Toward molecular switches and biochemical detectors employing adaptive femtosecond-scale laser pulses
|
|
|
- Amberly Lee
- 5 years ago
- Views:
Transcription
1 Toward molecular switches and biochemical detectors employing adaptive femtosecond-scale laser pulses Petra Sauer and Roland E. Allen, Institute for Quantum Studies and Physics Department, Texas A&M University Cis to trans (and trans to cis) photoisomerization of azobenzene, with nuclear motion allowing extra electronic transitions for pulse durations > about 50 fs UV or visible UV or visible Photoinduced ring-opening (and ring-closing) in a model dithienylethene visible UV! Response of dipicolinic acid to femtosecond-scale laser pulses, including excited states and nuclear motion (with Yuri Rostovtsev)







2 Representative applications: azobenzene Holographic data storage Light-driven molecular switch Photoelectrochemical Storage Liquid Crystal Display Instigator for Protein Folding Nanomechanical Device Droplet Driver Control of Protein Activity 2
3 One major result from these simulations -- we find that photochemical reactions involve: multiple steps (e.g., first S 0 S 1, and subsequently S 0 S 2 because nuclei move, or first S 2 S 1 and then S 1 S 0 for later de-excitation of the molecule) multiple excitations (e.g., linear combination of S 0, S 1, S 2, S 3 ) multiple configuration coordinates (e.g., rotational dihedral angle and length of central bond) In the present picture, with configuration interaction neglected, S 0 is the ground state S 1 is the excitation of one electron from HOMO to LUMO (highest occupied molecular orbital to lowest unoccupied molecular orbital) S 2 is the excitation of one electron from HOMO-1 to LUMO (from next-to-highest occupied molecular orbital) S 3 is the excitation of two electrons from HOMO to LUMO 3
4 We call our technique semiclassical electron-radiation-ion dynamics (SERID) to emphasize the limiting approximations: The radiation field and nuclear motion are both treated classically, and the ion cores are regarded as inert (with only the valence electrons included in the dynamics). Recall, however, that one still observes n-photon and n-phonon processes in a semiclassical treatment. Also, the nuclear motion is treated correctly on reasonably short time scales (e.g., picoseconds). List of approximations in present calculations: 1. The atomic nuclei are treated as positive point charges. 2. The treatment is nonrelativistic. 3. The nuclear motion is treated classically. 4. The radiation field is treated classically. 5. The electrons are treated in a time-dependent mean-field picture, with many-body effects neglected. 6. Only the valence electrons are treated, with the inner electrons plus nucleus represented as an inert core. 7. The basis functions are atomic orbitals. 8. The spin-orbit interaction, exchange interaction, and other spin-dependent interactions are neglected. 9. The calculations are not explicitly self-consistent,and there is no explicit Coulomb repulsion. 10. The Hamiltonian matrix elements and core-core repulsion are determined from density-functional calculations involving pairs of atoms. 11. The Peierls substitution is used to couple the electrons to the radiation field The influence of a larger environment is not included.
5 Density-functional-based parameterization of the electronic Hamiltonian H and the effective repulsive potential U: D. Porezag, Th. Frauenheim, et al., Phys. Rev B 51, (1991) P. Sauer and R. E. Allen, J. Mod. Optics 53, 2619 (2006) 5
6 6
7 Ehrenfest's theorem states that M d 2 X! dt 2 =! "H "X! where X! is the operator corresponding to any nuclear coordinate, M is the corresponding mass, and H is the full Hamiltonian for all particles in the molecule. If the electrons are treated in a time-dependent self-consistent field picture, and the one-electron wavefunctions are represented in terms of localized basis functions, one obtains M d 2 X! =! "# $H $#" % t " dt 2 j ( t ( ) )# $X! #" "# ( j t )! $U rep $X!. j Since quantum fluctuations, in the present context, are typically smaller than 0.05 Å for C and smaller than 0.10 Å even for H, it is a reasonable approximation to expand in a Taylor series about the expectation value:!x $ =!H!"# " t $ #!X $!H!"# ( t) ( ) % ' +!H!"# " t $ &!X $ X $ = X $ # ( ) ( ) ) + O ( X $ ( X $ ) 2 + % ' X $ ( X $ & X $ = X $ With X! X!, and with terms that are second-order in the quantum fluctuations neglected, we then take the expectation value to obtain the equation used for the nuclear motion: ( ) M d 2 X =!!" $H #"! % t " dt 2 j ( t)# $X #"!" j t j * ( )! $U rep $X., -.. 7
 ~ 1.00 ev C 2!")
8 photoisomerization of azobenzene ΔΕ cis to trans ~ 0.50 ev cis E(TS) ~ 1.00 ev C 2!! trans C 2h UV or visible UV or visible 8
9 What is already known about the isomerization process: Schematic of electronic energy levels for cis and trans forms S 2 S 1 n!! *!!! * S 0 Photoisomerization quantum yields for S 1 >> than for S 2 [P. Bortolus and S. Monti, J. Phys. Chem. 83, 648 (1979)]. Dominant isomerization product from cis state forms within 170 fs, and from trans state within 320 fs [T. Naegele et al., Chem. Phys. Lett. 272, 489 (1997)]. First configuration interaction calculations indicated an inversion pathway for S 1 excitation (which involves opening a CNN bond angle, with planar motion) and a rotation pathway for S 2 excitation (which involves rotation through the central CNNC dihedral angle) [S. Monti et al, Chem. Phys. 71, 87 (1982)]. Later calculations suggest that rotation pathway is most probable mechanism for both [T. Ishikawa et al, J. Chem Phys. 115, (2001)].
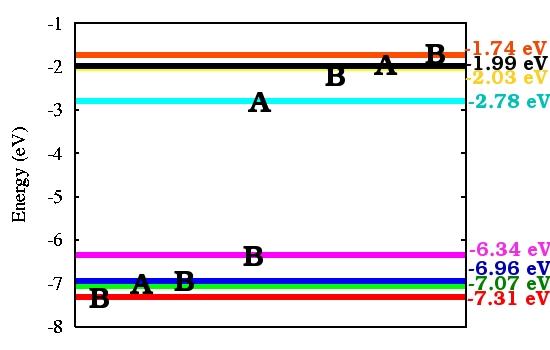
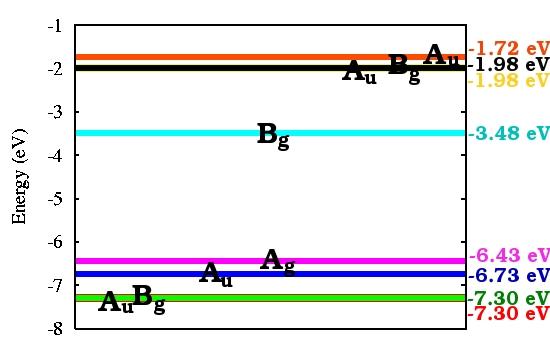


10 SERID calculated molecular orbital structure CIS LUMO HOMO HOMO-1 LUMO TRANS HOMO-1 HOMO 10
11 SERID results for S 1 excitation: electron dynamics during excitation Prepare state with an ultrashort 10 fs (FWHM) pulse, so that ion core motion during excitation is minimal, and transition is thus predominantly HOMO to LUMO. molecular orbital population molecular orbital energy laser pulse duration (FWHM) = 10 fs, photon energy = 3.55 ev, fluence = 0.28 kj/m 2, T = 0 K 11

12 Electron dynamics following HOMO to LUMO excitation molecular orbital population molecular orbital energy 75 fs Between ~ 50 and 100 fs, a series of avoided crossings leads to the transfer of ~ 0.6 electrons downward from LUMO to HOMO. 12

13 SERID results for S 1 excitation: nuclear dynamics The SERID simulations indicate rotational pathway rather than inversion pathway for S 1 excitation. 13
14 SERID results for S 2 excitation: electron dynamics during excitation Prepare excited state with an ultrashort 10 fs (FWHM) pulse, so that nuclear motion during excitation is minimal, and transition is predominantly HOMO-1 to LUMO. molecular orbital population molecular orbital energy laser pulse duration (FWHM) = 10 fs, photon energy = 4.18 ev, fluence = 0.15 kj/m 2, T = 0 K 14


15 Electron dynamics following HOMO-1 to LUMO excitation molecular orbital population molecular orbital energy 780 fs 900 fs At about 780 fs, a HOMO-1/HOMO avoided crossing leads to a transfer of roughly 0.60 holes up from the HOMO-1 into the HOMO. A subsequent HOMO/LUMO avoided crossing at about 900 fs then 15 transfers ~ 0.7 electrons down from the LUMO into the HOMO.

16 SERID results for S 2 excitation: nuclear dynamics The SERID simulations indicate rotational mechanism for S 2 excitation, occurring in the excited state, followed by return to the ground state. 16
17 Excitation with 100 fs (FWHM) pulse: electron dynamics during excitation Longer pulse initially excites electrons from HOMO to LUMO. As nuclei move, electrons are excited from HOMO-1 to LUMO. molecular orbital population molecular orbital energy laser pulse duration (FWHM) = 100 fs, photon energy = 3.55 ev, fluence = 0.47 kj/m 2, T = 0K 17
18 Excitation with 100 fs (FWHM) pulse: nuclear dynamics during excitation 18


19 Excitation with 100 fs (FWHM) pulse: electron dynamics after excitation molecular orbital population molecular orbital energy 260 fs 340 fs At about 260 fs, an avoided crossing between HOMO-1 and HOMO leads to a transfer of roughly 0.70 holes up into the HOMO. A subsequent series of HOMO-LUMO avoided crossings, between 340 fs and 550 fs, transfer ~ 0.3 electrons down into the HOMO. 19

20 Excitation with 100 fs (FWHM) pulse: nuclear dynamics after excitation Depopulation of the excited states occurs as the isomerization proceeds along a rotational pathway. 20
21 Summary for azobenzene isomerization An ultrashort 10 fs (FWHM) pulse matched to the HOMO-LUMO energy gap excites electrons from HOMO to LUMO. Isomerization proceeds along a rotational pathway. An avoided crossing between HOMO and LUMO, approximately halfway between cis and trans geometries, transfers the electronic population back into the ground state. An ultrashort 10 fs (FWHM) pulse, matched to the HOMO-1 to LUMO energy gap, excites electrons predominantly from HOMO-1 to LUMO. Isomerization again proceeds along a rotational pathway. An avoided crossing between HOMO-1 and HOMO near the trans configuration transfers hole population from HOMO-1 to HOMO. A later avoided crossing between HOMO and LUMO transfers electrons to the HOMO. A longer 100 fs (FWHM) laser pulse matched to the HOMO-LUMO energy gap excites electrons from both HOMO-1 and HOMO into the LUMO. The second excitation is due to nuclear motion during pulse excitation. An avoided crossing between HOMO-1 and HOMO near the cis configuration transfers hole population from HOMO-1 to HOMO. A subsequent HOMO-LUMO avoided crossing halfway between cis and trans configurations transfers electron population back into the ground state. 21

22 Lower fluence no isomerization: electron dynamics population during excitation molecular orbital energy during excitation population after excitation molecular orbital energy after excitation No HOMO-1/HOMO avoided crossing => no transfer of holes from HOMO-1 to HOMO. Then high occupancy of HOMO => no transfer of electrons from 22 LUMO to HOMO, despite HOMO/LUMO avoided crossings.
23 Lower fluence no isomerization: nuclear dynamics C a -N-N-C a during excitation C b -C a -N-N during excitation C a -N-N-C a after excitation C b -C a -N-N after excitation Similar behavior as with more intense laser pulse, except amplitude of oscillations not as large. Molecule never reaches geometry 23 required for HOMO-1/HOMO avoided crossing.
24 dithienylethene model system Both forms have C 2 symmetry. closed open barrier height much larger than openclosed energy difference visible UV 24



25 SERID calculated molecular orbital structure CLOSED ΔΕ g =1.53 ev HOMO LUMO OPEN ΔΕ g =3.14 ev HOMO LUMO 25

26 Electron dynamics during excitation Initial excitation is from HOMO into LUMO. Nuclear motion subsequently permits additional excitations out of HOMO-1 and into LUMO+1. orbital population orbital energy pulse duration (FWHM) = 150 fs,!! = 1.8 ev, F = 0.44 kj/m 2, T = 300 K 26

27 Nuclear dynamics during excitation C c -C c bond length C b -C a -C a -C b angle C a -C a bond length C c -C C c bondlength a -C b bond length 27


28 Electron dynamics leading to bond breakage Orbital Population Orbital Energy At ~ 785 fs: electron transfer from LUMO+1 to LUMO At ~ 960 fs: hole transfer from HOMO-1 to HOMO and electron transfer from LUMO+1 to LUMO 28


29 Electron dynamics following bond breakage orbital population orbital energy Between ~ 1 ps and 4 ps, a series of avoided crossings between HOMO and LUMO return molecule to the ground state. 29
30 Nuclear dynamics following excitation C c -C c bond length C b -C a -C a -C b angle Bond breaks at ~ 1 ps Ring opening, followed by rotation about central dihedral angle, until molecule returns to ground state at ~ 4 ps. 30


31 Nuclear dynamics following excitation (continued) C a -C a bond length C a -C b bond length tendency toward double bond after final HOMO-LUMO exchange of electron occupancy tendency toward single bond after final HOMO-LUMO exchange 31
32 Summary of ring-opening mechanism Laser pulse, matched to the HOMO-LUMO energy difference, initially excites electrons from HOMO to LUMO. Nuclear motion subsequently permits excitations out of HOMO-1 and into LUMO+1. After laser pulse excitation, an avoided crossing between HOMO-1 and HOMO transfers holes into the HOMO. An avoided crossing between LUMO+1 and LUMO transfers electrons into LUMO. Following these population transfers, ring-opening occurs at ~ 1 ps. A series of HOMO-LUMO avoided crossings, which occur between ~ 1ps and 4 ps, transfer electronic population back to the ground state. 32
33 dipicolinic acid (DPA): electronic and vibrational response 33

34 Bacterial Spore Cortex Coat Core S. Kozuka, Y. Yasud and K. Tochikubo, J. Bacteriol. 162, 1250 (1985) Between ~ 5 % and 15 % of dry spore weight (in compounds) 34
35 Overview of the parameterization scheme for nitrogen and oxygen We scale the density-functional-based hydrogen and carbon pairwise interactions to include interactions with nitrogen and oxygen. I. Electronic Properties II. Parameterization scheme consists of two parts: Diagonal Hamiltonian matrix elements (H ii ) scaled to the s and p atomic energies of the new atom Diagonal elements of the overlap matrix (S ii ) unchanged (equal to 1) Off-diagonal elements of the Hamiltonian scaled so that the energy eigenvalue difference of the active space region equals the energy difference calculated using DFT. The off-diagonal elements of the overlap matrix are left unchanged. Structural Properties The repulsive potential is scaled to match the structural properties (i.e. bond lengths and vibrational frequencies) of the diatomic molecule corresponding to a given pairwise interaction. 35
36 Parameterization scheme: electronic properties Schematic illustration for parameterizing nitrogen-nitrogen interactions starting with carbon-carbon interactions: C 2 (N 2 ) H ij = i H j and S ij = i j eigenvalues of!"# #" $ #" H!" = # S!" 36
37 Parameterization scheme: structural properties We then scale the effective nuclear repulsive potential: V new (r new ) = (c 0 + c 1 r)v old (r old ). The constants c 0 and c 1 are determined from the equilibrium conditions ( de rep dr + de elec dr ) r =r 0 = 0 ( d 2 E rep and dr 2 + d 2 E elec dr 2 ) r =r0 = M! 2 where the derivatives of the electronic energy are determined numerically from the scaled Hamiltonian and the overlap matrix:!e elec!x = % " n (!H!x # $!S n!x )" f (n) n n - /! 2 E elec = 2 /!x 2 / /./, n % ' ' ' ' & 2 %!H " n!x # $!S ( ( n & '!x ) * ", m * $ n # $ f (n) * m * * ) m+n,$m +$ n %! 2 H + " n!x # 2!$ n, & ' 2!x n!s!x # $ n 0 2! 2 S (!x 2 ) * " f (n) 2 n
38 Results of the parameterization for various diatomics Molecule Scale Factor Property Exp. SERID Molecule Scale Factor Property Exp. SERID N 2 CN O 2 CO NO r o (Å) ω (cm -1 ) D 0 (ev) ΔE g (ev) r o (Å) ω (cm -1 ) D 0 (ev) ΔE g (ev) r o (Å) ω (cm -1 ) D 0 (ev) ΔE g (ev) r o (Å) ω (cm -1 ) D 0 (ev) ΔE g (ev) r o (Å) ω (cm -1 ) D 0 (ev) ΔE g (ev) NH OH r o (Å) ω (cm -1 ) D 0 (ev) ΔE g (ev) r o (Å) ω (cm -1 ) D 0 (ev) ΔE g (ev) In all cases the experimental bond lengths and frequencies were taken from the CRC Handbook of Chem. and Phys. (2004) online, the binding energies from Herzberg, Spectra of Diatomic Molecules (1989) and the excitation energies from a B3LYP//augccpVQZ calculation, except for the hydride bond lengths, which were taken from Sun et al, J. Chem Phys. 74, 6842 (1981), and the excitation energy of N2, which was calculated without diffuse functions. 38
39 Parameterization applied to the equilibrium states of the azabenzenes Molecule Bond Exp. (Å) SERID (Å) pyrazine 1 C-C C-N C-H pyrazine pyridine pyridazine pyrimidine pyridazine 2 pyridine 2 pyrimidine 1 N-N N-C a C a -C b C b -C b C a -H C b -H N-C a C a -C b C b -C c C a -H a C b -H b C c -H c C a -N C a -N C b -C c C a -H a C b -H b C c -H c Cradoc et. al., J. Am. Chem. Soc. (1988). 2 Hannay et. al., J. Chem. Phys. (1999). 3 Haettig et. al., J. Chem. Phys. (1998). 4 Osamura et. al., J. Am. Chem. Soc. (1987). tetrazine 3 C-N N-N C-H tetrazine s-triazine triazine 4 C-N C-H
40 Parameterization applied to the equilibrium states of various oxygen-containing molecules Molecule Bond SERID (Å) Exp. (Å) CO 2 1 C-O acetaldehyde acetone acetic acid ethanol H 2 O 1 Acetaldehyde 1 Acetic Acid 2 Furan 1 Acetone 1 O-H C a -O C a -C b C a -H C b -H C-C C-O a C-O b C-H O b -H C b -C b C a -C b C a -O C-H C-C C-O C-H CRC Handbook of Chem. and Physics (2004) online. 2 Herzberg, Spectra of Diatomic Molecules (1989) furan Ethanol 2 C a -C b O-H C a -O C a -H C b -H
41 Summary of Parameterization Scheme Hamiltonian and overlap matrix elements scaled to the electronic properties for the relevant pairwise interaction. Effective nuclear repulsion then scaled to the equilibrium bond lengths and vibrational frequencies of the diatomic molecule corresponding to the desired pairwise interaction. Parameterization scheme yields bond lengths for larger molecules that are normally within 2-3% of the experimental bond lengths (and within 5% in every case). 41
42 Equilibration Method for DPA y Equilibration of the Total Energy x Equilibration of the C a -C b Bonds We obtained the equilibrium configuration of DPA through a 2000 fs simulation in which the velocity of each atom was reduced by 0.3% at each time step. 42
43 Nuclear equilibrium properties Bond Bond lengths SERID (Å) MCSCF/ 6-21G (Å) * N-C a C a -C b C b -C c C a -C d C d -O a C d -O b H a -O b H b -C b H c -C c Key Normal Modes Excited During Laser Pulse Simulation Description Ring stretch Ring breathe Ring breathe Ring shear Ring stretch SERID (cm -1 ) B3LYP/ cc-pvdz (cm -1 ) * Hameka et al. J. Mol. Structure, 365 (1996)
44 Electronic equilibrium properties Level SERID (ev) B3LYP/ cc-pvdz (ev) HOMO HOMO HOMO HOMO SERID: LUMO ΔE gap = 4.03 ev HOMO-2, HOMO-1, and HOMO nearly degenerate LUMO+1 LUMO LUMO and LUMO+1 nearly degenerate LUMO
45 DPA subjected to a (010) polarized pulse matched to the HOMO-2 to LUMO+1 transition frequency Applied Pulse Shape: Laser Pulse Parameters: t pulse (FWHM) = 5 fs A 0 = 0.13 G cm E = 4.61 ev (010) polarization #!(t) =! " cos! $ %!(t " tpulse / 2) & ' ( sin()t) tpulse 45
46 Normalized frequency spectrum after completion of laser pulse 1036 cm -1 The two peaks with the largest intensity correspond to two ring breathing modes cm -1 46
47 Absorption spectrum Experiment B3LYP/6-311G+(2df,2pd) ~ 270 nm R. Nudelman, B. V. Bronk, and S. Ferima, Appl. Spect. 54 (2000) 445. DFT calculation after J. R. Xie, V. H. Smith, and R. E. Allen, Chem. Phys. (2005). 47
 = & \" $ % i i=1 ( ) 2 $ # i 2 f i = oscillator strength Ω i = transition frequency Γ i = half-width of transition n all transitions from filled to unfilled orbitals")
48 Molecular orbitals involved in transitions π to π* at 231 nm (y-dipole) π to π* at 246 nm (x-dipole) Artificially broadened spectrum: n f i # i! (" ) = & " $ % i i=1 ( ) 2 $ # i 2 f i = oscillator strength Ω i = transition frequency Γ i = half-width of transition n all transitions from filled to unfilled orbitals z n to π* at 298 nm y 48 x
49 SERID calculated transitions Choose short pulse in order to excite a wide possible range of transitions. E = 4.03 ev t pulse (FWHM)=5 fs polarization is (100) T = 0 K transition from HOMO-2 to LUMO (E = 4.34 ev, λ = 286 nm) E = 4.03 ev t pulse (FWHM)=5 fs polarization is (010) T = 0 K transition from HOMO-2 to LUMO+1 (E = 4.61 ev, λ = 269 nm) 49
50 SERID excitation spectrum with frozen nuclei (1 0 0) (0 1 0) ( ) = 1.130e #! " $!" # ev' % & ev ( ) 2 ( ) = e #! " $!" # 4.613eV' % & ev ( ) 2 A t ( )! cos I t # % $ # % $! 2t pulse ( )! cos 2! 2t pulse # $ % t " t pulse 2 # $ % t " t pulse 2 & & ' ( ( ' & & ' ( ( '!t ~ t pulse = 25 fs 4!E!t! "!E! 0.026eV 50
51 SERID calculated excitation spectrum with nuclear motion at 0 K fluence = kj/m 2 and (1 0 0) polarization! (" ) = e # $!" # ev' % & 0.033eV ( ) 2 Average Transition Energy As absorption increases, the transition energy decreases, causing shift to lower frequencies plateau region for higher frequencies abrupt cut-off for lower frequencies 51
52 SERID calculated excitation spectrum about 4.34 ev with T = 300 K fluence = kj/m 2 and (1 0 0) polarization Low energy sidebands due to excitation out of HOMO-1 and HOMO orbitals High energy sidebands due to excitations to LUMO+1 orbital.! (" ) = 0.323e # $!" # 4.301eV' % & ev ( ) 2 52
53 Comparison of excitation spectra Nuclear motion decreases, shifts, and broadens the absorption spectra. 53
54 SERID calculated excitation spectrum about 4.61 ev with T = 300 K fluence = kj/m 2 and (0 1 0) polarization Steady increase in absorption with increasing frequency, due to excitations from orbitals below HOMO-2.! (" ) = e # $!" # 4.645eV' % & ev ( ) 2 54
55 Full SERID calculated excitation spectrum 288 nm 270 nm R. Nudelman, B. V. Bronk and S. Ferima, Appl. Spect. 54, 445 (2000). Gaussian fit (sum of x- and y- polarized fits):! (") = 0.10 e $ " # 270.1nm ' # % & nm ( ) 2 $ " # 288.3nm ' # % & 6.43nm ( ) e 2 55
56 Conclusions for DPA spectrum Predominant feature of the theoretical absorption spectrum of DPA consists of two closely spaced π to π* transitions, one with an electric dipole moment in the x-direction and one in the y- direction. Our SERID method qualitatively reproduces key features of the absorption spectrum, as seen experimentally and as calculated with density functional theory. Width of excitation spectrum is due to motion of the nuclei, which cause a change in the excitation energy, and additionally permit excitation to and from other molecular orbitals. 56
57 A final reminder of our motivation: Although real applications (such as molecular switches and biochemical detectors) will involve adaptive techniques -- with femtosecondscale laser pulses whose durations, photon energies, fluences, shapes, etc. are tailored for specific applications -- as well as larger systems, one needs an understanding of the rich interplay of electronic and nuclear dynamics to guide more empirical approaches. This understanding can be obtained through detailed studies of the kind reported here. Summary of the processes studied Cis to trans (and trans to cis) photoisomerization of azobenzene, with nuclear motion allowing extra electronic transitions for pulse durations > about 50 fs UV or visible UV or visible Photoinduced ring-opening (and ring-closing) in a model dithienylethene visible UV! Response of dipicolinic acid to femtosecond-scale laser pulses, including excited states and nuclear motion 57
Dynamics of the photoinduced ring-opening of stilbene, a prototypical diarylethene
 Chemical Physics Letters 434 (2007) 260 264 www.elsevier.com/locate/cplett Dynamics of the photoinduced ring-opening of stilbene, a prototypical diarylethene Petra Sauer, Roland E. Allen * Department of
Chemical Physics Letters 434 (2007) 260 264 www.elsevier.com/locate/cplett Dynamics of the photoinduced ring-opening of stilbene, a prototypical diarylethene Petra Sauer, Roland E. Allen * Department of
College Station, TX USA. To link to this article: DOI: / URL:
 This article was downloaded by:[texas A & M University] [Texas A & M University] On: 30 January 2007 Access Details: [subscription number 762304754] Publisher: Taylor & Francis Informa Ltd Registered in
This article was downloaded by:[texas A & M University] [Texas A & M University] On: 30 January 2007 Access Details: [subscription number 762304754] Publisher: Taylor & Francis Informa Ltd Registered in
The intricate dance of electrons, photons, and ions when matter responds to light. Yusheng Dou and Roland E. Allen Texas A&M University
 The intricate dance of electrons, photons, and ions when matter responds to light Yusheng Dou and Roland E. Allen Texas A&M University This talk is based on papers at our web site, http://lightandmatter.net.
The intricate dance of electrons, photons, and ions when matter responds to light Yusheng Dou and Roland E. Allen Texas A&M University This talk is based on papers at our web site, http://lightandmatter.net.
Quantum Chemistry. NC State University. Lecture 5. The electronic structure of molecules Absorption spectroscopy Fluorescence spectroscopy
 Quantum Chemistry Lecture 5 The electronic structure of molecules Absorption spectroscopy Fluorescence spectroscopy NC State University 3.5 Selective absorption and emission by atmospheric gases (source:
Quantum Chemistry Lecture 5 The electronic structure of molecules Absorption spectroscopy Fluorescence spectroscopy NC State University 3.5 Selective absorption and emission by atmospheric gases (source:
Mechanisms for Laser Control of Chemical Reactions
 Mechanisms for Laser Control of Chemical Reactions Ben R. Torralva and Roland E. Allen Department of Physics, Texas A&M University, College Station, Texas 77843, USA Abstract During the past several years
Mechanisms for Laser Control of Chemical Reactions Ben R. Torralva and Roland E. Allen Department of Physics, Texas A&M University, College Station, Texas 77843, USA Abstract During the past several years
I 2 Vapor Absorption Experiment and Determination of Bond Dissociation Energy.
 I 2 Vapor Absorption Experiment and Determination of Bond Dissociation Energy. What determines the UV-Vis (i.e., electronic transitions) band appearance? Usually described by HOMO LUMO electron jump LUMO
I 2 Vapor Absorption Experiment and Determination of Bond Dissociation Energy. What determines the UV-Vis (i.e., electronic transitions) band appearance? Usually described by HOMO LUMO electron jump LUMO
CHEM1901/ J-5 June 2013
 CHEM1901/3 2013-J-5 June 2013 Oxygen exists in the troposphere as a diatomic molecule. 4 (a) Using arrows to indicate relative electron spin, fill the left-most valence orbital energy diagram for O 2,
CHEM1901/3 2013-J-5 June 2013 Oxygen exists in the troposphere as a diatomic molecule. 4 (a) Using arrows to indicate relative electron spin, fill the left-most valence orbital energy diagram for O 2,
Spectroscopic properties of dipicolinic acid and its dianion
 Chemical Physics 322 (2006) 254 268 www.elsevier.com/locate/chemphys Spectroscopic properties of dipicolinic acid and its dianion John Rui-Hua Xie a, Vedene H. Smith Jr. b, Roland E. Allen a, * a Department
Chemical Physics 322 (2006) 254 268 www.elsevier.com/locate/chemphys Spectroscopic properties of dipicolinic acid and its dianion John Rui-Hua Xie a, Vedene H. Smith Jr. b, Roland E. Allen a, * a Department
Practical 1: Structure and electronic properties of organic molecules. B/ Structure, electronic and vibrational properties of the water molecule
 D1CH9116 - MMCO Molecular Modelling applied to organic chemistry Practical 1: tructure and electronic properties of organic molecules B/ tructure, electronic and vibrational properties of the water molecule
D1CH9116 - MMCO Molecular Modelling applied to organic chemistry Practical 1: tructure and electronic properties of organic molecules B/ tructure, electronic and vibrational properties of the water molecule
The symmetry properties & relative energies of atomic orbitals determine how they react to form molecular orbitals. These molecular orbitals are then
 1 The symmetry properties & relative energies of atomic orbitals determine how they react to form molecular orbitals. These molecular orbitals are then filled with the available electrons according to
1 The symmetry properties & relative energies of atomic orbitals determine how they react to form molecular orbitals. These molecular orbitals are then filled with the available electrons according to
I 2 Vapor Absorption Experiment and Determination of Bond Dissociation Energy.
 I 2 Vapor Absorption Experiment and Determination of Bond Dissociation Energy. What determines the UV-Vis (i.e., electronic transitions) band appearance? Usually described by HOMO LUMO electron jump LUMO
I 2 Vapor Absorption Experiment and Determination of Bond Dissociation Energy. What determines the UV-Vis (i.e., electronic transitions) band appearance? Usually described by HOMO LUMO electron jump LUMO
Chemistry 3502/4502. Final Exam Part I. May 14, 2005
 Chemistry 3502/4502 Final Exam Part I May 14, 2005 1. For which of the below systems is = where H is the Hamiltonian operator and T is the kinetic-energy operator? (a) The free particle (e) The
Chemistry 3502/4502 Final Exam Part I May 14, 2005 1. For which of the below systems is = where H is the Hamiltonian operator and T is the kinetic-energy operator? (a) The free particle (e) The
Chemistry 3502/4502. Final Exam Part I. May 14, 2005
 Advocacy chit Chemistry 350/450 Final Exam Part I May 4, 005. For which of the below systems is = where H is the Hamiltonian operator and T is the kinetic-energy operator? (a) The free particle
Advocacy chit Chemistry 350/450 Final Exam Part I May 4, 005. For which of the below systems is = where H is the Hamiltonian operator and T is the kinetic-energy operator? (a) The free particle
Chemistry 543--Final Exam--Keiderling May 5, pm SES
 Chemistry 543--Final Exam--Keiderling May 5,1992 -- 1-5pm -- 174 SES Please answer all questions in the answer book provided. Make sure your name is clearly indicated and that the answers are clearly numbered,
Chemistry 543--Final Exam--Keiderling May 5,1992 -- 1-5pm -- 174 SES Please answer all questions in the answer book provided. Make sure your name is clearly indicated and that the answers are clearly numbered,
Last Name or Student ID
 11/05/18, Chem433 Exam # 2 Last ame or Student ID 1. (2 pts) 2. (9 pts) 3. (2 pts) 4. (2 pts) 5. (2 pts) 6. (2 pts) 7. (2 pts) 8. (4 pts) 9. (14 pts) 10. (10 pts) 11. (26/31 pts) 12. (25/27 pts) Extra
11/05/18, Chem433 Exam # 2 Last ame or Student ID 1. (2 pts) 2. (9 pts) 3. (2 pts) 4. (2 pts) 5. (2 pts) 6. (2 pts) 7. (2 pts) 8. (4 pts) 9. (14 pts) 10. (10 pts) 11. (26/31 pts) 12. (25/27 pts) Extra
Molecular spectroscopy Multispectral imaging (FAFF 020, FYST29) fall 2017
 fall 2017") Molecular spectroscopy Multispectral imaging (FAFF 00, FYST9) fall 017 Lecture prepared by Joakim Bood joakim.bood@forbrf.lth.se Molecular structure Electronic structure Rotational structure Vibrational
Molecular spectroscopy Multispectral imaging (FAFF 00, FYST9) fall 017 Lecture prepared by Joakim Bood joakim.bood@forbrf.lth.se Molecular structure Electronic structure Rotational structure Vibrational
Lecture 9 Electronic Spectroscopy
 Lecture 9 Electronic Spectroscopy Molecular Orbital Theory: A Review - LCAO approximaton & AO overlap - Variation Principle & Secular Determinant - Homonuclear Diatomic MOs - Energy Levels, Bond Order
Lecture 9 Electronic Spectroscopy Molecular Orbital Theory: A Review - LCAO approximaton & AO overlap - Variation Principle & Secular Determinant - Homonuclear Diatomic MOs - Energy Levels, Bond Order
Molecular Orbital Theory. Molecular Orbital Theory: Electrons are located in the molecule, not held in discrete regions between two bonded atoms
 Molecular Orbital Theory Valence Bond Theory: Electrons are located in discrete pairs between specific atoms Molecular Orbital Theory: Electrons are located in the molecule, not held in discrete regions
Molecular Orbital Theory Valence Bond Theory: Electrons are located in discrete pairs between specific atoms Molecular Orbital Theory: Electrons are located in the molecule, not held in discrete regions
Molecular Physics. Attraction between the ions causes the chemical bond.
 Molecular Physics A molecule is a stable configuration of electron(s) and more than one nucleus. Two types of bonds: covalent and ionic (two extremes of same process) Covalent Bond Electron is in a molecular
Molecular Physics A molecule is a stable configuration of electron(s) and more than one nucleus. Two types of bonds: covalent and ionic (two extremes of same process) Covalent Bond Electron is in a molecular
In this lecture we will understand how the molecular orbitals are formed from the interaction of atomic orbitals.
 Lecture 7 Title: Understanding of Molecular Orbital Page-1 In this lecture we will understand how the molecular orbitals are formed from the interaction of atomic orbitals. We will see how the electrons
Lecture 7 Title: Understanding of Molecular Orbital Page-1 In this lecture we will understand how the molecular orbitals are formed from the interaction of atomic orbitals. We will see how the electrons
Vibronic Coupling in Quantum Wires: Applications to Polydiacetylene
 Vibronic Coupling in Quantum Wires: Applications to Polydiacetylene An Exhaustively Researched Report by Will Bassett and Cole Johnson Overall Goal In order to elucidate the absorbance spectra of different
Vibronic Coupling in Quantum Wires: Applications to Polydiacetylene An Exhaustively Researched Report by Will Bassett and Cole Johnson Overall Goal In order to elucidate the absorbance spectra of different
Electronic structure of correlated electron systems. Lecture 2
 Electronic structure of correlated electron systems Lecture 2 Band Structure approach vs atomic Band structure Delocalized Bloch states Fill up states with electrons starting from the lowest energy No
Electronic structure of correlated electron systems Lecture 2 Band Structure approach vs atomic Band structure Delocalized Bloch states Fill up states with electrons starting from the lowest energy No
Chapter 6. Electronic spectra and HOMO-LUMO studies on Nickel, copper substituted Phthalocyanine for solar cell applications
 Chapter 6 Electronic spectra and HOMO-LUMO studies on Nickel, copper substituted Phthalocyanine for solar cell applications 6.1 Structures of Ni, Cu substituted Phthalocyanine Almost all of the metals
Chapter 6 Electronic spectra and HOMO-LUMO studies on Nickel, copper substituted Phthalocyanine for solar cell applications 6.1 Structures of Ni, Cu substituted Phthalocyanine Almost all of the metals
( ) x10 8 m. The energy in a mole of 400 nm photons is calculated by: ' & sec( ) ( & % ) 6.022x10 23 photons' E = h! = hc & 6.
 x10 8 m. The energy in a mole of 400 nm photons is calculated by: ' & sec( ) ( & % ) 6.022x10 23 photons' E = h! = hc & 6.") Introduction to Spectroscopy Spectroscopic techniques are widely used to detect molecules, to measure the concentration of a species in solution, and to determine molecular structure. For proteins, most
Introduction to Spectroscopy Spectroscopic techniques are widely used to detect molecules, to measure the concentration of a species in solution, and to determine molecular structure. For proteins, most
Introduction to Computational Chemistry
 Introduction to Computational Chemistry Vesa Hänninen Laboratory of Physical Chemistry Chemicum 4th floor vesa.hanninen@helsinki.fi September 10, 2013 Lecture 3. Electron correlation methods September
Introduction to Computational Chemistry Vesa Hänninen Laboratory of Physical Chemistry Chemicum 4th floor vesa.hanninen@helsinki.fi September 10, 2013 Lecture 3. Electron correlation methods September
Analysis of the ultrafast dynamics of the silver trimer upon photodetachment
 J. Phys. B: At. Mol. Opt. Phys. 29 (1996) L545 L549. Printed in the UK LETTER TO THE EDITOR Analysis of the ultrafast dynamics of the silver trimer upon photodetachment H O Jeschke, M E Garcia and K H
J. Phys. B: At. Mol. Opt. Phys. 29 (1996) L545 L549. Printed in the UK LETTER TO THE EDITOR Analysis of the ultrafast dynamics of the silver trimer upon photodetachment H O Jeschke, M E Garcia and K H
SUPPLEMENTARY INFORMATION
 SUPPLEMENTARY INFORMATION DOI: 10.1038/NPHYS2210 Femtosecond torsional relaxation Theoretical methodology: J. Clark, S. Tretiak, T. Nelson, G. Cirmi & G. Lanzani To model non-adiabatic excited state dynamics
SUPPLEMENTARY INFORMATION DOI: 10.1038/NPHYS2210 Femtosecond torsional relaxation Theoretical methodology: J. Clark, S. Tretiak, T. Nelson, G. Cirmi & G. Lanzani To model non-adiabatic excited state dynamics
CHAPTER 13 Molecular Spectroscopy 2: Electronic Transitions
 CHAPTER 13 Molecular Spectroscopy 2: Electronic Transitions I. General Features of Electronic spectroscopy. A. Visible and ultraviolet photons excite electronic state transitions. ε photon = 120 to 1200
CHAPTER 13 Molecular Spectroscopy 2: Electronic Transitions I. General Features of Electronic spectroscopy. A. Visible and ultraviolet photons excite electronic state transitions. ε photon = 120 to 1200
Nanotechnology and Solar Energy. Solar Electricity Photovoltaics. Fuel from the Sun Photosynthesis Biofuels Split Water Fuel Cells
 Nanotechnology and Solar Energy Solar Electricity Photovoltaics Fuel from the Sun Photosynthesis Biofuels Split Water Fuel Cells Solar cell A photon from the Sun generates an electron-hole pair in a semiconductor.
Nanotechnology and Solar Energy Solar Electricity Photovoltaics Fuel from the Sun Photosynthesis Biofuels Split Water Fuel Cells Solar cell A photon from the Sun generates an electron-hole pair in a semiconductor.
3: Many electrons. Orbital symmetries. l =2 1. m l
 3: Many electrons Orbital symmetries Atomic orbitals are labelled according to the principal quantum number, n, and the orbital angular momentum quantum number, l. Electrons in a diatomic molecule experience
3: Many electrons Orbital symmetries Atomic orbitals are labelled according to the principal quantum number, n, and the orbital angular momentum quantum number, l. Electrons in a diatomic molecule experience
Lecture 10. Born-Oppenheimer approximation LCAO-MO application to H + The potential energy surface MOs for diatomic molecules. NC State University
 Chemistry 431 Lecture 10 Diatomic molecules Born-Oppenheimer approximation LCAO-MO application to H + 2 The potential energy surface MOs for diatomic molecules NC State University Born-Oppenheimer approximation
Chemistry 431 Lecture 10 Diatomic molecules Born-Oppenheimer approximation LCAO-MO application to H + 2 The potential energy surface MOs for diatomic molecules NC State University Born-Oppenheimer approximation
As a partial differential equation, the Helmholtz equation does not lend itself easily to analytical
 Aaron Rury Research Prospectus 21.6.2009 Introduction: The Helmhlotz equation, ( 2 +k 2 )u(r)=0 1, serves as the basis for much of optical physics. As a partial differential equation, the Helmholtz equation
Aaron Rury Research Prospectus 21.6.2009 Introduction: The Helmhlotz equation, ( 2 +k 2 )u(r)=0 1, serves as the basis for much of optical physics. As a partial differential equation, the Helmholtz equation
CHEM J-5 June 2014
 CHEM1101 2014-J-5 June 2014 The molecular orbital energy level diagrams for H 2, H 2 +, H 2 and O 2 are shown below. Fill in the valence electrons for each species in its ground state and label the types
CHEM1101 2014-J-5 June 2014 The molecular orbital energy level diagrams for H 2, H 2 +, H 2 and O 2 are shown below. Fill in the valence electrons for each species in its ground state and label the types
(002)(110) (004)(220) (222) (112) (211) (202) (200) * * 2θ (degree)
(110) (004)(220) (222) (112) (211) (202) (200) * * 2θ (degree)") Supplementary Figures. (002)(110) Tetragonal I4/mcm Intensity (a.u) (004)(220) 10 (112) (211) (202) 20 Supplementary Figure 1. X-ray diffraction (XRD) pattern of the sample. The XRD characterization indicates
Supplementary Figures. (002)(110) Tetragonal I4/mcm Intensity (a.u) (004)(220) 10 (112) (211) (202) 20 Supplementary Figure 1. X-ray diffraction (XRD) pattern of the sample. The XRD characterization indicates
Lecture 19: Building Atoms and Molecules
 Lecture 19: Building Atoms and Molecules +e r n = 3 n = 2 n = 1 +e +e r y even Lecture 19, p 1 Today Nuclear Magnetic Resonance Using RF photons to drive transitions between nuclear spin orientations in
Lecture 19: Building Atoms and Molecules +e r n = 3 n = 2 n = 1 +e +e r y even Lecture 19, p 1 Today Nuclear Magnetic Resonance Using RF photons to drive transitions between nuclear spin orientations in
Coulomb pairing and double-photoionization in aromatic hydrocarbons
 Coulomb pairing and double-photoionization in aromatic hydrocarbons D. L. Huber * Physics Department, University of Wisconsin-Madison, Madison, WI 53706, USA Abstract Recently reported anomalies in the
Coulomb pairing and double-photoionization in aromatic hydrocarbons D. L. Huber * Physics Department, University of Wisconsin-Madison, Madison, WI 53706, USA Abstract Recently reported anomalies in the
Chem 1140; Molecular Modeling
 P. Wipf 1 Chem 1140 $E = -! (q a + q b )" ab S ab + ab!k
P. Wipf 1 Chem 1140 $E = -! (q a + q b )" ab S ab + ab!k
CHEMISTRY 4021/8021 MIDTERM EXAM 1 SPRING 2014
 CHEMISTRY 4021/8021 Q1) Propose a simple, united-atom molecular mechanics force-field needed to generate a potential energy surface for an isolated molecule of acetone (Me 2 CO). I.e., provide an energy
CHEMISTRY 4021/8021 Q1) Propose a simple, united-atom molecular mechanics force-field needed to generate a potential energy surface for an isolated molecule of acetone (Me 2 CO). I.e., provide an energy
Supplementary Materials
 Supplementary Materials Sample characterization The presence of Si-QDs is established by Transmission Electron Microscopy (TEM), by which the average QD diameter of d QD 2.2 ± 0.5 nm has been determined
Supplementary Materials Sample characterization The presence of Si-QDs is established by Transmission Electron Microscopy (TEM), by which the average QD diameter of d QD 2.2 ± 0.5 nm has been determined
What happens when light falls on a material? Transmission Reflection Absorption Luminescence. Elastic Scattering Inelastic Scattering
 Raman Spectroscopy What happens when light falls on a material? Transmission Reflection Absorption Luminescence Elastic Scattering Inelastic Scattering Raman, Fluorescence and IR Scattering Absorption
Raman Spectroscopy What happens when light falls on a material? Transmission Reflection Absorption Luminescence Elastic Scattering Inelastic Scattering Raman, Fluorescence and IR Scattering Absorption
Lecture 3: Optical Properties of Insulators, Semiconductors, and Metals. 5 nm
 Metals Lecture 3: Optical Properties of Insulators, Semiconductors, and Metals 5 nm Course Info Next Week (Sept. 5 and 7) no classes First H/W is due Sept. 1 The Previous Lecture Origin frequency dependence
Metals Lecture 3: Optical Properties of Insulators, Semiconductors, and Metals 5 nm Course Info Next Week (Sept. 5 and 7) no classes First H/W is due Sept. 1 The Previous Lecture Origin frequency dependence
Theoretical Chemistry - Level II - Practical Class Molecular Orbitals in Diatomics
 Theoretical Chemistry - Level II - Practical Class Molecular Orbitals in Diatomics Problem 1 Draw molecular orbital diagrams for O 2 and O 2 +. E / ev dioxygen molecule, O 2 dioxygenyl cation, O 2 + 25
Theoretical Chemistry - Level II - Practical Class Molecular Orbitals in Diatomics Problem 1 Draw molecular orbital diagrams for O 2 and O 2 +. E / ev dioxygen molecule, O 2 dioxygenyl cation, O 2 + 25
Literature values: ΔH f, gas = % error Source: ΔH f, solid = % error. For comparison, your experimental value was ΔH f = phase:
 1 Molecular Calculations Lab: Some guideline given at the bottom of page 3. 1. Use the semi-empirical AM1 method to calculate ΔH f for the compound you used in the heat of combustion experiment. Be sure
1 Molecular Calculations Lab: Some guideline given at the bottom of page 3. 1. Use the semi-empirical AM1 method to calculate ΔH f for the compound you used in the heat of combustion experiment. Be sure
5.80 Small-Molecule Spectroscopy and Dynamics
 MIT OpenCourseWare http://ocw.mit.edu 5.80 Small-Molecule Spectroscopy and Dynamics Fall 2008 For information about citing these materials or our Terms of Use, visit: http://ocw.mit.edu/terms. 5.80 Lecture
MIT OpenCourseWare http://ocw.mit.edu 5.80 Small-Molecule Spectroscopy and Dynamics Fall 2008 For information about citing these materials or our Terms of Use, visit: http://ocw.mit.edu/terms. 5.80 Lecture
Lecture 19: Building Atoms and Molecules
 Lecture 19: Building Atoms and Molecules +e r n = 3 n = 2 n = 1 +e +e r ψ even Lecture 19, p 1 Today Nuclear Magnetic Resonance Using RF photons to drive transitions between nuclear spin orientations in
Lecture 19: Building Atoms and Molecules +e r n = 3 n = 2 n = 1 +e +e r ψ even Lecture 19, p 1 Today Nuclear Magnetic Resonance Using RF photons to drive transitions between nuclear spin orientations in
QUANTUM CHEMISTRY FOR TRANSITION METALS
 QUANTUM CHEMISTRY FOR TRANSITION METALS Outline I Introduction II Correlation Static correlation effects MC methods DFT III Relativity Generalities From 4 to 1 components Effective core potential Outline
QUANTUM CHEMISTRY FOR TRANSITION METALS Outline I Introduction II Correlation Static correlation effects MC methods DFT III Relativity Generalities From 4 to 1 components Effective core potential Outline
Journal of Theoretical Physics
 1 Journal of Theoretical Physics Founded and Edited by M. Apostol 53 (2000) ISSN 1453-4428 Ionization potential for metallic clusters L. C. Cune and M. Apostol Department of Theoretical Physics, Institute
1 Journal of Theoretical Physics Founded and Edited by M. Apostol 53 (2000) ISSN 1453-4428 Ionization potential for metallic clusters L. C. Cune and M. Apostol Department of Theoretical Physics, Institute
2m dx 2. The particle in a one dimensional box (of size L) energy levels are
 energy levels are") Name: Chem 3322 test #1 solutions, out of 40 marks I want complete, detailed answers to the questions. Show all your work to get full credit. indefinite integral : sin 2 (ax)dx = x 2 sin(2ax) 4a (1) with
Name: Chem 3322 test #1 solutions, out of 40 marks I want complete, detailed answers to the questions. Show all your work to get full credit. indefinite integral : sin 2 (ax)dx = x 2 sin(2ax) 4a (1) with
CHEM3023: Spins, Atoms and Molecules
 CHEM3023: Spins, Atoms and Molecules Lecture 3 The Born-Oppenheimer approximation C.-K. Skylaris Learning outcomes Separate molecular Hamiltonians to electronic and nuclear parts according to the Born-Oppenheimer
CHEM3023: Spins, Atoms and Molecules Lecture 3 The Born-Oppenheimer approximation C.-K. Skylaris Learning outcomes Separate molecular Hamiltonians to electronic and nuclear parts according to the Born-Oppenheimer
Quantum Chemical Simulations and Descriptors. Dr. Antonio Chana, Dr. Mosè Casalegno
 Quantum Chemical Simulations and Descriptors Dr. Antonio Chana, Dr. Mosè Casalegno Classical Mechanics: basics It models real-world objects as point particles, objects with negligible size. The motion
Quantum Chemical Simulations and Descriptors Dr. Antonio Chana, Dr. Mosè Casalegno Classical Mechanics: basics It models real-world objects as point particles, objects with negligible size. The motion
Andrew Rosen *Note: If you can rotate a molecule to have one isomer equal to another, they are both the same
 *Note: If you can rotate a molecule to have one isomer equal to another, they are both the same *Note: For hybridization, if an SP 2 is made, there is one unhybridized p orbital (because p usually has
*Note: If you can rotate a molecule to have one isomer equal to another, they are both the same *Note: For hybridization, if an SP 2 is made, there is one unhybridized p orbital (because p usually has
Calculate a rate given a species concentration change.
 Kinetics Define a rate for a given process. Change in concentration of a reagent with time. A rate is always positive, and is usually referred to with only magnitude (i.e. no sign) Reaction rates can be
Kinetics Define a rate for a given process. Change in concentration of a reagent with time. A rate is always positive, and is usually referred to with only magnitude (i.e. no sign) Reaction rates can be
Tuning Color Through Substitution
 1 Tuning Color Through Substitution Introduction In this experiment, the effect of substituents on the absorbance spectra of molecules will be related to the structure of the molecular orbitals involved
1 Tuning Color Through Substitution Introduction In this experiment, the effect of substituents on the absorbance spectra of molecules will be related to the structure of the molecular orbitals involved
CHEM 344 Molecular Modeling
 CHEM 344 Molecular Modeling The Use of Computational Chemistry to Support Experimental Organic Chemistry Day 1 all calculation data obtained from Gaussian09 using B3LYP/6-31G(d) unless otherwise noted.
CHEM 344 Molecular Modeling The Use of Computational Chemistry to Support Experimental Organic Chemistry Day 1 all calculation data obtained from Gaussian09 using B3LYP/6-31G(d) unless otherwise noted.
5.61 Physical Chemistry Final Exam 12/16/09. MASSACHUSETTS INSTITUTE OF TECHNOLOGY Department of Chemistry Chemistry Physical Chemistry
 5.6 Physical Chemistry Final Exam 2/6/09 MASSACHUSETTS INSTITUTE OF TECHNOLOGY Department of Chemistry Chemistry - 5.6 Physical Chemistry Final Examination () PRINT your name on the cover page. (2) It
5.6 Physical Chemistry Final Exam 2/6/09 MASSACHUSETTS INSTITUTE OF TECHNOLOGY Department of Chemistry Chemistry - 5.6 Physical Chemistry Final Examination () PRINT your name on the cover page. (2) It
Organic Electronic Devices
 Organic Electronic Devices Week 2: Electronic Structure Lecture 2.1: Atomic and Molecular Orbitals Bryan W. Boudouris Chemical Engineering Purdue University 1 Lecture Overview and Learning Objectives Concepts
Organic Electronic Devices Week 2: Electronic Structure Lecture 2.1: Atomic and Molecular Orbitals Bryan W. Boudouris Chemical Engineering Purdue University 1 Lecture Overview and Learning Objectives Concepts
Ψ t = ih Ψ t t. Time Dependent Wave Equation Quantum Mechanical Description. Hamiltonian Static/Time-dependent. Time-dependent Energy operator
 Time Dependent Wave Equation Quantum Mechanical Description Hamiltonian Static/Time-dependent Time-dependent Energy operator H 0 + H t Ψ t = ih Ψ t t The Hamiltonian and wavefunction are time-dependent
Time Dependent Wave Equation Quantum Mechanical Description Hamiltonian Static/Time-dependent Time-dependent Energy operator H 0 + H t Ψ t = ih Ψ t t The Hamiltonian and wavefunction are time-dependent
Nonadiabatic dynamics simulations of singlet fission in 2,5-bis(fluorene-9-ylidene)-2,5-dihydrothiophene crystals. Supporting Information.
-2,5-dihydrothiophene crystals. Supporting Information.") Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 18 Nonadiabatic dynamics simulations of singlet fission in,5-bis(fluorene-9-ylidene)-,5-dihydrothiophene
Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 18 Nonadiabatic dynamics simulations of singlet fission in,5-bis(fluorene-9-ylidene)-,5-dihydrothiophene
Physical Chemistry Lab II CHEM 4644 Spring 2011 Final Exam 5 questions at 3 points each equals 15 total points possible.
 Physical Chemistry Lab II Name: KEY CHEM 4644 Spring 2011 Final Exam 5 questions at 3 points each equals 15 total points possible. Constants: c = 3.00 10 8 m/s h = 6.63 10-34 J s 1 Hartree = 4.36 10-18
Physical Chemistry Lab II Name: KEY CHEM 4644 Spring 2011 Final Exam 5 questions at 3 points each equals 15 total points possible. Constants: c = 3.00 10 8 m/s h = 6.63 10-34 J s 1 Hartree = 4.36 10-18
ATMO 551a Fall Resonant Electromagnetic (EM) Interactions in Planetary atmospheres. Electron transition between different electron orbits
 Interactions in Planetary atmospheres. Electron transition between different electron orbits") Resonant Electromagnetic (EM) Interactions in Planetary atmospheres There are three classes of energy states that interact with EM radiation that we are interested in to understand how light (EM radiation)
Resonant Electromagnetic (EM) Interactions in Planetary atmospheres There are three classes of energy states that interact with EM radiation that we are interested in to understand how light (EM radiation)
 This article was originally published in a journal published by Elsevier, and the attached copy is provided by Elsevier for the author s benefit and for the benefit of the author s institution, for non-commercial
This article was originally published in a journal published by Elsevier, and the attached copy is provided by Elsevier for the author s benefit and for the benefit of the author s institution, for non-commercial
Introduction to Franck-Condon Factors
 Introduction to Franck-Condon Factors Theresa Julia Zielinski Monmouth University Department of Chemistry, Medical Technology, and Physics West Long Branch, NJ 07764 tzielins@monmouth.edu and La Salle
Introduction to Franck-Condon Factors Theresa Julia Zielinski Monmouth University Department of Chemistry, Medical Technology, and Physics West Long Branch, NJ 07764 tzielins@monmouth.edu and La Salle
2m 2 Ze2. , where δ. ) 2 l,n is the quantum defect (of order one but larger
 2 l,n is the quantum defect (of order one but larger") PHYS 402, Atomic and Molecular Physics Spring 2017, final exam, solutions 1. Hydrogenic atom energies: Consider a hydrogenic atom or ion with nuclear charge Z and the usual quantum states φ nlm. (a) (2
PHYS 402, Atomic and Molecular Physics Spring 2017, final exam, solutions 1. Hydrogenic atom energies: Consider a hydrogenic atom or ion with nuclear charge Z and the usual quantum states φ nlm. (a) (2
Introduction to DFTB. Marcus Elstner. July 28, 2006
 Introduction to DFTB Marcus Elstner July 28, 2006 I. Non-selfconsistent solution of the KS equations DFT can treat up to 100 atoms in routine applications, sometimes even more and about several ps in MD
Introduction to DFTB Marcus Elstner July 28, 2006 I. Non-selfconsistent solution of the KS equations DFT can treat up to 100 atoms in routine applications, sometimes even more and about several ps in MD
Electronic Spectroscopy of Polyatomics
 Electronic Spectroscopy of Polyatomics We shall discuss the electronic spectroscopy of the following types of polyatomic molecules: 1. general AH 2 molecules, A = first-row element 2. formaldehyde 3. benzene
Electronic Spectroscopy of Polyatomics We shall discuss the electronic spectroscopy of the following types of polyatomic molecules: 1. general AH 2 molecules, A = first-row element 2. formaldehyde 3. benzene
Quantum Condensed Matter Physics Lecture 9
 Quantum Condensed Matter Physics Lecture 9 David Ritchie QCMP Lent/Easter 2018 http://www.sp.phy.cam.ac.uk/drp2/home 9.1 Quantum Condensed Matter Physics 1. Classical and Semi-classical models for electrons
Quantum Condensed Matter Physics Lecture 9 David Ritchie QCMP Lent/Easter 2018 http://www.sp.phy.cam.ac.uk/drp2/home 9.1 Quantum Condensed Matter Physics 1. Classical and Semi-classical models for electrons
MD simulation: output
 Properties MD simulation: output Trajectory of atoms positions: e. g. diffusion, mass transport velocities: e. g. v-v autocorrelation spectrum Energies temperature displacement fluctuations Mean square
Properties MD simulation: output Trajectory of atoms positions: e. g. diffusion, mass transport velocities: e. g. v-v autocorrelation spectrum Energies temperature displacement fluctuations Mean square
Valence bond theory accounts, at least qualitatively, for the stability of the covalent bond in terms of overlapping atomic orbitals.
 Molecular Orbital Theory Valence bond theory accounts, at least qualitatively, for the stability of the covalent bond in terms of overlapping atomic orbitals. Using the concept of hybridization, valence
Molecular Orbital Theory Valence bond theory accounts, at least qualitatively, for the stability of the covalent bond in terms of overlapping atomic orbitals. Using the concept of hybridization, valence
Molecular orbitals, potential energy surfaces and symmetry
 Molecular orbitals, potential energy surfaces and symmetry mathematical presentation of molecular symmetry group theory spectroscopy valence theory molecular orbitals Wave functions Hamiltonian: electronic,
Molecular orbitals, potential energy surfaces and symmetry mathematical presentation of molecular symmetry group theory spectroscopy valence theory molecular orbitals Wave functions Hamiltonian: electronic,
Calculations of band structures
 Chemistry and Physics at Albany Planning for the Future Calculations of band structures using wave-function based correlation methods Elke Pahl Centre of Theoretical Chemistry and Physics Institute of
Chemistry and Physics at Albany Planning for the Future Calculations of band structures using wave-function based correlation methods Elke Pahl Centre of Theoretical Chemistry and Physics Institute of
DENSITY FUNCTIONAL THEORY FOR NON-THEORISTS JOHN P. PERDEW DEPARTMENTS OF PHYSICS AND CHEMISTRY TEMPLE UNIVERSITY
 DENSITY FUNCTIONAL THEORY FOR NON-THEORISTS JOHN P. PERDEW DEPARTMENTS OF PHYSICS AND CHEMISTRY TEMPLE UNIVERSITY A TUTORIAL FOR PHYSICAL SCIENTISTS WHO MAY OR MAY NOT HATE EQUATIONS AND PROOFS REFERENCES
DENSITY FUNCTIONAL THEORY FOR NON-THEORISTS JOHN P. PERDEW DEPARTMENTS OF PHYSICS AND CHEMISTRY TEMPLE UNIVERSITY A TUTORIAL FOR PHYSICAL SCIENTISTS WHO MAY OR MAY NOT HATE EQUATIONS AND PROOFS REFERENCES
Conjugated Systems, Orbital Symmetry and UV Spectroscopy
 Conjugated Systems, Orbital Symmetry and UV Spectroscopy Introduction There are several possible arrangements for a molecule which contains two double bonds (diene): Isolated: (two or more single bonds
Conjugated Systems, Orbital Symmetry and UV Spectroscopy Introduction There are several possible arrangements for a molecule which contains two double bonds (diene): Isolated: (two or more single bonds
From Last Time Important new Quantum Mechanical Concepts. Atoms and Molecules. Today. Symmetry. Simple molecules.
 Today From Last Time Important new Quantum Mechanical Concepts Indistinguishability: Symmetries of the wavefunction: Symmetric and Antisymmetric Pauli exclusion principle: only one fermion per state Spin
Today From Last Time Important new Quantum Mechanical Concepts Indistinguishability: Symmetries of the wavefunction: Symmetric and Antisymmetric Pauli exclusion principle: only one fermion per state Spin
Modeling Ultrafast Deactivation in Oligothiophenes via Nonadiabatic Dynamics
 Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 2015 Supplementary Data for Modeling Ultrafast Deactivation in Oligothiophenes via Nonadiabatic
Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 2015 Supplementary Data for Modeling Ultrafast Deactivation in Oligothiophenes via Nonadiabatic
CHEM 344 Molecular Modeling
 CHEM 344 Molecular Modeling The Use of Computational Chemistry to Support Experimental Organic Chemistry Part 1: Molecular Orbital Theory, Hybridization, & Formal Charge * all calculation data obtained
CHEM 344 Molecular Modeling The Use of Computational Chemistry to Support Experimental Organic Chemistry Part 1: Molecular Orbital Theory, Hybridization, & Formal Charge * all calculation data obtained
Section 3 Electronic Configurations, Term Symbols, and States
 Section 3 Electronic Configurations, Term Symbols, and States Introductory Remarks- The Orbital, Configuration, and State Pictures of Electronic Structure One of the goals of quantum chemistry is to allow
Section 3 Electronic Configurations, Term Symbols, and States Introductory Remarks- The Orbital, Configuration, and State Pictures of Electronic Structure One of the goals of quantum chemistry is to allow
Chem120a : Exam 3 (Chem Bio) Solutions
 Solutions") Chem10a : Exam 3 (Chem Bio) Solutions November 7, 006 Problem 1 This problem will basically involve us doing two Hückel calculations: one for the linear geometry, and one for the triangular geometry. We
Chem10a : Exam 3 (Chem Bio) Solutions November 7, 006 Problem 1 This problem will basically involve us doing two Hückel calculations: one for the linear geometry, and one for the triangular geometry. We
Be H. Delocalized Bonding. Localized Bonding. σ 2. σ 1. Two (sp-1s) Be-H σ bonds. The two σ bonding MO s in BeH 2. MO diagram for BeH 2
 Be-H σ bonds. The two σ bonding MO s in BeH 2. MO diagram for BeH 2") The Delocalized Approach to Bonding: The localized models for bonding we have examined (Lewis and VBT) assume that all electrons are restricted to specific bonds between atoms or in lone pairs. In contrast,
The Delocalized Approach to Bonding: The localized models for bonding we have examined (Lewis and VBT) assume that all electrons are restricted to specific bonds between atoms or in lone pairs. In contrast,
Excited States Calculations for Protonated PAHs
 52 Chapter 3 Excited States Calculations for Protonated PAHs 3.1 Introduction Protonated PAHs are closed shell ions. Their electronic structure should therefore be similar to that of neutral PAHs, but
52 Chapter 3 Excited States Calculations for Protonated PAHs 3.1 Introduction Protonated PAHs are closed shell ions. Their electronic structure should therefore be similar to that of neutral PAHs, but
DFT calculations of NMR indirect spin spin coupling constants
 DFT calculations of NMR indirect spin spin coupling constants Dalton program system Program capabilities Density functional theory Kohn Sham theory LDA, GGA and hybrid theories Indirect NMR spin spin coupling
DFT calculations of NMR indirect spin spin coupling constants Dalton program system Program capabilities Density functional theory Kohn Sham theory LDA, GGA and hybrid theories Indirect NMR spin spin coupling
16.1 Molecular Vibrations
 16.1 Molecular Vibrations molecular degrees of freedom are used to predict the number of vibrational modes vibrations occur as coordinated movement among many nuclei the harmonic oscillator approximation
16.1 Molecular Vibrations molecular degrees of freedom are used to predict the number of vibrational modes vibrations occur as coordinated movement among many nuclei the harmonic oscillator approximation
Review of Optical Properties of Materials
 Review of Optical Properties of Materials Review of optics Absorption in semiconductors: qualitative discussion Derivation of Optical Absorption Coefficient in Direct Semiconductors Photons When dealing
Review of Optical Properties of Materials Review of optics Absorption in semiconductors: qualitative discussion Derivation of Optical Absorption Coefficient in Direct Semiconductors Photons When dealing
Born-Oppenheimer Approximation
 Born-Oppenheimer Approximation Adiabatic Assumption: Nuclei move so much more slowly than electron that the electrons that the electrons are assumed to be obtained if the nuclear kinetic energy is ignored,
Born-Oppenheimer Approximation Adiabatic Assumption: Nuclei move so much more slowly than electron that the electrons that the electrons are assumed to be obtained if the nuclear kinetic energy is ignored,
van Quantum tot Molecuul
 10 HC10: Molecular and vibrational spectroscopy van Quantum tot Molecuul Dr Juan Rojo VU Amsterdam and Nikhef Theory Group http://www.juanrojo.com/ j.rojo@vu.nl Molecular and Vibrational Spectroscopy Based
10 HC10: Molecular and vibrational spectroscopy van Quantum tot Molecuul Dr Juan Rojo VU Amsterdam and Nikhef Theory Group http://www.juanrojo.com/ j.rojo@vu.nl Molecular and Vibrational Spectroscopy Based
σ u * 1s g - gerade u - ungerade * - antibonding σ g 1s
 One of these two states is a repulsive (dissociative) state. Other excited states can be constructed using linear combinations of other orbitals. Some will be binding and others will be repulsive. Thus
One of these two states is a repulsive (dissociative) state. Other excited states can be constructed using linear combinations of other orbitals. Some will be binding and others will be repulsive. Thus
HW posted on web page HW10: Chap 14 Concept 8,20,24,26 Prob. 4,8. From Last Time
 HW posted on web page HW10: Chap 14 Concept 8,20,24,26 Prob. 4,8 From Last Time Philosophical effects in quantum mechanics Interpretation of the wave function: Calculation using the basic premises of quantum
HW posted on web page HW10: Chap 14 Concept 8,20,24,26 Prob. 4,8 From Last Time Philosophical effects in quantum mechanics Interpretation of the wave function: Calculation using the basic premises of quantum
l ; Y l,l-1, Y l,-1+1; etc., plus a single non-degenerate function Y l,0, in axial symmetry.
 Chapter 6 Along "Reaction Paths", Orbitals Can be Connected One-to-One According to Their Symmetries and Energies. This is the Origin of the Woodward-offmann Rules I. Reduction in Symmetry As fragments
Chapter 6 Along "Reaction Paths", Orbitals Can be Connected One-to-One According to Their Symmetries and Energies. This is the Origin of the Woodward-offmann Rules I. Reduction in Symmetry As fragments
Same idea for polyatomics, keep track of identical atom e.g. NH 3 consider only valence electrons F(2s,2p) H(1s)
 H(1s)") XIII 63 Polyatomic bonding -09 -mod, Notes (13) Engel 16-17 Balance: nuclear repulsion, positive e-n attraction, neg. united atom AO ε i applies to all bonding, just more nuclei repulsion biggest at low
XIII 63 Polyatomic bonding -09 -mod, Notes (13) Engel 16-17 Balance: nuclear repulsion, positive e-n attraction, neg. united atom AO ε i applies to all bonding, just more nuclei repulsion biggest at low
CHEM 2010 Symmetry, Electronic Structure and Bonding Winter Numbering of Chapters and Assigned Problems
 CHEM 2010 Symmetry, Electronic Structure and Bonding Winter 2011 Numbering of Chapters and Assigned Problems The following table shows the correspondence between the chapter numbers in the full book (Physical
CHEM 2010 Symmetry, Electronic Structure and Bonding Winter 2011 Numbering of Chapters and Assigned Problems The following table shows the correspondence between the chapter numbers in the full book (Physical
An Introduction to Quantum Chemistry and Potential Energy Surfaces. Benjamin G. Levine
 An Introduction to Quantum Chemistry and Potential Energy Surfaces Benjamin G. Levine This Week s Lecture Potential energy surfaces What are they? What are they good for? How do we use them to solve chemical
An Introduction to Quantum Chemistry and Potential Energy Surfaces Benjamin G. Levine This Week s Lecture Potential energy surfaces What are they? What are they good for? How do we use them to solve chemical
( ) electron gives S = 1/2 and L = l 1
 electron gives S = 1/2 and L = l 1") Practice Modern Physics II, W018, Set 1 Question 1 Energy Level Diagram of Boron ion B + For neutral B, Z = 5 (A) Draw the fine-structure diagram of B + that includes all n = 3 states Label the states
Practice Modern Physics II, W018, Set 1 Question 1 Energy Level Diagram of Boron ion B + For neutral B, Z = 5 (A) Draw the fine-structure diagram of B + that includes all n = 3 states Label the states
Valence electronic structure of isopropyl iodide investigated by electron momentum spectroscopy. --- Influence of intramolecular interactions
 Valence electronic structure of isopropyl iodide investigated by electron momentum spectroscopy --- Influence of intramolecular interactions Minfu Zhao, Xu Shan, Shanshan Niu, Yaguo Tang, Zhaohui Liu,
Valence electronic structure of isopropyl iodide investigated by electron momentum spectroscopy --- Influence of intramolecular interactions Minfu Zhao, Xu Shan, Shanshan Niu, Yaguo Tang, Zhaohui Liu,
Beyond the Hartree-Fock Approximation: Configuration Interaction
 Beyond the Hartree-Fock Approximation: Configuration Interaction The Hartree-Fock (HF) method uses a single determinant (single electronic configuration) description of the electronic wavefunction. For
Beyond the Hartree-Fock Approximation: Configuration Interaction The Hartree-Fock (HF) method uses a single determinant (single electronic configuration) description of the electronic wavefunction. For
Problem 1. Anthracene and a chiral derivative of anthracene
 Molecular Photophysics 330 Physical rganic Chemistry 6C50 Thursday November 5 004, 4.00-7.00 h This exam consists of four problems that have an equal weight in the final score Most problems are composed
Molecular Photophysics 330 Physical rganic Chemistry 6C50 Thursday November 5 004, 4.00-7.00 h This exam consists of four problems that have an equal weight in the final score Most problems are composed
ELEMENTARY BAND THEORY
 ELEMENTARY BAND THEORY PHYSICIST Solid state band Valence band, VB Conduction band, CB Fermi energy, E F Bloch orbital, delocalized n-doping p-doping Band gap, E g Direct band gap Indirect band gap Phonon
ELEMENTARY BAND THEORY PHYSICIST Solid state band Valence band, VB Conduction band, CB Fermi energy, E F Bloch orbital, delocalized n-doping p-doping Band gap, E g Direct band gap Indirect band gap Phonon
CHAPTER 13 LECTURE NOTES
 CHAPTER 13 LECTURE NOTES Spectroscopy is concerned with the measurement of (a) the wavelengths (or frequencies) at which molecules absorb/emit energy, and (b) the amount of radiation absorbed at these
CHAPTER 13 LECTURE NOTES Spectroscopy is concerned with the measurement of (a) the wavelengths (or frequencies) at which molecules absorb/emit energy, and (b) the amount of radiation absorbed at these
Models for Time-Dependent Phenomena
 Models for Time-Dependent Phenomena I. Phenomena in laser-matter interaction: atoms II. Phenomena in laser-matter interaction: molecules III. Model systems and TDDFT Manfred Lein p.1 Outline Phenomena
Models for Time-Dependent Phenomena I. Phenomena in laser-matter interaction: atoms II. Phenomena in laser-matter interaction: molecules III. Model systems and TDDFT Manfred Lein p.1 Outline Phenomena
one ν im: transition state saddle point
 Hypothetical Potential Energy Surface Ethane conformations Hartree-Fock theory, basis set stationary points all ν s >0: minimum eclipsed one ν im: transition state saddle point multiple ν im: hilltop 1
Hypothetical Potential Energy Surface Ethane conformations Hartree-Fock theory, basis set stationary points all ν s >0: minimum eclipsed one ν im: transition state saddle point multiple ν im: hilltop 1
CD Basis Set of Spectra that is used is that derived from comparing the spectra of globular proteins whose secondary structures are known from X-ray
 CD Basis Set of Spectra that is used is that derived from comparing the spectra of globular proteins whose secondary structures are known from X-ray crystallography An example of the use of CD Modeling
CD Basis Set of Spectra that is used is that derived from comparing the spectra of globular proteins whose secondary structures are known from X-ray crystallography An example of the use of CD Modeling
Figure 1: Transition State, Saddle Point, Reaction Pathway
 Computational Chemistry Workshops West Ridge Research Building-UAF Campus 9:00am-4:00pm, Room 009 Electronic Structure - July 19-21, 2016 Molecular Dynamics - July 26-28, 2016 Potential Energy Surfaces
Computational Chemistry Workshops West Ridge Research Building-UAF Campus 9:00am-4:00pm, Room 009 Electronic Structure - July 19-21, 2016 Molecular Dynamics - July 26-28, 2016 Potential Energy Surfaces