Moreover, the circular logic
|
|
|
- Reginald Rich
- 5 years ago
- Views:
Transcription
1 Moreover, the circular logic How do we know what is the right distance without a good alignment? And how do we construct a good alignment without knowing what substitutions were made previously? ATGCGT--GCAAGT --GCGGCGGCAGTT ATGCGT--GCAAGT- --GCGGCGGC-AGTT Which is better? Less gaps but 7matches More gaps but 8 matches
2 Substitution matrices for proteins We need to compare, DNA or protein sequences, estimate their distances etc (e.g. Helpful for inferring molecular function by finding similarity to a sequence with known function). For that purpose we need: Good alignment of sequences. For that purpose we need: A measure for judging the quality of an alignment in relation to other possible alignments, a scoring system.
3
4
5 A practical aspect as well, since we don t study very diverged DNA sequences
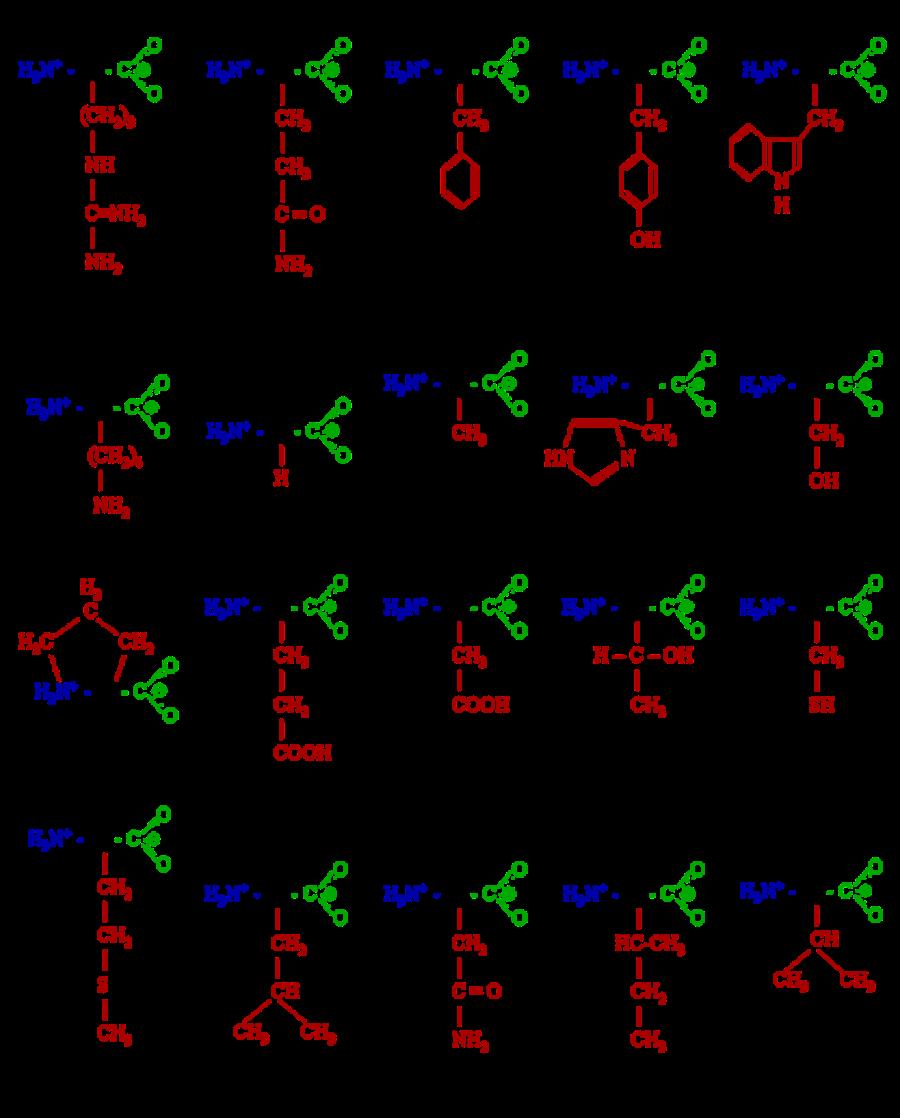
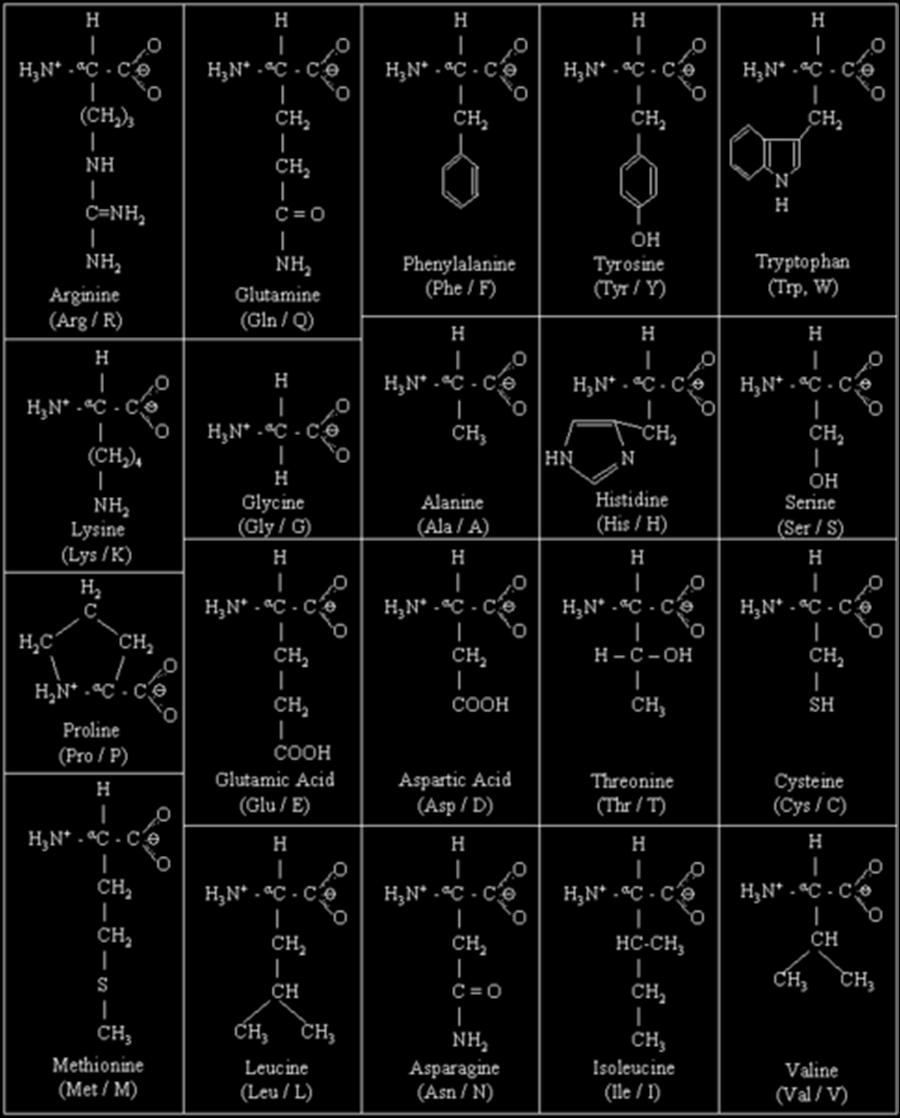
6
7
8
9

10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25 Commonly multiplied by a power of 10 to deal with decimals
26
27
28 A sample substitution matrix 134 LQQGELDLVMTSDILPRSELHYSPMFDFEVRLVLAPDHPLASKTQITPEDLASETLLI 137 LDSNSVDLVLMGVPPRNVEVEAEAFMDNPLVVIAPPDHPLAGERAISLARLAEETFVM BLOSUM62 D:D = +6 C 9 S -1 4 T P A G D:R = -2 N D E Q H R K M I L V F Y W C S T P A G N D E Q H R K M I L V F Y W
29 BLOSUM Brief Overview Based purely on counts and alignment BLOSUM65 < BLOSUM45 in evolutionary distance a set of sequences S = S...S, where S = s...s, ij 20 j i j i i= 1 j= 1 i= 1 1 k i i1 in s is the j-th amino acid in sequence S. The probability of observing each amino acid X =p(x) p(x) cx (, S)/ cx (, S) where cx (, S k = j i k ) is the count of amino acid X in sequence S PX X random= px px orpx ifl= m 2 ( l, m ) 2 ( l). ( m) ( l), i j i
30 In general log-odds for alignment PX (, X data) = # of X, X combinations/all possible pairwise combinations l m l m finally take the log2-odds likelihood ratio and multiply by 2 for easy representation { } Note: all possible pairwise combinations=k. nn ( 1)/2 Blosum does use pseudo count to accomadate for combinations not observed: add 1 in numerator, add 210 (20*19+20) in denominator ie P(X,X data) = l m count(x l,x m) k. ( 1) / { nn } P(X l,x m data) λ.log = subs. matrix P (X,X random) l m λ being a scaling factor for representation
31 BLOSUM vs PAM vs others? BLOSUM: Based on a range of evolutionary periods -- Each matrix constructed separately Indirectly accounts for interdependence of residues Range of sequences, range of replacements PAM Based on extrapolation from a short evolutionary period -- Errors in PAM1 are magnified through PAM250 Assumes Markov process too much extrapolation Rare replacements too infrequent to be represented accurately Others Incorporation of secondary structure/structural alignments Use of structural alignments Transmemberane protein-specific matrices
32 How do we use the matrices for sequence alignment? AGGCTATCACCTGACCTCCAGGCCGATGCCC TAGCTATCACGACCGCGGTCGATTTGCCCGAC -AGGCTATCACCTGACCTCCAGGCCGA--TGCCC--- TAG-CTATCAC--GACCGC--GGTCGATTTGCCCGAC Definition Given two strings x = x 1 x 2...x M, y = y 1 y 2 y N, An alignment of two sequences x and y is an arrangement of x and y by position, where a and b can be padded with gap symbols to achieve the same length.
33 Why do we bother aligning? Finding important molecular regions that are conserved across species Given a new sequence, infer its function based on similarity to another Seq. Determining the evolutionary constraints at work Mutations in a population or a family of genes Find similar looking sequences in a database Inferring the secondary or tertiary structure of a sequence of interest -Molecular modeling using a template (homology modeling)

34 Alignment is a path in the alignment matrix X: ACACACTA Y: AGCACACA X: AGCACAC-A Y: A-CACACTA m+ n! mn!! m+ n 2 Alignments possible!
35 Dynamic programming for global alignment simple case Consider two sequences: x[1.. M], and y[1.. N] for a new position to be aligned we have ONLY three choices 1. x i aligns to y j x 1 x i-1 x i Align x[1.. i] with y[1.. j] y 1 y j-1 y j 2. x i aligns to a gap x 1 x i-1 x i y 1 y j - 3. y j aligns to a gap x 1 x i - y 1 y j-1 y j x[1..(i-1)] is already aligned with y[1..(j)], so align x[i] with a gap in y x[1.. i] is already aligned with y[1.. (j-1)], so align a gap in x to y[j]
36 1. x i aligns to y j x 1 x i-1 y 1 y j-1 x i y j F (i,j) = F (i-1, j-1) + s(i,j) 2. x i aligns to a gap x 1 x i-1 x i y 1 y j - 3. y j aligns to a gap x 1 x i - y 1 y j-1 y j F (i,j) = F (i-1, j) gap_open_penalty (go) F (i,j) = F (i, j-1) gap_open_penalty (go)
37 If we could make F (i, j-1), F (i-1, j), F (i-1, j-1) optimal, then we can make the next ones optimal as well F (i-1, j-1) + s (x i, y j ) F (i, j) = max F (i-1, j) go F (i, j-1) go Where s (x i, y j ) = Score for a match, if x i = y j ; score for a mismatch, if x i y j ;
38 The Needleman-Wunsch Algorithm pioneering application of DP to biological sequences 1. Initialization. a. F(0, 0) = 0 b. F(0, j) = - j go c. F(i, 0) = - i go 2. Main Iteration. Filling-in partial alignments For each i = 1 M For each j = 1 N F(i-1,j-1) + s(x i, y j ) [case 1] F(i, j) = max F(i-1, j) go [case 2] F(i, j-1) go [case 3] DIAG, if [case 1] Ptr(i,j) = LEFT, if [case 2] UP, if [case 3] 3. Termination. F(M, N) is the optimal score, and from Ptr(M, N) can trace back optimal alignment
39 An example T C G C A F (i-1, j-1) + s (x i, y j ) F (i, j) = max F (i-1, j) go F (i, j-1) go T s (x i, y j )=1 for match, -1 for mismatch go=2 C C A Trace back arrows from lower right to upper left and Maximize the scores Alignment score = 2 T C G C A T C - C A
40 Are gaps that bad? Current model: Gap of length n incurs penalty n.go γ(n) Bunch of gaps are better than individual gaps Convex (saturating) gap penalty function: γ(n): for all n, γ(n + 1) - γ(n) γ(n) - γ(n 1) A common function is Affine gap penalty γ(n) = go + (n 1) ge gap gap open extend γ(n)
41 Convex gap dynamic programming Initialization: same as before Iteration: F(i-1, j-1) + s(x i, y j ) F(i, j) = max max k=0 i-1 F(k,j) γ(i-k) max k=0 j-1 F(i,k) γ(j-k) Termination: same Running Time: O(N 2 M) Space: O(NM) (assume N>M)
42 Fast implementation of affine gap penalty We need three matrices for tracking scores Matrix-1: a[i,j]= to store maximum score of an alignment that ends in x[i] matched to y[j] Matrix-2: b[i,j]= to store maximum score of an alignment that ends in gap matched to y[j] Matrix-3: c[i,j]= to store maximum score of an alignment that ends in gap matched to x[i] x i y j - y j.x i -
43 Implementation Cont d ai [ 1, j 1] ai [, j] = max bi [ 1, j 1] + si (, j) ci [ 1, j 1] ai [, j 1] + go bi [, j] = max bi [, j 1] + ge ci [, j 1] + go ai [ 1, j] + go ci [, j] = max bi [ 1, j] + go ci [ 1, j] + ge Pointer-matrices: Three matrices to figure out which state within each score matrix maximization was used to obtain the optimal alignment of position i,j. Of course you need to know which matrix yielded the best score in the end (m,n) as well Covered by assignment!


44 For checking results of your code
45 Pair HMMs for alignment HMM for sequence alignment, which incorporates affine gap scores. Match (M) Insertion in x (X) insertion in y (Y) Hidden States Observation Symbols Match (M): {(a,b) a,b in }. Insertion in x (X): {(a,-) a in }. Insertion in y (Y): {(-,a) a in }.
46 Simple representation 1-δ M p xiyj 1-ε 1-ε X q xi δ δ Y q yj ε ε M X Y M 1-2δ 1- ε 1- ε X δ ε 0 Y δ 0 ε
47 Full representation δ ε Begin 1-2δ-τ M p xiyj δ X q xi 1-ε-τ τ τ End 1-2δ-τ δ δ 1-ε-τ Y q yj ε τ τ
48 Initialization: v M (0, 0) = 1. v X (0, 0) = v Y (0, 0) = 0 v * (-1, j) = v * (i, -1) = 0. Recurrence: i = 0,,n, j = 0,,m, except for(0,0); Termination: = = = 1), ( 1), ( max ), ( ) 1, ( ) 1, ( max ), ( 1) 1, ( ) (1 1) 1, ( ) (1 1) 1, ( ) 2 (1 max ), ( X M Y X M X Y X M M j i v j i v q j i v j i v j i v q j i v j i v j i v j i v p j i v j i j i y x y x ε δ ε δ τ ε τ ε τ δ )), ( ),, ( ),, ( max( Y X M E m n v m n v m n v v τ = Algorithm: Viterbi algorithm for pair HMMs
49 Viterbi Pair HMM is equivalent to global dynamic programming with affine gap! Proof (sketch) Add the following missing details to DEKM s summary Take the viterbi matrices and divide by the terms for random sequence probability, and take the log of resulting transformation i.e, (1 2 δ τ) v ( i 1, j 1). p q q (1 η) v i j v i j p q q M 2 xiyj xi yi M X 2 (, ) = max (1 ε τ) ( 1, 1). xiyj xi yi (1 η) Y 2 (1 ε τ) v ( i 1, j 1). pxiyj qxiqyi (1 η) M δv ( i 1, j). qxi qxi (1 η) (, ) = max i X εv ( i 1, j). qxi qxi (1 η) X v i j qx
 = 5 NN ( -1) 2 Now consider one position being G, rest")
50 Multiple Sequence Alignment (MSA) Si () = sx ( i, yi) all possible pairs How to score? and How to align? Scoring, common approach: Sum of pairs or its variants for each column Consider L aligned at all N positions, LL score =5 total score, Si ( ) = 5 NN ( -1) 2 Now consider one position being G, rest being L GL score=-4 ΔSi ( ) 5( N 1) + 4( N 1) 9( N 1) 9 = = = Si () Si () 2.5 NN ( 1) 2.5N Relative evidence decreases with N, opposed to having increased confidence in L being the "correct" residue at that position
51 Clustal W Pairwise alignment: calculation of distance matrix Rooted nj tree (guide tree) and calculation of sequence weights Progressive alignment following the guide tree
52 Step 1-Calculation of Distance Matrix using pairwise alignment Use the Distance Matrix to create a Guide Tree to determine the order of the sequences. Hbb-Hu 1 - Hbb-Ho Hba-Hu Hba-Ho Myg-Ph Gib-Pe Lgb-Lu D = 1 (I) D = Difference score I = # of identical aa s in pairwise global alignment total number of aa s in shortest sequence
53 Step 2-Create Rooted Tree and calculate weights Neighbor joining algorithm simple, to be discussed later Weight Alignment Order of alignment: 1 Hba-Hu vs Hba-Ho 2 Hbb-Hu vs Hbb-Ho 3 A vs B 4 Myg-Ph vs C 5 Gib-Pe vs D 6 Lgh-Lu vs E
54 Step 3-Progressive alignment
55 Step 3-Progressive alignment Scoring during progressive alignment
56 Recommended MSA Programs MUSCLE (fast and accurate) MAVID (genome-scale alignment) SAM ( hidden markov, powerful and wide range of options)
Evolution. CT Amemiya et al. Nature 496, (2013) doi: /nature12027
 doi: /nature12027") Sequence Alignment Evolution CT Amemiya et al. Nature 496, 311-316 (2013) doi:10.1038/nature12027 Evolutionary Rates next generation OK OK OK X X Still OK? Sequence conservation implies function Alignment
Sequence Alignment Evolution CT Amemiya et al. Nature 496, 311-316 (2013) doi:10.1038/nature12027 Evolutionary Rates next generation OK OK OK X X Still OK? Sequence conservation implies function Alignment
Quantifying sequence similarity
 Quantifying sequence similarity Bas E. Dutilh Systems Biology: Bioinformatic Data Analysis Utrecht University, February 16 th 2016 After this lecture, you can define homology, similarity, and identity
Quantifying sequence similarity Bas E. Dutilh Systems Biology: Bioinformatic Data Analysis Utrecht University, February 16 th 2016 After this lecture, you can define homology, similarity, and identity
Practical considerations of working with sequencing data
 Practical considerations of working with sequencing data File Types Fastq ->aligner -> reference(genome) coordinates Coordinate files SAM/BAM most complete, contains all of the info in fastq and more!
Practical considerations of working with sequencing data File Types Fastq ->aligner -> reference(genome) coordinates Coordinate files SAM/BAM most complete, contains all of the info in fastq and more!
Pairwise alignment using HMMs
 Pairwise alignment using HMMs The states of an HMM fulfill the Markov property: probability of transition depends only on the last state. CpG islands and casino example: HMMs emit sequence of symbols (nucleotides
Pairwise alignment using HMMs The states of an HMM fulfill the Markov property: probability of transition depends only on the last state. CpG islands and casino example: HMMs emit sequence of symbols (nucleotides
Local Alignment: Smith-Waterman algorithm
 Local Alignment: Smith-Waterman algorithm Example: a shared common domain of two protein sequences; extended sections of genomic DNA sequence. Sensitive to detect similarity in highly diverged sequences.
Local Alignment: Smith-Waterman algorithm Example: a shared common domain of two protein sequences; extended sections of genomic DNA sequence. Sensitive to detect similarity in highly diverged sequences.
Bioinformatics for Computer Scientists (Part 2 Sequence Alignment) Sepp Hochreiter
 Sepp Hochreiter") Bioinformatics for Computer Scientists (Part 2 Sequence Alignment) Institute of Bioinformatics Johannes Kepler University, Linz, Austria Sequence Alignment 2. Sequence Alignment Sequence Alignment 2.1
Bioinformatics for Computer Scientists (Part 2 Sequence Alignment) Institute of Bioinformatics Johannes Kepler University, Linz, Austria Sequence Alignment 2. Sequence Alignment Sequence Alignment 2.1
Pairwise sequence alignment
 Department of Evolutionary Biology Example Alignment between very similar human alpha- and beta globins: GSAQVKGHGKKVADALTNAVAHVDDMPNALSALSDLHAHKL G+ +VK+HGKKV A+++++AH+D++ +++++LS+LH KL GNPKVKAHGKKVLGAFSDGLAHLDNLKGTFATLSELHCDKL
Department of Evolutionary Biology Example Alignment between very similar human alpha- and beta globins: GSAQVKGHGKKVADALTNAVAHVDDMPNALSALSDLHAHKL G+ +VK+HGKKV A+++++AH+D++ +++++LS+LH KL GNPKVKAHGKKVLGAFSDGLAHLDNLKGTFATLSELHCDKL
Algorithms in Bioinformatics FOUR Pairwise Sequence Alignment. Pairwise Sequence Alignment. Convention: DNA Sequences 5. Sequence Alignment
 Algorithms in Bioinformatics FOUR Sami Khuri Department of Computer Science San José State University Pairwise Sequence Alignment Homology Similarity Global string alignment Local string alignment Dot
Algorithms in Bioinformatics FOUR Sami Khuri Department of Computer Science San José State University Pairwise Sequence Alignment Homology Similarity Global string alignment Local string alignment Dot
Sequence analysis and Genomics
 Sequence analysis and Genomics October 12 th November 23 rd 2 PM 5 PM Prof. Peter Stadler Dr. Katja Nowick Katja: group leader TFome and Transcriptome Evolution Bioinformatics group Paul-Flechsig-Institute
Sequence analysis and Genomics October 12 th November 23 rd 2 PM 5 PM Prof. Peter Stadler Dr. Katja Nowick Katja: group leader TFome and Transcriptome Evolution Bioinformatics group Paul-Flechsig-Institute
HMM applications. Applications of HMMs. Gene finding with HMMs. Using the gene finder
 HMM applications Applications of HMMs Gene finding Pairwise alignment (pair HMMs) Characterizing protein families (profile HMMs) Predicting membrane proteins, and membrane protein topology Gene finding
HMM applications Applications of HMMs Gene finding Pairwise alignment (pair HMMs) Characterizing protein families (profile HMMs) Predicting membrane proteins, and membrane protein topology Gene finding
First generation sequencing and pairwise alignment (High-tech, not high throughput) Analysis of Biological Sequences
 Analysis of Biological Sequences") First generation sequencing and pairwise alignment (High-tech, not high throughput) Analysis of Biological Sequences 140.638 where do sequences come from? DNA is not hard to extract (getting DNA from a
First generation sequencing and pairwise alignment (High-tech, not high throughput) Analysis of Biological Sequences 140.638 where do sequences come from? DNA is not hard to extract (getting DNA from a
THEORY. Based on sequence Length According to the length of sequence being compared it is of following two types
 Exp 11- THEORY Sequence Alignment is a process of aligning two sequences to achieve maximum levels of identity between them. This help to derive functional, structural and evolutionary relationships between
Exp 11- THEORY Sequence Alignment is a process of aligning two sequences to achieve maximum levels of identity between them. This help to derive functional, structural and evolutionary relationships between
Bioinformatics (GLOBEX, Summer 2015) Pairwise sequence alignment
 Pairwise sequence alignment") Bioinformatics (GLOBEX, Summer 2015) Pairwise sequence alignment Substitution score matrices, PAM, BLOSUM Needleman-Wunsch algorithm (Global) Smith-Waterman algorithm (Local) BLAST (local, heuristic) E-value
Bioinformatics (GLOBEX, Summer 2015) Pairwise sequence alignment Substitution score matrices, PAM, BLOSUM Needleman-Wunsch algorithm (Global) Smith-Waterman algorithm (Local) BLAST (local, heuristic) E-value
Sara C. Madeira. Universidade da Beira Interior. (Thanks to Ana Teresa Freitas, IST for useful resources on this subject)
") Bioinformática Sequence Alignment Pairwise Sequence Alignment Universidade da Beira Interior (Thanks to Ana Teresa Freitas, IST for useful resources on this subject) 1 16/3/29 & 23/3/29 27/4/29 Outline
Bioinformática Sequence Alignment Pairwise Sequence Alignment Universidade da Beira Interior (Thanks to Ana Teresa Freitas, IST for useful resources on this subject) 1 16/3/29 & 23/3/29 27/4/29 Outline
Large-Scale Genomic Surveys
 Bioinformatics Subtopics Fold Recognition Secondary Structure Prediction Docking & Drug Design Protein Geometry Protein Flexibility Homology Modeling Sequence Alignment Structure Classification Gene Prediction
Bioinformatics Subtopics Fold Recognition Secondary Structure Prediction Docking & Drug Design Protein Geometry Protein Flexibility Homology Modeling Sequence Alignment Structure Classification Gene Prediction
Algorithms in Bioinformatics
 Algorithms in Bioinformatics Sami Khuri Department of omputer Science San José State University San José, alifornia, USA khuri@cs.sjsu.edu www.cs.sjsu.edu/faculty/khuri Pairwise Sequence Alignment Homology
Algorithms in Bioinformatics Sami Khuri Department of omputer Science San José State University San José, alifornia, USA khuri@cs.sjsu.edu www.cs.sjsu.edu/faculty/khuri Pairwise Sequence Alignment Homology
CISC 889 Bioinformatics (Spring 2004) Sequence pairwise alignment (I)
 Sequence pairwise alignment (I)") CISC 889 Bioinformatics (Spring 2004) Sequence pairwise alignment (I) Contents Alignment algorithms Needleman-Wunsch (global alignment) Smith-Waterman (local alignment) Heuristic algorithms FASTA BLAST
CISC 889 Bioinformatics (Spring 2004) Sequence pairwise alignment (I) Contents Alignment algorithms Needleman-Wunsch (global alignment) Smith-Waterman (local alignment) Heuristic algorithms FASTA BLAST
Minimum Edit Distance. Defini'on of Minimum Edit Distance
 Minimum Edit Distance Defini'on of Minimum Edit Distance How similar are two strings? Spell correc'on The user typed graffe Which is closest? graf gra@ grail giraffe Computa'onal Biology Align two sequences
Minimum Edit Distance Defini'on of Minimum Edit Distance How similar are two strings? Spell correc'on The user typed graffe Which is closest? graf gra@ grail giraffe Computa'onal Biology Align two sequences
Welcome to CS262: Computational Genomics
 Instructor: Welcome to CS262: Computational Genomics Serafim Batzoglou TA: Paul Chen email: cs262-win2015-staff@lists.stanford.edu Tuesdays & Thursdays 12:50-2:05pm Clark S361 http://cs262.stanford.edu
Instructor: Welcome to CS262: Computational Genomics Serafim Batzoglou TA: Paul Chen email: cs262-win2015-staff@lists.stanford.edu Tuesdays & Thursdays 12:50-2:05pm Clark S361 http://cs262.stanford.edu
Pair Hidden Markov Models
 Pair Hidden Markov Models Scribe: Rishi Bedi Lecturer: Serafim Batzoglou January 29, 2015 1 Recap of HMMs alphabet: Σ = {b 1,...b M } set of states: Q = {1,..., K} transition probabilities: A = [a ij ]
Pair Hidden Markov Models Scribe: Rishi Bedi Lecturer: Serafim Batzoglou January 29, 2015 1 Recap of HMMs alphabet: Σ = {b 1,...b M } set of states: Q = {1,..., K} transition probabilities: A = [a ij ]
Sequence Alignments. Dynamic programming approaches, scoring, and significance. Lucy Skrabanek ICB, WMC January 31, 2013
 Sequence Alignments Dynamic programming approaches, scoring, and significance Lucy Skrabanek ICB, WMC January 31, 213 Sequence alignment Compare two (or more) sequences to: Find regions of conservation
Sequence Alignments Dynamic programming approaches, scoring, and significance Lucy Skrabanek ICB, WMC January 31, 213 Sequence alignment Compare two (or more) sequences to: Find regions of conservation
Statistical Machine Learning Methods for Bioinformatics II. Hidden Markov Model for Biological Sequences
 Statistical Machine Learning Methods for Bioinformatics II. Hidden Markov Model for Biological Sequences Jianlin Cheng, PhD Department of Computer Science University of Missouri 2008 Free for Academic
Statistical Machine Learning Methods for Bioinformatics II. Hidden Markov Model for Biological Sequences Jianlin Cheng, PhD Department of Computer Science University of Missouri 2008 Free for Academic
HMMs and biological sequence analysis
 HMMs and biological sequence analysis Hidden Markov Model A Markov chain is a sequence of random variables X 1, X 2, X 3,... That has the property that the value of the current state depends only on the
HMMs and biological sequence analysis Hidden Markov Model A Markov chain is a sequence of random variables X 1, X 2, X 3,... That has the property that the value of the current state depends only on the
Pairwise Alignment. Guan-Shieng Huang. Dept. of CSIE, NCNU. Pairwise Alignment p.1/55
 Pairwise Alignment Guan-Shieng Huang shieng@ncnu.edu.tw Dept. of CSIE, NCNU Pairwise Alignment p.1/55 Approach 1. Problem definition 2. Computational method (algorithms) 3. Complexity and performance Pairwise
Pairwise Alignment Guan-Shieng Huang shieng@ncnu.edu.tw Dept. of CSIE, NCNU Pairwise Alignment p.1/55 Approach 1. Problem definition 2. Computational method (algorithms) 3. Complexity and performance Pairwise
EECS730: Introduction to Bioinformatics
 EECS730: Introduction to Bioinformatics Lecture 03: Edit distance and sequence alignment Slides adapted from Dr. Shaojie Zhang (University of Central Florida) KUMC visit How many of you would like to attend
EECS730: Introduction to Bioinformatics Lecture 03: Edit distance and sequence alignment Slides adapted from Dr. Shaojie Zhang (University of Central Florida) KUMC visit How many of you would like to attend
InDel 3-5. InDel 8-9. InDel 3-5. InDel 8-9. InDel InDel 8-9
 Lecture 5 Alignment I. Introduction. For sequence data, the process of generating an alignment establishes positional homologies; that is, alignment provides the identification of homologous phylogenetic
Lecture 5 Alignment I. Introduction. For sequence data, the process of generating an alignment establishes positional homologies; that is, alignment provides the identification of homologous phylogenetic
Biochemistry 324 Bioinformatics. Pairwise sequence alignment
 Biochemistry 324 Bioinformatics Pairwise sequence alignment How do we compare genes/proteins? When we have sequenced a genome, we try and identify the function of unknown genes by finding a similar gene
Biochemistry 324 Bioinformatics Pairwise sequence alignment How do we compare genes/proteins? When we have sequenced a genome, we try and identify the function of unknown genes by finding a similar gene
Statistical Machine Learning Methods for Biomedical Informatics II. Hidden Markov Model for Biological Sequences
 Statistical Machine Learning Methods for Biomedical Informatics II. Hidden Markov Model for Biological Sequences Jianlin Cheng, PhD William and Nancy Thompson Missouri Distinguished Professor Department
Statistical Machine Learning Methods for Biomedical Informatics II. Hidden Markov Model for Biological Sequences Jianlin Cheng, PhD William and Nancy Thompson Missouri Distinguished Professor Department
Hidden Markov Models. x 1 x 2 x 3 x K
 Hidden Markov Models 1 1 1 1 2 2 2 2 K K K K x 1 x 2 x 3 x K Viterbi, Forward, Backward VITERBI FORWARD BACKWARD Initialization: V 0 (0) = 1 V k (0) = 0, for all k > 0 Initialization: f 0 (0) = 1 f k (0)
Hidden Markov Models 1 1 1 1 2 2 2 2 K K K K x 1 x 2 x 3 x K Viterbi, Forward, Backward VITERBI FORWARD BACKWARD Initialization: V 0 (0) = 1 V k (0) = 0, for all k > 0 Initialization: f 0 (0) = 1 f k (0)
CONCEPT OF SEQUENCE COMPARISON. Natapol Pornputtapong 18 January 2018
 CONCEPT OF SEQUENCE COMPARISON Natapol Pornputtapong 18 January 2018 SEQUENCE ANALYSIS - A ROSETTA STONE OF LIFE Sequence analysis is the process of subjecting a DNA, RNA or peptide sequence to any of
CONCEPT OF SEQUENCE COMPARISON Natapol Pornputtapong 18 January 2018 SEQUENCE ANALYSIS - A ROSETTA STONE OF LIFE Sequence analysis is the process of subjecting a DNA, RNA or peptide sequence to any of
Bio nformatics. Lecture 3. Saad Mneimneh
 Bio nformatics Lecture 3 Sequencing As before, DNA is cut into small ( 0.4KB) fragments and a clone library is formed. Biological experiments allow to read a certain number of these short fragments per
Bio nformatics Lecture 3 Sequencing As before, DNA is cut into small ( 0.4KB) fragments and a clone library is formed. Biological experiments allow to read a certain number of these short fragments per
5. MULTIPLE SEQUENCE ALIGNMENT BIOINFORMATICS COURSE MTAT
 5. MULTIPLE SEQUENCE ALIGNMENT BIOINFORMATICS COURSE MTAT.03.239 03.10.2012 ALIGNMENT Alignment is the task of locating equivalent regions of two or more sequences to maximize their similarity. Homology:
5. MULTIPLE SEQUENCE ALIGNMENT BIOINFORMATICS COURSE MTAT.03.239 03.10.2012 ALIGNMENT Alignment is the task of locating equivalent regions of two or more sequences to maximize their similarity. Homology:
Sequence Alignment: Scoring Schemes. COMP 571 Luay Nakhleh, Rice University
 Sequence Alignment: Scoring Schemes COMP 571 Luay Nakhleh, Rice University Scoring Schemes Recall that an alignment score is aimed at providing a scale to measure the degree of similarity (or difference)
Sequence Alignment: Scoring Schemes COMP 571 Luay Nakhleh, Rice University Scoring Schemes Recall that an alignment score is aimed at providing a scale to measure the degree of similarity (or difference)
20 Grundlagen der Bioinformatik, SS 08, D. Huson, May 27, Global and local alignment of two sequences using dynamic programming
 20 Grundlagen der Bioinformatik, SS 08, D. Huson, May 27, 2008 4 Pairwise alignment We will discuss: 1. Strings 2. Dot matrix method for comparing sequences 3. Edit distance 4. Global and local alignment
20 Grundlagen der Bioinformatik, SS 08, D. Huson, May 27, 2008 4 Pairwise alignment We will discuss: 1. Strings 2. Dot matrix method for comparing sequences 3. Edit distance 4. Global and local alignment
Week 10: Homology Modelling (II) - HHpred
 - HHpred") Week 10: Homology Modelling (II) - HHpred Course: Tools for Structural Biology Fabian Glaser BKU - Technion 1 2 Identify and align related structures by sequence methods is not an easy task All comparative
Week 10: Homology Modelling (II) - HHpred Course: Tools for Structural Biology Fabian Glaser BKU - Technion 1 2 Identify and align related structures by sequence methods is not an easy task All comparative
Sequence comparison: Score matrices
 Sequence comparison: Score matrices http://facultywashingtonedu/jht/gs559_2013/ Genome 559: Introduction to Statistical and omputational Genomics Prof James H Thomas FYI - informal inductive proof of best
Sequence comparison: Score matrices http://facultywashingtonedu/jht/gs559_2013/ Genome 559: Introduction to Statistical and omputational Genomics Prof James H Thomas FYI - informal inductive proof of best
Tools and Algorithms in Bioinformatics
 Tools and Algorithms in Bioinformatics GCBA815, Fall 2015 Week-4 BLAST Algorithm Continued Multiple Sequence Alignment Babu Guda, Ph.D. Department of Genetics, Cell Biology & Anatomy Bioinformatics and
Tools and Algorithms in Bioinformatics GCBA815, Fall 2015 Week-4 BLAST Algorithm Continued Multiple Sequence Alignment Babu Guda, Ph.D. Department of Genetics, Cell Biology & Anatomy Bioinformatics and
Pairwise & Multiple sequence alignments
 Pairwise & Multiple sequence alignments Urmila Kulkarni-Kale Bioinformatics Centre 411 007 urmila@bioinfo.ernet.in Basis for Sequence comparison Theory of evolution: gene sequences have evolved/derived
Pairwise & Multiple sequence alignments Urmila Kulkarni-Kale Bioinformatics Centre 411 007 urmila@bioinfo.ernet.in Basis for Sequence comparison Theory of evolution: gene sequences have evolved/derived
Sequence comparison: Score matrices. Genome 559: Introduction to Statistical and Computational Genomics Prof. James H. Thomas
 Sequence comparison: Score matrices Genome 559: Introduction to Statistical and omputational Genomics Prof James H Thomas FYI - informal inductive proof of best alignment path onsider the last step in
Sequence comparison: Score matrices Genome 559: Introduction to Statistical and omputational Genomics Prof James H Thomas FYI - informal inductive proof of best alignment path onsider the last step in
Sequence comparison: Score matrices. Genome 559: Introduction to Statistical and Computational Genomics Prof. James H. Thomas
 Sequence comparison: Score matrices Genome 559: Introduction to Statistical and omputational Genomics Prof James H Thomas Informal inductive proof of best alignment path onsider the last step in the best
Sequence comparison: Score matrices Genome 559: Introduction to Statistical and omputational Genomics Prof James H Thomas Informal inductive proof of best alignment path onsider the last step in the best
Lecture 4: Evolutionary Models and Substitution Matrices (PAM and BLOSUM)
") Bioinformatics II Probability and Statistics Universität Zürich and ETH Zürich Spring Semester 2009 Lecture 4: Evolutionary Models and Substitution Matrices (PAM and BLOSUM) Dr Fraser Daly adapted from
Bioinformatics II Probability and Statistics Universität Zürich and ETH Zürich Spring Semester 2009 Lecture 4: Evolutionary Models and Substitution Matrices (PAM and BLOSUM) Dr Fraser Daly adapted from
Introduction to Bioinformatics Algorithms Homework 3 Solution
 Introduction to Bioinformatics Algorithms Homework 3 Solution Saad Mneimneh Computer Science Hunter College of CUNY Problem 1: Concave penalty function We have seen in class the following recurrence for
Introduction to Bioinformatics Algorithms Homework 3 Solution Saad Mneimneh Computer Science Hunter College of CUNY Problem 1: Concave penalty function We have seen in class the following recurrence for
Single alignment: Substitution Matrix. 16 march 2017
 Single alignment: Substitution Matrix 16 march 2017 BLOSUM Matrix BLOSUM Matrix [2] (Blocks Amino Acid Substitution Matrices ) It is based on the amino acids substitutions observed in ~2000 conserved block
Single alignment: Substitution Matrix 16 march 2017 BLOSUM Matrix BLOSUM Matrix [2] (Blocks Amino Acid Substitution Matrices ) It is based on the amino acids substitutions observed in ~2000 conserved block
Homology Modeling (Comparative Structure Modeling) GBCB 5874: Problem Solving in GBCB
 GBCB 5874: Problem Solving in GBCB") Homology Modeling (Comparative Structure Modeling) Aims of Structural Genomics High-throughput 3D structure determination and analysis To determine or predict the 3D structures of all the proteins encoded
Homology Modeling (Comparative Structure Modeling) Aims of Structural Genomics High-throughput 3D structure determination and analysis To determine or predict the 3D structures of all the proteins encoded
EECS730: Introduction to Bioinformatics
 EECS730: Introduction to Bioinformatics Lecture 07: profile Hidden Markov Model http://bibiserv.techfak.uni-bielefeld.de/sadr2/databasesearch/hmmer/profilehmm.gif Slides adapted from Dr. Shaojie Zhang
EECS730: Introduction to Bioinformatics Lecture 07: profile Hidden Markov Model http://bibiserv.techfak.uni-bielefeld.de/sadr2/databasesearch/hmmer/profilehmm.gif Slides adapted from Dr. Shaojie Zhang
Bioinformatics. Scoring Matrices. David Gilbert Bioinformatics Research Centre
 Bioinformatics Scoring Matrices David Gilbert Bioinformatics Research Centre www.brc.dcs.gla.ac.uk Department of Computing Science, University of Glasgow Learning Objectives To explain the requirement
Bioinformatics Scoring Matrices David Gilbert Bioinformatics Research Centre www.brc.dcs.gla.ac.uk Department of Computing Science, University of Glasgow Learning Objectives To explain the requirement
3. SEQUENCE ANALYSIS BIOINFORMATICS COURSE MTAT
 3. SEQUENCE ANALYSIS BIOINFORMATICS COURSE MTAT.03.239 25.09.2012 SEQUENCE ANALYSIS IS IMPORTANT FOR... Prediction of function Gene finding the process of identifying the regions of genomic DNA that encode
3. SEQUENCE ANALYSIS BIOINFORMATICS COURSE MTAT.03.239 25.09.2012 SEQUENCE ANALYSIS IS IMPORTANT FOR... Prediction of function Gene finding the process of identifying the regions of genomic DNA that encode
Sequence Analysis '17- lecture 8. Multiple sequence alignment
 Sequence Analysis '17- lecture 8 Multiple sequence alignment Ex5 explanation How many random database search scores have e-values 10? (Answer: 10!) Why? e-value of x = m*p(s x), where m is the database
Sequence Analysis '17- lecture 8 Multiple sequence alignment Ex5 explanation How many random database search scores have e-values 10? (Answer: 10!) Why? e-value of x = m*p(s x), where m is the database
Multiple Sequence Alignment using Profile HMM
 Multiple Sequence Alignment using Profile HMM. based on Chapter 5 and Section 6.5 from Biological Sequence Analysis by R. Durbin et al., 1998 Acknowledgements: M.Sc. students Beatrice Miron, Oana Răţoi,
Multiple Sequence Alignment using Profile HMM. based on Chapter 5 and Section 6.5 from Biological Sequence Analysis by R. Durbin et al., 1998 Acknowledgements: M.Sc. students Beatrice Miron, Oana Răţoi,
Hidden Markov Models
 Hidden Markov Models Slides revised and adapted to Bioinformática 55 Engª Biomédica/IST 2005 Ana Teresa Freitas Forward Algorithm For Markov chains we calculate the probability of a sequence, P(x) How
Hidden Markov Models Slides revised and adapted to Bioinformática 55 Engª Biomédica/IST 2005 Ana Teresa Freitas Forward Algorithm For Markov chains we calculate the probability of a sequence, P(x) How
Scoring Matrices. Shifra Ben-Dor Irit Orr
 Scoring Matrices Shifra Ben-Dor Irit Orr Scoring matrices Sequence alignment and database searching programs compare sequences to each other as a series of characters. All algorithms (programs) for comparison
Scoring Matrices Shifra Ben-Dor Irit Orr Scoring matrices Sequence alignment and database searching programs compare sequences to each other as a series of characters. All algorithms (programs) for comparison
An Introduction to Bioinformatics Algorithms Hidden Markov Models
 Hidden Markov Models Outline 1. CG-Islands 2. The Fair Bet Casino 3. Hidden Markov Model 4. Decoding Algorithm 5. Forward-Backward Algorithm 6. Profile HMMs 7. HMM Parameter Estimation 8. Viterbi Training
Hidden Markov Models Outline 1. CG-Islands 2. The Fair Bet Casino 3. Hidden Markov Model 4. Decoding Algorithm 5. Forward-Backward Algorithm 6. Profile HMMs 7. HMM Parameter Estimation 8. Viterbi Training
Sequence analysis and comparison
 The aim with sequence identification: Sequence analysis and comparison Marjolein Thunnissen Lund September 2012 Is there any known protein sequence that is homologous to mine? Are there any other species
The aim with sequence identification: Sequence analysis and comparison Marjolein Thunnissen Lund September 2012 Is there any known protein sequence that is homologous to mine? Are there any other species
Module: Sequence Alignment Theory and Applications Session: Introduction to Searching and Sequence Alignment
 Module: Sequence Alignment Theory and Applications Session: Introduction to Searching and Sequence Alignment Introduction to Bioinformatics online course : IBT Jonathan Kayondo Learning Objectives Understand
Module: Sequence Alignment Theory and Applications Session: Introduction to Searching and Sequence Alignment Introduction to Bioinformatics online course : IBT Jonathan Kayondo Learning Objectives Understand
Sequence Alignment: A General Overview. COMP Fall 2010 Luay Nakhleh, Rice University
 Sequence Alignment: A General Overview COMP 571 - Fall 2010 Luay Nakhleh, Rice University Life through Evolution All living organisms are related to each other through evolution This means: any pair of
Sequence Alignment: A General Overview COMP 571 - Fall 2010 Luay Nakhleh, Rice University Life through Evolution All living organisms are related to each other through evolution This means: any pair of
Hidden Markov Models
 Hidden Markov Models Outline 1. CG-Islands 2. The Fair Bet Casino 3. Hidden Markov Model 4. Decoding Algorithm 5. Forward-Backward Algorithm 6. Profile HMMs 7. HMM Parameter Estimation 8. Viterbi Training
Hidden Markov Models Outline 1. CG-Islands 2. The Fair Bet Casino 3. Hidden Markov Model 4. Decoding Algorithm 5. Forward-Backward Algorithm 6. Profile HMMs 7. HMM Parameter Estimation 8. Viterbi Training
Hidden Markov Models. x 1 x 2 x 3 x K
 Hidden Markov Models 1 1 1 1 2 2 2 2 K K K K x 1 x 2 x 3 x K HiSeq X & NextSeq Viterbi, Forward, Backward VITERBI FORWARD BACKWARD Initialization: V 0 (0) = 1 V k (0) = 0, for all k > 0 Initialization:
Hidden Markov Models 1 1 1 1 2 2 2 2 K K K K x 1 x 2 x 3 x K HiSeq X & NextSeq Viterbi, Forward, Backward VITERBI FORWARD BACKWARD Initialization: V 0 (0) = 1 V k (0) = 0, for all k > 0 Initialization:
Today s Lecture: HMMs
 Today s Lecture: HMMs Definitions Examples Probability calculations WDAG Dynamic programming algorithms: Forward Viterbi Parameter estimation Viterbi training 1 Hidden Markov Models Probability models
Today s Lecture: HMMs Definitions Examples Probability calculations WDAG Dynamic programming algorithms: Forward Viterbi Parameter estimation Viterbi training 1 Hidden Markov Models Probability models
Pairwise sequence alignments
 Pairwise sequence alignments Volker Flegel VI, October 2003 Page 1 Outline Introduction Definitions Biological context of pairwise alignments Computing of pairwise alignments Some programs VI, October
Pairwise sequence alignments Volker Flegel VI, October 2003 Page 1 Outline Introduction Definitions Biological context of pairwise alignments Computing of pairwise alignments Some programs VI, October
Lecture 2: Pairwise Alignment. CG Ron Shamir
 Lecture 2: Pairwise Alignment 1 Main source 2 Why compare sequences? Human hexosaminidase A vs Mouse hexosaminidase A 3 www.mathworks.com/.../jan04/bio_genome.html Sequence Alignment עימוד רצפים The problem:
Lecture 2: Pairwise Alignment 1 Main source 2 Why compare sequences? Human hexosaminidase A vs Mouse hexosaminidase A 3 www.mathworks.com/.../jan04/bio_genome.html Sequence Alignment עימוד רצפים The problem:
An Introduction to Sequence Similarity ( Homology ) Searching
 Searching") An Introduction to Sequence Similarity ( Homology ) Searching Gary D. Stormo 1 UNIT 3.1 1 Washington University, School of Medicine, St. Louis, Missouri ABSTRACT Homologous sequences usually have the same,
An Introduction to Sequence Similarity ( Homology ) Searching Gary D. Stormo 1 UNIT 3.1 1 Washington University, School of Medicine, St. Louis, Missouri ABSTRACT Homologous sequences usually have the same,
Hidden Markov Models
 Hidden Markov Models Outline CG-islands The Fair Bet Casino Hidden Markov Model Decoding Algorithm Forward-Backward Algorithm Profile HMMs HMM Parameter Estimation Viterbi training Baum-Welch algorithm
Hidden Markov Models Outline CG-islands The Fair Bet Casino Hidden Markov Model Decoding Algorithm Forward-Backward Algorithm Profile HMMs HMM Parameter Estimation Viterbi training Baum-Welch algorithm
Lecture 8 Learning Sequence Motif Models Using Expectation Maximization (EM) Colin Dewey February 14, 2008
 Colin Dewey February 14, 2008") Lecture 8 Learning Sequence Motif Models Using Expectation Maximization (EM) Colin Dewey February 14, 2008 1 Sequence Motifs what is a sequence motif? a sequence pattern of biological significance typically
Lecture 8 Learning Sequence Motif Models Using Expectation Maximization (EM) Colin Dewey February 14, 2008 1 Sequence Motifs what is a sequence motif? a sequence pattern of biological significance typically
Substitution matrices
 Introduction to Bioinformatics Substitution matrices Jacques van Helden Jacques.van-Helden@univ-amu.fr Université d Aix-Marseille, France Lab. Technological Advances for Genomics and Clinics (TAGC, INSERM
Introduction to Bioinformatics Substitution matrices Jacques van Helden Jacques.van-Helden@univ-amu.fr Université d Aix-Marseille, France Lab. Technological Advances for Genomics and Clinics (TAGC, INSERM
Sequence Analysis 17: lecture 5. Substitution matrices Multiple sequence alignment
 Sequence Analysis 17: lecture 5 Substitution matrices Multiple sequence alignment Substitution matrices Used to score aligned positions, usually of amino acids. Expressed as the log-likelihood ratio of
Sequence Analysis 17: lecture 5 Substitution matrices Multiple sequence alignment Substitution matrices Used to score aligned positions, usually of amino acids. Expressed as the log-likelihood ratio of
Computational Biology
 Computational Biology Lecture 6 31 October 2004 1 Overview Scoring matrices (Thanks to Shannon McWeeney) BLAST algorithm Start sequence alignment 2 1 What is a homologous sequence? A homologous sequence,
Computational Biology Lecture 6 31 October 2004 1 Overview Scoring matrices (Thanks to Shannon McWeeney) BLAST algorithm Start sequence alignment 2 1 What is a homologous sequence? A homologous sequence,
Pairwise sequence alignments. Vassilios Ioannidis (From Volker Flegel )
") Pairwise sequence alignments Vassilios Ioannidis (From Volker Flegel ) Outline Introduction Definitions Biological context of pairwise alignments Computing of pairwise alignments Some programs Importance
Pairwise sequence alignments Vassilios Ioannidis (From Volker Flegel ) Outline Introduction Definitions Biological context of pairwise alignments Computing of pairwise alignments Some programs Importance
Introduction to Bioinformatics
 Introduction to Bioinformatics Jianlin Cheng, PhD Department of Computer Science Informatics Institute 2011 Topics Introduction Biological Sequence Alignment and Database Search Analysis of gene expression
Introduction to Bioinformatics Jianlin Cheng, PhD Department of Computer Science Informatics Institute 2011 Topics Introduction Biological Sequence Alignment and Database Search Analysis of gene expression
Hidden Markov Models. Main source: Durbin et al., Biological Sequence Alignment (Cambridge, 98)
") Hidden Markov Models Main source: Durbin et al., Biological Sequence Alignment (Cambridge, 98) 1 The occasionally dishonest casino A P A (1) = P A (2) = = 1/6 P A->B = P B->A = 1/10 B P B (1)=0.1... P
Hidden Markov Models Main source: Durbin et al., Biological Sequence Alignment (Cambridge, 98) 1 The occasionally dishonest casino A P A (1) = P A (2) = = 1/6 P A->B = P B->A = 1/10 B P B (1)=0.1... P
Alignment principles and homology searching using (PSI-)BLAST. Jaap Heringa Centre for Integrative Bioinformatics VU (IBIVU)
BLAST. Jaap Heringa Centre for Integrative Bioinformatics VU (IBIVU)") Alignment principles and homology searching using (PSI-)BLAST Jaap Heringa Centre for Integrative Bioinformatics VU (IBIVU) http://ibivu.cs.vu.nl Bioinformatics Nothing in Biology makes sense except in
Alignment principles and homology searching using (PSI-)BLAST Jaap Heringa Centre for Integrative Bioinformatics VU (IBIVU) http://ibivu.cs.vu.nl Bioinformatics Nothing in Biology makes sense except in
Stephen Scott.
 1 / 21 sscott@cse.unl.edu 2 / 21 Introduction Designed to model (profile) a multiple alignment of a protein family (e.g., Fig. 5.1) Gives a probabilistic model of the proteins in the family Useful for
1 / 21 sscott@cse.unl.edu 2 / 21 Introduction Designed to model (profile) a multiple alignment of a protein family (e.g., Fig. 5.1) Gives a probabilistic model of the proteins in the family Useful for
Pairwise sequence alignment and pair hidden Markov models
 Pairwise sequence alignment and pair hidden Markov models Martin C. Frith April 13, 2012 ntroduction Pairwise alignment and pair hidden Markov models (phmms) are basic textbook fare [2]. However, there
Pairwise sequence alignment and pair hidden Markov models Martin C. Frith April 13, 2012 ntroduction Pairwise alignment and pair hidden Markov models (phmms) are basic textbook fare [2]. However, there
Chapter 5. Proteomics and the analysis of protein sequence Ⅱ
 Proteomics Chapter 5. Proteomics and the analysis of protein sequence Ⅱ 1 Pairwise similarity searching (1) Figure 5.5: manual alignment One of the amino acids in the top sequence has no equivalent and
Proteomics Chapter 5. Proteomics and the analysis of protein sequence Ⅱ 1 Pairwise similarity searching (1) Figure 5.5: manual alignment One of the amino acids in the top sequence has no equivalent and
Multiple Sequence Alignment, Gunnar Klau, December 9, 2005, 17:
 Multiple Sequence Alignment, Gunnar Klau, December 9, 2005, 17:50 5001 5 Multiple Sequence Alignment The first part of this exposition is based on the following sources, which are recommended reading:
Multiple Sequence Alignment, Gunnar Klau, December 9, 2005, 17:50 5001 5 Multiple Sequence Alignment The first part of this exposition is based on the following sources, which are recommended reading:
In-Depth Assessment of Local Sequence Alignment
 2012 International Conference on Environment Science and Engieering IPCBEE vol.3 2(2012) (2012)IACSIT Press, Singapoore In-Depth Assessment of Local Sequence Alignment Atoosa Ghahremani and Mahmood A.
2012 International Conference on Environment Science and Engieering IPCBEE vol.3 2(2012) (2012)IACSIT Press, Singapoore In-Depth Assessment of Local Sequence Alignment Atoosa Ghahremani and Mahmood A.
Markov Chains and Hidden Markov Models. = stochastic, generative models
 Markov Chains and Hidden Markov Models = stochastic, generative models (Drawing heavily from Durbin et al., Biological Sequence Analysis) BCH339N Systems Biology / Bioinformatics Spring 2016 Edward Marcotte,
Markov Chains and Hidden Markov Models = stochastic, generative models (Drawing heavily from Durbin et al., Biological Sequence Analysis) BCH339N Systems Biology / Bioinformatics Spring 2016 Edward Marcotte,
Copyright 2000 N. AYDIN. All rights reserved. 1
 Introduction to Bioinformatics Prof. Dr. Nizamettin AYDIN naydin@yildiz.edu.tr Multiple Sequence Alignment Outline Multiple sequence alignment introduction to msa methods of msa progressive global alignment
Introduction to Bioinformatics Prof. Dr. Nizamettin AYDIN naydin@yildiz.edu.tr Multiple Sequence Alignment Outline Multiple sequence alignment introduction to msa methods of msa progressive global alignment
Lecture 14: Multiple Sequence Alignment (Gene Finding, Conserved Elements) Scribe: John Ekins
 Scribe: John Ekins") Lecture 14: Multiple Sequence Alignment (Gene Finding, Conserved Elements) 2 19 2015 Scribe: John Ekins Multiple Sequence Alignment Given N sequences x 1, x 2,, x N : Insert gaps in each of the sequences
Lecture 14: Multiple Sequence Alignment (Gene Finding, Conserved Elements) 2 19 2015 Scribe: John Ekins Multiple Sequence Alignment Given N sequences x 1, x 2,, x N : Insert gaps in each of the sequences
Sequence Analysis, '18 -- lecture 9. Families and superfamilies. Sequence weights. Profiles. Logos. Building a representative model for a gene.
 Sequence Analysis, '18 -- lecture 9 Families and superfamilies. Sequence weights. Profiles. Logos. Building a representative model for a gene. How can I represent thousands of homolog sequences in a compact
Sequence Analysis, '18 -- lecture 9 Families and superfamilies. Sequence weights. Profiles. Logos. Building a representative model for a gene. How can I represent thousands of homolog sequences in a compact
Sequence Analysis '17 -- lecture 7
 Sequence Analysis '17 -- lecture 7 Significance E-values How significant is that? Please give me a number for......how likely the data would not have been the result of chance,......as opposed to......a
Sequence Analysis '17 -- lecture 7 Significance E-values How significant is that? Please give me a number for......how likely the data would not have been the result of chance,......as opposed to......a
Sequence Bioinformatics. Multiple Sequence Alignment Waqas Nasir
 Sequence Bioinformatics Multiple Sequence Alignment Waqas Nasir 2010-11-12 Multiple Sequence Alignment One amino acid plays coy; a pair of homologous sequences whisper; many aligned sequences shout out
Sequence Bioinformatics Multiple Sequence Alignment Waqas Nasir 2010-11-12 Multiple Sequence Alignment One amino acid plays coy; a pair of homologous sequences whisper; many aligned sequences shout out
Lecture 2, 5/12/2001: Local alignment the Smith-Waterman algorithm. Alignment scoring schemes and theory: substitution matrices and gap models
 Lecture 2, 5/12/2001: Local alignment the Smith-Waterman algorithm Alignment scoring schemes and theory: substitution matrices and gap models 1 Local sequence alignments Local sequence alignments are necessary
Lecture 2, 5/12/2001: Local alignment the Smith-Waterman algorithm Alignment scoring schemes and theory: substitution matrices and gap models 1 Local sequence alignments Local sequence alignments are necessary
1.5 Sequence alignment
 1.5 Sequence alignment The dramatic increase in the number of sequenced genomes and proteomes has lead to development of various bioinformatic methods and algorithms for extracting information (data mining)
1.5 Sequence alignment The dramatic increase in the number of sequenced genomes and proteomes has lead to development of various bioinformatic methods and algorithms for extracting information (data mining)
Collected Works of Charles Dickens
 Collected Works of Charles Dickens A Random Dickens Quote If there were no bad people, there would be no good lawyers. Original Sentence It was a dark and stormy night; the night was dark except at sunny
Collected Works of Charles Dickens A Random Dickens Quote If there were no bad people, there would be no good lawyers. Original Sentence It was a dark and stormy night; the night was dark except at sunny
HMM for modeling aligned multiple sequences: phylo-hmm & multivariate HMM
 I529: Machine Learning in Bioinformatics (Spring 2017) HMM for modeling aligned multiple sequences: phylo-hmm & multivariate HMM Yuzhen Ye School of Informatics and Computing Indiana University, Bloomington
I529: Machine Learning in Bioinformatics (Spring 2017) HMM for modeling aligned multiple sequences: phylo-hmm & multivariate HMM Yuzhen Ye School of Informatics and Computing Indiana University, Bloomington
Lecture 4: Hidden Markov Models: An Introduction to Dynamic Decision Making. November 11, 2010
 Hidden Lecture 4: Hidden : An Introduction to Dynamic Decision Making November 11, 2010 Special Meeting 1/26 Markov Model Hidden When a dynamical system is probabilistic it may be determined by the transition
Hidden Lecture 4: Hidden : An Introduction to Dynamic Decision Making November 11, 2010 Special Meeting 1/26 Markov Model Hidden When a dynamical system is probabilistic it may be determined by the transition
Homology Modeling. Roberto Lins EPFL - summer semester 2005
 Homology Modeling Roberto Lins EPFL - summer semester 2005 Disclaimer: course material is mainly taken from: P.E. Bourne & H Weissig, Structural Bioinformatics; C.A. Orengo, D.T. Jones & J.M. Thornton,
Homology Modeling Roberto Lins EPFL - summer semester 2005 Disclaimer: course material is mainly taken from: P.E. Bourne & H Weissig, Structural Bioinformatics; C.A. Orengo, D.T. Jones & J.M. Thornton,
BIO 285/CSCI 285/MATH 285 Bioinformatics Programming Lecture 8 Pairwise Sequence Alignment 2 And Python Function Instructor: Lei Qian Fisk University
 BIO 285/CSCI 285/MATH 285 Bioinformatics Programming Lecture 8 Pairwise Sequence Alignment 2 And Python Function Instructor: Lei Qian Fisk University Measures of Sequence Similarity Alignment with dot
BIO 285/CSCI 285/MATH 285 Bioinformatics Programming Lecture 8 Pairwise Sequence Alignment 2 And Python Function Instructor: Lei Qian Fisk University Measures of Sequence Similarity Alignment with dot
Comparative Gene Finding. BMI/CS 776 Spring 2015 Colin Dewey
 Comparative Gene Finding BMI/CS 776 www.biostat.wisc.edu/bmi776/ Spring 2015 Colin Dewey cdewey@biostat.wisc.edu Goals for Lecture the key concepts to understand are the following: using related genomes
Comparative Gene Finding BMI/CS 776 www.biostat.wisc.edu/bmi776/ Spring 2015 Colin Dewey cdewey@biostat.wisc.edu Goals for Lecture the key concepts to understand are the following: using related genomes
3/1/17. Content. TWINSCAN model. Example. TWINSCAN algorithm. HMM for modeling aligned multiple sequences: phylo-hmm & multivariate HMM
 I529: Machine Learning in Bioinformatics (Spring 2017) Content HMM for modeling aligned multiple sequences: phylo-hmm & multivariate HMM Yuzhen Ye School of Informatics and Computing Indiana University,
I529: Machine Learning in Bioinformatics (Spring 2017) Content HMM for modeling aligned multiple sequences: phylo-hmm & multivariate HMM Yuzhen Ye School of Informatics and Computing Indiana University,
Bioinformatics and BLAST
 Bioinformatics and BLAST Overview Recap of last time Similarity discussion Algorithms: Needleman-Wunsch Smith-Waterman BLAST Implementation issues and current research Recap from Last Time Genome consists
Bioinformatics and BLAST Overview Recap of last time Similarity discussion Algorithms: Needleman-Wunsch Smith-Waterman BLAST Implementation issues and current research Recap from Last Time Genome consists
Pairwise alignment. 2.1 Introduction GSAQVKGHGKKVADALTNAVAHVDDMPNALSALSD----LHAHKL
 2 Pairwise alignment 2.1 Introduction The most basic sequence analysis task is to ask if two sequences are related. This is usually done by first aligning the sequences (or parts of them) and then deciding
2 Pairwise alignment 2.1 Introduction The most basic sequence analysis task is to ask if two sequences are related. This is usually done by first aligning the sequences (or parts of them) and then deciding
8 Grundlagen der Bioinformatik, SS 09, D. Huson, April 28, 2009
 8 Grundlagen der Bioinformatik, SS 09, D. Huson, April 28, 2009 2 Pairwise alignment We will discuss: 1. Strings 2. Dot matrix method for comparing sequences 3. Edit distance and alignment 4. The number
8 Grundlagen der Bioinformatik, SS 09, D. Huson, April 28, 2009 2 Pairwise alignment We will discuss: 1. Strings 2. Dot matrix method for comparing sequences 3. Edit distance and alignment 4. The number
Lecture 1, 31/10/2001: Introduction to sequence alignment. The Needleman-Wunsch algorithm for global sequence alignment: description and properties
 Lecture 1, 31/10/2001: Introduction to sequence alignment The Needleman-Wunsch algorithm for global sequence alignment: description and properties 1 Computational sequence-analysis The major goal of computational
Lecture 1, 31/10/2001: Introduction to sequence alignment The Needleman-Wunsch algorithm for global sequence alignment: description and properties 1 Computational sequence-analysis The major goal of computational
C E N T R. Introduction to bioinformatics 2007 E B I O I N F O R M A T I C S V U F O R I N T. Lecture 5 G R A T I V. Pair-wise Sequence Alignment
 C E N T R E F O R I N T E G R A T I V E B I O I N F O R M A T I C S V U Introduction to bioinformatics 2007 Lecture 5 Pair-wise Sequence Alignment Bioinformatics Nothing in Biology makes sense except in
C E N T R E F O R I N T E G R A T I V E B I O I N F O R M A T I C S V U Introduction to bioinformatics 2007 Lecture 5 Pair-wise Sequence Alignment Bioinformatics Nothing in Biology makes sense except in
Sequence Alignment Techniques and Their Uses
 Sequence Alignment Techniques and Their Uses Sarah Fiorentino Since rapid sequencing technology and whole genomes sequencing, the amount of sequence information has grown exponentially. With all of this
Sequence Alignment Techniques and Their Uses Sarah Fiorentino Since rapid sequencing technology and whole genomes sequencing, the amount of sequence information has grown exponentially. With all of this
Global alignments - review
 Global alignments - review Take two sequences: X[j] and Y[j] M[i-1, j-1] ± 1 M[i, j] = max M[i, j-1] 2 M[i-1, j] 2 The best alignment for X[1 i] and Y[1 j] is called M[i, j] X[j] Initiation: M[,]= pply
Global alignments - review Take two sequences: X[j] and Y[j] M[i-1, j-1] ± 1 M[i, j] = max M[i, j-1] 2 M[i-1, j] 2 The best alignment for X[1 i] and Y[1 j] is called M[i, j] X[j] Initiation: M[,]= pply
Alignment & BLAST. By: Hadi Mozafari KUMS
 Alignment & BLAST By: Hadi Mozafari KUMS SIMILARITY - ALIGNMENT Comparison of primary DNA or protein sequences to other primary or secondary sequences Expecting that the function of the similar sequence
Alignment & BLAST By: Hadi Mozafari KUMS SIMILARITY - ALIGNMENT Comparison of primary DNA or protein sequences to other primary or secondary sequences Expecting that the function of the similar sequence
Lecture 4: Evolutionary models and substitution matrices (PAM and BLOSUM).
.") 1 Bioinformatics: In-depth PROBABILITY & STATISTICS Spring Semester 2011 University of Zürich and ETH Zürich Lecture 4: Evolutionary models and substitution matrices (PAM and BLOSUM). Dr. Stefanie Muff
1 Bioinformatics: In-depth PROBABILITY & STATISTICS Spring Semester 2011 University of Zürich and ETH Zürich Lecture 4: Evolutionary models and substitution matrices (PAM and BLOSUM). Dr. Stefanie Muff
Similarity searching summary (2)
") Similarity searching / sequence alignment summary Biol4230 Thurs, February 22, 2016 Bill Pearson wrp@virginia.edu 4-2818 Pinn 6-057 What have we covered? Homology excess similiarity but no excess similarity
Similarity searching / sequence alignment summary Biol4230 Thurs, February 22, 2016 Bill Pearson wrp@virginia.edu 4-2818 Pinn 6-057 What have we covered? Homology excess similiarity but no excess similarity