Ab Initio Molecular Dynamics
|
|
|
- Julian Park
- 5 years ago
- Views:
Transcription
1
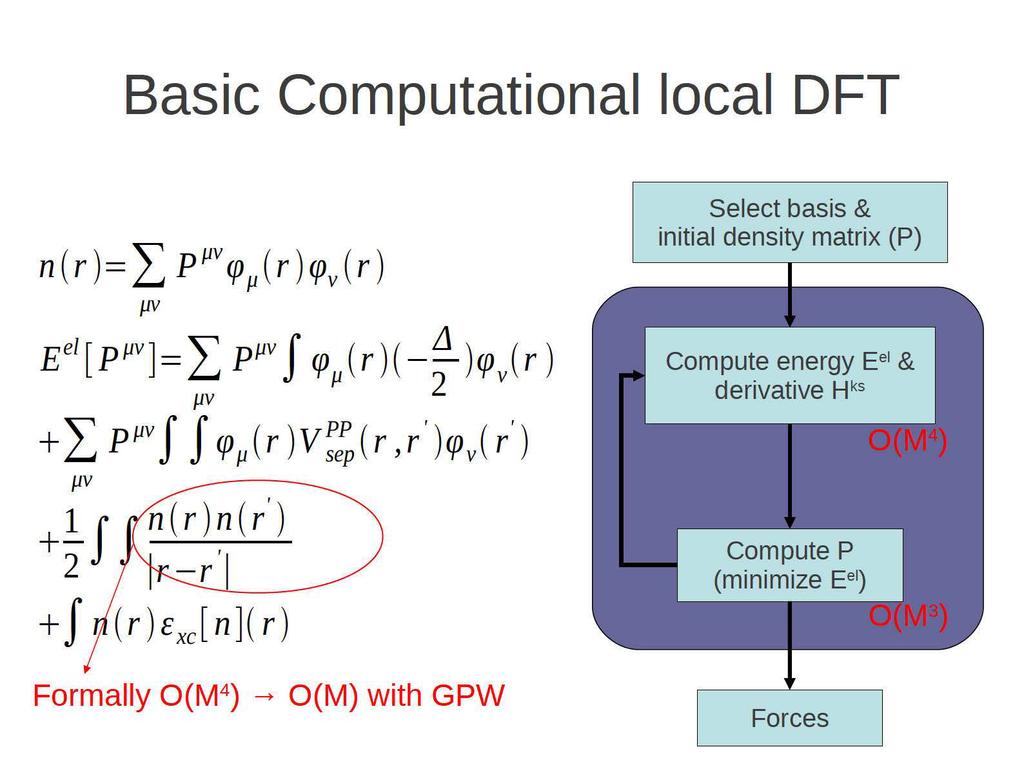
2 Ab Initio Molecular Dynamics Molecular Dynamics Classical equations of motion for the ions: F=ma NVE and other ensembles (NVT, NPT) Crucial for systems including liquids 'Ab Initio' or 'First Principles' Forces on atoms derive from a non-empirical description of the electronic structure 99% DFT


3 An chemist's example: onba ortho- Nitrobenzaldehyde Donten M; Hamm P; VandeVondele J; JPC B 115(5): (2011)
4

5 Interfaces at ambient conditions We want to build and study models that realistically include solids, liquids, molecules An example interface, as found in Dye sensitized solar cells. Schiffmann, VandeVondele, Hutter, Wirz, Urakawa, Hutter, baiker, J. Phys. Chem. C, 2010, 114 (18), pp
6 AIMD is GGA dominated In order for AIMD to be practical, the SCF + forces (BOMD) need to be computed in O(60sec) for typical systems O(100 atoms) MD steps of 0.5 fs * 60sec / step = 2 weeks for 10 ps Highly efficient schemes for GGAs are needed Highly robust methods are essential
7 AIMD is for short timescales 10ps 100ps is the typical lengthscale of AIMD Only very fast processes can be observed spontaneously Activated events (reactions) or slow dynamics (diffusion) need more advanced techniques Free energy methods (constraints, enhanced sampling, etc... needed).
8 Efficient GGA DFT CP2K
9 Gaussian and plane waves: GPW in CP2K Primary basis: Gaussians - compact - sparse H ks (and P) - Many terms analytic Auxiliary basis: Plane waves - regular grid for e - density - FFT for Poisson equation - No four center integrals needed Chemistry Physics The GPW algorithm : compute the GGA Kohn-Sham matrix in O(N) time, PBC are natural. J. VandeVondele, M. Krack, F. Mohamed, M. Parrinello, T. Chassaing and J. Hutter, Comp. Phys. Comm. 167, 103 (2005). Lippert, G; Hutter, J; Parrinello, M. Mol. Phys., 92 (3), (1997).
10
φ ν (r ) RS n(r ) FFT n(g ) V H (G )= 4πn(G ) G 2, E H =Ω G n cc (G )V H (G ) FFT V H ( r) V μν = V H (r )φ μ (r )φ ν (r ) RS A")
11 A closer look at GPW Real space (rs) density mapping and integration Fourier space (FFT) for the coulomb problem O(N) Only a linear number of terms have a non-zero contribution to the sum. Each term affects only a constant volume in space μν P μν φ μ (r )φ ν (r ) RS n(r ) FFT n(g ) V H (G )= 4πn(G ) G 2, E H =Ω G n cc (G )V H (G ) FFT V H ( r) V μν = V H (r )φ μ (r )φ ν (r ) RS A Gaussian basis allows for a very efficient procedure to compute the density on the grid
12 Orbital transformations (OT) A cubic, very robust algorithm avoiding the of traditional diagonalization New variables C X =C 0 cos X T SX X sin X T SX X T SC 0 =0 X T SX C X T SC X =1 X Direct minimization of E ks [{X}] Linear constraint -> guaranteed convergence! J. VandeVondele, J. Hutter, J. Chem. Phys., 2003, Vol. 118 No. 10,
 electronic")
. Sulpizi, M.; Raugei, S.; VandeVondele, J.; Carloni, P.")
13 DFT for large systems DNA crystal Solvated metallo-protein 2388 atoms 2825 atoms Linear scaling construction of the Kohn-Sham matrix, robust and efficient O(N 3 ) electronic minimization Parameter free & out-of-the-box for H-Rn J. VandeVondele, J. Hutter, JCP 118, (2003). Sulpizi, M.; Raugei, S.; VandeVondele, J.; Carloni, P.; Sprik, M. JPCB 111, 3969 (2007).

 2 COCl in")

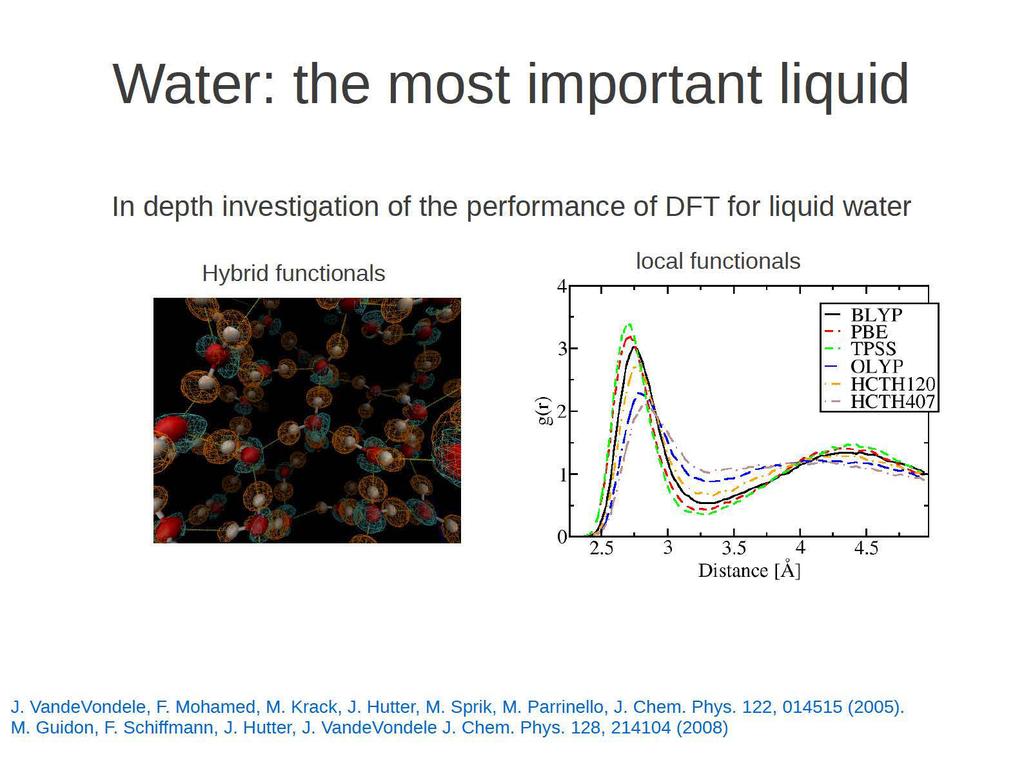
14 Robust Ab initio MD Complex Interfaces 'Simple' liquids Solutes in explicit solvent Hydrogen bonding in H2O Ru(bpy) 2 COCl in acetonitrile, [21.43Å] 3 or 620 Atoms e.g. Redox properties In situ IR spectroscopy (1300 atoms)
15
16
17

18 CP2K: science (I) Electronic structure of nanoparticles

19 CP2K: science (II) Structure prediction of metal organic frameworks

20 CP2K: science (III) Disordered and frustrated materials

21 CP2K: science (IV) Functionalized solid/liquid interfaces
 ~10% efficiency,short")

22 Dye sensitized solar cells (DSSC) Sustainable energy production converting sunlight into electricity Grätzel, Nature (1991,2001) ~10% efficiency,short energy-payback, tolerant to impurities, inexpensive technologies, cheap materials, wide temperature range, diffuse light OK, various colors (semi-transparent), flexible

 I - /I 3 - in Acetonitrile")

23 DSSC: atomistic engineering Prototypical high performance cells: Mesoporous TiO 2 (Anatase) Ruthenium Dye (N3) I - /I 3 - in Acetonitrile Grätzel, Nature (1991,2001) Taylor the interface to improve efficiency, stability and cost

24 N3 binding on anatase(101) Relative binding energies are indicative: 2-3 carboxylate groups interact 2 bpy ligands involved Monodendate and bridged binding possible Protonation strongly influences relative stability
25 IR spectra: Theory & Experiment Theory & Experiment Exp. difference spectrum N3/N712 Promising agreement (+ 30 cm -1 ), a unique binding geometry is not identified. Theory provides assignments Conclusions: base-induced desorption of dye adsorbed in the presence of protons change in binding mode depending on protonation
 Experimental data consistent with self-assembly: Dense")
 F. Schiffmann, J. VandeVondele, J. Hutter, R. Wirz, A. Urakawa, A. Baiker, J.")
26 Suggested binding mode Proposed monomer binding geometries Equilibrium between two Configurations (ph dependent) Experimental data consistent with self-assembly: Dense packing Dimers in STM (rutile) Desorption with base Computed self-assembly of I 2 on anatase(101) F. Schiffmann, J. VandeVondele, J. Hutter, R. Wirz, A. Urakawa, A. Baiker, J. Phys. Chem. C

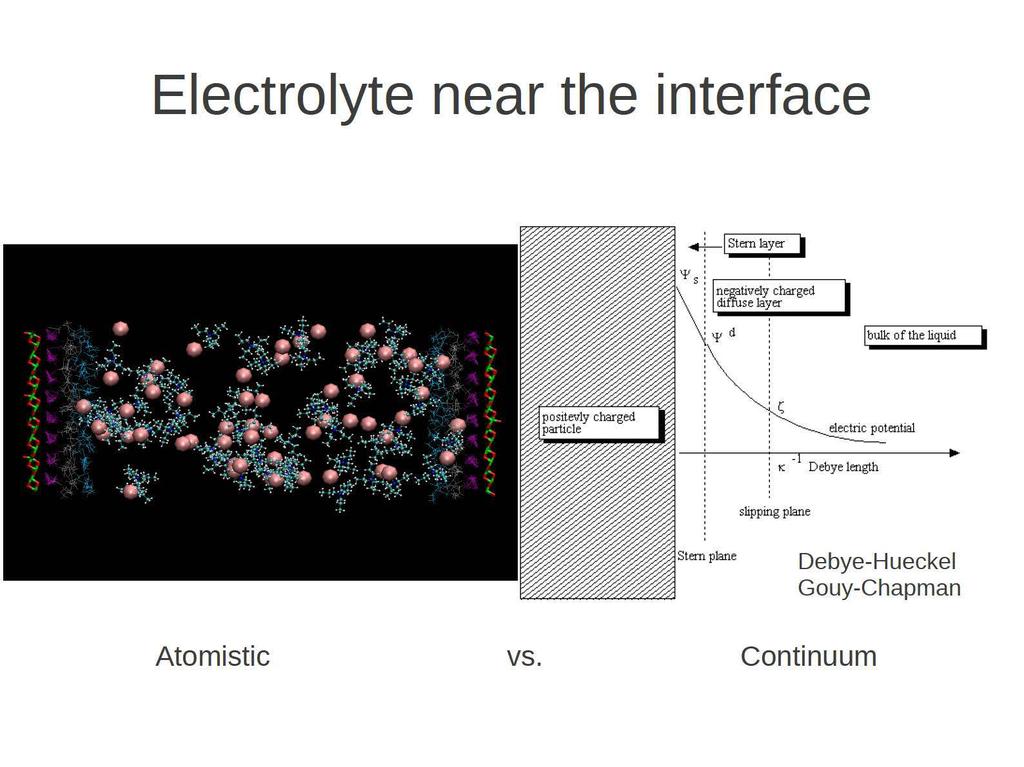
27 Including the electrolyte
28
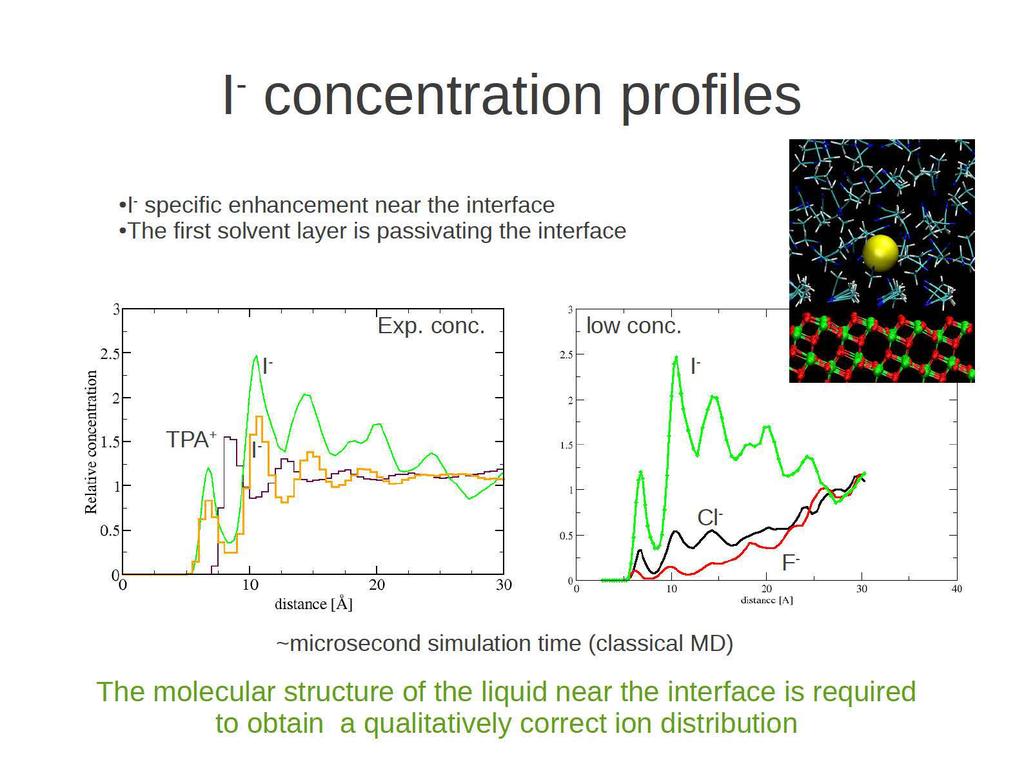
29

30 Dye - Iodide interaction DFT calculations of complex formation. Free energies of binding in explicit solution. I -, I 2 - form stable complexes in solution SCN - group essential in complex formation
31
32
33 Recent developments Full linear scaling GGA DFT Efficiently using hybrid functionals
34 Linear scaling GGA DFT
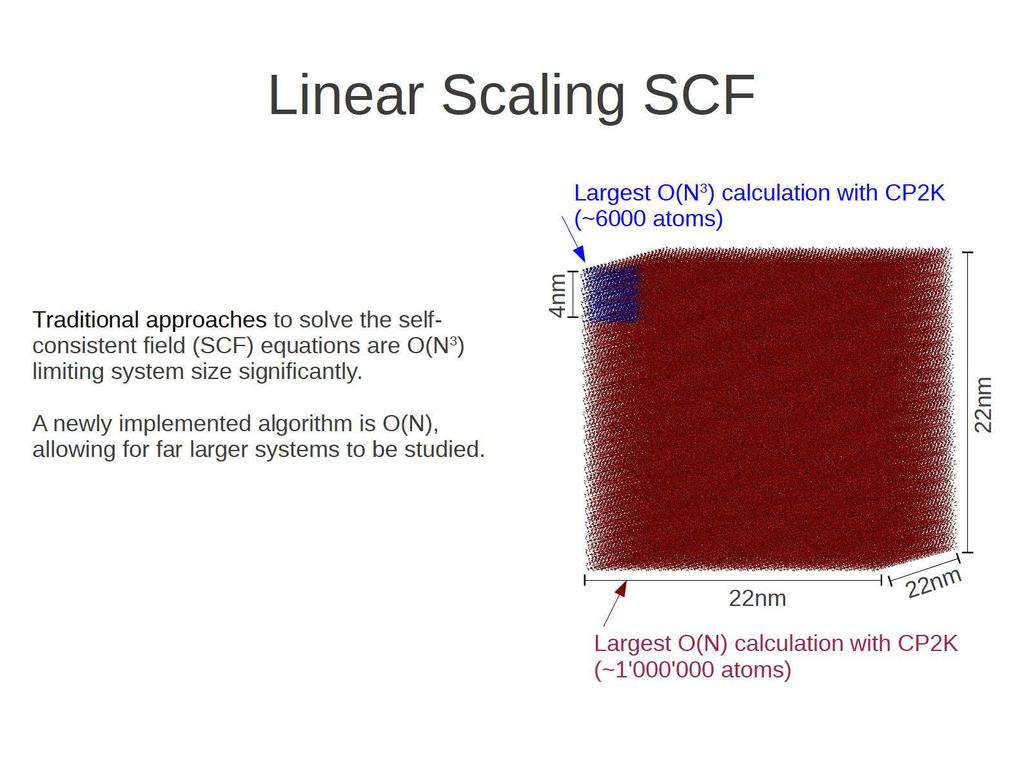
35




36 Linear Scaling SCF New regime: small devices, heterostructures, interfaces, nano-particles, a small virus. Gate-all-around FET Solvated STMV: 1M With Mathieu Luisier 1.5M atoms Anatase nanocrystal Caplovicova et al. App. Cat. B, 224, 117
37 Sign matrix iterations The density matrix (P) is function of H A simple iterative scheme (Newton-Schultz) gives sign(a): Using only sparse matrix matrix multiplies (not SPMV!) linear scaling can be obtained A dedicated sparse matrix multiply library is extremely important This library is being ported to GPUs
38
39
40 Towards O(1) : constant walltime with proportional resources Total time Stringent test: Small blocks, large overhead Very sparse matrices Running with 200 atoms / MPI task Overhead Local multiplies Communication Local multiplies constant (OK!). Overhead & Communication Grows with sqrt(n) Needs a replacement for Cannon Work is underway to replace the Cannon algorithm with something new! Retain the sqrt(n) max comm, yield constant comm in the limit.
41 Hybrid functionals
 13, 6670-6688.")
42 Advances in DFT Exchange and correlation functionals of improving can be constructed by adding new ingredients: Dispersion / van der Waals corrected functionals (Screened) Hybrid functionals Goerigk, L. and Grimme, S. Phys. Chem. Chem. Phys. (2011) 13, Mundy, Kathmann, Rousseau, Schenter, VandeVondele, Hutter, SCIDAC reviews (spring 2010).

43 Hartree-Fock exchange An easy term in Gaussian basis sets, but brute force scaling as O(N 4 ) 2'825 atoms 31'247 basis functions 976'375'009 elements in P 953'308'158'199'750'081 integrals Exa-Pet-Ter-Gig-Meg-Kil
44
45 O(N) HFX: measurements Linear scaling is key... thousands of molecules possible On 'standard' cluster hardware in minutes.
46 In-core integral compression Almost all simulations are performed using an in-core algorithm 10x speedup is observed. Highly efficient scheme: index free and lossy compression Guidon, M; Schiffmann, F; Hutter, J; VandeVondele, J ; JCP 128(21):
47
48 Robust Hartree-Fock exchange in the condensed phase How to treat this expression, k=k' is only conditionally convergent for g(r)=1/r? This 'difficult' point is integrable, but what for a Gamma-point code (k=k'=0)? Avoids spurious self-exchange interactions with images in other cells. This can be implemented robustly in a simple way. Truncated Coulomb: Needs, Alavi
49
50 The curse of HFX: The Basis Good quality calculations need good quality basis sets Increasing the quality of the basis steeply [O(N 4 )] increases the cost of HFX 1 st HFX step for 20 water molecules on 128 cores Most commonly used basis 'good' results 'Converged' results
51 Auxiliary Density Matrix Methods (ADMM) For certain density matrices HFX can be computed very efficiently (e.g. small basis sets or increased sparsity) Transform an expensive matrix into a cheap one, use a GGA for estimating the difference One example: wavefunction fitting, using an auxiliary basis Guidon M; Hutter J; VandeVondele J; JCTC 6(8):
52 ADMM: accuracy GTMKN24 (>1000 datapoints) using FIT3 and pfit3 auxiliary basis sets Error wrt to Exp. : identical (5.0 kcal/mol) Error wrt to Ref. : < 1 kcal/mol BSSE tests BSSE reduced by 5x compared to no ADMM H 2 O dimer: 0.5 kcal/mol FIT3, 0.2 kcal/mol aug-fit3


53 ADMM: performance STD DZVP-MOLOPT-SR-GTH ADMM: MOLOPT/FIT3 ADMM: MOLOPT/EMBED A fully solvated protein computed within minutes using hybrid functionals Guidon M; Hutter J; VandeVondele J; JCTC 6(8):

54 IR spectroscopy from AIMD with hybrid functionals H 2 PO4 - H 2 PO 4 - in explicit water ~ 100 ps AIMD / 64 waters Both Hybrids and GGA capture the main effect of solvation. Hybrid give 2x more accurate spectra VandeVondele et al. JPCA
55 Phosphate in solution Analyzing the amount of charge transfer in H 2 PO 4 - and HPO 4 2- : Reduced charge transfer is observed with hybrid functionals, but only for the double anion. The effect is partially electronic, partially geometric VandeVondele et al. Submitted

56
Dye Sensitized Solar Cells
 Large Scale simulations with CP2K Dye Sensitized Solar Cells Florian Schiffmann 2nd CP2K tutorial f.schiffmann@ucl.ac.uk University College London Dye sensitized solar cells Grätzel, Nature (1991,2001)
Large Scale simulations with CP2K Dye Sensitized Solar Cells Florian Schiffmann 2nd CP2K tutorial f.schiffmann@ucl.ac.uk University College London Dye sensitized solar cells Grätzel, Nature (1991,2001)
Large Scale Electronic Structure Calculations
 Large Scale Electronic Structure Calculations Jürg Hutter University of Zurich 8. September, 2008 / Speedup08 CP2K Program System GNU General Public License Community Developers Platform on "Berlios" (cp2k.berlios.de)
Large Scale Electronic Structure Calculations Jürg Hutter University of Zurich 8. September, 2008 / Speedup08 CP2K Program System GNU General Public License Community Developers Platform on "Berlios" (cp2k.berlios.de)
DFT with Hybrid Functionals
 DFT with Hybrid Functionals Sanliang Ling University College London 4th CP2K Tutorial, 31st August 4th September 2015, Zurich What are hybrid functionals? Hybrid functionals: mixing non-local Hartree-Fock
DFT with Hybrid Functionals Sanliang Ling University College London 4th CP2K Tutorial, 31st August 4th September 2015, Zurich What are hybrid functionals? Hybrid functionals: mixing non-local Hartree-Fock
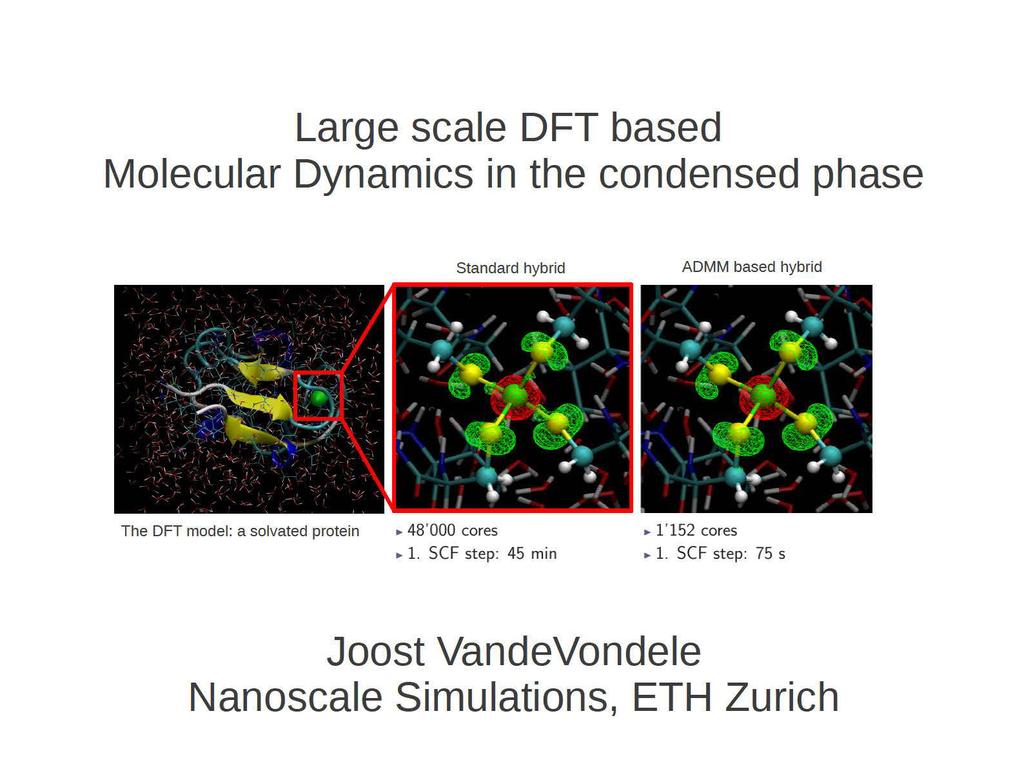
CP2K: summary and new developments. Joost VandeVondele Nanoscale Simulations, ETH Zurich
 CP2K: summary and new developments Standard hybrid ADMM based hybrid The DFT model: a solvated protein Joost VandeVondele Nanoscale Simulations, ETH Zurich What is CP2K? CP2K is a freely available program
CP2K: summary and new developments Standard hybrid ADMM based hybrid The DFT model: a solvated protein Joost VandeVondele Nanoscale Simulations, ETH Zurich What is CP2K? CP2K is a freely available program
Post Hartree-Fock: MP2 and RPA in CP2K
 Post Hartree-Fock: MP2 and RPA in CP2K A tutorial Jan Wilhelm jan.wilhelm@chem.uzh.ch 4 September 2015 Further reading MP2 and RPA by Mauro Del Ben, Jürg Hutter, Joost VandeVondele Del Ben, M; Hutter,
Post Hartree-Fock: MP2 and RPA in CP2K A tutorial Jan Wilhelm jan.wilhelm@chem.uzh.ch 4 September 2015 Further reading MP2 and RPA by Mauro Del Ben, Jürg Hutter, Joost VandeVondele Del Ben, M; Hutter,
CP2K: Selected Developments
 CP2K: Selected Developments Jürg Hutter Department of Chemistry University of Zurich Outline Introduction History and Performance Current and Future Developments Post-Hartree-Fock Methods GW Methods RPA
CP2K: Selected Developments Jürg Hutter Department of Chemistry University of Zurich Outline Introduction History and Performance Current and Future Developments Post-Hartree-Fock Methods GW Methods RPA
Is there a future for quantum chemistry on supercomputers? Jürg Hutter Physical-Chemistry Institute, University of Zurich
 Is there a future for quantum chemistry on supercomputers? Jürg Hutter Physical-Chemistry Institute, University of Zurich Chemistry Chemistry is the science of atomic matter, especially its chemical reactions,
Is there a future for quantum chemistry on supercomputers? Jürg Hutter Physical-Chemistry Institute, University of Zurich Chemistry Chemistry is the science of atomic matter, especially its chemical reactions,
Supporting Information: Surface Polarons Reducing Overpotentials in. the Oxygen Evolution Reaction
 Supporting Information: Surface Polarons Reducing Overpotentials in the Oxygen Evolution Reaction Patrick Gono Julia Wiktor Francesco Ambrosio and Alfredo Pasquarello Chaire de Simulation à l Echelle Atomique
Supporting Information: Surface Polarons Reducing Overpotentials in the Oxygen Evolution Reaction Patrick Gono Julia Wiktor Francesco Ambrosio and Alfredo Pasquarello Chaire de Simulation à l Echelle Atomique
CP2K: Past, Present, Future. Jürg Hutter Department of Chemistry, University of Zurich
 CP2K: Past, Present, Future Jürg Hutter Department of Chemistry, University of Zurich Outline Past History of CP2K Development of features Present Quickstep DFT code Post-HF methods (RPA, MP2) Libraries
CP2K: Past, Present, Future Jürg Hutter Department of Chemistry, University of Zurich Outline Past History of CP2K Development of features Present Quickstep DFT code Post-HF methods (RPA, MP2) Libraries
Day 1 : Introduction to AIMD and PIMD
 Day 1 : Introduction to AIMD and PIMD Aug 2018 In today s exercise, we perform ab-initio molecular dynamics (AIMD) and path integral molecular dynamics (PIMD) using CP2K[?]. We will use the Zundel s cation
Day 1 : Introduction to AIMD and PIMD Aug 2018 In today s exercise, we perform ab-initio molecular dynamics (AIMD) and path integral molecular dynamics (PIMD) using CP2K[?]. We will use the Zundel s cation
Ab-initio modeling of opto-electronic properties of molecules in solvents and in proximity to a semiconductor nanoparticle
 Ab-initio modeling of opto-electronic properties of molecules in solvents and in proximity to a semiconductor nanoparticle Alain Delgado (a,b), Stefano Corni (b), Carlo Andrea Rozzi (b) Stefano Pittalis
Ab-initio modeling of opto-electronic properties of molecules in solvents and in proximity to a semiconductor nanoparticle Alain Delgado (a,b), Stefano Corni (b), Carlo Andrea Rozzi (b) Stefano Pittalis
Zurich Open Repository and Archive. Gaussian basis sets for accurate calculations on molecular systems in gas and condensed phases
 University of Zurich Zurich Open Repository and Archive Winterthurerstr. 190 CH-8057 Zurich http://www.zora.uzh.ch Year: 2007 Gaussian basis sets for accurate calculations on molecular systems in gas and
University of Zurich Zurich Open Repository and Archive Winterthurerstr. 190 CH-8057 Zurich http://www.zora.uzh.ch Year: 2007 Gaussian basis sets for accurate calculations on molecular systems in gas and
CCSD(T) benchmarks of non-equilibrium water clusters: the importance of monomer deformation
 benchmarks of non-equilibrium water clusters: the importance of monomer deformation") CCSD(T) benchmarks of non-equilibrium water clusters: the importance of monomer deformation Biswajit Santra 1, Angelos Michaelides 1,2, and Matthias Scheffler 1 1 Fritz-Haber-Institut der MPG, Berlin,
CCSD(T) benchmarks of non-equilibrium water clusters: the importance of monomer deformation Biswajit Santra 1, Angelos Michaelides 1,2, and Matthias Scheffler 1 1 Fritz-Haber-Institut der MPG, Berlin,
CP2K. New Frontiers. ab initio Molecular Dynamics
 CP2K New Frontiers in ab initio Molecular Dynamics Jürg Hutter, Joost VandeVondele, Valery Weber Physical-Chemistry Institute, University of Zurich Ab Initio Molecular Dynamics Molecular Dynamics Sampling
CP2K New Frontiers in ab initio Molecular Dynamics Jürg Hutter, Joost VandeVondele, Valery Weber Physical-Chemistry Institute, University of Zurich Ab Initio Molecular Dynamics Molecular Dynamics Sampling
Electronic structure simulations of water solid interfaces
 Electronic structure simulations of water solid interfaces Angelos Michaelides London Centre for Nanotechnology & Department of Chemistry, University College London www.chem.ucl.ac.uk/ice Main co-workers:
Electronic structure simulations of water solid interfaces Angelos Michaelides London Centre for Nanotechnology & Department of Chemistry, University College London www.chem.ucl.ac.uk/ice Main co-workers:
Accelerated Quantum Molecular Dynamics
 Accelerated Quantum Molecular Dynamics Enrique Martinez, Christian Negre, Marc J. Cawkwell, Danny Perez, Arthur F. Voter and Anders M. N. Niklasson Outline Quantum MD Current approaches Challenges Extended
Accelerated Quantum Molecular Dynamics Enrique Martinez, Christian Negre, Marc J. Cawkwell, Danny Perez, Arthur F. Voter and Anders M. N. Niklasson Outline Quantum MD Current approaches Challenges Extended
Hybrid Functionals, van der Waals Corrections and ASE Interface to CP2K
 Hybrid Functionals, van der Waals Corrections and ASE Interface to CP2K Sanliang Ling University College London CP2K Summer School, 23rd - 26th August 2016, King's College London Part I: Hybrid Functionals
Hybrid Functionals, van der Waals Corrections and ASE Interface to CP2K Sanliang Ling University College London CP2K Summer School, 23rd - 26th August 2016, King's College London Part I: Hybrid Functionals
Ab initio molecular dynamics
 Ab initio molecular dynamics Kari Laasonen, Physical Chemistry, Aalto University, Espoo, Finland (Atte Sillanpää, Jaakko Saukkoriipi, Giorgio Lanzani, University of Oulu) Computational chemistry is a field
Ab initio molecular dynamics Kari Laasonen, Physical Chemistry, Aalto University, Espoo, Finland (Atte Sillanpää, Jaakko Saukkoriipi, Giorgio Lanzani, University of Oulu) Computational chemistry is a field
CP2K: the gaussian plane wave (GPW) method
 method") CP2K: the gaussian plane wave (GPW) method Basis sets and Kohn-Sham energy calculation R. Vuilleumier Département de chimie Ecole normale supérieure Paris Tutorial CPMD-CP2K CPMD and CP2K CPMD CP2K http://www.cpmd.org
CP2K: the gaussian plane wave (GPW) method Basis sets and Kohn-Sham energy calculation R. Vuilleumier Département de chimie Ecole normale supérieure Paris Tutorial CPMD-CP2K CPMD and CP2K CPMD CP2K http://www.cpmd.org
Advanced Electronic Structure Theory Density functional theory. Dr Fred Manby
 Advanced Electronic Structure Theory Density functional theory Dr Fred Manby fred.manby@bris.ac.uk http://www.chm.bris.ac.uk/pt/manby/ 6 Strengths of DFT DFT is one of many theories used by (computational)
Advanced Electronic Structure Theory Density functional theory Dr Fred Manby fred.manby@bris.ac.uk http://www.chm.bris.ac.uk/pt/manby/ 6 Strengths of DFT DFT is one of many theories used by (computational)
DENSITY FUNCTIONAL THEORY FOR NON-THEORISTS JOHN P. PERDEW DEPARTMENTS OF PHYSICS AND CHEMISTRY TEMPLE UNIVERSITY
 DENSITY FUNCTIONAL THEORY FOR NON-THEORISTS JOHN P. PERDEW DEPARTMENTS OF PHYSICS AND CHEMISTRY TEMPLE UNIVERSITY A TUTORIAL FOR PHYSICAL SCIENTISTS WHO MAY OR MAY NOT HATE EQUATIONS AND PROOFS REFERENCES
DENSITY FUNCTIONAL THEORY FOR NON-THEORISTS JOHN P. PERDEW DEPARTMENTS OF PHYSICS AND CHEMISTRY TEMPLE UNIVERSITY A TUTORIAL FOR PHYSICAL SCIENTISTS WHO MAY OR MAY NOT HATE EQUATIONS AND PROOFS REFERENCES
Efficient use of CP2K
 Efficient use of CP2K Fawzi Mohamed 1 1 Department of Chemisty Humboldt University of Berlin June 10, 2009 cp2k Fortran 95 opensource (GPL) general framework for different methods DFT no official release
Efficient use of CP2K Fawzi Mohamed 1 1 Department of Chemisty Humboldt University of Berlin June 10, 2009 cp2k Fortran 95 opensource (GPL) general framework for different methods DFT no official release
The Electronic Structure of Dye- Sensitized TiO 2 Clusters from Many- Body Perturbation Theory
 The Electronic Structure of Dye- Sensitized TiO 2 Clusters from Many- Body Perturbation Theory Noa Marom Center for Computational Materials Institute for Computational Engineering and Sciences The University
The Electronic Structure of Dye- Sensitized TiO 2 Clusters from Many- Body Perturbation Theory Noa Marom Center for Computational Materials Institute for Computational Engineering and Sciences The University
Electronic structure theory: Fundamentals to frontiers. 2. Density functional theory
 Electronic structure theory: Fundamentals to frontiers. 2. Density functional theory MARTIN HEAD-GORDON, Department of Chemistry, University of California, and Chemical Sciences Division, Lawrence Berkeley
Electronic structure theory: Fundamentals to frontiers. 2. Density functional theory MARTIN HEAD-GORDON, Department of Chemistry, University of California, and Chemical Sciences Division, Lawrence Berkeley
Exchange Correlation Functional Investigation of RT-TDDFT on a Sodium Chloride. Dimer. Philip Straughn
 Exchange Correlation Functional Investigation of RT-TDDFT on a Sodium Chloride Dimer Philip Straughn Abstract Charge transfer between Na and Cl ions is an important problem in physical chemistry. However,
Exchange Correlation Functional Investigation of RT-TDDFT on a Sodium Chloride Dimer Philip Straughn Abstract Charge transfer between Na and Cl ions is an important problem in physical chemistry. However,
SUPPLEMENTARY INFORMATION
 Calculations predict a stable molecular crystal of N 8 : Barak Hirshberg a, R. Benny Gerber a,b, and Anna I. Krylov c a Institute of Chemistry and The Fritz Haber Center for Molecular Dynamics, The Hebrew
Calculations predict a stable molecular crystal of N 8 : Barak Hirshberg a, R. Benny Gerber a,b, and Anna I. Krylov c a Institute of Chemistry and The Fritz Haber Center for Molecular Dynamics, The Hebrew
Ligand-Stabilized Reduced-Dimensionality Perovskites
 Ligand-Stabilized Reduced-Dimensionality Perovskites Supporting Information Li Na Quan, 1,2 Mingjian Yuan, 1 Riccardo Comin, 1 Oleksandr Voznyy, 1 Eric M. Beauregard, 1 Sjoerd Hoogland, 1 Andrei Buin,
Ligand-Stabilized Reduced-Dimensionality Perovskites Supporting Information Li Na Quan, 1,2 Mingjian Yuan, 1 Riccardo Comin, 1 Oleksandr Voznyy, 1 Eric M. Beauregard, 1 Sjoerd Hoogland, 1 Andrei Buin,
Structure of Cement Phases from ab initio Modeling Crystalline C-S-HC
 Structure of Cement Phases from ab initio Modeling Crystalline C-S-HC Sergey V. Churakov sergey.churakov@psi.ch Paul Scherrer Institute Switzerland Cement Phase Composition C-S-H H Solid Solution Model
Structure of Cement Phases from ab initio Modeling Crystalline C-S-HC Sergey V. Churakov sergey.churakov@psi.ch Paul Scherrer Institute Switzerland Cement Phase Composition C-S-H H Solid Solution Model
Computer simulation methods (2) Dr. Vania Calandrini
 Dr. Vania Calandrini") Computer simulation methods (2) Dr. Vania Calandrini in the previous lecture: time average versus ensemble average MC versus MD simulations equipartition theorem (=> computing T) virial theorem (=> computing
Computer simulation methods (2) Dr. Vania Calandrini in the previous lecture: time average versus ensemble average MC versus MD simulations equipartition theorem (=> computing T) virial theorem (=> computing
Supporting Information
 alladium monophosphine d(h 3 ): does it really exist in solution? ietro Vidossich*, Gregori Ujaque,* and Agustí Lledós* Supporting Information Contents: 1. Schemes S1 and S2 2. Computational details 3.
alladium monophosphine d(h 3 ): does it really exist in solution? ietro Vidossich*, Gregori Ujaque,* and Agustí Lledós* Supporting Information Contents: 1. Schemes S1 and S2 2. Computational details 3.
Introduction to DFTB. Marcus Elstner. July 28, 2006
 Introduction to DFTB Marcus Elstner July 28, 2006 I. Non-selfconsistent solution of the KS equations DFT can treat up to 100 atoms in routine applications, sometimes even more and about several ps in MD
Introduction to DFTB Marcus Elstner July 28, 2006 I. Non-selfconsistent solution of the KS equations DFT can treat up to 100 atoms in routine applications, sometimes even more and about several ps in MD
Supporting Information for. Ab Initio Metadynamics Study of VO + 2 /VO2+ Redox Reaction Mechanism at the Graphite. Edge Water Interface
 Supporting Information for Ab Initio Metadynamics Study of VO + 2 /VO2+ Redox Reaction Mechanism at the Graphite Edge Water Interface Zhen Jiang, Konstantin Klyukin, and Vitaly Alexandrov,, Department
Supporting Information for Ab Initio Metadynamics Study of VO + 2 /VO2+ Redox Reaction Mechanism at the Graphite Edge Water Interface Zhen Jiang, Konstantin Klyukin, and Vitaly Alexandrov,, Department
Ari P Seitsonen CNRS & Université Pierre et Marie Curie, Paris
 Self-organisation on noble metal surfaces Ari P Seitsonen CNRS & Université Pierre et Marie Curie, Paris Collaborations Alexandre Dmitriev, Nian Lin, Johannes Barth, Klaus Kern,... Thomas Greber, Jürg
Self-organisation on noble metal surfaces Ari P Seitsonen CNRS & Université Pierre et Marie Curie, Paris Collaborations Alexandre Dmitriev, Nian Lin, Johannes Barth, Klaus Kern,... Thomas Greber, Jürg
The Molecular Dynamics Method
 The Molecular Dynamics Method Thermal motion of a lipid bilayer Water permeation through channels Selective sugar transport Potential Energy (hyper)surface What is Force? Energy U(x) F = d dx U(x) Conformation
The Molecular Dynamics Method Thermal motion of a lipid bilayer Water permeation through channels Selective sugar transport Potential Energy (hyper)surface What is Force? Energy U(x) F = d dx U(x) Conformation
Density Functional Theory
 Density Functional Theory Iain Bethune EPCC ibethune@epcc.ed.ac.uk Overview Background Classical Atomistic Simulation Essential Quantum Mechanics DFT: Approximations and Theory DFT: Implementation using
Density Functional Theory Iain Bethune EPCC ibethune@epcc.ed.ac.uk Overview Background Classical Atomistic Simulation Essential Quantum Mechanics DFT: Approximations and Theory DFT: Implementation using
Hyeyoung Shin a, Tod A. Pascal ab, William A. Goddard III abc*, and Hyungjun Kim a* Korea
 The Scaled Effective Solvent Method for Predicting the Equilibrium Ensemble of Structures with Analysis of Thermodynamic Properties of Amorphous Polyethylene Glycol-Water Mixtures Hyeyoung Shin a, Tod
The Scaled Effective Solvent Method for Predicting the Equilibrium Ensemble of Structures with Analysis of Thermodynamic Properties of Amorphous Polyethylene Glycol-Water Mixtures Hyeyoung Shin a, Tod
Electrochemistry project, Chemistry Department, November Ab-initio Molecular Dynamics Simulation
 Electrochemistry project, Chemistry Department, November 2006 Ab-initio Molecular Dynamics Simulation Outline Introduction Ab-initio concepts Total energy concepts Adsorption energy calculation Project
Electrochemistry project, Chemistry Department, November 2006 Ab-initio Molecular Dynamics Simulation Outline Introduction Ab-initio concepts Total energy concepts Adsorption energy calculation Project
Mesoporous titanium dioxide electrolyte bulk heterojunction
 Mesoporous titanium dioxide electrolyte bulk heterojunction The term "bulk heterojunction" is used to describe a heterojunction composed of two different materials acting as electron- and a hole- transporters,
Mesoporous titanium dioxide electrolyte bulk heterojunction The term "bulk heterojunction" is used to describe a heterojunction composed of two different materials acting as electron- and a hole- transporters,
CHE3935. Lecture 4 Quantum Mechanical Simulation Methods Continued
 CHE3935 Lecture 4 Quantum Mechanical Simulation Methods Continued 1 OUTLINE Review Introduction to CPMD MD and ensembles The functionals of density functional theory Return to ab initio methods Binding
CHE3935 Lecture 4 Quantum Mechanical Simulation Methods Continued 1 OUTLINE Review Introduction to CPMD MD and ensembles The functionals of density functional theory Return to ab initio methods Binding
Hands-on : Model Potential Molecular Dynamics
 Hands-on : Model Potential Molecular Dynamics OUTLINE 0. DL_POLY code introduction 0.a Input files 1. THF solvent molecule 1.a Geometry optimization 1.b NVE/NVT dynamics 2. Liquid THF 2.a Equilibration
Hands-on : Model Potential Molecular Dynamics OUTLINE 0. DL_POLY code introduction 0.a Input files 1. THF solvent molecule 1.a Geometry optimization 1.b NVE/NVT dynamics 2. Liquid THF 2.a Equilibration
Calculating Vibrational Spectra from Molecular Dynamics
 Calculating Vibrational Spectra from Molecular Dynamics A Simulating a Trajectory with Wannier Centers To calculate IR spectra from Molecular Dynamics, it is necessary to have dipole information for the
Calculating Vibrational Spectra from Molecular Dynamics A Simulating a Trajectory with Wannier Centers To calculate IR spectra from Molecular Dynamics, it is necessary to have dipole information for the
Intermolecular Forces in Density Functional Theory
 Intermolecular Forces in Density Functional Theory Problems of DFT Peter Pulay at WATOC2005: There are 3 problems with DFT 1. Accuracy does not converge 2. Spin states of open shell systems often incorrect
Intermolecular Forces in Density Functional Theory Problems of DFT Peter Pulay at WATOC2005: There are 3 problems with DFT 1. Accuracy does not converge 2. Spin states of open shell systems often incorrect
Electronic Supporting Information Topological design of porous organic molecules
 Electronic Supplementary Material (ESI) for Nanoscale. This journal is The Royal Society of Chemistry 2017 Electronic Supporting Information Topological design of porous organic molecules Valentina Santolini,
Electronic Supplementary Material (ESI) for Nanoscale. This journal is The Royal Society of Chemistry 2017 Electronic Supporting Information Topological design of porous organic molecules Valentina Santolini,
Wavelets for density functional calculations: Four families and three. applications
 Wavelets for density functional calculations: Four families and three Haar wavelets Daubechies wavelets: BigDFT code applications Stefan Goedecker Stefan.Goedecker@unibas.ch http://comphys.unibas.ch/ Interpolating
Wavelets for density functional calculations: Four families and three Haar wavelets Daubechies wavelets: BigDFT code applications Stefan Goedecker Stefan.Goedecker@unibas.ch http://comphys.unibas.ch/ Interpolating
Molecular Mechanics. I. Quantum mechanical treatment of molecular systems
 Molecular Mechanics I. Quantum mechanical treatment of molecular systems The first principle approach for describing the properties of molecules, including proteins, involves quantum mechanics. For example,
Molecular Mechanics I. Quantum mechanical treatment of molecular systems The first principle approach for describing the properties of molecules, including proteins, involves quantum mechanics. For example,
Opportunities from Accurate and Efficient Density Functional Theory Calculations for Large Systems
 Seminar CENTRE FOR PREDICTIVE MODELLING, WARWICK Opportunities from Accurate and Efficient Density Functional Theory Calculations for Large Systems Luigi Genovese L_Sim CEA Grenoble October 30, 2017 http://bigdft.org
Seminar CENTRE FOR PREDICTIVE MODELLING, WARWICK Opportunities from Accurate and Efficient Density Functional Theory Calculations for Large Systems Luigi Genovese L_Sim CEA Grenoble October 30, 2017 http://bigdft.org
Supporting Information for. Dynamics Study"
 Supporting Information for "CO 2 Adsorption and Reactivity on Rutile TiO 2 (110) in Water: An Ab Initio Molecular Dynamics Study" Konstantin Klyukin and Vitaly Alexandrov,, Department of Chemical and Biomolecular
Supporting Information for "CO 2 Adsorption and Reactivity on Rutile TiO 2 (110) in Water: An Ab Initio Molecular Dynamics Study" Konstantin Klyukin and Vitaly Alexandrov,, Department of Chemical and Biomolecular
Bridging Scales Through Wavefunction Analysis
 Bridging Scales Through Wavefunction Analysis Felix Plasser Institute for Theoretical Chemistry, University of Vienna Excited States Bridging Scales Marseille, November 7 10, 2016 F. Plasser Wavefunction
Bridging Scales Through Wavefunction Analysis Felix Plasser Institute for Theoretical Chemistry, University of Vienna Excited States Bridging Scales Marseille, November 7 10, 2016 F. Plasser Wavefunction
New Perspective on structure and bonding in water using XAS and XRS
 New Perspective on structure and bonding in water using XAS and XRS Anders Nilsson Stanford Synchrotron Radiation Laboratory (SSRL) and Stockholm University, Sweden R. Ludwig Angew. Chem. 40, 1808 (2001)
New Perspective on structure and bonding in water using XAS and XRS Anders Nilsson Stanford Synchrotron Radiation Laboratory (SSRL) and Stockholm University, Sweden R. Ludwig Angew. Chem. 40, 1808 (2001)
Supplementary Information for Electronic signature of the instantaneous asymmetry in the first coordination shell in liquid water
 Supplementary Information for Electronic signature of the instantaneous asymmetry in the first coordination shell in liquid water Thomas D. Kühne 1, 2 and Rustam Z. Khaliullin 1, 1 Institute of Physical
Supplementary Information for Electronic signature of the instantaneous asymmetry in the first coordination shell in liquid water Thomas D. Kühne 1, 2 and Rustam Z. Khaliullin 1, 1 Institute of Physical
Outline. Introduction: graphene. Adsorption on graphene: - Chemisorption - Physisorption. Summary
 Outline Introduction: graphene Adsorption on graphene: - Chemisorption - Physisorption Summary 1 Electronic band structure: Electronic properties K Γ M v F = 10 6 ms -1 = c/300 massless Dirac particles!
Outline Introduction: graphene Adsorption on graphene: - Chemisorption - Physisorption Summary 1 Electronic band structure: Electronic properties K Γ M v F = 10 6 ms -1 = c/300 massless Dirac particles!
Ab initio Molecular Dynamics Born Oppenheimer and beyond
 Ab initio Molecular Dynamics Born Oppenheimer and beyond Reminder, reliability of MD MD trajectories are chaotic (exponential divergence with respect to initial conditions), BUT... With a good integrator
Ab initio Molecular Dynamics Born Oppenheimer and beyond Reminder, reliability of MD MD trajectories are chaotic (exponential divergence with respect to initial conditions), BUT... With a good integrator
Ab-initio molecular dynamics: from the basics up to quantum effects Roberto Car Princeton University
 Ab-initio molecular dynamics: from the basics up to quantum effects Roberto Car Princeton University Hands-on Tutorial Workshop on Ab-Initio Molecular Simulations, Fritz- Haber-Institut, Berlin, July 12-21,
Ab-initio molecular dynamics: from the basics up to quantum effects Roberto Car Princeton University Hands-on Tutorial Workshop on Ab-Initio Molecular Simulations, Fritz- Haber-Institut, Berlin, July 12-21,
Computational Chemistry I
 Computational Chemistry I Text book Cramer: Essentials of Quantum Chemistry, Wiley (2 ed.) Chapter 3. Post Hartree-Fock methods (Cramer: chapter 7) There are many ways to improve the HF method. Most of
Computational Chemistry I Text book Cramer: Essentials of Quantum Chemistry, Wiley (2 ed.) Chapter 3. Post Hartree-Fock methods (Cramer: chapter 7) There are many ways to improve the HF method. Most of
th International Workshop on Computational Physics and Materials Science: Total Energy and Force Methods January 2011
 2220-4 15th International Workshop on Computational Physics and Materials Science: Total Energy and Force Methods 13-15 January 2011 Computational study of optical and structural properties of an organic
2220-4 15th International Workshop on Computational Physics and Materials Science: Total Energy and Force Methods 13-15 January 2011 Computational study of optical and structural properties of an organic
Design of Efficient Catalysts with Double Transition Metal. Atoms on C 2 N Layer
 Supporting Information Design of Efficient Catalysts with Double Transition Metal Atoms on C 2 N Layer Xiyu Li, 1, Wenhui Zhong, 2, Peng Cui, 1 Jun Li, 1 Jun Jiang 1, * 1 Hefei National Laboratory for
Supporting Information Design of Efficient Catalysts with Double Transition Metal Atoms on C 2 N Layer Xiyu Li, 1, Wenhui Zhong, 2, Peng Cui, 1 Jun Li, 1 Jun Jiang 1, * 1 Hefei National Laboratory for
Free energy sampling for electrochemical systems
 Free energy sampling for electrochemical systems Mira Todorova, Anoop Kishore Vatti, Suhyun Yoo and Jörg Neugebauer Department of Computational Materials Design Düsseldorf, Germany m.todorova@mpie.de IPAM,
Free energy sampling for electrochemical systems Mira Todorova, Anoop Kishore Vatti, Suhyun Yoo and Jörg Neugebauer Department of Computational Materials Design Düsseldorf, Germany m.todorova@mpie.de IPAM,
Non-covalent force fields computed ab initio
 Non-covalent force fields computed ab initio Supermolecule calculations Symmetry-adapted perturbation theory (SAPT) Supermolecule calculations Requirements: E = E AB E A E B. Include electron correlation,
Non-covalent force fields computed ab initio Supermolecule calculations Symmetry-adapted perturbation theory (SAPT) Supermolecule calculations Requirements: E = E AB E A E B. Include electron correlation,
Compression Algorithms for Electronic Structure Computations
 Compression Algorithms for Electronic Structure Computations François Gygi University of California, Davis fgygi@ucdavis.edu http://eslab.ucdavis.edu http://www.quantum-simulation.org IPAM Workshop, Computation
Compression Algorithms for Electronic Structure Computations François Gygi University of California, Davis fgygi@ucdavis.edu http://eslab.ucdavis.edu http://www.quantum-simulation.org IPAM Workshop, Computation
CLIMBING THE LADDER OF DENSITY FUNCTIONAL APPROXIMATIONS JOHN P. PERDEW DEPARTMENT OF PHYSICS TEMPLE UNIVERSITY PHILADELPHIA, PA 19122
 CLIMBING THE LADDER OF DENSITY FUNCTIONAL APPROXIMATIONS JOHN P. PERDEW DEPARTMENT OF PHYSICS TEMPLE UNIVERSITY PHILADELPHIA, PA 191 THANKS TO MANY COLLABORATORS, INCLUDING SY VOSKO DAVID LANGRETH ALEX
CLIMBING THE LADDER OF DENSITY FUNCTIONAL APPROXIMATIONS JOHN P. PERDEW DEPARTMENT OF PHYSICS TEMPLE UNIVERSITY PHILADELPHIA, PA 191 THANKS TO MANY COLLABORATORS, INCLUDING SY VOSKO DAVID LANGRETH ALEX
Representing High-Dimensional Potential-Energy Surfaces by Artificial Neural Networks
 Energy Landscapes, Chemnitz 200 Representing High-Dimensional Potential-Energy Surfaces by Artificial Neural Networks Jörg Behler Lehrstuhl für Theoretische Chemie Ruhr-Universität Bochum D-44780 Bochum,
Energy Landscapes, Chemnitz 200 Representing High-Dimensional Potential-Energy Surfaces by Artificial Neural Networks Jörg Behler Lehrstuhl für Theoretische Chemie Ruhr-Universität Bochum D-44780 Bochum,
NWChem: Hartree-Fock, Density Functional Theory, Time-Dependent Density Functional Theory
 NWChem: Hartree-Fock, Density Functional Theory, Time-Depent Density Functional Theory Hartree-Fock! Functionality! Input! Wavefunctions! Initial MO vectors! Direct and semidirect algorithms! Convergence,
NWChem: Hartree-Fock, Density Functional Theory, Time-Depent Density Functional Theory Hartree-Fock! Functionality! Input! Wavefunctions! Initial MO vectors! Direct and semidirect algorithms! Convergence,
Au-C Au-Au. g(r) r/a. Supplementary Figures
 r/a. Supplementary Figures") g(r) Supplementary Figures 60 50 40 30 20 10 0 Au-C Au-Au 2 4 r/a 6 8 Supplementary Figure 1 Radial bond distributions for Au-C and Au-Au bond. The zero density regime between the first two peaks in g
g(r) Supplementary Figures 60 50 40 30 20 10 0 Au-C Au-Au 2 4 r/a 6 8 Supplementary Figure 1 Radial bond distributions for Au-C and Au-Au bond. The zero density regime between the first two peaks in g
Anion-π and π-π cooperative interactions
 Chapter 5 Anion-π and π-π cooperative interactions 5.1 Introduction The design of selective receptors of anionic species is a very active area of research within supramolecular chemistry due to the potential
Chapter 5 Anion-π and π-π cooperative interactions 5.1 Introduction The design of selective receptors of anionic species is a very active area of research within supramolecular chemistry due to the potential
Ab initio molecular dynamics with nuclear quantum effects at classical cost: ring polymer contraction for density functional theory
 Ab initio molecular dynamics with nuclear quantum effects at classical cost: ring polymer contraction for density functional theory Ondrej Marsalek 1 1, a) and Thomas E. Markland Department of Chemistry,
Ab initio molecular dynamics with nuclear quantum effects at classical cost: ring polymer contraction for density functional theory Ondrej Marsalek 1 1, a) and Thomas E. Markland Department of Chemistry,
Computational Modeling of Protein-Ligand Interactions
 Computational Modeling of Protein-Ligand Interactions Steven R. Gwaltney Department of Chemistry Mississippi State University Mississippi State, MS 39762 Auguste Comte, 1830 Every attempt to refer chemical
Computational Modeling of Protein-Ligand Interactions Steven R. Gwaltney Department of Chemistry Mississippi State University Mississippi State, MS 39762 Auguste Comte, 1830 Every attempt to refer chemical
Advanced in silico drug design
 Advanced in silico drug design RNDr. Martin Lepšík, Ph.D. Lecture: Advanced scoring Palacky University, Olomouc 2016 1 Outline 1. Scoring Definition, Types 2. Physics-based Scoring: Master Equation Terms
Advanced in silico drug design RNDr. Martin Lepšík, Ph.D. Lecture: Advanced scoring Palacky University, Olomouc 2016 1 Outline 1. Scoring Definition, Types 2. Physics-based Scoring: Master Equation Terms
Potential Energy (hyper)surface
surface") The Molecular Dynamics Method Thermal motion of a lipid bilayer Water permeation through channels Selective sugar transport Potential Energy (hyper)surface What is Force? Energy U(x) F = " d dx U(x) Conformation
The Molecular Dynamics Method Thermal motion of a lipid bilayer Water permeation through channels Selective sugar transport Potential Energy (hyper)surface What is Force? Energy U(x) F = " d dx U(x) Conformation
Basic introduction of NWChem software
 Basic introduction of NWChem software Background NWChem is part of the Molecular Science Software Suite Designed and developed to be a highly efficient and portable Massively Parallel computational chemistry
Basic introduction of NWChem software Background NWChem is part of the Molecular Science Software Suite Designed and developed to be a highly efficient and portable Massively Parallel computational chemistry
Accuracy benchmarking of DFT results, domain libraries for electrostatics, hybrid functional and solvation
 Accuracy benchmarking of DFT results, domain libraries for electrostatics, hybrid functional and solvation Stefan Goedecker Stefan.Goedecker@unibas.ch http://comphys.unibas.ch/ Multi-wavelets: High accuracy
Accuracy benchmarking of DFT results, domain libraries for electrostatics, hybrid functional and solvation Stefan Goedecker Stefan.Goedecker@unibas.ch http://comphys.unibas.ch/ Multi-wavelets: High accuracy
New Algorithms for Conventional and. Jing Kong ( 孔静 ) Q-Chem Inc. Pittsburgh, PA
 Q-Chem Inc. Pittsburgh, PA") New Algorithms for Conventional and Nondynamic DFT Jing Kong ( 孔静 ) Q-Chem Inc. Pittsburgh, PA XC Numerical Integration mrxc: multiresolution exchage-correlation Chunming Chang Nick Russ Phys. Rev. A.,
New Algorithms for Conventional and Nondynamic DFT Jing Kong ( 孔静 ) Q-Chem Inc. Pittsburgh, PA XC Numerical Integration mrxc: multiresolution exchage-correlation Chunming Chang Nick Russ Phys. Rev. A.,
XYZ file format Protein Data Bank (pdb) file format Z - matrix
 file format Z - matrix") Chemistry block (exercise 1) In this exercise, students will be introduced how to preform simple quantum chemical calculations. Input files for Gaussian09. Output file structure. Geometry optimization,
Chemistry block (exercise 1) In this exercise, students will be introduced how to preform simple quantum chemical calculations. Input files for Gaussian09. Output file structure. Geometry optimization,
ab initio Electronic Structure Calculations
 ab initio Electronic Structure Calculations New scalability frontiers using the BG/L Supercomputer C. Bekas, A. Curioni and W. Andreoni IBM, Zurich Research Laboratory Rueschlikon 8803, Switzerland ab
ab initio Electronic Structure Calculations New scalability frontiers using the BG/L Supercomputer C. Bekas, A. Curioni and W. Andreoni IBM, Zurich Research Laboratory Rueschlikon 8803, Switzerland ab
Nanotechnology and Solar Energy. Solar Electricity Photovoltaics. Fuel from the Sun Photosynthesis Biofuels Split Water Fuel Cells
 Nanotechnology and Solar Energy Solar Electricity Photovoltaics Fuel from the Sun Photosynthesis Biofuels Split Water Fuel Cells Solar cell A photon from the Sun generates an electron-hole pair in a semiconductor.
Nanotechnology and Solar Energy Solar Electricity Photovoltaics Fuel from the Sun Photosynthesis Biofuels Split Water Fuel Cells Solar cell A photon from the Sun generates an electron-hole pair in a semiconductor.
Chemistry 4560/5560 Molecular Modeling Fall 2014
 Final Exam Name:. User s guide: 1. Read questions carefully and make sure you understand them before answering (if not, ask). 2. Answer only the question that is asked, not a different question. 3. Unless
Final Exam Name:. User s guide: 1. Read questions carefully and make sure you understand them before answering (if not, ask). 2. Answer only the question that is asked, not a different question. 3. Unless
Walter Kohn was awarded with the Nobel Prize in Chemistry in 1998 for his development of the density functional theory.
 Walter Kohn was awarded with the Nobel Prize in Chemistry in 1998 for his development of the density functional theory. Walter Kohn receiving his Nobel Prize from His Majesty the King at the Stockholm
Walter Kohn was awarded with the Nobel Prize in Chemistry in 1998 for his development of the density functional theory. Walter Kohn receiving his Nobel Prize from His Majesty the King at the Stockholm
Compressed representation of Kohn-Sham orbitals via selected columns of the density matrix
 Lin Lin Compressed Kohn-Sham Orbitals 1 Compressed representation of Kohn-Sham orbitals via selected columns of the density matrix Lin Lin Department of Mathematics, UC Berkeley; Computational Research
Lin Lin Compressed Kohn-Sham Orbitals 1 Compressed representation of Kohn-Sham orbitals via selected columns of the density matrix Lin Lin Department of Mathematics, UC Berkeley; Computational Research
The Ab Initio Nanoreactor: Discovering Chemical Reaction Networks Todd J. Martínez Department of Chemistry & The PULSE Institute Stanford University
 The Ab Initio Nanoreactor: Discovering Chemical Reaction Networks Todd J. Martínez Department of Chemistry & The PULSE Institute Stanford University Traditional Approach to Reaction Mechanisms Traditional
The Ab Initio Nanoreactor: Discovering Chemical Reaction Networks Todd J. Martínez Department of Chemistry & The PULSE Institute Stanford University Traditional Approach to Reaction Mechanisms Traditional
André Schleife Department of Materials Science and Engineering
 André Schleife Department of Materials Science and Engineering Yesterday you (should have) learned this: http://upload.wikimedia.org/wikipedia/commons/e/ea/ Simple_Harmonic_Motion_Orbit.gif 1. deterministic
André Schleife Department of Materials Science and Engineering Yesterday you (should have) learned this: http://upload.wikimedia.org/wikipedia/commons/e/ea/ Simple_Harmonic_Motion_Orbit.gif 1. deterministic
Dispersion Interactions from the Exchange-Hole Dipole Moment
 Dispersion Interactions from the Exchange-Hole Dipole Moment Erin R. Johnson and Alberto Otero-de-la-Roza Chemistry and Chemical Biology, University of California, Merced E. R. Johnson (UC Merced) Dispersion
Dispersion Interactions from the Exchange-Hole Dipole Moment Erin R. Johnson and Alberto Otero-de-la-Roza Chemistry and Chemical Biology, University of California, Merced E. R. Johnson (UC Merced) Dispersion
Fast and accurate Coulomb calculation with Gaussian functions
 Fast and accurate Coulomb calculation with Gaussian functions László Füsti-Molnár and Jing Kong Q-CHEM Inc., Pittsburgh, Pennysylvania 15213 THE JOURNAL OF CHEMICAL PHYSICS 122, 074108 2005 Received 8
Fast and accurate Coulomb calculation with Gaussian functions László Füsti-Molnár and Jing Kong Q-CHEM Inc., Pittsburgh, Pennysylvania 15213 THE JOURNAL OF CHEMICAL PHYSICS 122, 074108 2005 Received 8
Effect of the High-k Dielectric/Semiconductor Interface on Electronic Properties in Ultra-thin Channels
 Effect of the High-k Dielectric/Semiconductor Interface on Electronic Properties in Ultra-thin Channels Evan Wilson, Daniel Valencia, Mark J. W. Rodwell, Gerhard Klimeck and Michael Povolotskyi Electrical
Effect of the High-k Dielectric/Semiconductor Interface on Electronic Properties in Ultra-thin Channels Evan Wilson, Daniel Valencia, Mark J. W. Rodwell, Gerhard Klimeck and Michael Povolotskyi Electrical
Accurate van der Waals interactions from ground state electron density
 Accurate van der Waals interactions from ground state electron density Alexandre Tkatchenko Theory Department, Fritz Haber Institut der MPG Berlin, Germany tkatchen@fhi berlin.mpg.de Haber Institute EXCITCM09,
Accurate van der Waals interactions from ground state electron density Alexandre Tkatchenko Theory Department, Fritz Haber Institut der MPG Berlin, Germany tkatchen@fhi berlin.mpg.de Haber Institute EXCITCM09,
Session 1. Introduction to Computational Chemistry. Computational (chemistry education) and/or (Computational chemistry) education
 and/or (Computational chemistry) education") Session 1 Introduction to Computational Chemistry 1 Introduction to Computational Chemistry Computational (chemistry education) and/or (Computational chemistry) education First one: Use computational tools
Session 1 Introduction to Computational Chemistry 1 Introduction to Computational Chemistry Computational (chemistry education) and/or (Computational chemistry) education First one: Use computational tools
Molecular dynamics simulation of Aquaporin-1. 4 nm
 Molecular dynamics simulation of Aquaporin-1 4 nm Molecular Dynamics Simulations Schrödinger equation i~@ t (r, R) =H (r, R) Born-Oppenheimer approximation H e e(r; R) =E e (R) e(r; R) Nucleic motion described
Molecular dynamics simulation of Aquaporin-1 4 nm Molecular Dynamics Simulations Schrödinger equation i~@ t (r, R) =H (r, R) Born-Oppenheimer approximation H e e(r; R) =E e (R) e(r; R) Nucleic motion described
Functionalized Carbon Nanotubes a key to nanotechnology?
 1 27th Max Born Symposium Multiscale Modeling of Real Materials Wroclaw, Sep 19, 2010 Functionalized Carbon Nanotubes a key to nanotechnology? Karolina Milowska, Magda Birowska & Jacek A. Majewski Faculty
1 27th Max Born Symposium Multiscale Modeling of Real Materials Wroclaw, Sep 19, 2010 Functionalized Carbon Nanotubes a key to nanotechnology? Karolina Milowska, Magda Birowska & Jacek A. Majewski Faculty
Many electrons: Density functional theory Part II. Bedřich Velický VI.
 Many electrons: Density functional theory Part II. Bedřich Velický velicky@karlov.mff.cuni.cz VI. NEVF 514 Surface Physics Winter Term 013-014 Troja 1 st November 013 This class is the second devoted to
Many electrons: Density functional theory Part II. Bedřich Velický velicky@karlov.mff.cuni.cz VI. NEVF 514 Surface Physics Winter Term 013-014 Troja 1 st November 013 This class is the second devoted to
Yuan Ping 1,2,3*, Robert J. Nielsen 1,2, William A. Goddard III 1,2*
 Supporting Information for the Reaction Mechanism with Free Energy Barriers at Constant Potentials for the Oxygen Evolution Reaction at the IrO2 (110) Surface Yuan Ping 1,2,3*, Robert J. Nielsen 1,2, William
Supporting Information for the Reaction Mechanism with Free Energy Barriers at Constant Potentials for the Oxygen Evolution Reaction at the IrO2 (110) Surface Yuan Ping 1,2,3*, Robert J. Nielsen 1,2, William
3rd Advanced in silico Drug Design KFC/ADD Molecular mechanics intro Karel Berka, Ph.D. Martin Lepšík, Ph.D. Pavel Polishchuk, Ph.D.
 3rd Advanced in silico Drug Design KFC/ADD Molecular mechanics intro Karel Berka, Ph.D. Martin Lepšík, Ph.D. Pavel Polishchuk, Ph.D. Thierry Langer, Ph.D. Jana Vrbková, Ph.D. UP Olomouc, 23.1.-26.1. 2018
3rd Advanced in silico Drug Design KFC/ADD Molecular mechanics intro Karel Berka, Ph.D. Martin Lepšík, Ph.D. Pavel Polishchuk, Ph.D. Thierry Langer, Ph.D. Jana Vrbková, Ph.D. UP Olomouc, 23.1.-26.1. 2018
OVERVIEW OF QUANTUM CHEMISTRY METHODS
 OVERVIEW OF QUANTUM CHEMISTRY METHODS Outline I Generalities Correlation, basis sets Spin II Wavefunction methods Hartree-Fock Configuration interaction Coupled cluster Perturbative methods III Density
OVERVIEW OF QUANTUM CHEMISTRY METHODS Outline I Generalities Correlation, basis sets Spin II Wavefunction methods Hartree-Fock Configuration interaction Coupled cluster Perturbative methods III Density
Probing the Origins of Intermolecular Vibrational and Relaxational Dynamics in Organic Solids with CP2K
 Probing the Origins of Intermolecular Vibrational and Relaxational Dynamics in Organic Solids with CP2K Michael Ruggiero Department of Chemical Engineering and Biotechnology, University of Cambridge CP2K
Probing the Origins of Intermolecular Vibrational and Relaxational Dynamics in Organic Solids with CP2K Michael Ruggiero Department of Chemical Engineering and Biotechnology, University of Cambridge CP2K
Fragmentation methods
 Fragmentation methods Scaling of QM Methods HF, DFT scale as N 4 MP2 scales as N 5 CC methods scale as N 7 What if we could freeze the value of N regardless of the size of the system? Then each method
Fragmentation methods Scaling of QM Methods HF, DFT scale as N 4 MP2 scales as N 5 CC methods scale as N 7 What if we could freeze the value of N regardless of the size of the system? Then each method
Pancake Bonding in 1,2,3,5-dithiadiazolyl and 1,2,3,5-diselenadiazolyl Radical Dimers and their Derivatives
 This journal is The Owner Societies 12 Bonds or Not Bonds? Electronic Supplementary Information Pancake Bonding in 1,2,3,-dithiadiazolyl and 1,2,3,-diselenadiazolyl Radical Dimers and their Derivatives
This journal is The Owner Societies 12 Bonds or Not Bonds? Electronic Supplementary Information Pancake Bonding in 1,2,3,-dithiadiazolyl and 1,2,3,-diselenadiazolyl Radical Dimers and their Derivatives
Introduction to Computer Simulations of Soft Matter Methodologies and Applications Boulder July, 19-20, 2012
 Introduction to Computer Simulations of Soft Matter Methodologies and Applications Boulder July, 19-20, 2012 K. Kremer Max Planck Institute for Polymer Research, Mainz Overview Simulations, general considerations
Introduction to Computer Simulations of Soft Matter Methodologies and Applications Boulder July, 19-20, 2012 K. Kremer Max Planck Institute for Polymer Research, Mainz Overview Simulations, general considerations
Ab Ini'o Molecular Dynamics (MD) Simula?ons
 Simula?ons") Ab Ini'o Molecular Dynamics (MD) Simula?ons Rick Remsing ICMS, CCDM, Temple University, Philadelphia, PA What are Molecular Dynamics (MD) Simulations? Technique to compute statistical and transport properties
Ab Ini'o Molecular Dynamics (MD) Simula?ons Rick Remsing ICMS, CCDM, Temple University, Philadelphia, PA What are Molecular Dynamics (MD) Simulations? Technique to compute statistical and transport properties
16 years ago TODAY (9/11) at 8:46, the first tower was hit at 9:03, the second tower was hit. Lecture 2 (9/11/17)
 at 8:46, the first tower was hit at 9:03, the second tower was hit. Lecture 2 (9/11/17)") 16 years ago TODAY (9/11) at 8:46, the first tower was hit at 9:03, the second tower was hit By Anthony Quintano - https://www.flickr.com/photos/quintanomedia/15071865580, CC BY 2.0, https://commons.wikimedia.org/w/index.php?curid=38538291
16 years ago TODAY (9/11) at 8:46, the first tower was hit at 9:03, the second tower was hit By Anthony Quintano - https://www.flickr.com/photos/quintanomedia/15071865580, CC BY 2.0, https://commons.wikimedia.org/w/index.php?curid=38538291
Joint ICTP-IAEA Workshop on Fusion Plasma Modelling using Atomic and Molecular Data January 2012
 2327-3 Joint ICTP-IAEA Workshop on Fusion Plasma Modelling using Atomic and Molecular Data 23-27 January 2012 Qunatum Methods for Plasma-Facing Materials Alain ALLOUCHE Univ.de Provence, Lab.de la Phys.
2327-3 Joint ICTP-IAEA Workshop on Fusion Plasma Modelling using Atomic and Molecular Data 23-27 January 2012 Qunatum Methods for Plasma-Facing Materials Alain ALLOUCHE Univ.de Provence, Lab.de la Phys.
Supporting information. Realizing Two-Dimensional Magnetic Semiconductors with. Enhanced Curie Temperature by Antiaromatic Ring Based
 Supporting information Realizing Two-Dimensional Magnetic Semiconductors with Enhanced Curie Temperature by Antiaromatic Ring Based Organometallic Frameworks Xingxing Li and Jinlong Yang* Department of
Supporting information Realizing Two-Dimensional Magnetic Semiconductors with Enhanced Curie Temperature by Antiaromatic Ring Based Organometallic Frameworks Xingxing Li and Jinlong Yang* Department of
Density Functional Theory: from theory to Applications
 Density Functional Theory: from theory to Applications Uni Mainz January 17, 2011 CP-PAW/COSMO Interface Mixed quantum/ classical molecular dynamics (QM/MM) Quantum mechanics is computationally expensive
Density Functional Theory: from theory to Applications Uni Mainz January 17, 2011 CP-PAW/COSMO Interface Mixed quantum/ classical molecular dynamics (QM/MM) Quantum mechanics is computationally expensive
Car-Parrinello Molecular Dynamics
 Car-Parrinello Molecular Dynamics Eric J. Bylaska HPCC Group Moral: A man dreams of a miracle and wakes up with loaves of bread Erich Maria Remarque Molecular Dynamics Loop (1) Compute Forces on atoms,
Car-Parrinello Molecular Dynamics Eric J. Bylaska HPCC Group Moral: A man dreams of a miracle and wakes up with loaves of bread Erich Maria Remarque Molecular Dynamics Loop (1) Compute Forces on atoms,