AN INTRODUCTION TO MCSCF: PART 2
|
|
|
- Isaac Edwards
- 5 years ago
- Views:
Transcription
1 AN INTRODUCTION TO MCSCF: PART 2
2 ORBITAL APPROXIMATION Hartree product (hp) expressed as a product of spinorbitals ψ ι = φ i σ i φ i = space orbital, σ i = spin function (α,β) Pauli Principle requires antisymmetry: Closed Shells:
3 ORBITAL APPROXIMATION For more complex species (one or more open shells) antisymmetric wavefunction is generally expressed as a linear combination of Slater determinants For example, consider simple excited state represented by excitation φ i ->φ a out of closed shell:
4 For more complex open shell species (e.g., low-spin open shells with multiple partially filled orbitals, such as s 1 d 7 Fe) wavefunctions are linear combinations of several determinants. But, the coefficients on these determinants are determined by spin and symmetry, not by the Variational Principle
5 HARTREE-FOCK METHOD Optimization of the orbitals (minimization of the energy with respect to all orbitals), based on the Variational Principle) leads to Hartree- Fock equations (closed shells): For open shells, there are multiple Fock operators, one for each type of orbital occupancy; e.g. UHF:
6 LCAO METHOD Generally solve HF problem by LCAO expansion: expand φ i as linear combination of basis functions (AOs), χ µ : The C µi are expansion coefficients obtained via the Variational Principle FC = SCε HFR matrix equation, solved iteratively
7 MCSCF METHOD Hartree-Fock (or DFT) is most common zeroth order wavefunction, but Many problems are not well represented by single configuration wavefunctions: Diradicals (broadly defined) Excited states Transition states (frequently) Unsaturated transition metals High energy species Generally, any system with near degeneracies
8 In such cases, the correct zeroth order wavefunction is MCSCF: Φ is the MCSCF wavefunction Ψ K is a configuration wavefunction Can be a single determinant Could be a linear combination of determinants in order to be spin-correct Generally called configuration state function (CSF), meaning spin-correct, symmetrycorrect configuration wavefunction
9 Generally, two approaches to treating Φ in computer codes: Expand in terms of CSFs Most commonly GUGA (graphical unitary group approach) Made feasible by Shavitt, Schaefer Expand directly in terms of determinants Can be faster code More determinants to deal with Each determinant not spin-correct, but easily dealt with Not as robust: GUGA code is generally preferred Some applications only available for determinant code Both available in GAMESS
10 A K are CI expansion coefficients Determined variationally using linear variation theory Solution of this (non-iterative) matrix eigenvalue equation yields MCSCF energies E M for each electronic state CI coefficients A KM corresponding to state M
11 MCSCF METHOD Solution of MCSCF problem requires two sets of iterations to solve for two sets of coefficients For each set of CI coefficients A K, solve for LCAO coefficients C µi (micro-iterations) For given set of C µi, solve CI equations for new A K Continue until self-consistency
12 MCSCF METHOD Most common implementation is FORS (fully optimized reaction space)/casscf (complete active space) SCF Define active space in terms of orbitals and electrons Perform full CI within active space Very chemical approach Can be computationally demanding Ideal active space is full valence Not always feasible; upper limit is ~(16,16) Sometimes tricky to choose active space
13 Two sets of coefficient optimizations CI coefficients optimized by solving linear variation secular equation Orbital optimization analogous to, but more complex than, simple HF solutions Need to optimize mixing between sets of subspaces:core, active, virtual Core-active Active-virtual Core-virtual Cf., HF high-spin open shell: Fock operators for Doubly occupied-singly occupied Doubly occupied-virtual Singly occupied virtual
14 Orbital optimizations As for HF, each subspace invariant to internal mixing Only mixing between subspaces will change energy Exception: if MCSCF is not FORS/ CASSCF (CI is not Full CI), must also optimize active-active mixing: FORS simpler although more demanding computationally Non-FORS less robust, more difficult to converge Can think of optimization variables as rotation angles connecting orbitals in different subspaces (recall UHF)
15 Orbital optimizations Taylor expansion of orbital gradient g(x) = E (x) = g(x 0 ) + g (x 0 ) (x-x 0 ) + g = E = orbital hessian - second derivative of energy wrt orbital rotations x. So, at optimal E E (x) = 0 = E (x 0 ) + E (X 0 ) (x-x 0 ), ignoring higher order terms. Rearranging, x = x 0 - E (x 0 )/E (x 0 ): Newton-Raphson equation In many dimensions, x is vector Completely analogous to geometry opt Exact calc of orbital hessian (FULLNR=.T.) Takes much more AO to MO 4-label integral transformation time (need 2 virtual indices as in [vo vo], v = virtual, o = occupied More memory required
16 As in geom opt, alternative to FULLNR is approximate updating of orbital hessian SOSCF=.T.: calc diagonal, guess off-diagonal Takes more iterations, but less time. Convergence less robust Easily can do 750 basis functions on workstation Alternatives are JACOBI: simple pairwise rotations, similar to SCFDM FOCAS: uses only orbital gradients, not even diagonal hessian elements as in SOSCF. Each iteration is faster, but many more required Best strategy Start with SOSCF Use FULLNR as backup
17 CHOOSING ACTIVE SPACES Full valence active space Occupied orbitals are usually easy: choose all of them. Virtual orbitals not always easy: # of orbitals wanted = minimal valence basis set # of available virtuals generally much larger Virtuals are generally more diffuse and not easy to identify, especially with Large basis sets, especially diffuse functions Transition metals High symmetry
18 Strategies for full valence active space MVOQ in $SCF Since virtual MOs are typically diffuse, ease of identification is improved if they are made more compact MVOQ = n removes n electrons from SCF calculation Generates a cation with +n charge - pulls orbitals in Easier to find correct virtuals for active space Improved convergence
19 Strategies for full valence active space Localized orbitals (LMOs) Specify LOCAL=BOYS or RUDNBERG in $CONTRL Transforms orbitals to bonds, lone pairs Easier to understand occupied FV space Can use these to construct virtual part of FV active space Disadvantage: LMOs destroy symmetry, so the size of the problem (# of determinants) increases Partial solution: symmetry localized orbitals can be specified using SYMLOC=.T. in $LOCAL Localizes orbitals only within each irrep Sometimes not localized enough
20 Strategies for less than FV active space Need to identify chemically important orbitals Orbitals directly involved in the chemical process Orbitals that may interact strongly with reacting orbitals Examples Recall H 2 : Active space includes H-H bonding orbital and H-H* FORS(2,2): 2 electrons in 2 orbitals Internal rotation in ethylene Full Valence active space is (12,12) Minimum active space includes only CC σ,π,π,σ : (4,4) The two active spaces give ~same internal rotation barrier This active space cannot account for other processes, such as C-H bond cleavage
21 More Examples Internal rotation in H 2 C=NH Start with analogous active space to ethylene: CN (4,4) Recognize that N lone pair will interact with π system as internal rotation takes place Add N lone pair to active space: (6,5), 6 electrons in 5 orbitals Also correctly describes dissociation to H 2 C + NH: NH fragment will be correctly described by σ 2 π x1 π y 1 Dissociation of H 2 C=O -> H 2 C + O Again, start with CO (4,4) Recognize O has two lone pairs, one 2s, one 2p Recognize that 2s lone pair has low energy & likely inactive Including 2p lone pair [(6,5) active space] ensures three 2p orbitals are treated equally in dissociated oxygen Isomerization to HCOH requires additional (4,4) from CH/OH
22 Important to consider both reactant and product when choosing active space Ensures number of active electrons & orbitals are same Verifies reactant orbitals will be able to convert smoothly into product orbitals. Transition state orbitals can help make this transition smooth
23 Consider isomerization of bicyclobutane to 1,3- butadiene Superficially only need to break two bonds: FORS(4,4) But, to treat all peripheral bonds equally, need all of them in active space: FORS(10,10) Now, consider isoelectronic NO dimer, N 2 O 2
24 Replace two bridge CH groups with nitrogens Replace two peripheral CH 2 groups with oxygens Very high energy species: important HEDM compound First guess at good active space might be (10,10) But, one O lone pair on each O interacts strongly and must be included in active space for smooth PES Correct active space is (14,12) Pay attention to orbitals along reaction path!
25 MULTI-REFERENCE DYNAMIC CORRELATION Multi-reference CI: MRCI CI from set of MCSCF configurations Most commonly stops at singles and doubles MR(SD)CI: Very demanding ~ very difficult to go past 16 electrons in 16 orbitals Multi-reference perturbation theory Several flavors: CASPT2, MRMP2, GVVPT2 Mostly second order (except CASPT3) More efficient than MRCI Not usually as accurate as MRCI Can blow up if perturbation is too big
26 MULTI-REFERENCE DYNAMIC CORRELATION MRCI, MRPT generally not size-consistent +Q correction can make MRCI nearly size consistent MRPT developers like to say the method is nearly size-consistent Not really true Cf., GN methods are slightly empirical
27 STRATEGIES FOR INCONSISTENT ACTIVE SPACES Sometimes different parts of PES require different active spaces. Strategies Optimize geometries, obtain frequencies with separate active spaces Final MRPT or MRCI with composite active space If composite active space is too large Optimize geometries with separate active spaces Use MRPT with separate active spaces to correlate all electrons
28 NATURAL ORBITAL ANALYSIS Complex wavefunctions like MCSCF are very useful, but qualitative interpretations are important Two useful tools are Natural orbitals Localized orbitals Natural orbitals introduced by Löwdin in 1955 Diagonalize the 1st order density matrix ρ Simply the HF orbitals for HF theory
29 NATURAL ORBITAL ANALYSIS For fully variational methods (HF, MCSCF), 1st order density matrix is simply obtained from ΨΨ For other methods (MPn, CC, MRMP), must also calculate non-hellmann-feynman contribution: requires gradient of energy Eigenvectors of 1st order density matrix are natural orbitals Eigenvalues are natural orbital occupation numbers (NOON): λ i
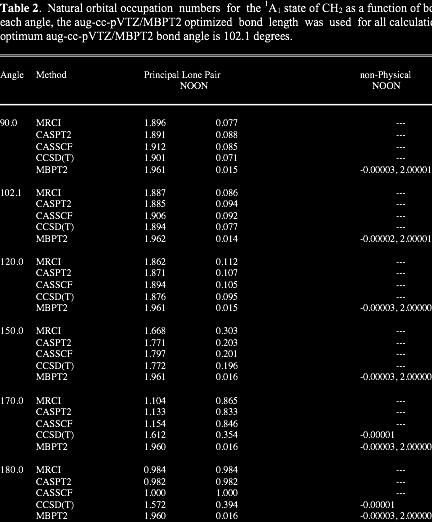
30 NATURAL ORBITAL ANALYSIS For RHF & ROHF, NOON are integers: 2,1,0 For other methods,noon are not integers Deviation from 2 (occupied orbitals) or 0 (virtual orbitals) indicate importance of configurational mixing For H 2, λ 1 ~2, λ 2 0 near R e ; λ 1, λ 2 1 near dissociation NOON are also good diagnostic for need for MCSCF zeroth order wavefunction NOON for single reference assume non-physical values when such methods start to break down. Examples
31

32

33
34 MCSCF/LMO/CI METHOD See Gordon&Cundari, Coord Chem Rev., 147, (1996) Choose active space for particular bond type Determine MCSCF LMOs within active space These are atom-like in nature Perform CI within LMO MCSCF space Applied to analyze TM-MG double bonds TM=transition metal (or Tom) MG=main group (or Mark Gordon)
35 Possible resonance contributors Straight line = covalent structure, electrons shared Arrow = ionic structure, both electrons on atom at base of arrow Lower arrow = σ, upper arrow = π
36 Table 1. Percent contributors of covalent and ionic resonance structures in H 2 M=EH 2 compounds. Nucleophilic structures are defined as those with M + E - ionicity, electrophilic means M - E Ti Zr Nb Ta --- Si C Si C Si C Si C A B C D E F G H I Neut Nucl Elec This method 1st to show σ ylide structure D is an important resonance contributor
37 STEPS TO RUN CASSCF RUN HF TO GET STARTING ORBITALS Grab $vec group from.dat & insert in input file Set up MCSCF input file
38 H 2 INPUT FILE $CONTRL SCFTYP=MCSCF RUNTYP=ENERGY MULT=1 $END $BASIS GBASIS=N31 NGAUSS=6 NDFUNC=1 $END $GUESS GUESS=MOREAD NORB=4 $END $DATA RHF/6-31G(d) H2 DNH 2 HYDROGEN $END --- CLOSED SHELL ORBITALS --- GENERATED AT Mon Aug 25 10:56: RHF/6-31G(d) H2 E(RHF)= , E(NUC)= , 7 ITERS $VEC E E E E E E E E E E E E E E E E+00 $END $DRT NMCC=0 NDOC=1 NVAL=1 FORS=.T. GROUP=D2H $END $MCSCF CISTEP=GUGA FORS=.T. $END
39 H 2 LOG FILE
40 $CONTRL SCFTYP=MCSCF RUNTYP=ENERGY NZVAR=3 COORD=ZMT $END $SYSTEM TIMLIM=5 MEMORY= $END $BASIS GBASIS=STO NGAUSS=3 $END $DATA Methylene...1-A-1 state...mcscf/sto-3g Cnv 2 C H 1 rch H 1 rch 2 ahoh rch=1.09 ahoh=130.0 $END $GUESS GUESS=MOREAD NORB=7 $END $MCSCF CISTEP=GUGA $END $DRT NMCC=3 NDOC=1 NVAL=1 FORS=.T. GROUP=C2V $END --- RHF ORBITALS --- GENERATED AT 21:48: Methylene...1-A-1 state...mcscf/sto-2g E(RHF)= , E(NUC)= , 8 ITERS $VEC E E E E E E E E E E E E E E E E E E E E E E E E E E E E E E E E E E E E E E E E E E E E E E E E E-01 $END
41 ! EXAM06.! 1-A-1 CH2 MCSCF methylene geometry optimization.!! At the initial geometry:! The initial energy is ,! the FINAL E= after 14 iterations,! the RMS gradient is !! After 4 steps,! FINAL E= , RMS gradient= ,! r(ch)= , ang(hch)= ! $CONTRL SCFTYP=MCSCF RUNTYP=OPTIMIZE NZVAR=3 COORD=ZMT $END $SYSTEM TIMLIM=5 MEMORY= $END $BASIS GBASIS=STO NGAUSS=2 $END $DATA Methylene...1-A-1 state...mcscf/sto-2g Cnv 2 C H 1 rch H 1 rch 2 ahoh rch=1.09 ahoh=99.0 $END $ZMAT ZMAT(1)=1,1,2, 1,1,3, 2,2,1,3 $END!! Normally one starts a MCSCF run with converged SCF orbitals $GUESS GUESS=HUCKEL $END!! two active electrons in two active orbitals.! must find at least two roots since ground state is 3-B-1! $DET NCORE=3 NACT=2 NELS=2 NSTATE=2 $END
42 ALTERNATIVES TO MCSCF SINGLE REFERENCE METHODS Spin-Flip: Anna Krylov Completely renormalized CCSD(T): Piecuch Adds a denominator to CCSD(T) that permits correct single bond breaking Potentially huge impact: Treats diradicals correctly Open or closed shells CCTYP=CR-CCL in GAMESS
43
44
45
46 RECENT DEVELOPMENTS ORMAS (Joe Ivanic) Occupation restricted multiple active spaces Method for expanding size of MCSCF Identify several smaller subspaces Also ORMAS-PT2 Eliminating deadwood from MCSCF, CI Ruedenberg, Ivanic, Bytautas Extrapolate to complete basis set Interpolate to exact Full CI Allows full CI on much larger systems Parallel MCSCF, CI
AN INTRODUCTION TO QUANTUM CHEMISTRY. Mark S. Gordon Iowa State University
 AN INTRODUCTION TO QUANTUM CHEMISTRY Mark S. Gordon Iowa State University 1 OUTLINE Theoretical Background in Quantum Chemistry Overview of GAMESS Program Applications 2 QUANTUM CHEMISTRY In principle,
AN INTRODUCTION TO QUANTUM CHEMISTRY Mark S. Gordon Iowa State University 1 OUTLINE Theoretical Background in Quantum Chemistry Overview of GAMESS Program Applications 2 QUANTUM CHEMISTRY In principle,
Computational chemistry with GAMESS: a very brief overview with examples
 Computational chemistry with GAMESS: a very brief overview with examples PHY-6120 Molecular Physics (Spring 2015), UConn Phys. Dept. Feb 17 th 2015 H = ħ2 2μ i Intro: V(R) for diatomic molecules + k Z
Computational chemistry with GAMESS: a very brief overview with examples PHY-6120 Molecular Physics (Spring 2015), UConn Phys. Dept. Feb 17 th 2015 H = ħ2 2μ i Intro: V(R) for diatomic molecules + k Z
Beyond the Hartree-Fock Approximation: Configuration Interaction
 Beyond the Hartree-Fock Approximation: Configuration Interaction The Hartree-Fock (HF) method uses a single determinant (single electronic configuration) description of the electronic wavefunction. For
Beyond the Hartree-Fock Approximation: Configuration Interaction The Hartree-Fock (HF) method uses a single determinant (single electronic configuration) description of the electronic wavefunction. For
Molecular Simulation I
 Molecular Simulation I Quantum Chemistry Classical Mechanics E = Ψ H Ψ ΨΨ U = E bond +E angle +E torsion +E non-bond Jeffry D. Madura Department of Chemistry & Biochemistry Center for Computational Sciences
Molecular Simulation I Quantum Chemistry Classical Mechanics E = Ψ H Ψ ΨΨ U = E bond +E angle +E torsion +E non-bond Jeffry D. Madura Department of Chemistry & Biochemistry Center for Computational Sciences
THE CONSTRUCTION AND INTERPRETATION OF MCSCF WAVEFUNCTIONS
 Annu. Rev. Phys. Chem. 1998. 49:233 66 Copyright c 1998 by Annual Reviews. All rights reserved THE CONSTRUCTION AND INTERPRETATION OF MCSCF WAVEFUNCTIONS Michael W. Schmidt and Mark S. Gordon Department
Annu. Rev. Phys. Chem. 1998. 49:233 66 Copyright c 1998 by Annual Reviews. All rights reserved THE CONSTRUCTION AND INTERPRETATION OF MCSCF WAVEFUNCTIONS Michael W. Schmidt and Mark S. Gordon Department
Introduction to Computational Chemistry
 Introduction to Computational Chemistry Vesa Hänninen Laboratory of Physical Chemistry Chemicum 4th floor vesa.hanninen@helsinki.fi September 10, 2013 Lecture 3. Electron correlation methods September
Introduction to Computational Chemistry Vesa Hänninen Laboratory of Physical Chemistry Chemicum 4th floor vesa.hanninen@helsinki.fi September 10, 2013 Lecture 3. Electron correlation methods September
Electron Correlation Methods
 Electron Correlation Methods HF method: electron-electron interaction is replaced by an average interaction E HF c = E 0 E HF E 0 exact ground state energy E HF HF energy for a given basis set HF E c
Electron Correlation Methods HF method: electron-electron interaction is replaced by an average interaction E HF c = E 0 E HF E 0 exact ground state energy E HF HF energy for a given basis set HF E c
Multiconfigurational Quantum Chemistry. Björn O. Roos as told by RL Department of Theoretical Chemistry Chemical Center Lund University Sweden
 Multiconfigurational Quantum Chemistry Björn O. Roos as told by RL Department of Theoretical Chemistry Chemical Center Lund University Sweden April 20, 2009 1 The Slater determinant Using the spin-orbitals,
Multiconfigurational Quantum Chemistry Björn O. Roos as told by RL Department of Theoretical Chemistry Chemical Center Lund University Sweden April 20, 2009 1 The Slater determinant Using the spin-orbitals,
4 Post-Hartree Fock Methods: MPn and Configuration Interaction
 4 Post-Hartree Fock Methods: MPn and Configuration Interaction In the limit of a complete basis, the Hartree-Fock (HF) energy in the complete basis set limit (ECBS HF ) yields an upper boundary to the
4 Post-Hartree Fock Methods: MPn and Configuration Interaction In the limit of a complete basis, the Hartree-Fock (HF) energy in the complete basis set limit (ECBS HF ) yields an upper boundary to the
Practical Issues on the Use of the CASPT2/CASSCF Method in Modeling Photochemistry: the Selection and Protection of an Active Space
 Practical Issues on the Use of the CASPT2/CASSCF Method in Modeling Photochemistry: the Selection and Protection of an Active Space Roland Lindh Dept. of Chemistry Ångström The Theoretical Chemistry Programme
Practical Issues on the Use of the CASPT2/CASSCF Method in Modeling Photochemistry: the Selection and Protection of an Active Space Roland Lindh Dept. of Chemistry Ångström The Theoretical Chemistry Programme
Practical Advice for Quantum Chemistry Computations. C. David Sherrill School of Chemistry and Biochemistry Georgia Institute of Technology
 Practical Advice for Quantum Chemistry Computations C. David Sherrill School of Chemistry and Biochemistry Georgia Institute of Technology Choice of Basis Set STO-3G is too small 6-31G* or 6-31G** 6 probably
Practical Advice for Quantum Chemistry Computations C. David Sherrill School of Chemistry and Biochemistry Georgia Institute of Technology Choice of Basis Set STO-3G is too small 6-31G* or 6-31G** 6 probably
Lec20 Fri 3mar17
 564-17 Lec20 Fri 3mar17 [PDF]GAUSSIAN 09W TUTORIAL www.molcalx.com.cn/wp-content/uploads/2015/01/gaussian09w_tutorial.pdf by A Tomberg - Cited by 8 - Related articles GAUSSIAN 09W TUTORIAL. AN INTRODUCTION
564-17 Lec20 Fri 3mar17 [PDF]GAUSSIAN 09W TUTORIAL www.molcalx.com.cn/wp-content/uploads/2015/01/gaussian09w_tutorial.pdf by A Tomberg - Cited by 8 - Related articles GAUSSIAN 09W TUTORIAL. AN INTRODUCTION
Jack Simons, Henry Eyring Scientist and Professor Chemistry Department University of Utah
 1. Born-Oppenheimer approx.- energy surfaces 2. Mean-field (Hartree-Fock) theory- orbitals 3. Pros and cons of HF- RHF, UHF 4. Beyond HF- why? 5. First, one usually does HF-how? 6. Basis sets and notations
1. Born-Oppenheimer approx.- energy surfaces 2. Mean-field (Hartree-Fock) theory- orbitals 3. Pros and cons of HF- RHF, UHF 4. Beyond HF- why? 5. First, one usually does HF-how? 6. Basis sets and notations
Chemistry 4560/5560 Molecular Modeling Fall 2014
 Final Exam Name:. User s guide: 1. Read questions carefully and make sure you understand them before answering (if not, ask). 2. Answer only the question that is asked, not a different question. 3. Unless
Final Exam Name:. User s guide: 1. Read questions carefully and make sure you understand them before answering (if not, ask). 2. Answer only the question that is asked, not a different question. 3. Unless
Electron Correlation - Methods beyond Hartree-Fock
 Electron Correlation - Methods beyond Hartree-Fock how to approach chemical accuracy Alexander A. Auer Max-Planck-Institute for Chemical Energy Conversion, Mülheim September 4, 2014 MMER Summerschool 2014
Electron Correlation - Methods beyond Hartree-Fock how to approach chemical accuracy Alexander A. Auer Max-Planck-Institute for Chemical Energy Conversion, Mülheim September 4, 2014 MMER Summerschool 2014
The MCSCF Method *, Molecular Orbitals, Reference Spaces and COLUMBUS Input
 The MCSCF Method *, Molecular Orbitals, Reference Spaces and COLUMBUS Input Hans Lischka University of Vienna *Excerpt of a course presented by R. Shepard, Argonne National Laboratory, at the Workshop
The MCSCF Method *, Molecular Orbitals, Reference Spaces and COLUMBUS Input Hans Lischka University of Vienna *Excerpt of a course presented by R. Shepard, Argonne National Laboratory, at the Workshop
This is called a singlet or spin singlet, because the so called multiplicity, or number of possible orientations of the total spin, which is
 9. Open shell systems The derivation of Hartree-Fock equations (Chapter 7) was done for a special case of a closed shell systems. Closed shell means that each MO is occupied by two electrons with the opposite
9. Open shell systems The derivation of Hartree-Fock equations (Chapter 7) was done for a special case of a closed shell systems. Closed shell means that each MO is occupied by two electrons with the opposite
QUANTUM CHEMISTRY FOR TRANSITION METALS
 QUANTUM CHEMISTRY FOR TRANSITION METALS Outline I Introduction II Correlation Static correlation effects MC methods DFT III Relativity Generalities From 4 to 1 components Effective core potential Outline
QUANTUM CHEMISTRY FOR TRANSITION METALS Outline I Introduction II Correlation Static correlation effects MC methods DFT III Relativity Generalities From 4 to 1 components Effective core potential Outline
Introduction to multiconfigurational quantum chemistry. Emmanuel Fromager
 Institut de Chimie, Strasbourg, France Page 1 Emmanuel Fromager Institut de Chimie de Strasbourg - Laboratoire de Chimie Quantique - Université de Strasbourg /CNRS M2 lecture, Strasbourg, France. Notations
Institut de Chimie, Strasbourg, France Page 1 Emmanuel Fromager Institut de Chimie de Strasbourg - Laboratoire de Chimie Quantique - Université de Strasbourg /CNRS M2 lecture, Strasbourg, France. Notations
Introduction to Electronic Structure Theory
 CSC/PRACE Spring School in Computational Chemistry 2017 Introduction to Electronic Structure Theory Mikael Johansson http://www.iki.fi/~mpjohans Objective: To get familiarised with the, subjectively chosen,
CSC/PRACE Spring School in Computational Chemistry 2017 Introduction to Electronic Structure Theory Mikael Johansson http://www.iki.fi/~mpjohans Objective: To get familiarised with the, subjectively chosen,
Exercise 1: Structure and dipole moment of a small molecule
 Introduction to computational chemistry Exercise 1: Structure and dipole moment of a small molecule Vesa Hänninen 1 Introduction In this exercise the equilibrium structure and the dipole moment of a small
Introduction to computational chemistry Exercise 1: Structure and dipole moment of a small molecule Vesa Hänninen 1 Introduction In this exercise the equilibrium structure and the dipole moment of a small
2~:J~ -ryej- r- 2 Jr. A - f3. sr(djk nv~tor rn~ +~ rvjs (::-CJ) ::;-1-.'--~ -. rhd. ('-.Ji.L.~ )- r'-d)c, -r/~ JJr - 2~d ~2-Jr fn'6.
 ::;-1-.'--~ -. rhd. ('-.Ji.L.~ )- r'-d)c, -r/~ JJr - 2~d ~2-Jr fn'6.") .~, ~ I, sr(djk nv~tor rn~ +~ rvjs (::-CJ) ::;-1-.'--~ -. rhd. ('-.Ji.L.~ )- r'-d)c, -r/~ JJr - 2~d ~2-Jr fn'6.)1e'" 21t-ol Je C'...-------- lj-vi, J? Jr Jr \Ji 2~:J~ -ryej- r- 2 Jr A - f3 c _,~,= ~,.,w._..._.
.~, ~ I, sr(djk nv~tor rn~ +~ rvjs (::-CJ) ::;-1-.'--~ -. rhd. ('-.Ji.L.~ )- r'-d)c, -r/~ JJr - 2~d ~2-Jr fn'6.)1e'" 21t-ol Je C'...-------- lj-vi, J? Jr Jr \Ji 2~:J~ -ryej- r- 2 Jr A - f3 c _,~,= ~,.,w._..._.
Lecture 4: methods and terminology, part II
 So theory guys have got it made in rooms free of pollution. Instead of problems with the reflux, they have only solutions... In other words, experimentalists will likely die of cancer From working hard,
So theory guys have got it made in rooms free of pollution. Instead of problems with the reflux, they have only solutions... In other words, experimentalists will likely die of cancer From working hard,
3: Many electrons. Orbital symmetries. l =2 1. m l
 3: Many electrons Orbital symmetries Atomic orbitals are labelled according to the principal quantum number, n, and the orbital angular momentum quantum number, l. Electrons in a diatomic molecule experience
3: Many electrons Orbital symmetries Atomic orbitals are labelled according to the principal quantum number, n, and the orbital angular momentum quantum number, l. Electrons in a diatomic molecule experience
Be H. Delocalized Bonding. Localized Bonding. σ 2. σ 1. Two (sp-1s) Be-H σ bonds. The two σ bonding MO s in BeH 2. MO diagram for BeH 2
 Be-H σ bonds. The two σ bonding MO s in BeH 2. MO diagram for BeH 2") The Delocalized Approach to Bonding: The localized models for bonding we have examined (Lewis and VBT) assume that all electrons are restricted to specific bonds between atoms or in lone pairs. In contrast,
The Delocalized Approach to Bonding: The localized models for bonding we have examined (Lewis and VBT) assume that all electrons are restricted to specific bonds between atoms or in lone pairs. In contrast,
Chemistry Publications
 Chemistry Publications Chemistry 2007 Accurate Ab Initio Potential Energy Curve of F2. I. Nonrelativistic Full Valence Configuration Interaction Energies Using the Correlation Energy Extrapolation by Intrinsic
Chemistry Publications Chemistry 2007 Accurate Ab Initio Potential Energy Curve of F2. I. Nonrelativistic Full Valence Configuration Interaction Energies Using the Correlation Energy Extrapolation by Intrinsic
Handbook of Computational Quantum Chemistry. DAVID B. COOK The Department of Chemistry, University of Sheffield
 Handbook of Computational Quantum Chemistry DAVID B. COOK The Department of Chemistry, University of Sheffield Oxford New York Tokyo OXFORD UNIVERSITY PRESS 1998 CONTENTS 1 Mechanics and molecules 1 1.1
Handbook of Computational Quantum Chemistry DAVID B. COOK The Department of Chemistry, University of Sheffield Oxford New York Tokyo OXFORD UNIVERSITY PRESS 1998 CONTENTS 1 Mechanics and molecules 1 1.1
C:\Users\Leonardo\Desktop\README_ELMO_NOTES.txt Montag, 7. Juli :48
 *********** * Input * *********** Besides the normal GAMESS-UK input directives, to perform an ELMO calculation one needs to specify the elmo keyword and to provide additional instructions that are contained
*********** * Input * *********** Besides the normal GAMESS-UK input directives, to perform an ELMO calculation one needs to specify the elmo keyword and to provide additional instructions that are contained
Density Functional Theory
 Chemistry 380.37 Fall 2015 Dr. Jean M. Standard October 28, 2015 Density Functional Theory What is a Functional? A functional is a general mathematical quantity that represents a rule to convert a function
Chemistry 380.37 Fall 2015 Dr. Jean M. Standard October 28, 2015 Density Functional Theory What is a Functional? A functional is a general mathematical quantity that represents a rule to convert a function
Jack Simons, Henry Eyring Scientist and Professor Chemistry Department University of Utah
 1. Born-Oppenheimer approx.- energy surfaces 2. Mean-field (Hartree-Fock) theory- orbitals 3. Pros and cons of HF- RHF, UHF 4. Beyond HF- why? 5. First, one usually does HF-how? 6. Basis sets and notations
1. Born-Oppenheimer approx.- energy surfaces 2. Mean-field (Hartree-Fock) theory- orbitals 3. Pros and cons of HF- RHF, UHF 4. Beyond HF- why? 5. First, one usually does HF-how? 6. Basis sets and notations
Introduction to computational chemistry Exercise I: Structure and electronic energy of a small molecule. Vesa Hänninen
 Introduction to computational chemistry Exercise I: Structure and electronic energy of a small molecule Vesa Hänninen 1 Introduction In this exercise the equilibrium structure and the electronic energy
Introduction to computational chemistry Exercise I: Structure and electronic energy of a small molecule Vesa Hänninen 1 Introduction In this exercise the equilibrium structure and the electronic energy
Hartree, Hartree-Fock and post-hf methods
 Hartree, Hartree-Fock and post-hf methods MSE697 fall 2015 Nicolas Onofrio School of Materials Engineering DLR 428 Purdue University nonofrio@purdue.edu 1 The curse of dimensionality Let s consider a multi
Hartree, Hartree-Fock and post-hf methods MSE697 fall 2015 Nicolas Onofrio School of Materials Engineering DLR 428 Purdue University nonofrio@purdue.edu 1 The curse of dimensionality Let s consider a multi
Jack Simons, Henry Eyring Scientist and Professor Chemistry Department University of Utah
 1. Born-Oppenheimer approx.- energy surfaces 2. Mean-field (Hartree-Fock) theory- orbitals 3. Pros and cons of HF- RHF, UHF 4. Beyond HF- why? 5. First, one usually does HF-how? 6. Basis sets and notations
1. Born-Oppenheimer approx.- energy surfaces 2. Mean-field (Hartree-Fock) theory- orbitals 3. Pros and cons of HF- RHF, UHF 4. Beyond HF- why? 5. First, one usually does HF-how? 6. Basis sets and notations
Chemistry 334 Part 2: Computational Quantum Chemistry
 Chemistry 334 Part 2: Computational Quantum Chemistry 1. Definition Louis Scudiero, Ben Shepler and Kirk Peterson Washington State University January 2006 Computational chemistry is an area of theoretical
Chemistry 334 Part 2: Computational Quantum Chemistry 1. Definition Louis Scudiero, Ben Shepler and Kirk Peterson Washington State University January 2006 Computational chemistry is an area of theoretical
Computational Material Science Part II. Ito Chao ( ) Institute of Chemistry Academia Sinica
 Institute of Chemistry Academia Sinica") Computational Material Science Part II Ito Chao ( ) Institute of Chemistry Academia Sinica Ab Initio Implementations of Hartree-Fock Molecular Orbital Theory Fundamental assumption of HF theory: each electron
Computational Material Science Part II Ito Chao ( ) Institute of Chemistry Academia Sinica Ab Initio Implementations of Hartree-Fock Molecular Orbital Theory Fundamental assumption of HF theory: each electron
On the Uniqueness of Molecular Orbitals and limitations of the MO-model.
 On the Uniqueness of Molecular Orbitals and limitations of the MO-model. The purpose of these notes is to make clear that molecular orbitals are a particular way to represent many-electron wave functions.
On the Uniqueness of Molecular Orbitals and limitations of the MO-model. The purpose of these notes is to make clear that molecular orbitals are a particular way to represent many-electron wave functions.
Ab initio calculations for potential energy surfaces. D. Talbi GRAAL- Montpellier
 Ab initio calculations for potential energy surfaces D. Talbi GRAAL- Montpellier A theoretical study of a reaction is a two step process I-Electronic calculations : techniques of quantum chemistry potential
Ab initio calculations for potential energy surfaces D. Talbi GRAAL- Montpellier A theoretical study of a reaction is a two step process I-Electronic calculations : techniques of quantum chemistry potential
LUMO + 1 LUMO. Tómas Arnar Guðmundsson Report 2 Reikniefnafræði G
 Q1: Display all the MOs for N2 in your report and classify each one of them as bonding, antibonding or non-bonding, and say whether the symmetry of the orbital is σ or π. Sketch a molecular orbital diagram
Q1: Display all the MOs for N2 in your report and classify each one of them as bonding, antibonding or non-bonding, and say whether the symmetry of the orbital is σ or π. Sketch a molecular orbital diagram
Electric properties of molecules
 Electric properties of molecules For a molecule in a uniform electric fielde the Hamiltonian has the form: Ĥ(E) = Ĥ + E ˆµ x where we assume that the field is directed along the x axis and ˆµ x is the
Electric properties of molecules For a molecule in a uniform electric fielde the Hamiltonian has the form: Ĥ(E) = Ĥ + E ˆµ x where we assume that the field is directed along the x axis and ˆµ x is the
I. CSFs Are Used to Express the Full N-Electron Wavefunction
 Chapter 11 One Must be Able to Evaluate the Matrix Elements Among Properly Symmetry Adapted N- Electron Configuration Functions for Any Operator, the Electronic Hamiltonian in Particular. The Slater-Condon
Chapter 11 One Must be Able to Evaluate the Matrix Elements Among Properly Symmetry Adapted N- Electron Configuration Functions for Any Operator, the Electronic Hamiltonian in Particular. The Slater-Condon
Chapter: 22. Visualization: Making INPUT File and Processing of Output Results
 Chapter: 22 Visualization: Making INPUT File and Processing of Output Results Keywords: visualization, input and output structure, molecular orbital, electron density. In the previous chapters, we have
Chapter: 22 Visualization: Making INPUT File and Processing of Output Results Keywords: visualization, input and output structure, molecular orbital, electron density. In the previous chapters, we have
Advanced Electronic Structure Theory Density functional theory. Dr Fred Manby
 Advanced Electronic Structure Theory Density functional theory Dr Fred Manby fred.manby@bris.ac.uk http://www.chm.bris.ac.uk/pt/manby/ 6 Strengths of DFT DFT is one of many theories used by (computational)
Advanced Electronic Structure Theory Density functional theory Dr Fred Manby fred.manby@bris.ac.uk http://www.chm.bris.ac.uk/pt/manby/ 6 Strengths of DFT DFT is one of many theories used by (computational)
Same idea for polyatomics, keep track of identical atom e.g. NH 3 consider only valence electrons F(2s,2p) H(1s)
 H(1s)") XIII 63 Polyatomic bonding -09 -mod, Notes (13) Engel 16-17 Balance: nuclear repulsion, positive e-n attraction, neg. united atom AO ε i applies to all bonding, just more nuclei repulsion biggest at low
XIII 63 Polyatomic bonding -09 -mod, Notes (13) Engel 16-17 Balance: nuclear repulsion, positive e-n attraction, neg. united atom AO ε i applies to all bonding, just more nuclei repulsion biggest at low
We also deduced that transformations between Slater determinants are always of the form
 .3 Hartree-Fock The Hartree-Fock method assumes that the true N-body ground state wave function can be approximated by a single Slater determinant and minimizes the energy among all possible choices of
.3 Hartree-Fock The Hartree-Fock method assumes that the true N-body ground state wave function can be approximated by a single Slater determinant and minimizes the energy among all possible choices of
σ u * 1s g - gerade u - ungerade * - antibonding σ g 1s
 One of these two states is a repulsive (dissociative) state. Other excited states can be constructed using linear combinations of other orbitals. Some will be binding and others will be repulsive. Thus
One of these two states is a repulsive (dissociative) state. Other excited states can be constructed using linear combinations of other orbitals. Some will be binding and others will be repulsive. Thus
Chapter 10: Chemical Bonding II. Bonding Theories
 Chapter 10: Chemical Bonding II Dr. Chris Kozak Memorial University of Newfoundland, Canada Bonding Theories Previously, we saw how the shapes of molecules can be predicted from the orientation of electron
Chapter 10: Chemical Bonding II Dr. Chris Kozak Memorial University of Newfoundland, Canada Bonding Theories Previously, we saw how the shapes of molecules can be predicted from the orientation of electron
Performance of Hartree-Fock and Correlated Methods
 Chemistry 460 Fall 2017 Dr. Jean M. Standard December 4, 2017 Performance of Hartree-Fock and Correlated Methods Hartree-Fock Methods Hartree-Fock methods generally yield optimized geomtries and molecular
Chemistry 460 Fall 2017 Dr. Jean M. Standard December 4, 2017 Performance of Hartree-Fock and Correlated Methods Hartree-Fock Methods Hartree-Fock methods generally yield optimized geomtries and molecular
Vol. 9 COMPUTATIONAL CHEMISTRY 319
 Vol. 9 COMPUTATIONAL CHEMISTRY 319 COMPUTATIONAL QUANTUM CHEMISTRY FOR FREE-RADICAL POLYMERIZATION Introduction Chemistry is traditionally thought of as an experimental science, but recent rapid and continuing
Vol. 9 COMPUTATIONAL CHEMISTRY 319 COMPUTATIONAL QUANTUM CHEMISTRY FOR FREE-RADICAL POLYMERIZATION Introduction Chemistry is traditionally thought of as an experimental science, but recent rapid and continuing
Example questions for Molecular modelling (Level 4) Dr. Adrian Mulholland
 Dr. Adrian Mulholland") Example questions for Molecular modelling (Level 4) Dr. Adrian Mulholland 1) Question. Two methods which are widely used for the optimization of molecular geometies are the Steepest descents and Newton-Raphson
Example questions for Molecular modelling (Level 4) Dr. Adrian Mulholland 1) Question. Two methods which are widely used for the optimization of molecular geometies are the Steepest descents and Newton-Raphson
$ +! j. % i PERTURBATION THEORY AND SUBGROUPS (REVISED 11/15/08)
") PERTURBATION THEORY AND SUBGROUPS REVISED 11/15/08) The use of groups and their subgroups is of much importance when perturbation theory is employed in understanding molecular orbital theory and spectroscopy
PERTURBATION THEORY AND SUBGROUPS REVISED 11/15/08) The use of groups and their subgroups is of much importance when perturbation theory is employed in understanding molecular orbital theory and spectroscopy
Molecular Orbital Theory This means that the coefficients in the MO will not be the same!
 Diatomic molecules: Heteronuclear molecules In heteronuclear diatomic molecules, the relative contribution of atomic orbitals to each MO is not equal. Some MO s will have more contribution from AO s on
Diatomic molecules: Heteronuclear molecules In heteronuclear diatomic molecules, the relative contribution of atomic orbitals to each MO is not equal. Some MO s will have more contribution from AO s on
CASSCF and NEVPT2 calculations: Ground and excited states of multireference systems. A case study of Ni(CO)4 and the magnetic system NArO
4 and the magnetic system NArO") CASSCF and NEVPT2 calculations: Ground and excited states of multireference systems. A case study of Ni(CO)4 and the magnetic system NArO The ground states of many molecules are often well described by
CASSCF and NEVPT2 calculations: Ground and excited states of multireference systems. A case study of Ni(CO)4 and the magnetic system NArO The ground states of many molecules are often well described by
Handbook of Computational Quantum Chemistry
 Handbook of Computational Quantum Chemistry David B. Cook Dept. of Chemistry University of Sheffield DOVER PUBLICATIONS, INC. Mineola, New York F Contents 1 Mechanics and molecules 1 1.1 1.2 1.3 1.4 1.5
Handbook of Computational Quantum Chemistry David B. Cook Dept. of Chemistry University of Sheffield DOVER PUBLICATIONS, INC. Mineola, New York F Contents 1 Mechanics and molecules 1 1.1 1.2 1.3 1.4 1.5
Ionic Versus Covalent Bonding
 Ionic Versus Covalent Bonding Ionic compounds are formed when electrons are transferred from one atom to another The transfer of electrons forms ions Each ion is isoelectronic with a noble gas Electrostatic
Ionic Versus Covalent Bonding Ionic compounds are formed when electrons are transferred from one atom to another The transfer of electrons forms ions Each ion is isoelectronic with a noble gas Electrostatic
The Hartree-Fock approximation
 Contents The Born-Oppenheimer approximation Literature Quantum mechanics 2 - Lecture 7 November 21, 2012 Contents The Born-Oppenheimer approximation Literature 1 The Born-Oppenheimer approximation 2 3
Contents The Born-Oppenheimer approximation Literature Quantum mechanics 2 - Lecture 7 November 21, 2012 Contents The Born-Oppenheimer approximation Literature 1 The Born-Oppenheimer approximation 2 3
An Introduction to Quantum Chemistry and Potential Energy Surfaces. Benjamin G. Levine
 An Introduction to Quantum Chemistry and Potential Energy Surfaces Benjamin G. Levine This Week s Lecture Potential energy surfaces What are they? What are they good for? How do we use them to solve chemical
An Introduction to Quantum Chemistry and Potential Energy Surfaces Benjamin G. Levine This Week s Lecture Potential energy surfaces What are they? What are they good for? How do we use them to solve chemical
Electron Correlation
 Electron Correlation Levels of QM Theory HΨ=EΨ Born-Oppenheimer approximation Nuclear equation: H n Ψ n =E n Ψ n Electronic equation: H e Ψ e =E e Ψ e Single determinant SCF Semi-empirical methods Correlation
Electron Correlation Levels of QM Theory HΨ=EΨ Born-Oppenheimer approximation Nuclear equation: H n Ψ n =E n Ψ n Electronic equation: H e Ψ e =E e Ψ e Single determinant SCF Semi-empirical methods Correlation
A One-Slide Summary of Quantum Mechanics
 A One-Slide Summary of Quantum Mechanics Fundamental Postulate: O! = a! What is!?! is an oracle! operator wave function (scalar) observable Where does! come from?! is refined Variational Process H! = E!
A One-Slide Summary of Quantum Mechanics Fundamental Postulate: O! = a! What is!?! is an oracle! operator wave function (scalar) observable Where does! come from?! is refined Variational Process H! = E!
Electronic structure theory: Fundamentals to frontiers. 1. Hartree-Fock theory
 Electronic structure theory: Fundamentals to frontiers. 1. Hartree-Fock theory MARTIN HEAD-GORDON, Department of Chemistry, University of California, and Chemical Sciences Division, Lawrence Berkeley National
Electronic structure theory: Fundamentals to frontiers. 1. Hartree-Fock theory MARTIN HEAD-GORDON, Department of Chemistry, University of California, and Chemical Sciences Division, Lawrence Berkeley National
Quantum Chemistry Methods
 1 Quantum Chemistry Methods T. Helgaker, Department of Chemistry, University of Oslo, Norway The electronic Schrödinger equation Hartree Fock theory self-consistent field theory basis functions and basis
1 Quantum Chemistry Methods T. Helgaker, Department of Chemistry, University of Oslo, Norway The electronic Schrödinger equation Hartree Fock theory self-consistent field theory basis functions and basis
Building a wavefunction within the Complete-Active. Cluster with Singles and Doubles formalism: straightforward description of quasidegeneracy
 Building a wavefunction within the Complete-Active Active-Space Coupled-Cluster Cluster with Singles and Doubles formalism: straightforward description of quasidegeneracy Dmitry I. Lyakh (Karazin Kharkiv
Building a wavefunction within the Complete-Active Active-Space Coupled-Cluster Cluster with Singles and Doubles formalism: straightforward description of quasidegeneracy Dmitry I. Lyakh (Karazin Kharkiv
Electronic Structure of the Active Site of Truncated Hemoglobin N in MCSCF Approach
 Mathematical Biology & Bioinformatics. 2011. V. 6. 1. P. 23 38. URL: http://www.matbio.org/2011/tulub_en2011(6_23).pdf =========== PROCEEDINGS OF THE III INTERNATIONAL CONFERENCE ========= ===========
Mathematical Biology & Bioinformatics. 2011. V. 6. 1. P. 23 38. URL: http://www.matbio.org/2011/tulub_en2011(6_23).pdf =========== PROCEEDINGS OF THE III INTERNATIONAL CONFERENCE ========= ===========
Jack Simons, Henry Eyring Scientist and Professor Chemistry Department University of Utah
 1. Born-Oppenheimer approx.- energy surfaces 2. Mean-field (Hartree-Fock) theory- orbitals 3. Pros and cons of HF- RHF, UHF 4. Beyond HF- why? 5. First, one usually does HF-how? 6. Basis sets and notations
1. Born-Oppenheimer approx.- energy surfaces 2. Mean-field (Hartree-Fock) theory- orbitals 3. Pros and cons of HF- RHF, UHF 4. Beyond HF- why? 5. First, one usually does HF-how? 6. Basis sets and notations
Molecular Orbital Theory. Molecular Orbital Theory: Electrons are located in the molecule, not held in discrete regions between two bonded atoms
 Molecular Orbital Theory Valence Bond Theory: Electrons are located in discrete pairs between specific atoms Molecular Orbital Theory: Electrons are located in the molecule, not held in discrete regions
Molecular Orbital Theory Valence Bond Theory: Electrons are located in discrete pairs between specific atoms Molecular Orbital Theory: Electrons are located in the molecule, not held in discrete regions
Chemistry 433 Computational Chemistry Fall Semester 2002 Dr. Rainer Glaser
 Chemistry 433 Computational Chemistry Fall Semester 2002 Dr. Rainer Glaser Second 1-Hour Examination Electron Correlation Methods Wednesday, November 6, 2002, 11:00-11:50 Name: Answer Key Question 1. Electron
Chemistry 433 Computational Chemistry Fall Semester 2002 Dr. Rainer Glaser Second 1-Hour Examination Electron Correlation Methods Wednesday, November 6, 2002, 11:00-11:50 Name: Answer Key Question 1. Electron
Chapter 1 Chemical Bonding
 Chapter 1 Chemical Bonding 1.1 Atoms, Electrons, and Orbitals Atoms are composed of + Protons positively charged mass = 1.6726 X 10-27 kg Neutrons neutral mass = 1.6750 X 10-27 kg Electrons negatively
Chapter 1 Chemical Bonding 1.1 Atoms, Electrons, and Orbitals Atoms are composed of + Protons positively charged mass = 1.6726 X 10-27 kg Neutrons neutral mass = 1.6750 X 10-27 kg Electrons negatively
The successful wavefunction can be written as a determinant: # 1 (2) # 2 (2) Electrons. This can be generalized to our 2N-electron wavefunction:
 # 2 (2) Electrons. This can be generalized to our 2N-electron wavefunction:") T2. CNDO to AM1: The Semiempirical Molecular Orbital Models The discussion in sections T2.1 T2.3 applies also to ab initio molecular orbital calculations. T2.1 Slater Determinants Consider the general
T2. CNDO to AM1: The Semiempirical Molecular Orbital Models The discussion in sections T2.1 T2.3 applies also to ab initio molecular orbital calculations. T2.1 Slater Determinants Consider the general
Lecture 12. Symmetry Operations. NC State University
 Chemistry 431 Lecture 12 Group Theory Symmetry Operations NC State University Wave functions as the basis for irreducible representations The energy of the system will not change when symmetry Operations
Chemistry 431 Lecture 12 Group Theory Symmetry Operations NC State University Wave functions as the basis for irreducible representations The energy of the system will not change when symmetry Operations
Lecture 5: More about one- Final words about the Hartree-Fock theory. First step above it by the Møller-Plesset perturbation theory.
 Lecture 5: More about one- determinant wave functions Final words about the Hartree-Fock theory. First step above it by the Møller-Plesset perturbation theory. Items from Lecture 4 Could the Koopmans theorem
Lecture 5: More about one- determinant wave functions Final words about the Hartree-Fock theory. First step above it by the Møller-Plesset perturbation theory. Items from Lecture 4 Could the Koopmans theorem
ATOMIC STRUCTURE. Atomic Structure. Atomic orbitals and their energies (a) Hydrogenic radial wavefunctions
 Hydrogenic radial wavefunctions") ATOMIC STRUCTURE Atomic orbitals and their energies (a) Hydrogenic radial wavefunctions Bundet Boekfa Chem Div, Fac Lib Arts & Sci Kasetsart University Kamphaeng Saen Campus 1 2 Atomic orbitals and their
ATOMIC STRUCTURE Atomic orbitals and their energies (a) Hydrogenic radial wavefunctions Bundet Boekfa Chem Div, Fac Lib Arts & Sci Kasetsart University Kamphaeng Saen Campus 1 2 Atomic orbitals and their
NMR and IR spectra & vibrational analysis
 Lab 5: NMR and IR spectra & vibrational analysis A brief theoretical background 1 Some of the available chemical quantum methods for calculating NMR chemical shifts are based on the Hartree-Fock self-consistent
Lab 5: NMR and IR spectra & vibrational analysis A brief theoretical background 1 Some of the available chemical quantum methods for calculating NMR chemical shifts are based on the Hartree-Fock self-consistent
Hartree-Fock-Roothan Self-Consistent Field Method
 Hartree-Fock-Roothan Self-Consistent Field Method 1. Helium Here is a summary of the derivation of the Hartree-Fock equations presented in class. First consider the ground state of He and start with with
Hartree-Fock-Roothan Self-Consistent Field Method 1. Helium Here is a summary of the derivation of the Hartree-Fock equations presented in class. First consider the ground state of He and start with with
Coupled-Cluster Perturbative Triples for Bond Breaking
 Coupled-Cluster Perturbative Triples for Bond Breaking Andrew G. Taube and Rodney J. Bartlett Quantum Theory Project University of Florida INT CC Meeting Seattle July 8, 2008 Why does chemistry need triples?
Coupled-Cluster Perturbative Triples for Bond Breaking Andrew G. Taube and Rodney J. Bartlett Quantum Theory Project University of Florida INT CC Meeting Seattle July 8, 2008 Why does chemistry need triples?
Lecture 4: Hartree-Fock Theory
 Lecture 4: Hartree-Fock Theory One determinant to rule them all, One determinant to find them, One determinant to bring them all and in the darkness bind them Second quantization rehearsal The formalism
Lecture 4: Hartree-Fock Theory One determinant to rule them all, One determinant to find them, One determinant to bring them all and in the darkness bind them Second quantization rehearsal The formalism
7: Hückel theory for polyatomic molecules
 7: ückel theory for polyatomic molecules Introduction Approximate treatment of π electron systems in organic molecules: 1 Approximations 3 4 5 6 1. π and σ frameworks completely separated. Trial wavefunctions
7: ückel theory for polyatomic molecules Introduction Approximate treatment of π electron systems in organic molecules: 1 Approximations 3 4 5 6 1. π and σ frameworks completely separated. Trial wavefunctions
Computational Chemistry I
 Computational Chemistry I Text book Cramer: Essentials of Quantum Chemistry, Wiley (2 ed.) Chapter 3. Post Hartree-Fock methods (Cramer: chapter 7) There are many ways to improve the HF method. Most of
Computational Chemistry I Text book Cramer: Essentials of Quantum Chemistry, Wiley (2 ed.) Chapter 3. Post Hartree-Fock methods (Cramer: chapter 7) There are many ways to improve the HF method. Most of
NWChem: Coupled Cluster Method (Tensor Contraction Engine)
") NWChem: Coupled Cluster Method (Tensor Contraction Engine) Why CC is important?! Correlation effects are important!! CC is size-extensive theory: can be used to describe dissociation processes.! Higher-order
NWChem: Coupled Cluster Method (Tensor Contraction Engine) Why CC is important?! Correlation effects are important!! CC is size-extensive theory: can be used to describe dissociation processes.! Higher-order
The wavefunction that describes a bonding pair of electrons:
 4.2. Molecular Properties from VB Theory a) Bonding and Bond distances The wavefunction that describes a bonding pair of electrons: Ψ b = a(h 1 ) + b(h 2 ) where h 1 and h 2 are HAOs on adjacent atoms
4.2. Molecular Properties from VB Theory a) Bonding and Bond distances The wavefunction that describes a bonding pair of electrons: Ψ b = a(h 1 ) + b(h 2 ) where h 1 and h 2 are HAOs on adjacent atoms
Stationarity of electron distribution in ground-state molecular systems
 J Math Chem (2013) 51:1388 1396 DOI 10.1007/s10910-013-0153-8 ORIGINAL PAPER Stationarity of electron distribution in ground-state molecular systems Dariusz Szczepanik Janusz Mrozek Received: 10 December
J Math Chem (2013) 51:1388 1396 DOI 10.1007/s10910-013-0153-8 ORIGINAL PAPER Stationarity of electron distribution in ground-state molecular systems Dariusz Szczepanik Janusz Mrozek Received: 10 December
ELECTRONIC STRUCTURE CALCULATIONS OF OPEN-SHELL MOLECULES. Lyudmila V. Slipchenko. A Dissertation Presented to the FACULTY OF THE GRADUATE SCHOOL
 ELECTRONIC STRUCTURE CALCULATIONS OF OPEN-SHELL MOLECULES by Lyudmila V. Slipchenko A Dissertation Presented to the FACULTY OF THE GRADUATE SCHOOL UNIVERSITY OF SOUTHERN CALIFORNIA In Partial Fulfillment
ELECTRONIC STRUCTURE CALCULATIONS OF OPEN-SHELL MOLECULES by Lyudmila V. Slipchenko A Dissertation Presented to the FACULTY OF THE GRADUATE SCHOOL UNIVERSITY OF SOUTHERN CALIFORNIA In Partial Fulfillment
Size-extensive wave functions for QMC A linear-scaling GVB approach
 Size-extensive wave functions for QMC A linear-scaling GVB approach Claudia Filippi, University of Twente, The Netherlands Francesco Fracchia, University of Pisa, Italy Claudio Amovilli, University of
Size-extensive wave functions for QMC A linear-scaling GVB approach Claudia Filippi, University of Twente, The Netherlands Francesco Fracchia, University of Pisa, Italy Claudio Amovilli, University of
Quantum mechanics can be used to calculate any property of a molecule. The energy E of a wavefunction Ψ evaluated for the Hamiltonian H is,
 Chapter : Molecules Quantum mechanics can be used to calculate any property of a molecule The energy E of a wavefunction Ψ evaluated for the Hamiltonian H is, E = Ψ H Ψ Ψ Ψ 1) At first this seems like
Chapter : Molecules Quantum mechanics can be used to calculate any property of a molecule The energy E of a wavefunction Ψ evaluated for the Hamiltonian H is, E = Ψ H Ψ Ψ Ψ 1) At first this seems like
COPYRIGHTED MATERIAL. Quantum Mechanics for Organic Chemistry &CHAPTER 1
 &CHAPTER 1 Quantum Mechanics for Organic Chemistry Computational chemistry, as explored in this book, will be restricted to quantum mechanical descriptions of the molecules of interest. This should not
&CHAPTER 1 Quantum Mechanics for Organic Chemistry Computational chemistry, as explored in this book, will be restricted to quantum mechanical descriptions of the molecules of interest. This should not
Chem 4502 Introduction to Quantum Mechanics and Spectroscopy 3 Credits Fall Semester 2014 Laura Gagliardi. Lecture 28, December 08, 2014
 Chem 4502 Introduction to Quantum Mechanics and Spectroscopy 3 Credits Fall Semester 2014 Laura Gagliardi Lecture 28, December 08, 2014 Solved Homework Water, H 2 O, involves 2 hydrogen atoms and an oxygen
Chem 4502 Introduction to Quantum Mechanics and Spectroscopy 3 Credits Fall Semester 2014 Laura Gagliardi Lecture 28, December 08, 2014 Solved Homework Water, H 2 O, involves 2 hydrogen atoms and an oxygen
Chem 442 Review for Exam 2. Exact separation of the Hamiltonian of a hydrogenic atom into center-of-mass (3D) and relative (3D) components.
 and relative (3D) components.") Chem 44 Review for Exam Hydrogenic atoms: The Coulomb energy between two point charges Ze and e: V r Ze r Exact separation of the Hamiltonian of a hydrogenic atom into center-of-mass (3D) and relative
Chem 44 Review for Exam Hydrogenic atoms: The Coulomb energy between two point charges Ze and e: V r Ze r Exact separation of the Hamiltonian of a hydrogenic atom into center-of-mass (3D) and relative
COMPUTATIONAL MODELING OF SMALL MOLECULES. Rebecca J. Weber, B.S. Dissertation Prepared for the Degree of DOCTOR OF PHILOSOPHY
 COMPUTATIONAL MODELING OF SMALL MOLECULES Rebecca J. Weber, B.S. Dissertation Prepared for the Degree of DOCTOR OF PHILOSOPHY UNIVERSITY OF NORTH TEXAS December 2015 APPROVED: Angela K. Wilson, Major Professor
COMPUTATIONAL MODELING OF SMALL MOLECULES Rebecca J. Weber, B.S. Dissertation Prepared for the Degree of DOCTOR OF PHILOSOPHY UNIVERSITY OF NORTH TEXAS December 2015 APPROVED: Angela K. Wilson, Major Professor
Ψ = Σ m C i,m φ 1...φ m...φ N + φ 1...φ i...φ N.
 Chapter 19 Corrections to the mean-field model are needed to describe the instantaneous Coulombic interactions among the electrons. This is achieved by including more than one Slater determinant in the
Chapter 19 Corrections to the mean-field model are needed to describe the instantaneous Coulombic interactions among the electrons. This is achieved by including more than one Slater determinant in the
OVERVIEW OF QUANTUM CHEMISTRY METHODS
 OVERVIEW OF QUANTUM CHEMISTRY METHODS Outline I Generalities Correlation, basis sets Spin II Wavefunction methods Hartree-Fock Configuration interaction Coupled cluster Perturbative methods III Density
OVERVIEW OF QUANTUM CHEMISTRY METHODS Outline I Generalities Correlation, basis sets Spin II Wavefunction methods Hartree-Fock Configuration interaction Coupled cluster Perturbative methods III Density
MRCI calculations in MOLPRO
 1 MRCI calculations in MOLPRO Molpro is a software package written in Fortran and maintained by H.J. Werner and P.J. Knowles. It is often used for performing sophisticated electronic structure calculations,
1 MRCI calculations in MOLPRO Molpro is a software package written in Fortran and maintained by H.J. Werner and P.J. Knowles. It is often used for performing sophisticated electronic structure calculations,
Lecture 16 February 20 Transition metals, Pd and Pt
 Lecture 16 February 20 Transition metals, Pd and Pt Nature of the Chemical Bond with applications to catalysis, materials science, nanotechnology, surface science, bioinorganic chemistry, and energy Course
Lecture 16 February 20 Transition metals, Pd and Pt Nature of the Chemical Bond with applications to catalysis, materials science, nanotechnology, surface science, bioinorganic chemistry, and energy Course
β(n α +1)φ 2 α(2)!φ 1
φ 2 α(2)!φ 1") Spin Contamination in Unrestricted Hartree-Fock Wavefunctions An unrestricted Hartree-Fock (UHF) wavefuntion with α α spins and β β spins has the form ψ (1,,!, ) = Aϕ 1 α(1)ϕ α()!ϕ α α( α )φ 1 β( α +1)φ
Spin Contamination in Unrestricted Hartree-Fock Wavefunctions An unrestricted Hartree-Fock (UHF) wavefuntion with α α spins and β β spins has the form ψ (1,,!, ) = Aϕ 1 α(1)ϕ α()!ϕ α α( α )φ 1 β( α +1)φ
4 Diatomic molecules
 s manual for Burrows et.al. Chemistry 3 Third edition 4 Diatomic molecules Answers to worked examples WE 4.1 The Lewis model (on p. 174 in Chemistry 3 ) Use the Lewis model to describe the bonding in (a)
s manual for Burrows et.al. Chemistry 3 Third edition 4 Diatomic molecules Answers to worked examples WE 4.1 The Lewis model (on p. 174 in Chemistry 3 ) Use the Lewis model to describe the bonding in (a)
CHEM J-5 June 2014
 CHEM1101 2014-J-5 June 2014 The molecular orbital energy level diagrams for H 2, H 2 +, H 2 and O 2 are shown below. Fill in the valence electrons for each species in its ground state and label the types
CHEM1101 2014-J-5 June 2014 The molecular orbital energy level diagrams for H 2, H 2 +, H 2 and O 2 are shown below. Fill in the valence electrons for each species in its ground state and label the types
This is a very succinct primer intended as supplementary material for an undergraduate course in physical chemistry.
 1 Computational Chemistry (Quantum Chemistry) Primer This is a very succinct primer intended as supplementary material for an undergraduate course in physical chemistry. TABLE OF CONTENTS Methods...1 Basis
1 Computational Chemistry (Quantum Chemistry) Primer This is a very succinct primer intended as supplementary material for an undergraduate course in physical chemistry. TABLE OF CONTENTS Methods...1 Basis
v(r i r j ) = h(r i )+ 1 N
 = h(r i )+ 1 N") Chapter 1 Hartree-Fock Theory 1.1 Formalism For N electrons in an external potential V ext (r), the many-electron Hamiltonian can be written as follows: N H = [ p i i=1 m +V ext(r i )]+ 1 N N v(r i r j
Chapter 1 Hartree-Fock Theory 1.1 Formalism For N electrons in an external potential V ext (r), the many-electron Hamiltonian can be written as follows: N H = [ p i i=1 m +V ext(r i )]+ 1 N N v(r i r j
O(3P) + C2H4 Potential Energy Surface: Study at the Multireference Level
 + C2H4 Potential Energy Surface: Study at the Multireference Level") Chemistry Publications Chemistry 10-2009 O(3P) + C2H4 Potential Energy Surface: Study at the Multireference Level Aaron C. West Iowa State University, westac@iastate.edu Joshua S. Kretchmer University
Chemistry Publications Chemistry 10-2009 O(3P) + C2H4 Potential Energy Surface: Study at the Multireference Level Aaron C. West Iowa State University, westac@iastate.edu Joshua S. Kretchmer University
Lecture 11 January 30, Transition metals, Pd and Pt
 Lecture 11 January 30, 2011 Nature of the Chemical Bond with applications to catalysis, materials science, nanotechnology, surface science, bioinorganic chemistry, and energy Course number: Ch120a Hours:
Lecture 11 January 30, 2011 Nature of the Chemical Bond with applications to catalysis, materials science, nanotechnology, surface science, bioinorganic chemistry, and energy Course number: Ch120a Hours:
0 belonging to the unperturbed Hamiltonian H 0 are known
 Time Independent Perturbation Theory D Perturbation theory is used in two qualitatively different contexts in quantum chemistry. It allows one to estimate (because perturbation theory is usually employed
Time Independent Perturbation Theory D Perturbation theory is used in two qualitatively different contexts in quantum chemistry. It allows one to estimate (because perturbation theory is usually employed
5.4. Electronic structure of water
 5.4. Electronic structure of water Water belongs to C 2v point group, we have discussed the corresponding character table. Here it is again: C 2v E C 2 σ v (yz) σ v (xz) A 1 1 1 1 1 A 2 1 1-1 -1 B 1 1-1
5.4. Electronic structure of water Water belongs to C 2v point group, we have discussed the corresponding character table. Here it is again: C 2v E C 2 σ v (yz) σ v (xz) A 1 1 1 1 1 A 2 1 1-1 -1 B 1 1-1
Lecture 9 Electronic Spectroscopy
 Lecture 9 Electronic Spectroscopy Molecular Orbital Theory: A Review - LCAO approximaton & AO overlap - Variation Principle & Secular Determinant - Homonuclear Diatomic MOs - Energy Levels, Bond Order
Lecture 9 Electronic Spectroscopy Molecular Orbital Theory: A Review - LCAO approximaton & AO overlap - Variation Principle & Secular Determinant - Homonuclear Diatomic MOs - Energy Levels, Bond Order