The MCSCF Method *, Molecular Orbitals, Reference Spaces and COLUMBUS Input
|
|
|
- Joel Horton
- 5 years ago
- Views:
Transcription
1 The MCSCF Method *, Molecular Orbitals, Reference Spaces and COLUMBUS Input Hans Lischka University of Vienna *Excerpt of a course presented by R. Shepard, Argonne National Laboratory, at the Workshop on Theoretical Chemistry, Feb. 2004, Mariapfarr, Austria 1
2 MCSCF Approach Single-configuration wave function: Multi-configuration wave function: Ψ Ψ SR MR = = Φ Φ 1 1 K c I Ψ Φ Φ I i i { Φ } i KΦ n Φ n In a MCSCF calculation two types of optimizations have to be performed: (i) (ii) w.r.t. the expansion coefficients of the CSFs and w.r.t. the MO coefficients 2
3 Orbital Variation Exponential parametrization of the transformation matrix Φ = Φ 0 U=χC 0 U with fixed C 0 such that C 0T SC 0 =1 U must be orthogonal Let U=exp(K) with K= K T => N(N-1)/2 independent unconstrained parameters K pq (e.g. for p>q) Kˆ = = r, s r> s K K rs rs E Tˆ rs rs = r> s K mc; Φ = exp( Kˆ ) mc; Φ rs ( E E ) 0 rs sr 3
4 CSF Variation Ncsf 1 P ˆ = p ( n n 0 0 n ) = p ˆ n n=1 Ncsf Ncsf 1 n=1 0 = c n0 n, m = c nm n with m 0 = 0 n=1 mc = exp( ˆ P )0 Ncsf n=1 P n ρ = mc ˆ H mc = 0 exp( ˆ P ) ˆ H exp( ˆ P )0 4
5 Combined Orbital and CSF Variation mc;φ = exp( ˆ K ) mc;φ 0 = exp( ˆ K )exp( ˆ P )0;φ 0 ρ = mc;φ ˆ H mc;φ = mc;φ 0 exp( ˆ K ) ˆ H exp( ˆ K ) mc;φ 0 = 0;φ 0 exp( ˆ P )exp( ˆ K ) ˆ H exp( ˆ K )exp( ˆ P )0;φ 0 5
6 Energy Variation First derivatives ρ K rs K= 0 p= 0 ρ p n K= 0 p= 0 w rs = 0;φ 0 v n = 0;φ 0 [ H ˆ, T ˆ rs ]0;φ 0 [ H ˆ, P ˆ n ]0;φ 0 6
7 Energy Variation II Second derivatives 2 ρ K pq K rs K= 0 2 ρ p= 0 p m p n K= 0 p= 0 [ T ] + ˆ rs B pq,rs = 1 0;φ 0 [ H ˆ, T ˆ 2 ], ˆ pq [ P ] + ˆ n M mn = 1 0;φ 0 [ H ˆ, P ˆ 2 ], ˆ m [ H, T ˆ ], T ˆ ] 0;φ 0 rs pq [ H, P ˆ ], P ˆ ] 0;φ 0 n m 2 ρ K pq p n K= 0 p= 0 C pq,n = 0;φ 0 [ H ˆ, T ˆ ], P ˆ ] 0;φ 0 pq n 7
8 8 Energy Derivatives III ( ) ( ) ( ) = = p k M C C B p k v w p k Cp k Mp p Bk k v p w k p k T T T T T T T T T T E E,,, ρ Newton-Raphson type procedure (second order convergence) Improvements of convergence radius (estimate of higher order terms)
9 Molecular Orbitals and MCSCF The correct form of the MOs is crucial for the subsequent CI calculation Balancing several electronic states simultaneously state averaging (SA) Choice of the orbital and CSF spaces Complete Active Space (CAS) all possible CSFs are constructed within a given space Restricted Active Space (RAS) only n-tuple holes are allowed (n-fold excitations from doubly occupied space) Auxiliary space (AUX) n-fold excitations into an origianlly empty orbital space 9
10 Orbital Spaces AUX CAS n-electrons RAS n-tuple excitations 10
11 Orbital Spaces II Restricted direct product spaces Generalized Valence Bond Perfect Pairing (GVB-PP) wavefunction 2 el. in 2 orb. n Each electron pair is restricted to singlet coupling localized orbitals 11



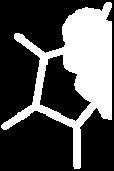
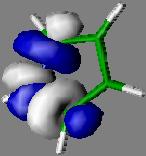

12 Pyrrole 12

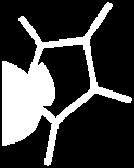







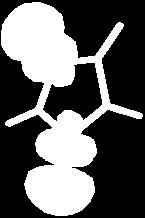






13 Pyrrole II 13
14 $COLUMBUS/colinp: Columbus Input COLUMBUS INPUT FACILITY main menu options -> 1) Integral program input (for argos/dalton/turbocol) 2) SCF input 3) MCSCF input 4) CI input 5) Set up job control 6) Exit the input facility 14
15 colinp input I Integral input: Cartesian geometry, choice of basis set from library, select Dalton integral program SCF input: number of occupied orbital per irred. rep. MCSCF input: Enter number of DRTS [1-8]: The distinct row table describes the wave function in the unitary group approach (UGA). For each class of wavefunctions (e.g. for each symmetry, for each multiplicity) we need one DRT. number of electrons for DRT #1 multiplicity for DRT #1 spatial symmetry for DRT #1: irreps are numbered (according to ordering in AO integral section) 15
16 colinp input II excitation level (cas,ras)->aux: 0 excitation level ras->(cas,aux): 0 number of doubly occupied orbitals per irrep:?? number of CAS orbitals per irrep:?? Apply add. group restrictions for DRT 1 [y n]: n Input for characterization of wavefunction finished Input for MCSCF procedure: state-averaging! 16
17 Ethylene CAS(2,2) Two SCF orbitals (one closed shell, occupied and one virtual orbital) are moved to the CAS CAS(2el,2orb) Closed shell SCF CAS(2el,2orb): 1b 2g 1b 3u π* orbital: virtual π orbital σ orbitals: (14 el., 7 orb.) Doubly occ. (DOCC) 1b 2g 1b 3u π-cas DOCC (14el,7orb) 17








18 Ethylene Orbitals 1a g 1b 1u 2a g 1b 2u 2b 1u y 3a g z 18

")
 19")
19 Ethylene Orbitals II 1b 3g 1b 3u (π) 1b 2g (π*) 19
20 Representation as symmetry table for input into COLUMBUS SM1 Orbital occupation of ethylene CAS(2,2), D2h System: C2H4, Point Group: D2h, N. Electrons: 16, Multiplicity: 1 Level: MR-CISD/SA-3-CAS(2,2) IRREP a g b 3u b 2u b 1g b 1u b 2g b 3g a u SCF DOCC 3 1 (π) MCSCF DOCC RAS CAS 0 1 (π) (π*) 0 0 AUX MRCI FC FV DOCC ACT AUX INT
21 State Multiplicity N. electrons Symmetry (A g ) (π) (B 1u ) (π) 1 (π ) (A g ) (π ) 2 Number of distinct row tables (DRTs): 2 21
22 colinp input III 1) Def. of CI wave function (data from MCSCF) 2) Def. of CI wave function for geom.opt (data from MCSCF) 3) Def. of CI wave function - one-drt case 4) Def. of CI wave function - one-drt case (geom.opt.) 5) Definition of CI wave function - multiple-drt case 6) Definition of CI wave function - multiple-drt case (NAD coupl.) 7) Skip DRT input (old input files in the current directory) 22
23 colinp input IV Input for the reference wavefunction: procedure similar to MCSCF case number of frozen core orbitals per irrep number of frozen virt. orbitals per irrep: 0 0 number of internal(=docc+active+aux) orbitals per irrep ref doubly occ orbitals per irrep auxiliary internal orbitals per irrep Enter the excitation level (0,1,2): 2 Generalized interacting space restrictions [y n]: y Enter the allowed reference symmetries: accept default Apply additional group restrictions for DRT [y n]: n 23
24 Ethylene: MR-CISD input: CAS(2,2) reference space This reference space is identical to the CAS(2,2) of the MCSCF calculation. Core orbitals (1a g and 1b 1u are moved to the frozen core (FC) space All single and double excitations are constructed from all references IRREP a g b 3u b 2u b 1g b 1u b 2g b 3g a u SCF DOCC 3 1 (π) MCSCF DOCC RAS CAS 0 1 (π) (π*) 0 0 AUX MRCI FC FV DOCC ACT 0 1 (π) (π*) 0 0 AUX INT
25 colinp input V Type of calculation: CI [Y] AQCC [N] AQCC-LRT [N] LRT shift: LRTSHIFT [0 ] State(s) to be optimized NROOT [1 ] ROOT TO FOLLOW [0] Reference space diagonalization INCORE[Y] NITER [ ] RTOL [ ] Bk-procedure: NITER [1 ] MINSUB [1 ] MAXSUB [6 RTOL [1e-3 ] CI/AQCC procedure: NITER [20 ] MINSUB [1 ] MAXSUB [6 ] RTOL [1e-3 ] FINISHED [ ] 25
26 colinp input VI 5) Set up job control: 1) Job control for single point or gradient calculation 2) Potential energy curve for one int. coordinate 3) Vibrational frequencies and force constants 4) Exit 1) single point calculation 26
27 colinp input VII 1) (Done with selections) 2) [ ] SCF 3) [ ] MCSCF 4) [ ] transition moments for MCSCF 5) [ ] standard MR-CISD with one DRT (ciudg) 6) [ ] standard MR-CISD with several DRTs (ciudg) 7) [ ] transition moments for MR-CISD 8) [ ] <L> value calculation for MR-CISD 9) [ ] parallel MR-CISD (pciudg) 10) [ ] sequential version of pciudg (sciudg) 11) [ ] one-electron properties for all methods 12) [ ] convert MOs into molden format 13) [ ] finite field calculation for all methods 27
28 Executing Columbus & Results $COLUMBUS/runc m >&runls & This command will execute a Columbus calculation according to the specifications given in the colinp input. Note: Columbus consists of a collection of individual programs communicating via files. Getting results: see directories LISTINGS, MOLDEN and MOCOEFS (see WORK for actual calculation) Each program step usually has its own listing: e.g. mcscfls, mcscfsm; ciudgls, ciudgsm, More information under: ntation_main.html item usage 28
A TUTORIAL FOR COLUMBUS
 A TUTORIAL FOR COLUMBUS Bernhard Sellner, Jaroslaw Szymczak, Felix Plasser, Reed Nieman, Mario Barbatti Institute for Theoretical Chemistry University of Vienna www.univie.ac.at/columbus/tutorial Vienna,
A TUTORIAL FOR COLUMBUS Bernhard Sellner, Jaroslaw Szymczak, Felix Plasser, Reed Nieman, Mario Barbatti Institute for Theoretical Chemistry University of Vienna www.univie.ac.at/columbus/tutorial Vienna,
The COLUMBUS Project - General Purpose Ab Initio Quantum Chemistry. I. Background and Overview
 The COLUMBUS Project - General Purpose Ab Initio Quantum Chemistry I. Background and Overview Ron Shepard Chemistry Division Argonne National Laboratory CScADS Workshop, Snowbird, Utah, July 23, 2007 Quantum
The COLUMBUS Project - General Purpose Ab Initio Quantum Chemistry I. Background and Overview Ron Shepard Chemistry Division Argonne National Laboratory CScADS Workshop, Snowbird, Utah, July 23, 2007 Quantum
Electron Correlation Methods
 Electron Correlation Methods HF method: electron-electron interaction is replaced by an average interaction E HF c = E 0 E HF E 0 exact ground state energy E HF HF energy for a given basis set HF E c
Electron Correlation Methods HF method: electron-electron interaction is replaced by an average interaction E HF c = E 0 E HF E 0 exact ground state energy E HF HF energy for a given basis set HF E c
Multiconfigurational Quantum Chemistry. Björn O. Roos as told by RL Department of Theoretical Chemistry Chemical Center Lund University Sweden
 Multiconfigurational Quantum Chemistry Björn O. Roos as told by RL Department of Theoretical Chemistry Chemical Center Lund University Sweden April 20, 2009 1 The Slater determinant Using the spin-orbitals,
Multiconfigurational Quantum Chemistry Björn O. Roos as told by RL Department of Theoretical Chemistry Chemical Center Lund University Sweden April 20, 2009 1 The Slater determinant Using the spin-orbitals,
COLUMBUS in RIO. A Quantum Chemistry Course on Multireference Methods, Energy Surfaces, Excited States, and Dynamics. Nov Dec.
 COLUMBUS in RIO A Quantum Chemistry Course on Multireference Methods, Energy Surfaces, Excited States, and Dynamics Nov. 27 - Dec. 02, 2005 Instituto Militar de Engenharia Rio de Janeiro - Brazil http://www.univie.ac.at/columbus/rio
COLUMBUS in RIO A Quantum Chemistry Course on Multireference Methods, Energy Surfaces, Excited States, and Dynamics Nov. 27 - Dec. 02, 2005 Instituto Militar de Engenharia Rio de Janeiro - Brazil http://www.univie.ac.at/columbus/rio
MRCI calculations in MOLPRO
 1 MRCI calculations in MOLPRO Molpro is a software package written in Fortran and maintained by H.J. Werner and P.J. Knowles. It is often used for performing sophisticated electronic structure calculations,
1 MRCI calculations in MOLPRO Molpro is a software package written in Fortran and maintained by H.J. Werner and P.J. Knowles. It is often used for performing sophisticated electronic structure calculations,
Introduction to multiconfigurational quantum chemistry. Emmanuel Fromager
 Institut de Chimie, Strasbourg, France Page 1 Emmanuel Fromager Institut de Chimie de Strasbourg - Laboratoire de Chimie Quantique - Université de Strasbourg /CNRS M2 lecture, Strasbourg, France. Notations
Institut de Chimie, Strasbourg, France Page 1 Emmanuel Fromager Institut de Chimie de Strasbourg - Laboratoire de Chimie Quantique - Université de Strasbourg /CNRS M2 lecture, Strasbourg, France. Notations
CASSCF and NEVPT2 calculations: Ground and excited states of multireference systems. A case study of Ni(CO)4 and the magnetic system NArO
4 and the magnetic system NArO") CASSCF and NEVPT2 calculations: Ground and excited states of multireference systems. A case study of Ni(CO)4 and the magnetic system NArO The ground states of many molecules are often well described by
CASSCF and NEVPT2 calculations: Ground and excited states of multireference systems. A case study of Ni(CO)4 and the magnetic system NArO The ground states of many molecules are often well described by
Extended Wavefunction Analysis for Multireference Methods
 Extended Wavefunction Analysis for Multireference Methods Felix Plasser González Research Group Institute for Theoretical Chemistry, University of Vienna, Austria Vienna, 1 st April 2016 Introduction Analysis
Extended Wavefunction Analysis for Multireference Methods Felix Plasser González Research Group Institute for Theoretical Chemistry, University of Vienna, Austria Vienna, 1 st April 2016 Introduction Analysis
AN INTRODUCTION TO QUANTUM CHEMISTRY. Mark S. Gordon Iowa State University
 AN INTRODUCTION TO QUANTUM CHEMISTRY Mark S. Gordon Iowa State University 1 OUTLINE Theoretical Background in Quantum Chemistry Overview of GAMESS Program Applications 2 QUANTUM CHEMISTRY In principle,
AN INTRODUCTION TO QUANTUM CHEMISTRY Mark S. Gordon Iowa State University 1 OUTLINE Theoretical Background in Quantum Chemistry Overview of GAMESS Program Applications 2 QUANTUM CHEMISTRY In principle,
Advanced usage of MOLCAS. Ex. 1. Usage of Symmetry in Molcas
 Advanced usage of MOLCAS Ex 1. Symmetry in Molcas (20 min). Ex 2. Transition state optimization (30 min). Ex 3. Ways to run RASSCF program (30 min). Ex 4. Optional. Using EXPBAS module (20 min) Ex 5. Optional.
Advanced usage of MOLCAS Ex 1. Symmetry in Molcas (20 min). Ex 2. Transition state optimization (30 min). Ex 3. Ways to run RASSCF program (30 min). Ex 4. Optional. Using EXPBAS module (20 min) Ex 5. Optional.
Accurate description of potential energy surfaces by ab initio methods : a review and application to ozone
 Accurate description of potential energy surfaces by ab initio methods : a review and application to ozone Péter G. Szalay Laboratory of Theoretical Chemistry Institute of Chemistry Eötvös Loránd University,
Accurate description of potential energy surfaces by ab initio methods : a review and application to ozone Péter G. Szalay Laboratory of Theoretical Chemistry Institute of Chemistry Eötvös Loránd University,
Special 5: Wavefunction Analysis and Visualization
 Special 5: Wavefunction Analysis and Visualization Felix Plasser Institute for Theoretical Chemistry, University of Vienna COLUMBUS in China Tianjin, October 10 14, 2016 F. Plasser Wavefunction Analysis
Special 5: Wavefunction Analysis and Visualization Felix Plasser Institute for Theoretical Chemistry, University of Vienna COLUMBUS in China Tianjin, October 10 14, 2016 F. Plasser Wavefunction Analysis
Appendix D Simulating Spectroscopic Bands Using Gaussian and PGopher
 429 Appendix D Simulating Spectroscopic Bands Using Gaussian and PGopher This appendix contains methods for using Gaussian 09 121 and PGopher 120 to simulate vibrational and electronic bands of molecules.
429 Appendix D Simulating Spectroscopic Bands Using Gaussian and PGopher This appendix contains methods for using Gaussian 09 121 and PGopher 120 to simulate vibrational and electronic bands of molecules.
AN INTRODUCTION TO MCSCF: PART 2
 AN INTRODUCTION TO MCSCF: PART 2 ORBITAL APPROXIMATION Hartree product (hp) expressed as a product of spinorbitals ψ ι = φ i σ i φ i = space orbital, σ i = spin function (α,β) Pauli Principle requires
AN INTRODUCTION TO MCSCF: PART 2 ORBITAL APPROXIMATION Hartree product (hp) expressed as a product of spinorbitals ψ ι = φ i σ i φ i = space orbital, σ i = spin function (α,β) Pauli Principle requires
Introduction to Computational Chemistry
 Introduction to Computational Chemistry Vesa Hänninen Laboratory of Physical Chemistry Chemicum 4th floor vesa.hanninen@helsinki.fi September 10, 2013 Lecture 3. Electron correlation methods September
Introduction to Computational Chemistry Vesa Hänninen Laboratory of Physical Chemistry Chemicum 4th floor vesa.hanninen@helsinki.fi September 10, 2013 Lecture 3. Electron correlation methods September
Practical Issues on the Use of the CASPT2/CASSCF Method in Modeling Photochemistry: the Selection and Protection of an Active Space
 Practical Issues on the Use of the CASPT2/CASSCF Method in Modeling Photochemistry: the Selection and Protection of an Active Space Roland Lindh Dept. of Chemistry Ångström The Theoretical Chemistry Programme
Practical Issues on the Use of the CASPT2/CASSCF Method in Modeling Photochemistry: the Selection and Protection of an Active Space Roland Lindh Dept. of Chemistry Ångström The Theoretical Chemistry Programme
Using Web-Based Computations in Organic Chemistry
 10/30/2017 1 Using Web-Based Computations in Organic Chemistry John Keller UAF Department of Chemistry & Biochemistry The UAF WebMO site Practical aspects of computational chemistry theory and nomenclature
10/30/2017 1 Using Web-Based Computations in Organic Chemistry John Keller UAF Department of Chemistry & Biochemistry The UAF WebMO site Practical aspects of computational chemistry theory and nomenclature
4 Post-Hartree Fock Methods: MPn and Configuration Interaction
 4 Post-Hartree Fock Methods: MPn and Configuration Interaction In the limit of a complete basis, the Hartree-Fock (HF) energy in the complete basis set limit (ECBS HF ) yields an upper boundary to the
4 Post-Hartree Fock Methods: MPn and Configuration Interaction In the limit of a complete basis, the Hartree-Fock (HF) energy in the complete basis set limit (ECBS HF ) yields an upper boundary to the
Lecture 4: Hartree-Fock Theory
 Lecture 4: Hartree-Fock Theory One determinant to rule them all, One determinant to find them, One determinant to bring them all and in the darkness bind them Second quantization rehearsal The formalism
Lecture 4: Hartree-Fock Theory One determinant to rule them all, One determinant to find them, One determinant to bring them all and in the darkness bind them Second quantization rehearsal The formalism
Performance of Hartree-Fock and Correlated Methods
 Chemistry 460 Fall 2017 Dr. Jean M. Standard December 4, 2017 Performance of Hartree-Fock and Correlated Methods Hartree-Fock Methods Hartree-Fock methods generally yield optimized geomtries and molecular
Chemistry 460 Fall 2017 Dr. Jean M. Standard December 4, 2017 Performance of Hartree-Fock and Correlated Methods Hartree-Fock Methods Hartree-Fock methods generally yield optimized geomtries and molecular
Multi-reference Density Functional Theory. COLUMBUS Workshop Argonne National Laboratory 15 August 2005
 Multi-reference Density Functional Theory COLUMBUS Workshop Argonne National Laboratory 15 August 2005 Capt Eric V. Beck Air Force Institute of Technology Department of Engineering Physics 2950 Hobson
Multi-reference Density Functional Theory COLUMBUS Workshop Argonne National Laboratory 15 August 2005 Capt Eric V. Beck Air Force Institute of Technology Department of Engineering Physics 2950 Hobson
Ab initio calculations for potential energy surfaces. D. Talbi GRAAL- Montpellier
 Ab initio calculations for potential energy surfaces D. Talbi GRAAL- Montpellier A theoretical study of a reaction is a two step process I-Electronic calculations : techniques of quantum chemistry potential
Ab initio calculations for potential energy surfaces D. Talbi GRAAL- Montpellier A theoretical study of a reaction is a two step process I-Electronic calculations : techniques of quantum chemistry potential
I. CSFs Are Used to Express the Full N-Electron Wavefunction
 Chapter 11 One Must be Able to Evaluate the Matrix Elements Among Properly Symmetry Adapted N- Electron Configuration Functions for Any Operator, the Electronic Hamiltonian in Particular. The Slater-Condon
Chapter 11 One Must be Able to Evaluate the Matrix Elements Among Properly Symmetry Adapted N- Electron Configuration Functions for Any Operator, the Electronic Hamiltonian in Particular. The Slater-Condon
Lec20 Fri 3mar17
 564-17 Lec20 Fri 3mar17 [PDF]GAUSSIAN 09W TUTORIAL www.molcalx.com.cn/wp-content/uploads/2015/01/gaussian09w_tutorial.pdf by A Tomberg - Cited by 8 - Related articles GAUSSIAN 09W TUTORIAL. AN INTRODUCTION
564-17 Lec20 Fri 3mar17 [PDF]GAUSSIAN 09W TUTORIAL www.molcalx.com.cn/wp-content/uploads/2015/01/gaussian09w_tutorial.pdf by A Tomberg - Cited by 8 - Related articles GAUSSIAN 09W TUTORIAL. AN INTRODUCTION
QUANTUM CHEMISTRY FOR TRANSITION METALS
 QUANTUM CHEMISTRY FOR TRANSITION METALS Outline I Introduction II Correlation Static correlation effects MC methods DFT III Relativity Generalities From 4 to 1 components Effective core potential Outline
QUANTUM CHEMISTRY FOR TRANSITION METALS Outline I Introduction II Correlation Static correlation effects MC methods DFT III Relativity Generalities From 4 to 1 components Effective core potential Outline
Handbook of Computational Quantum Chemistry. DAVID B. COOK The Department of Chemistry, University of Sheffield
 Handbook of Computational Quantum Chemistry DAVID B. COOK The Department of Chemistry, University of Sheffield Oxford New York Tokyo OXFORD UNIVERSITY PRESS 1998 CONTENTS 1 Mechanics and molecules 1 1.1
Handbook of Computational Quantum Chemistry DAVID B. COOK The Department of Chemistry, University of Sheffield Oxford New York Tokyo OXFORD UNIVERSITY PRESS 1998 CONTENTS 1 Mechanics and molecules 1 1.1
Other methods to consider electron correlation: Coupled-Cluster and Perturbation Theory
 Other methods to consider electron correlation: Coupled-Cluster and Perturbation Theory Péter G. Szalay Eötvös Loránd University Institute of Chemistry H-1518 Budapest, P.O.Box 32, Hungary szalay@chem.elte.hu
Other methods to consider electron correlation: Coupled-Cluster and Perturbation Theory Péter G. Szalay Eötvös Loránd University Institute of Chemistry H-1518 Budapest, P.O.Box 32, Hungary szalay@chem.elte.hu
Project 1 Report File: Chem4PB3_project_1_2017-solutions last changed: 02-Feb-2017
 Project 1 Report File: Chem4PB3_project_1_2017-solutions last changed: 02-Feb-2017 1. Formaldehyde comparison of results from different methods Yellow shaded boxes are closest to experimental Method #e-
Project 1 Report File: Chem4PB3_project_1_2017-solutions last changed: 02-Feb-2017 1. Formaldehyde comparison of results from different methods Yellow shaded boxes are closest to experimental Method #e-
Electron Correlation - Methods beyond Hartree-Fock
 Electron Correlation - Methods beyond Hartree-Fock how to approach chemical accuracy Alexander A. Auer Max-Planck-Institute for Chemical Energy Conversion, Mülheim September 4, 2014 MMER Summerschool 2014
Electron Correlation - Methods beyond Hartree-Fock how to approach chemical accuracy Alexander A. Auer Max-Planck-Institute for Chemical Energy Conversion, Mülheim September 4, 2014 MMER Summerschool 2014
Introduction to Hartree-Fock calculations in Spartan
 EE5 in 2008 Hannes Jónsson Introduction to Hartree-Fock calculations in Spartan In this exercise, you will get to use state of the art software for carrying out calculations of wavefunctions for molecues,
EE5 in 2008 Hannes Jónsson Introduction to Hartree-Fock calculations in Spartan In this exercise, you will get to use state of the art software for carrying out calculations of wavefunctions for molecues,
Molecular Simulation I
 Molecular Simulation I Quantum Chemistry Classical Mechanics E = Ψ H Ψ ΨΨ U = E bond +E angle +E torsion +E non-bond Jeffry D. Madura Department of Chemistry & Biochemistry Center for Computational Sciences
Molecular Simulation I Quantum Chemistry Classical Mechanics E = Ψ H Ψ ΨΨ U = E bond +E angle +E torsion +E non-bond Jeffry D. Madura Department of Chemistry & Biochemistry Center for Computational Sciences
IFM Chemistry Computational Chemistry 2010, 7.5 hp LAB2. Computer laboratory exercise 1 (LAB2): Quantum chemical calculations
: Quantum chemical calculations") Computer laboratory exercise 1 (LAB2): Quantum chemical calculations Introduction: The objective of the second computer laboratory exercise is to get acquainted with a program for performing quantum chemical
Computer laboratory exercise 1 (LAB2): Quantum chemical calculations Introduction: The objective of the second computer laboratory exercise is to get acquainted with a program for performing quantum chemical
This is called a singlet or spin singlet, because the so called multiplicity, or number of possible orientations of the total spin, which is
 9. Open shell systems The derivation of Hartree-Fock equations (Chapter 7) was done for a special case of a closed shell systems. Closed shell means that each MO is occupied by two electrons with the opposite
9. Open shell systems The derivation of Hartree-Fock equations (Chapter 7) was done for a special case of a closed shell systems. Closed shell means that each MO is occupied by two electrons with the opposite
Let me go through the basic steps of an MREOM calculation in ORCA. On chem440a/b the calculations can be found under ~nooijen/orca_examples_2017/mreom
 MREOM calculations in ORCA. The procedure to run MREOM calculations in ORCA is similar to running MREOM calculations in ACES. There are essentially three steps. 1. Determine a good set of CASSCF reference
MREOM calculations in ORCA. The procedure to run MREOM calculations in ORCA is similar to running MREOM calculations in ACES. There are essentially three steps. 1. Determine a good set of CASSCF reference
ABC of DFT: Hands-on session 1 Introduction into calculations on molecules
 ABC of DFT: Hands-on session 1 Introduction into calculations on molecules Tutor: Alexej Bagrets Wann? 09.11.2012, 11:30-13:00 Wo? KIT Campus Nord, Flachbau Physik, Geb. 30.22, Computerpool, Raum FE-6
ABC of DFT: Hands-on session 1 Introduction into calculations on molecules Tutor: Alexej Bagrets Wann? 09.11.2012, 11:30-13:00 Wo? KIT Campus Nord, Flachbau Physik, Geb. 30.22, Computerpool, Raum FE-6
Building a wavefunction within the Complete-Active. Cluster with Singles and Doubles formalism: straightforward description of quasidegeneracy
 Building a wavefunction within the Complete-Active Active-Space Coupled-Cluster Cluster with Singles and Doubles formalism: straightforward description of quasidegeneracy Dmitry I. Lyakh (Karazin Kharkiv
Building a wavefunction within the Complete-Active Active-Space Coupled-Cluster Cluster with Singles and Doubles formalism: straightforward description of quasidegeneracy Dmitry I. Lyakh (Karazin Kharkiv
Lecture 4: methods and terminology, part II
 So theory guys have got it made in rooms free of pollution. Instead of problems with the reflux, they have only solutions... In other words, experimentalists will likely die of cancer From working hard,
So theory guys have got it made in rooms free of pollution. Instead of problems with the reflux, they have only solutions... In other words, experimentalists will likely die of cancer From working hard,
Computational chemistry with GAMESS: a very brief overview with examples
 Computational chemistry with GAMESS: a very brief overview with examples PHY-6120 Molecular Physics (Spring 2015), UConn Phys. Dept. Feb 17 th 2015 H = ħ2 2μ i Intro: V(R) for diatomic molecules + k Z
Computational chemistry with GAMESS: a very brief overview with examples PHY-6120 Molecular Physics (Spring 2015), UConn Phys. Dept. Feb 17 th 2015 H = ħ2 2μ i Intro: V(R) for diatomic molecules + k Z
NMR and IR spectra & vibrational analysis
 Lab 5: NMR and IR spectra & vibrational analysis A brief theoretical background 1 Some of the available chemical quantum methods for calculating NMR chemical shifts are based on the Hartree-Fock self-consistent
Lab 5: NMR and IR spectra & vibrational analysis A brief theoretical background 1 Some of the available chemical quantum methods for calculating NMR chemical shifts are based on the Hartree-Fock self-consistent
Example: H 2 O (the car file)
") Example: H 2 O (the car file) As a practical example of DFT methods we calculate the energy and electronic properties of the water molecule. In order to carry out the DFT calculation you will need a set
Example: H 2 O (the car file) As a practical example of DFT methods we calculate the energy and electronic properties of the water molecule. In order to carry out the DFT calculation you will need a set
Exercise 1: Structure and dipole moment of a small molecule
 Introduction to computational chemistry Exercise 1: Structure and dipole moment of a small molecule Vesa Hänninen 1 Introduction In this exercise the equilibrium structure and the dipole moment of a small
Introduction to computational chemistry Exercise 1: Structure and dipole moment of a small molecule Vesa Hänninen 1 Introduction In this exercise the equilibrium structure and the dipole moment of a small
Handbook of Computational Quantum Chemistry
 Handbook of Computational Quantum Chemistry David B. Cook Dept. of Chemistry University of Sheffield DOVER PUBLICATIONS, INC. Mineola, New York F Contents 1 Mechanics and molecules 1 1.1 1.2 1.3 1.4 1.5
Handbook of Computational Quantum Chemistry David B. Cook Dept. of Chemistry University of Sheffield DOVER PUBLICATIONS, INC. Mineola, New York F Contents 1 Mechanics and molecules 1 1.1 1.2 1.3 1.4 1.5
Methods for Treating Electron Correlation CHEM 430
 Methods for Treating Electron Correlation CHEM 430 Electron Correlation Energy in the Hartree-Fock approximation, each electron sees the average density of all of the other electrons two electrons cannot
Methods for Treating Electron Correlation CHEM 430 Electron Correlation Energy in the Hartree-Fock approximation, each electron sees the average density of all of the other electrons two electrons cannot
We also deduced that transformations between Slater determinants are always of the form
 .3 Hartree-Fock The Hartree-Fock method assumes that the true N-body ground state wave function can be approximated by a single Slater determinant and minimizes the energy among all possible choices of
.3 Hartree-Fock The Hartree-Fock method assumes that the true N-body ground state wave function can be approximated by a single Slater determinant and minimizes the energy among all possible choices of
Multiconfigurational methods in MOLCAS
 Multiconfigurational methods in MOLCAS Valera Veryazov Valera.Veryazov@teokem.lu.se Division of Theoretical Chemistry Lund University Sweden CASSCF//CASPT2 p. 1/?? Overview What is correlation? CASPT2
Multiconfigurational methods in MOLCAS Valera Veryazov Valera.Veryazov@teokem.lu.se Division of Theoretical Chemistry Lund University Sweden CASSCF//CASPT2 p. 1/?? Overview What is correlation? CASPT2
Computational Methods. Chem 561
 Computational Methods Chem 561 Lecture Outline 1. Ab initio methods a) HF SCF b) Post-HF methods 2. Density Functional Theory 3. Semiempirical methods 4. Molecular Mechanics Computational Chemistry " Computational
Computational Methods Chem 561 Lecture Outline 1. Ab initio methods a) HF SCF b) Post-HF methods 2. Density Functional Theory 3. Semiempirical methods 4. Molecular Mechanics Computational Chemistry " Computational
Lecture 9. Hartree Fock Method and Koopman s Theorem
 Lecture 9 Hartree Fock Method and Koopman s Theorem Ψ(N) is approximated as a single slater determinant Φ of N orthogonal One electron spin-orbitals. One electron orbital φ i = φ i (r) χ i (σ) χ i (σ)
Lecture 9 Hartree Fock Method and Koopman s Theorem Ψ(N) is approximated as a single slater determinant Φ of N orthogonal One electron spin-orbitals. One electron orbital φ i = φ i (r) χ i (σ) χ i (σ)
Beyond the Hartree-Fock Approximation: Configuration Interaction
 Beyond the Hartree-Fock Approximation: Configuration Interaction The Hartree-Fock (HF) method uses a single determinant (single electronic configuration) description of the electronic wavefunction. For
Beyond the Hartree-Fock Approximation: Configuration Interaction The Hartree-Fock (HF) method uses a single determinant (single electronic configuration) description of the electronic wavefunction. For
THE CONSTRUCTION AND INTERPRETATION OF MCSCF WAVEFUNCTIONS
 Annu. Rev. Phys. Chem. 1998. 49:233 66 Copyright c 1998 by Annual Reviews. All rights reserved THE CONSTRUCTION AND INTERPRETATION OF MCSCF WAVEFUNCTIONS Michael W. Schmidt and Mark S. Gordon Department
Annu. Rev. Phys. Chem. 1998. 49:233 66 Copyright c 1998 by Annual Reviews. All rights reserved THE CONSTRUCTION AND INTERPRETATION OF MCSCF WAVEFUNCTIONS Michael W. Schmidt and Mark S. Gordon Department
Basis sets for electron correlation
 Basis sets for electron correlation Trygve Helgaker Centre for Theoretical and Computational Chemistry Department of Chemistry, University of Oslo, Norway The 12th Sostrup Summer School Quantum Chemistry
Basis sets for electron correlation Trygve Helgaker Centre for Theoretical and Computational Chemistry Department of Chemistry, University of Oslo, Norway The 12th Sostrup Summer School Quantum Chemistry
XMVB 3.0 A Right Way To Do Valence Bond Calculations. Zhenhua Chen Xiamen University
 XMVB 3.0 A Right Way To Do Valence Bond Calculations Zhenhua Chen Xiamen University XMVB 3.0 Xiamen Valence Bond An Ab Initio Non-orthogonal Valence Bond Program Version 3.0 Lingchun Song, Zhenhua Chen,
XMVB 3.0 A Right Way To Do Valence Bond Calculations Zhenhua Chen Xiamen University XMVB 3.0 Xiamen Valence Bond An Ab Initio Non-orthogonal Valence Bond Program Version 3.0 Lingchun Song, Zhenhua Chen,
Hints on Using the Orca Program
 Computational Chemistry Workshops West Ridge Research Building-UAF Campus 9:00am-4:00pm, Room 009 Electronic Structure - July 19-21, 2016 Molecular Dynamics - July 26-28, 2016 Hints on Using the Orca Program
Computational Chemistry Workshops West Ridge Research Building-UAF Campus 9:00am-4:00pm, Room 009 Electronic Structure - July 19-21, 2016 Molecular Dynamics - July 26-28, 2016 Hints on Using the Orca Program
Recent Advances in the MRexpT approach
 Recent Advances in the MRexpT approach Michael Hanrath University of Cologne Institute for Theoretical Chemistry July 8, 008 Michael Hanrath, Univ. of Cologne, Theor. Chemistry Advances MRexpT July 8,
Recent Advances in the MRexpT approach Michael Hanrath University of Cologne Institute for Theoretical Chemistry July 8, 008 Michael Hanrath, Univ. of Cologne, Theor. Chemistry Advances MRexpT July 8,
Ethene. Introduction. The ethene molecule is planar (i.e. all the six atoms lie in the same plane) and has a high degree of symmetry:
 and has a high degree of symmetry:") FY1006 Innføring i kvantefysikk og TFY4215 Kjemisk fysikk og kvantemekanikk Spring 2012 Chemical Physics Exercise 1 To be delivered by Friday 27.04.12 Introduction Ethene. Ethylene, C 2 H 4, or ethene,
FY1006 Innføring i kvantefysikk og TFY4215 Kjemisk fysikk og kvantemekanikk Spring 2012 Chemical Physics Exercise 1 To be delivered by Friday 27.04.12 Introduction Ethene. Ethylene, C 2 H 4, or ethene,
Tutorial I: IQ MOL and Basic DFT and MP2 Calculations 1 / 30
 Tutorial I: IQ MOL and Basic DFT and MP2 Calculations Q-Chem User Workshop, Denver March 21, 2015 1 / 30 2 / 30 Introduction to IQMOL DFT and MP2 Calculations 3 / 30 IQMOL and Q-CHEM IQMOL is an open-source
Tutorial I: IQ MOL and Basic DFT and MP2 Calculations Q-Chem User Workshop, Denver March 21, 2015 1 / 30 2 / 30 Introduction to IQMOL DFT and MP2 Calculations 3 / 30 IQMOL and Q-CHEM IQMOL is an open-source
5.4. Electronic structure of water
 5.4. Electronic structure of water Water belongs to C 2v point group, we have discussed the corresponding character table. Here it is again: C 2v E C 2 σ v (yz) σ v (xz) A 1 1 1 1 1 A 2 1 1-1 -1 B 1 1-1
5.4. Electronic structure of water Water belongs to C 2v point group, we have discussed the corresponding character table. Here it is again: C 2v E C 2 σ v (yz) σ v (xz) A 1 1 1 1 1 A 2 1 1-1 -1 B 1 1-1
Lecture 3: Electron statistics in a solid
 Lecture 3: Electron statistics in a solid Contents Density of states. DOS in a 3D uniform solid.................... 3.2 DOS for a 2D solid........................ 4.3 DOS for a D solid........................
Lecture 3: Electron statistics in a solid Contents Density of states. DOS in a 3D uniform solid.................... 3.2 DOS for a 2D solid........................ 4.3 DOS for a D solid........................
Lecture 10. Born-Oppenheimer approximation LCAO-MO application to H + The potential energy surface MOs for diatomic molecules. NC State University
 Chemistry 431 Lecture 10 Diatomic molecules Born-Oppenheimer approximation LCAO-MO application to H + 2 The potential energy surface MOs for diatomic molecules NC State University Born-Oppenheimer approximation
Chemistry 431 Lecture 10 Diatomic molecules Born-Oppenheimer approximation LCAO-MO application to H + 2 The potential energy surface MOs for diatomic molecules NC State University Born-Oppenheimer approximation
Size-extensive wave functions for QMC A linear-scaling GVB approach
 Size-extensive wave functions for QMC A linear-scaling GVB approach Claudia Filippi, University of Twente, The Netherlands Francesco Fracchia, University of Pisa, Italy Claudio Amovilli, University of
Size-extensive wave functions for QMC A linear-scaling GVB approach Claudia Filippi, University of Twente, The Netherlands Francesco Fracchia, University of Pisa, Italy Claudio Amovilli, University of
Gaussian: Basic Tutorial
 Input file: # hf sto-g pop=full Water - Single Point Energy 0 H.0 H.0 H 04.5 Route Section Start with # Contains the keywords Gaussian: Basic Tutorial Route Section Title Section Charge-Multiplicity Molecule
Input file: # hf sto-g pop=full Water - Single Point Energy 0 H.0 H.0 H 04.5 Route Section Start with # Contains the keywords Gaussian: Basic Tutorial Route Section Title Section Charge-Multiplicity Molecule
Electronically Excited States
 Electronically Excited States Absorption of light (photon) by molecule Wavelength range visible and UV (ca. 800 200 nm) Molecule accepts the energy from the electromagnetic field promoted to an electronically
Electronically Excited States Absorption of light (photon) by molecule Wavelength range visible and UV (ca. 800 200 nm) Molecule accepts the energy from the electromagnetic field promoted to an electronically
Electron Correlation
 Electron Correlation Levels of QM Theory HΨ=EΨ Born-Oppenheimer approximation Nuclear equation: H n Ψ n =E n Ψ n Electronic equation: H e Ψ e =E e Ψ e Single determinant SCF Semi-empirical methods Correlation
Electron Correlation Levels of QM Theory HΨ=EΨ Born-Oppenheimer approximation Nuclear equation: H n Ψ n =E n Ψ n Electronic equation: H e Ψ e =E e Ψ e Single determinant SCF Semi-empirical methods Correlation
Wavefunction and electronic struture in solids: Bloch functions, Fermi level and other concepts.
 Wavefunction and electronic struture in solids: Bloch functions, Fermi level and other concepts. Silvia Casassa Università degli Studi di Torino July 12, 2017 Minnesota Workshop on ab initio MSC Symmetry
Wavefunction and electronic struture in solids: Bloch functions, Fermi level and other concepts. Silvia Casassa Università degli Studi di Torino July 12, 2017 Minnesota Workshop on ab initio MSC Symmetry
Michal Dallos, Hans Lischka
 Theor Chem Acc (2004) 112: 16 26 DOI 10.1007/s00214-003-0557-9 Regular article A systematic theoretical investigation of the lowest valence- and Rydberg-excited singlet states of trans-butadiene. The character
Theor Chem Acc (2004) 112: 16 26 DOI 10.1007/s00214-003-0557-9 Regular article A systematic theoretical investigation of the lowest valence- and Rydberg-excited singlet states of trans-butadiene. The character
Theoretical UV/VIS Spectroscopy
 Theoretical UV/VIS Spectroscopy Why is a Ruby Red When Chromium Oxide is Green? How Does a Ruby Laser Work? Goals of this Exercise: - Calculation of the energy of electronically excited states - Understanding
Theoretical UV/VIS Spectroscopy Why is a Ruby Red When Chromium Oxide is Green? How Does a Ruby Laser Work? Goals of this Exercise: - Calculation of the energy of electronically excited states - Understanding
Translation Symmetry, Space Groups, Bloch functions, Fermi energy
 Translation Symmetry, Space Groups, Bloch functions, Fermi energy Roberto Orlando and Silvia Casassa Università degli Studi di Torino July 20, 2015 School Ab initio Modelling of Solids (UniTo) Symmetry
Translation Symmetry, Space Groups, Bloch functions, Fermi energy Roberto Orlando and Silvia Casassa Università degli Studi di Torino July 20, 2015 School Ab initio Modelling of Solids (UniTo) Symmetry
( R)Ψ el ( r;r) = E el ( R)Ψ el ( r;r)
Ψ el ( r;r) = E el ( R)Ψ el ( r;r)") Born Oppenheimer Approximation: Ĥ el ( R)Ψ el ( r;r) = E el ( R)Ψ el ( r;r) For a molecule with N electrons and M nuclei: Ĥ el What is E el (R)? s* potential surface Reaction Barrier Unstable intermediate
Born Oppenheimer Approximation: Ĥ el ( R)Ψ el ( r;r) = E el ( R)Ψ el ( r;r) For a molecule with N electrons and M nuclei: Ĥ el What is E el (R)? s* potential surface Reaction Barrier Unstable intermediate
SUPPLEMENTARY INFORMATION
 DOI: 10.1038/NCHEM.1677 Entangled quantum electronic wavefunctions of the Mn 4 CaO 5 cluster in photosystem II Yuki Kurashige 1 *, Garnet Kin-Lic Chan 2, Takeshi Yanai 1 1 Department of Theoretical and
DOI: 10.1038/NCHEM.1677 Entangled quantum electronic wavefunctions of the Mn 4 CaO 5 cluster in photosystem II Yuki Kurashige 1 *, Garnet Kin-Lic Chan 2, Takeshi Yanai 1 1 Department of Theoretical and
Algorithmic Challenges in Photodynamics Simulations on HPC systems
 Algorithmic Challenges in Photodynamics Simulations on HPC systems Felix Plasser González Research Group Institute for Theoretical Chemistry, University of Vienna, Austria Bratislava, 21 st March 2016
Algorithmic Challenges in Photodynamics Simulations on HPC systems Felix Plasser González Research Group Institute for Theoretical Chemistry, University of Vienna, Austria Bratislava, 21 st March 2016
Reactivity and Organocatalysis. (Adalgisa Sinicropi and Massimo Olivucci)
") Reactivity and Organocatalysis (Adalgisa Sinicropi and Massimo Olivucci) The Aldol Reaction - O R 1 O R 1 + - O O OH * * H R 2 R 1 R 2 The (1957) Zimmerman-Traxler Transition State Counterion (e.g. Li
Reactivity and Organocatalysis (Adalgisa Sinicropi and Massimo Olivucci) The Aldol Reaction - O R 1 O R 1 + - O O OH * * H R 2 R 1 R 2 The (1957) Zimmerman-Traxler Transition State Counterion (e.g. Li
arxiv: v2 [physics.chem-ph] 17 Sep 2017
![arxiv: v2 [physics.chem-ph] 17 Sep 2017](/thumbs/91/105444484.jpg "arxiv: v2 [physics.chem-ph] 17 Sep 2017") arxiv:1602.07302v2 [physics.chem-ph] 17 Sep 2017 GPView: A Program for Wave Function Analysis and Visualization Tian Shi a,, Ping Wang b a Department of Chemistry, Wayne State University, Detroit, Michigan
arxiv:1602.07302v2 [physics.chem-ph] 17 Sep 2017 GPView: A Program for Wave Function Analysis and Visualization Tian Shi a,, Ping Wang b a Department of Chemistry, Wayne State University, Detroit, Michigan
Figure 1: Transition State, Saddle Point, Reaction Pathway
 Computational Chemistry Workshops West Ridge Research Building-UAF Campus 9:00am-4:00pm, Room 009 Electronic Structure - July 19-21, 2016 Molecular Dynamics - July 26-28, 2016 Potential Energy Surfaces
Computational Chemistry Workshops West Ridge Research Building-UAF Campus 9:00am-4:00pm, Room 009 Electronic Structure - July 19-21, 2016 Molecular Dynamics - July 26-28, 2016 Potential Energy Surfaces
Lecture 12. Symmetry Operations. NC State University
 Chemistry 431 Lecture 12 Group Theory Symmetry Operations NC State University Wave functions as the basis for irreducible representations The energy of the system will not change when symmetry Operations
Chemistry 431 Lecture 12 Group Theory Symmetry Operations NC State University Wave functions as the basis for irreducible representations The energy of the system will not change when symmetry Operations
Symmetry Adapted Orbitals
 Symmetry Adapted Orbitals z B x y e a Figure Symmetry adapted fragment orbitals for 3 L= ML 3 M C is isolobal with P 3 P Figure 2 Isolobal relationship Introduction so far the fragments used in forming
Symmetry Adapted Orbitals z B x y e a Figure Symmetry adapted fragment orbitals for 3 L= ML 3 M C is isolobal with P 3 P Figure 2 Isolobal relationship Introduction so far the fragments used in forming
NWChem: Coupled Cluster Method (Tensor Contraction Engine)
") NWChem: Coupled Cluster Method (Tensor Contraction Engine) Why CC is important?! Correlation effects are important!! CC is size-extensive theory: can be used to describe dissociation processes.! Higher-order
NWChem: Coupled Cluster Method (Tensor Contraction Engine) Why CC is important?! Correlation effects are important!! CC is size-extensive theory: can be used to describe dissociation processes.! Higher-order
General Physical Chemistry II
 General Physical Chemistry II Lecture 10 Aleksey Kocherzhenko October 7, 2014" Last time " promotion" Promotion and hybridization" [He] 2s 2 2p x 1 2p y 1 2p z0 " 2 unpaired electrons" [He] 2s 1 2p x 1
General Physical Chemistry II Lecture 10 Aleksey Kocherzhenko October 7, 2014" Last time " promotion" Promotion and hybridization" [He] 2s 2 2p x 1 2p y 1 2p z0 " 2 unpaired electrons" [He] 2s 1 2p x 1
Chm 331 Fall 2015, Exercise Set 4 NMR Review Problems
 Chm 331 Fall 015, Exercise Set 4 NMR Review Problems Mr. Linck Version.0. Compiled December 1, 015 at 11:04:44 4.1 Diagonal Matrix Elements for the nmr H 0 Find the diagonal matrix elements for H 0 (the
Chm 331 Fall 015, Exercise Set 4 NMR Review Problems Mr. Linck Version.0. Compiled December 1, 015 at 11:04:44 4.1 Diagonal Matrix Elements for the nmr H 0 Find the diagonal matrix elements for H 0 (the
Jack Simons, Henry Eyring Scientist and Professor Chemistry Department University of Utah
 1. Born-Oppenheimer approx.- energy surfaces 2. Mean-field (Hartree-Fock) theory- orbitals 3. Pros and cons of HF- RHF, UHF 4. Beyond HF- why? 5. First, one usually does HF-how? 6. Basis sets and notations
1. Born-Oppenheimer approx.- energy surfaces 2. Mean-field (Hartree-Fock) theory- orbitals 3. Pros and cons of HF- RHF, UHF 4. Beyond HF- why? 5. First, one usually does HF-how? 6. Basis sets and notations
PAPER:2, PHYSICAL CHEMISTRY-I QUANTUM CHEMISTRY. Module No. 34. Hückel Molecular orbital Theory Application PART IV
 Subject PHYSICAL Paper No and Title TOPIC Sub-Topic (if any), PHYSICAL -II QUANTUM Hückel Molecular orbital Theory Module No. 34 TABLE OF CONTENTS 1. Learning outcomes. Hückel Molecular Orbital (HMO) Theory
Subject PHYSICAL Paper No and Title TOPIC Sub-Topic (if any), PHYSICAL -II QUANTUM Hückel Molecular orbital Theory Module No. 34 TABLE OF CONTENTS 1. Learning outcomes. Hückel Molecular Orbital (HMO) Theory
DFTB Parameter Development and Implementation in Gaussian Development Version Program
 DFTB Parameter Development and Implementation in Gaussian Development Version Program Guishan Zheng a*, ichael Frisch b,keiji orokuma a a Cherry L. Emerson Center for Scientific Computation and Department
DFTB Parameter Development and Implementation in Gaussian Development Version Program Guishan Zheng a*, ichael Frisch b,keiji orokuma a a Cherry L. Emerson Center for Scientific Computation and Department
Chapter: 22. Visualization: Making INPUT File and Processing of Output Results
 Chapter: 22 Visualization: Making INPUT File and Processing of Output Results Keywords: visualization, input and output structure, molecular orbital, electron density. In the previous chapters, we have
Chapter: 22 Visualization: Making INPUT File and Processing of Output Results Keywords: visualization, input and output structure, molecular orbital, electron density. In the previous chapters, we have
Exchange coupling can frequently be understood using a simple molecular orbital approach.
 6.4 Exchange Coupling, a different perspective So far, we ve only been looking at the effects of J on the magnetic susceptibility but haven t said anything about how one might predict the sign and magnitude
6.4 Exchange Coupling, a different perspective So far, we ve only been looking at the effects of J on the magnetic susceptibility but haven t said anything about how one might predict the sign and magnitude
Introduction to the Unitary Group Approach
 Introduction to the Unitary Group Approach Isaiah Shavitt Department of Chemistry University of Illinois at Urbana Champaign This tutorial introduces the concepts and results underlying the unitary group
Introduction to the Unitary Group Approach Isaiah Shavitt Department of Chemistry University of Illinois at Urbana Champaign This tutorial introduces the concepts and results underlying the unitary group
MO Calculation for a Diatomic Molecule. /4 0 ) i=1 j>i (1/r ij )
 i=1 j>i (1/r ij )") MO Calculation for a Diatomic Molecule Introduction The properties of any molecular system can in principle be found by looking at the solutions to the corresponding time independent Schrodinger equation
MO Calculation for a Diatomic Molecule Introduction The properties of any molecular system can in principle be found by looking at the solutions to the corresponding time independent Schrodinger equation
Introduction to Ab Initio Quantum Chemical Computation
 c:\374-17\computation\computation17.doc for 9mar17 Prof. Patrik Callis 8mar17 Introduction to Ab Initio Quantum Chemical Computation Purpose: 1. To become acquainted with basic concepts of ab initio quantum
c:\374-17\computation\computation17.doc for 9mar17 Prof. Patrik Callis 8mar17 Introduction to Ab Initio Quantum Chemical Computation Purpose: 1. To become acquainted with basic concepts of ab initio quantum
Multiconfiguration Self-Consistent Field and Multireference Configuration Interaction Methods and Applications
 pubs.acs.org/cr Multiconfiguration Self-Consistent Field and Multireference Configuration Interaction Methods and Applications Peter G. Szalay Laboratory for Theoretical Chemistry, Institute of Chemistry,
pubs.acs.org/cr Multiconfiguration Self-Consistent Field and Multireference Configuration Interaction Methods and Applications Peter G. Szalay Laboratory for Theoretical Chemistry, Institute of Chemistry,
Nature of the Chemical Bond with applications to catalysis, materials science, nanotechnology, surface science, bioinorganic chemistry, and energy
 Lecture 12, October 21, 2016 Transition metals Heme-Fe Nature of the Chemical Bond with applications to catalysis, materials science, nanotechnology, surface science, bioinorganic chemistry, and energy
Lecture 12, October 21, 2016 Transition metals Heme-Fe Nature of the Chemical Bond with applications to catalysis, materials science, nanotechnology, surface science, bioinorganic chemistry, and energy
Lecture 16 February 20 Transition metals, Pd and Pt
 Lecture 16 February 20 Transition metals, Pd and Pt Nature of the Chemical Bond with applications to catalysis, materials science, nanotechnology, surface science, bioinorganic chemistry, and energy Course
Lecture 16 February 20 Transition metals, Pd and Pt Nature of the Chemical Bond with applications to catalysis, materials science, nanotechnology, surface science, bioinorganic chemistry, and energy Course
Consider a s ystem with 2 parts with well defined transformation properties
 Direct Product of Representations Further important developments of the theory of symmetry are needed for systems that consist of parts (e.g. two electrons, spin and orbit of an electron, one electron
Direct Product of Representations Further important developments of the theory of symmetry are needed for systems that consist of parts (e.g. two electrons, spin and orbit of an electron, one electron
Problem Set 5 Solutions
 Chemistry 362 Dr Jean M Standard Problem Set 5 Solutions ow many vibrational modes do the following molecules or ions possess? [int: Drawing Lewis structures may be useful in some cases] In all of the
Chemistry 362 Dr Jean M Standard Problem Set 5 Solutions ow many vibrational modes do the following molecules or ions possess? [int: Drawing Lewis structures may be useful in some cases] In all of the
Introduction to DFTB. Marcus Elstner. July 28, 2006
 Introduction to DFTB Marcus Elstner July 28, 2006 I. Non-selfconsistent solution of the KS equations DFT can treat up to 100 atoms in routine applications, sometimes even more and about several ps in MD
Introduction to DFTB Marcus Elstner July 28, 2006 I. Non-selfconsistent solution of the KS equations DFT can treat up to 100 atoms in routine applications, sometimes even more and about several ps in MD
Lecture 5: More about one- Final words about the Hartree-Fock theory. First step above it by the Møller-Plesset perturbation theory.
 Lecture 5: More about one- determinant wave functions Final words about the Hartree-Fock theory. First step above it by the Møller-Plesset perturbation theory. Items from Lecture 4 Could the Koopmans theorem
Lecture 5: More about one- determinant wave functions Final words about the Hartree-Fock theory. First step above it by the Møller-Plesset perturbation theory. Items from Lecture 4 Could the Koopmans theorem
A Computer Study of Molecular Electronic Structure
 A Computer Study of Molecular Electronic Structure The following exercises are designed to give you a brief introduction to some of the types of information that are now readily accessible from electronic
A Computer Study of Molecular Electronic Structure The following exercises are designed to give you a brief introduction to some of the types of information that are now readily accessible from electronic
Molecular Orbitals for Ozone
 Molecular Orbitals for Ozone Purpose: In this exercise you will do semi-empirical molecular orbital calculations on ozone with the goal of understanding the molecular orbital print out provided by Spartan
Molecular Orbitals for Ozone Purpose: In this exercise you will do semi-empirical molecular orbital calculations on ozone with the goal of understanding the molecular orbital print out provided by Spartan
Technical Note Calculations of Orbital Overlap Range Function EDR( r ; d) and Overlap Distance D(r )using Multiwfn
 and Overlap Distance D(r )using Multiwfn") Technical Note Calculations of Orbital Overlap Range Function EDR( r ; d) and Overlap Distance D(r )using Multiwfn Abstract The orbital overlap range function EDR( r; d) (J. Chem. Phys. 2014, 141, 144104)
Technical Note Calculations of Orbital Overlap Range Function EDR( r ; d) and Overlap Distance D(r )using Multiwfn Abstract The orbital overlap range function EDR( r; d) (J. Chem. Phys. 2014, 141, 144104)
NWChem: Coupled Cluster Method (Tensor Contraction Engine)
") NWChem: Coupled Cluster Method (ensor Contraction Engine) What we want to solve H Ψ = E Ψ Many Particle Systems Molecular/Atomic Physics, Quantum Chemistry (electronic Schrödinger equations) Solid State
NWChem: Coupled Cluster Method (ensor Contraction Engine) What we want to solve H Ψ = E Ψ Many Particle Systems Molecular/Atomic Physics, Quantum Chemistry (electronic Schrödinger equations) Solid State
Lecture 9 Electronic Spectroscopy
 Lecture 9 Electronic Spectroscopy Molecular Orbital Theory: A Review - LCAO approximaton & AO overlap - Variation Principle & Secular Determinant - Homonuclear Diatomic MOs - Energy Levels, Bond Order
Lecture 9 Electronic Spectroscopy Molecular Orbital Theory: A Review - LCAO approximaton & AO overlap - Variation Principle & Secular Determinant - Homonuclear Diatomic MOs - Energy Levels, Bond Order
Yingwei Wang Computational Quantum Chemistry 1 Hartree energy 2. 2 Many-body system 2. 3 Born-Oppenheimer approximation 2
 Purdue University CHM 67300 Computational Quantum Chemistry REVIEW Yingwei Wang October 10, 2013 Review: Prof Slipchenko s class, Fall 2013 Contents 1 Hartree energy 2 2 Many-body system 2 3 Born-Oppenheimer
Purdue University CHM 67300 Computational Quantum Chemistry REVIEW Yingwei Wang October 10, 2013 Review: Prof Slipchenko s class, Fall 2013 Contents 1 Hartree energy 2 2 Many-body system 2 3 Born-Oppenheimer
Supporting information
 Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 2014 Supporting information Resonant Raman Spectra of Molecules with Diradical Character:
Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 2014 Supporting information Resonant Raman Spectra of Molecules with Diradical Character: