Quantum Mechanics - Molecular Mechanics (QM/MM) CHEM 430
|
|
|
- Ami Kathleen Ward
- 5 years ago
- Views:
Transcription
1 Quantum Mechanics - Molecular Mechanics (QM/MM), CHEM 430


2
3 Quantum Mechanics In theory, a very accurate treatment of the system Largely ab initio, i.e. parameter-free Very expensive typically scales as O(N 4 ) or worse Limited to very small systems at high accuracy (e.g. DFT) Can be used for larger systems at lower accuracy (e.g. semi-empirical) Entire proteins cannot be simulated without enormous supercomputer power
4 Molecular Mechanics Treats the electrons implicitly no handling of polarization or electron transfer Bonds, angles, and dihedrals are held by a parameterized force field V total = V bond + V angle + V dihed bonds angles dihed. + impr. V impr + i j (V vdw + V elec ) Can be used to simulate very large systems e.g. transmembrane proteins Cannot handle bond breaking or formation, so cannot be used to simulate chemical reactions
5 The QM/MM Modelling Approach Couple quantum mechanics and molecular mechanics approaches QM treatment of the active site reacting centre excited state processes (e.g. spectroscopy) problem structures (e.g. complex transition metal centre) Classical MM treatment of environment enzyme structure zeolite framework explicit solvent molecules bulky organometallic ligands
6 The QM/MM Idea Multi-layered method Monard, et. al, Acc. Chem. Res., 32, 904 (1999)
7 Hybrid QM/MM Combines quantum mechanical and molecular mechanical methods Treats just the reacting part of the system quantum mechanically, and uses MM for the surroundings Uses a combined Hamiltonian for the system: Ĥ total = ĤQM + ĤMM + ĤQM/MM QM region Ĥ QM QM/MM interaction potential Ĥ QM/MM MM region Boundary region Ĥ MM
8 Example Systems Study of serine protease deacylation reaction a Catalyzed isomerization of methylmalonyl-coa b a Topf, M., Várnai, P. and Richards, W. G. J. Am. Chem. Soc. 2002, 124, b Loferer, M., Webb, B., Grant, G, and Liedl, K. J. Am. Chem. Soc. 2003, 125, 1072
9 QM/MM Partitioning E = E QM + E MM + E QM/MM The tough part how do QM and MM interact? E QM = ψ Hˆ ψ ψψ Energy of MM subsystem Warshel and Levitt, J. Mol. Biol. Field, Bash and Karplus, J. Comp. Chem.
10 QM Region What should be used in the QM region? Ab Initio DFT Semiempirical Usually, the answer to this is dictated by cost. Most QM/MM simulations to date have used semiempirical QM regions Why? QM/MM interaction term can be problematic it is not good to have this boundary close to the chemistry of interest
11 Pitfalls in QM/MM V( R) = k r r + k + i bonds ( eq ) ( eq θ θ ) ibond, ibond, ibond, iangle, iangle, iangle, i angles qq + + V r r ( ) 1 i j 1 ij 2 2 LJ i j i j ri rj i j i, j atoms i, j atoms Not clear which force fields to use much experience with expected accuracy of ab initio methods alone and MM methods alone, but not much with QM/MM No direct map from wavefunction to parameters
 Charge perturbations (none, charge")
12 Hybrid Computational Schemes QM/MM Couplings M 2 M 1 L Q 2 Q 1 M 2 Q 2 Unpolarised or mechanical embedding Polarisation of QM region electrostatic embedding MM polarisation shell model or dipole polarisabilities Termination Scheme Link Atoms, or Boundary zone Chemical type (hydrogen atoms, pseudopotentials adjusted connection atoms, localised orbitals) Charge perturbations (none, charge deletion, charge shift, selection of 1e integrals, double link atoms) Total Energy Expression Additive, Uncorrected E(M,MM) + E(QL,QM) +E(QM/MM) Additive, Boundary corrected E(M,MM) + E(QL,QM) - E(L,MM) + Subtractive E(MQ,MM) + E(QL,QM) - E(QL,MM)
13 Mechanical Embedding Crudest level of QM/MM Include only Van der Waals in E QM/MM Useful to impose only steric constraints Can take advantage of this to isolate effects VdW HQM / MM = Vij ( ri, rj ) i MM j QM
14 Electrostatic Embedding Include electrostatic interaction in H QM/MM Many possible implementations best is to evaluate integrals over continuous QM charge density and discrete MM charge density ρ () QM r H = H + q dr r r mechanical QM / MM QM / MM i i MM Oft-used approximation (questionable): mechanical HQM / MM = HQM / MM + qq i j ( ρqm ) i MM j QM i
15 Atomic Charge Schemes Atoms are not well-defined in molecules there is no quantum mechanical operator corresponding to an atom. This leads to ambiguity in the definition of an atomic charge Population Analysis Schemes Basically, sum over all electrons using the basis functions of a given atom Depends on the atom-centered nature of the basis set Breaks down as the basis functions become more delocalized results do not usually converge with increasing basis set!
16 Charge Schemes Atoms-in-molecules Atoms are defined by critical points of the charge density More stable than Mulliken/Lowdin schemes with respect to basis set expansion Implemented in Gaussian Not clear whether stable= correct
17 Charge Schemes ESP-Fitting Determine charges which reproduce the electrostatic potential generated by the molecule If using charges in an MM potential, this appears to be the right way But, equations have many solutions, especially when molecule has an interior Charge for solvated ion will be essentially undetermined
18 Charge Schemes Restricted ESP-Fitting (RESP) Attempts to avoid unphysical solutions of ESPcharges Requires user guidance in imposing reasonable values of charges
19 Boundary Treatment How do we deal with bonds between the QM and MM regions? The valence of the QM region must be satisfied MM bond, angle, dihedral terms need a partner atom to act on, in order to maintain the geometry of the system QM/MM is often used to simulate a solute quantum mechanically, with explicit solvent treated with MM in this instance, the problem of QM-MM bonds is avoided
20 Covalent Embedding Most difficult embedding cutting across covalent bonds Almost always required in biological context Many strategies; still not clear which is best or whether any of them work
21 Link Atoms Conventional solution: link atoms (usually hydrogen atoms, but sometimes halogens or even methyl groups) are added along the bond a The link atom satisfies the valence of the QM region The QM atom is used for calculation of all MM bond terms For nonbond (electrostatic terms), originally the link atom did not interact with any MM atom (termed a QQ link in CHARMM parlance) Better properties are usually obtained if the link atom interacts with the entire MM region ( HQ link) Poor handling of electron density a Singh, U. and Kollman, P. J. Comput. Chem. 1986, 7, 718
22 Covalent Embedding Potential Problems with Link Atom Idea Extra degrees of freedom which somehow need to be removed; i.e. the link atom somehow needs to be connected to the MM part of the simulation Electronic structure at boundary will be very different if H and the atom it replaces do not have similar electronegativities
23 Covalent Embedding Thiel Adjust electronegativity of link atom to be equivalent to target atom. Also adjust size of atom Can only do this easily with semiempirical models Still can cause problems, especially with electronically excited states the 2s-3s transition of H-like atom is much lower than the 1s-2s transition!
24 Covalent Embedding Frozen orbital ideas:
 b includes the QM-MM orbitals in the SCF a Warshel, A. and Levitt, M. J. Mol. Biol. 1976, 103, 227 b Gao, J. et. al. J. Phys. Chem.")
25 Improved Bond Treatments Local Self-Consistent Field (LSCF) a uses a parameterized frozen orbital along the QM-MM bond, which is not optimized in the SCF Generalized Hybrid Orbital (GHO) b includes the QM-MM orbitals in the SCF a Warshel, A. and Levitt, M. J. Mol. Biol. 1976, 103, 227 b Gao, J. et. al. J. Phys. Chem. A 1998, 102, 4714




26 General QM/MM Methodology Two main strategies: Additive Method Subtractive Method
27 Link Atoms QM/MM with extra Link Atoms (L) to terminate broken covalent bonds L Inner L Outer L
28 Boundary Regions Boundary region approaches introduce no new atoms, e.g. Solid State Systems with ionic character Link atoms are inappropriate Inner Outer Boundary Range of representations within QM code inclide modified ab-initio atom with model potential, Semi-empirical parameterisation, Frozen orbitals, Design atoms Often associated with re-parameterised MM potentials
29 Additive Schemes Energy Expressions Without link atom correction E(O,MM) + E(I,QM) + E(IO,QM/MM) Link atom correction E(O,MM) + E(IL,QM) + E(IO,QM/MM) - E(L,MM) Boundary methods E(OB,MM) + E(IB,QM) + E(IBO,QM/MM) Highly variable in implementation QM/MM couplings, QM termination etc Advantages No requirement for forcefield for reacting centre Can naturally build in electrostatic polarisation of QM region - effects of environment of excitations etc Disadvantages Electrostatic coupling of the two regions, E(IO,QM/MM) is problematic with link atoms Need for boundary atom parameterisation
30 Subtractive Schemes Energy Expression E(OI,MM) + E(IL,QM) - E(IL,MM) includes link atom correction can treat polarisation of both the MM and QM regions at the force-field level Termination Any (provided a force field model for IL is available) Advantages Potentially highly accurate and free from artefacts Can also be used for QM/QM schemes (e.g. IMOMO, Morokuma et al) Disadvantages Need for accurate forcefields (mismatch of QM and MM models can generate catastrophes on potential energy surface) Usually no electrostatic influence on QM wavefunction included (e.g. QMPot), (but can be extended to electrostatic embedding: ONIOM-EE)
31 Other Approaches ONIOM a divides the system into the real (full) system and the model (subset) and treats the model at high level, and the real at low level, giving the total energy as E(high, real) E(low, real)+e(high, model) E(low, model) which relies on the approximation E(high, model) E(low, model) E(high, real) E(low, real) The model system still has to be properly terminated Extension to three level systems is relatively straightforward (e.g. ab initio core, semi-empirical boundary, MM surroundings) a Svensson, M. et. al. J. Phys. Chem. 1996, 100, 19357
32 Other Approaches Empirical Valence Bond method a treats any point on a reaction surface as a combination of two or more valence bond structures Parameterization is made from QM or experimental data An effective method, but must be carefully set up for each system Effective Fragment Potential b adds fragments to a standard QM treatment, which are fully polarizable and are parameterized from separate ab initio calculations Treatment of bonds between the true QM region and the fragments is still problematic a Warshel, A. and Weiss, M. J. Am. Chem. Soc. 1980, 102, 6218 b Webb, P. and Gordon, M. J. Phys. Chem. 1999, 103, 1265
33 Adaptive QM/MM Schemes (i) Change of the QM region during the simulation Potential for discontinuity in energy and forces Generally based on principle that forces on atoms in a buffer region are interpolated between QM values and MM values, depending on the distance from QM zone Rode s Hot Spot method Kerdcharoen, Liedl, and Rode, Chem Phys 1996, 211, 313. ONIOM-XS Kerdcharoen, and Morokuma, Chem Phys Lett 2002, 355, 257. LOTF schemes (MM with on-the-fly parameterisation) Csanyi, Albaret, Payne, De Vita, Phys. Rev. Lett. 2004, 93,
34 Adaptive QM/MM Schemes (ii) Truhlar s schemes define a conserved potential energy by performing multiple QM/MM calculations (permuting boundary molecules between QM and MM zones) with geometry dependent weights to apply interpolation Number of possible contributions is 2 N (where N molecules in the boundary zone) Schemes linear in N are also possible, weighting functions are quite complex Heyden, Lin, and Truhlar, J. Phys. Chem. B, 2007, 111, Bulo, Ensing, Sikkema, and Visscher, J. Chem. Theory Comput. 2009, 5, Takenaka, Kitamura, Koyano and Nagaoka, M. Chem Phys Lett 2012, 524, 56.
35 Summary of Current Approaches
36 Dynamics Chemical reactions are often simulated by molecular dynamics, e.g. with umbrella sampling Dynamics of a QM/MM system are almost identical to those of an MM system: Forces are calculated from first derivatives on each atom The QM nuclei are treated identically to the MM partial charges The system is propagated by standard Newtonian dynamics
37 Monte Carlo QM/MM can also be used in conjunction with Monte Carlo methods A complication: the MM atoms affect the QM electron density, so an SCF is required for every Monte Carlo move Workaround: approximate the energy change of the QM region by first-order perturbation theory ( Perturbative QM/MC ) as long as moves are far enough away from the QM region a a Truong, T. and Stefanovich, E. Chem. Phys. Lett. 1996, 256, 348
38 Drawbacks of QM/MM Some parameterization is still required for the boundary treatment The choice of the size of the QM region is still something of an art Although the QM region polarizes in response to the MM partial charges, the reverse is not also true (although fully polarizable QM/MM methods are being developed) The free energy of a QM system can be determined via frequency calculation; however, this is rather inaccurate when applied to QM/MM systems (second derivatives are poorly determined, e.g. due to the harmonic approximation)
39 Cautions Most force fields do not include polarizability, but QM region will This can lead to imbalance and amplification of errors All covalent embedding schemes should be treated with caution it is surely possible to break almost every implemented scheme One needs to test carefully the dependence of the results on the QM/MM partitioning
40 Coarser than QM/MM? Continuum solvation models treat solvent as a dielectric continuum (PCM=Polarizable Continuum Model; SCRF=Self- Consistent Reaction Field)
41 Continuum Solvation Algorithm: Compute reaction field polarization of dielectric continuum which generates electric field acting on solute Compute electronic wavefunction in presence of new solvent-generated field Loop until reaction field does not change Issues: Shape of cavity (spherical and ellipsoidal are rarely acceptable at present) Dielectric of solvent zero vs infinite frequency? H-bonding between solvent and solute will not be properly represented Atomic radii used to generate cavity

42 Supermolecule Approach Explicit solvent molecules in first solvation shell Surround with dielectric continuum Expensive, but can be very accurate Not feasible if solute is very large Related approach QM/MM/PCM


43 Chorismate Mutase Plays a key role in the shikimate pathway of bacteria, fungi, and other higher plants
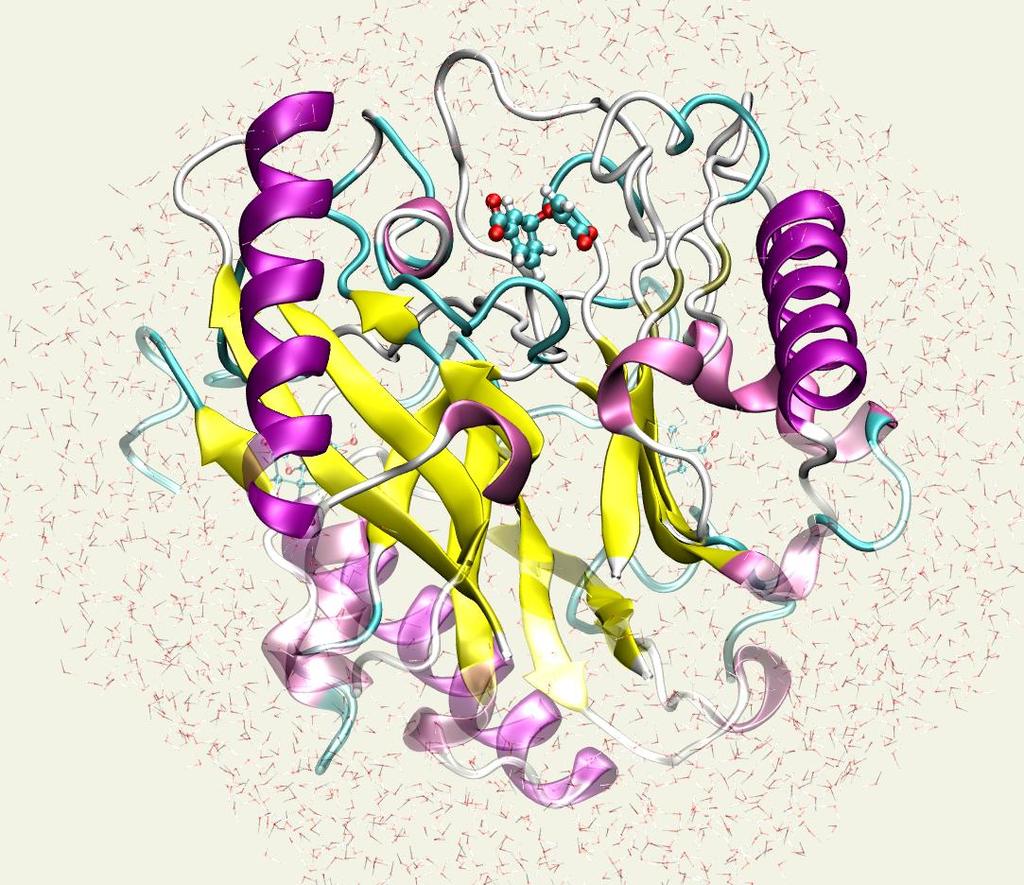
44 Chorismate Mutase



45 DG 1 DG DG RXN DG 2

46 Reaction Path Methods Eigenvector Following Methods: Typically require transition state to be known a priori Too expensive for high dimensional systems Reaction Coordinate Driving: Cons: Predetermined reaction coordinate Usually some linear combination of distances Gradually changed Difficult or impossible to define reaction coordinate Hysteresis: requires repeated walks to resolve Sequential method: inefficient use of modern computational resources Chain-of-replica Methods: Path is defined as discrete structures from reactant to product Removes predetermination of reaction coordinate Restraints are applied to force points to be minima in all directions except path Can take advantage of parallel computers (i.e. Beowulf cluster)add an outline
47 The Replica Path Method


48 The Replica Path Method


49 The Replica Path Method Define X number of steps to describe the pathway of interest

50

51


52 H = 6.1 H = -18.5


53 H = 6.0 H = -18.9
54 What Next? Need to compute free energies! Methodology? Can we use the Replica Path Method? Simulation methods? Harmonic methods? new methods to explore this...
55 Off-Path Simulation Method for Computing Free Energy Barriers
56 Off-Path Simulation Results: Butane at 300K
57 Off-Path Simulation Results: Maltose at 300K
58 Conclusions Replica Path Method Chorismate Mutase reaction profile Examined methodological dependence Showed the role of Arg63 in Chorismate Mutase is NOT catalytic Same Environment, Multiple State Method (SEMS) Vibrational Subsystem Analysis (VSA) Off-Path Simulation Method Butane: quantitative agreement between OPS PMF and brute force PMF Maltose: Good agreement between OPS and umbrella sampling Additional Developments...
59 What Next? Need to compute free energies! Methodology? Can we use the Replica Path Method? Simulation methods? Harmonic methods? new methods to explore this...

60 Off-Path Simulation Method for Computing Free Energy Barriers







61 Off-Path Simulation Results: Butane at 300K

62 Off-Path Simulation Results: Maltose at 300K
63 Conclusions Replica Path Method Chorismate Mutase reaction profile Examined methodological dependence Showed the role of Arg63 in Chorismate Mutase is NOT catalytic Same Environment, Multiple State Method (SEMS) Vibrational Subsystem Analysis (VSA) Off-Path Simulation Method Butane: quantitative agreement between OPS PMF and brute force PMF Maltose: Good agreement between OPS and umbrella sampling Additional Developments...
A new extension of QM/MM methods: the adaptive buffered-force QM/MM method
 A new extension of QM/MM methods: the adaptive buffered-force QM/MM method Letif Mones Engineering Department, University of Cambridge lam81@cam.ac.uk Overview Basic concept of the QM/MM methods MM vs.
A new extension of QM/MM methods: the adaptive buffered-force QM/MM method Letif Mones Engineering Department, University of Cambridge lam81@cam.ac.uk Overview Basic concept of the QM/MM methods MM vs.
Multiscale Materials Modeling
 Multiscale Materials Modeling Lecture 09 Quantum Mechanics/Molecular Mechanics (QM/MM) Techniques Fundamentals of Sustainable Technology These notes created by David Keffer, University of Tennessee, Knoxville,
Multiscale Materials Modeling Lecture 09 Quantum Mechanics/Molecular Mechanics (QM/MM) Techniques Fundamentals of Sustainable Technology These notes created by David Keffer, University of Tennessee, Knoxville,
QM/MM: what have we learned, where are we, and where do we go from here?
 Theor Chem Acc (2007) 117: 185 199 DOI 10.1007/s00214-006-0143-z FEATURE ARTICLE Hai Lin Donald G. Truhlar QM/MM: what have we learned, where are we, and where do we go from here? Received: 25 May 2005
Theor Chem Acc (2007) 117: 185 199 DOI 10.1007/s00214-006-0143-z FEATURE ARTICLE Hai Lin Donald G. Truhlar QM/MM: what have we learned, where are we, and where do we go from here? Received: 25 May 2005
Free Energy Simulation Methods
 Free Energy Simulation Methods Free energy simulation methods Many methods have been developed to compute (relative) free energies on the basis of statistical mechanics Free energy perturbation Thermodynamic
Free Energy Simulation Methods Free energy simulation methods Many methods have been developed to compute (relative) free energies on the basis of statistical mechanics Free energy perturbation Thermodynamic
Multiscale Materials Modeling
 Multiscale Materials Modeling Lecture 10 Adaptive Resolution Fundamentals of Sustainable Technology These notes created by David Keffer, University of Tennessee, Knoxville, 2012. Reminder In the previous
Multiscale Materials Modeling Lecture 10 Adaptive Resolution Fundamentals of Sustainable Technology These notes created by David Keffer, University of Tennessee, Knoxville, 2012. Reminder In the previous
Session 1. Introduction to Computational Chemistry. Computational (chemistry education) and/or (Computational chemistry) education
 and/or (Computational chemistry) education") Session 1 Introduction to Computational Chemistry 1 Introduction to Computational Chemistry Computational (chemistry education) and/or (Computational chemistry) education First one: Use computational tools
Session 1 Introduction to Computational Chemistry 1 Introduction to Computational Chemistry Computational (chemistry education) and/or (Computational chemistry) education First one: Use computational tools
Modeling in Solution. Important Observations. Energy Concepts
 Modeling in Solution 1 Important Observations Various electrostatic effects for a solvated molecule are often less important than for an isolated gaseous molecule when the molecule is dissolved in a solvent
Modeling in Solution 1 Important Observations Various electrostatic effects for a solvated molecule are often less important than for an isolated gaseous molecule when the molecule is dissolved in a solvent
Dihedral Angles. Homayoun Valafar. Department of Computer Science and Engineering, USC 02/03/10 CSCE 769
 Dihedral Angles Homayoun Valafar Department of Computer Science and Engineering, USC The precise definition of a dihedral or torsion angle can be found in spatial geometry Angle between to planes Dihedral
Dihedral Angles Homayoun Valafar Department of Computer Science and Engineering, USC The precise definition of a dihedral or torsion angle can be found in spatial geometry Angle between to planes Dihedral
Structural Bioinformatics (C3210) Molecular Mechanics
 Molecular Mechanics") Structural Bioinformatics (C3210) Molecular Mechanics How to Calculate Energies Calculation of molecular energies is of key importance in protein folding, molecular modelling etc. There are two main computational
Structural Bioinformatics (C3210) Molecular Mechanics How to Calculate Energies Calculation of molecular energies is of key importance in protein folding, molecular modelling etc. There are two main computational
QM/MM Theory and Examples with ORCA
 QM/MM Theory and Examples with ORCA Marius Retegan Max Planck Institute for Chemical Energy Conversion Stiftstr. 34-36 Mülheim an der Ruhr QM/MM: an historical overview 1976: Warshell and Levitt Theoretical
QM/MM Theory and Examples with ORCA Marius Retegan Max Planck Institute for Chemical Energy Conversion Stiftstr. 34-36 Mülheim an der Ruhr QM/MM: an historical overview 1976: Warshell and Levitt Theoretical
Molecular Mechanics. I. Quantum mechanical treatment of molecular systems
 Molecular Mechanics I. Quantum mechanical treatment of molecular systems The first principle approach for describing the properties of molecules, including proteins, involves quantum mechanics. For example,
Molecular Mechanics I. Quantum mechanical treatment of molecular systems The first principle approach for describing the properties of molecules, including proteins, involves quantum mechanics. For example,
Combining Quantum Mechanics Methods with Molecular Mechanics Methods in ONIOM
 J. Chem. Theory Comput. 2006, 2, 815-826 815 Combining Quantum Mechanics Methods with Molecular Mechanics Methods in ONIOM Thom Vreven,*,, K. Suzie Byun, István Komáromi,, Stefan Dapprich, John A. Montgomery,
J. Chem. Theory Comput. 2006, 2, 815-826 815 Combining Quantum Mechanics Methods with Molecular Mechanics Methods in ONIOM Thom Vreven,*,, K. Suzie Byun, István Komáromi,, Stefan Dapprich, John A. Montgomery,
Biomolecular modeling I
 2016, December 6 Biomolecular structure Structural elements of life Biomolecules proteins, nucleic acids, lipids, carbohydrates... Biomolecular structure Biomolecules biomolecular complexes aggregates...
2016, December 6 Biomolecular structure Structural elements of life Biomolecules proteins, nucleic acids, lipids, carbohydrates... Biomolecular structure Biomolecules biomolecular complexes aggregates...
Citation for the original published paper (version of record):
:") http://www.diva-portal.org Postprint This is the accepted version of a paper published in Journal of Computational Chemistry. This paper has been peer-reviewed but does not include the final publisher
http://www.diva-portal.org Postprint This is the accepted version of a paper published in Journal of Computational Chemistry. This paper has been peer-reviewed but does not include the final publisher
Density Functional Theory: from theory to Applications
 Density Functional Theory: from theory to Applications Uni Mainz January 17, 2011 CP-PAW/COSMO Interface Mixed quantum/ classical molecular dynamics (QM/MM) Quantum mechanics is computationally expensive
Density Functional Theory: from theory to Applications Uni Mainz January 17, 2011 CP-PAW/COSMO Interface Mixed quantum/ classical molecular dynamics (QM/MM) Quantum mechanics is computationally expensive
Example questions for Molecular modelling (Level 4) Dr. Adrian Mulholland
 Dr. Adrian Mulholland") Example questions for Molecular modelling (Level 4) Dr. Adrian Mulholland 1) Question. Two methods which are widely used for the optimization of molecular geometies are the Steepest descents and Newton-Raphson
Example questions for Molecular modelling (Level 4) Dr. Adrian Mulholland 1) Question. Two methods which are widely used for the optimization of molecular geometies are the Steepest descents and Newton-Raphson
QUANTUM MECHANICAL METHODS FOR ENZYME KINETICS
 Annu. Rev. Phys. Chem. 2002. 53:467 505 First published as a Review In Advance on 20 December 2001. DOI: 10.1146/annurev.physchem.53.091301.150114 Copyright c 2002 by Annual Reviews. All rights reserved
Annu. Rev. Phys. Chem. 2002. 53:467 505 First published as a Review In Advance on 20 December 2001. DOI: 10.1146/annurev.physchem.53.091301.150114 Copyright c 2002 by Annual Reviews. All rights reserved
k θ (θ θ 0 ) 2 angles r i j r i j
 2 angles r i j r i j") 1 Force fields 1.1 Introduction The term force field is slightly misleading, since it refers to the parameters of the potential used to calculate the forces (via gradient) in molecular dynamics simulations.
1 Force fields 1.1 Introduction The term force field is slightly misleading, since it refers to the parameters of the potential used to calculate the forces (via gradient) in molecular dynamics simulations.
All-atom Molecular Mechanics. Trent E. Balius AMS 535 / CHE /27/2010
 All-atom Molecular Mechanics Trent E. Balius AMS 535 / CHE 535 09/27/2010 Outline Molecular models Molecular mechanics Force Fields Potential energy function functional form parameters and parameterization
All-atom Molecular Mechanics Trent E. Balius AMS 535 / CHE 535 09/27/2010 Outline Molecular models Molecular mechanics Force Fields Potential energy function functional form parameters and parameterization
Scuola di Chimica Computazionale
 Societa Chimica Italiana Gruppo Interdivisionale di Chimica Computazionale Scuola di Chimica Computazionale Introduzione, per Esercizi, all Uso del Calcolatore in Chimica Organica e Biologica Modellistica
Societa Chimica Italiana Gruppo Interdivisionale di Chimica Computazionale Scuola di Chimica Computazionale Introduzione, per Esercizi, all Uso del Calcolatore in Chimica Organica e Biologica Modellistica
Computational Methods. Chem 561
 Computational Methods Chem 561 Lecture Outline 1. Ab initio methods a) HF SCF b) Post-HF methods 2. Density Functional Theory 3. Semiempirical methods 4. Molecular Mechanics Computational Chemistry " Computational
Computational Methods Chem 561 Lecture Outline 1. Ab initio methods a) HF SCF b) Post-HF methods 2. Density Functional Theory 3. Semiempirical methods 4. Molecular Mechanics Computational Chemistry " Computational
Interatomic Potentials. The electronic-structure problem
 Interatomic Potentials Before we can start a simulation, we need the model! Interaction between atoms and molecules is determined by quantum mechanics: Schrödinger Equation + Born-Oppenheimer approximation
Interatomic Potentials Before we can start a simulation, we need the model! Interaction between atoms and molecules is determined by quantum mechanics: Schrödinger Equation + Born-Oppenheimer approximation
Computational Chemistry. An Introduction to Molecular Dynamic Simulations
 Computational Chemistry An Introduction to Molecular Dynamic Simulations Computational chemistry simulates chemical structures and reactions numerically, based in full or in part on the fundamental laws
Computational Chemistry An Introduction to Molecular Dynamic Simulations Computational chemistry simulates chemical structures and reactions numerically, based in full or in part on the fundamental laws
An introduction to Molecular Dynamics. EMBO, June 2016
 An introduction to Molecular Dynamics EMBO, June 2016 What is MD? everything that living things do can be understood in terms of the jiggling and wiggling of atoms. The Feynman Lectures in Physics vol.
An introduction to Molecular Dynamics EMBO, June 2016 What is MD? everything that living things do can be understood in terms of the jiggling and wiggling of atoms. The Feynman Lectures in Physics vol.
Computational Studies of the Photoreceptor Rhodopsin. Scott E. Feller Wabash College
 Computational Studies of the Photoreceptor Rhodopsin Scott E. Feller Wabash College Rhodopsin Photocycle Dark-adapted Rhodopsin hn Isomerize retinal Photorhodopsin ~200 fs Bathorhodopsin Meta-II ms timescale
Computational Studies of the Photoreceptor Rhodopsin Scott E. Feller Wabash College Rhodopsin Photocycle Dark-adapted Rhodopsin hn Isomerize retinal Photorhodopsin ~200 fs Bathorhodopsin Meta-II ms timescale
Why Is Molecular Interaction Important in Our Life
 Why Is Molecular Interaction Important in ur Life QuLiS and Graduate School of Science iroshima University http://www.nabit.hiroshima-u.ac.jp/iwatasue/indexe.htm Suehiro Iwata Sept. 29, 2007 Department
Why Is Molecular Interaction Important in ur Life QuLiS and Graduate School of Science iroshima University http://www.nabit.hiroshima-u.ac.jp/iwatasue/indexe.htm Suehiro Iwata Sept. 29, 2007 Department
Chemistry 4560/5560 Molecular Modeling Fall 2014
 Final Exam Name:. User s guide: 1. Read questions carefully and make sure you understand them before answering (if not, ask). 2. Answer only the question that is asked, not a different question. 3. Unless
Final Exam Name:. User s guide: 1. Read questions carefully and make sure you understand them before answering (if not, ask). 2. Answer only the question that is asked, not a different question. 3. Unless
Molecular mechanics. classical description of molecules. Marcus Elstner and Tomáš Kubař. April 29, 2016
 classical description of molecules April 29, 2016 Chemical bond Conceptual and chemical basis quantum effect solution of the SR numerically expensive (only small molecules can be treated) approximations
classical description of molecules April 29, 2016 Chemical bond Conceptual and chemical basis quantum effect solution of the SR numerically expensive (only small molecules can be treated) approximations
Available online at ScienceDirect. Procedia Materials Science 6 (2014 )
") Available online at www.sciencedirect.com ScienceDirect Procedia Materials Science 6 (2014 ) 256 264 3rd International Conference on Materials Processing and Characterisation (ICMPC 2014) A new approach
Available online at www.sciencedirect.com ScienceDirect Procedia Materials Science 6 (2014 ) 256 264 3rd International Conference on Materials Processing and Characterisation (ICMPC 2014) A new approach
Lecture 11: Potential Energy Functions
 Lecture 11: Potential Energy Functions Dr. Ronald M. Levy ronlevy@temple.edu Originally contributed by Lauren Wickstrom (2011) Microscopic/Macroscopic Connection The connection between microscopic interactions
Lecture 11: Potential Energy Functions Dr. Ronald M. Levy ronlevy@temple.edu Originally contributed by Lauren Wickstrom (2011) Microscopic/Macroscopic Connection The connection between microscopic interactions
Free energy simulations
 Free energy simulations Marcus Elstner and Tomáš Kubař January 14, 2013 Motivation a physical quantity that is of most interest in chemistry? free energies Helmholtz F or Gibbs G holy grail of computational
Free energy simulations Marcus Elstner and Tomáš Kubař January 14, 2013 Motivation a physical quantity that is of most interest in chemistry? free energies Helmholtz F or Gibbs G holy grail of computational
Close agreement between the orientation dependence of hydrogen bonds observed in protein structures and quantum mechanical calculations
 Close agreement between the orientation dependence of hydrogen bonds observed in protein structures and quantum mechanical calculations Alexandre V. Morozov, Tanja Kortemme, Kiril Tsemekhman, David Baker
Close agreement between the orientation dependence of hydrogen bonds observed in protein structures and quantum mechanical calculations Alexandre V. Morozov, Tanja Kortemme, Kiril Tsemekhman, David Baker
Methods and Code Integration. Ryan M. Olson University of Minnesota
 Methods and Code Integration Ryan M. Olson University of Minnesota Overview Methods Electrostatically Embedded Many-Body Method Adaptive Partitioning Configurational-Biased Grand Canonical Monte Carlo
Methods and Code Integration Ryan M. Olson University of Minnesota Overview Methods Electrostatically Embedded Many-Body Method Adaptive Partitioning Configurational-Biased Grand Canonical Monte Carlo
Advanced Quantum Chemistry III: Part 6
 Advanced Quantum Chemistry III: Part 6 Norio Yoshida Kyushu University Last updated 16-1-6 2015 Winter Term 1 Quantum Chemistry for Condensed Phase Liquid phase Solid phase Biological systems 2 Divide
Advanced Quantum Chemistry III: Part 6 Norio Yoshida Kyushu University Last updated 16-1-6 2015 Winter Term 1 Quantum Chemistry for Condensed Phase Liquid phase Solid phase Biological systems 2 Divide
We can model covalent bonding in molecules in essentially two ways:
 CHEM 2060 Lecture 22: VB Theory L22-1 PART FIVE: The Covalent Bond We can model covalent bonding in molecules in essentially two ways: 1. Localized Bonds (retains electron pair concept of Lewis Structures)
CHEM 2060 Lecture 22: VB Theory L22-1 PART FIVE: The Covalent Bond We can model covalent bonding in molecules in essentially two ways: 1. Localized Bonds (retains electron pair concept of Lewis Structures)
Can a continuum solvent model reproduce the free energy landscape of a β-hairpin folding in water?
 Can a continuum solvent model reproduce the free energy landscape of a β-hairpin folding in water? Ruhong Zhou 1 and Bruce J. Berne 2 1 IBM Thomas J. Watson Research Center; and 2 Department of Chemistry,
Can a continuum solvent model reproduce the free energy landscape of a β-hairpin folding in water? Ruhong Zhou 1 and Bruce J. Berne 2 1 IBM Thomas J. Watson Research Center; and 2 Department of Chemistry,
2/28/2011. Chapter 6 An Overview of Organic Reactions. Organic Chemical Reactions. 6.1 Kinds of Organic Reactions
 John E. McMurry http://www.cengage.com/chemistry/mcmurry Chapter 6 An Overview of Organic Reactions CHP 6 Problems: 6.1-13, 17-36. Richard Morrison University of Georgia, Athens Organic Chemical Reactions
John E. McMurry http://www.cengage.com/chemistry/mcmurry Chapter 6 An Overview of Organic Reactions CHP 6 Problems: 6.1-13, 17-36. Richard Morrison University of Georgia, Athens Organic Chemical Reactions
Interfacing Q-Chem and CHARMM to Perform QM/MM Reaction Path Calculations*
 Interfacing Q-Chem and CHARMM to Perform QM/MM Reaction Path Calculations* H. LEE WOODCOCK III, 1 MILAN HODO S CEK, 2 ANDREW T. B. GILBERT, 3 PETER M. W. GILL, 3 HENRY F. SCHAEFER III, 4 BERNARD R. BROOKS
Interfacing Q-Chem and CHARMM to Perform QM/MM Reaction Path Calculations* H. LEE WOODCOCK III, 1 MILAN HODO S CEK, 2 ANDREW T. B. GILBERT, 3 PETER M. W. GILL, 3 HENRY F. SCHAEFER III, 4 BERNARD R. BROOKS
Molecular Mechanics. Yohann Moreau. November 26, 2015
 Molecular Mechanics Yohann Moreau yohann.moreau@ujf-grenoble.fr November 26, 2015 Yohann Moreau (UJF) Molecular Mechanics, Label RFCT 2015 November 26, 2015 1 / 29 Introduction A so-called Force-Field
Molecular Mechanics Yohann Moreau yohann.moreau@ujf-grenoble.fr November 26, 2015 Yohann Moreau (UJF) Molecular Mechanics, Label RFCT 2015 November 26, 2015 1 / 29 Introduction A so-called Force-Field
Solutions and Non-Covalent Binding Forces
 Chapter 3 Solutions and Non-Covalent Binding Forces 3.1 Solvent and solution properties Molecules stick together using the following forces: dipole-dipole, dipole-induced dipole, hydrogen bond, van der
Chapter 3 Solutions and Non-Covalent Binding Forces 3.1 Solvent and solution properties Molecules stick together using the following forces: dipole-dipole, dipole-induced dipole, hydrogen bond, van der
Q-Chem 5: Facilitating Worldwide Scientific Breakthroughs
 Q-Chem 5: Facilitating Worldwide Scientific Breakthroughs Founded in 1993, Q-Chem strives to bring its customers state-ofthe-art methods and algorithms for performing quantum chemistry calculations. Cutting-edge
Q-Chem 5: Facilitating Worldwide Scientific Breakthroughs Founded in 1993, Q-Chem strives to bring its customers state-ofthe-art methods and algorithms for performing quantum chemistry calculations. Cutting-edge
Intermolecular Forces in Density Functional Theory
 Intermolecular Forces in Density Functional Theory Problems of DFT Peter Pulay at WATOC2005: There are 3 problems with DFT 1. Accuracy does not converge 2. Spin states of open shell systems often incorrect
Intermolecular Forces in Density Functional Theory Problems of DFT Peter Pulay at WATOC2005: There are 3 problems with DFT 1. Accuracy does not converge 2. Spin states of open shell systems often incorrect
An Introduction to Quantum Chemistry and Potential Energy Surfaces. Benjamin G. Levine
 An Introduction to Quantum Chemistry and Potential Energy Surfaces Benjamin G. Levine This Week s Lecture Potential energy surfaces What are they? What are they good for? How do we use them to solve chemical
An Introduction to Quantum Chemistry and Potential Energy Surfaces Benjamin G. Levine This Week s Lecture Potential energy surfaces What are they? What are they good for? How do we use them to solve chemical
TD!DFT applied to biological problems. Marcus Elstner TU Braunschweig
 TD!DFT applied to biological problems Marcus Elstner TU Braunschweig 0) biological structures Biological structures: proteins, DNA, lipids N lipids Amino acids: 20 DNA bases: 4 (A,C,G,T) Proteins - contain
TD!DFT applied to biological problems Marcus Elstner TU Braunschweig 0) biological structures Biological structures: proteins, DNA, lipids N lipids Amino acids: 20 DNA bases: 4 (A,C,G,T) Proteins - contain
DEVELOPMENT OF MULTISCALE MODELS FOR COMPLEX CHEMICAL SYSTEMS
 9 OCTOBER 2013 Scientific Background on the Nobel Prize in Chemistry 2013 DEVELOPMENT OF MULTISCALE MODELS FOR COMPLEX CHEMICAL SYSTEMS THE ROYAL SWEDISH ACADEMY OF SCIENCES has as its aim to promote the
9 OCTOBER 2013 Scientific Background on the Nobel Prize in Chemistry 2013 DEVELOPMENT OF MULTISCALE MODELS FOR COMPLEX CHEMICAL SYSTEMS THE ROYAL SWEDISH ACADEMY OF SCIENCES has as its aim to promote the
Advanced Quantum Chemistry III: Part 6
 Advanced Quantum Chemistry III: Part 6 Norio Yoshida Kyushu University Last updated 14-1-15 2013 Winter Term 1 Quantum Chemistry for Condensed Phase Liquid phase Solid phase Biological systems 2 Divide
Advanced Quantum Chemistry III: Part 6 Norio Yoshida Kyushu University Last updated 14-1-15 2013 Winter Term 1 Quantum Chemistry for Condensed Phase Liquid phase Solid phase Biological systems 2 Divide
ONETEP PB/SA: Application to G-Quadruplex DNA Stability. Danny Cole
 ONETEP PB/SA: Application to G-Quadruplex DNA Stability Danny Cole Introduction Historical overview of structure and free energy calculation of complex molecules using molecular mechanics and continuum
ONETEP PB/SA: Application to G-Quadruplex DNA Stability Danny Cole Introduction Historical overview of structure and free energy calculation of complex molecules using molecular mechanics and continuum
Statistical Mechanics for Proteins
 The Partition Function From Q all relevant thermodynamic properties can be obtained by differentiation of the free energy F: = kt q p E q pd d h T V Q ), ( exp 1! 1 ),, ( 3 3 3 ),, ( ln ),, ( T V Q kt
The Partition Function From Q all relevant thermodynamic properties can be obtained by differentiation of the free energy F: = kt q p E q pd d h T V Q ), ( exp 1! 1 ),, ( 3 3 3 ),, ( ln ),, ( T V Q kt
Theory of electron transfer. Winterschool for Theoretical Chemistry and Spectroscopy Han-sur-Lesse, Belgium, December 2011
 Theory of electron transfer Winterschool for Theoretical Chemistry and Spectroscopy Han-sur-Lesse, Belgium, 12-16 December 2011 Electron transfer Electrolyse Battery Anode (oxidation): 2 H2O(l) O2(g) +
Theory of electron transfer Winterschool for Theoretical Chemistry and Spectroscopy Han-sur-Lesse, Belgium, 12-16 December 2011 Electron transfer Electrolyse Battery Anode (oxidation): 2 H2O(l) O2(g) +
Fragmentation methods
 Fragmentation methods Scaling of QM Methods HF, DFT scale as N 4 MP2 scales as N 5 CC methods scale as N 7 What if we could freeze the value of N regardless of the size of the system? Then each method
Fragmentation methods Scaling of QM Methods HF, DFT scale as N 4 MP2 scales as N 5 CC methods scale as N 7 What if we could freeze the value of N regardless of the size of the system? Then each method
74 these states cannot be reliably obtained from experiments. In addition, the barriers between the local minima can also not be obtained reliably fro
 73 Chapter 5 Development of Adiabatic Force Field for Polyvinyl Chloride (PVC) and Chlorinated PVC (CPVC) 5.1 Introduction Chlorinated polyvinyl chloride has become an important specialty polymer due to
73 Chapter 5 Development of Adiabatic Force Field for Polyvinyl Chloride (PVC) and Chlorinated PVC (CPVC) 5.1 Introduction Chlorinated polyvinyl chloride has become an important specialty polymer due to
Integrated Tools for Computational Chemical Dynamics
 Integrated Tools for Computational Chemical Dynamics Research Tools Design Consortium Department of Chemistry, UM Chemical & Materials Sciences Div. and Environmental Molecular Sci. Lab Participants U
Integrated Tools for Computational Chemical Dynamics Research Tools Design Consortium Department of Chemistry, UM Chemical & Materials Sciences Div. and Environmental Molecular Sci. Lab Participants U
Molecular Mechanics Based on the Combined Density
 Prepared for J. Chem. Theory Comput. Electrostatically Embedded Multiconfiguration Revised 02/13/2008 Molecular Mechanics Based on the Combined Density Functional and Molecular Mechanical Method Masahiro
Prepared for J. Chem. Theory Comput. Electrostatically Embedded Multiconfiguration Revised 02/13/2008 Molecular Mechanics Based on the Combined Density Functional and Molecular Mechanical Method Masahiro
1. Reactions can be followed by measuring changes in concentration, mass and volume of reactants and products.
 Higher Chemistry - Traffic Lights Unit 1 CHEMICAL CHANGES AND STRUCTURE I know: Controlling the rate Collision theory and relative rates 1. Reactions can be followed by measuring changes in concentration,
Higher Chemistry - Traffic Lights Unit 1 CHEMICAL CHANGES AND STRUCTURE I know: Controlling the rate Collision theory and relative rates 1. Reactions can be followed by measuring changes in concentration,
Theoretical models for the solvent effect
 Theoretical models for the solvent effect Benedetta Mennucci Dipartimento di Chimica e Chimica Industriale Web: http://benedetta.dcci.unipi.it Email: bene@dcci.unipi.it 8. Excited electronic states in
Theoretical models for the solvent effect Benedetta Mennucci Dipartimento di Chimica e Chimica Industriale Web: http://benedetta.dcci.unipi.it Email: bene@dcci.unipi.it 8. Excited electronic states in
Chemical Bonding. The Octet Rule
 Chemical Bonding There are basically two types of chemical bonds: 1. Covalent bonds electrons are shared by more than one nucleus 2. Ionic bonds electrostatic attraction between ions creates chemical bond
Chemical Bonding There are basically two types of chemical bonds: 1. Covalent bonds electrons are shared by more than one nucleus 2. Ionic bonds electrostatic attraction between ions creates chemical bond
A practical guide to modelling enzyme-catalysed reactions
 Chem Soc Rev View Article Online / Journal Homepage / Table of Contents for this issue Dynamic Article Links Cite this: Chem. Soc. Rev., 2012, 41, 3025 3038 www.rsc.org/csr TUTORIAL REVIEW A practical
Chem Soc Rev View Article Online / Journal Homepage / Table of Contents for this issue Dynamic Article Links Cite this: Chem. Soc. Rev., 2012, 41, 3025 3038 www.rsc.org/csr TUTORIAL REVIEW A practical
Chapter 7: Chemical Bonding and Molecular Structure
 Chapter 7: Chemical Bonding and Molecular Structure Ionic Bond Covalent Bond Electronegativity and Bond Polarity Lewis Structures Orbital Overlap Hybrid Orbitals The Shapes of Molecules (VSEPR Model) Molecular
Chapter 7: Chemical Bonding and Molecular Structure Ionic Bond Covalent Bond Electronegativity and Bond Polarity Lewis Structures Orbital Overlap Hybrid Orbitals The Shapes of Molecules (VSEPR Model) Molecular
Water models in classical simulations
 Water models in classical simulations Maria Fyta Institut für Computerphysik, Universität Stuttgart Stuttgart, Germany Water transparent, odorless, tasteless and ubiquitous really simple: two H atoms attached
Water models in classical simulations Maria Fyta Institut für Computerphysik, Universität Stuttgart Stuttgart, Germany Water transparent, odorless, tasteless and ubiquitous really simple: two H atoms attached
Introduction to Computational Chemistry Computational (chemistry education) and/or. (Computational chemistry) education
 and/or. (Computational chemistry) education") Introduction to Computational Chemistry Computational (chemistry education) and/or (Computational chemistry) education First one: Use computational tools to help increase student understanding of material
Introduction to Computational Chemistry Computational (chemistry education) and/or (Computational chemistry) education First one: Use computational tools to help increase student understanding of material
Development of a ReaxFF Reactive Force Field for Glycine and Application to Solvent Effect and Tautomerization
 J. Phys. Chem. B 2011, 115, 249 261 249 Development of a ReaxFF Reactive Force Field for Glycine and Application to Solvent Effect and Tautomerization Obaidur Rahaman, Adri C. T. van Duin, William A. Goddard
J. Phys. Chem. B 2011, 115, 249 261 249 Development of a ReaxFF Reactive Force Field for Glycine and Application to Solvent Effect and Tautomerization Obaidur Rahaman, Adri C. T. van Duin, William A. Goddard
Advanced in silico drug design
 Advanced in silico drug design RNDr. Martin Lepšík, Ph.D. Lecture: Advanced scoring Palacky University, Olomouc 2016 1 Outline 1. Scoring Definition, Types 2. Physics-based Scoring: Master Equation Terms
Advanced in silico drug design RNDr. Martin Lepšík, Ph.D. Lecture: Advanced scoring Palacky University, Olomouc 2016 1 Outline 1. Scoring Definition, Types 2. Physics-based Scoring: Master Equation Terms
Computational Modeling of Protein-Ligand Interactions
 Computational Modeling of Protein-Ligand Interactions Steven R. Gwaltney Department of Chemistry Mississippi State University Mississippi State, MS 39762 Auguste Comte, 1830 Every attempt to refer chemical
Computational Modeling of Protein-Ligand Interactions Steven R. Gwaltney Department of Chemistry Mississippi State University Mississippi State, MS 39762 Auguste Comte, 1830 Every attempt to refer chemical
Hybrid Ab-Initio/Empirical Molecular Dynamics: Combining the ONIOM Scheme with the Atom-Centered Density Matrix Propagation (ADMP) Approach
 Approach") 4210 J. Phys. Chem. B 2004, 108, 4210-4220 Hybrid Ab-Initio/Empirical Molecular Dynamics: Combining the ONIOM Scheme with the Atom-Centered Density Matrix Propagation (ADMP) Approach Nadia Rega, Srinivasan
4210 J. Phys. Chem. B 2004, 108, 4210-4220 Hybrid Ab-Initio/Empirical Molecular Dynamics: Combining the ONIOM Scheme with the Atom-Centered Density Matrix Propagation (ADMP) Approach Nadia Rega, Srinivasan
Development and Application of Combined Quantum Mechanical and Molecular Mechanical Methods
 University of Nebraska - Lincoln DigitalCommons@University of Nebraska - Lincoln Student Research Projects, Dissertations, and Theses - Chemistry Department Chemistry, Department of Fall 12-2014 Development
University of Nebraska - Lincoln DigitalCommons@University of Nebraska - Lincoln Student Research Projects, Dissertations, and Theses - Chemistry Department Chemistry, Department of Fall 12-2014 Development
Tools for QM studies of large systems
 Tools for QM studies of large systems Automated, hessian-free saddle point search & characterization QM/MM implementation for zeolites Shaama Mallikarjun Sharada Advisors: Prof. Alexis T Bell, Prof. Martin
Tools for QM studies of large systems Automated, hessian-free saddle point search & characterization QM/MM implementation for zeolites Shaama Mallikarjun Sharada Advisors: Prof. Alexis T Bell, Prof. Martin
23 The Born-Oppenheimer approximation, the Many Electron Hamiltonian and the molecular Schrödinger Equation M I
 23 The Born-Oppenheimer approximation, the Many Electron Hamiltonian and the molecular Schrödinger Equation 1. Now we will write down the Hamiltonian for a molecular system comprising N nuclei and n electrons.
23 The Born-Oppenheimer approximation, the Many Electron Hamiltonian and the molecular Schrödinger Equation 1. Now we will write down the Hamiltonian for a molecular system comprising N nuclei and n electrons.
Exploring chemical reactivity in biological systems with hybrid QM-MM. MM methods. Darío o A. Estrin University of Buenos Aires ARGENTINA
 SMR.1824-16 13th International Workshop on Computational Physics and Materials Science: Total Energy and Force Methods 11-13 January 2007 ------------------------------------------------------------------------------------------------------------------------
SMR.1824-16 13th International Workshop on Computational Physics and Materials Science: Total Energy and Force Methods 11-13 January 2007 ------------------------------------------------------------------------------------------------------------------------
Computational Biology & Computational Medicine
 Computational Biology & Computational Medicine Homayoun Valafar Outline Why proteins? What are proteins? How do we compute them? How do we use computational approaches? Why Proteins? Molecular basis of
Computational Biology & Computational Medicine Homayoun Valafar Outline Why proteins? What are proteins? How do we compute them? How do we use computational approaches? Why Proteins? Molecular basis of
DFT calculations of NMR indirect spin spin coupling constants
 DFT calculations of NMR indirect spin spin coupling constants Dalton program system Program capabilities Density functional theory Kohn Sham theory LDA, GGA and hybrid theories Indirect NMR spin spin coupling
DFT calculations of NMR indirect spin spin coupling constants Dalton program system Program capabilities Density functional theory Kohn Sham theory LDA, GGA and hybrid theories Indirect NMR spin spin coupling
CE 530 Molecular Simulation
 1 CE 530 Molecular Simulation Lecture 14 Molecular Models David A. Kofke Department of Chemical Engineering SUNY Buffalo kofke@eng.buffalo.edu 2 Review Monte Carlo ensemble averaging, no dynamics easy
1 CE 530 Molecular Simulation Lecture 14 Molecular Models David A. Kofke Department of Chemical Engineering SUNY Buffalo kofke@eng.buffalo.edu 2 Review Monte Carlo ensemble averaging, no dynamics easy
Molecular Mechanics / ReaxFF
 Molecular Dynamics simulations Lecture 09: Molecular Mechanics / ReaxFF Dr. Olli Pakarinen University of Helsinki Fall 2012 Lecture notes based on notes by Dr. Jani Kotakoski, 2010 CONTENTS Molecular mechanics
Molecular Dynamics simulations Lecture 09: Molecular Mechanics / ReaxFF Dr. Olli Pakarinen University of Helsinki Fall 2012 Lecture notes based on notes by Dr. Jani Kotakoski, 2010 CONTENTS Molecular mechanics
pk a Calculations in Solution and Proteins with QM/MM Free Energy Perturbation Simulations: A Quantitative Test of QM/MM Protocols
 J. Phys. Chem. B 2005, 109, 17715-17733 17715 pk a Calculations in Solution and Proteins with QM/MM Free Energy Perturbation Simulations: A Quantitative Test of QM/MM Protocols Demian Riccardi, Patricia
J. Phys. Chem. B 2005, 109, 17715-17733 17715 pk a Calculations in Solution and Proteins with QM/MM Free Energy Perturbation Simulations: A Quantitative Test of QM/MM Protocols Demian Riccardi, Patricia
Dimer Dissociation of a Photoreceptor Protein from QM/MM and MD Simulations
 Dimer Dissociation of a Photoreceptor Protein from QM/MM and MD Simulations IMA University of Minnesota Minneapolis, MN, July 20, 2015 Haisheng Ren Advisor: Prof. Jiali Gao Department of Chemistry, University
Dimer Dissociation of a Photoreceptor Protein from QM/MM and MD Simulations IMA University of Minnesota Minneapolis, MN, July 20, 2015 Haisheng Ren Advisor: Prof. Jiali Gao Department of Chemistry, University
Coulson's. Valence. ROY McWEENY THIRD EDITION OXFORD UNIVERSITY PRESS
 Coulson's Valence ROY McWEENY THIRD EDITION OXFORD UNIVERSITY PRESS Contents 1. THEORIES OFVALENCE 1 1.1. Essentialsofany theory of valence 1 1.2. Electronic character of valence 2 1.3. Importance of the
Coulson's Valence ROY McWEENY THIRD EDITION OXFORD UNIVERSITY PRESS Contents 1. THEORIES OFVALENCE 1 1.1. Essentialsofany theory of valence 1 1.2. Electronic character of valence 2 1.3. Importance of the
The Chemical Basis of Life
 The Chemical Basis of Life Chapter 2 Objectives Identify the four elements that make up 96% of living matter. Distinguish between the following pairs of terms: neutron and proton, atomic number and mass
The Chemical Basis of Life Chapter 2 Objectives Identify the four elements that make up 96% of living matter. Distinguish between the following pairs of terms: neutron and proton, atomic number and mass
AN INTRODUCTION TO QUANTUM CHEMISTRY. Mark S. Gordon Iowa State University
 AN INTRODUCTION TO QUANTUM CHEMISTRY Mark S. Gordon Iowa State University 1 OUTLINE Theoretical Background in Quantum Chemistry Overview of GAMESS Program Applications 2 QUANTUM CHEMISTRY In principle,
AN INTRODUCTION TO QUANTUM CHEMISTRY Mark S. Gordon Iowa State University 1 OUTLINE Theoretical Background in Quantum Chemistry Overview of GAMESS Program Applications 2 QUANTUM CHEMISTRY In principle,
Walter Kohn was awarded with the Nobel Prize in Chemistry in 1998 for his development of the density functional theory.
 Walter Kohn was awarded with the Nobel Prize in Chemistry in 1998 for his development of the density functional theory. Walter Kohn receiving his Nobel Prize from His Majesty the King at the Stockholm
Walter Kohn was awarded with the Nobel Prize in Chemistry in 1998 for his development of the density functional theory. Walter Kohn receiving his Nobel Prize from His Majesty the King at the Stockholm
Non-covalent force fields computed ab initio
 Non-covalent force fields computed ab initio Supermolecule calculations Symmetry-adapted perturbation theory (SAPT) Supermolecule calculations Requirements: E = E AB E A E B. Include electron correlation,
Non-covalent force fields computed ab initio Supermolecule calculations Symmetry-adapted perturbation theory (SAPT) Supermolecule calculations Requirements: E = E AB E A E B. Include electron correlation,
Lecture 4: methods and terminology, part II
 So theory guys have got it made in rooms free of pollution. Instead of problems with the reflux, they have only solutions... In other words, experimentalists will likely die of cancer From working hard,
So theory guys have got it made in rooms free of pollution. Instead of problems with the reflux, they have only solutions... In other words, experimentalists will likely die of cancer From working hard,
Fondamenti di Chimica Farmaceutica. Computer Chemistry in Drug Research: Introduction
 Fondamenti di Chimica Farmaceutica Computer Chemistry in Drug Research: Introduction Introduction Introduction Introduction Computer Chemistry in Drug Design Drug Discovery: Target identification Lead
Fondamenti di Chimica Farmaceutica Computer Chemistry in Drug Research: Introduction Introduction Introduction Introduction Computer Chemistry in Drug Design Drug Discovery: Target identification Lead
Multi-scale modelling of transition metal enzymes
 Degree Project C in Chemistry, 15 c Multi-scale modelling of transition metal enzymes Nathalie Proos Vedin 18th June 2015 Supervisor: Marcus Lundberg Uppsala University Department of Chemistry Ångström
Degree Project C in Chemistry, 15 c Multi-scale modelling of transition metal enzymes Nathalie Proos Vedin 18th June 2015 Supervisor: Marcus Lundberg Uppsala University Department of Chemistry Ångström
Free Energy Change and Activation Barrier for a Menshutkin Reaction Including Effects of the Solvent
 Proposed Exercise for the Physical Chemistry Section of the Teaching with Cache Workbook: Free Energy Change and Activation Barrier for a Menshutkin Reaction Including Effects of the Solvent Contributed
Proposed Exercise for the Physical Chemistry Section of the Teaching with Cache Workbook: Free Energy Change and Activation Barrier for a Menshutkin Reaction Including Effects of the Solvent Contributed
Chemistry, Physics and the Born- Oppenheimer Approximation
 Chemistry, Physics and the Born- Oppenheimer Approximation Hendrik J. Monkhorst Quantum Theory Project University of Florida Gainesville, FL 32611-8435 Outline 1. Structure of Matter 2. Shell Models 3.
Chemistry, Physics and the Born- Oppenheimer Approximation Hendrik J. Monkhorst Quantum Theory Project University of Florida Gainesville, FL 32611-8435 Outline 1. Structure of Matter 2. Shell Models 3.
CHAPTER 2 INTERATOMIC FORCES. atoms together in a solid?
 CHAPTER 2 INTERATOMIC FORCES What kind of force holds the atoms together in a solid? Interatomic Binding All of the mechanisms which cause bonding between the atoms derive from electrostatic interaction
CHAPTER 2 INTERATOMIC FORCES What kind of force holds the atoms together in a solid? Interatomic Binding All of the mechanisms which cause bonding between the atoms derive from electrostatic interaction
The chemical bond The topological approach
 The chemical bond The topological approach Julia Contreras-García 1. Why and how studying chemical bonds? 2. Topology a) critical points b) topological partitions 3. Chemical functions a) electron density
The chemical bond The topological approach Julia Contreras-García 1. Why and how studying chemical bonds? 2. Topology a) critical points b) topological partitions 3. Chemical functions a) electron density
Oslo node. Highly accurate calculations benchmarking and extrapolations
 Oslo node Highly accurate calculations benchmarking and extrapolations Torgeir Ruden, with A. Halkier, P. Jørgensen, J. Olsen, W. Klopper, J. Gauss, P. Taylor Explicitly correlated methods Pål Dahle, collaboration
Oslo node Highly accurate calculations benchmarking and extrapolations Torgeir Ruden, with A. Halkier, P. Jørgensen, J. Olsen, W. Klopper, J. Gauss, P. Taylor Explicitly correlated methods Pål Dahle, collaboration
Potentials, periodicity
 Potentials, periodicity Lecture 2 1/23/18 1 Survey responses 2 Topic requests DFT (10), Molecular dynamics (7), Monte Carlo (5) Machine Learning (4), High-throughput, Databases (4) NEB, phonons, Non-equilibrium
Potentials, periodicity Lecture 2 1/23/18 1 Survey responses 2 Topic requests DFT (10), Molecular dynamics (7), Monte Carlo (5) Machine Learning (4), High-throughput, Databases (4) NEB, phonons, Non-equilibrium
Semiempirical, empirical and hybrid methods
 Semiempirical, empirical and hybrid methods Gérald MONARD Théorie - Modélisation - Simulation UMR 7565 CNRS - Université de Lorraine Faculté des Sciences - B.P. 7239 5456 Vandœuvre-les-Nancy Cedex - FRANCE
Semiempirical, empirical and hybrid methods Gérald MONARD Théorie - Modélisation - Simulation UMR 7565 CNRS - Université de Lorraine Faculté des Sciences - B.P. 7239 5456 Vandœuvre-les-Nancy Cedex - FRANCE
Chapter 1: The Biochemical Basis of life pg : The Fundamental Chemistry of Life pg. 8 18
 UNIT 1: Biochemistry Chapter 1: The Biochemical Basis of life pg. 6 69 1.1: The Fundamental Chemistry of Life pg. 8 18 The properties of life are based on the hierarchical arrangement of chemical parts.
UNIT 1: Biochemistry Chapter 1: The Biochemical Basis of life pg. 6 69 1.1: The Fundamental Chemistry of Life pg. 8 18 The properties of life are based on the hierarchical arrangement of chemical parts.
Biochemistry,530:,, Introduc5on,to,Structural,Biology, Autumn,Quarter,2015,
 Biochemistry,530:,, Introduc5on,to,Structural,Biology, Autumn,Quarter,2015, Course,Informa5on, BIOC%530% GraduateAlevel,discussion,of,the,structure,,func5on,,and,chemistry,of,proteins,and, nucleic,acids,,control,of,enzyma5c,reac5ons.,please,see,the,course,syllabus,and,
Biochemistry,530:,, Introduc5on,to,Structural,Biology, Autumn,Quarter,2015, Course,Informa5on, BIOC%530% GraduateAlevel,discussion,of,the,structure,,func5on,,and,chemistry,of,proteins,and, nucleic,acids,,control,of,enzyma5c,reac5ons.,please,see,the,course,syllabus,and,
From Small Molecules to Biological Molecules: Modelling Interactions. Dr. Antonio Chana Milano, 22 nd April 2008
 From Small Molecules to Biological Molecules: Modelling Interactions Dr. Antonio Chana Milano, 22 nd April 2008 Computational Methods & Applicability 2 *Diagram taken from S. C. Glotzer The University
From Small Molecules to Biological Molecules: Modelling Interactions Dr. Antonio Chana Milano, 22 nd April 2008 Computational Methods & Applicability 2 *Diagram taken from S. C. Glotzer The University
Density Functional Theory
 Density Functional Theory March 26, 2009 ? DENSITY FUNCTIONAL THEORY is a method to successfully describe the behavior of atomic and molecular systems and is used for instance for: structural prediction
Density Functional Theory March 26, 2009 ? DENSITY FUNCTIONAL THEORY is a method to successfully describe the behavior of atomic and molecular systems and is used for instance for: structural prediction
Alek Marenich Cramer -Truhlar Group. University of Minnesota
 and Computational Electrochemistry Alek Marenich Cramer -Truhlar Group University of Minnesota Outline Solvation and charge models Energetics and dynamics in solution Computational electrochemistry Future
and Computational Electrochemistry Alek Marenich Cramer -Truhlar Group University of Minnesota Outline Solvation and charge models Energetics and dynamics in solution Computational electrochemistry Future
10/6/2010. Chapter 6 An Overview of Organic Reactions. Organic Chemical Reactions. 6.1 Kinds of Organic Reactions
 John E. McMurry http://www.cengage.com/chemistry/mcmurry Chapter 6 An Overview of Organic Reactions Richard Morrison University of Georgia, Athens Organic Chemical Reactions Organic chemical reactions
John E. McMurry http://www.cengage.com/chemistry/mcmurry Chapter 6 An Overview of Organic Reactions Richard Morrison University of Georgia, Athens Organic Chemical Reactions Organic chemical reactions
Multi-scale approaches in description and design of enzymes
 Multi-scale approaches in description and design of enzymes Anastassia Alexandrova and Manuel Sparta UCLA & CNSI Catalysis: it is all about the barrier The inside-out protocol: Big Aim: development of
Multi-scale approaches in description and design of enzymes Anastassia Alexandrova and Manuel Sparta UCLA & CNSI Catalysis: it is all about the barrier The inside-out protocol: Big Aim: development of
Chapter 9. Chemical Bonding I: The Lewis Model. HIV-Protease. Lecture Presentation
 Lecture Presentation Chapter 9 Chemical Bonding I: The Lewis Model HIV-Protease HIV-protease is a protein synthesized by the human immunodeficiency virus (HIV). This particular protein is crucial to the
Lecture Presentation Chapter 9 Chemical Bonding I: The Lewis Model HIV-Protease HIV-protease is a protein synthesized by the human immunodeficiency virus (HIV). This particular protein is crucial to the
Semi Empirical Force Fields and Their Limitations. Potential Energy Surface (PES)
") Semi Empirical Force Fields and Their Limitations Ioan Kosztin Beckman Institute University of Illinois at Urbana-Champaign Potential Energy Surface (PES) Schrödinger equation: H T Ψ( r, = E Ψ( r, H =
Semi Empirical Force Fields and Their Limitations Ioan Kosztin Beckman Institute University of Illinois at Urbana-Champaign Potential Energy Surface (PES) Schrödinger equation: H T Ψ( r, = E Ψ( r, H =
Ab initio calculations for potential energy surfaces. D. Talbi GRAAL- Montpellier
 Ab initio calculations for potential energy surfaces D. Talbi GRAAL- Montpellier A theoretical study of a reaction is a two step process I-Electronic calculations : techniques of quantum chemistry potential
Ab initio calculations for potential energy surfaces D. Talbi GRAAL- Montpellier A theoretical study of a reaction is a two step process I-Electronic calculations : techniques of quantum chemistry potential
362 Lecture 6 and 7. Spring 2017 Monday, Jan 30
 362 Lecture 6 and 7 Spring 2017 Monday, Jan 30 Quantum Numbers n is the principal quantum number, indicates the size of the orbital, has all positive integer values of 1 to (infinity) l is the angular
362 Lecture 6 and 7 Spring 2017 Monday, Jan 30 Quantum Numbers n is the principal quantum number, indicates the size of the orbital, has all positive integer values of 1 to (infinity) l is the angular