goals to define what a multiple sequence alignment is and how it is generated; to describe profile HMMs to introduce databases of multiple
Multiple Sequence Alignment: A Critical Comparison of Four Popular Programs
 Multiple Sequence Alignment: A Critical Comparison of Four Popular Programs Shirley Sutton, Biochemistry 218 Final Project, March 14, 2008 Introduction For both the computational biologist and the research
Multiple Sequence Alignment: A Critical Comparison of Four Popular Programs Shirley Sutton, Biochemistry 218 Final Project, March 14, 2008 Introduction For both the computational biologist and the research
Estimating Phylogenies (Evolutionary Trees) II. Biol4230 Thurs, March 2, 2017 Bill Pearson Jordan 6-057
 II. Biol4230 Thurs, March 2, 2017 Bill Pearson Jordan 6-057") Estimating Phylogenies (Evolutionary Trees) II Biol4230 Thurs, March 2, 2017 Bill Pearson wrp@virginia.edu 4-2818 Jordan 6-057 Tree estimation strategies: Parsimony?no model, simply count minimum number
Estimating Phylogenies (Evolutionary Trees) II Biol4230 Thurs, March 2, 2017 Bill Pearson wrp@virginia.edu 4-2818 Jordan 6-057 Tree estimation strategies: Parsimony?no model, simply count minimum number
Sequence Alignment: A General Overview. COMP Fall 2010 Luay Nakhleh, Rice University
 Sequence Alignment: A General Overview COMP 571 - Fall 2010 Luay Nakhleh, Rice University Life through Evolution All living organisms are related to each other through evolution This means: any pair of
Sequence Alignment: A General Overview COMP 571 - Fall 2010 Luay Nakhleh, Rice University Life through Evolution All living organisms are related to each other through evolution This means: any pair of
Biochemistry 324 Bioinformatics. Pairwise sequence alignment
 Biochemistry 324 Bioinformatics Pairwise sequence alignment How do we compare genes/proteins? When we have sequenced a genome, we try and identify the function of unknown genes by finding a similar gene
Biochemistry 324 Bioinformatics Pairwise sequence alignment How do we compare genes/proteins? When we have sequenced a genome, we try and identify the function of unknown genes by finding a similar gene
Dr. Amira A. AL-Hosary
 Phylogenetic analysis Amira A. AL-Hosary PhD of infectious diseases Department of Animal Medicine (Infectious Diseases) Faculty of Veterinary Medicine Assiut University-Egypt Phylogenetic Basics: Biological
Phylogenetic analysis Amira A. AL-Hosary PhD of infectious diseases Department of Animal Medicine (Infectious Diseases) Faculty of Veterinary Medicine Assiut University-Egypt Phylogenetic Basics: Biological
Lecture 4: Evolutionary Models and Substitution Matrices (PAM and BLOSUM)
") Bioinformatics II Probability and Statistics Universität Zürich and ETH Zürich Spring Semester 2009 Lecture 4: Evolutionary Models and Substitution Matrices (PAM and BLOSUM) Dr Fraser Daly adapted from
Bioinformatics II Probability and Statistics Universität Zürich and ETH Zürich Spring Semester 2009 Lecture 4: Evolutionary Models and Substitution Matrices (PAM and BLOSUM) Dr Fraser Daly adapted from
Module: Sequence Alignment Theory and Applications Session: Introduction to Searching and Sequence Alignment
 Module: Sequence Alignment Theory and Applications Session: Introduction to Searching and Sequence Alignment Introduction to Bioinformatics online course : IBT Jonathan Kayondo Learning Objectives Understand
Module: Sequence Alignment Theory and Applications Session: Introduction to Searching and Sequence Alignment Introduction to Bioinformatics online course : IBT Jonathan Kayondo Learning Objectives Understand
Algorithms in Bioinformatics
 Algorithms in Bioinformatics Sami Khuri Department of omputer Science San José State University San José, alifornia, USA khuri@cs.sjsu.edu www.cs.sjsu.edu/faculty/khuri Pairwise Sequence Alignment Homology
Algorithms in Bioinformatics Sami Khuri Department of omputer Science San José State University San José, alifornia, USA khuri@cs.sjsu.edu www.cs.sjsu.edu/faculty/khuri Pairwise Sequence Alignment Homology
Comparison of Cost Functions in Sequence Alignment. Ryan Healey
 Comparison of Cost Functions in Sequence Alignment Ryan Healey Use of Cost Functions Used to score and align sequences Mathematically model how sequences mutate and evolve. Evolution and mutation can be
Comparison of Cost Functions in Sequence Alignment Ryan Healey Use of Cost Functions Used to score and align sequences Mathematically model how sequences mutate and evolve. Evolution and mutation can be
Phylogenies Scores for Exhaustive Maximum Likelihood and Parsimony Scores Searches
 Int. J. Bioinformatics Research and Applications, Vol. x, No. x, xxxx Phylogenies Scores for Exhaustive Maximum Likelihood and s Searches Hyrum D. Carroll, Perry G. Ridge, Mark J. Clement, Quinn O. Snell
Int. J. Bioinformatics Research and Applications, Vol. x, No. x, xxxx Phylogenies Scores for Exhaustive Maximum Likelihood and s Searches Hyrum D. Carroll, Perry G. Ridge, Mark J. Clement, Quinn O. Snell
A greedy, graph-based algorithm for the alignment of multiple homologous gene lists
 A greedy, graph-based algorithm for the alignment of multiple homologous gene lists Jan Fostier, Sebastian Proost, Bart Dhoedt, Yvan Saeys, Piet Demeester, Yves Van de Peer, and Klaas Vandepoele Bioinformatics
A greedy, graph-based algorithm for the alignment of multiple homologous gene lists Jan Fostier, Sebastian Proost, Bart Dhoedt, Yvan Saeys, Piet Demeester, Yves Van de Peer, and Klaas Vandepoele Bioinformatics
Pairwise Alignment. Guan-Shieng Huang. Dept. of CSIE, NCNU. Pairwise Alignment p.1/55
 Pairwise Alignment Guan-Shieng Huang shieng@ncnu.edu.tw Dept. of CSIE, NCNU Pairwise Alignment p.1/55 Approach 1. Problem definition 2. Computational method (algorithms) 3. Complexity and performance Pairwise
Pairwise Alignment Guan-Shieng Huang shieng@ncnu.edu.tw Dept. of CSIE, NCNU Pairwise Alignment p.1/55 Approach 1. Problem definition 2. Computational method (algorithms) 3. Complexity and performance Pairwise
17 Non-collinear alignment Motivation A B C A B C A B C A B C D A C. This exposition is based on:
 17 Non-collinear alignment This exposition is based on: 1. Darling, A.E., Mau, B., Perna, N.T. (2010) progressivemauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5(6):e11147.
17 Non-collinear alignment This exposition is based on: 1. Darling, A.E., Mau, B., Perna, N.T. (2010) progressivemauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5(6):e11147.
CMPS 6630: Introduction to Computational Biology and Bioinformatics. Structure Comparison
 CMPS 6630: Introduction to Computational Biology and Bioinformatics Structure Comparison Protein Structure Comparison Motivation Understand sequence and structure variability Understand Domain architecture
CMPS 6630: Introduction to Computational Biology and Bioinformatics Structure Comparison Protein Structure Comparison Motivation Understand sequence and structure variability Understand Domain architecture
Copyright 2000 N. AYDIN. All rights reserved. 1
 Introduction to Bioinformatics Prof. Dr. Nizamettin AYDIN naydin@yildiz.edu.tr Multiple Sequence Alignment Outline Multiple sequence alignment introduction to msa methods of msa progressive global alignment
Introduction to Bioinformatics Prof. Dr. Nizamettin AYDIN naydin@yildiz.edu.tr Multiple Sequence Alignment Outline Multiple sequence alignment introduction to msa methods of msa progressive global alignment
Pairwise & Multiple sequence alignments
 Pairwise & Multiple sequence alignments Urmila Kulkarni-Kale Bioinformatics Centre 411 007 urmila@bioinfo.ernet.in Basis for Sequence comparison Theory of evolution: gene sequences have evolved/derived
Pairwise & Multiple sequence alignments Urmila Kulkarni-Kale Bioinformatics Centre 411 007 urmila@bioinfo.ernet.in Basis for Sequence comparison Theory of evolution: gene sequences have evolved/derived
EVOLUTIONARY DISTANCES
 EVOLUTIONARY DISTANCES FROM STRINGS TO TREES Luca Bortolussi 1 1 Dipartimento di Matematica ed Informatica Università degli studi di Trieste luca@dmi.units.it Trieste, 14 th November 2007 OUTLINE 1 STRINGS:
EVOLUTIONARY DISTANCES FROM STRINGS TO TREES Luca Bortolussi 1 1 Dipartimento di Matematica ed Informatica Università degli studi di Trieste luca@dmi.units.it Trieste, 14 th November 2007 OUTLINE 1 STRINGS:
Sequence analysis and Genomics
 Sequence analysis and Genomics October 12 th November 23 rd 2 PM 5 PM Prof. Peter Stadler Dr. Katja Nowick Katja: group leader TFome and Transcriptome Evolution Bioinformatics group Paul-Flechsig-Institute
Sequence analysis and Genomics October 12 th November 23 rd 2 PM 5 PM Prof. Peter Stadler Dr. Katja Nowick Katja: group leader TFome and Transcriptome Evolution Bioinformatics group Paul-Flechsig-Institute
Using phylogenetics to estimate species divergence times... Basics and basic issues for Bayesian inference of divergence times (plus some digression)
") Using phylogenetics to estimate species divergence times... More accurately... Basics and basic issues for Bayesian inference of divergence times (plus some digression) "A comparison of the structures
Using phylogenetics to estimate species divergence times... More accurately... Basics and basic issues for Bayesian inference of divergence times (plus some digression) "A comparison of the structures
Bioinformatics for Computer Scientists (Part 2 Sequence Alignment) Sepp Hochreiter
 Sepp Hochreiter") Bioinformatics for Computer Scientists (Part 2 Sequence Alignment) Institute of Bioinformatics Johannes Kepler University, Linz, Austria Sequence Alignment 2. Sequence Alignment Sequence Alignment 2.1
Bioinformatics for Computer Scientists (Part 2 Sequence Alignment) Institute of Bioinformatics Johannes Kepler University, Linz, Austria Sequence Alignment 2. Sequence Alignment Sequence Alignment 2.1
Amira A. AL-Hosary PhD of infectious diseases Department of Animal Medicine (Infectious Diseases) Faculty of Veterinary Medicine Assiut
 Faculty of Veterinary Medicine Assiut") Amira A. AL-Hosary PhD of infectious diseases Department of Animal Medicine (Infectious Diseases) Faculty of Veterinary Medicine Assiut University-Egypt Phylogenetic analysis Phylogenetic Basics: Biological
Amira A. AL-Hosary PhD of infectious diseases Department of Animal Medicine (Infectious Diseases) Faculty of Veterinary Medicine Assiut University-Egypt Phylogenetic analysis Phylogenetic Basics: Biological
NJMerge: A generic technique for scaling phylogeny estimation methods and its application to species trees
 NJMerge: A generic technique for scaling phylogeny estimation methods and its application to species trees Erin Molloy and Tandy Warnow {emolloy2, warnow}@illinois.edu University of Illinois at Urbana
NJMerge: A generic technique for scaling phylogeny estimation methods and its application to species trees Erin Molloy and Tandy Warnow {emolloy2, warnow}@illinois.edu University of Illinois at Urbana
Sequence Analysis '17- lecture 8. Multiple sequence alignment
 Sequence Analysis '17- lecture 8 Multiple sequence alignment Ex5 explanation How many random database search scores have e-values 10? (Answer: 10!) Why? e-value of x = m*p(s x), where m is the database
Sequence Analysis '17- lecture 8 Multiple sequence alignment Ex5 explanation How many random database search scores have e-values 10? (Answer: 10!) Why? e-value of x = m*p(s x), where m is the database
Lecture 4: Evolutionary models and substitution matrices (PAM and BLOSUM).
.") 1 Bioinformatics: In-depth PROBABILITY & STATISTICS Spring Semester 2011 University of Zürich and ETH Zürich Lecture 4: Evolutionary models and substitution matrices (PAM and BLOSUM). Dr. Stefanie Muff
1 Bioinformatics: In-depth PROBABILITY & STATISTICS Spring Semester 2011 University of Zürich and ETH Zürich Lecture 4: Evolutionary models and substitution matrices (PAM and BLOSUM). Dr. Stefanie Muff
Phylogenetic Tree Reconstruction
 I519 Introduction to Bioinformatics, 2011 Phylogenetic Tree Reconstruction Yuzhen Ye (yye@indiana.edu) School of Informatics & Computing, IUB Evolution theory Speciation Evolution of new organisms is driven
I519 Introduction to Bioinformatics, 2011 Phylogenetic Tree Reconstruction Yuzhen Ye (yye@indiana.edu) School of Informatics & Computing, IUB Evolution theory Speciation Evolution of new organisms is driven
Multiple Sequence Alignment
 Multiple equence lignment Four ami Khuri Dept of omputer cience an José tate University Multiple equence lignment v Progressive lignment v Guide Tree v lustalw v Toffee v Muscle v MFFT * 20 * 0 * 60 *
Multiple equence lignment Four ami Khuri Dept of omputer cience an José tate University Multiple equence lignment v Progressive lignment v Guide Tree v lustalw v Toffee v Muscle v MFFT * 20 * 0 * 60 *
Large Grain Size Stochastic Optimization Alignment
 Brigham Young University BYU ScholarsArchive All Faculty Publications 2006-10-01 Large Grain Size Stochastic Optimization Alignment Hyrum Carroll hyrumcarroll@gmail.com Mark J. Clement clement@cs.byu.edu
Brigham Young University BYU ScholarsArchive All Faculty Publications 2006-10-01 Large Grain Size Stochastic Optimization Alignment Hyrum Carroll hyrumcarroll@gmail.com Mark J. Clement clement@cs.byu.edu
Multiple Sequence Alignment, Gunnar Klau, December 9, 2005, 17:
 Multiple Sequence Alignment, Gunnar Klau, December 9, 2005, 17:50 5001 5 Multiple Sequence Alignment The first part of this exposition is based on the following sources, which are recommended reading:
Multiple Sequence Alignment, Gunnar Klau, December 9, 2005, 17:50 5001 5 Multiple Sequence Alignment The first part of this exposition is based on the following sources, which are recommended reading:
Phylogenetic Tree Estimation With and Without Alignment: New Distance Methods and Benchmarking
 Syst. Biol. 0(0):1 14, 2016 The Author(s) 2016. Published by Oxford University Press, on behalf of the Society of Systematic Biologists. All rights reserved. For Permissions, please email: journals.permissions@oup.com
Syst. Biol. 0(0):1 14, 2016 The Author(s) 2016. Published by Oxford University Press, on behalf of the Society of Systematic Biologists. All rights reserved. For Permissions, please email: journals.permissions@oup.com
Multiple Sequence Alignment using Profile HMM
 Multiple Sequence Alignment using Profile HMM. based on Chapter 5 and Section 6.5 from Biological Sequence Analysis by R. Durbin et al., 1998 Acknowledgements: M.Sc. students Beatrice Miron, Oana Răţoi,
Multiple Sequence Alignment using Profile HMM. based on Chapter 5 and Section 6.5 from Biological Sequence Analysis by R. Durbin et al., 1998 Acknowledgements: M.Sc. students Beatrice Miron, Oana Răţoi,
Evaluation Measures of Multiple Sequence Alignments. Gaston H. Gonnet, *Chantal Korostensky and Steve Benner. Institute for Scientic Computing
 Evaluation Measures of Multiple Sequence Alignments Gaston H. Gonnet, *Chantal Korostensky and Steve Benner Institute for Scientic Computing ETH Zurich, 8092 Zuerich, Switzerland phone: ++41 1 632 74 79
Evaluation Measures of Multiple Sequence Alignments Gaston H. Gonnet, *Chantal Korostensky and Steve Benner Institute for Scientic Computing ETH Zurich, 8092 Zuerich, Switzerland phone: ++41 1 632 74 79
POPULATION GENETICS Winter 2005 Lecture 17 Molecular phylogenetics
 POPULATION GENETICS Winter 2005 Lecture 17 Molecular phylogenetics - in deriving a phylogeny our goal is simply to reconstruct the historical relationships between a group of taxa. - before we review the
POPULATION GENETICS Winter 2005 Lecture 17 Molecular phylogenetics - in deriving a phylogeny our goal is simply to reconstruct the historical relationships between a group of taxa. - before we review the
Molecular Modeling Lecture 7. Homology modeling insertions/deletions manual realignment
 Molecular Modeling 2018-- Lecture 7 Homology modeling insertions/deletions manual realignment Homology modeling also called comparative modeling Sequences that have similar sequence have similar structure.
Molecular Modeling 2018-- Lecture 7 Homology modeling insertions/deletions manual realignment Homology modeling also called comparative modeling Sequences that have similar sequence have similar structure.
Inferring phylogeny. Constructing phylogenetic trees. Tõnu Margus. Bioinformatics MTAT
 Inferring phylogeny Constructing phylogenetic trees Tõnu Margus Contents What is phylogeny? How/why it is possible to infer it? Representing evolutionary relationships on trees What type questions questions
Inferring phylogeny Constructing phylogenetic trees Tõnu Margus Contents What is phylogeny? How/why it is possible to infer it? Representing evolutionary relationships on trees What type questions questions
Bioinformatics (GLOBEX, Summer 2015) Pairwise sequence alignment
 Pairwise sequence alignment") Bioinformatics (GLOBEX, Summer 2015) Pairwise sequence alignment Substitution score matrices, PAM, BLOSUM Needleman-Wunsch algorithm (Global) Smith-Waterman algorithm (Local) BLAST (local, heuristic) E-value
Bioinformatics (GLOBEX, Summer 2015) Pairwise sequence alignment Substitution score matrices, PAM, BLOSUM Needleman-Wunsch algorithm (Global) Smith-Waterman algorithm (Local) BLAST (local, heuristic) E-value
Multiple Alignment. Anders Gorm Pedersen Molecular Evolution Group Center for Biological Sequence Analysis
 Multiple Alignment Anders Gorm Pedersen Molecular Evolution Group Center for Biological Sequence Analysis gorm@cbs.dtu.dk Refresher: pairwise alignments 43.2% identity; Global alignment score: 374 10 20
Multiple Alignment Anders Gorm Pedersen Molecular Evolution Group Center for Biological Sequence Analysis gorm@cbs.dtu.dk Refresher: pairwise alignments 43.2% identity; Global alignment score: 374 10 20
Overview Multiple Sequence Alignment
 Overview Multiple Sequence Alignment Inge Jonassen Bioinformatics group Dept. of Informatics, UoB Inge.Jonassen@ii.uib.no Definition/examples Use of alignments The alignment problem scoring alignments
Overview Multiple Sequence Alignment Inge Jonassen Bioinformatics group Dept. of Informatics, UoB Inge.Jonassen@ii.uib.no Definition/examples Use of alignments The alignment problem scoring alignments
Phylogenetic Analysis. Han Liang, Ph.D. Assistant Professor of Bioinformatics and Computational Biology UT MD Anderson Cancer Center
 Phylogenetic Analysis Han Liang, Ph.D. Assistant Professor of Bioinformatics and Computational Biology UT MD Anderson Cancer Center Outline Basic Concepts Tree Construction Methods Distance-based methods
Phylogenetic Analysis Han Liang, Ph.D. Assistant Professor of Bioinformatics and Computational Biology UT MD Anderson Cancer Center Outline Basic Concepts Tree Construction Methods Distance-based methods
Single alignment: Substitution Matrix. 16 march 2017
 Single alignment: Substitution Matrix 16 march 2017 BLOSUM Matrix BLOSUM Matrix [2] (Blocks Amino Acid Substitution Matrices ) It is based on the amino acids substitutions observed in ~2000 conserved block
Single alignment: Substitution Matrix 16 march 2017 BLOSUM Matrix BLOSUM Matrix [2] (Blocks Amino Acid Substitution Matrices ) It is based on the amino acids substitutions observed in ~2000 conserved block
Sequence Alignment Techniques and Their Uses
 Sequence Alignment Techniques and Their Uses Sarah Fiorentino Since rapid sequencing technology and whole genomes sequencing, the amount of sequence information has grown exponentially. With all of this
Sequence Alignment Techniques and Their Uses Sarah Fiorentino Since rapid sequencing technology and whole genomes sequencing, the amount of sequence information has grown exponentially. With all of this
Phylogenetics. BIOL 7711 Computational Bioscience
 Consortium for Comparative Genomics! University of Colorado School of Medicine Phylogenetics BIOL 7711 Computational Bioscience Biochemistry and Molecular Genetics Computational Bioscience Program Consortium
Consortium for Comparative Genomics! University of Colorado School of Medicine Phylogenetics BIOL 7711 Computational Bioscience Biochemistry and Molecular Genetics Computational Bioscience Program Consortium
Sequence analysis and comparison
 The aim with sequence identification: Sequence analysis and comparison Marjolein Thunnissen Lund September 2012 Is there any known protein sequence that is homologous to mine? Are there any other species
The aim with sequence identification: Sequence analysis and comparison Marjolein Thunnissen Lund September 2012 Is there any known protein sequence that is homologous to mine? Are there any other species
C3020 Molecular Evolution. Exercises #3: Phylogenetics
 C3020 Molecular Evolution Exercises #3: Phylogenetics Consider the following sequences for five taxa 1-5 and the known outgroup O, which has the ancestral states (note that sequence 3 has changed from
C3020 Molecular Evolution Exercises #3: Phylogenetics Consider the following sequences for five taxa 1-5 and the known outgroup O, which has the ancestral states (note that sequence 3 has changed from
Sequence Analysis 17: lecture 5. Substitution matrices Multiple sequence alignment
 Sequence Analysis 17: lecture 5 Substitution matrices Multiple sequence alignment Substitution matrices Used to score aligned positions, usually of amino acids. Expressed as the log-likelihood ratio of
Sequence Analysis 17: lecture 5 Substitution matrices Multiple sequence alignment Substitution matrices Used to score aligned positions, usually of amino acids. Expressed as the log-likelihood ratio of
Phylogenomics, Multiple Sequence Alignment, and Metagenomics. Tandy Warnow University of Illinois at Urbana-Champaign
 Phylogenomics, Multiple Sequence Alignment, and Metagenomics Tandy Warnow University of Illinois at Urbana-Champaign Phylogeny (evolutionary tree) Orangutan Gorilla Chimpanzee Human From the Tree of the
Phylogenomics, Multiple Sequence Alignment, and Metagenomics Tandy Warnow University of Illinois at Urbana-Champaign Phylogeny (evolutionary tree) Orangutan Gorilla Chimpanzee Human From the Tree of the
Handling Rearrangements in DNA Sequence Alignment
 Handling Rearrangements in DNA Sequence Alignment Maneesh Bhand 12/5/10 1 Introduction Sequence alignment is one of the core problems of bioinformatics, with a broad range of applications such as genome
Handling Rearrangements in DNA Sequence Alignment Maneesh Bhand 12/5/10 1 Introduction Sequence alignment is one of the core problems of bioinformatics, with a broad range of applications such as genome
First generation sequencing and pairwise alignment (High-tech, not high throughput) Analysis of Biological Sequences
 Analysis of Biological Sequences") First generation sequencing and pairwise alignment (High-tech, not high throughput) Analysis of Biological Sequences 140.638 where do sequences come from? DNA is not hard to extract (getting DNA from a
First generation sequencing and pairwise alignment (High-tech, not high throughput) Analysis of Biological Sequences 140.638 where do sequences come from? DNA is not hard to extract (getting DNA from a
Alignment principles and homology searching using (PSI-)BLAST. Jaap Heringa Centre for Integrative Bioinformatics VU (IBIVU)
BLAST. Jaap Heringa Centre for Integrative Bioinformatics VU (IBIVU)") Alignment principles and homology searching using (PSI-)BLAST Jaap Heringa Centre for Integrative Bioinformatics VU (IBIVU) http://ibivu.cs.vu.nl Bioinformatics Nothing in Biology makes sense except in
Alignment principles and homology searching using (PSI-)BLAST Jaap Heringa Centre for Integrative Bioinformatics VU (IBIVU) http://ibivu.cs.vu.nl Bioinformatics Nothing in Biology makes sense except in
Lecture 7 Sequence analysis. Hidden Markov Models
 Lecture 7 Sequence analysis. Hidden Markov Models Nicolas Lartillot may 2012 Nicolas Lartillot (Universite de Montréal) BIN6009 may 2012 1 / 60 1 Motivation 2 Examples of Hidden Markov models 3 Hidden
Lecture 7 Sequence analysis. Hidden Markov Models Nicolas Lartillot may 2012 Nicolas Lartillot (Universite de Montréal) BIN6009 may 2012 1 / 60 1 Motivation 2 Examples of Hidden Markov models 3 Hidden
Local Alignment Statistics
 Local Alignment Statistics Stephen Altschul National Center for Biotechnology Information National Library of Medicine National Institutes of Health Bethesda, MD Central Issues in Biological Sequence Comparison
Local Alignment Statistics Stephen Altschul National Center for Biotechnology Information National Library of Medicine National Institutes of Health Bethesda, MD Central Issues in Biological Sequence Comparison
Pairwise sequence alignments
 Pairwise sequence alignments Volker Flegel VI, October 2003 Page 1 Outline Introduction Definitions Biological context of pairwise alignments Computing of pairwise alignments Some programs VI, October
Pairwise sequence alignments Volker Flegel VI, October 2003 Page 1 Outline Introduction Definitions Biological context of pairwise alignments Computing of pairwise alignments Some programs VI, October
Computational Biology
 Computational Biology Lecture 6 31 October 2004 1 Overview Scoring matrices (Thanks to Shannon McWeeney) BLAST algorithm Start sequence alignment 2 1 What is a homologous sequence? A homologous sequence,
Computational Biology Lecture 6 31 October 2004 1 Overview Scoring matrices (Thanks to Shannon McWeeney) BLAST algorithm Start sequence alignment 2 1 What is a homologous sequence? A homologous sequence,
BMI/CS 776 Lecture #20 Alignment of whole genomes. Colin Dewey (with slides adapted from those by Mark Craven)
") BMI/CS 776 Lecture #20 Alignment of whole genomes Colin Dewey (with slides adapted from those by Mark Craven) 2007.03.29 1 Multiple whole genome alignment Input set of whole genome sequences genomes diverged
BMI/CS 776 Lecture #20 Alignment of whole genomes Colin Dewey (with slides adapted from those by Mark Craven) 2007.03.29 1 Multiple whole genome alignment Input set of whole genome sequences genomes diverged
CS 394C Algorithms for Computational Biology. Tandy Warnow Spring 2012
 CS 394C Algorithms for Computational Biology Tandy Warnow Spring 2012 Biology: 21st Century Science! When the human genome was sequenced seven years ago, scientists knew that most of the major scientific
CS 394C Algorithms for Computational Biology Tandy Warnow Spring 2012 Biology: 21st Century Science! When the human genome was sequenced seven years ago, scientists knew that most of the major scientific
Pairwise sequence alignments. Vassilios Ioannidis (From Volker Flegel )
") Pairwise sequence alignments Vassilios Ioannidis (From Volker Flegel ) Outline Introduction Definitions Biological context of pairwise alignments Computing of pairwise alignments Some programs Importance
Pairwise sequence alignments Vassilios Ioannidis (From Volker Flegel ) Outline Introduction Definitions Biological context of pairwise alignments Computing of pairwise alignments Some programs Importance
Sequence Alignment (chapter 6)
") Sequence lignment (chapter 6) he biological problem lobal alignment Local alignment Multiple alignment Introduction to bioinformatics, utumn 6 Background: comparative genomics Basic question in biology:
Sequence lignment (chapter 6) he biological problem lobal alignment Local alignment Multiple alignment Introduction to bioinformatics, utumn 6 Background: comparative genomics Basic question in biology:
Research Proposal. Title: Multiple Sequence Alignment used to investigate the co-evolving positions in OxyR Protein family.
 Research Proposal Title: Multiple Sequence Alignment used to investigate the co-evolving positions in OxyR Protein family. Name: Minjal Pancholi Howard University Washington, DC. June 19, 2009 Research
Research Proposal Title: Multiple Sequence Alignment used to investigate the co-evolving positions in OxyR Protein family. Name: Minjal Pancholi Howard University Washington, DC. June 19, 2009 Research
Sequence comparison: Score matrices. Genome 559: Introduction to Statistical and Computational Genomics Prof. James H. Thomas
 Sequence comparison: Score matrices Genome 559: Introduction to Statistical and omputational Genomics Prof James H Thomas Informal inductive proof of best alignment path onsider the last step in the best
Sequence comparison: Score matrices Genome 559: Introduction to Statistical and omputational Genomics Prof James H Thomas Informal inductive proof of best alignment path onsider the last step in the best
Algorithms in Bioinformatics
 Algorithms in Bioinformatics Sami Khuri Department of Computer Science San José State University San José, California, USA khuri@cs.sjsu.edu www.cs.sjsu.edu/faculty/khuri Distance Methods Character Methods
Algorithms in Bioinformatics Sami Khuri Department of Computer Science San José State University San José, California, USA khuri@cs.sjsu.edu www.cs.sjsu.edu/faculty/khuri Distance Methods Character Methods
Network alignment and querying
 Network biology minicourse (part 4) Algorithmic challenges in genomics Network alignment and querying Roded Sharan School of Computer Science, Tel Aviv University Multiple Species PPI Data Rapid growth
Network biology minicourse (part 4) Algorithmic challenges in genomics Network alignment and querying Roded Sharan School of Computer Science, Tel Aviv University Multiple Species PPI Data Rapid growth
EECS730: Introduction to Bioinformatics
 EECS730: Introduction to Bioinformatics Lecture 07: profile Hidden Markov Model http://bibiserv.techfak.uni-bielefeld.de/sadr2/databasesearch/hmmer/profilehmm.gif Slides adapted from Dr. Shaojie Zhang
EECS730: Introduction to Bioinformatics Lecture 07: profile Hidden Markov Model http://bibiserv.techfak.uni-bielefeld.de/sadr2/databasesearch/hmmer/profilehmm.gif Slides adapted from Dr. Shaojie Zhang
Statistical Machine Learning Methods for Bioinformatics II. Hidden Markov Model for Biological Sequences
 Statistical Machine Learning Methods for Bioinformatics II. Hidden Markov Model for Biological Sequences Jianlin Cheng, PhD Department of Computer Science University of Missouri 2008 Free for Academic
Statistical Machine Learning Methods for Bioinformatics II. Hidden Markov Model for Biological Sequences Jianlin Cheng, PhD Department of Computer Science University of Missouri 2008 Free for Academic
Bayesian Inference using Markov Chain Monte Carlo in Phylogenetic Studies
 Bayesian Inference using Markov Chain Monte Carlo in Phylogenetic Studies 1 What is phylogeny? Essay written for the course in Markov Chains 2004 Torbjörn Karfunkel Phylogeny is the evolutionary development
Bayesian Inference using Markov Chain Monte Carlo in Phylogenetic Studies 1 What is phylogeny? Essay written for the course in Markov Chains 2004 Torbjörn Karfunkel Phylogeny is the evolutionary development
Page 1. References. Hidden Markov models and multiple sequence alignment. Markov chains. Probability review. Example. Markovian sequence
 Page Hidden Markov models and multiple sequence alignment Russ B Altman BMI 4 CS 74 Some slides borrowed from Scott C Schmidler (BMI graduate student) References Bioinformatics Classic: Krogh et al (994)
Page Hidden Markov models and multiple sequence alignment Russ B Altman BMI 4 CS 74 Some slides borrowed from Scott C Schmidler (BMI graduate student) References Bioinformatics Classic: Krogh et al (994)
Homology Modeling. Roberto Lins EPFL - summer semester 2005
 Homology Modeling Roberto Lins EPFL - summer semester 2005 Disclaimer: course material is mainly taken from: P.E. Bourne & H Weissig, Structural Bioinformatics; C.A. Orengo, D.T. Jones & J.M. Thornton,
Homology Modeling Roberto Lins EPFL - summer semester 2005 Disclaimer: course material is mainly taken from: P.E. Bourne & H Weissig, Structural Bioinformatics; C.A. Orengo, D.T. Jones & J.M. Thornton,
Motivating the need for optimal sequence alignments...
 1 Motivating the need for optimal sequence alignments... 2 3 Note that this actually combines two objectives of optimal sequence alignments: (i) use the score of the alignment o infer homology; (ii) use
1 Motivating the need for optimal sequence alignments... 2 3 Note that this actually combines two objectives of optimal sequence alignments: (i) use the score of the alignment o infer homology; (ii) use
An Introduction to Bioinformatics Algorithms Hidden Markov Models
 Hidden Markov Models Outline 1. CG-Islands 2. The Fair Bet Casino 3. Hidden Markov Model 4. Decoding Algorithm 5. Forward-Backward Algorithm 6. Profile HMMs 7. HMM Parameter Estimation 8. Viterbi Training
Hidden Markov Models Outline 1. CG-Islands 2. The Fair Bet Casino 3. Hidden Markov Model 4. Decoding Algorithm 5. Forward-Backward Algorithm 6. Profile HMMs 7. HMM Parameter Estimation 8. Viterbi Training
BINF6201/8201. Molecular phylogenetic methods
 BINF60/80 Molecular phylogenetic methods 0-7-06 Phylogenetics Ø According to the evolutionary theory, all life forms on this planet are related to one another by descent. Ø Traditionally, phylogenetics
BINF60/80 Molecular phylogenetic methods 0-7-06 Phylogenetics Ø According to the evolutionary theory, all life forms on this planet are related to one another by descent. Ø Traditionally, phylogenetics
Hidden Markov models in population genetics and evolutionary biology
 Hidden Markov models in population genetics and evolutionary biology Gerton Lunter Wellcome Trust Centre for Human Genetics Oxford, UK April 29, 2013 Topics for today Markov chains Hidden Markov models
Hidden Markov models in population genetics and evolutionary biology Gerton Lunter Wellcome Trust Centre for Human Genetics Oxford, UK April 29, 2013 Topics for today Markov chains Hidden Markov models
Phylogenetic hypotheses and the utility of multiple sequence alignment
 Phylogenetic hypotheses and the utility of multiple sequence alignment Ward C. Wheeler 1 and Gonzalo Giribet 2 1 Division of Invertebrate Zoology, American Museum of Natural History Central Park West at
Phylogenetic hypotheses and the utility of multiple sequence alignment Ward C. Wheeler 1 and Gonzalo Giribet 2 1 Division of Invertebrate Zoology, American Museum of Natural History Central Park West at
Whole Genome Alignments and Synteny Maps
 Whole Genome Alignments and Synteny Maps IINTRODUCTION It was not until closely related organism genomes have been sequenced that people start to think about aligning genomes and chromosomes instead of
Whole Genome Alignments and Synteny Maps IINTRODUCTION It was not until closely related organism genomes have been sequenced that people start to think about aligning genomes and chromosomes instead of
Sequence Analysis, '18 -- lecture 9. Families and superfamilies. Sequence weights. Profiles. Logos. Building a representative model for a gene.
 Sequence Analysis, '18 -- lecture 9 Families and superfamilies. Sequence weights. Profiles. Logos. Building a representative model for a gene. How can I represent thousands of homolog sequences in a compact
Sequence Analysis, '18 -- lecture 9 Families and superfamilies. Sequence weights. Profiles. Logos. Building a representative model for a gene. How can I represent thousands of homolog sequences in a compact