Humans have two copies of each chromosome. Inherited from mother and father. Genotyping technologies do not maintain the phase
|
|
|
- Gyles Hardy
- 5 years ago
- Views:
Transcription
1
2 Humans have two copies of each chromosome Inherited from mother and father. Genotyping technologies do not maintain the phase
3 Genotyping technologies do not maintain the phase
4 Recall that proximal SNPs are in LD. Few haplotypes must be observed Ex:T G G C is the best phasing of the 1st two columns This phasing is most reliable for short regions LD decays at 10kb
 Father Mother")
5 If: child is heterozygous, and a parent is homozygous, we know which allele comes from which parent. Location 1: A from mother, T from father Location 2: C from father, G from mother Child haplotypes (TC,AG) Father Mother A/T----C/C A/A----C/G A/T----C/G Child

6


7
8 A G A G C T T A G T A - - T G G G T C T A G A T - A T G C A A T G T G A T G A G A G C T A G C A T G A C T T T T G G T T C G C G The fragments are alligned to the unphased reference Uninformative fragments and columns are removed Tri- allelic SNP columns are removed. Relabel the two alleles using 0/1
9 A G A G C T A G C A T G A C T T T T G G T T C G C G A G A G C T T A G T A - - T G G G T C T A G A T - A T G C A A T G T G A T G A G A G C T A G C A T G A C T T T T G G T T C G C G Goal: Reconstruct the phased binary string, and its complement, given substrings
10 Consider a binary string, (and its complement)
11 The string is revealed to us only through a collection of substrings of the string, and its complement. Given the substrings, can the string be reconstructed?
12 The error free reconstruction is unique. The problem becomes much harder if some of the substrings have errors (do not match the consensus) DeYine MEC: Minimum # calls that need to be corrected for a match MEC reconstruction: Find the string that minimizes the MEC error. MEC reconstruction is NP- hard even when all fragments have length 2!
13 Greedily select a fragment that extends the current haplotype
14 Some fragments will not match without error. These are assigned & corrected greedily
15 A Greedy scheme such as this was employed for JCV s genome. MEC: The minimum number of base- calls that need to be Ylipped for an error free assignment. Goal: reconstruct a haplotype that minimizes MEC X! 1-0 X! X! X!
16 The Greedy approach often leads to suboptimal solutions A local Ylipping of the current haplotype might improve the MEC X! X! X! 1 - X! 0 X!
17 The haplotype change also involves a reassignment of fragments. Here, the MEC error reduced to 2. This suggests a generic strategy Start with a haplotype, and move to a new one if it can improve the MEC! X! X!
18
19 Error probability q We can compute the likelihood of X, given H,q Pr(X H,q) = The goal is to either compute H that maximizes likelihood, OR To sample H from Pr(X H,q) i Pr(X i H,q) H=(h,h) Haplotype H h: h: X 1 : X 2 : X 3 : X n : Fragment matrix X
20 H $ Pr[H H'] min& 1, % H Pr(X H',q) Pr(X H,q) ' ) (
21 A simple neighborhood is deyined by Ylipping one column at a time (Ex: col. 11) Waterman and Churchill S = {11}
22 While this Ylip- update markov chain has the right stationary distribution, it does not converge fast.
23 n columns, each spanned by d fragments. Two haplotypes (H 1,H 2 ) are equally likely Hard to move from one good haplotype to another d n columns
24 Let p=1- q. Mixing time bound based on conductance arguments (Jerrum & Sinclair 92) Similar empirical results for hitting time. Even modest values of n,d are problematic. # # Thm : Mixing time is Ω n p2 + q 2 % % $ $ 2 pq n=20 & ( ' d & (, '
, independent of d.")
25 If we modify the neighborhood to include the Yirst n/2 columns Thm: the mixing time of the Markov Chain is O(n 6 ), independent of d. Proof uses: Canonical Paths
26
27 The theoretical analysis on examples tells us: Choice of neighborhood (subsets of columns that are Ylipped) is important. Unfortunately, it is not easy to predict what the correct neighborhood should be for an arbitrary example
28 Each column is a node. (x,y) is an edge if there is a fragment touching columns x and y
29
30
31
32
33
34
35
36
37 The Hapcut algorithm allows us (in a heuristic sense) to escape a current local minimum. It can be modiyied to sample from the Haplotypes, instead of a single haplotype output.
38
39 1.856M variants used for haplotype assembly of huref (using an earlier version of the algorithm described here) Chr 22 stats 25K variant sites, 53K useful rows ~7 variant per fragment 609 disjoint haplotypes (largest contains 1008 variants) N50 haplotype length=350kb (50% of the variant sites lie in haplotypes 350kb or greater) Paired end sequencing is critical.

40 Greedy Hapcut HASH HASH/Hapcut have nearly identical performances HASH is slower but allows for sampling of multiple haplotypes Both offer >20% improvement over the naïve method

41

42 Errors were simulated on Huref sequences Switch error in reconstruction is low, and decreases with increased depth The computed MEC error tracks simulated errors

43 A switch w.r.t hapmap phasing is illustrative of either low LD, or HASH error
44 HASH mismatch rate is low ( ) Adjusted rate ( ) Hapmap error rate is (CEU) to 0.02 (YRI) even with trios Without trios, error is 0.05

45 Haplotyping results would be very poor with Next gen sequencing If 250,000 long reads were available, could we solve the assembly problem? Adjusted N50 200, , ,000 AN , Read Length Conclusion: Read- length helps, but we need very long reads to achieve reasonable N Read Length
46 Accuracy of haplotypes solved using computation (HASH/ Hapcut) Length of haplotypes: Described by the number of nodes in the graph Primarily a technology issue. Sanger sequencing: long haplotypes, prohibitively expensive Next Gen sequencing: Affordable, short haplotypes Single molecule sequencing longer reads reads, but, Ability to switch sequencing ON/ OFF in a strobe mode
47 Fix: Read Length, Coverage, Max Insert Size to keep cost constant Varying insert improves haplotype length AN Varying Advance Lengths % of advance lengths = 9000bp (100-x)% of advance lengths = 3000bp
48 Accuracy of haplotypes solved using computation (HASH/ Hapcut) Length of haplotypes: Described by the number of nodes in the graph Primarily a technology issue. Sanger sequencing: long haplotypes, prohibitively expensive Next Gen sequencing: Affordable, short haplotypes Single molecule sequencing longer reads reads, but, Ability to switch sequencing ON/ OFF in a strobe mode
49 We need to Yind a distribution (pdf) f(a) on advance length a that will optimize AN50 Q: Compute the shape that maximizes AN50 f(a) Advance length a
50 Optimization over different shapes is difyicult. Our solution: Use the beta distribution Pros: Only two parameters Cons: Considers a limited sampling of the space.



51 Choosing the right distribution of advance lengths Varying α, β parameters of the β- distribution Distribution of advance length for a=1.6 b= Distribution of advance length for a=0.6 b= AN Distribution of advance length for a=1 b= alpha beta
 in each tube Test each tube for heterozygozity Genotype each homozygous tube")
52 Isolate a single cell from suspension Use protease digestion to release all DNA from the cytoplasm (46 chromosomes) Randomly partition chromosomes into 48 chambers Multiple strand displacement ampliyication of single genomes (This is the tricky part) in each tube Test each tube for heterozygozity Genotype each homozygous tube
53 Tubes chr1 chr 1 X X Loci A small PCR based test is used to see which chromosome is in which tube (marked by X) How many marks per locus should we see? We can combine the content of two tubes if they do not contain homologous pairs. Finally, we genotype the remaining pairs/ sequence them.
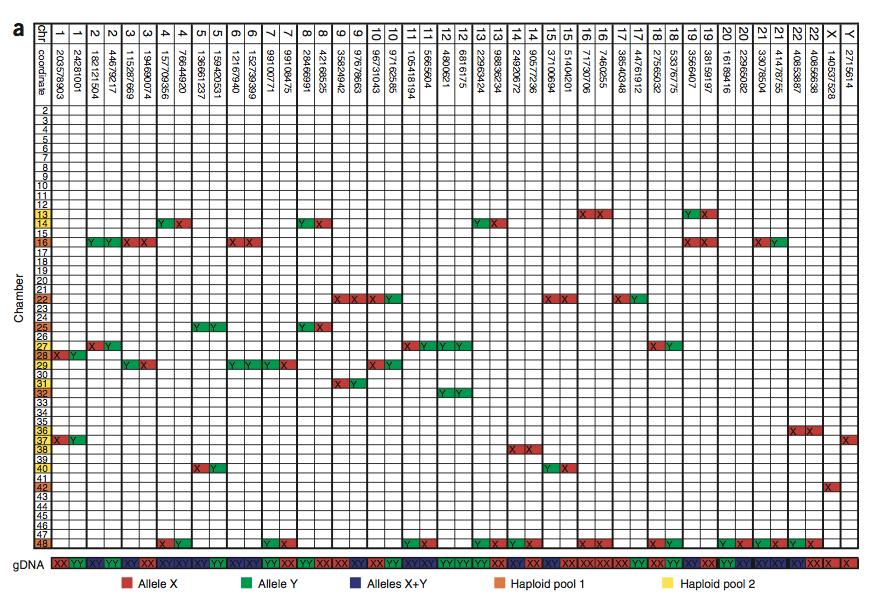
54
55 1. We started (and Yinished) by considering sources of variation 2. Models of evolution of population under natural assumptions 1. HW/Linkage equlibrium 2. EfYicient simulation of populations via coalescent theory 3. Detecting structural variation 4. Detection of regions under selection 5. Association testing 6. Population sub- structure 7. Haplotype phasing
56 1. Phylogeny reconstruction (perfect phylogeny, distance- based methods) 2. Optimization using (Integer- ) Linear programming, simulated annealing and other paradigms. Maximum Ylows. 3. Greedy algorithms, dynamic programming 4. Stochastic sampling methods (MCMC, Gibbs sampling) 5. Statistical tests for signiyicance 6. EfYicient simulation techniques 1. Coalescent for populations, including recombination, selection, etc. 2. Simulating genotype/phenotype associations
57 1. We started (and Yinished) by considering sources of variation 2. Models of evolution of population under natural assumptions 1. HW/Linkage equlibrium 2. EfYicient simulation of populations via coalescent theory 3. Detecting structural variation 4. Detection of regions under selection 5. Association testing 6. Population sub- structure 7. Evolution under recombinations/recombination hot- spot detection via counting of recombination events (partially) 8. Haplotype phasing (partially)
58 1. Phylogeny reconstruction (perfect phylogeny, distance- based methods) 2. Optimization using (Integer- ) Linear programming, simulated annealing and other paradigms 3. Greedy algorithms, dynamic programming 4. Stochastic sampling methods (MCMC, Gibbs sampling) 5. Statistical tests for signiyicance 6. EfYicient simulation techniques 1. Coalescent for populations 2. Simulating genotype/phenotype associations 3. Pairwise analysis
1.5.1 ESTIMATION OF HAPLOTYPE FREQUENCIES:
 .5. ESTIMATION OF HAPLOTYPE FREQUENCIES: Chapter - 8 For SNPs, alleles A j,b j at locus j there are 4 haplotypes: A A, A B, B A and B B frequencies q,q,q 3,q 4. Assume HWE at haplotype level. Only the
.5. ESTIMATION OF HAPLOTYPE FREQUENCIES: Chapter - 8 For SNPs, alleles A j,b j at locus j there are 4 haplotypes: A A, A B, B A and B B frequencies q,q,q 3,q 4. Assume HWE at haplotype level. Only the
Estimating Recombination Rates. LRH selection test, and recombination
 Estimating Recombination Rates LRH selection test, and recombination Recall that LRH tests for selection by looking at frequencies of specific haplotypes. Clearly the test is dependent on the recombination
Estimating Recombination Rates LRH selection test, and recombination Recall that LRH tests for selection by looking at frequencies of specific haplotypes. Clearly the test is dependent on the recombination
Genotype Imputation. Class Discussion for January 19, 2016
 Genotype Imputation Class Discussion for January 19, 2016 Intuition Patterns of genetic variation in one individual guide our interpretation of the genomes of other individuals Imputation uses previously
Genotype Imputation Class Discussion for January 19, 2016 Intuition Patterns of genetic variation in one individual guide our interpretation of the genomes of other individuals Imputation uses previously
Complexity and Approximation of the Minimum Recombination Haplotype Configuration Problem
 Complexity and Approximation of the Minimum Recombination Haplotype Configuration Problem Lan Liu 1, Xi Chen 3, Jing Xiao 3, and Tao Jiang 1,2 1 Department of Computer Science and Engineering, University
Complexity and Approximation of the Minimum Recombination Haplotype Configuration Problem Lan Liu 1, Xi Chen 3, Jing Xiao 3, and Tao Jiang 1,2 1 Department of Computer Science and Engineering, University
Solutions to Even-Numbered Exercises to accompany An Introduction to Population Genetics: Theory and Applications Rasmus Nielsen Montgomery Slatkin
 Solutions to Even-Numbered Exercises to accompany An Introduction to Population Genetics: Theory and Applications Rasmus Nielsen Montgomery Slatkin CHAPTER 1 1.2 The expected homozygosity, given allele
Solutions to Even-Numbered Exercises to accompany An Introduction to Population Genetics: Theory and Applications Rasmus Nielsen Montgomery Slatkin CHAPTER 1 1.2 The expected homozygosity, given allele
Computational statistics
 Computational statistics Combinatorial optimization Thierry Denœux February 2017 Thierry Denœux Computational statistics February 2017 1 / 37 Combinatorial optimization Assume we seek the maximum of f
Computational statistics Combinatorial optimization Thierry Denœux February 2017 Thierry Denœux Computational statistics February 2017 1 / 37 Combinatorial optimization Assume we seek the maximum of f
Haplotyping as Perfect Phylogeny: A direct approach
 Haplotyping as Perfect Phylogeny: A direct approach Vineet Bafna Dan Gusfield Giuseppe Lancia Shibu Yooseph February 7, 2003 Abstract A full Haplotype Map of the human genome will prove extremely valuable
Haplotyping as Perfect Phylogeny: A direct approach Vineet Bafna Dan Gusfield Giuseppe Lancia Shibu Yooseph February 7, 2003 Abstract A full Haplotype Map of the human genome will prove extremely valuable
Phasing via the Expectation Maximization (EM) Algorithm
 Algorithm") Computing Haplotype Frequencies and Haplotype Phasing via the Expectation Maximization (EM) Algorithm Department of Computer Science Brown University, Providence sorin@cs.brown.edu September 14, 2010 Outline
Computing Haplotype Frequencies and Haplotype Phasing via the Expectation Maximization (EM) Algorithm Department of Computer Science Brown University, Providence sorin@cs.brown.edu September 14, 2010 Outline
theta H H H H H H H H H H H K K K K K K K K K K centimorgans
 Linkage Phase Recall that the recombination fraction ρ for two loci denotes the probability of a recombination event between those two loci. For loci on different chromosomes, ρ = 1=2. For loci on the
Linkage Phase Recall that the recombination fraction ρ for two loci denotes the probability of a recombination event between those two loci. For loci on different chromosomes, ρ = 1=2. For loci on the
The E-M Algorithm in Genetics. Biostatistics 666 Lecture 8
 The E-M Algorithm in Genetics Biostatistics 666 Lecture 8 Maximum Likelihood Estimation of Allele Frequencies Find parameter estimates which make observed data most likely General approach, as long as
The E-M Algorithm in Genetics Biostatistics 666 Lecture 8 Maximum Likelihood Estimation of Allele Frequencies Find parameter estimates which make observed data most likely General approach, as long as
On the Fixed Parameter Tractability and Approximability of the Minimum Error Correction problem
 On the Fixed Parameter Tractability and Approximability of the Minimum Error Correction problem Paola Bonizzoni, Riccardo Dondi, Gunnar W. Klau, Yuri Pirola, Nadia Pisanti and Simone Zaccaria DISCo, computer
On the Fixed Parameter Tractability and Approximability of the Minimum Error Correction problem Paola Bonizzoni, Riccardo Dondi, Gunnar W. Klau, Yuri Pirola, Nadia Pisanti and Simone Zaccaria DISCo, computer
Calculation of IBD probabilities
 Calculation of IBD probabilities David Evans University of Bristol This Session Identity by Descent (IBD) vs Identity by state (IBS) Why is IBD important? Calculating IBD probabilities Lander-Green Algorithm
Calculation of IBD probabilities David Evans University of Bristol This Session Identity by Descent (IBD) vs Identity by state (IBS) Why is IBD important? Calculating IBD probabilities Lander-Green Algorithm
CSci 8980: Advanced Topics in Graphical Models Analysis of Genetic Variation
 CSci 8980: Advanced Topics in Graphical Models Analysis of Genetic Variation Instructor: Arindam Banerjee November 26, 2007 Genetic Polymorphism Single nucleotide polymorphism (SNP) Genetic Polymorphism
CSci 8980: Advanced Topics in Graphical Models Analysis of Genetic Variation Instructor: Arindam Banerjee November 26, 2007 Genetic Polymorphism Single nucleotide polymorphism (SNP) Genetic Polymorphism
1. Understand the methods for analyzing population structure in genomes
 MSCBIO 2070/02-710: Computational Genomics, Spring 2016 HW3: Population Genetics Due: 24:00 EST, April 4, 2016 by autolab Your goals in this assignment are to 1. Understand the methods for analyzing population
MSCBIO 2070/02-710: Computational Genomics, Spring 2016 HW3: Population Genetics Due: 24:00 EST, April 4, 2016 by autolab Your goals in this assignment are to 1. Understand the methods for analyzing population
Introduction to Linkage Disequilibrium
 Introduction to September 10, 2014 Suppose we have two genes on a single chromosome gene A and gene B such that each gene has only two alleles Aalleles : A 1 and A 2 Balleles : B 1 and B 2 Suppose we have
Introduction to September 10, 2014 Suppose we have two genes on a single chromosome gene A and gene B such that each gene has only two alleles Aalleles : A 1 and A 2 Balleles : B 1 and B 2 Suppose we have
Allen Holder - Trinity University
 Haplotyping - Trinity University Population Problems - joint with Courtney Davis, University of Utah Single Individuals - joint with John Louie, Carrol College, and Lena Sherbakov, Williams University
Haplotyping - Trinity University Population Problems - joint with Courtney Davis, University of Utah Single Individuals - joint with John Louie, Carrol College, and Lena Sherbakov, Williams University
Population Genetics. with implications for Linkage Disequilibrium. Chiara Sabatti, Human Genetics 6357a Gonda
 1 Population Genetics with implications for Linkage Disequilibrium Chiara Sabatti, Human Genetics 6357a Gonda csabatti@mednet.ucla.edu 2 Hardy-Weinberg Hypotheses: infinite populations; no inbreeding;
1 Population Genetics with implications for Linkage Disequilibrium Chiara Sabatti, Human Genetics 6357a Gonda csabatti@mednet.ucla.edu 2 Hardy-Weinberg Hypotheses: infinite populations; no inbreeding;
Homework Assignment, Evolutionary Systems Biology, Spring Homework Part I: Phylogenetics:
 Homework Assignment, Evolutionary Systems Biology, Spring 2009. Homework Part I: Phylogenetics: Introduction. The objective of this assignment is to understand the basics of phylogenetic relationships
Homework Assignment, Evolutionary Systems Biology, Spring 2009. Homework Part I: Phylogenetics: Introduction. The objective of this assignment is to understand the basics of phylogenetic relationships
2. Map genetic distance between markers
 Chapter 5. Linkage Analysis Linkage is an important tool for the mapping of genetic loci and a method for mapping disease loci. With the availability of numerous DNA markers throughout the human genome,
Chapter 5. Linkage Analysis Linkage is an important tool for the mapping of genetic loci and a method for mapping disease loci. With the availability of numerous DNA markers throughout the human genome,
Genotype Imputation. Biostatistics 666
 Genotype Imputation Biostatistics 666 Previously Hidden Markov Models for Relative Pairs Linkage analysis using affected sibling pairs Estimation of pairwise relationships Identity-by-Descent Relatives
Genotype Imputation Biostatistics 666 Previously Hidden Markov Models for Relative Pairs Linkage analysis using affected sibling pairs Estimation of pairwise relationships Identity-by-Descent Relatives
Approximate Counting and Markov Chain Monte Carlo
 Approximate Counting and Markov Chain Monte Carlo A Randomized Approach Arindam Pal Department of Computer Science and Engineering Indian Institute of Technology Delhi March 18, 2011 April 8, 2011 Arindam
Approximate Counting and Markov Chain Monte Carlo A Randomized Approach Arindam Pal Department of Computer Science and Engineering Indian Institute of Technology Delhi March 18, 2011 April 8, 2011 Arindam
The Quantitative TDT
 The Quantitative TDT (Quantitative Transmission Disequilibrium Test) Warren J. Ewens NUS, Singapore 10 June, 2009 The initial aim of the (QUALITATIVE) TDT was to test for linkage between a marker locus
The Quantitative TDT (Quantitative Transmission Disequilibrium Test) Warren J. Ewens NUS, Singapore 10 June, 2009 The initial aim of the (QUALITATIVE) TDT was to test for linkage between a marker locus
Tutorial Session 2. MCMC for the analysis of genetic data on pedigrees:
 MCMC for the analysis of genetic data on pedigrees: Tutorial Session 2 Elizabeth Thompson University of Washington Genetic mapping and linkage lod scores Monte Carlo likelihood and likelihood ratio estimation
MCMC for the analysis of genetic data on pedigrees: Tutorial Session 2 Elizabeth Thompson University of Washington Genetic mapping and linkage lod scores Monte Carlo likelihood and likelihood ratio estimation
A Phylogenetic Network Construction due to Constrained Recombination
 A Phylogenetic Network Construction due to Constrained Recombination Mohd. Abdul Hai Zahid Research Scholar Research Supervisors: Dr. R.C. Joshi Dr. Ankush Mittal Department of Electronics and Computer
A Phylogenetic Network Construction due to Constrained Recombination Mohd. Abdul Hai Zahid Research Scholar Research Supervisors: Dr. R.C. Joshi Dr. Ankush Mittal Department of Electronics and Computer
Linkage and Linkage Disequilibrium
 Linkage and Linkage Disequilibrium Summer Institute in Statistical Genetics 2014 Module 10 Topic 3 Linkage in a simple genetic cross Linkage In the early 1900 s Bateson and Punnet conducted genetic studies
Linkage and Linkage Disequilibrium Summer Institute in Statistical Genetics 2014 Module 10 Topic 3 Linkage in a simple genetic cross Linkage In the early 1900 s Bateson and Punnet conducted genetic studies
Effect of Genetic Divergence in Identifying Ancestral Origin using HAPAA
 Effect of Genetic Divergence in Identifying Ancestral Origin using HAPAA Andreas Sundquist*, Eugene Fratkin*, Chuong B. Do, Serafim Batzoglou Department of Computer Science, Stanford University, Stanford,
Effect of Genetic Divergence in Identifying Ancestral Origin using HAPAA Andreas Sundquist*, Eugene Fratkin*, Chuong B. Do, Serafim Batzoglou Department of Computer Science, Stanford University, Stanford,
Calculation of IBD probabilities
 Calculation of IBD probabilities David Evans and Stacey Cherny University of Oxford Wellcome Trust Centre for Human Genetics This Session IBD vs IBS Why is IBD important? Calculating IBD probabilities
Calculation of IBD probabilities David Evans and Stacey Cherny University of Oxford Wellcome Trust Centre for Human Genetics This Session IBD vs IBS Why is IBD important? Calculating IBD probabilities
Optimal Haplotype Assembly via a Branch-and-Bound Algorithm
 DRAFT: OPTIMAL HAPLOTYPE ASSEMBLY VIA A BRANCH-AND-BOUND ALGORITHM 1 Optimal Haplotype Assembly via a Branch-and-Bound Algorithm Shreepriya Das and Haris Vikalo, Senior Member, IEEE Abstract Haplotype
DRAFT: OPTIMAL HAPLOTYPE ASSEMBLY VIA A BRANCH-AND-BOUND ALGORITHM 1 Optimal Haplotype Assembly via a Branch-and-Bound Algorithm Shreepriya Das and Haris Vikalo, Senior Member, IEEE Abstract Haplotype
3.4 Relaxations and bounds
 3.4 Relaxations and bounds Consider a generic Discrete Optimization problem z = min{c(x) : x X} with an optimal solution x X. In general, the algorithms generate not only a decreasing sequence of upper
3.4 Relaxations and bounds Consider a generic Discrete Optimization problem z = min{c(x) : x X} with an optimal solution x X. In general, the algorithms generate not only a decreasing sequence of upper
Statistical issues in QTL mapping in mice
 Statistical issues in QTL mapping in mice Karl W Broman Department of Biostatistics Johns Hopkins University http://www.biostat.jhsph.edu/~kbroman Outline Overview of QTL mapping The X chromosome Mapping
Statistical issues in QTL mapping in mice Karl W Broman Department of Biostatistics Johns Hopkins University http://www.biostat.jhsph.edu/~kbroman Outline Overview of QTL mapping The X chromosome Mapping
The genomes of recombinant inbred lines
 The genomes of recombinant inbred lines Karl W Broman Department of Biostatistics Johns Hopkins University http://www.biostat.jhsph.edu/~kbroman C57BL/6 2 1 Recombinant inbred lines (by sibling mating)
The genomes of recombinant inbred lines Karl W Broman Department of Biostatistics Johns Hopkins University http://www.biostat.jhsph.edu/~kbroman C57BL/6 2 1 Recombinant inbred lines (by sibling mating)
Efficient Haplotype Inference with Boolean Satisfiability
 Efficient Haplotype Inference with Boolean Satisfiability Joao Marques-Silva 1 and Ines Lynce 2 1 School of Electronics and Computer Science University of Southampton 2 INESC-ID/IST Technical University
Efficient Haplotype Inference with Boolean Satisfiability Joao Marques-Silva 1 and Ines Lynce 2 1 School of Electronics and Computer Science University of Southampton 2 INESC-ID/IST Technical University
Supplementary Information for Discovery and characterization of indel and point mutations
 Supplementary Information for Discovery and characterization of indel and point mutations using DeNovoGear Avinash Ramu 1 Michiel J. Noordam 1 Rachel S. Schwartz 2 Arthur Wuster 3 Matthew E. Hurles 3 Reed
Supplementary Information for Discovery and characterization of indel and point mutations using DeNovoGear Avinash Ramu 1 Michiel J. Noordam 1 Rachel S. Schwartz 2 Arthur Wuster 3 Matthew E. Hurles 3 Reed
Chapter 6 Linkage Disequilibrium & Gene Mapping (Recombination)
") 12/5/14 Chapter 6 Linkage Disequilibrium & Gene Mapping (Recombination) Linkage Disequilibrium Genealogical Interpretation of LD Association Mapping 1 Linkage and Recombination v linkage equilibrium ²
12/5/14 Chapter 6 Linkage Disequilibrium & Gene Mapping (Recombination) Linkage Disequilibrium Genealogical Interpretation of LD Association Mapping 1 Linkage and Recombination v linkage equilibrium ²
A graph contains a set of nodes (vertices) connected by links (edges or arcs)
 connected by links (edges or arcs)") BOLTZMANN MACHINES Generative Models Graphical Models A graph contains a set of nodes (vertices) connected by links (edges or arcs) In a probabilistic graphical model, each node represents a random variable,
BOLTZMANN MACHINES Generative Models Graphical Models A graph contains a set of nodes (vertices) connected by links (edges or arcs) In a probabilistic graphical model, each node represents a random variable,
Hidden Markov Dirichlet Process: Modeling Genetic Recombination in Open Ancestral Space
 Hidden Markov Dirichlet Process: Modeling Genetic Recombination in Open Ancestral Space Kyung-Ah Sohn School of Computer Science Carnegie Mellon University Pittsburgh, PA 15213 ksohn@cs.cmu.edu Eric P.
Hidden Markov Dirichlet Process: Modeling Genetic Recombination in Open Ancestral Space Kyung-Ah Sohn School of Computer Science Carnegie Mellon University Pittsburgh, PA 15213 ksohn@cs.cmu.edu Eric P.
Linear Regression (1/1/17)
") STA613/CBB540: Statistical methods in computational biology Linear Regression (1/1/17) Lecturer: Barbara Engelhardt Scribe: Ethan Hada 1. Linear regression 1.1. Linear regression basics. Linear regression
STA613/CBB540: Statistical methods in computational biology Linear Regression (1/1/17) Lecturer: Barbara Engelhardt Scribe: Ethan Hada 1. Linear regression 1.1. Linear regression basics. Linear regression
SNP Association Studies with Case-Parent Trios
 SNP Association Studies with Case-Parent Trios Department of Biostatistics Johns Hopkins Bloomberg School of Public Health September 3, 2009 Population-based Association Studies Balding (2006). Nature
SNP Association Studies with Case-Parent Trios Department of Biostatistics Johns Hopkins Bloomberg School of Public Health September 3, 2009 Population-based Association Studies Balding (2006). Nature
Chromosome Chr Duplica Duplic t a ion Pixley
 Chromosome Duplication Pixley Figure 4-6 Molecular Biology of the Cell ( Garland Science 2008) Figure 4-72 Molecular Biology of the Cell ( Garland Science 2008) Interphase During mitosis (cell division),
Chromosome Duplication Pixley Figure 4-6 Molecular Biology of the Cell ( Garland Science 2008) Figure 4-72 Molecular Biology of the Cell ( Garland Science 2008) Interphase During mitosis (cell division),
Nature Genetics: doi:0.1038/ng.2768
 Supplementary Figure 1: Graphic representation of the duplicated region at Xq28 in each one of the 31 samples as revealed by acgh. Duplications are represented in red and triplications in blue. Top: Genomic
Supplementary Figure 1: Graphic representation of the duplicated region at Xq28 in each one of the 31 samples as revealed by acgh. Duplications are represented in red and triplications in blue. Top: Genomic
De novo assembly and genotyping of variants using colored de Bruijn graphs
 De novo assembly and genotyping of variants using colored de Bruijn graphs Iqbal et al. 2012 Kolmogorov Mikhail 2013 Challenges Detecting genetic variants that are highly divergent from a reference Detecting
De novo assembly and genotyping of variants using colored de Bruijn graphs Iqbal et al. 2012 Kolmogorov Mikhail 2013 Challenges Detecting genetic variants that are highly divergent from a reference Detecting
Supporting Information
 Supporting Information Hammer et al. 10.1073/pnas.1109300108 SI Materials and Methods Two-Population Model. Estimating demographic parameters. For each pair of sub-saharan African populations we consider
Supporting Information Hammer et al. 10.1073/pnas.1109300108 SI Materials and Methods Two-Population Model. Estimating demographic parameters. For each pair of sub-saharan African populations we consider
Case-Control Association Testing. Case-Control Association Testing
 Introduction Association mapping is now routinely being used to identify loci that are involved with complex traits. Technological advances have made it feasible to perform case-control association studies
Introduction Association mapping is now routinely being used to identify loci that are involved with complex traits. Technological advances have made it feasible to perform case-control association studies
Stochastic processes and
 Stochastic processes and Markov chains (part II) Wessel van Wieringen w.n.van.wieringen@vu.nl wieringen@vu nl Department of Epidemiology and Biostatistics, VUmc & Department of Mathematics, VU University
Stochastic processes and Markov chains (part II) Wessel van Wieringen w.n.van.wieringen@vu.nl wieringen@vu nl Department of Epidemiology and Biostatistics, VUmc & Department of Mathematics, VU University
Frequency Spectra and Inference in Population Genetics
 Frequency Spectra and Inference in Population Genetics Although coalescent models have come to play a central role in population genetics, there are some situations where genealogies may not lead to efficient
Frequency Spectra and Inference in Population Genetics Although coalescent models have come to play a central role in population genetics, there are some situations where genealogies may not lead to efficient
Sampling Good Motifs with Markov Chains
 Sampling Good Motifs with Markov Chains Chris Peikert December 10, 2004 Abstract Markov chain Monte Carlo (MCMC) techniques have been used with some success in bioinformatics [LAB + 93]. However, these
Sampling Good Motifs with Markov Chains Chris Peikert December 10, 2004 Abstract Markov chain Monte Carlo (MCMC) techniques have been used with some success in bioinformatics [LAB + 93]. However, these
6 Markov Chain Monte Carlo (MCMC)
") 6 Markov Chain Monte Carlo (MCMC) The underlying idea in MCMC is to replace the iid samples of basic MC methods, with dependent samples from an ergodic Markov chain, whose limiting (stationary) distribution
6 Markov Chain Monte Carlo (MCMC) The underlying idea in MCMC is to replace the iid samples of basic MC methods, with dependent samples from an ergodic Markov chain, whose limiting (stationary) distribution
Lesson 4: Understanding Genetics
 Lesson 4: Understanding Genetics 1 Terms Alleles Chromosome Co dominance Crossover Deoxyribonucleic acid DNA Dominant Genetic code Genome Genotype Heredity Heritability Heritability estimate Heterozygous
Lesson 4: Understanding Genetics 1 Terms Alleles Chromosome Co dominance Crossover Deoxyribonucleic acid DNA Dominant Genetic code Genome Genotype Heredity Heritability Heritability estimate Heterozygous
Fei Lu. Post doctoral Associate Cornell University
 Fei Lu Post doctoral Associate Cornell University http://www.maizegenetics.net Genotyping by sequencing (GBS) is simple and cost effective 1. Digest DNA 2. Ligate adapters with barcodes 3. Pool DNAs 4.
Fei Lu Post doctoral Associate Cornell University http://www.maizegenetics.net Genotyping by sequencing (GBS) is simple and cost effective 1. Digest DNA 2. Ligate adapters with barcodes 3. Pool DNAs 4.
Algorithms in Bioinformatics FOUR Pairwise Sequence Alignment. Pairwise Sequence Alignment. Convention: DNA Sequences 5. Sequence Alignment
 Algorithms in Bioinformatics FOUR Sami Khuri Department of Computer Science San José State University Pairwise Sequence Alignment Homology Similarity Global string alignment Local string alignment Dot
Algorithms in Bioinformatics FOUR Sami Khuri Department of Computer Science San José State University Pairwise Sequence Alignment Homology Similarity Global string alignment Local string alignment Dot
Markov Chains and MCMC
 Markov Chains and MCMC Markov chains Let S = {1, 2,..., N} be a finite set consisting of N states. A Markov chain Y 0, Y 1, Y 2,... is a sequence of random variables, with Y t S for all points in time
Markov Chains and MCMC Markov chains Let S = {1, 2,..., N} be a finite set consisting of N states. A Markov chain Y 0, Y 1, Y 2,... is a sequence of random variables, with Y t S for all points in time
CS 781 Lecture 9 March 10, 2011 Topics: Local Search and Optimization Metropolis Algorithm Greedy Optimization Hopfield Networks Max Cut Problem Nash
 CS 781 Lecture 9 March 10, 2011 Topics: Local Search and Optimization Metropolis Algorithm Greedy Optimization Hopfield Networks Max Cut Problem Nash Equilibrium Price of Stability Coping With NP-Hardness
CS 781 Lecture 9 March 10, 2011 Topics: Local Search and Optimization Metropolis Algorithm Greedy Optimization Hopfield Networks Max Cut Problem Nash Equilibrium Price of Stability Coping With NP-Hardness
The Wright-Fisher Model and Genetic Drift
 The Wright-Fisher Model and Genetic Drift January 22, 2015 1 1 Hardy-Weinberg Equilibrium Our goal is to understand the dynamics of allele and genotype frequencies in an infinite, randomlymating population
The Wright-Fisher Model and Genetic Drift January 22, 2015 1 1 Hardy-Weinberg Equilibrium Our goal is to understand the dynamics of allele and genotype frequencies in an infinite, randomlymating population
Quiz Section 4 Molecular analysis of inheritance: An amphibian puzzle
 Genome 371, Autumn 2018 Quiz Section 4 Molecular analysis of inheritance: An amphibian puzzle Goals: To illustrate how molecular tools can be used to track inheritance. In this particular example, we will
Genome 371, Autumn 2018 Quiz Section 4 Molecular analysis of inheritance: An amphibian puzzle Goals: To illustrate how molecular tools can be used to track inheritance. In this particular example, we will
Chapter 8: Introduction to Evolutionary Computation
 Computational Intelligence: Second Edition Contents Some Theories about Evolution Evolution is an optimization process: the aim is to improve the ability of an organism to survive in dynamically changing
Computational Intelligence: Second Edition Contents Some Theories about Evolution Evolution is an optimization process: the aim is to improve the ability of an organism to survive in dynamically changing
Gene mapping in model organisms
 Gene mapping in model organisms Karl W Broman Department of Biostatistics Johns Hopkins University http://www.biostat.jhsph.edu/~kbroman Goal Identify genes that contribute to common human diseases. 2
Gene mapping in model organisms Karl W Broman Department of Biostatistics Johns Hopkins University http://www.biostat.jhsph.edu/~kbroman Goal Identify genes that contribute to common human diseases. 2
Chapter 13 Meiosis and Sexual Reproduction
 Biology 110 Sec. 11 J. Greg Doheny Chapter 13 Meiosis and Sexual Reproduction Quiz Questions: 1. What word do you use to describe a chromosome or gene allele that we inherit from our Mother? From our Father?
Biology 110 Sec. 11 J. Greg Doheny Chapter 13 Meiosis and Sexual Reproduction Quiz Questions: 1. What word do you use to describe a chromosome or gene allele that we inherit from our Mother? From our Father?
Integer Programming in Computational Biology. D. Gusfield University of California, Davis Presented December 12, 2016.!
 Integer Programming in Computational Biology D. Gusfield University of California, Davis Presented December 12, 2016. There are many important phylogeny problems that depart from simple tree models: Missing
Integer Programming in Computational Biology D. Gusfield University of California, Davis Presented December 12, 2016. There are many important phylogeny problems that depart from simple tree models: Missing
Bayesian Inference of Interactions and Associations
 Bayesian Inference of Interactions and Associations Jun Liu Department of Statistics Harvard University http://www.fas.harvard.edu/~junliu Based on collaborations with Yu Zhang, Jing Zhang, Yuan Yuan,
Bayesian Inference of Interactions and Associations Jun Liu Department of Statistics Harvard University http://www.fas.harvard.edu/~junliu Based on collaborations with Yu Zhang, Jing Zhang, Yuan Yuan,
Modelling Genetic Variations with Fragmentation-Coagulation Processes
 Modelling Genetic Variations with Fragmentation-Coagulation Processes Yee Whye Teh, Charles Blundell, Lloyd Elliott Gatsby Computational Neuroscience Unit, UCL Genetic Variations in Populations Inferring
Modelling Genetic Variations with Fragmentation-Coagulation Processes Yee Whye Teh, Charles Blundell, Lloyd Elliott Gatsby Computational Neuroscience Unit, UCL Genetic Variations in Populations Inferring
CMPS 6630: Introduction to Computational Biology and Bioinformatics. Sequence Assembly
 CMPS 6630: Introduction to Computational Biology and Bioinformatics Sequence Assembly Why Genome Sequencing? Sanger (1982) introduced chaintermination sequencing. Main idea: Obtain fragments of all possible
CMPS 6630: Introduction to Computational Biology and Bioinformatics Sequence Assembly Why Genome Sequencing? Sanger (1982) introduced chaintermination sequencing. Main idea: Obtain fragments of all possible
Population Genetics I. Bio
 Population Genetics I. Bio5488-2018 Don Conrad dconrad@genetics.wustl.edu Why study population genetics? Functional Inference Demographic inference: History of mankind is written in our DNA. We can learn
Population Genetics I. Bio5488-2018 Don Conrad dconrad@genetics.wustl.edu Why study population genetics? Functional Inference Demographic inference: History of mankind is written in our DNA. We can learn
CSE 190, Great ideas in algorithms: Pairwise independent hash functions
 CSE 190, Great ideas in algorithms: Pairwise independent hash functions 1 Hash functions The goal of hash functions is to map elements from a large domain to a small one. Typically, to obtain the required
CSE 190, Great ideas in algorithms: Pairwise independent hash functions 1 Hash functions The goal of hash functions is to map elements from a large domain to a small one. Typically, to obtain the required
For 5% confidence χ 2 with 1 degree of freedom should exceed 3.841, so there is clear evidence for disequilibrium between S and M.
 STAT 550 Howework 6 Anton Amirov 1. This question relates to the same study you saw in Homework-4, by Dr. Arno Motulsky and coworkers, and published in Thompson et al. (1988; Am.J.Hum.Genet, 42, 113-124).
STAT 550 Howework 6 Anton Amirov 1. This question relates to the same study you saw in Homework-4, by Dr. Arno Motulsky and coworkers, and published in Thompson et al. (1988; Am.J.Hum.Genet, 42, 113-124).
Processes of Evolution
 15 Processes of Evolution Forces of Evolution Concept 15.4 Selection Can Be Stabilizing, Directional, or Disruptive Natural selection can act on quantitative traits in three ways: Stabilizing selection
15 Processes of Evolution Forces of Evolution Concept 15.4 Selection Can Be Stabilizing, Directional, or Disruptive Natural selection can act on quantitative traits in three ways: Stabilizing selection
The Monte Carlo Method
 The Monte Carlo Method Example: estimate the value of π. Choose X and Y independently and uniformly at random in [0, 1]. Let Pr(Z = 1) = π 4. 4E[Z] = π. { 1 if X Z = 2 + Y 2 1, 0 otherwise, Let Z 1,...,
The Monte Carlo Method Example: estimate the value of π. Choose X and Y independently and uniformly at random in [0, 1]. Let Pr(Z = 1) = π 4. 4E[Z] = π. { 1 if X Z = 2 + Y 2 1, 0 otherwise, Let Z 1,...,
Properties of normal phylogenetic networks
 Properties of normal phylogenetic networks Stephen J. Willson Department of Mathematics Iowa State University Ames, IA 50011 USA swillson@iastate.edu August 13, 2009 Abstract. A phylogenetic network is
Properties of normal phylogenetic networks Stephen J. Willson Department of Mathematics Iowa State University Ames, IA 50011 USA swillson@iastate.edu August 13, 2009 Abstract. A phylogenetic network is
Probabilistic Methods for Single Individual Haplotype Reconstruction: HapTree and HapTree-X. Emily Rita Berger
 Probabilistic Methods for Single Individual Haplotype Reconstruction: HapTree and HapTree-X by Emily Rita Berger Adissertationsubmittedinpartialsatisfactionofthe requirements for the degree of Doctor of
Probabilistic Methods for Single Individual Haplotype Reconstruction: HapTree and HapTree-X by Emily Rita Berger Adissertationsubmittedinpartialsatisfactionofthe requirements for the degree of Doctor of
CPSC 540: Machine Learning
 CPSC 540: Machine Learning More Approximate Inference Mark Schmidt University of British Columbia Winter 2018 Last Time: Approximate Inference We ve been discussing graphical models for density estimation,
CPSC 540: Machine Learning More Approximate Inference Mark Schmidt University of British Columbia Winter 2018 Last Time: Approximate Inference We ve been discussing graphical models for density estimation,
EM algorithm. Rather than jumping into the details of the particular EM algorithm, we ll look at a simpler example to get the idea of how it works
 EM algorithm The example in the book for doing the EM algorithm is rather difficult, and was not available in software at the time that the authors wrote the book, but they implemented a SAS macro to implement
EM algorithm The example in the book for doing the EM algorithm is rather difficult, and was not available in software at the time that the authors wrote the book, but they implemented a SAS macro to implement
Statistical Genetics I: STAT/BIOST 550 Spring Quarter, 2014
 Overview - 1 Statistical Genetics I: STAT/BIOST 550 Spring Quarter, 2014 Elizabeth Thompson University of Washington Seattle, WA, USA MWF 8:30-9:20; THO 211 Web page: www.stat.washington.edu/ thompson/stat550/
Overview - 1 Statistical Genetics I: STAT/BIOST 550 Spring Quarter, 2014 Elizabeth Thompson University of Washington Seattle, WA, USA MWF 8:30-9:20; THO 211 Web page: www.stat.washington.edu/ thompson/stat550/
Stat 516, Homework 1
 Stat 516, Homework 1 Due date: October 7 1. Consider an urn with n distinct balls numbered 1,..., n. We sample balls from the urn with replacement. Let N be the number of draws until we encounter a ball
Stat 516, Homework 1 Due date: October 7 1. Consider an urn with n distinct balls numbered 1,..., n. We sample balls from the urn with replacement. Let N be the number of draws until we encounter a ball
Life Cycles, Meiosis and Genetic Variability24/02/2015 2:26 PM
 Life Cycles, Meiosis and Genetic Variability iclicker: 1. A chromosome just before mitosis contains two double stranded DNA molecules. 2. This replicated chromosome contains DNA from only one of your parents
Life Cycles, Meiosis and Genetic Variability iclicker: 1. A chromosome just before mitosis contains two double stranded DNA molecules. 2. This replicated chromosome contains DNA from only one of your parents
Learning gene regulatory networks Statistical methods for haplotype inference Part I
 Learning gene regulatory networks Statistical methods for haplotype inference Part I Input: Measurement of mrn levels of all genes from microarray or rna sequencing Samples (e.g. 200 patients with lung
Learning gene regulatory networks Statistical methods for haplotype inference Part I Input: Measurement of mrn levels of all genes from microarray or rna sequencing Samples (e.g. 200 patients with lung
I519 Introduction to Bioinformatics, Genome Comparison. Yuzhen Ye School of Informatics & Computing, IUB
 I519 Introduction to Bioinformatics, 2011 Genome Comparison Yuzhen Ye (yye@indiana.edu) School of Informatics & Computing, IUB Whole genome comparison/alignment Build better phylogenies Identify polymorphism
I519 Introduction to Bioinformatics, 2011 Genome Comparison Yuzhen Ye (yye@indiana.edu) School of Informatics & Computing, IUB Whole genome comparison/alignment Build better phylogenies Identify polymorphism
CINQA Workshop Probability Math 105 Silvia Heubach Department of Mathematics, CSULA Thursday, September 6, 2012
 CINQA Workshop Probability Math 105 Silvia Heubach Department of Mathematics, CSULA Thursday, September 6, 2012 Silvia Heubach/CINQA 2012 Workshop Objectives To familiarize biology faculty with one of
CINQA Workshop Probability Math 105 Silvia Heubach Department of Mathematics, CSULA Thursday, September 6, 2012 Silvia Heubach/CINQA 2012 Workshop Objectives To familiarize biology faculty with one of
The Pure Parsimony Problem
 Haplotyping and Minimum Diversity Graphs Courtney Davis - University of Utah - Trinity University Some Genetics Mother Paired Gene Representation Physical Trait ABABBA AAABBB Physical Trait ABA AAA Mother
Haplotyping and Minimum Diversity Graphs Courtney Davis - University of Utah - Trinity University Some Genetics Mother Paired Gene Representation Physical Trait ABABBA AAABBB Physical Trait ABA AAA Mother
This course is about VARIATION: its causes, effects, and history.
 This course is about VARIATION: its causes, effects, and history. For thousands of years, western thought had accepted the Platonic view that an object s ultimate reality was its essence or ideal type.
This course is about VARIATION: its causes, effects, and history. For thousands of years, western thought had accepted the Platonic view that an object s ultimate reality was its essence or ideal type.
Computational Methods for Learning Population History from Large Scale Genetic Variation Datasets
 Computational Methods for Learning Population History from Large Scale Genetic Variation Datasets Ming-Chi Tsai CMU-CB-13-102 July 2, 2013 School of Computer Science Carnegie Mellon University Pittsburgh,
Computational Methods for Learning Population History from Large Scale Genetic Variation Datasets Ming-Chi Tsai CMU-CB-13-102 July 2, 2013 School of Computer Science Carnegie Mellon University Pittsburgh,
Patterns of inheritance
 Patterns of inheritance Learning goals By the end of this material you would have learnt about: How traits and characteristics are passed on from one generation to another The different patterns of inheritance
Patterns of inheritance Learning goals By the end of this material you would have learnt about: How traits and characteristics are passed on from one generation to another The different patterns of inheritance
An Overview of Combinatorial Methods for Haplotype Inference
 An Overview of Combinatorial Methods for Haplotype Inference Dan Gusfield 1 Department of Computer Science, University of California, Davis Davis, CA. 95616 Abstract A current high-priority phase of human
An Overview of Combinatorial Methods for Haplotype Inference Dan Gusfield 1 Department of Computer Science, University of California, Davis Davis, CA. 95616 Abstract A current high-priority phase of human
Sampling from Bayes Nets
 from Bayes Nets http://www.youtube.com/watch?v=mvrtaljp8dm http://www.youtube.com/watch?v=geqip_0vjec Paper reviews Should be useful feedback for the authors A critique of the paper No paper is perfect!
from Bayes Nets http://www.youtube.com/watch?v=mvrtaljp8dm http://www.youtube.com/watch?v=geqip_0vjec Paper reviews Should be useful feedback for the authors A critique of the paper No paper is perfect!
Notes on Population Genetics
 Notes on Population Genetics Graham Coop 1 1 Department of Evolution and Ecology & Center for Population Biology, University of California, Davis. To whom correspondence should be addressed: gmcoop@ucdavis.edu
Notes on Population Genetics Graham Coop 1 1 Department of Evolution and Ecology & Center for Population Biology, University of California, Davis. To whom correspondence should be addressed: gmcoop@ucdavis.edu
STA 4273H: Statistical Machine Learning
 STA 4273H: Statistical Machine Learning Russ Salakhutdinov Department of Statistics! rsalakhu@utstat.toronto.edu! http://www.utstat.utoronto.ca/~rsalakhu/ Sidney Smith Hall, Room 6002 Lecture 11 Project
STA 4273H: Statistical Machine Learning Russ Salakhutdinov Department of Statistics! rsalakhu@utstat.toronto.edu! http://www.utstat.utoronto.ca/~rsalakhu/ Sidney Smith Hall, Room 6002 Lecture 11 Project
Learning ancestral genetic processes using nonparametric Bayesian models
 Learning ancestral genetic processes using nonparametric Bayesian models Kyung-Ah Sohn October 31, 2011 Committee Members: Eric P. Xing, Chair Zoubin Ghahramani Russell Schwartz Kathryn Roeder Matthew
Learning ancestral genetic processes using nonparametric Bayesian models Kyung-Ah Sohn October 31, 2011 Committee Members: Eric P. Xing, Chair Zoubin Ghahramani Russell Schwartz Kathryn Roeder Matthew
An Efficient and Accurate Graph-Based Approach to Detect Population Substructure
 An Efficient and Accurate Graph-Based Approach to Detect Population Substructure Srinath Sridhar, Satish Rao and Eran Halperin Abstract. Currently, large-scale projects are underway to perform whole genome
An Efficient and Accurate Graph-Based Approach to Detect Population Substructure Srinath Sridhar, Satish Rao and Eran Halperin Abstract. Currently, large-scale projects are underway to perform whole genome
Meiosis and Mendel. Chapter 6
 Meiosis and Mendel Chapter 6 6.1 CHROMOSOMES AND MEIOSIS Key Concept Gametes have half the number of chromosomes that body cells have. Body Cells vs. Gametes You have body cells and gametes body cells
Meiosis and Mendel Chapter 6 6.1 CHROMOSOMES AND MEIOSIS Key Concept Gametes have half the number of chromosomes that body cells have. Body Cells vs. Gametes You have body cells and gametes body cells
A Markov Chain Model for Haplotype Assembly from SNP Fragments
 162 Genome Informatics 17(2): 162{171 (2006) A Markov Chain Model for Haplotype Assembly from SNP Fragments Rui-Sheng Wang 1;2 Ling-Yun Wu 3 wangrsh@amss.ac.cn wlyun@amt.ac.cn Xiang-Sun Zhang 3* Luonan
162 Genome Informatics 17(2): 162{171 (2006) A Markov Chain Model for Haplotype Assembly from SNP Fragments Rui-Sheng Wang 1;2 Ling-Yun Wu 3 wangrsh@amss.ac.cn wlyun@amt.ac.cn Xiang-Sun Zhang 3* Luonan
On Approximating An Implicit Cover Problem in Biology
 On Approximating An Implicit Cover Problem in Biology Mary V. Ashley 1, Tanya Y. Berger-Wolf 2, Wanpracha Chaovalitwongse 3, Bhaskar DasGupta 2, Ashfaq Khokhar 2, and Saad Sheikh 2 1 Department of Biological
On Approximating An Implicit Cover Problem in Biology Mary V. Ashley 1, Tanya Y. Berger-Wolf 2, Wanpracha Chaovalitwongse 3, Bhaskar DasGupta 2, Ashfaq Khokhar 2, and Saad Sheikh 2 1 Department of Biological
Sequence comparison by compression
 Sequence comparison by compression Motivation similarity as a marker for homology. And homology is used to infer function. Sometimes, we are only interested in a numerical distance between two sequences.
Sequence comparison by compression Motivation similarity as a marker for homology. And homology is used to infer function. Sometimes, we are only interested in a numerical distance between two sequences.
Affected Sibling Pairs. Biostatistics 666
 Affected Sibling airs Biostatistics 666 Today Discussion of linkage analysis using affected sibling pairs Our exploration will include several components we have seen before: A simple disease model IBD
Affected Sibling airs Biostatistics 666 Today Discussion of linkage analysis using affected sibling pairs Our exploration will include several components we have seen before: A simple disease model IBD
Haploid & diploid recombination and their evolutionary impact
 Haploid & diploid recombination and their evolutionary impact W. Garrett Mitchener College of Charleston Mathematics Department MitchenerG@cofc.edu http://mitchenerg.people.cofc.edu Introduction The basis
Haploid & diploid recombination and their evolutionary impact W. Garrett Mitchener College of Charleston Mathematics Department MitchenerG@cofc.edu http://mitchenerg.people.cofc.edu Introduction The basis
Fragment Assembly of DNA
 Wright State University CORE Scholar Computer Science and Engineering Faculty Publications Computer Science and Engineering 2003 Fragment Assembly of DNA Dan E. Krane Wright State University - Main Campus,
Wright State University CORE Scholar Computer Science and Engineering Faculty Publications Computer Science and Engineering 2003 Fragment Assembly of DNA Dan E. Krane Wright State University - Main Campus,
Name Class Date. KEY CONCEPT Gametes have half the number of chromosomes that body cells have.
 Section 1: Chromosomes and Meiosis KEY CONCEPT Gametes have half the number of chromosomes that body cells have. VOCABULARY somatic cell autosome fertilization gamete sex chromosome diploid homologous
Section 1: Chromosomes and Meiosis KEY CONCEPT Gametes have half the number of chromosomes that body cells have. VOCABULARY somatic cell autosome fertilization gamete sex chromosome diploid homologous
An applied statistician does probability:
 An applied statistician does probability: It s not pretty Karl W Broman Department of Biostatistics & Medical Informatics University of Wisconsin Madison www.biostat.wisc.edu/~kbroman An applied statistician
An applied statistician does probability: It s not pretty Karl W Broman Department of Biostatistics & Medical Informatics University of Wisconsin Madison www.biostat.wisc.edu/~kbroman An applied statistician
A PARSIMONY APPROACH TO ANALYSIS OF HUMAN SEGMENTAL DUPLICATIONS
 A PARSIMONY APPROACH TO ANALYSIS OF HUMAN SEGMENTAL DUPLICATIONS CRYSTAL L. KAHN and BENJAMIN J. RAPHAEL Box 1910, Brown University Department of Computer Science & Center for Computational Molecular Biology
A PARSIMONY APPROACH TO ANALYSIS OF HUMAN SEGMENTAL DUPLICATIONS CRYSTAL L. KAHN and BENJAMIN J. RAPHAEL Box 1910, Brown University Department of Computer Science & Center for Computational Molecular Biology
CMPS 6630: Introduction to Computational Biology and Bioinformatics. Structure Comparison
 CMPS 6630: Introduction to Computational Biology and Bioinformatics Structure Comparison Protein Structure Comparison Motivation Understand sequence and structure variability Understand Domain architecture
CMPS 6630: Introduction to Computational Biology and Bioinformatics Structure Comparison Protein Structure Comparison Motivation Understand sequence and structure variability Understand Domain architecture
Equivalence relations
 Equivalence relations R A A is an equivalence relation if R is 1. reflexive (a, a) R 2. symmetric, and (a, b) R (b, a) R 3. transitive. (a, b), (b, c) R (a, c) R Example: Let S be a relation on people
Equivalence relations R A A is an equivalence relation if R is 1. reflexive (a, a) R 2. symmetric, and (a, b) R (b, a) R 3. transitive. (a, b), (b, c) R (a, c) R Example: Let S be a relation on people
CSCE 750 Final Exam Answer Key Wednesday December 7, 2005
 CSCE 750 Final Exam Answer Key Wednesday December 7, 2005 Do all problems. Put your answers on blank paper or in a test booklet. There are 00 points total in the exam. You have 80 minutes. Please note
CSCE 750 Final Exam Answer Key Wednesday December 7, 2005 Do all problems. Put your answers on blank paper or in a test booklet. There are 00 points total in the exam. You have 80 minutes. Please note
Full file at CHAPTER 2 Genetics
 CHAPTER 2 Genetics MULTIPLE CHOICE 1. Chromosomes are a. small linear bodies. b. contained in cells. c. replicated during cell division. 2. A cross between true-breeding plants bearing yellow seeds produces
CHAPTER 2 Genetics MULTIPLE CHOICE 1. Chromosomes are a. small linear bodies. b. contained in cells. c. replicated during cell division. 2. A cross between true-breeding plants bearing yellow seeds produces