Atomistic Modeling of DNA and Protein Structures
|
|
|
- Quentin Haynes
- 5 years ago
- Views:
Transcription
1 Atomistic Modeling of DNA and Protein Structures George C. Schatz Introduction In this lecture we introduce one of the most commonly used computational tools for studying protein and DNA structure, the use of empirical potential energy functions, and we describe their use in molecular mechanics and molecular dynamics calculations. As an application, we present results of a molecular mechanics study of the mechanical properties (mechanical pulling) of a small protein. We also describe modeling of DNA hairpin structures.
2 References: Computer Simulation of Liquids, M. P. Allen and D. J. Tildesley, Clarendon Press, Oxford, 1987; Introduction to Modern Statistical Mechanics, D. Chandler, Oxford, New York, 1987; Understanding Molecular Simulations: from Algorithms to Applications, D. Frenkel and B. Smit, Academic Press; San Diego, Molecular Modeling: Principles and Applications, (2 nd ed) Andrew Leach, Prentice Hall, Englewood Cliffs, 2001.
3 What is a force field? This is the potential energy function V(R) that determines the interactions between the atoms in a molecule or solid. The derivatives of this function (or more technically the gradients) give the forces that these atoms exert against each other. Thus Newton s equations say that F=ma, where the force F is related to V via F=-dV/dR, m is the mass and a is the acceleration. A simple example of a force field is a Lennard-Jones, or 6-12 potential. This describes the interaction of two argon atoms pretty well, and it is often used to describe the interaction of two methane molecules or even, to a lesser degree of rigor, two water molecules. 7 6 Lennard-Jones potential σ σ V= 4ε R R 12 6 energy/well depth r/σ
4 For a liquid composed of argon, or methane, one can approximate that the overall potential is simply the sum of all pair-wise interactions: V= 4ε 12 6 σ ij σ ij ij ij Rij Rij This neglects three-body and higher interactions, which works pretty well for argon, but is a problem for more complex materials.
5 Where do force fields come from? The correct force fields can be determined by electronic structure calculations, where one calculates the energies of the molecule or material as a function of the positions of the nuclei by solving the Schrödinger equation for the molecular orbitals. We will learn about this process in a later lecture, but suffice it for now to realize that this is a lot of work, and often it is not feasible to do. As an alternative, one can guess a force field, such as the 6-12 potential that we just looked at. This has a little science in it, as the correct interatomic potential at long range looks like -1/R 6, however the 1/R 12 part that describes the short range repulsion is just made up. The correct potential looks more like exp(-r), but it is numerically simpler to use a power law, and 1/R 12 is mathematically convenient. The 6-12 potential has two parameters, the well depth and the equilibrium distance. These are found by fitting results to experiment. You might think that for modeling liquid argon, ε and σ should be obtained by fitting the exact potential for the Ar dimer, but in fact it is better to fit properties of the liquid (such as density, boiling temperature, etc) if you want to obtain realistic estimates of the property of the liquid.
6 What is a protein? Proteins are large molecules that are composed of peptide chains and sometimes other components (heme groups, sugars, nucleotides). The peptide is a polymer of amino acids. Amino acids have the general formula: NH 2 H C COOH R where R is one of 20 possible organic side chains that occur in biological systems. Actually in solution at neutral ph, all amino acids are substantially ionized into zwitterions: however we ll ignore this here. NH 3 + H C COO - R
7 For the naturally occuring amino acids, there are 20 possibilities for side chains (the R group). Examples are (we ll use these later): Name abbr. abbr. formula Alanine Ala A CH 3 Aspartic acid Asp D CH 2 COOH Arginine Arg R (CH 2 ) 3 NHC(NH 2 ) 2 Proline Pro P CH 2 CH 2 CH 2 - Phenylalanine Phe F CH 2 -phenyl Peptides are formed by linking many amino acids together to form a polymer: R 1 R 2 R 3 NH 3+ -C -CO -N -C -CO -N -C -COO - H H H
8 A simple protein: BPTI (bovine pancreatic trypsin inhibitor) This consists of a single peptide having 58 amino acids. These are: ARG PRO ASP PHE CYS LEU GLU PRO PRO TYR THR GLY PRO CYS LYS ALA ARG ILE ILE ARG TYR PHE TYR ASN ALA LYS ALA GLY LEU CYS GLN THR PHE VAL TYR GLY GLY CYS ARG ALA LYS ARG ASN ASN PHE LYS SER ALA GLU ASP CYS MET ARG THR CYS GLY GLY ALA

9 The structure of the first four residues (leaving out hydrogens) is: Color map: red oxygen, blue nitrogen, cyan carbon. ASP PRO ARG PHE

10 The BPTI structure: Ribbon represents the backbone of protein.

11 Note that this structure consists of a jumble of several α-helices, where the general structure of each helix is:
12 How does one describe force fields for proteins? There is no unique procedure, but the general idea is to write the potential as a sum of terms as follows: 1) Atoms that are directly bonded together are represented as a harmonic oscillator in terms of the interatomic separation 2) Three atoms that are bonded can have a bending potential (an oscillator function) as a function of the internal angle 3) Four atoms that form a bonded dihedral angle might have an oscillator function in terms of that angle. 4) In addition, there might be electrostatic interactions between partially charged atoms. The charges are determined by electronic structure methods (perhaps with empirical adjustments). 5) Some force fields include hydrogen bonds for O-H O, O-H N, etc. 6) Also, there is a 6-12 Lennard-Jones interaction between all atoms. This is especially important for describing non-bonded interactions.
13 CHARMM (Chemistry at HARvard using Molecular Mechanics: ``CHARMM: A Program for Macromolecular Energy, Minimization, and Dynamics Calculations'',J. Comp. Chem. 1983, 4, ; A. D. MacKerell, Jr., et al. ``An All-Atom Empirical Energy Function for the Simulation of Nucleic Acids'', J. Am. Chem. Soc. 1995, 117, ) k ( r r ) k b θ ( ) ,3 0 bonds angles 1,3 V= + θ θ + K (S S ) + k k cos( nφ ) + k ( ω ω ) proper dihedrals φ φ ω improper dihedrals e 2 A B qq + + ij ij i j 12 6 pairs, i j rij rij εrij
14 Dirty Laundry: (as described in the literature) Charges for the parameter sets were determined such that gas-phase molecule-water interaction energies and geometries were reproduced as well as dipole moments and heats of sublimation of the compounds. Bond, angle, and dihedral force constants were set so as to match geometries and vibrational spectra for crystal structures, IR and raman intensities and 6-31G* gas-phase calculated properties. A proper balance between solvent-solvent, solute-solvent and solutesolute interaction energies was sought with reference to the TIP3P water molecule. Nucleic acid parameters were tested for their abilities to reproduce acidbase crystals with respect to lattice parameters, nonbonded parameters, and heats of sublimation. vdw parameters were determined empirically
15 AMBER (Assisted Model Building with Energy Refinement): S. J. Weiner, et al. ``A New Force Field for Molecular Mechanical Simulation of Nucleic Acids and Proteins'', J. Am. Chem. Soc., 1982, 106, ; S. J. Weiner, et al. ``An All Atom Force Field for Simulations of Proteins and Nucleic Acids'', J. Comp. Chem. 1986, 7, ; W. D. Cornell, et al. ``A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules'', J. Am. Chem. Soc. 1995, 117, ) V ( ) θ ( ) 2 2 Kr r req K eq + n [ 1+ cos( nφ γ) ] bonds angles dihedrals 2 = + θ θ V + A ij Bij i< j Rij Rij qq i εr ij j + C ij H bonds Rij D ij Rij
16 What do you do with a protein force field? 1) If you know the structure (say from X-Ray measurements) you can build the protein and study its properties using molecular mechanics, molecular dynamics, and other methods. The standard source for protein structures is the Protein Data Bank (pdb). This is available at the web site: 2) If you don t know the structure, but you know the sequence (say from genome data or from sequencing of an unknown protein), you can attempt to build the protein and determine its threedimensional structure. This is the protein folding problem! Unfortunately this is an unsolved process and it is still an active area of research.
17 Classical Molecular Dynamics If we have N particles, each with mass M, then the energy of the system is: 1 E Mv V 2 = kα + kα 2 Here v kα is the velocity of the αth component (α = x, y, z) of the kth particle, and V is the potential energy function. Newton s equations of motion for this system are: 2 dxkα dvkα V M = M = = F 2 dt dt X k α These define a set of 3N differential equations for the coordinates X kα. To solve these equations, we have to specify initial values of the coordinates and momenta of the particles, and then we numerically integrate the differential equations. kα
18 Integrating the classical equations of motion: There are many approaches to numerically integrating the classical equations of motion. Since these are coupled nonlinear ordinary differential equations, stability can be a problem. One example of a numerical integration method is called the leap-frog algorithm. This algorithm (which is closely related to what is often called the velocity Verlet method) defines the velocities v kα and coordinates X kα as follows: v kα(t+ δ t) = v kα(t δ t) + ( δt)m F kα(t) X kα(t+δ t) = X kα(t) + ( δ t)v kα(t+ δt) 2 In this algorithm, the energy at time t is evaluated using the following expression for the velocity at time t: v kα(t) = [v kα(t δ t) + v kα(t + δt)] 2 2 2
19 Molecular Mechanics: This is energy minimization, i.e., from a given starting structure, the goal is to locate the lowest possible energy structure, ideally the global minimum. In most protein structures, it is hard to locate the true local minimum, so inevitably one finds a local minimum that one hopes is at least similar to the global minimum. Finding minima can be done in many ways. One way is to follow the gradient downhill. Since this will inevitably lead to local minima, one usually performs a series of MM and MD calculations in which the MD part is used to explore structures.
20 Software for doing MM and MD calculations for proteins based on empirical force fields There are many programs available that enable one to study the properties of biomolecules. Many have the same names as the force fields that they were originally designed for: CHARMM, Amber, GROMOS. However the force field is usually a separate product from the code, and is mostly public domain. Most simulation codes are commercial although some are cheap to academic users. One code that is entirely public domain is Tinker. Yes there is a Tinker force field as well, but this is not commonly used. However the Tinker simulation code is very popular. It has the feature that it can use all of the commonly available (public domain) force fields, including some, like MM3, that are more sophisticated than CHARMM or Amber (MM3 is usually not used for proteins as the sophistication makes applications run more slowly.) Tinker was developed by Jay Ponder, at Washington University in St. Louis. It is available at:
21 BRIEF OVERVIEW OF TINKER SIMULATION PACKAGE TINKER is a system of programs and routines for molecular mechanics and dynamics as well as other energy-based and structural manipulation calculations. Rather than incorporating all the functionality in one monolithic program, TINKER provides a set of relatively small programs that interact to perform complex computations. The series of major programs included in the distribution system perform the following core tasks: (1) build protein and nucleic acid models from sequence (2) energy minimization and structural optimization (3) analysis of energy distribution within a structure (4) molecular dynamics and stochastic dynamics (5) simulated annealing with a choice of cooling schedules (6) normal modes and vibrational frequencies (7) conformational search and global optimization (8) transition state location and conformational pathways (9) fitting of energy parameters to crystal data (10) Distances and geometries (11) molecular volumes and surface areas (12) free energy changes for structural mutations
22 BRIEF OVERVIEW OF AMBER SIMULATION PACKAGE The Amber simulation package (not to be confused with the Amber force field) consists of around 50 programs for applying molecular mechanics, normal mode analysis, molecular dynamics and free energy calculations to simulate the structural and energetic properties of organic and biological molecules such as DNA and protein, and analysis of the structures, non-covalent interactions, and dynamic trajectories. In order to speed up computing of large molecules, the Amber codes have been optimized for explicit solvent periodic calculations and parallel computing. Amber was originally developed by Peter Kollman at UCSF, and is now administered by David Case at Scripps. Web site:
23 CAPABILITIES of AMBER PACKAGE (1) Pre-defined libraries containing common bio-residues such as amino acids and nucleic acids (2) Molecular graphic interface to facilitate building and visualizing molecules (3) Energy minimization and structural optimization (4) Molecular dynamics with parallel computing ability (5) Particle-mesh Ewald (PME) procedure is used to handle long-range electrostatic interactions. (6) Rectangular and truncated octahedron periodic boundaries simulations as well as non-periodic simulations. (7) Explicit solvation and implicit solvation models (8) Simulated annealing with a choice of cooling schedules (9) A variety of constraints for NMR structure refinement calculations (10) Free energy calculations using thermodynamic integration (11) QM/MM calculations (12) Normal modes and vibrational frequencies (13) Tools for analyzing and processing trajectory or coordinate (14) Analysis of energy distribution within a structure
24 Application of Amber force field to the determination of the mechanical unfolding of BPTI As an application of the Amber force field (and the Amber program), we show how to simulate the mechanical unfolding of the BPTI protein. This mimics AFM experiments in which proteins are pulled apart by attaching them to AFM tips (see next slide). This also shows up in the real world, leading to the mechanical stability of abalone shells (see second slide), or the elasticity of muscle. BPTI is not related to any of these studies, but it is convenient for simulation. References to mechanical unfolding experiments and theory include: Stretching Single Protein Molecules: Titin is a Weird Spring, H. P. Erickson, Science, 276, 1090 (1997) Molecular mechanistic origin of the toughness of natural adhesives, fibers and composites, Smith, B. L.; Schaffer, T. E.; Viani, M.; Thompson, J. B.; Frederick, N. A.; Kindt, J.; Belcher, A.; Stucky, G. D.; Morse, D. E.; Hansma, P. K. Nature 1999, 399, 761. Ubiquitin-like Protein Domains Show High Resistance to Mechanical Unfolding Similar to That of the I27 Domain in Titin: Evidence from Simulations. Li, Pai- Chi; Makarov, Dmitrii E. Journal of Physical Chemistry B (2004), 108(2),

,")
25 Journal of Molecular Biology (2003), 333(5),

26 Smith, B. L.; Schaffer, T. E.; Viani, M.; Thompson, J. B.; Frederick, N. A.; Kindt, J.; Belcher, A.; Stucky, G. D.; Morse, D. E.; Hansma, P. K. Nature 1999, 399, 761.
27 Details of BPTI calculation First, we download the 6PTI.pdb file of bovine pancreatic trypsin inhibitor (BPTI) from Protein Data Bank at following link: The X-Ray structures usually have a few problems including missing or illdefined residue structures. In the present case we had to define we had to define the conformation of residues 39 and 50, changed the cysteine residue name CYS to CYX, and deleted the CONECT records. We also have to decide what to do about the solvent in which the structure is going to be immersed. Explicit solvent means that we surround the protein with lots of water molecules, while Implicit solvent means that we add additional terms to the protein force field that mimics the effect of interaction with the solvent. Here we chose to use the generalized Born implicit solvation model.

28 To study mechanical properties, it is necessary to exert force against portions of the molecule. Here we chose to pull on one end of the peptide chain while the other end is fixed. To do this we bonded two dummy atoms to the C- and N- terminal of the protein with a force constant of 100 kcal/mol Å 2 and a bond length of 1Å. All other force field parameters of the dummy atom such as angle, torsion, van der Waals and mass are all zero. (Note: Since we only did energy minimizations, a dummy atom with zero mass is acceptable. However, in a molecular dynamic simulation, the mass of dummy atoms must be some nonzero number, otherwise, simulation will crash.) The BPTI protein with the dummy atoms is shown to the right. Color map: red oxygen, white hydrogen, blue nitrogen, cyan carbon, yellow sulfur, green dummy atom
29 Next, we fixed the dummy atoms and performed a step energy minimization on this protein using steepest descent method and generalized Born solvation model. After the minimization, we manually modified the minimized structure to move the N- terminal dummy atom 0.5 Å in the direction from the nitrogen atom to the dummy atom. Then we did energy minimization again with both dummy atoms fixed. We repeated moving the N- terminal dummy atom 0.5 Å in the same direction, followed by minimization. This process was terminated when two residues of the N-terminal part of protein were pulled out.


30 Initial Final
31 The energy and force versus dummy atom displacement for this process are shown below. There are small peaks on the curves, which occur when hydrogen bonds are broken. For example, the peak at 8A is from breaking hydrogen bonds between the N-terminal arginine residue and nearby residues (shown in next page).

32 The peak at 8A is from breaking hydrogen bonds between the N-terminal arginine residue and nearby residues

33 Molecular Dynamics Studies of DNA Structures Hai Long and George C. Schatz Northwestern University

34 B-Type DNA Structure Right handed double helix DNA is highly charged 36º per base pair 10 residues per turn 3.38 Å between base pairs or 33.8 Å between one turn Radius ~10 Å Contains a major and a minor groove
35 DNA Hairpins Stilbenedicarboxamide Frederick D. Lewis, Xiaoyang Liu, Yansheng Wu, and Xiaobing Zuo, J. Am. Chem. Soc., 125 (42), , 2003.

36 Properties of the Hairpin DNAs Stepwise evolution of circular dichroism (CD) spectra CD Spectra depends on distance as well as the angle between stilbene chromophore 3 vectors:µ i,µ j, and R ij Can we calculate CD Spectra by MD simulations?
37 Experimental CD spectra of Sa(n)Sa , Wavelength, nm

38 Molecular Dynamic (MD) Simulation of DNA Force Field used in MD simulation CHARMM GROMOS AMBER
39 MD Simulation of Hairpin DNAs Partial charge of atoms in the Sa residue: Calculated by GAMESS at 6-31G* level and fit by RESP method Explicit water simulation After 1ns equilibrating run, begin sampling trajectories for another 3ns Family of the Hairpin DNAs: Sa1Sa, Sa2Sa, Sa3Sa, Sa4Sa, Sa4Sa, Sa5Sa, Sa6Sa, Sa8Sa, Sa11Sa

40 Snapshots of MD simulation of DNA Hairpins
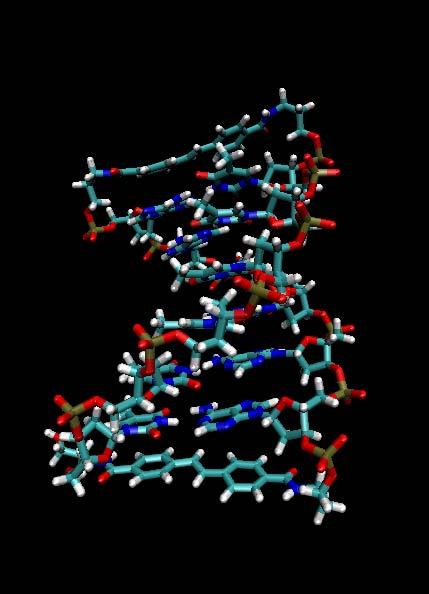
41 MD Simulation
42 Angle Correlation When X=1, Y=30 The average angle between one Sa and one DNA base pair is 15º The angles between the Sa residues as a function of number of DNA base pairs. The linear fitting result is with R= y = ( 4 ± 5) + (34.8 ± 0.8)x
43 Distance Correlation When X=1, Y=7.4A The average height between one Sa and one DNA base pair is 3.7A The distances between the Sa residues as a function of number of DNA base pairs. The linear fitting result is y = ( 4.0 ± 0.2) + (3.36 ± 0.03)x with R=0.9997
44 Calculation of CD Spectra from MD Simulation Results Calculated by Xiaobing Zuo Calculated CD Spectra using average angle and distance from simulations Experimental CD spectra of Sa(n)Sa. ε n=1 n=2 n=3 n=4 n=5 n=6 n=7 n=8 n= Wavelength (nm) ,4 8 Wavelength, nm
Sa.")
45 Calculation of CD Spectra from MD Simulation Trajectories CD spectra for conjugates 2 (a) and 5 (b) calculated from snapshots of MD simulated geometries Calculated by Xiaobing Zuo Experimental CD spectra of Sa(n)Sa λ, nm
Why Proteins Fold? (Parts of this presentation are based on work of Ashok Kolaskar) CS490B: Introduction to Bioinformatics Mar.
 CS490B: Introduction to Bioinformatics Mar.") Why Proteins Fold? (Parts of this presentation are based on work of Ashok Kolaskar) CS490B: Introduction to Bioinformatics Mar. 25, 2002 Molecular Dynamics: Introduction At physiological conditions, the
Why Proteins Fold? (Parts of this presentation are based on work of Ashok Kolaskar) CS490B: Introduction to Bioinformatics Mar. 25, 2002 Molecular Dynamics: Introduction At physiological conditions, the
Potential Energy (hyper)surface
surface") The Molecular Dynamics Method Thermal motion of a lipid bilayer Water permeation through channels Selective sugar transport Potential Energy (hyper)surface What is Force? Energy U(x) F = " d dx U(x) Conformation
The Molecular Dynamics Method Thermal motion of a lipid bilayer Water permeation through channels Selective sugar transport Potential Energy (hyper)surface What is Force? Energy U(x) F = " d dx U(x) Conformation
All-atom Molecular Mechanics. Trent E. Balius AMS 535 / CHE /27/2010
 All-atom Molecular Mechanics Trent E. Balius AMS 535 / CHE 535 09/27/2010 Outline Molecular models Molecular mechanics Force Fields Potential energy function functional form parameters and parameterization
All-atom Molecular Mechanics Trent E. Balius AMS 535 / CHE 535 09/27/2010 Outline Molecular models Molecular mechanics Force Fields Potential energy function functional form parameters and parameterization
Using Higher Calculus to Study Biologically Important Molecules Julie C. Mitchell
 Using Higher Calculus to Study Biologically Important Molecules Julie C. Mitchell Mathematics and Biochemistry University of Wisconsin - Madison 0 There Are Many Kinds Of Proteins The word protein comes
Using Higher Calculus to Study Biologically Important Molecules Julie C. Mitchell Mathematics and Biochemistry University of Wisconsin - Madison 0 There Are Many Kinds Of Proteins The word protein comes
Example questions for Molecular modelling (Level 4) Dr. Adrian Mulholland
 Dr. Adrian Mulholland") Example questions for Molecular modelling (Level 4) Dr. Adrian Mulholland 1) Question. Two methods which are widely used for the optimization of molecular geometies are the Steepest descents and Newton-Raphson
Example questions for Molecular modelling (Level 4) Dr. Adrian Mulholland 1) Question. Two methods which are widely used for the optimization of molecular geometies are the Steepest descents and Newton-Raphson
Figure 1. Molecules geometries of 5021 and Each neutral group in CHARMM topology was grouped in dash circle.
 Project I Chemistry 8021, Spring 2005/2/23 This document was turned in by a student as a homework paper. 1. Methods First, the cartesian coordinates of 5021 and 8021 molecules (Fig. 1) are generated, in
Project I Chemistry 8021, Spring 2005/2/23 This document was turned in by a student as a homework paper. 1. Methods First, the cartesian coordinates of 5021 and 8021 molecules (Fig. 1) are generated, in
Bioengineering 215. An Introduction to Molecular Dynamics for Biomolecules
 Bioengineering 215 An Introduction to Molecular Dynamics for Biomolecules David Parker May 18, 2007 ntroduction A principal tool to study biological molecules is molecular dynamics simulations (MD). MD
Bioengineering 215 An Introduction to Molecular Dynamics for Biomolecules David Parker May 18, 2007 ntroduction A principal tool to study biological molecules is molecular dynamics simulations (MD). MD
Dihedral Angles. Homayoun Valafar. Department of Computer Science and Engineering, USC 02/03/10 CSCE 769
 Dihedral Angles Homayoun Valafar Department of Computer Science and Engineering, USC The precise definition of a dihedral or torsion angle can be found in spatial geometry Angle between to planes Dihedral
Dihedral Angles Homayoun Valafar Department of Computer Science and Engineering, USC The precise definition of a dihedral or torsion angle can be found in spatial geometry Angle between to planes Dihedral
Lecture 11: Potential Energy Functions
 Lecture 11: Potential Energy Functions Dr. Ronald M. Levy ronlevy@temple.edu Originally contributed by Lauren Wickstrom (2011) Microscopic/Macroscopic Connection The connection between microscopic interactions
Lecture 11: Potential Energy Functions Dr. Ronald M. Levy ronlevy@temple.edu Originally contributed by Lauren Wickstrom (2011) Microscopic/Macroscopic Connection The connection between microscopic interactions
Molecular Mechanics, Dynamics & Docking
 Molecular Mechanics, Dynamics & Docking Lawrence Hunter, Ph.D. Director, Computational Bioscience Program University of Colorado School of Medicine Larry.Hunter@uchsc.edu http://compbio.uchsc.edu/hunter
Molecular Mechanics, Dynamics & Docking Lawrence Hunter, Ph.D. Director, Computational Bioscience Program University of Colorado School of Medicine Larry.Hunter@uchsc.edu http://compbio.uchsc.edu/hunter
Force Fields for Classical Molecular Dynamics simulations of Biomolecules. Emad Tajkhorshid
 Force Fields for Classical Molecular Dynamics simulations of Biomolecules Emad Tajkhorshid Theoretical and Computational Biophysics Group, Beckman Institute Departments of Biochemistry and Pharmacology,
Force Fields for Classical Molecular Dynamics simulations of Biomolecules Emad Tajkhorshid Theoretical and Computational Biophysics Group, Beckman Institute Departments of Biochemistry and Pharmacology,
The Molecular Dynamics Method
 The Molecular Dynamics Method Thermal motion of a lipid bilayer Water permeation through channels Selective sugar transport Potential Energy (hyper)surface What is Force? Energy U(x) F = d dx U(x) Conformation
The Molecular Dynamics Method Thermal motion of a lipid bilayer Water permeation through channels Selective sugar transport Potential Energy (hyper)surface What is Force? Energy U(x) F = d dx U(x) Conformation
Structural Bioinformatics (C3210) Molecular Mechanics
 Molecular Mechanics") Structural Bioinformatics (C3210) Molecular Mechanics How to Calculate Energies Calculation of molecular energies is of key importance in protein folding, molecular modelling etc. There are two main computational
Structural Bioinformatics (C3210) Molecular Mechanics How to Calculate Energies Calculation of molecular energies is of key importance in protein folding, molecular modelling etc. There are two main computational
Biomolecules are dynamic no single structure is a perfect model
 Molecular Dynamics Simulations of Biomolecules References: A. R. Leach Molecular Modeling Principles and Applications Prentice Hall, 2001. M. P. Allen and D. J. Tildesley "Computer Simulation of Liquids",
Molecular Dynamics Simulations of Biomolecules References: A. R. Leach Molecular Modeling Principles and Applications Prentice Hall, 2001. M. P. Allen and D. J. Tildesley "Computer Simulation of Liquids",
Oxygen Binding in Hemocyanin
 Supporting Information for Quantum Mechanics/Molecular Mechanics Study of Oxygen Binding in Hemocyanin Toru Saito and Walter Thiel* Max-Planck-Institut für Kohlenforschung, Kaiser-Wilhelm-Platz 1, D-45470
Supporting Information for Quantum Mechanics/Molecular Mechanics Study of Oxygen Binding in Hemocyanin Toru Saito and Walter Thiel* Max-Planck-Institut für Kohlenforschung, Kaiser-Wilhelm-Platz 1, D-45470
Viewing and Analyzing Proteins, Ligands and their Complexes 2
 2 Viewing and Analyzing Proteins, Ligands and their Complexes 2 Overview Viewing the accessible surface Analyzing the properties of proteins containing thousands of atoms is best accomplished by representing
2 Viewing and Analyzing Proteins, Ligands and their Complexes 2 Overview Viewing the accessible surface Analyzing the properties of proteins containing thousands of atoms is best accomplished by representing
Dominant Paths in Protein Folding
 Dominant Paths in Protein Folding Henri Orland SPhT, CEA-Saclay France work in collaboration with P. Faccioli, F. Pederiva, M. Sega University of Trento Henri Orland Annecy meeting 2006 Outline Basic notions
Dominant Paths in Protein Folding Henri Orland SPhT, CEA-Saclay France work in collaboration with P. Faccioli, F. Pederiva, M. Sega University of Trento Henri Orland Annecy meeting 2006 Outline Basic notions
Introduction to Comparative Protein Modeling. Chapter 4 Part I
 Introduction to Comparative Protein Modeling Chapter 4 Part I 1 Information on Proteins Each modeling study depends on the quality of the known experimental data. Basis of the model Search in the literature
Introduction to Comparative Protein Modeling Chapter 4 Part I 1 Information on Proteins Each modeling study depends on the quality of the known experimental data. Basis of the model Search in the literature
Packing of Secondary Structures
 7.88 Lecture Notes - 4 7.24/7.88J/5.48J The Protein Folding and Human Disease Professor Gossard Retrieving, Viewing Protein Structures from the Protein Data Base Helix helix packing Packing of Secondary
7.88 Lecture Notes - 4 7.24/7.88J/5.48J The Protein Folding and Human Disease Professor Gossard Retrieving, Viewing Protein Structures from the Protein Data Base Helix helix packing Packing of Secondary
Biochemistry,530:,, Introduc5on,to,Structural,Biology, Autumn,Quarter,2015,
 Biochemistry,530:,, Introduc5on,to,Structural,Biology, Autumn,Quarter,2015, Course,Informa5on, BIOC%530% GraduateAlevel,discussion,of,the,structure,,func5on,,and,chemistry,of,proteins,and, nucleic,acids,,control,of,enzyma5c,reac5ons.,please,see,the,course,syllabus,and,
Biochemistry,530:,, Introduc5on,to,Structural,Biology, Autumn,Quarter,2015, Course,Informa5on, BIOC%530% GraduateAlevel,discussion,of,the,structure,,func5on,,and,chemistry,of,proteins,and, nucleic,acids,,control,of,enzyma5c,reac5ons.,please,see,the,course,syllabus,and,
Other Methods for Generating Ions 1. MALDI matrix assisted laser desorption ionization MS 2. Spray ionization techniques 3. Fast atom bombardment 4.
 Other Methods for Generating Ions 1. MALDI matrix assisted laser desorption ionization MS 2. Spray ionization techniques 3. Fast atom bombardment 4. Field Desorption 5. MS MS techniques Matrix assisted
Other Methods for Generating Ions 1. MALDI matrix assisted laser desorption ionization MS 2. Spray ionization techniques 3. Fast atom bombardment 4. Field Desorption 5. MS MS techniques Matrix assisted
Ranjit P. Bahadur Assistant Professor Department of Biotechnology Indian Institute of Technology Kharagpur, India. 1 st November, 2013
 Hydration of protein-rna recognition sites Ranjit P. Bahadur Assistant Professor Department of Biotechnology Indian Institute of Technology Kharagpur, India 1 st November, 2013 Central Dogma of life DNA
Hydration of protein-rna recognition sites Ranjit P. Bahadur Assistant Professor Department of Biotechnology Indian Institute of Technology Kharagpur, India 1 st November, 2013 Central Dogma of life DNA
Lecture 2 and 3: Review of forces (ctd.) and elementary statistical mechanics. Contributions to protein stability
 and elementary statistical mechanics. Contributions to protein stability") Lecture 2 and 3: Review of forces (ctd.) and elementary statistical mechanics. Contributions to protein stability Part I. Review of forces Covalent bonds Non-covalent Interactions: Van der Waals Interactions
Lecture 2 and 3: Review of forces (ctd.) and elementary statistical mechanics. Contributions to protein stability Part I. Review of forces Covalent bonds Non-covalent Interactions: Van der Waals Interactions
Properties of amino acids in proteins
 Properties of amino acids in proteins one of the primary roles of DNA (but not the only one!) is to code for proteins A typical bacterium builds thousands types of proteins, all from ~20 amino acids repeated
Properties of amino acids in proteins one of the primary roles of DNA (but not the only one!) is to code for proteins A typical bacterium builds thousands types of proteins, all from ~20 amino acids repeated
Section Week 3. Junaid Malek, M.D.
 Section Week 3 Junaid Malek, M.D. Biological Polymers DA 4 monomers (building blocks), limited structure (double-helix) RA 4 monomers, greater flexibility, multiple structures Proteins 20 Amino Acids,
Section Week 3 Junaid Malek, M.D. Biological Polymers DA 4 monomers (building blocks), limited structure (double-helix) RA 4 monomers, greater flexibility, multiple structures Proteins 20 Amino Acids,
Proteins: Characteristics and Properties of Amino Acids
 SBI4U:Biochemistry Macromolecules Eachaminoacidhasatleastoneamineandoneacidfunctionalgroupasthe nameimplies.thedifferentpropertiesresultfromvariationsinthestructuresof differentrgroups.thergroupisoftenreferredtoastheaminoacidsidechain.
SBI4U:Biochemistry Macromolecules Eachaminoacidhasatleastoneamineandoneacidfunctionalgroupasthe nameimplies.thedifferentpropertiesresultfromvariationsinthestructuresof differentrgroups.thergroupisoftenreferredtoastheaminoacidsidechain.
Homology modeling. Dinesh Gupta ICGEB, New Delhi 1/27/2010 5:59 PM
 Homology modeling Dinesh Gupta ICGEB, New Delhi Protein structure prediction Methods: Homology (comparative) modelling Threading Ab-initio Protein Homology modeling Homology modeling is an extrapolation
Homology modeling Dinesh Gupta ICGEB, New Delhi Protein structure prediction Methods: Homology (comparative) modelling Threading Ab-initio Protein Homology modeling Homology modeling is an extrapolation
Protein Structure Bioinformatics Introduction
 1 Swiss Institute of Bioinformatics Protein Structure Bioinformatics Introduction Basel, 27. September 2004 Torsten Schwede Biozentrum - Universität Basel Swiss Institute of Bioinformatics Klingelbergstr
1 Swiss Institute of Bioinformatics Protein Structure Bioinformatics Introduction Basel, 27. September 2004 Torsten Schwede Biozentrum - Universität Basel Swiss Institute of Bioinformatics Klingelbergstr
Molecular Mechanics. I. Quantum mechanical treatment of molecular systems
 Molecular Mechanics I. Quantum mechanical treatment of molecular systems The first principle approach for describing the properties of molecules, including proteins, involves quantum mechanics. For example,
Molecular Mechanics I. Quantum mechanical treatment of molecular systems The first principle approach for describing the properties of molecules, including proteins, involves quantum mechanics. For example,
Advanced Molecular Dynamics
 Advanced Molecular Dynamics Introduction May 2, 2017 Who am I? I am an associate professor at Theoretical Physics Topics I work on: Algorithms for (parallel) molecular simulations including GPU acceleration
Advanced Molecular Dynamics Introduction May 2, 2017 Who am I? I am an associate professor at Theoretical Physics Topics I work on: Algorithms for (parallel) molecular simulations including GPU acceleration
Physiochemical Properties of Residues
 Physiochemical Properties of Residues Various Sources C N Cα R Slide 1 Conformational Propensities Conformational Propensity is the frequency in which a residue adopts a given conformation (in a polypeptide)
Physiochemical Properties of Residues Various Sources C N Cα R Slide 1 Conformational Propensities Conformational Propensity is the frequency in which a residue adopts a given conformation (in a polypeptide)
Semi Empirical Force Fields and Their Limitations. Potential Energy Surface (PES)
") Semi Empirical Force Fields and Their Limitations Ioan Kosztin Beckman Institute University of Illinois at Urbana-Champaign Potential Energy Surface (PES) Schrödinger equation: H T Ψ( r, = E Ψ( r, H =
Semi Empirical Force Fields and Their Limitations Ioan Kosztin Beckman Institute University of Illinois at Urbana-Champaign Potential Energy Surface (PES) Schrödinger equation: H T Ψ( r, = E Ψ( r, H =
The Molecular Dynamics Method
 H-bond energy (kcal/mol) - 4.0 The Molecular Dynamics Method Fibronectin III_1, a mechanical protein that glues cells together in wound healing and in preventing tumor metastasis 0 ATPase, a molecular
H-bond energy (kcal/mol) - 4.0 The Molecular Dynamics Method Fibronectin III_1, a mechanical protein that glues cells together in wound healing and in preventing tumor metastasis 0 ATPase, a molecular
Why study protein dynamics?
 Why study protein dynamics? Protein flexibility is crucial for function. One average structure is not enough. Proteins constantly sample configurational space. Transport - binding and moving molecules
Why study protein dynamics? Protein flexibility is crucial for function. One average structure is not enough. Proteins constantly sample configurational space. Transport - binding and moving molecules
Sequential resonance assignments in (small) proteins: homonuclear method 2º structure determination
 proteins: homonuclear method 2º structure determination") Lecture 9 M230 Feigon Sequential resonance assignments in (small) proteins: homonuclear method 2º structure determination Reading resources v Roberts NMR of Macromolecules, Chap 4 by Christina Redfield
Lecture 9 M230 Feigon Sequential resonance assignments in (small) proteins: homonuclear method 2º structure determination Reading resources v Roberts NMR of Macromolecules, Chap 4 by Christina Redfield
Gromacs Workshop Spring CSC
 Gromacs Workshop Spring 2007 @ CSC Erik Lindahl Center for Biomembrane Research Stockholm University, Sweden David van der Spoel Dept. Cell & Molecular Biology Uppsala University, Sweden Berk Hess Max-Planck-Institut
Gromacs Workshop Spring 2007 @ CSC Erik Lindahl Center for Biomembrane Research Stockholm University, Sweden David van der Spoel Dept. Cell & Molecular Biology Uppsala University, Sweden Berk Hess Max-Planck-Institut
Molecular Mechanics. Yohann Moreau. November 26, 2015
 Molecular Mechanics Yohann Moreau yohann.moreau@ujf-grenoble.fr November 26, 2015 Yohann Moreau (UJF) Molecular Mechanics, Label RFCT 2015 November 26, 2015 1 / 29 Introduction A so-called Force-Field
Molecular Mechanics Yohann Moreau yohann.moreau@ujf-grenoble.fr November 26, 2015 Yohann Moreau (UJF) Molecular Mechanics, Label RFCT 2015 November 26, 2015 1 / 29 Introduction A so-called Force-Field
CE 530 Molecular Simulation
 1 CE 530 Molecular Simulation Lecture 14 Molecular Models David A. Kofke Department of Chemical Engineering SUNY Buffalo kofke@eng.buffalo.edu 2 Review Monte Carlo ensemble averaging, no dynamics easy
1 CE 530 Molecular Simulation Lecture 14 Molecular Models David A. Kofke Department of Chemical Engineering SUNY Buffalo kofke@eng.buffalo.edu 2 Review Monte Carlo ensemble averaging, no dynamics easy
Introduction to molecular dynamics
 1 Introduction to molecular dynamics Yves Lansac Université François Rabelais, Tours, France Visiting MSE, GIST for the summer Molecular Simulation 2 Molecular simulation is a computational experiment.
1 Introduction to molecular dynamics Yves Lansac Université François Rabelais, Tours, France Visiting MSE, GIST for the summer Molecular Simulation 2 Molecular simulation is a computational experiment.
Molecular Simulation II. Classical Mechanical Treatment
 Molecular Simulation II Quantum Chemistry Classical Mechanics E = Ψ H Ψ ΨΨ U = E bond +E angle +E torsion +E non-bond Jeffry D. Madura Department of Chemistry & Biochemistry Center for Computational Sciences
Molecular Simulation II Quantum Chemistry Classical Mechanics E = Ψ H Ψ ΨΨ U = E bond +E angle +E torsion +E non-bond Jeffry D. Madura Department of Chemistry & Biochemistry Center for Computational Sciences
Computational Biology & Computational Medicine
 Computational Biology & Computational Medicine Homayoun Valafar Outline Why proteins? What are proteins? How do we compute them? How do we use computational approaches? Why Proteins? Molecular basis of
Computational Biology & Computational Medicine Homayoun Valafar Outline Why proteins? What are proteins? How do we compute them? How do we use computational approaches? Why Proteins? Molecular basis of
Applications of Molecular Dynamics
 June 4, 0 Molecular Modeling and Simulation Applications of Molecular Dynamics Agricultural Bioinformatics Research Unit, Graduate School of Agricultural and Life Sciences, The University of Tokyo Tohru
June 4, 0 Molecular Modeling and Simulation Applications of Molecular Dynamics Agricultural Bioinformatics Research Unit, Graduate School of Agricultural and Life Sciences, The University of Tokyo Tohru
UNIT TWELVE. a, I _,o "' I I I. I I.P. l'o. H-c-c. I ~o I ~ I / H HI oh H...- I II I II 'oh. HO\HO~ I "-oh
 UNT TWELVE PROTENS : PEPTDE BONDNG AND POLYPEPTDES 12 CONCEPTS Many proteins are important in biological structure-for example, the keratin of hair, collagen of skin and leather, and fibroin of silk. Other
UNT TWELVE PROTENS : PEPTDE BONDNG AND POLYPEPTDES 12 CONCEPTS Many proteins are important in biological structure-for example, the keratin of hair, collagen of skin and leather, and fibroin of silk. Other
Molecular Dynamics Simulations. Dr. Noelia Faginas Lago Dipartimento di Chimica,Biologia e Biotecnologie Università di Perugia
 Molecular Dynamics Simulations Dr. Noelia Faginas Lago Dipartimento di Chimica,Biologia e Biotecnologie Università di Perugia 1 An Introduction to Molecular Dynamics Simulations Macroscopic properties
Molecular Dynamics Simulations Dr. Noelia Faginas Lago Dipartimento di Chimica,Biologia e Biotecnologie Università di Perugia 1 An Introduction to Molecular Dynamics Simulations Macroscopic properties
Computational Structural Biology and Molecular Simulation. Introduction to VMD Molecular Visualization and Analysis
 Computational Structural Biology and Molecular Simulation Introduction to VMD Molecular Visualization and Analysis Emad Tajkhorshid Department of Biochemistry, Beckman Institute, Center for Computational
Computational Structural Biology and Molecular Simulation Introduction to VMD Molecular Visualization and Analysis Emad Tajkhorshid Department of Biochemistry, Beckman Institute, Center for Computational
An introduction to Molecular Dynamics. EMBO, June 2016
 An introduction to Molecular Dynamics EMBO, June 2016 What is MD? everything that living things do can be understood in terms of the jiggling and wiggling of atoms. The Feynman Lectures in Physics vol.
An introduction to Molecular Dynamics EMBO, June 2016 What is MD? everything that living things do can be understood in terms of the jiggling and wiggling of atoms. The Feynman Lectures in Physics vol.
Molecular Modelling. part of Bioinformatik von RNA- und Proteinstrukturen. Sonja Prohaska. Leipzig, SS Computational EvoDevo University Leipzig
 part of Bioinformatik von RNA- und Proteinstrukturen Computational EvoDevo University Leipzig Leipzig, SS 2011 Protein Structure levels or organization Primary structure: sequence of amino acids (from
part of Bioinformatik von RNA- und Proteinstrukturen Computational EvoDevo University Leipzig Leipzig, SS 2011 Protein Structure levels or organization Primary structure: sequence of amino acids (from
Biochemistry Quiz Review 1I. 1. Of the 20 standard amino acids, only is not optically active. The reason is that its side chain.
 Biochemistry Quiz Review 1I A general note: Short answer questions are just that, short. Writing a paragraph filled with every term you can remember from class won t improve your answer just answer clearly,
Biochemistry Quiz Review 1I A general note: Short answer questions are just that, short. Writing a paragraph filled with every term you can remember from class won t improve your answer just answer clearly,
Peptide folding in non-aqueous environments investigated with molecular dynamics simulations Soto Becerra, Patricia
 University of Groningen Peptide folding in non-aqueous environments investigated with molecular dynamics simulations Soto Becerra, Patricia IMPORTANT NOTE: You are advised to consult the publisher's version
University of Groningen Peptide folding in non-aqueous environments investigated with molecular dynamics simulations Soto Becerra, Patricia IMPORTANT NOTE: You are advised to consult the publisher's version
Secondary Structure. Bioch/BIMS 503 Lecture 2. Structure and Function of Proteins. Further Reading. Φ, Ψ angles alone determine protein structure
 Bioch/BIMS 503 Lecture 2 Structure and Function of Proteins August 28, 2008 Robert Nakamoto rkn3c@virginia.edu 2-0279 Secondary Structure Φ Ψ angles determine protein structure Φ Ψ angles are restricted
Bioch/BIMS 503 Lecture 2 Structure and Function of Proteins August 28, 2008 Robert Nakamoto rkn3c@virginia.edu 2-0279 Secondary Structure Φ Ψ angles determine protein structure Φ Ψ angles are restricted
Subject of the Lecture:
 Subject of the Lecture: Conceptual basis for the development of force fields. Implementation/validation Water - a worked example Extensions - combining molecular mechanics and quantum mechanics (QM/MM)
Subject of the Lecture: Conceptual basis for the development of force fields. Implementation/validation Water - a worked example Extensions - combining molecular mechanics and quantum mechanics (QM/MM)
Scuola di Chimica Computazionale
 Societa Chimica Italiana Gruppo Interdivisionale di Chimica Computazionale Scuola di Chimica Computazionale Introduzione, per Esercizi, all Uso del Calcolatore in Chimica Organica e Biologica Modellistica
Societa Chimica Italiana Gruppo Interdivisionale di Chimica Computazionale Scuola di Chimica Computazionale Introduzione, per Esercizi, all Uso del Calcolatore in Chimica Organica e Biologica Modellistica
Lecture 15: Realities of Genome Assembly Protein Sequencing
 Lecture 15: Realities of Genome Assembly Protein Sequencing Study Chapter 8.10-8.15 1 Euler s Theorems A graph is balanced if for every vertex the number of incoming edges equals to the number of outgoing
Lecture 15: Realities of Genome Assembly Protein Sequencing Study Chapter 8.10-8.15 1 Euler s Theorems A graph is balanced if for every vertex the number of incoming edges equals to the number of outgoing
Model Mélange. Physical Models of Peptides and Proteins
 Model Mélange Physical Models of Peptides and Proteins In the Model Mélange activity, you will visit four different stations each featuring a variety of different physical models of peptides or proteins.
Model Mélange Physical Models of Peptides and Proteins In the Model Mélange activity, you will visit four different stations each featuring a variety of different physical models of peptides or proteins.
NMR parameters intensity chemical shift coupling constants 1D 1 H spectra of nucleic acids and proteins
 Lecture #2 M230 NMR parameters intensity chemical shift coupling constants Juli Feigon 1D 1 H spectra of nucleic acids and proteins NMR Parameters A. Intensity (area) 1D NMR spectrum: integrated intensity
Lecture #2 M230 NMR parameters intensity chemical shift coupling constants Juli Feigon 1D 1 H spectra of nucleic acids and proteins NMR Parameters A. Intensity (area) 1D NMR spectrum: integrated intensity
LS1a Fall 2014 Problem Set #2 Due Monday 10/6 at 6 pm in the drop boxes on the Science Center 2 nd Floor
 LS1a Fall 2014 Problem Set #2 Due Monday 10/6 at 6 pm in the drop boxes on the Science Center 2 nd Floor Note: Adequate space is given for each answer. Questions that require a brief explanation should
LS1a Fall 2014 Problem Set #2 Due Monday 10/6 at 6 pm in the drop boxes on the Science Center 2 nd Floor Note: Adequate space is given for each answer. Questions that require a brief explanation should
Computer simulations of protein folding with a small number of distance restraints
 Vol. 49 No. 3/2002 683 692 QUARTERLY Computer simulations of protein folding with a small number of distance restraints Andrzej Sikorski 1, Andrzej Kolinski 1,2 and Jeffrey Skolnick 2 1 Department of Chemistry,
Vol. 49 No. 3/2002 683 692 QUARTERLY Computer simulations of protein folding with a small number of distance restraints Andrzej Sikorski 1, Andrzej Kolinski 1,2 and Jeffrey Skolnick 2 1 Department of Chemistry,
Hands-on Course in Computational Structural Biology and Molecular Simulation BIOP590C/MCB590C. Course Details
 Hands-on Course in Computational Structural Biology and Molecular Simulation BIOP590C/MCB590C Emad Tajkhorshid Center for Computational Biology and Biophysics Email: emad@life.uiuc.edu or tajkhors@uiuc.edu
Hands-on Course in Computational Structural Biology and Molecular Simulation BIOP590C/MCB590C Emad Tajkhorshid Center for Computational Biology and Biophysics Email: emad@life.uiuc.edu or tajkhors@uiuc.edu
Supplementary Information
 Supplementary Information Resveratrol Serves as a Protein-Substrate Interaction Stabilizer in Human SIRT1 Activation Xuben Hou,, David Rooklin, Hao Fang *,,, Yingkai Zhang Department of Medicinal Chemistry
Supplementary Information Resveratrol Serves as a Protein-Substrate Interaction Stabilizer in Human SIRT1 Activation Xuben Hou,, David Rooklin, Hao Fang *,,, Yingkai Zhang Department of Medicinal Chemistry
Molecular Dynamics, Monte Carlo and Docking. Lecture 21. Introduction to Bioinformatics MNW2
 Molecular Dynamics, Monte Carlo and Docking Lecture 21 Introduction to Bioinformatics MNW2 If you throw up a stone, it is Physics. If you throw up a stone, it is Physics. If it lands on your head, it is
Molecular Dynamics, Monte Carlo and Docking Lecture 21 Introduction to Bioinformatics MNW2 If you throw up a stone, it is Physics. If you throw up a stone, it is Physics. If it lands on your head, it is
Molecular dynamics simulation. CS/CME/BioE/Biophys/BMI 279 Oct. 5 and 10, 2017 Ron Dror
 Molecular dynamics simulation CS/CME/BioE/Biophys/BMI 279 Oct. 5 and 10, 2017 Ron Dror 1 Outline Molecular dynamics (MD): The basic idea Equations of motion Key properties of MD simulations Sample applications
Molecular dynamics simulation CS/CME/BioE/Biophys/BMI 279 Oct. 5 and 10, 2017 Ron Dror 1 Outline Molecular dynamics (MD): The basic idea Equations of motion Key properties of MD simulations Sample applications
Structure Investigation of Fam20C, a Golgi Casein Kinase
 Structure Investigation of Fam20C, a Golgi Casein Kinase Sharon Grubner National Taiwan University, Dr. Jung-Hsin Lin University of California San Diego, Dr. Rommie Amaro Abstract This research project
Structure Investigation of Fam20C, a Golgi Casein Kinase Sharon Grubner National Taiwan University, Dr. Jung-Hsin Lin University of California San Diego, Dr. Rommie Amaro Abstract This research project
Exam I Answer Key: Summer 2006, Semester C
 1. Which of the following tripeptides would migrate most rapidly towards the negative electrode if electrophoresis is carried out at ph 3.0? a. gly-gly-gly b. glu-glu-asp c. lys-glu-lys d. val-asn-lys
1. Which of the following tripeptides would migrate most rapidly towards the negative electrode if electrophoresis is carried out at ph 3.0? a. gly-gly-gly b. glu-glu-asp c. lys-glu-lys d. val-asn-lys
PROTEIN STRUCTURE AMINO ACIDS H R. Zwitterion (dipolar ion) CO 2 H. PEPTIDES Formal reactions showing formation of peptide bond by dehydration:
 CO 2 H. PEPTIDES Formal reactions showing formation of peptide bond by dehydration:") PTEI STUTUE ydrolysis of proteins with aqueous acid or base yields a mixture of free amino acids. Each type of protein yields a characteristic mixture of the ~ 20 amino acids. AMI AIDS Zwitterion (dipolar
PTEI STUTUE ydrolysis of proteins with aqueous acid or base yields a mixture of free amino acids. Each type of protein yields a characteristic mixture of the ~ 20 amino acids. AMI AIDS Zwitterion (dipolar
CHMI 2227 EL. Biochemistry I. Test January Prof : Eric R. Gauthier, Ph.D.
 CHMI 2227 EL Biochemistry I Test 1 26 January 2007 Prof : Eric R. Gauthier, Ph.D. Guidelines: 1) Duration: 55 min 2) 14 questions, on 7 pages. For 70 marks (5 marks per question). Worth 15 % of the final
CHMI 2227 EL Biochemistry I Test 1 26 January 2007 Prof : Eric R. Gauthier, Ph.D. Guidelines: 1) Duration: 55 min 2) 14 questions, on 7 pages. For 70 marks (5 marks per question). Worth 15 % of the final
A. Two of the common amino acids are analyzed. Amino acid X and amino acid Y both have an isoionic point in the range of
 Questions with Answers- Amino Acids & Peptides A. Two of the common amino acids are analyzed. Amino acid X and amino acid Y both have an isoionic point in the range of 5.0-6.5 (Questions 1-4) 1. Which
Questions with Answers- Amino Acids & Peptides A. Two of the common amino acids are analyzed. Amino acid X and amino acid Y both have an isoionic point in the range of 5.0-6.5 (Questions 1-4) 1. Which
Force Fields for MD simulations
 Force Fields for MD simulations Topology/parameter files Where do the numbers an MD code uses come from? ow to make topology files for ligands, cofactors, special amino acids, ow to obtain/develop missing
Force Fields for MD simulations Topology/parameter files Where do the numbers an MD code uses come from? ow to make topology files for ligands, cofactors, special amino acids, ow to obtain/develop missing
Solutions to Assignment #4 Getting Started with HyperChem
 Solutions to Assignment #4 Getting Started with HyperChem 1. This first exercise is meant to familiarize you with the different methods for visualizing molecules available in HyperChem. (a) Create a molecule
Solutions to Assignment #4 Getting Started with HyperChem 1. This first exercise is meant to familiarize you with the different methods for visualizing molecules available in HyperChem. (a) Create a molecule
Molecular Dynamics. A very brief introduction
 Molecular Dynamics A very brief introduction Sander Pronk Dept. of Theoretical Physics KTH Royal Institute of Technology & Science For Life Laboratory Stockholm, Sweden Why computer simulations? Two primary
Molecular Dynamics A very brief introduction Sander Pronk Dept. of Theoretical Physics KTH Royal Institute of Technology & Science For Life Laboratory Stockholm, Sweden Why computer simulations? Two primary
Molecular Dynamics, Monte Carlo and Docking. Lecture 21. Introduction to Bioinformatics MNW2
 Molecular Dynamics, Monte Carlo and Docking Lecture 21 Introduction to Bioinformatics MNW2 Allowed phi-psi angles Red areas are preferred, yellow areas are allowed, and white is avoided 2.3a Hamiltonian
Molecular Dynamics, Monte Carlo and Docking Lecture 21 Introduction to Bioinformatics MNW2 Allowed phi-psi angles Red areas are preferred, yellow areas are allowed, and white is avoided 2.3a Hamiltonian
NH 2. Biochemistry I, Fall Term Sept 9, Lecture 5: Amino Acids & Peptides Assigned reading in Campbell: Chapter
 Biochemistry I, Fall Term Sept 9, 2005 Lecture 5: Amino Acids & Peptides Assigned reading in Campbell: Chapter 3.1-3.4. Key Terms: ptical Activity, Chirality Peptide bond Condensation reaction ydrolysis
Biochemistry I, Fall Term Sept 9, 2005 Lecture 5: Amino Acids & Peptides Assigned reading in Campbell: Chapter 3.1-3.4. Key Terms: ptical Activity, Chirality Peptide bond Condensation reaction ydrolysis
THE UNIVERSITY OF MANITOBA. PAPER NO: _1_ LOCATION: 173 Robert Schultz Theatre PAGE NO: 1 of 5 DEPARTMENT & COURSE NO: CHEM / MBIO 2770 TIME: 1 HOUR
 THE UNIVERSITY OF MANITOBA 1 November 1, 2016 Mid-Term EXAMINATION PAPER NO: _1_ LOCATION: 173 Robert Schultz Theatre PAGE NO: 1 of 5 DEPARTMENT & COURSE NO: CHEM / MBIO 2770 TIME: 1 HOUR EXAMINATION:
THE UNIVERSITY OF MANITOBA 1 November 1, 2016 Mid-Term EXAMINATION PAPER NO: _1_ LOCATION: 173 Robert Schultz Theatre PAGE NO: 1 of 5 DEPARTMENT & COURSE NO: CHEM / MBIO 2770 TIME: 1 HOUR EXAMINATION:
Molecular dynamics simulation of Aquaporin-1. 4 nm
 Molecular dynamics simulation of Aquaporin-1 4 nm Molecular Dynamics Simulations Schrödinger equation i~@ t (r, R) =H (r, R) Born-Oppenheimer approximation H e e(r; R) =E e (R) e(r; R) Nucleic motion described
Molecular dynamics simulation of Aquaporin-1 4 nm Molecular Dynamics Simulations Schrödinger equation i~@ t (r, R) =H (r, R) Born-Oppenheimer approximation H e e(r; R) =E e (R) e(r; R) Nucleic motion described
Exam III. Please read through each question carefully, and make sure you provide all of the requested information.
 09-107 onors Chemistry ame Exam III Please read through each question carefully, and make sure you provide all of the requested information. 1. A series of octahedral metal compounds are made from 1 mol
09-107 onors Chemistry ame Exam III Please read through each question carefully, and make sure you provide all of the requested information. 1. A series of octahedral metal compounds are made from 1 mol
Principles of Physical Biochemistry
 Principles of Physical Biochemistry Kensal E. van Hold e W. Curtis Johnso n P. Shing Ho Preface x i PART 1 MACROMOLECULAR STRUCTURE AND DYNAMICS 1 1 Biological Macromolecules 2 1.1 General Principles
Principles of Physical Biochemistry Kensal E. van Hold e W. Curtis Johnso n P. Shing Ho Preface x i PART 1 MACROMOLECULAR STRUCTURE AND DYNAMICS 1 1 Biological Macromolecules 2 1.1 General Principles
Energy Minimization of Protein Tertiary Structure by Parallel Simulated Annealing using Genetic Crossover
 Minimization of Protein Tertiary Structure by Parallel Simulated Annealing using Genetic Crossover Tomoyuki Hiroyasu, Mitsunori Miki, Shinya Ogura, Keiko Aoi, Takeshi Yoshida, Yuko Okamoto Jack Dongarra
Minimization of Protein Tertiary Structure by Parallel Simulated Annealing using Genetic Crossover Tomoyuki Hiroyasu, Mitsunori Miki, Shinya Ogura, Keiko Aoi, Takeshi Yoshida, Yuko Okamoto Jack Dongarra
Close agreement between the orientation dependence of hydrogen bonds observed in protein structures and quantum mechanical calculations
 Close agreement between the orientation dependence of hydrogen bonds observed in protein structures and quantum mechanical calculations Alexandre V. Morozov, Tanja Kortemme, Kiril Tsemekhman, David Baker
Close agreement between the orientation dependence of hydrogen bonds observed in protein structures and quantum mechanical calculations Alexandre V. Morozov, Tanja Kortemme, Kiril Tsemekhman, David Baker
Journal of Pharmacology and Experimental Therapy-JPET#172536
 A NEW NON-PEPTIDIC INHIBITOR OF THE 14-3-3 DOCKING SITE INDUCES APOPTOTIC CELL DEATH IN CHRONIC MYELOID LEUKEMIA SENSITIVE OR RESISTANT TO IMATINIB Manuela Mancini, Valentina Corradi, Sara Petta, Enza
A NEW NON-PEPTIDIC INHIBITOR OF THE 14-3-3 DOCKING SITE INDUCES APOPTOTIC CELL DEATH IN CHRONIC MYELOID LEUKEMIA SENSITIVE OR RESISTANT TO IMATINIB Manuela Mancini, Valentina Corradi, Sara Petta, Enza
This semester. Books
 Models mostly proteins from detailed to more abstract models Some simulation methods This semester Books None necessary for my group and Prof Rarey Molecular Modelling: Principles and Applications Leach,
Models mostly proteins from detailed to more abstract models Some simulation methods This semester Books None necessary for my group and Prof Rarey Molecular Modelling: Principles and Applications Leach,
Biological Macromolecules
 Introduction for Chem 493 Chemistry of Biological Macromolecules Dr. L. Luyt January 2008 Dr. L. Luyt Chem 493-2008 1 Biological macromolecules are the molecules of life allow for organization serve a
Introduction for Chem 493 Chemistry of Biological Macromolecules Dr. L. Luyt January 2008 Dr. L. Luyt Chem 493-2008 1 Biological macromolecules are the molecules of life allow for organization serve a
Protein Fragment Search Program ver Overview: Contents:
 Protein Fragment Search Program ver 1.1.1 Developed by: BioPhysics Laboratory, Faculty of Life and Environmental Science, Shimane University 1060 Nishikawatsu-cho, Matsue-shi, Shimane, 690-8504, Japan
Protein Fragment Search Program ver 1.1.1 Developed by: BioPhysics Laboratory, Faculty of Life and Environmental Science, Shimane University 1060 Nishikawatsu-cho, Matsue-shi, Shimane, 690-8504, Japan
7.012 Problem Set 1. i) What are two main differences between prokaryotic cells and eukaryotic cells?
 What are two main differences between prokaryotic cells and eukaryotic cells?") ame 7.01 Problem Set 1 Section Question 1 a) What are the four major types of biological molecules discussed in lecture? Give one important function of each type of biological molecule in the cell? b)
ame 7.01 Problem Set 1 Section Question 1 a) What are the four major types of biological molecules discussed in lecture? Give one important function of each type of biological molecule in the cell? b)
3rd Advanced in silico Drug Design KFC/ADD Molecular mechanics intro Karel Berka, Ph.D. Martin Lepšík, Ph.D. Pavel Polishchuk, Ph.D.
 3rd Advanced in silico Drug Design KFC/ADD Molecular mechanics intro Karel Berka, Ph.D. Martin Lepšík, Ph.D. Pavel Polishchuk, Ph.D. Thierry Langer, Ph.D. Jana Vrbková, Ph.D. UP Olomouc, 23.1.-26.1. 2018
3rd Advanced in silico Drug Design KFC/ADD Molecular mechanics intro Karel Berka, Ph.D. Martin Lepšík, Ph.D. Pavel Polishchuk, Ph.D. Thierry Langer, Ph.D. Jana Vrbková, Ph.D. UP Olomouc, 23.1.-26.1. 2018
Rotamers in the CHARMM19 Force Field
 Appendix A Rotamers in the CHARMM19 Force Field The people may be made to follow a path of action, but they may not be made to understand it. Confucius (551 BC - 479 BC) ( ) V r 1 (j),r 2 (j),r 3 (j),...,r
Appendix A Rotamers in the CHARMM19 Force Field The people may be made to follow a path of action, but they may not be made to understand it. Confucius (551 BC - 479 BC) ( ) V r 1 (j),r 2 (j),r 3 (j),...,r
What makes a good graphene-binding peptide? Adsorption of amino acids and peptides at aqueous graphene interfaces: Electronic Supplementary
 Electronic Supplementary Material (ESI) for Journal of Materials Chemistry B. This journal is The Royal Society of Chemistry 21 What makes a good graphene-binding peptide? Adsorption of amino acids and
Electronic Supplementary Material (ESI) for Journal of Materials Chemistry B. This journal is The Royal Society of Chemistry 21 What makes a good graphene-binding peptide? Adsorption of amino acids and
BCH 4053 Exam I Review Spring 2017
 BCH 4053 SI - Spring 2017 Reed BCH 4053 Exam I Review Spring 2017 Chapter 1 1. Calculate G for the reaction A + A P + Q. Assume the following equilibrium concentrations: [A] = 20mM, [Q] = [P] = 40fM. Assume
BCH 4053 SI - Spring 2017 Reed BCH 4053 Exam I Review Spring 2017 Chapter 1 1. Calculate G for the reaction A + A P + Q. Assume the following equilibrium concentrations: [A] = 20mM, [Q] = [P] = 40fM. Assume
Chapter 4: Amino Acids
 Chapter 4: Amino Acids All peptides and polypeptides are polymers of alpha-amino acids. lipid polysaccharide enzyme 1940s 1980s. Lipids membrane 1960s. Polysaccharide Are energy metabolites and many of
Chapter 4: Amino Acids All peptides and polypeptides are polymers of alpha-amino acids. lipid polysaccharide enzyme 1940s 1980s. Lipids membrane 1960s. Polysaccharide Are energy metabolites and many of
Analysis of the simulation
 Analysis of the simulation Marcus Elstner and Tomáš Kubař January 7, 2014 Thermodynamic properties time averages of thermodynamic quantites correspond to ensemble averages (ergodic theorem) some quantities
Analysis of the simulation Marcus Elstner and Tomáš Kubař January 7, 2014 Thermodynamic properties time averages of thermodynamic quantites correspond to ensemble averages (ergodic theorem) some quantities
k θ (θ θ 0 ) 2 angles r i j r i j
 2 angles r i j r i j") 1 Force fields 1.1 Introduction The term force field is slightly misleading, since it refers to the parameters of the potential used to calculate the forces (via gradient) in molecular dynamics simulations.
1 Force fields 1.1 Introduction The term force field is slightly misleading, since it refers to the parameters of the potential used to calculate the forces (via gradient) in molecular dynamics simulations.
Force Fields for Classical Molecular Dynamics simulations of Biomolecules. Emad Tajkhorshid
 Force Fields for Classical Molecular Dynamics simulations of Biomolecules Emad Tajkhorshid Beckman Institute Departments of Biochemistry Center for Biophysics and Computational Biology University of Illinois
Force Fields for Classical Molecular Dynamics simulations of Biomolecules Emad Tajkhorshid Beckman Institute Departments of Biochemistry Center for Biophysics and Computational Biology University of Illinois
Protein structure. Protein structure. Amino acid residue. Cell communication channel. Bioinformatics Methods
 Cell communication channel Bioinformatics Methods Iosif Vaisman Email: ivaisman@gmu.edu SEQUENCE STRUCTURE DNA Sequence Protein Sequence Protein Structure Protein structure ATGAAATTTGGAAACTTCCTTCTCACTTATCAGCCACCT...
Cell communication channel Bioinformatics Methods Iosif Vaisman Email: ivaisman@gmu.edu SEQUENCE STRUCTURE DNA Sequence Protein Sequence Protein Structure Protein structure ATGAAATTTGGAAACTTCCTTCTCACTTATCAGCCACCT...
Multiscale Materials Modeling
 Multiscale Materials Modeling Lecture 09 Quantum Mechanics/Molecular Mechanics (QM/MM) Techniques Fundamentals of Sustainable Technology These notes created by David Keffer, University of Tennessee, Knoxville,
Multiscale Materials Modeling Lecture 09 Quantum Mechanics/Molecular Mechanics (QM/MM) Techniques Fundamentals of Sustainable Technology These notes created by David Keffer, University of Tennessee, Knoxville,
Contents. xiii. Preface v
 Contents Preface Chapter 1 Biological Macromolecules 1.1 General PrincipIes 1.1.1 Macrornolecules 1.2 1.1.2 Configuration and Conformation Molecular lnteractions in Macromolecular Structures 1.2.1 Weak
Contents Preface Chapter 1 Biological Macromolecules 1.1 General PrincipIes 1.1.1 Macrornolecules 1.2 1.1.2 Configuration and Conformation Molecular lnteractions in Macromolecular Structures 1.2.1 Weak
Problem Set 1
 2006 7.012 Problem Set 1 Due before 5 PM on FRIDAY, September 15, 2006. Turn answers in to the box outside of 68-120. PLEASE WRITE YOUR ANSWERS ON THIS PRINTOUT. 1. For each of the following parts, pick
2006 7.012 Problem Set 1 Due before 5 PM on FRIDAY, September 15, 2006. Turn answers in to the box outside of 68-120. PLEASE WRITE YOUR ANSWERS ON THIS PRINTOUT. 1. For each of the following parts, pick
Structure and evolution of the spliceosomal peptidyl-prolyl cistrans isomerase Cwc27
 Acta Cryst. (2014). D70, doi:10.1107/s1399004714021695 Supporting information Volume 70 (2014) Supporting information for article: Structure and evolution of the spliceosomal peptidyl-prolyl cistrans isomerase
Acta Cryst. (2014). D70, doi:10.1107/s1399004714021695 Supporting information Volume 70 (2014) Supporting information for article: Structure and evolution of the spliceosomal peptidyl-prolyl cistrans isomerase
SIMPLE MICROMECHANICAL MODEL OF PROTEIN CRYSTALS FOR THEIR MECHANICAL CHARACTERIZATIONS
 EPJ Web of Conferences 6, 6 51 (21) DOI:1.151/epjconf/21651 Owned by the authors, published by EDP Sciences, 21 SIMPLE MICROMECHANICAL MODEL OF PROTEIN CRYSTALS FOR THEIR MECHANICAL CHARACTERIZATIONS Gwonchan
EPJ Web of Conferences 6, 6 51 (21) DOI:1.151/epjconf/21651 Owned by the authors, published by EDP Sciences, 21 SIMPLE MICROMECHANICAL MODEL OF PROTEIN CRYSTALS FOR THEIR MECHANICAL CHARACTERIZATIONS Gwonchan
NMR study of complexes between low molecular mass inhibitors and the West Nile virus NS2B-NS3 protease
 University of Wollongong Research Online Faculty of Science - Papers (Archive) Faculty of Science, Medicine and Health 2009 NMR study of complexes between low molecular mass inhibitors and the West Nile
University of Wollongong Research Online Faculty of Science - Papers (Archive) Faculty of Science, Medicine and Health 2009 NMR study of complexes between low molecular mass inhibitors and the West Nile
Examples of Protein Modeling. Protein Modeling. Primary Structure. Protein Structure Description. Protein Sequence Sources. Importing Sequences to MOE
 Examples of Protein Modeling Protein Modeling Visualization Examination of an experimental structure to gain insight about a research question Dynamics To examine the dynamics of protein structures To
Examples of Protein Modeling Protein Modeling Visualization Examination of an experimental structure to gain insight about a research question Dynamics To examine the dynamics of protein structures To
Cooperativity and Specificity of Cys 2 His 2 Zinc Finger Protein-DNA Interactions: A Molecular Dynamics Simulation Study
 7662 J. Phys. Chem. B 2010, 114, 7662 7671 Cooperativity and Specificity of Cys 2 His 2 Zinc Finger Protein-DNA Interactions: A Molecular Dynamics Simulation Study Juyong Lee, Jin-Soo Kim, and Chaok Seok*
7662 J. Phys. Chem. B 2010, 114, 7662 7671 Cooperativity and Specificity of Cys 2 His 2 Zinc Finger Protein-DNA Interactions: A Molecular Dynamics Simulation Study Juyong Lee, Jin-Soo Kim, and Chaok Seok*
Chemistry Chapter 22
 hemistry 2100 hapter 22 Proteins Proteins serve many functions, including the following. 1. Structure: ollagen and keratin are the chief constituents of skin, bone, hair, and nails. 2. atalysts: Virtually
hemistry 2100 hapter 22 Proteins Proteins serve many functions, including the following. 1. Structure: ollagen and keratin are the chief constituents of skin, bone, hair, and nails. 2. atalysts: Virtually