Functional Annotation
|
|
|
- Rudolph Leonard
- 5 years ago
- Views:
Transcription
1 Functional Annotation
2 Outline Introduction Strategy Pipeline Databases
3 Now, what s next?

4 Functional Annotation Adding the layers of analysis and interpretation necessary to extract its biological significance and place it into the context of our understanding of biological processes. (Lincoln Stein, 2001)


5 Levels of annotation 1) Protein-Level Annotationto name the protein and assign them a function 2) Process-Level Annotationto connect genes to biological processes
6 Extrinsic Approach 3-Approaches Homolog search/information Transfer search Information (mostly) based on proteins with known function Intrinsic Approach ab initio Intrinsic characteristics of gene/protein features Give additional, but limited information to unknown function proteins Pathways Biological processes, metabolic pathways Connection of pathways and processes with genes and proteins

7 From the computer to the lab Multi domain proteins Proteins with no match Integrating results Well annotated genomes Data make sense Integrate bioinformatics and experimental biologist

8 BLASTP Annotation Strategy
9 BLASTP First level of annotation Function prediction Second level of annotation Pathway and Operon prediction
10 First level of annotation Function prediction
11 CONSENSUS GENES GENES NON-CONS GENES INTRINSIC APPROACH PROTEINS EXTRINSIC APPROACH BLASTP INTERPRO SCAN BLASTP INTERPRO SCAN NeMeSyS LipoP TMHMM Signal P Swiss-Prot SMART HAMAP PROFILES SUPERFAMILY GENE3D PRINT S PFAM PRODOM PROSITE PIR Superfamily TIGRFAMs CONSENSUS SCRIPT ANNOTATION GENE ONTOLOGY IF NO ANNOTATION BLASTP UniRef 90 FIRST LEVEL OF ANNOTION
12 CONSENSUS GENES GENES NON-CONS GENES A B C INTRINSIC APPROACH PROTEINS EXTRINSIC APPROACH BLASTP INTERPRO SCAN BLASTP INTERPRO SCAN NeMeSyS LipoP TMHMM Signal P Swiss-Prot SMART HAMAP PROFILES SUPERFAMILY GENE3D PRINT S PFAM PRODOM PROSITE PIR Superfamily TIGRFAMs D CONSENSUS SCRIPT ANNOTATION GENE ONTOLOGY IF NO ANNOTATION BLASTP UniRef 90 FIRST LEVEL OF ANNOTION
13 CONSENSUS GENES GENES NON-CONS GENES A B C INTRINSIC APPROACH PROTEINS EXTRINSIC APPROACH BLASTP INTERPRO SCAN BLASTP INTERPRO SCAN NeMeSyS LipoP TMHMM Signal P Swiss-Prot SMART HAMAP PROFILES SUPERFAMILY GENE3D PRINT S PFAM PRODOM PROSITE PIR Superfamily TIGRFAMs D CONSENSUS SCRIPT ANNOTATION GENE ONTOLOGY IF NO ANNOTATION BLASTP UniRef 90 FIRST LEVEL OF ANNOTION
14 NeMesys Neisseria meningiditis database Manually annotated
15 Why?? The main aim of NeMeSys is to facilitate the identification of gene function, notably the discover of genes essential for meningococcal pathogenesis and/or viability And Ultimately to narrow the gap between sequences and function in the meningococcus.
16 How?? Manually annotated using MicroScope(platform for microbial genome annotation). Was created sequencing N.meningiditis8013serogrupC (Central and Eastern Europe, and 4500 transposon mutants). To maximize the potential of NeMesysfor functional analysis, they manually (re)annotatedmultiple strains: N.meningiditis MC58 (ST-32, Serogroup C) N.meningiditis Z2491(ST-4, Serogroup A) N.meningiditis FAM18 (ST-11, Serogroup C) N.meningiditis O53442 (ST-4821, Serogroup C) N.meningiditis α14 (Unencapsulated, carrier strain) N.lactamica (comensal) N.gonorrhoeae FA 1090 (clinical isolate) N.gonorrhoeae NCCP11945(clinical isolate)
17 Data Sharing All information is store within PkDGB (Prokariotic Genome Database) in a thematic subdatabase named as NesseriaScope The user has unlimited access to the whole array of exploratory tools and libraries. Up to 10 mutants can be requested
18 MAGE interface A B C D E
19 CONSENSUS GENES GENES NON-CONS GENES A B C INTRINSIC APPROACH PROTEINS EXTRINSIC APPROACH BLASTP INTERPRO SCAN BLASTP INTERPRO SCAN NeMeSyS LipoP TMHMM Signal P Swiss-Prot SMART HAMAP PROFILES SUPERFAMILY GENE3D PRINT S PFAM PRODOM PROSITE PIR Superfamily TIGRFAMs D CONSENSUS SCRIPT ANNOTATION GENE ONTOLOGY IF NO ANNOTATION BLASTP UniRef 90 FIRST LEVEL OF ANNOTION
20 INTRINSIC APPROACH Ab initio approaches not based on homology or any kind of similarity Based on NN or HMM Try to find the function or sub cellular location to better classify Propose to use 3 programs: SignalP for signal peptides LipoP for lipoproteins TmHMM for transmembrane helices
21 Why do we need so much information? Predicting destination/location of protein gives more information regarding function Sub cellular location is very important in determining function Signal peptides and transmembrane proteins are potential vaccine and drug targets
22 SIGNAL PEPTIDES Based on a combination of several artificial neural networks [sliding window] and Hidden Markov Models Predicts the presence and location of signal peptide cleavage sites in amino acid sequences [c-score] The method incorporates a prediction of cleavage sites and a signal peptide/nonsignal peptide prediction [s-score]

23
24 LIPOPROTEINS LipoP used to make predictions about lipoproteins Discriminates between lipoproteins and other signal peptides. HMM based High confidence results with low false positives
25 TRANS-MEMBRANE HELICES TmHMM to find the trans-membrane helices that span through the membrane Trans-membrane helices have diverse functions Based on HMM Uses scores generated intrinsically for each query protein Results comparable to experimentally discovered TM proteins
26 CONSENSUS GENES GENES NON-CONS GENES A B C INTRINSIC APPROACH PROTEINS EXTRINSIC APPROACH BLASTP INTERPRO SCAN BLASTP INTERPRO SCAN NeMeSyS LipoP TMHMM Signal P Swiss-Prot SMART HAMAP PROFILES SUPERFAMILY GENE3D PRINT S PFAM PRODOM PROSITE PIR Superfamily TIGRFAMs D CONSENSUS SCRIPT ANNOTATION GENE ONTOLOGY IF NO ANNOTATION BLASTP UniRef 90 FIRST LEVEL OF ANNOTION
27 Universal Protein Resource (UNIPROT)
28 Homology Search against UniProt BLASTP against UniProt Statistically significant Blast hits usually signify sequence homology Protein sequence analysis allows protein classification As many as 1,736 genes (approx 90%) are shared Genes encode proteins displaying at least 30% amino acid identityover at least 80% of their lengthand are in synteny
29 Central resource for storing and interconnecting information from large and disparate sources Most comprehensive catalogue of protein sequence and functional annotation UniProt Protein data only Curated in Swiss-Prot, not TrEMBL GenBank & RefSeq Protein and nucleic acid data Curated in RefSeq, not GenBanl UniProt: Merger of European Bioinformatics Institute (EBI), Protein Information Resource (PIR), Swiss Institute of Bioinformatics(SIB) 3 database components, each addressing a key need in protein bioinformatics
30 UniProt NREF Clustering at at 100%,90%, 50% 100, 90, 50% Automated Annotation Automated Automated merging of sequences Merging of Sequences UniProt Knowledgebase UniProt Archive Literature-Based Annotation Annotation Swiss - Prot TrEMBL PIR-PSD RefSeq EMBL/DDBJ GenBank/ / EnsEMBLPDB Patent EMBL/DDBJ Data Other Data
31 o UniProtKB Expertly curated, comprehensive protein database Central access point for integrated protein information with cross-references to multiple sources Targets - Relevant literature, numerous Cross-references UniProtKB/Swiss-Prot : Manually annotated records with information extracted from literature and curator-evaluated computational analysis UniProtKB/TrEMBL : High quality computationally analyzed records enriched with automatic annotation and classification
32 InterProScan
33 What is InterPro InterPro is a collaborative project aimed at providing an integrated layer on top of the most commonly used signature databases by creating a unique, non-redundant characterization of a given protein family, domain or functional site.
34 InterPro member databases No. Member database Short description 1 PROSITE Patterns Biologically significant amino acid patterns stored as regex. 2 PROSITE Profiles Stored as weight matrices (profiles) for detection of domains 3 HAMAP Profiles Similar to PROSITE profiles, specifically for Bacteria and Archae 4 PRINTS Collection of protein family fingerprints 5 PFAM A Curated database of protein families 6 PRODOM Database of protein domain families obtained through UniProt 7 SMART Allows identification and annotation of genetically mobile 8 TIGRFAMs domains Collection of protein families with curatedmultiple sequence alignments and HMMs 9 PIR Superfamily Classification based on evolutionary relationship of whole proteins 10 SUPEFAMILY Library of Profile HMMs that represent all SCOP proteins 11 GENE3D Protein family database, supplementary to CATH 12 PANTHER Classify proteins to facilitate high-throughput analysis
35 InterPro important Applications Applications ProfileScan FingerPRINTScan HMMpfam BLASTProDom.pl SuperFamily HMMPIR Short description Scans against PROSITE profiles. These profiles are based on weight matrices and are more sensitive for the detection of divergent protein families. Scans against the fingerprints in the PRINTS database. These fingerprints are groups of motifs that together are more potent than single motifs by making use of the biological context inherent in a multiple motif method. Scans the hidden markov models (HMMs) that are present in the protein domain databases Pfam, TIGRFAMMs and SMART. Scans the families in the ProDom database. ProDom is a comprehensive set of protein domain families automatically generated from the UniProtKB/Swiss-Prot sequence database using psi-blast. In InterProScan the BLASTpgb program is used to scan the database. SUPERFAMILY is a library of profile hidden Markov models that represent all proteins of known structure. Scans the hidden markov models (HMMs) that are present in the PIR Protein Sequence Database (PSD) of functionally annotated protein sequences, PIR-PSD.
36 CONSENSUS GENES GENES NON-CONS GENES A B C INTRINSIC APPROACH PROTEINS EXTRINSIC APPROACH BLASTP INTERPRO SCAN BLASTP INTERPRO SCAN NeMeSyS LipoP TMHMM Signal P Swiss-Prot SMART HAMAP PROFILES SUPERFAMILY GENE3D PRINT S PFAM PRODOM PROSITE PIR Superfamily TIGRFAMs D CONSENSUS SCRIPT ANNOTATION GENE ONTOLOGY IF NO ANNOTATION BLASTP UniRef 90 FIRST LEVEL OF ANNOTION
37 Second level of annotation Pathway and Operon prediction
38 FIRST LEVEL OF ANNOTION Pathways OPERONS BLASTP BLASTP OPERON_DB KEGG DOORS CONSENSUS SCRIPT SECOD LEVEL OF ANNOTION
39 FIRST LEVEL OF ANNOTION E F Pathways OPERONS BLASTP BLASTP OPERON_DB KEGG DOORS CONSENSUS SCRIPT SECOD LEVEL OF ANNOTION
40 FIRST LEVEL OF ANNOTION E F Pathways OPERONS BLASTP BLASTP OPERON_DB KEGG DOORS CONSENSUS SCRIPT SECOD LEVEL OF ANNOTION
41 KEGG Kyoto Encyclopedia of Genes and Genomes KEGG/KAAS KEGG Orthology KEGG GENES KEGG PATHWAYS Applications
42 KEGG Visualizes the functions of enzymes in a genome by mapping them onto biosynthetic pathways Consists of many databases of which the most relevant to our purposes are: GENES PATHWAY
43 KAAS KAAS: KEGG Automatic Annotation Server KAAS input is a fasta file Provides annotations through BLAST comparisons against the KEGG GENES database KEGG GENES: database of func. annotated genes GENE contains 5.3 million genes from various genomes: 129 Eukaryotic 971 Bacterial 74 Archaeal
44 Enzyme Commission (EC) numbers EC numbers identify enzyme-catalyzed reactions NOT enzymes If several enzymes catalyze the same reaction, they receive the same EC number Can be used to identify proteins on a metabolic pathway, but not on reference pathways such as cellular processes.
45 KEGG Orthology(KO) Orthologs genes with common ancestry, with same function in different organisms Tend to have a similar sequence and location in genomes Using orthologidentifiers leads to a more specific classification system, since many related enzymes share the same EC number. Link information in GENES and PATHWAY databses
46 KEGG GENES Category Definition Example ENTRY KO number K00033 NAME Enzyme reference names E , PGD, gnd DEFINITION Enzyme identity and EC number 6-phosphogluconate dehydrogenase [EC: ] PATHWAY Pathways involved in ko00030 Pentose phosphate pathway MODULE Chemical reactions involved in and compounds acted on M00006 Pentose phosphate pathway, oxidative phase, glucose 6P => ribulose 5P CLASS Type of pathway Metabolism; Carbohydrate Metabolism; Pentose phosphate pathway DBLINKS GENES Link to other databases entries for the enzyme Gene information for a particular organism COG: COG0362 HSA: 5226(PGD)
47
48 KEGG PATHWAY database Graphical representations of biosynthetic pathways Predicts pathways by comparing the enzymes found in a genome with reference pathways. Information in GENES databases is linked to the information in PATHWAY database through KO identifiers. If pathways are incomplete, then the missing enzymes are visually detectable Both metabolic and reference pathways mapped
49 Sample KEGG Pathway Map
50

51 Two-Component Systems NarXphosphorylatesNarLupon binding to nitrate and nitrite. This causes a downregulation of Fumerate reductase expression and an upregulation of Formate dehydrogenase expression.
52 KEGG Why use it? It s a tool for visualizing proteins of a genome in biosynthetic pathways Aids in checking annotations Visually detect missing proteins Can aid the comparative genomics group Comparisons of protein pathways between genomes
53 FIRST LEVEL OF ANNOTION E F Pathways OPERONS BLASTP BLASTP OPERON_DB KEGG DOORS CONSENSUS SCRIPT SECOD LEVEL OF ANNOTION
54 What is Operon? Operon family of co-regulated genes Adjacent Same orientation Not separated by promoters/terminators Related functions Strong selective pressure, conserved Knowledge of operon -> FUNCTION
55 Operon DB: Database of predicted operons Microbial Genomes Computer prediction of operon structures. 500 genomes.
56 Computational Approach Gene pair adjacent, same strand, intergenic length separation P(gene pair in operon) = 1 P(conserved D)X P(SN S)-P chance P(conserved S) D,S = sets of gene pairs P chance = P(conserved S has homologsin other genomes)
57 Algorithm Identification of conserved pairs Identification of orthologs using BLAST Finding conserved gene clusters Homology Teams software. x Evolutionary distance D(G 1,G 2 ) = n(g 1 )+n(g 2 ) h(g 1 ;G 2 ) h(g 2 ;G 1 ) n(g 1 ) + n(g 2 ) Larger dist = greater prob of conservation
58 Alternative possibilities considered Functionally unrelated genes may have the same order due simply because they were adjacent in a common ancestor. Genes may be adjacent in two genomes by chance alone, or due to horizontal transfer of the gene pair.
.")
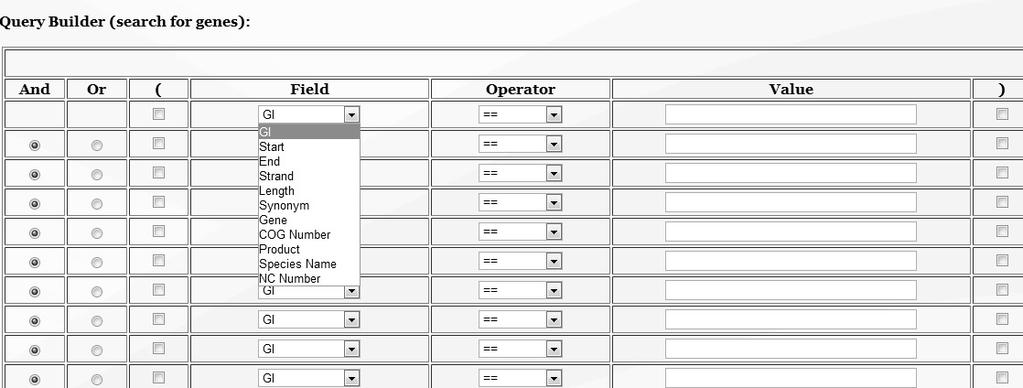
59 Interface 1). List of genomes 2). List of gene pairs
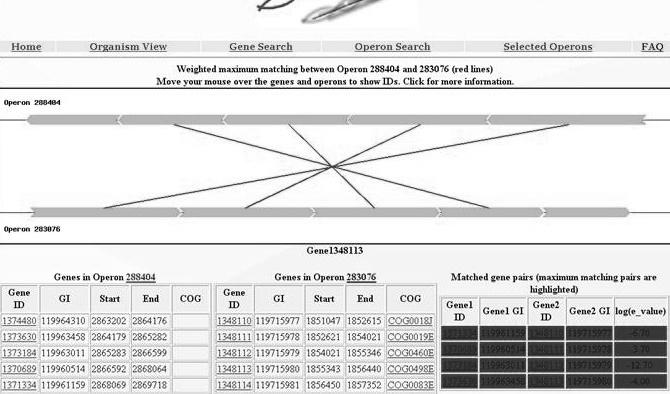
60 Sample output
61 DOOR Universal, genome specific prediction Reliability α Intergenic distances Different features (scored) Intergenicdistance Conservation of gene pairs(neighborhood) Phylogenetic patterns Ratio between lengths of two genes Frequencies of specific DNA motifs in the intergenic regions.(meme & CUBIC)
62

63
64
65 Doubts? No stand-alone version/code available Can t be automated New query runs??
66 QUESTIONS?
67 Mechanism RAW FASTA EMBL USER SPECIFIED
68 Features Pure Perl, hence modular organization makes implementation efficient for bulk sequence analysis. Indexes corresponding databases, hence fast retrieval. User specified post-processing and cut-offs are made possible to filter final results.
69 RAW FORMAT OUTPUT NF A5FDCE74AB7C3AD 272 HMMPIR PIRSF Prephenate dehydratase e 141 T 06-Aug-2005 IPR Prephenatedehydratasewith ACT region Molecular Function:prephenate dehydratase activity (GO: ), Biological Process:Lphenylalanine biosynthesis (GO: ) Where NF : is the id of the input sequence. 27A9BBAC0587AB84: is the crc64 (checksum) of the protein sequence (supposed to be unique). 272: is the length of the sequence (in AA). HMMPIR: is the analysis method launched. PIRSF001424: is the database members entry for this match. Prephenate dehydratase: is the database member description for the entry. 1: is the start of the domain match. 270: is the end of the domain match. 6.5e-141: is the e-value of the match (reported by member database method). T: is the status of the match (T: true,?: unknown). 06-Aug-2005: is the date of the run. IPR008237: is the corresponding InterPro entry (if iprlookup requested by the user). Prephenate dehydratase with ACT region: is the description of the InterPro entry. Molecular Function:prephenatedehydrataseactivity (GO: ): is the GO (gene ontology) description for the InterPro entry.
 Biological process (WHICH?) Molecular function (WHAT?")

70 Gene Ontology Classification Cellular component (WHERE?) Biological process (WHICH?) Molecular function (WHAT?) Associated with Located in Active in Performs Gene product = cytochrome c WHERE?- Mitochondrial matrix and inner membrane WHAT?-oxidoreductase activity WHICH?- oxidative phospholyration and induction of cell death
71 Gene Ontology Classification Specific function- specific name and gene symbol Likely (or unlikely) function- putative or homolog Generic function- protein family Unknown function- hypothetical, unknown function From: JCVI Gene Naming and Annotation Conventions
-max_target_seqs: maximum number of targets to report
 Review of exercise 1 tblastn -num_threads 2 -db contig -query DH10B.fasta -out blastout.xls -evalue 1e-10 -outfmt "6 qseqid sseqid qstart qend sstart send length nident pident evalue" Other options: -max_target_seqs:
Review of exercise 1 tblastn -num_threads 2 -db contig -query DH10B.fasta -out blastout.xls -evalue 1e-10 -outfmt "6 qseqid sseqid qstart qend sstart send length nident pident evalue" Other options: -max_target_seqs:
We have: We will: Assembled six genomes Made predictions of most likely gene locations. Add a layers of biological meaning to the sequences
 Recap We have: Assembled six genomes Made predictions of most likely gene locations We will: Add a layers of biological meaning to the sequences Start with Biology This will motivate the choices we make
Recap We have: Assembled six genomes Made predictions of most likely gene locations We will: Add a layers of biological meaning to the sequences Start with Biology This will motivate the choices we make
Genome Annotation. Bioinformatics and Computational Biology. Genome sequencing Assembly. Gene prediction. Protein targeting.
 Genome Annotation Bioinformatics and Computational Biology Genome Annotation Frank Oliver Glöckner 1 Genome Analysis Roadmap Genome sequencing Assembly Gene prediction Protein targeting trna prediction
Genome Annotation Bioinformatics and Computational Biology Genome Annotation Frank Oliver Glöckner 1 Genome Analysis Roadmap Genome sequencing Assembly Gene prediction Protein targeting trna prediction
CSCE555 Bioinformatics. Protein Function Annotation
 CSCE555 Bioinformatics Protein Function Annotation Why we need to do function annotation? Fig from: Network-based prediction of protein function. Molecular Systems Biology 3:88. 2007 What s function? The
CSCE555 Bioinformatics Protein Function Annotation Why we need to do function annotation? Fig from: Network-based prediction of protein function. Molecular Systems Biology 3:88. 2007 What s function? The
EBI web resources II: Ensembl and InterPro
 EBI web resources II: Ensembl and InterPro Yanbin Yin http://www.ebi.ac.uk/training/online/course/ 1 Homework 3 Go to http://www.ebi.ac.uk/interpro/training.htmland finish the second online training course
EBI web resources II: Ensembl and InterPro Yanbin Yin http://www.ebi.ac.uk/training/online/course/ 1 Homework 3 Go to http://www.ebi.ac.uk/interpro/training.htmland finish the second online training course
EBI web resources II: Ensembl and InterPro. Yanbin Yin Spring 2013
 EBI web resources II: Ensembl and InterPro Yanbin Yin Spring 2013 1 Outline Intro to genome annotation Protein family/domain databases InterPro, Pfam, Superfamily etc. Genome browser Ensembl Hands on Practice
EBI web resources II: Ensembl and InterPro Yanbin Yin Spring 2013 1 Outline Intro to genome annotation Protein family/domain databases InterPro, Pfam, Superfamily etc. Genome browser Ensembl Hands on Practice
Bioinformatics. Dept. of Computational Biology & Bioinformatics
 Bioinformatics Dept. of Computational Biology & Bioinformatics 3 Bioinformatics - play with sequences & structures Dept. of Computational Biology & Bioinformatics 4 ORGANIZATION OF LIFE ROLE OF BIOINFORMATICS
Bioinformatics Dept. of Computational Biology & Bioinformatics 3 Bioinformatics - play with sequences & structures Dept. of Computational Biology & Bioinformatics 4 ORGANIZATION OF LIFE ROLE OF BIOINFORMATICS
Protein Bioinformatics. Rickard Sandberg Dept. of Cell and Molecular Biology Karolinska Institutet sandberg.cmb.ki.
 Protein Bioinformatics Rickard Sandberg Dept. of Cell and Molecular Biology Karolinska Institutet rickard.sandberg@ki.se sandberg.cmb.ki.se Outline Protein features motifs patterns profiles signals 2 Protein
Protein Bioinformatics Rickard Sandberg Dept. of Cell and Molecular Biology Karolinska Institutet rickard.sandberg@ki.se sandberg.cmb.ki.se Outline Protein features motifs patterns profiles signals 2 Protein
Chapter 5. Proteomics and the analysis of protein sequence Ⅱ
 Proteomics Chapter 5. Proteomics and the analysis of protein sequence Ⅱ 1 Pairwise similarity searching (1) Figure 5.5: manual alignment One of the amino acids in the top sequence has no equivalent and
Proteomics Chapter 5. Proteomics and the analysis of protein sequence Ⅱ 1 Pairwise similarity searching (1) Figure 5.5: manual alignment One of the amino acids in the top sequence has no equivalent and
Christian Sigrist. November 14 Protein Bioinformatics: Sequence-Structure-Function 2018 Basel
 Christian Sigrist General Definition on Conserved Regions Conserved regions in proteins can be classified into 5 different groups: Domains: specific combination of secondary structures organized into a
Christian Sigrist General Definition on Conserved Regions Conserved regions in proteins can be classified into 5 different groups: Domains: specific combination of secondary structures organized into a
CS612 - Algorithms in Bioinformatics
 Fall 2017 Databases and Protein Structure Representation October 2, 2017 Molecular Biology as Information Science > 12, 000 genomes sequenced, mostly bacterial (2013) > 5x10 6 unique sequences available
Fall 2017 Databases and Protein Structure Representation October 2, 2017 Molecular Biology as Information Science > 12, 000 genomes sequenced, mostly bacterial (2013) > 5x10 6 unique sequences available
Homology. and. Information Gathering and Domain Annotation for Proteins
 Homology and Information Gathering and Domain Annotation for Proteins Outline WHAT IS HOMOLOGY? HOW TO GATHER KNOWN PROTEIN INFORMATION? HOW TO ANNOTATE PROTEIN DOMAINS? EXAMPLES AND EXERCISES Homology
Homology and Information Gathering and Domain Annotation for Proteins Outline WHAT IS HOMOLOGY? HOW TO GATHER KNOWN PROTEIN INFORMATION? HOW TO ANNOTATE PROTEIN DOMAINS? EXAMPLES AND EXERCISES Homology
Homology and Information Gathering and Domain Annotation for Proteins
 Homology and Information Gathering and Domain Annotation for Proteins Outline Homology Information Gathering for Proteins Domain Annotation for Proteins Examples and exercises The concept of homology The
Homology and Information Gathering and Domain Annotation for Proteins Outline Homology Information Gathering for Proteins Domain Annotation for Proteins Examples and exercises The concept of homology The
Protein function prediction based on sequence analysis
 Performing sequence searches Post-Blast analysis, Using profiles and pattern-matching Protein function prediction based on sequence analysis Slides from a lecture on MOL204 - Applied Bioinformatics 18-Oct-2005
Performing sequence searches Post-Blast analysis, Using profiles and pattern-matching Protein function prediction based on sequence analysis Slides from a lecture on MOL204 - Applied Bioinformatics 18-Oct-2005
Week 10: Homology Modelling (II) - HHpred
 - HHpred") Week 10: Homology Modelling (II) - HHpred Course: Tools for Structural Biology Fabian Glaser BKU - Technion 1 2 Identify and align related structures by sequence methods is not an easy task All comparative
Week 10: Homology Modelling (II) - HHpred Course: Tools for Structural Biology Fabian Glaser BKU - Technion 1 2 Identify and align related structures by sequence methods is not an easy task All comparative
Lecture 2. The Blast2GO annotation framework
 Lecture 2 The Blast2GO annotation framework Annotation steps Modulation of annotation intensity Export/Import Functions Sequence Selection Additional Tools Functional assignment Annotation Transference
Lecture 2 The Blast2GO annotation framework Annotation steps Modulation of annotation intensity Export/Import Functions Sequence Selection Additional Tools Functional assignment Annotation Transference
Motifs, Profiles and Domains. Michael Tress Protein Design Group Centro Nacional de Biotecnología, CSIC
 Motifs, Profiles and Domains Michael Tress Protein Design Group Centro Nacional de Biotecnología, CSIC Comparing Two Proteins Sequence Alignment Determining the pattern of evolution and identifying conserved
Motifs, Profiles and Domains Michael Tress Protein Design Group Centro Nacional de Biotecnología, CSIC Comparing Two Proteins Sequence Alignment Determining the pattern of evolution and identifying conserved
BMD645. Integration of Omics
 BMD645 Integration of Omics Shu-Jen Chen, Chang Gung University Dec. 11, 2009 1 Traditional Biology vs. Systems Biology Traditional biology : Single genes or proteins Systems biology: Simultaneously study
BMD645 Integration of Omics Shu-Jen Chen, Chang Gung University Dec. 11, 2009 1 Traditional Biology vs. Systems Biology Traditional biology : Single genes or proteins Systems biology: Simultaneously study
Functional Annotation & Comparative Genomics. Lu Wang, Georgia Tech
 Functional Annotation & Comparative Genomics Lu Wang, Georgia Tech Outline Functional annotation What is functional annotation? What needs to be annotated Approaches to functional annotation Pros/cons
Functional Annotation & Comparative Genomics Lu Wang, Georgia Tech Outline Functional annotation What is functional annotation? What needs to be annotated Approaches to functional annotation Pros/cons
FUNCTION ANNOTATION PRELIMINARY RESULTS
 FUNCTION ANNOTATION PRELIMINARY RESULTS FACTION I KAI YUAN KALYANI PATANKAR KIERA BERGER CAMILA MEDRANO HUBERT PAN JUNKE WANG YANXI CHEN AJAY RAMAKRISHNAN MRUNAL DEHANKAR OVERVIEW Introduction Previous
FUNCTION ANNOTATION PRELIMINARY RESULTS FACTION I KAI YUAN KALYANI PATANKAR KIERA BERGER CAMILA MEDRANO HUBERT PAN JUNKE WANG YANXI CHEN AJAY RAMAKRISHNAN MRUNAL DEHANKAR OVERVIEW Introduction Previous
DATA ACQUISITION FROM BIO-DATABASES AND BLAST. Natapol Pornputtapong 18 January 2018
 DATA ACQUISITION FROM BIO-DATABASES AND BLAST Natapol Pornputtapong 18 January 2018 DATABASE Collections of data To share multi-user interface To prevent data loss To make sure to get the right things
DATA ACQUISITION FROM BIO-DATABASES AND BLAST Natapol Pornputtapong 18 January 2018 DATABASE Collections of data To share multi-user interface To prevent data loss To make sure to get the right things
Some Problems from Enzyme Families
 Some Problems from Enzyme Families Greg Butler Department of Computer Science Concordia University, Montreal www.cs.concordia.ca/~faculty/gregb gregb@cs.concordia.ca Abstract I will discuss some problems
Some Problems from Enzyme Families Greg Butler Department of Computer Science Concordia University, Montreal www.cs.concordia.ca/~faculty/gregb gregb@cs.concordia.ca Abstract I will discuss some problems
Hands-On Nine The PAX6 Gene and Protein
 Hands-On Nine The PAX6 Gene and Protein Main Purpose of Hands-On Activity: Using bioinformatics tools to examine the sequences, homology, and disease relevance of the Pax6: a master gene of eye formation.
Hands-On Nine The PAX6 Gene and Protein Main Purpose of Hands-On Activity: Using bioinformatics tools to examine the sequences, homology, and disease relevance of the Pax6: a master gene of eye formation.
Networks & pathways. Hedi Peterson MTAT Bioinformatics
 Networks & pathways Hedi Peterson (peterson@quretec.com) MTAT.03.239 Bioinformatics 03.11.2010 Networks are graphs Nodes Edges Edges Directed, undirected, weighted Nodes Genes Proteins Metabolites Enzymes
Networks & pathways Hedi Peterson (peterson@quretec.com) MTAT.03.239 Bioinformatics 03.11.2010 Networks are graphs Nodes Edges Edges Directed, undirected, weighted Nodes Genes Proteins Metabolites Enzymes
SUPPLEMENTARY INFORMATION
 Supplementary information S1 (box). Supplementary Methods description. Prokaryotic Genome Database Archaeal and bacterial genome sequences were downloaded from the NCBI FTP site (ftp://ftp.ncbi.nlm.nih.gov/genomes/all/)
Supplementary information S1 (box). Supplementary Methods description. Prokaryotic Genome Database Archaeal and bacterial genome sequences were downloaded from the NCBI FTP site (ftp://ftp.ncbi.nlm.nih.gov/genomes/all/)
Syllabus of BIOINF 528 (2017 Fall, Bioinformatics Program)
") Syllabus of BIOINF 528 (2017 Fall, Bioinformatics Program) Course Name: Structural Bioinformatics Course Description: Instructor: This course introduces fundamental concepts and methods for structural
Syllabus of BIOINF 528 (2017 Fall, Bioinformatics Program) Course Name: Structural Bioinformatics Course Description: Instructor: This course introduces fundamental concepts and methods for structural
Bioinformatics methods COMPUTATIONAL WORKFLOW
 Bioinformatics methods COMPUTATIONAL WORKFLOW RAW READ PROCESSING: 1. FastQC on raw reads 2. Kraken on raw reads to ID and remove contaminants 3. SortmeRNA to filter out rrna 4. Trimmomatic to filter by
Bioinformatics methods COMPUTATIONAL WORKFLOW RAW READ PROCESSING: 1. FastQC on raw reads 2. Kraken on raw reads to ID and remove contaminants 3. SortmeRNA to filter out rrna 4. Trimmomatic to filter by
Protein bioinforma-cs. Åsa Björklund CMB/LICR
 Protein bioinforma-cs Åsa Björklund CMB/LICR asa.bjorklund@licr.ki.se In this lecture Protein structures and 3D structure predic-on Protein domains HMMs Protein networks Protein func-on annota-on / predic-on
Protein bioinforma-cs Åsa Björklund CMB/LICR asa.bjorklund@licr.ki.se In this lecture Protein structures and 3D structure predic-on Protein domains HMMs Protein networks Protein func-on annota-on / predic-on
Large-Scale Genomic Surveys
 Bioinformatics Subtopics Fold Recognition Secondary Structure Prediction Docking & Drug Design Protein Geometry Protein Flexibility Homology Modeling Sequence Alignment Structure Classification Gene Prediction
Bioinformatics Subtopics Fold Recognition Secondary Structure Prediction Docking & Drug Design Protein Geometry Protein Flexibility Homology Modeling Sequence Alignment Structure Classification Gene Prediction
Comprehensive genome analysis of 203 genomes provides structural genomics with new insights into protein family space
 Published online February 15, 26 166 18 Nucleic Acids Research, 26, Vol. 34, No. 3 doi:1.193/nar/gkj494 Comprehensive genome analysis of 23 genomes provides structural genomics with new insights into protein
Published online February 15, 26 166 18 Nucleic Acids Research, 26, Vol. 34, No. 3 doi:1.193/nar/gkj494 Comprehensive genome analysis of 23 genomes provides structural genomics with new insights into protein
Update on human genome completion and annotations: Protein information resource
 UPDATE ON GENOME COMPLETION AND ANNOTATIONS Update on human genome completion and annotations: Protein information resource Cathy Wu 1 and Daniel W. Nebert 2 * 1 Director of PIR, Department of Biochemistry
UPDATE ON GENOME COMPLETION AND ANNOTATIONS Update on human genome completion and annotations: Protein information resource Cathy Wu 1 and Daniel W. Nebert 2 * 1 Director of PIR, Department of Biochemistry
A Protein Ontology from Large-scale Textmining?
 A Protein Ontology from Large-scale Textmining? Protege-Workshop Manchester, 07-07-2003 Kai Kumpf, Juliane Fluck and Martin Hofmann Instructive mistakes: a narrative Aim: Protein ontology that supports
A Protein Ontology from Large-scale Textmining? Protege-Workshop Manchester, 07-07-2003 Kai Kumpf, Juliane Fluck and Martin Hofmann Instructive mistakes: a narrative Aim: Protein ontology that supports
Amino Acid Structures from Klug & Cummings. 10/7/2003 CAP/CGS 5991: Lecture 7 1
 Amino Acid Structures from Klug & Cummings 10/7/2003 CAP/CGS 5991: Lecture 7 1 Amino Acid Structures from Klug & Cummings 10/7/2003 CAP/CGS 5991: Lecture 7 2 Amino Acid Structures from Klug & Cummings
Amino Acid Structures from Klug & Cummings 10/7/2003 CAP/CGS 5991: Lecture 7 1 Amino Acid Structures from Klug & Cummings 10/7/2003 CAP/CGS 5991: Lecture 7 2 Amino Acid Structures from Klug & Cummings
Intro Secondary structure Transmembrane proteins Function End. Last time. Domains Hidden Markov Models
 Last time Domains Hidden Markov Models Today Secondary structure Transmembrane proteins Structure prediction NAD-specific glutamate dehydrogenase Hard Easy >P24295 DHE2_CLOSY MSKYVDRVIAEVEKKYADEPEFVQTVEEVL
Last time Domains Hidden Markov Models Today Secondary structure Transmembrane proteins Structure prediction NAD-specific glutamate dehydrogenase Hard Easy >P24295 DHE2_CLOSY MSKYVDRVIAEVEKKYADEPEFVQTVEEVL
In-Silico Approach for Hypothetical Protein Function Prediction
 In-Silico Approach for Hypothetical Protein Function Prediction Shabanam Khatoon Department of Computer Science, Faculty of Natural Sciences Jamia Millia Islamia, New Delhi Suraiya Jabin Department of
In-Silico Approach for Hypothetical Protein Function Prediction Shabanam Khatoon Department of Computer Science, Faculty of Natural Sciences Jamia Millia Islamia, New Delhi Suraiya Jabin Department of
Gene function annotation
 Gene function annotation Paul D. Thomas, Ph.D. University of Southern California What is function annotation? The formal answer to the question: what does this gene do? The association between: a description
Gene function annotation Paul D. Thomas, Ph.D. University of Southern California What is function annotation? The formal answer to the question: what does this gene do? The association between: a description
Bioinformatics. Proteins II. - Pattern, Profile, & Structure Database Searching. Robert Latek, Ph.D. Bioinformatics, Biocomputing
 Bioinformatics Proteins II. - Pattern, Profile, & Structure Database Searching Robert Latek, Ph.D. Bioinformatics, Biocomputing WIBR Bioinformatics Course, Whitehead Institute, 2002 1 Proteins I.-III.
Bioinformatics Proteins II. - Pattern, Profile, & Structure Database Searching Robert Latek, Ph.D. Bioinformatics, Biocomputing WIBR Bioinformatics Course, Whitehead Institute, 2002 1 Proteins I.-III.
RGP finder: prediction of Genomic Islands
 Training courses on MicroScope platform RGP finder: prediction of Genomic Islands Dynamics of bacterial genomes Gene gain Horizontal gene transfer Gene loss Deletion of one or several genes Duplication
Training courses on MicroScope platform RGP finder: prediction of Genomic Islands Dynamics of bacterial genomes Gene gain Horizontal gene transfer Gene loss Deletion of one or several genes Duplication
Today. Last time. Secondary structure Transmembrane proteins. Domains Hidden Markov Models. Structure prediction. Secondary structure
 Last time Today Domains Hidden Markov Models Structure prediction NAD-specific glutamate dehydrogenase Hard Easy >P24295 DHE2_CLOSY MSKYVDRVIAEVEKKYADEPEFVQTVEEVL SSLGPVVDAHPEYEEVALLERMVIPERVIE FRVPWEDDNGKVHVNTGYRVQFNGAIGPYK
Last time Today Domains Hidden Markov Models Structure prediction NAD-specific glutamate dehydrogenase Hard Easy >P24295 DHE2_CLOSY MSKYVDRVIAEVEKKYADEPEFVQTVEEVL SSLGPVVDAHPEYEEVALLERMVIPERVIE FRVPWEDDNGKVHVNTGYRVQFNGAIGPYK
Comparative genomics: Overview & Tools + MUMmer algorithm
 Comparative genomics: Overview & Tools + MUMmer algorithm Urmila Kulkarni-Kale Bioinformatics Centre University of Pune, Pune 411 007. urmila@bioinfo.ernet.in Genome sequence: Fact file 1995: The first
Comparative genomics: Overview & Tools + MUMmer algorithm Urmila Kulkarni-Kale Bioinformatics Centre University of Pune, Pune 411 007. urmila@bioinfo.ernet.in Genome sequence: Fact file 1995: The first
Computational methods for predicting protein-protein interactions
 Computational methods for predicting protein-protein interactions Tomi Peltola T-61.6070 Special course in bioinformatics I 3.4.2008 Outline Biological background Protein-protein interactions Computational
Computational methods for predicting protein-protein interactions Tomi Peltola T-61.6070 Special course in bioinformatics I 3.4.2008 Outline Biological background Protein-protein interactions Computational
Genome Annotation Project Presentation
 Halogeometricum borinquense Genome Annotation Project Presentation Loci Hbor_05620 & Hbor_05470 Presented by: Mohammad Reza Najaf Tomaraei Hbor_05620 Basic Information DNA Coordinates: 527,512 528,261
Halogeometricum borinquense Genome Annotation Project Presentation Loci Hbor_05620 & Hbor_05470 Presented by: Mohammad Reza Najaf Tomaraei Hbor_05620 Basic Information DNA Coordinates: 527,512 528,261
Computational approaches for functional genomics
 Computational approaches for functional genomics Kalin Vetsigian October 31, 2001 The rapidly increasing number of completely sequenced genomes have stimulated the development of new methods for finding
Computational approaches for functional genomics Kalin Vetsigian October 31, 2001 The rapidly increasing number of completely sequenced genomes have stimulated the development of new methods for finding
Sequence Alignment: A General Overview. COMP Fall 2010 Luay Nakhleh, Rice University
 Sequence Alignment: A General Overview COMP 571 - Fall 2010 Luay Nakhleh, Rice University Life through Evolution All living organisms are related to each other through evolution This means: any pair of
Sequence Alignment: A General Overview COMP 571 - Fall 2010 Luay Nakhleh, Rice University Life through Evolution All living organisms are related to each other through evolution This means: any pair of
BIOINFORMATICS: An Introduction
 BIOINFORMATICS: An Introduction What is Bioinformatics? The term was first coined in 1988 by Dr. Hwa Lim The original definition was : a collective term for data compilation, organisation, analysis and
BIOINFORMATICS: An Introduction What is Bioinformatics? The term was first coined in 1988 by Dr. Hwa Lim The original definition was : a collective term for data compilation, organisation, analysis and
NetAffx GPCR annotation database summary December 12, 2001
 NetAffx GPCR annotation database summary December 12, 2001 Introduction Only approximately 51% of the human proteome can be annotated by the standard motif-based recognition systems [1]. These systems,
NetAffx GPCR annotation database summary December 12, 2001 Introduction Only approximately 51% of the human proteome can be annotated by the standard motif-based recognition systems [1]. These systems,
Sifting through genomes with iterative-sequence clustering produces a large, phylogenetically diverse protein-family resource
 Sharpton et al. BMC Bioinformatics 2012, 13:264 RESEARCH ARTICLE Open Access Sifting through genomes with iterative-sequence clustering produces a large, phylogenetically diverse protein-family resource
Sharpton et al. BMC Bioinformatics 2012, 13:264 RESEARCH ARTICLE Open Access Sifting through genomes with iterative-sequence clustering produces a large, phylogenetically diverse protein-family resource
ATLAS of Biochemistry
 ATLAS of Biochemistry USER GUIDE http://lcsb-databases.epfl.ch/atlas/ CONTENT 1 2 3 GET STARTED Create your user account NAVIGATE Curated KEGG reactions ATLAS reactions Pathways Maps USE IT! Fill a gap
ATLAS of Biochemistry USER GUIDE http://lcsb-databases.epfl.ch/atlas/ CONTENT 1 2 3 GET STARTED Create your user account NAVIGATE Curated KEGG reactions ATLAS reactions Pathways Maps USE IT! Fill a gap
Bioinformatics Exercises
 Bioinformatics Exercises AP Biology Teachers Workshop Susan Cates, Ph.D. Evolution of Species Phylogenetic Trees show the relatedness of organisms Common Ancestor (Root of the tree) 1 Rooted vs. Unrooted
Bioinformatics Exercises AP Biology Teachers Workshop Susan Cates, Ph.D. Evolution of Species Phylogenetic Trees show the relatedness of organisms Common Ancestor (Root of the tree) 1 Rooted vs. Unrooted
Introductory course on Multiple Sequence Alignment Part I: Theoretical foundations
 Sequence Analysis and Structure Prediction Service Centro Nacional de Biotecnología CSIC 8-10 May, 2013 Introductory course on Multiple Sequence Alignment Part I: Theoretical foundations Course Notes Instructor:
Sequence Analysis and Structure Prediction Service Centro Nacional de Biotecnología CSIC 8-10 May, 2013 Introductory course on Multiple Sequence Alignment Part I: Theoretical foundations Course Notes Instructor:
Cross Discipline Analysis made possible with Data Pipelining. J.R. Tozer SciTegic
 Cross Discipline Analysis made possible with Data Pipelining J.R. Tozer SciTegic System Genesis Pipelining tool created to automate data processing in cheminformatics Modular system built with generic
Cross Discipline Analysis made possible with Data Pipelining J.R. Tozer SciTegic System Genesis Pipelining tool created to automate data processing in cheminformatics Modular system built with generic
Patterns and profiles applications of multiple alignments. Tore Samuelsson March 2013
 Patterns and profiles applications of multiple alignments Tore Samuelsson March 3 Protein patterns and the PROSITE database Proteins that bind the nucleotides ATP or GTP share a short sequence motif Entry
Patterns and profiles applications of multiple alignments Tore Samuelsson March 3 Protein patterns and the PROSITE database Proteins that bind the nucleotides ATP or GTP share a short sequence motif Entry
Structure to Function. Molecular Bioinformatics, X3, 2006
 Structure to Function Molecular Bioinformatics, X3, 2006 Structural GeNOMICS Structural Genomics project aims at determination of 3D structures of all proteins: - organize known proteins into families
Structure to Function Molecular Bioinformatics, X3, 2006 Structural GeNOMICS Structural Genomics project aims at determination of 3D structures of all proteins: - organize known proteins into families
CISC 636 Computational Biology & Bioinformatics (Fall 2016)
") CISC 636 Computational Biology & Bioinformatics (Fall 2016) Predicting Protein-Protein Interactions CISC636, F16, Lec22, Liao 1 Background Proteins do not function as isolated entities. Protein-Protein
CISC 636 Computational Biology & Bioinformatics (Fall 2016) Predicting Protein-Protein Interactions CISC636, F16, Lec22, Liao 1 Background Proteins do not function as isolated entities. Protein-Protein
Multiple sequence alignment
 Multiple sequence alignment Multiple sequence alignment: today s goals to define what a multiple sequence alignment is and how it is generated; to describe profile HMMs to introduce databases of multiple
Multiple sequence alignment Multiple sequence alignment: today s goals to define what a multiple sequence alignment is and how it is generated; to describe profile HMMs to introduce databases of multiple
Ensembl focuses on metazoan (animal) genomes. The genomes currently available at the Ensembl site are:
 genomes. The genomes currently available at the Ensembl site are:") Comparative genomics and proteomics Species available Ensembl focuses on metazoan (animal) genomes. The genomes currently available at the Ensembl site are: Vertebrates: human, chimpanzee, mouse, rat,
Comparative genomics and proteomics Species available Ensembl focuses on metazoan (animal) genomes. The genomes currently available at the Ensembl site are: Vertebrates: human, chimpanzee, mouse, rat,
SUPPLEMENTARY INFORMATION
 Supplementary information S3 (box) Methods Methods Genome weighting The currently available collection of archaeal and bacterial genomes has a highly biased distribution of isolates across taxa. For example,
Supplementary information S3 (box) Methods Methods Genome weighting The currently available collection of archaeal and bacterial genomes has a highly biased distribution of isolates across taxa. For example,
EBI web resources II: Ensembl and InterPro
 EBI web resources II: Ensembl and InterPro Yanbin Yin Fall 2015 h.p://www.ebi.ac.uk/training/online/course/ 1 Homework 3 Go to h.p://www.ebi.ac.uk/interpro/training.html and finish the second online training
EBI web resources II: Ensembl and InterPro Yanbin Yin Fall 2015 h.p://www.ebi.ac.uk/training/online/course/ 1 Homework 3 Go to h.p://www.ebi.ac.uk/interpro/training.html and finish the second online training
2MHR. Protein structure classification is important because it organizes the protein structure universe that is independent of sequence similarity.
 Protein structure classification is important because it organizes the protein structure universe that is independent of sequence similarity. A global picture of the protein universe will help us to understand
Protein structure classification is important because it organizes the protein structure universe that is independent of sequence similarity. A global picture of the protein universe will help us to understand
Protein structure alignments
 Protein structure alignments Proteins that fold in the same way, i.e. have the same fold are often homologs. Structure evolves slower than sequence Sequence is less conserved than structure If BLAST gives
Protein structure alignments Proteins that fold in the same way, i.e. have the same fold are often homologs. Structure evolves slower than sequence Sequence is less conserved than structure If BLAST gives
Browsing Genomic Information with Ensembl Plants
 Browsing Genomic Information with Ensembl Plants Etienne de Villiers, PhD (Adapted from slides by Bert Overduin EMBL-EBI) Outline of workshop Brief introduction to Ensembl Plants History Content Tutorial
Browsing Genomic Information with Ensembl Plants Etienne de Villiers, PhD (Adapted from slides by Bert Overduin EMBL-EBI) Outline of workshop Brief introduction to Ensembl Plants History Content Tutorial
Update on genome completion and annotations: Protein Information Resource
 UPDATE ON GENOME COMPLETION AND ANNOTATIONS Update on genome completion and annotations: Protein Information Resource Cathy Wu 1 and Daniel W. Nebert 2 * 1 Director of PIR, Department of Biochemistry and
UPDATE ON GENOME COMPLETION AND ANNOTATIONS Update on genome completion and annotations: Protein Information Resource Cathy Wu 1 and Daniel W. Nebert 2 * 1 Director of PIR, Department of Biochemistry and
Gene Ontology and overrepresentation analysis
 Gene Ontology and overrepresentation analysis Kjell Petersen J Express Microarray analysis course Oslo December 2009 Presentation adapted from Endre Anderssen and Vidar Beisvåg NMC Trondheim Overview How
Gene Ontology and overrepresentation analysis Kjell Petersen J Express Microarray analysis course Oslo December 2009 Presentation adapted from Endre Anderssen and Vidar Beisvåg NMC Trondheim Overview How
Hidden Markov Models (HMMs) and Profiles
 and Profiles") Hidden Markov Models (HMMs) and Profiles Swiss Institute of Bioinformatics (SIB) 26-30 November 2001 Markov Chain Models A Markov Chain Model is a succession of states S i (i = 0, 1,...) connected by transitions.
Hidden Markov Models (HMMs) and Profiles Swiss Institute of Bioinformatics (SIB) 26-30 November 2001 Markov Chain Models A Markov Chain Model is a succession of states S i (i = 0, 1,...) connected by transitions.
Sequence analysis and comparison
 The aim with sequence identification: Sequence analysis and comparison Marjolein Thunnissen Lund September 2012 Is there any known protein sequence that is homologous to mine? Are there any other species
The aim with sequence identification: Sequence analysis and comparison Marjolein Thunnissen Lund September 2012 Is there any known protein sequence that is homologous to mine? Are there any other species
Computational methods for the analysis of bacterial gene regulation Brouwer, Rutger Wubbe Willem
 University of Groningen Computational methods for the analysis of bacterial gene regulation Brouwer, Rutger Wubbe Willem IMPORTANT NOTE: You are advised to consult the publisher's version (publisher's
University of Groningen Computational methods for the analysis of bacterial gene regulation Brouwer, Rutger Wubbe Willem IMPORTANT NOTE: You are advised to consult the publisher's version (publisher's
Introduction to Bioinformatics Online Course: IBT
 Introduction to Bioinformatics Online Course: IBT Multiple Sequence Alignment Building Multiple Sequence Alignment Lec1 Building a Multiple Sequence Alignment Learning Outcomes 1- Understanding Why multiple
Introduction to Bioinformatics Online Course: IBT Multiple Sequence Alignment Building Multiple Sequence Alignment Lec1 Building a Multiple Sequence Alignment Learning Outcomes 1- Understanding Why multiple
CISC 889 Bioinformatics (Spring 2004) Sequence pairwise alignment (I)
 Sequence pairwise alignment (I)") CISC 889 Bioinformatics (Spring 2004) Sequence pairwise alignment (I) Contents Alignment algorithms Needleman-Wunsch (global alignment) Smith-Waterman (local alignment) Heuristic algorithms FASTA BLAST
CISC 889 Bioinformatics (Spring 2004) Sequence pairwise alignment (I) Contents Alignment algorithms Needleman-Wunsch (global alignment) Smith-Waterman (local alignment) Heuristic algorithms FASTA BLAST
Chemical Data Retrieval and Management
 Chemical Data Retrieval and Management ChEMBL, ChEBI, and the Chemistry Development Kit Stephan A. Beisken What is EMBL-EBI? Part of the European Molecular Biology Laboratory International, non-profit
Chemical Data Retrieval and Management ChEMBL, ChEBI, and the Chemistry Development Kit Stephan A. Beisken What is EMBL-EBI? Part of the European Molecular Biology Laboratory International, non-profit
SABIO-RK Integration and Curation of Reaction Kinetics Data Ulrike Wittig
 SABIO-RK Integration and Curation of Reaction Kinetics Data http://sabio.villa-bosch.de/sabiork Ulrike Wittig Overview Introduction /Motivation Database content /User interface Data integration Curation
SABIO-RK Integration and Curation of Reaction Kinetics Data http://sabio.villa-bosch.de/sabiork Ulrike Wittig Overview Introduction /Motivation Database content /User interface Data integration Curation
Alignment principles and homology searching using (PSI-)BLAST. Jaap Heringa Centre for Integrative Bioinformatics VU (IBIVU)
BLAST. Jaap Heringa Centre for Integrative Bioinformatics VU (IBIVU)") Alignment principles and homology searching using (PSI-)BLAST Jaap Heringa Centre for Integrative Bioinformatics VU (IBIVU) http://ibivu.cs.vu.nl Bioinformatics Nothing in Biology makes sense except in
Alignment principles and homology searching using (PSI-)BLAST Jaap Heringa Centre for Integrative Bioinformatics VU (IBIVU) http://ibivu.cs.vu.nl Bioinformatics Nothing in Biology makes sense except in
SCOP. all-β class. all-α class, 3 different folds. T4 endonuclease V. 4-helical cytokines. Globin-like
 SCOP all-β class 4-helical cytokines T4 endonuclease V all-α class, 3 different folds Globin-like TIM-barrel fold α/β class Profilin-like fold α+β class http://scop.mrc-lmb.cam.ac.uk/scop CATH Class, Architecture,
SCOP all-β class 4-helical cytokines T4 endonuclease V all-α class, 3 different folds Globin-like TIM-barrel fold α/β class Profilin-like fold α+β class http://scop.mrc-lmb.cam.ac.uk/scop CATH Class, Architecture,
Ch. 9 Multiple Sequence Alignment (MSA)
") Ch. 9 Multiple Sequence Alignment (MSA) - gather seqs. to make MSA - doing MSA with ClustalW - doing MSA with Tcoffee - comparing seqs. that cannot align Introduction - from pairwise alignment to MSA -
Ch. 9 Multiple Sequence Alignment (MSA) - gather seqs. to make MSA - doing MSA with ClustalW - doing MSA with Tcoffee - comparing seqs. that cannot align Introduction - from pairwise alignment to MSA -
Prediction of protein function from sequence analysis
 Prediction of protein function from sequence analysis Rita Casadio BIOCOMPUTING GROUP University of Bologna, Italy The omic era Genome Sequencing Projects: Archaea: 74 species In Progress:52 Bacteria:
Prediction of protein function from sequence analysis Rita Casadio BIOCOMPUTING GROUP University of Bologna, Italy The omic era Genome Sequencing Projects: Archaea: 74 species In Progress:52 Bacteria:
Comparing whole genomes
 BioNumerics Tutorial: Comparing whole genomes 1 Aim The Chromosome Comparison window in BioNumerics has been designed for large-scale comparison of sequences of unlimited length. In this tutorial you will
BioNumerics Tutorial: Comparing whole genomes 1 Aim The Chromosome Comparison window in BioNumerics has been designed for large-scale comparison of sequences of unlimited length. In this tutorial you will
Protein Families. João C. Setubal University of São Paulo Agosto /23/2012 J. C. Setubal
 Protein Families João C. Setubal University of São Paulo Agosto 2012 8/23/2012 J. C. Setubal 1 Motivation Phytophthora Science paper [Tyler et al., 2006] Comparison of the [P. sojae and P. ramorum] genomes
Protein Families João C. Setubal University of São Paulo Agosto 2012 8/23/2012 J. C. Setubal 1 Motivation Phytophthora Science paper [Tyler et al., 2006] Comparison of the [P. sojae and P. ramorum] genomes
Gene Ontology and Functional Enrichment. Genome 559: Introduction to Statistical and Computational Genomics Elhanan Borenstein
 Gene Ontology and Functional Enrichment Genome 559: Introduction to Statistical and Computational Genomics Elhanan Borenstein The parsimony principle: A quick review Find the tree that requires the fewest
Gene Ontology and Functional Enrichment Genome 559: Introduction to Statistical and Computational Genomics Elhanan Borenstein The parsimony principle: A quick review Find the tree that requires the fewest
Sequence Alignment Techniques and Their Uses
 Sequence Alignment Techniques and Their Uses Sarah Fiorentino Since rapid sequencing technology and whole genomes sequencing, the amount of sequence information has grown exponentially. With all of this
Sequence Alignment Techniques and Their Uses Sarah Fiorentino Since rapid sequencing technology and whole genomes sequencing, the amount of sequence information has grown exponentially. With all of this
Riboflavin Metabolism: A study to see if Mrub_1256 is Orthologous to E. coli b0415, and if Mrub_1254 is Orthologous to E.
 Augustana College Augustana Digital Commons Meiothermus ruber Genome Analysis Project Biology Winter 2-2016 Riboflavin Metabolism: A study to see if Mrub_1256 is Orthologous to E. coli b0415, and if Mrub_1254
Augustana College Augustana Digital Commons Meiothermus ruber Genome Analysis Project Biology Winter 2-2016 Riboflavin Metabolism: A study to see if Mrub_1256 is Orthologous to E. coli b0415, and if Mrub_1254
Homology Modeling. Roberto Lins EPFL - summer semester 2005
 Homology Modeling Roberto Lins EPFL - summer semester 2005 Disclaimer: course material is mainly taken from: P.E. Bourne & H Weissig, Structural Bioinformatics; C.A. Orengo, D.T. Jones & J.M. Thornton,
Homology Modeling Roberto Lins EPFL - summer semester 2005 Disclaimer: course material is mainly taken from: P.E. Bourne & H Weissig, Structural Bioinformatics; C.A. Orengo, D.T. Jones & J.M. Thornton,
Algorithms in Bioinformatics FOUR Pairwise Sequence Alignment. Pairwise Sequence Alignment. Convention: DNA Sequences 5. Sequence Alignment
 Algorithms in Bioinformatics FOUR Sami Khuri Department of Computer Science San José State University Pairwise Sequence Alignment Homology Similarity Global string alignment Local string alignment Dot
Algorithms in Bioinformatics FOUR Sami Khuri Department of Computer Science San José State University Pairwise Sequence Alignment Homology Similarity Global string alignment Local string alignment Dot
Statistical Machine Learning Methods for Bioinformatics II. Hidden Markov Model for Biological Sequences
 Statistical Machine Learning Methods for Bioinformatics II. Hidden Markov Model for Biological Sequences Jianlin Cheng, PhD Department of Computer Science University of Missouri 2008 Free for Academic
Statistical Machine Learning Methods for Bioinformatics II. Hidden Markov Model for Biological Sequences Jianlin Cheng, PhD Department of Computer Science University of Missouri 2008 Free for Academic
Meiothermus ruber Genome Analysis Project
 Augustana College Augustana Digital Commons Meiothermus ruber Genome Analysis Project Biology 2018 Predicted ortholog pairs between E. coli and M. ruber are b3456 and mrub_2379, b3457 and mrub_2378, b3456
Augustana College Augustana Digital Commons Meiothermus ruber Genome Analysis Project Biology 2018 Predicted ortholog pairs between E. coli and M. ruber are b3456 and mrub_2379, b3457 and mrub_2378, b3456
BIOINFORMATICS LAB AP BIOLOGY
 BIOINFORMATICS LAB AP BIOLOGY Bioinformatics is the science of collecting and analyzing complex biological data. Bioinformatics combines computer science, statistics and biology to allow scientists to
BIOINFORMATICS LAB AP BIOLOGY Bioinformatics is the science of collecting and analyzing complex biological data. Bioinformatics combines computer science, statistics and biology to allow scientists to
Grundlagen der Bioinformatik Summer semester Lecturer: Prof. Daniel Huson
 Grundlagen der Bioinformatik, SS 10, D. Huson, April 12, 2010 1 1 Introduction Grundlagen der Bioinformatik Summer semester 2010 Lecturer: Prof. Daniel Huson Office hours: Thursdays 17-18h (Sand 14, C310a)
Grundlagen der Bioinformatik, SS 10, D. Huson, April 12, 2010 1 1 Introduction Grundlagen der Bioinformatik Summer semester 2010 Lecturer: Prof. Daniel Huson Office hours: Thursdays 17-18h (Sand 14, C310a)
functional annotation preliminary results
 functional annotation preliminary results March 16, 216 Alicia Francis, Andrew Teng, Chen Guo, Devika Singh, Ellie Kim, Harshmi Shah, James Moore, Jose Jaimes, Nadav Topaz, Namrata Kalsi, Petar Penev,
functional annotation preliminary results March 16, 216 Alicia Francis, Andrew Teng, Chen Guo, Devika Singh, Ellie Kim, Harshmi Shah, James Moore, Jose Jaimes, Nadav Topaz, Namrata Kalsi, Petar Penev,
2 GENE FUNCTIONAL SIMILARITY. 2.1 Semantic values of GO terms
 Bioinformatics Advance Access published March 7, 2007 The Author (2007). Published by Oxford University Press. All rights reserved. For Permissions, please email: journals.permissions@oxfordjournals.org
Bioinformatics Advance Access published March 7, 2007 The Author (2007). Published by Oxford University Press. All rights reserved. For Permissions, please email: journals.permissions@oxfordjournals.org
Single alignment: Substitution Matrix. 16 march 2017
 Single alignment: Substitution Matrix 16 march 2017 BLOSUM Matrix BLOSUM Matrix [2] (Blocks Amino Acid Substitution Matrices ) It is based on the amino acids substitutions observed in ~2000 conserved block
Single alignment: Substitution Matrix 16 march 2017 BLOSUM Matrix BLOSUM Matrix [2] (Blocks Amino Acid Substitution Matrices ) It is based on the amino acids substitutions observed in ~2000 conserved block
METABOLIC PATHWAY PREDICTION/ALIGNMENT
 COMPUTATIONAL SYSTEMIC BIOLOGY METABOLIC PATHWAY PREDICTION/ALIGNMENT Hofestaedt R*, Chen M Bioinformatics / Medical Informatics, Technische Fakultaet, Universitaet Bielefeld Postfach 10 01 31, D-33501
COMPUTATIONAL SYSTEMIC BIOLOGY METABOLIC PATHWAY PREDICTION/ALIGNMENT Hofestaedt R*, Chen M Bioinformatics / Medical Informatics, Technische Fakultaet, Universitaet Bielefeld Postfach 10 01 31, D-33501
PROTEIN CLUSTERING AND CLASSIFICATION
 PROTEIN CLUSTERING AND CLASSIFICATION ori Sasson 1 and Michal Linial 2 1The School of Computer Science and Engeeniring and 2 The Life Science Institute, The Hebrew University of Jerusalem, Israel 1. Introduction
PROTEIN CLUSTERING AND CLASSIFICATION ori Sasson 1 and Michal Linial 2 1The School of Computer Science and Engeeniring and 2 The Life Science Institute, The Hebrew University of Jerusalem, Israel 1. Introduction
Bioinformatics in the post-sequence era
 Bioinformatics in the post-sequence era review Minoru Kanehisa 1 & Peer Bork 2 doi:10.1038/ng1109 In the past decade, bioinformatics has become an integral part of research and development in the biomedical
Bioinformatics in the post-sequence era review Minoru Kanehisa 1 & Peer Bork 2 doi:10.1038/ng1109 In the past decade, bioinformatics has become an integral part of research and development in the biomedical
Supplementary Information
 Supplementary Information Supplementary Figure 1. Schematic pipeline for single-cell genome assembly, cleaning and annotation. a. The assembly process was optimized to account for multiple cells putatively
Supplementary Information Supplementary Figure 1. Schematic pipeline for single-cell genome assembly, cleaning and annotation. a. The assembly process was optimized to account for multiple cells putatively
Protein Structure Prediction Using Neural Networks
 Protein Structure Prediction Using Neural Networks Martha Mercaldi Kasia Wilamowska Literature Review December 16, 2003 The Protein Folding Problem Evolution of Neural Networks Neural networks originally
Protein Structure Prediction Using Neural Networks Martha Mercaldi Kasia Wilamowska Literature Review December 16, 2003 The Protein Folding Problem Evolution of Neural Networks Neural networks originally
- conserved in Eukaryotes. - proteins in the cluster have identifiable conserved domains. - human gene should be included in the cluster.
 NCBI BLAST Services DELTA-BLAST BLAST (http://blast.ncbi.nlm.nih.gov/), Basic Local Alignment Search tool, is a suite of programs for finding similarities between biological sequences. DELTA-BLAST is a
NCBI BLAST Services DELTA-BLAST BLAST (http://blast.ncbi.nlm.nih.gov/), Basic Local Alignment Search tool, is a suite of programs for finding similarities between biological sequences. DELTA-BLAST is a
Agilent MassHunter Profinder: Solving the Challenge of Isotopologue Extraction for Qualitative Flux Analysis
 Agilent MassHunter Profinder: Solving the Challenge of Isotopologue Extraction for Qualitative Flux Analysis Technical Overview Introduction Metabolomics studies measure the relative abundance of metabolites
Agilent MassHunter Profinder: Solving the Challenge of Isotopologue Extraction for Qualitative Flux Analysis Technical Overview Introduction Metabolomics studies measure the relative abundance of metabolites
Mathangi Thiagarajan Rice Genome Annotation Workshop May 23rd, 2007
 -2 Transcript Alignment Assembly and Automated Gene Structure Improvements Using PASA-2 Mathangi Thiagarajan mathangi@jcvi.org Rice Genome Annotation Workshop May 23rd, 2007 About PASA PASA is an open
-2 Transcript Alignment Assembly and Automated Gene Structure Improvements Using PASA-2 Mathangi Thiagarajan mathangi@jcvi.org Rice Genome Annotation Workshop May 23rd, 2007 About PASA PASA is an open
Supplemental Materials
 JOURNAL OF MICROBIOLOGY & BIOLOGY EDUCATION, May 2013, p. 107-109 DOI: http://dx.doi.org/10.1128/jmbe.v14i1.496 Supplemental Materials for Engaging Students in a Bioinformatics Activity to Introduce Gene
JOURNAL OF MICROBIOLOGY & BIOLOGY EDUCATION, May 2013, p. 107-109 DOI: http://dx.doi.org/10.1128/jmbe.v14i1.496 Supplemental Materials for Engaging Students in a Bioinformatics Activity to Introduce Gene
GO annotation in InterPro: why stability does not indicate accuracy in a sea of changing annotations
 Database, 2016, 1 8 doi: 10.1093/database/baw027 Original article Original article GO annotation in InterPro: why stability does not indicate accuracy in a sea of changing annotations Amaia Sangrador-Vegas
Database, 2016, 1 8 doi: 10.1093/database/baw027 Original article Original article GO annotation in InterPro: why stability does not indicate accuracy in a sea of changing annotations Amaia Sangrador-Vegas
Amino Acid Structures from Klug & Cummings. Bioinformatics (Lec 12)
") Amino Acid Structures from Klug & Cummings 2/17/05 1 Amino Acid Structures from Klug & Cummings 2/17/05 2 Amino Acid Structures from Klug & Cummings 2/17/05 3 Amino Acid Structures from Klug & Cummings
Amino Acid Structures from Klug & Cummings 2/17/05 1 Amino Acid Structures from Klug & Cummings 2/17/05 2 Amino Acid Structures from Klug & Cummings 2/17/05 3 Amino Acid Structures from Klug & Cummings
Basic Local Alignment Search Tool
 Basic Local Alignment Search Tool Alignments used to uncover homologies between sequences combined with phylogenetic studies o can determine orthologous and paralogous relationships Local Alignment uses
Basic Local Alignment Search Tool Alignments used to uncover homologies between sequences combined with phylogenetic studies o can determine orthologous and paralogous relationships Local Alignment uses