ProtoNet 4.0: A hierarchical classification of one million protein sequences
|
|
|
- Arron Cannon
- 6 years ago
- Views:
Transcription
1 ProtoNet 4.0: A hierarchical classification of one million protein sequences Noam Kaplan 1*, Ori Sasson 2, Uri Inbar 2, Moriah Friedlich 2, Menachem Fromer 2, Hillel Fleischer 2, Elon Portugaly 2, Nathan Linial 2 and Michal Linial 1 1 Department of Biological Chemistry, Institute of Life Sciences, The Hebrew University of Jerusalem, Israel 2 School of Computer Science and Engineering, The Hebrew University of Jerusalem, Israel * Corresponding author Corresponding author details: Noam Kaplan kaplann@cc.huji.ac.il Address: The Hebrew University, Department of Biological Chemistry, Givat Ram Campus, Jerusalem, Israel, Telephone: ABSTRACT ProtoNet is an automatic hierarchical classification of the protein sequence space. In 2004 the ProtoNet (version 4.0) presents the analysis of over one million proteins merged from SwissProt and TrEMBL databases. In addition to rich visualization and analysis tools to navigate the clustering hierarchy, we incorporated several improvements that allow a simplified view of the scaffold of the proteins. An unsupervised biologically valid method that was developed resulted in a condensation of the ProtoNet hierarchy to only 12% of the clusters. A large portion of these clusters were automatically assigned high confidence biological names according to their correspondence with functional annotations. ProtoNet is available at: INTRODUCTION ProtoNet (1) (launched at 2002) is an automatic hierarchical clustering of the SwissProt and TrEMBL (2) protein databases. The clustering process is based on an all-against-all BLAST (3) similarity search. The similarities' E-score is used to perform a continuous bottom-up clustering process by joining at each step the two most similar protein clusters, resulting in a hierarchy of protein clusters at various degrees of biological granularity. This hierarchy is structured as a collection of trees, in which the root clusters contain all the proteins of the tree and the rest of the clusters
2 represent subdivisions of the proteins into smaller groups. Browsing this global hierarchical organization of the protein world provides several interesting biological insights about protein families and the evolution of structural and functional relations between proteins (4). Furthermore, ProtoNet can be used to assess the function of novel protein sequences, by finding the best matching cluster for the new sequence. We describe here several new developments in ProtoNet 4.0 including the increase of the sequences analyzed from 114,033 proteins in SwissProt (version 2.1) to 1,072,911 sequences (SwissProt and TrEMBL, version 4.0) and the improved methodology for simplification of the scaffold of the protein hierarchy. ProtoNet is available at: HIERARCHY CONDENSATION Due to the immense size of the ProtoNet hierarchy and the number of protein clusters, it would be very difficult to navigate in such a large hierarchy. Furthermore, it is obvious that many of the clusters are biologically irrelevant and uninteresting (for example huge root clusters containing hundreds of thousands non-related proteins). In order to get a condensed yet biologically relevant view of the hierarchy, we searched for some process-intrinsic parameter that would indicate which clusters are biologically relevant. The parameter found measures the stability of the cluster in the process, assuming that a stable cluster would be also more relevant biologically. We found this assumption to be correct, and that if we select a small subset of the clusters that show high stability, they would retain the biological validity of ProtoNet (Kaplan et al., in preparation). The default condensation of the ProtoNet hierarchy leaves 12% of the clusters. However, the ProtoNet website now offers an "advanced mode", in which advanced users can control the level of condensation and the method by which it is done, resulting in a larger or smaller set of clusters as required. Note that the condensation causes the trees to change from binary trees to non-binary trees, and some browsing options have been developed accordingly (see "Web Enhancements"). DATABASE UPDATES ProtoNet has gone through a major update of all database sources. Primarily, the protein database from which the ProtoNet tree is constructed has been updated and extended to include the TrEMBL protein database as well as SwissProt. This results in a leap from 114,033 to 1,072,911 protein sequences. Although TrEMBL is not manually validated by experts, it provides a much more extensive view of the protein world including whole genomes and thorough representation of several key organisms (see Table 1). Table 1 CLUSTER NAMES
3 Assigning a biological function to proteins is a major objective in bioinformatics. We have developed an automatic high-confidence method that assigns a functional annotation to ProtoNet protein clusters. The method finds a functional annotation from either InterPro (5), GO (6), SwissProt or ENZYME (7) databases that best fits the proteins of the cluster and assigns it a score relative to how well it fits the cluster. If this score is above a certain threshold, the annotation is assigned as the cluster's name. Understandably, not all protein clusters would have an existing annotation that fits them well. Clusters whose best fitting annotation does not pass the threshold remain nameless, possibly suggesting a novel functional group or clusters that are associated with mixed functions. By applying this method we were able to assign biological names to 78% of the clusters that contain 20 proteins or more. Due to the fact that a high threshold is used, the annotation can be assigned with high confidence to the cluster. WEBSITE ENHANCEMENTS Several enhancements have been made to the ProtoNet website in order to allow easier and more in-depth analysis of the ProtoNet trees. BROWSING CLUSTER NAMES Cluster names are extremely useful for quickly browsing the ProtoNet trees, eliminating the need to check the proteins of each cluster in order to get an impression of the hierarchy. Furthermore, the assignment of a biological function to clusters suggests an easy scheme of assigning function to proteins with unknown function: a protein can be assigned the function of all clusters to which it belongs. This scheme can be used not only on each of the 1,072,911 proteins from which the ProtoNet hierarchy is constructed, but also for new protein sequences given by the user (using the "Classify your protein" option in the website, which finds the most suitable cluster for a new protein sequence given by the user). BROWSING THE TREE Subtree View: In order to cope with the change to a non-binary tree, we have introduced the ProtoBrowser (Figure 1), which shows the tree in the vicinity of the cluster that is being displayed. Instead of presenting only the branch of the tree to which the cluster belonged to, the new display allows easy navigation to neighboring clusters and an enhanced global overview of biological protein families. Figure 1 Functionality View: PANDORA (8) is a web-based tool that allows in-depth biological analysis of large protein sets. When trying to biologically interpret large protein clusters that contain hundreds of proteins, PANDORA is a natural choice. ProtoNet now offers a direct link from its cluster page to PANDORA, providing the ability to easily understand what biological groups the cluster is built from and analyze them by several different biological aspects.
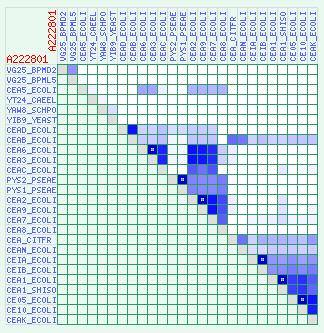
4 Similarity View: When viewing a protein cluster, it is sometimes helpful to obtain an in-depth look into the sequence similarity between the proteins of the cluster. This could allow the user to tell if there is a natural partitioning into subgroups or the cluster inner similarity is uniform, for example (see example in Shachar and Linial, 2004). In order to address this, the website offers a cluster similarity matrix (Figure 2), showing an all-against-all color coded matrix of all protein pairs in the cluster, colored according to the BLAST E-score between the two proteins. This also facilitates access to the BLAST result, which can be obtained simply by clicking on the appropriate cell in the matrix. Figure 2 MAINTENANCE AND UPDATING The ProtoNet source databases are generally updated twice a year. The next ProtoNet release is planned to include the UniProt Ref 100 protein database. Other future plans include allowing the users to select a subset of proteins from ProtoNet according to their needs (e.g. selecting for study only the proteins from the SwissProt database or only the proteins of a specific species) and expanding links to further biological databases such as OMIM (9) and DIP (10). ACKNOWLEDGEMENTS We thank the entire current and previous ProtoNet team for their endless support. Special thanks to Alex Savenok for the Web design as well as for the development of the visualization tools. We thank the fellowship support by the Sudarsky Center for Computational Biology (SCCB) to NK, UI, MF and MF. This study is partially supported by the EU NoE BioSapiens consortium and the CESG consortium supported by the NIH. REFERENCES 1. Sasson, O., Vaaknin, A., Fleischer, H., Portugaly, E., Bilu, Y., Linial, N. and Linial, M. (2003) ProtoNet: hierarchical classification of the protein space. Nucleic Acids Res., 31, Boeckmann, B., Bairoch, A., Apweiler, R., Blatter, M.C., Estreicher, A., Gasteiger, E., Martin, M.J., Michoud, K., O`Donovan, C., Phan, I. et al. (2003) The SWISS-PROT protein knowledgebase and its supplement TrEMBL in Nucleic Acids Res., 31, Altschul, S.F., Madden, T.L., Schaffer, A.A., Zhang, J., Zhang, Z., Miller, W. and Lipman, D.J. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res., 25, Shachar, O. and Linial, M. (2004) A robust method to detect structural and functional remote homologues. Proteins, in press. 5. Camon, E., Magrane, M., Barrell, D., Lee V., Dimmer, E., Maslen, J., Binns, D., Harte, N., Lopez, R. and Apweiler R. (2004) The Gene Ontology Annotation (GOA) Database: sharing knowledge in Uniprot with Gene Ontology. Nucleic Acids Res., 32, D262-D266.
5 6. Mulder, N.J., Apweiler, R., Attwood, T.K., Bairoch, A., Barrell, D., Bateman, A., Binns, D., Biswas, M., Bradley, P., Bork, P. et al. (2003) The InterPro Database, 2003 brings increased coverage and new features. Nucleic Acids Res., 31, Bairoch, A. (2000) The ENZYME database in Nucleic Acids Res., 28, Kaplan, N., Vaaknin, A. and Linial, M. (2003) PANDORA: keyword-based analysis of proteins sets by integration of annotation sources. Nucleic Acids Res., 31, McKusick, V.A. (1998) Mendelian Inheritance in Man. Johns Hopkins University Press, Baltimore. 10. Salwinski, L., Miller, C.S., Smith, A.J., Pettit, F.K., Bowie, J.U. and Eisenberg D. (2004) The Database of Interacting Proteins: 2004 update. Nucleic Acids Res., 32, D FIGURE LEGENDS Figure 1. The ProtoBrowser allows viewing the near vicinity of a cluster in the ProtoNet hierarchy. Blue triangle-shaped icons represent protein clusters. The cluster currently being viewed is the cluster A260586, which appears in the center in red. Clusters that include proteins with 3D solved structures as marked PDB. Figure 2. Example of a cluster similarity matrix. Colored cells represent different degrees of similarity, ranging from white (no similarity: BLAST E-score higher than 100) to dark blue (high similarity, BLAST E-score close to 0). It is evident that the cluster A is roughly divided into 3 subsets: in the upper left of the diagonal there are proteins which show no similarity to each other or to any protein in the cluster; in the center of the diagonal there is a subset of proteins that are similar to each other but to no other proteins; and at the bottom right of the diagonal there is another subset of proteins that are similar to each other but not to other proteins. TABLES Table 1. Species Proteins in ProtoNet 2.1 Proteins in ProtoNet 4.0 Homo sapiens 8,507 47,641 Mus musculus 5,678 41,813 Drosophila melanogaster 2,049 22,603 Arabidopsis thaliana 1,680 39,367 Plasmodium falciparum 153 8,434 FIGURES

6 Figure 1. Figure 2.
Functionally-Valid Unsupervised Compression of the Protein Space
 Functionally-Valid Unsupervised Compression of the Protein Space Noam Kaplan 1, Moriah Friedlich 2, Menachem Fromer 2 and Michal Linial 1* 1 Department of Biological Chemistry, Institute of Life Sciences,
Functionally-Valid Unsupervised Compression of the Protein Space Noam Kaplan 1, Moriah Friedlich 2, Menachem Fromer 2 and Michal Linial 1* 1 Department of Biological Chemistry, Institute of Life Sciences,
EVEREST: A collection of evolutionary conserved protein domains
 EVEREST: A collection of evolutionary conserved protein domains Elon Portugaly a, Nathan Linial a, Michal Linial b a School of Computer Science & Engineering. b Dept. of Biological Chemistry, Inst. of
EVEREST: A collection of evolutionary conserved protein domains Elon Portugaly a, Nathan Linial a, Michal Linial b a School of Computer Science & Engineering. b Dept. of Biological Chemistry, Inst. of
IMPORTANCE OF SECONDARY STRUCTURE ELEMENTS FOR PREDICTION OF GO ANNOTATIONS
 IMPORTANCE OF SECONDARY STRUCTURE ELEMENTS FOR PREDICTION OF GO ANNOTATIONS Aslı Filiz 1, Eser Aygün 2, Özlem Keskin 3 and Zehra Cataltepe 2 1 Informatics Institute and 2 Computer Engineering Department,
IMPORTANCE OF SECONDARY STRUCTURE ELEMENTS FOR PREDICTION OF GO ANNOTATIONS Aslı Filiz 1, Eser Aygün 2, Özlem Keskin 3 and Zehra Cataltepe 2 1 Informatics Institute and 2 Computer Engineering Department,
Fishing with (Proto)Net a principled approach to protein target selection
Net a principled approach to protein target selection") Comparative and Functional Genomics Comp Funct Genom 2003; 4: 542 548. Published online in Wiley InterScience (www.interscience.wiley.com). DOI: 10.1002/cfg.328 Conference Review Fishing with (Proto)Net
Comparative and Functional Genomics Comp Funct Genom 2003; 4: 542 548. Published online in Wiley InterScience (www.interscience.wiley.com). DOI: 10.1002/cfg.328 Conference Review Fishing with (Proto)Net
A robust method to detect structural and functional remote homologues
 A robust method to detect structural and functional remote homologues Ori Shachar 1 and Michal Linial 2,3 1 School of Computer Science and Engineering, 2 Department of Biological Chemistry, Institute of
A robust method to detect structural and functional remote homologues Ori Shachar 1 and Michal Linial 2,3 1 School of Computer Science and Engineering, 2 Department of Biological Chemistry, Institute of
PROTEIN FUNCTION PREDICTION WITH AMINO ACID SEQUENCE AND SECONDARY STRUCTURE ALIGNMENT SCORES
 PROTEIN FUNCTION PREDICTION WITH AMINO ACID SEQUENCE AND SECONDARY STRUCTURE ALIGNMENT SCORES Eser Aygün 1, Caner Kömürlü 2, Zafer Aydin 3 and Zehra Çataltepe 1 1 Computer Engineering Department and 2
PROTEIN FUNCTION PREDICTION WITH AMINO ACID SEQUENCE AND SECONDARY STRUCTURE ALIGNMENT SCORES Eser Aygün 1, Caner Kömürlü 2, Zafer Aydin 3 and Zehra Çataltepe 1 1 Computer Engineering Department and 2
Supplementary Materials for mplr-loc Web-server
 Supplementary Materials for mplr-loc Web-server Shibiao Wan and Man-Wai Mak email: shibiao.wan@connect.polyu.hk, enmwmak@polyu.edu.hk June 2014 Back to mplr-loc Server Contents 1 Introduction to mplr-loc
Supplementary Materials for mplr-loc Web-server Shibiao Wan and Man-Wai Mak email: shibiao.wan@connect.polyu.hk, enmwmak@polyu.edu.hk June 2014 Back to mplr-loc Server Contents 1 Introduction to mplr-loc
Estimation of Identification Methods of Gene Clusters Using GO Term Annotations from a Hierarchical Cluster Tree
 Estimation of Identification Methods of Gene Clusters Using GO Term Annotations from a Hierarchical Cluster Tree YOICHI YAMADA, YUKI MIYATA, MASANORI HIGASHIHARA*, KENJI SATOU Graduate School of Natural
Estimation of Identification Methods of Gene Clusters Using GO Term Annotations from a Hierarchical Cluster Tree YOICHI YAMADA, YUKI MIYATA, MASANORI HIGASHIHARA*, KENJI SATOU Graduate School of Natural
Update on human genome completion and annotations: Protein information resource
 UPDATE ON GENOME COMPLETION AND ANNOTATIONS Update on human genome completion and annotations: Protein information resource Cathy Wu 1 and Daniel W. Nebert 2 * 1 Director of PIR, Department of Biochemistry
UPDATE ON GENOME COMPLETION AND ANNOTATIONS Update on human genome completion and annotations: Protein information resource Cathy Wu 1 and Daniel W. Nebert 2 * 1 Director of PIR, Department of Biochemistry
Hands-On Nine The PAX6 Gene and Protein
 Hands-On Nine The PAX6 Gene and Protein Main Purpose of Hands-On Activity: Using bioinformatics tools to examine the sequences, homology, and disease relevance of the Pax6: a master gene of eye formation.
Hands-On Nine The PAX6 Gene and Protein Main Purpose of Hands-On Activity: Using bioinformatics tools to examine the sequences, homology, and disease relevance of the Pax6: a master gene of eye formation.
Supplementary Materials for R3P-Loc Web-server
 Supplementary Materials for R3P-Loc Web-server Shibiao Wan and Man-Wai Mak email: shibiao.wan@connect.polyu.hk, enmwmak@polyu.edu.hk June 2014 Back to R3P-Loc Server Contents 1 Introduction to R3P-Loc
Supplementary Materials for R3P-Loc Web-server Shibiao Wan and Man-Wai Mak email: shibiao.wan@connect.polyu.hk, enmwmak@polyu.edu.hk June 2014 Back to R3P-Loc Server Contents 1 Introduction to R3P-Loc
Truncated Profile Hidden Markov Models
 Boise State University ScholarWorks Electrical and Computer Engineering Faculty Publications and Presentations Department of Electrical and Computer Engineering 11-1-2005 Truncated Profile Hidden Markov
Boise State University ScholarWorks Electrical and Computer Engineering Faculty Publications and Presentations Department of Electrical and Computer Engineering 11-1-2005 Truncated Profile Hidden Markov
Genome Annotation. Bioinformatics and Computational Biology. Genome sequencing Assembly. Gene prediction. Protein targeting.
 Genome Annotation Bioinformatics and Computational Biology Genome Annotation Frank Oliver Glöckner 1 Genome Analysis Roadmap Genome sequencing Assembly Gene prediction Protein targeting trna prediction
Genome Annotation Bioinformatics and Computational Biology Genome Annotation Frank Oliver Glöckner 1 Genome Analysis Roadmap Genome sequencing Assembly Gene prediction Protein targeting trna prediction
Francisco M. Couto Mário J. Silva Pedro Coutinho
 Francisco M. Couto Mário J. Silva Pedro Coutinho DI FCUL TR 03 29 Departamento de Informática Faculdade de Ciências da Universidade de Lisboa Campo Grande, 1749 016 Lisboa Portugal Technical reports are
Francisco M. Couto Mário J. Silva Pedro Coutinho DI FCUL TR 03 29 Departamento de Informática Faculdade de Ciências da Universidade de Lisboa Campo Grande, 1749 016 Lisboa Portugal Technical reports are
A profile-based protein sequence alignment algorithm for a domain clustering database
 A profile-based protein sequence alignment algorithm for a domain clustering database Lin Xu,2 Fa Zhang and Zhiyong Liu 3, Key Laboratory of Computer System and architecture, the Institute of Computing
A profile-based protein sequence alignment algorithm for a domain clustering database Lin Xu,2 Fa Zhang and Zhiyong Liu 3, Key Laboratory of Computer System and architecture, the Institute of Computing
BIOINFORMATICS. The metric space of proteins comparative study of clustering algorithms. Ori Sasson 1, Nathan Linial 1 and Michal Linial 2
 BIOINFORMATICS Vol. 18 Suppl. 1 2002 Pages S14 S21 The metric space of proteins comparative study of clustering algorithms Ori Sasson 1, Nathan Linial 1 and Michal Linial 2 1 School of Computer Science
BIOINFORMATICS Vol. 18 Suppl. 1 2002 Pages S14 S21 The metric space of proteins comparative study of clustering algorithms Ori Sasson 1, Nathan Linial 1 and Michal Linial 2 1 School of Computer Science
Update on genome completion and annotations: Protein Information Resource
 UPDATE ON GENOME COMPLETION AND ANNOTATIONS Update on genome completion and annotations: Protein Information Resource Cathy Wu 1 and Daniel W. Nebert 2 * 1 Director of PIR, Department of Biochemistry and
UPDATE ON GENOME COMPLETION AND ANNOTATIONS Update on genome completion and annotations: Protein Information Resource Cathy Wu 1 and Daniel W. Nebert 2 * 1 Director of PIR, Department of Biochemistry and
BIOINFORMATICS. Efficient algorithms for exact hierarchical clustering of huge datasets: Tackling the entire protein space
 BIOINFORMATICS Vol. 00 no. 00 2005 Pages 1 10 Efficient algorithms for exact hierarchical clustering of huge datasets: Tackling the entire protein space Yaniv Loewenstein 1,, Elon Portugaly 1, Menachem
BIOINFORMATICS Vol. 00 no. 00 2005 Pages 1 10 Efficient algorithms for exact hierarchical clustering of huge datasets: Tackling the entire protein space Yaniv Loewenstein 1,, Elon Portugaly 1, Menachem
2MHR. Protein structure classification is important because it organizes the protein structure universe that is independent of sequence similarity.
 Protein structure classification is important because it organizes the protein structure universe that is independent of sequence similarity. A global picture of the protein universe will help us to understand
Protein structure classification is important because it organizes the protein structure universe that is independent of sequence similarity. A global picture of the protein universe will help us to understand
Bioinformatics. Dept. of Computational Biology & Bioinformatics
 Bioinformatics Dept. of Computational Biology & Bioinformatics 3 Bioinformatics - play with sequences & structures Dept. of Computational Biology & Bioinformatics 4 ORGANIZATION OF LIFE ROLE OF BIOINFORMATICS
Bioinformatics Dept. of Computational Biology & Bioinformatics 3 Bioinformatics - play with sequences & structures Dept. of Computational Biology & Bioinformatics 4 ORGANIZATION OF LIFE ROLE OF BIOINFORMATICS
EBI web resources II: Ensembl and InterPro. Yanbin Yin Spring 2013
 EBI web resources II: Ensembl and InterPro Yanbin Yin Spring 2013 1 Outline Intro to genome annotation Protein family/domain databases InterPro, Pfam, Superfamily etc. Genome browser Ensembl Hands on Practice
EBI web resources II: Ensembl and InterPro Yanbin Yin Spring 2013 1 Outline Intro to genome annotation Protein family/domain databases InterPro, Pfam, Superfamily etc. Genome browser Ensembl Hands on Practice
EBI web resources II: Ensembl and InterPro
 EBI web resources II: Ensembl and InterPro Yanbin Yin http://www.ebi.ac.uk/training/online/course/ 1 Homework 3 Go to http://www.ebi.ac.uk/interpro/training.htmland finish the second online training course
EBI web resources II: Ensembl and InterPro Yanbin Yin http://www.ebi.ac.uk/training/online/course/ 1 Homework 3 Go to http://www.ebi.ac.uk/interpro/training.htmland finish the second online training course
BIOINFORMATICS LAB AP BIOLOGY
 BIOINFORMATICS LAB AP BIOLOGY Bioinformatics is the science of collecting and analyzing complex biological data. Bioinformatics combines computer science, statistics and biology to allow scientists to
BIOINFORMATICS LAB AP BIOLOGY Bioinformatics is the science of collecting and analyzing complex biological data. Bioinformatics combines computer science, statistics and biology to allow scientists to
Sequence Alignment Techniques and Their Uses
 Sequence Alignment Techniques and Their Uses Sarah Fiorentino Since rapid sequencing technology and whole genomes sequencing, the amount of sequence information has grown exponentially. With all of this
Sequence Alignment Techniques and Their Uses Sarah Fiorentino Since rapid sequencing technology and whole genomes sequencing, the amount of sequence information has grown exponentially. With all of this
Methodologies for target selection in structural genomics
 Progress in Biophysics & Molecular Biology 73 (2000) 297 320 Review Methodologies for target selection in structural genomics Michal Linial a, *, Golan Yona b a Department of Biological Chemistry, Institute
Progress in Biophysics & Molecular Biology 73 (2000) 297 320 Review Methodologies for target selection in structural genomics Michal Linial a, *, Golan Yona b a Department of Biological Chemistry, Institute
Fast and accurate semi-supervised protein homology detection with large uncurated sequence databases
 Rutgers Computer Science Technical Report RU-DCS-TR634 May 2008 Fast and accurate semi-supervised protein homology detection with large uncurated sequence databases by Pai-Hsi Huang, Pavel Kuksa, Vladimir
Rutgers Computer Science Technical Report RU-DCS-TR634 May 2008 Fast and accurate semi-supervised protein homology detection with large uncurated sequence databases by Pai-Hsi Huang, Pavel Kuksa, Vladimir
PROTEIN CLUSTERING AND CLASSIFICATION
 PROTEIN CLUSTERING AND CLASSIFICATION ori Sasson 1 and Michal Linial 2 1The School of Computer Science and Engeeniring and 2 The Life Science Institute, The Hebrew University of Jerusalem, Israel 1. Introduction
PROTEIN CLUSTERING AND CLASSIFICATION ori Sasson 1 and Michal Linial 2 1The School of Computer Science and Engeeniring and 2 The Life Science Institute, The Hebrew University of Jerusalem, Israel 1. Introduction
Gene Ontology and overrepresentation analysis
 Gene Ontology and overrepresentation analysis Kjell Petersen J Express Microarray analysis course Oslo December 2009 Presentation adapted from Endre Anderssen and Vidar Beisvåg NMC Trondheim Overview How
Gene Ontology and overrepresentation analysis Kjell Petersen J Express Microarray analysis course Oslo December 2009 Presentation adapted from Endre Anderssen and Vidar Beisvåg NMC Trondheim Overview How
IMMERSIVE GRAPH-BASED VISUALIZATION AND EXPLORATION OF BIOLOGICAL DATA RELATIONSHIPS
 Data Science Journal, Volume 4, 31 December 2005 189 IMMERSIVE GRAPH-BASED VISUALIZATION AND EXPLORATION OF BIOLOGICAL DATA RELATIONSHIPS N Férey, PE Gros, J Hérisson, R Gherbi* *Bioinformatics team, Human-Computer
Data Science Journal, Volume 4, 31 December 2005 189 IMMERSIVE GRAPH-BASED VISUALIZATION AND EXPLORATION OF BIOLOGICAL DATA RELATIONSHIPS N Férey, PE Gros, J Hérisson, R Gherbi* *Bioinformatics team, Human-Computer
CSCE555 Bioinformatics. Protein Function Annotation
 CSCE555 Bioinformatics Protein Function Annotation Why we need to do function annotation? Fig from: Network-based prediction of protein function. Molecular Systems Biology 3:88. 2007 What s function? The
CSCE555 Bioinformatics Protein Function Annotation Why we need to do function annotation? Fig from: Network-based prediction of protein function. Molecular Systems Biology 3:88. 2007 What s function? The
Mathangi Thiagarajan Rice Genome Annotation Workshop May 23rd, 2007
 -2 Transcript Alignment Assembly and Automated Gene Structure Improvements Using PASA-2 Mathangi Thiagarajan mathangi@jcvi.org Rice Genome Annotation Workshop May 23rd, 2007 About PASA PASA is an open
-2 Transcript Alignment Assembly and Automated Gene Structure Improvements Using PASA-2 Mathangi Thiagarajan mathangi@jcvi.org Rice Genome Annotation Workshop May 23rd, 2007 About PASA PASA is an open
CS612 - Algorithms in Bioinformatics
 Fall 2017 Databases and Protein Structure Representation October 2, 2017 Molecular Biology as Information Science > 12, 000 genomes sequenced, mostly bacterial (2013) > 5x10 6 unique sequences available
Fall 2017 Databases and Protein Structure Representation October 2, 2017 Molecular Biology as Information Science > 12, 000 genomes sequenced, mostly bacterial (2013) > 5x10 6 unique sequences available
Ensembl focuses on metazoan (animal) genomes. The genomes currently available at the Ensembl site are:
 genomes. The genomes currently available at the Ensembl site are:") Comparative genomics and proteomics Species available Ensembl focuses on metazoan (animal) genomes. The genomes currently available at the Ensembl site are: Vertebrates: human, chimpanzee, mouse, rat,
Comparative genomics and proteomics Species available Ensembl focuses on metazoan (animal) genomes. The genomes currently available at the Ensembl site are: Vertebrates: human, chimpanzee, mouse, rat,
Supplementary Methods
 Supplementary Methods Homology modeling of the XylS N-terminal domain Identification of template The analysis was based on XylS from Pseudomonas putida (SwissProt (Boeckmann et al. 2003) sequence entry
Supplementary Methods Homology modeling of the XylS N-terminal domain Identification of template The analysis was based on XylS from Pseudomonas putida (SwissProt (Boeckmann et al. 2003) sequence entry
DATA ACQUISITION FROM BIO-DATABASES AND BLAST. Natapol Pornputtapong 18 January 2018
 DATA ACQUISITION FROM BIO-DATABASES AND BLAST Natapol Pornputtapong 18 January 2018 DATABASE Collections of data To share multi-user interface To prevent data loss To make sure to get the right things
DATA ACQUISITION FROM BIO-DATABASES AND BLAST Natapol Pornputtapong 18 January 2018 DATABASE Collections of data To share multi-user interface To prevent data loss To make sure to get the right things
Chapter 5. Proteomics and the analysis of protein sequence Ⅱ
 Proteomics Chapter 5. Proteomics and the analysis of protein sequence Ⅱ 1 Pairwise similarity searching (1) Figure 5.5: manual alignment One of the amino acids in the top sequence has no equivalent and
Proteomics Chapter 5. Proteomics and the analysis of protein sequence Ⅱ 1 Pairwise similarity searching (1) Figure 5.5: manual alignment One of the amino acids in the top sequence has no equivalent and
Syllabus of BIOINF 528 (2017 Fall, Bioinformatics Program)
") Syllabus of BIOINF 528 (2017 Fall, Bioinformatics Program) Course Name: Structural Bioinformatics Course Description: Instructor: This course introduces fundamental concepts and methods for structural
Syllabus of BIOINF 528 (2017 Fall, Bioinformatics Program) Course Name: Structural Bioinformatics Course Description: Instructor: This course introduces fundamental concepts and methods for structural
Introduction to Spark
 1 As you become familiar or continue to explore the Cresset technology and software applications, we encourage you to look through the user manual. This is accessible from the Help menu. However, don t
1 As you become familiar or continue to explore the Cresset technology and software applications, we encourage you to look through the user manual. This is accessible from the Help menu. However, don t
Cluster Analysis of Gene Expression Microarray Data. BIOL 495S/ CS 490B/ MATH 490B/ STAT 490B Introduction to Bioinformatics April 8, 2002
 Cluster Analysis of Gene Expression Microarray Data BIOL 495S/ CS 490B/ MATH 490B/ STAT 490B Introduction to Bioinformatics April 8, 2002 1 Data representations Data are relative measurements log 2 ( red
Cluster Analysis of Gene Expression Microarray Data BIOL 495S/ CS 490B/ MATH 490B/ STAT 490B Introduction to Bioinformatics April 8, 2002 1 Data representations Data are relative measurements log 2 ( red
Measuring Semantic Similarity between Gene Ontology Terms
 Measuring Semantic Similarity between Gene Ontology Terms Francisco M. Couto a Mário J. Silva a Pedro M. Coutinho b a Departamento de Informática, Faculdade de Ciências da Universidade de Lisboa, Portugal
Measuring Semantic Similarity between Gene Ontology Terms Francisco M. Couto a Mário J. Silva a Pedro M. Coutinho b a Departamento de Informática, Faculdade de Ciências da Universidade de Lisboa, Portugal
Investigation 3: Comparing DNA Sequences to Understand Evolutionary Relationships with BLAST
 Investigation 3: Comparing DNA Sequences to Understand Evolutionary Relationships with BLAST Introduction Bioinformatics is a powerful tool which can be used to determine evolutionary relationships and
Investigation 3: Comparing DNA Sequences to Understand Evolutionary Relationships with BLAST Introduction Bioinformatics is a powerful tool which can be used to determine evolutionary relationships and
Computational Structural Bioinformatics
 Computational Structural Bioinformatics ECS129 Instructor: Patrice Koehl http://koehllab.genomecenter.ucdavis.edu/teaching/ecs129 koehl@cs.ucdavis.edu Learning curve Math / CS Biology/ Chemistry Pre-requisite
Computational Structural Bioinformatics ECS129 Instructor: Patrice Koehl http://koehllab.genomecenter.ucdavis.edu/teaching/ecs129 koehl@cs.ucdavis.edu Learning curve Math / CS Biology/ Chemistry Pre-requisite
MegAlign Pro Pairwise Alignment Tutorials
 MegAlign Pro Pairwise Alignment Tutorials All demo data for the following tutorials can be found in the MegAlignProAlignments.zip archive here. Tutorial 1: Multiple versus pairwise alignments 1. Extract
MegAlign Pro Pairwise Alignment Tutorials All demo data for the following tutorials can be found in the MegAlignProAlignments.zip archive here. Tutorial 1: Multiple versus pairwise alignments 1. Extract
SUPPLEMENTARY INFORMATION
 Supplementary information S3 (box) Methods Methods Genome weighting The currently available collection of archaeal and bacterial genomes has a highly biased distribution of isolates across taxa. For example,
Supplementary information S3 (box) Methods Methods Genome weighting The currently available collection of archaeal and bacterial genomes has a highly biased distribution of isolates across taxa. For example,
Comprehensive genome analysis of 203 genomes provides structural genomics with new insights into protein family space
 Published online February 15, 26 166 18 Nucleic Acids Research, 26, Vol. 34, No. 3 doi:1.193/nar/gkj494 Comprehensive genome analysis of 23 genomes provides structural genomics with new insights into protein
Published online February 15, 26 166 18 Nucleic Acids Research, 26, Vol. 34, No. 3 doi:1.193/nar/gkj494 Comprehensive genome analysis of 23 genomes provides structural genomics with new insights into protein
Tutorial. Getting started. Sample to Insight. March 31, 2016
 Getting started March 31, 2016 Sample to Insight CLC bio, a QIAGEN Company Silkeborgvej 2 Prismet 8000 Aarhus C Denmark Telephone: +45 70 22 32 44 www.clcbio.com support-clcbio@qiagen.com Getting started
Getting started March 31, 2016 Sample to Insight CLC bio, a QIAGEN Company Silkeborgvej 2 Prismet 8000 Aarhus C Denmark Telephone: +45 70 22 32 44 www.clcbio.com support-clcbio@qiagen.com Getting started
A Browser for Pig Genome Data
 A Browser for Pig Genome Data Thomas Mailund January 2, 2004 This report briefly describe the blast and alignment data available at http://www.daimi.au.dk/ mailund/pig-genome/ hits.html. The report describes
A Browser for Pig Genome Data Thomas Mailund January 2, 2004 This report briefly describe the blast and alignment data available at http://www.daimi.au.dk/ mailund/pig-genome/ hits.html. The report describes
Small RNA in rice genome
 Vol. 45 No. 5 SCIENCE IN CHINA (Series C) October 2002 Small RNA in rice genome WANG Kai ( 1, ZHU Xiaopeng ( 2, ZHONG Lan ( 1,3 & CHEN Runsheng ( 1,2 1. Beijing Genomics Institute/Center of Genomics and
Vol. 45 No. 5 SCIENCE IN CHINA (Series C) October 2002 Small RNA in rice genome WANG Kai ( 1, ZHU Xiaopeng ( 2, ZHONG Lan ( 1,3 & CHEN Runsheng ( 1,2 1. Beijing Genomics Institute/Center of Genomics and
Improving Protein Function Prediction using the Hierarchical Structure of the Gene Ontology
 Improving Protein Function Prediction using the Hierarchical Structure of the Gene Ontology Roman Eisner, Brett Poulin, Duane Szafron, Paul Lu, Russ Greiner Department of Computing Science University of
Improving Protein Function Prediction using the Hierarchical Structure of the Gene Ontology Roman Eisner, Brett Poulin, Duane Szafron, Paul Lu, Russ Greiner Department of Computing Science University of
GENE ONTOLOGY (GO) Wilver Martínez Martínez Giovanny Silva Rincón
 Wilver Martínez Martínez Giovanny Silva Rincón") GENE ONTOLOGY (GO) Wilver Martínez Martínez Giovanny Silva Rincón What is GO? The Gene Ontology (GO) project is a collaborative effort to address the need for consistent descriptions of gene products in
GENE ONTOLOGY (GO) Wilver Martínez Martínez Giovanny Silva Rincón What is GO? The Gene Ontology (GO) project is a collaborative effort to address the need for consistent descriptions of gene products in
SUPPLEMENTARY INFORMATION
 Supplementary information S1 (box). Supplementary Methods description. Prokaryotic Genome Database Archaeal and bacterial genome sequences were downloaded from the NCBI FTP site (ftp://ftp.ncbi.nlm.nih.gov/genomes/all/)
Supplementary information S1 (box). Supplementary Methods description. Prokaryotic Genome Database Archaeal and bacterial genome sequences were downloaded from the NCBI FTP site (ftp://ftp.ncbi.nlm.nih.gov/genomes/all/)
Network by Weighted Graph Mining
 2012 4th International Conference on Bioinformatics and Biomedical Technology IPCBEE vol.29 (2012) (2012) IACSIT Press, Singapore + Prediction of Protein Function from Protein-Protein Interaction Network
2012 4th International Conference on Bioinformatics and Biomedical Technology IPCBEE vol.29 (2012) (2012) IACSIT Press, Singapore + Prediction of Protein Function from Protein-Protein Interaction Network
Procedure to Create NCBI KOGS
 Procedure to Create NCBI KOGS full details in: Tatusov et al (2003) BMC Bioinformatics 4:41. 1. Detect and mask typical repetitive domains Reason: masking prevents spurious lumping of non-orthologs based
Procedure to Create NCBI KOGS full details in: Tatusov et al (2003) BMC Bioinformatics 4:41. 1. Detect and mask typical repetitive domains Reason: masking prevents spurious lumping of non-orthologs based
Some Problems from Enzyme Families
 Some Problems from Enzyme Families Greg Butler Department of Computer Science Concordia University, Montreal www.cs.concordia.ca/~faculty/gregb gregb@cs.concordia.ca Abstract I will discuss some problems
Some Problems from Enzyme Families Greg Butler Department of Computer Science Concordia University, Montreal www.cs.concordia.ca/~faculty/gregb gregb@cs.concordia.ca Abstract I will discuss some problems
The Homology Kernel: A Biologically Motivated Sequence Embedding into Euclidean Space
 The Homology Kernel: A Biologically Motivated Sequence Embedding into Euclidean Space Eleazar Eskin Department of Computer Science Engineering University of California, San Diego eeskin@cs.ucsd.edu Sagi
The Homology Kernel: A Biologically Motivated Sequence Embedding into Euclidean Space Eleazar Eskin Department of Computer Science Engineering University of California, San Diego eeskin@cs.ucsd.edu Sagi
EasySDM: A Spatial Data Mining Platform
 EasySDM: A Spatial Data Mining Platform (User Manual) Authors: Amine Abdaoui and Mohamed Ala Al Chikha, Students at the National Computing Engineering School. Algiers. June 2013. 1. Overview EasySDM is
EasySDM: A Spatial Data Mining Platform (User Manual) Authors: Amine Abdaoui and Mohamed Ala Al Chikha, Students at the National Computing Engineering School. Algiers. June 2013. 1. Overview EasySDM is
Role of Duplicate Genes in Robustness against Deleterious Human Mutations
 Role of Duplicate Genes in Robustness against Deleterious Human Mutations Tzu-Lin Hsiao 1,2, Dennis Vitkup 1,2 * 1 Center for Computational Biology and Bioinformatics, Columbia University, New York, New
Role of Duplicate Genes in Robustness against Deleterious Human Mutations Tzu-Lin Hsiao 1,2, Dennis Vitkup 1,2 * 1 Center for Computational Biology and Bioinformatics, Columbia University, New York, New
CS570 Introduction to Data Mining
 CS570 Introduction to Data Mining Department of Mathematics and Computer Science Li Xiong Data Exploration and Data Preprocessing Data and Attributes Data exploration Data pre-processing Data cleaning
CS570 Introduction to Data Mining Department of Mathematics and Computer Science Li Xiong Data Exploration and Data Preprocessing Data and Attributes Data exploration Data pre-processing Data cleaning
Protein function prediction based on sequence analysis
 Performing sequence searches Post-Blast analysis, Using profiles and pattern-matching Protein function prediction based on sequence analysis Slides from a lecture on MOL204 - Applied Bioinformatics 18-Oct-2005
Performing sequence searches Post-Blast analysis, Using profiles and pattern-matching Protein function prediction based on sequence analysis Slides from a lecture on MOL204 - Applied Bioinformatics 18-Oct-2005
Supplementary text for the section Interactions conserved across species: can one select the conserved interactions?
 1 Supporting Information: What Evidence is There for the Homology of Protein-Protein Interactions? Anna C. F. Lewis, Nick S. Jones, Mason A. Porter, Charlotte M. Deane Supplementary text for the section
1 Supporting Information: What Evidence is There for the Homology of Protein-Protein Interactions? Anna C. F. Lewis, Nick S. Jones, Mason A. Porter, Charlotte M. Deane Supplementary text for the section
Prediction of protein function from sequence analysis
 Prediction of protein function from sequence analysis Rita Casadio BIOCOMPUTING GROUP University of Bologna, Italy The omic era Genome Sequencing Projects: Archaea: 74 species In Progress:52 Bacteria:
Prediction of protein function from sequence analysis Rita Casadio BIOCOMPUTING GROUP University of Bologna, Italy The omic era Genome Sequencing Projects: Archaea: 74 species In Progress:52 Bacteria:
PNmerger: a Cytoscape plugin to merge biological pathways and protein interaction networks
 PNmerger: a Cytoscape plugin to merge biological pathways and protein interaction networks http://www.hupo.org.cn/pnmerger Fuchu He E-mail: hefc@nic.bmi.ac.cn Tel: 86-10-68171208 FAX: 86-10-68214653 Yunping
PNmerger: a Cytoscape plugin to merge biological pathways and protein interaction networks http://www.hupo.org.cn/pnmerger Fuchu He E-mail: hefc@nic.bmi.ac.cn Tel: 86-10-68171208 FAX: 86-10-68214653 Yunping
Introduction to Evolutionary Concepts
 Introduction to Evolutionary Concepts and VMD/MultiSeq - Part I Zaida (Zan) Luthey-Schulten Dept. Chemistry, Beckman Institute, Biophysics, Institute of Genomics Biology, & Physics NIH Workshop 2009 VMD/MultiSeq
Introduction to Evolutionary Concepts and VMD/MultiSeq - Part I Zaida (Zan) Luthey-Schulten Dept. Chemistry, Beckman Institute, Biophysics, Institute of Genomics Biology, & Physics NIH Workshop 2009 VMD/MultiSeq
Shibiao Wan and Man-Wai Mak December 2013 Back to HybridGO-Loc Server
 Shibiao Wan and Man-Wai Mak December 2013 Back to HybridGO-Loc Server Contents 1 Functions of HybridGO-Loc Server 2 1.1 Webserver Interface....................................... 2 1.2 Inputing Protein
Shibiao Wan and Man-Wai Mak December 2013 Back to HybridGO-Loc Server Contents 1 Functions of HybridGO-Loc Server 2 1.1 Webserver Interface....................................... 2 1.2 Inputing Protein
Functional diversity within protein superfamilies
 Functional diversity within protein superfamilies James Casbon and Mansoor Saqi * Bioinformatics Group, The Genome Centre, Barts and The London, Queen Mary s School of Medicine and Dentistry, Charterhouse
Functional diversity within protein superfamilies James Casbon and Mansoor Saqi * Bioinformatics Group, The Genome Centre, Barts and The London, Queen Mary s School of Medicine and Dentistry, Charterhouse
Integration of functional genomics data
 Integration of functional genomics data Laboratoire Bordelais de Recherche en Informatique (UMR) Centre de Bioinformatique de Bordeaux (Plateforme) Rennes Oct. 2006 1 Observations and motivations Genomics
Integration of functional genomics data Laboratoire Bordelais de Recherche en Informatique (UMR) Centre de Bioinformatique de Bordeaux (Plateforme) Rennes Oct. 2006 1 Observations and motivations Genomics
BLAST Database Searching. BME 110: CompBio Tools Todd Lowe April 8, 2010
 BLAST Database Searching BME 110: CompBio Tools Todd Lowe April 8, 2010 Admin Reading: Read chapter 7, and the NCBI Blast Guide and tutorial http://www.ncbi.nlm.nih.gov/blast/why.shtml Read Chapter 8 for
BLAST Database Searching BME 110: CompBio Tools Todd Lowe April 8, 2010 Admin Reading: Read chapter 7, and the NCBI Blast Guide and tutorial http://www.ncbi.nlm.nih.gov/blast/why.shtml Read Chapter 8 for
Annotation Error in Public Databases ALEXANDRA SCHNOES UNIVERSITY OF CALIFORNIA, SAN FRANCISCO OCTOBER 25, 2010
 Annotation Error in Public Databases ALEXANDRA SCHNOES UNIVERSITY OF CALIFORNIA, SAN FRANCISCO OCTOBER 25, 2010 1 New genomes (and metagenomes) sequenced every day... 2 3 3 3 3 3 3 3 3 3 Computational
Annotation Error in Public Databases ALEXANDRA SCHNOES UNIVERSITY OF CALIFORNIA, SAN FRANCISCO OCTOBER 25, 2010 1 New genomes (and metagenomes) sequenced every day... 2 3 3 3 3 3 3 3 3 3 Computational
Chapter 7: Rapid alignment methods: FASTA and BLAST
 Chapter 7: Rapid alignment methods: FASTA and BLAST The biological problem Search strategies FASTA BLAST Introduction to bioinformatics, Autumn 2007 117 BLAST: Basic Local Alignment Search Tool BLAST (Altschul
Chapter 7: Rapid alignment methods: FASTA and BLAST The biological problem Search strategies FASTA BLAST Introduction to bioinformatics, Autumn 2007 117 BLAST: Basic Local Alignment Search Tool BLAST (Altschul
Homology Modeling. Roberto Lins EPFL - summer semester 2005
 Homology Modeling Roberto Lins EPFL - summer semester 2005 Disclaimer: course material is mainly taken from: P.E. Bourne & H Weissig, Structural Bioinformatics; C.A. Orengo, D.T. Jones & J.M. Thornton,
Homology Modeling Roberto Lins EPFL - summer semester 2005 Disclaimer: course material is mainly taken from: P.E. Bourne & H Weissig, Structural Bioinformatics; C.A. Orengo, D.T. Jones & J.M. Thornton,
Ensemble Non-negative Matrix Factorization Methods for Clustering Protein-Protein Interactions
 Belfield Campus Map Ensemble Non-negative Matrix Factorization Methods for Clustering Protein-Protein Interactions
Belfield Campus Map Ensemble Non-negative Matrix Factorization Methods for Clustering Protein-Protein Interactions
Sequence and Structure Alignment Z. Luthey-Schulten, UIUC Pittsburgh, 2006 VMD 1.8.5
 Sequence and Structure Alignment Z. Luthey-Schulten, UIUC Pittsburgh, 2006 VMD 1.8.5 Why Look at More Than One Sequence? 1. Multiple Sequence Alignment shows patterns of conservation 2. What and how many
Sequence and Structure Alignment Z. Luthey-Schulten, UIUC Pittsburgh, 2006 VMD 1.8.5 Why Look at More Than One Sequence? 1. Multiple Sequence Alignment shows patterns of conservation 2. What and how many
CMPS 6630: Introduction to Computational Biology and Bioinformatics. Structure Comparison
 CMPS 6630: Introduction to Computational Biology and Bioinformatics Structure Comparison Protein Structure Comparison Motivation Understand sequence and structure variability Understand Domain architecture
CMPS 6630: Introduction to Computational Biology and Bioinformatics Structure Comparison Protein Structure Comparison Motivation Understand sequence and structure variability Understand Domain architecture
Protein Structure and Evolutionary History Determine Sequence Space Topology. 44 Cummington Street, Boston MA, 02215
 Protein Structure and Evolutionary History Determine Sequence Space Topology Boris E. Shakhnovich 1, Eric Deeds 2, Charles Delisi 1 and Eugene Shakhnovich 3 1 Bioinformatics Program, Boston University
Protein Structure and Evolutionary History Determine Sequence Space Topology Boris E. Shakhnovich 1, Eric Deeds 2, Charles Delisi 1 and Eugene Shakhnovich 3 1 Bioinformatics Program, Boston University
A Novel Method for Similarity Analysis of Protein Sequences
 5th International Conference on Advanced Design and Manufacturing Engineering (ICADME 2015) A Novel Method for Similarity Analysis of Protein Sequences Longlong Liu 1, a, Tingting Zhao 1,b and Maojuan
5th International Conference on Advanced Design and Manufacturing Engineering (ICADME 2015) A Novel Method for Similarity Analysis of Protein Sequences Longlong Liu 1, a, Tingting Zhao 1,b and Maojuan
Comparing whole genomes
 BioNumerics Tutorial: Comparing whole genomes 1 Aim The Chromosome Comparison window in BioNumerics has been designed for large-scale comparison of sequences of unlimited length. In this tutorial you will
BioNumerics Tutorial: Comparing whole genomes 1 Aim The Chromosome Comparison window in BioNumerics has been designed for large-scale comparison of sequences of unlimited length. In this tutorial you will
Families of membranous proteins can be characterized by the amino acid composition of their transmembrane domains
 Families of membranous proteins can be characterized by the amino acid composition of their transmembrane domains Tali Sadka 1 and Michal Linial 1,2, * 1 School of Computer Science and Engineering and
Families of membranous proteins can be characterized by the amino acid composition of their transmembrane domains Tali Sadka 1 and Michal Linial 1,2, * 1 School of Computer Science and Engineering and
PA-GOSUB: A Searchable Database of Model Organism Protein Sequences With Their Predicted GO Molecular Function and Subcellular Localization
 PA-GOSUB: A Searchable Database of Model Organism Protein Sequences With Their Predicted GO Molecular Function and Subcellular Localization Paul Lu, Duane Szafron, Russell Greiner, David S. Wishart, Alona
PA-GOSUB: A Searchable Database of Model Organism Protein Sequences With Their Predicted GO Molecular Function and Subcellular Localization Paul Lu, Duane Szafron, Russell Greiner, David S. Wishart, Alona
BLAST. Varieties of BLAST
 BLAST Basic Local Alignment Search Tool (1990) Altschul, Gish, Miller, Myers, & Lipman Uses short-cuts or heuristics to improve search speed Like speed-reading, does not examine every nucleotide of database
BLAST Basic Local Alignment Search Tool (1990) Altschul, Gish, Miller, Myers, & Lipman Uses short-cuts or heuristics to improve search speed Like speed-reading, does not examine every nucleotide of database
Supplementary Information
 Supplementary Information For the article"comparable system-level organization of Archaea and ukaryotes" by J. Podani, Z. N. Oltvai, H. Jeong, B. Tombor, A.-L. Barabási, and. Szathmáry (reference numbers
Supplementary Information For the article"comparable system-level organization of Archaea and ukaryotes" by J. Podani, Z. N. Oltvai, H. Jeong, B. Tombor, A.-L. Barabási, and. Szathmáry (reference numbers
Computational approaches for functional genomics
 Computational approaches for functional genomics Kalin Vetsigian October 31, 2001 The rapidly increasing number of completely sequenced genomes have stimulated the development of new methods for finding
Computational approaches for functional genomics Kalin Vetsigian October 31, 2001 The rapidly increasing number of completely sequenced genomes have stimulated the development of new methods for finding
The MANTiS Manual. Contents. MANTiS Version 1.1
 The MANTiS Manual MANTiS Version 1.1 Contents Connection to the MANTiS database... 2 Memory settings... 2 Main functionalities... 2 Character Mapping View... 4 Genome content View... 5 Biological processes
The MANTiS Manual MANTiS Version 1.1 Contents Connection to the MANTiS database... 2 Memory settings... 2 Main functionalities... 2 Character Mapping View... 4 Genome content View... 5 Biological processes
An Introduction to Sequence Similarity ( Homology ) Searching
 Searching") An Introduction to Sequence Similarity ( Homology ) Searching Gary D. Stormo 1 UNIT 3.1 1 Washington University, School of Medicine, St. Louis, Missouri ABSTRACT Homologous sequences usually have the same,
An Introduction to Sequence Similarity ( Homology ) Searching Gary D. Stormo 1 UNIT 3.1 1 Washington University, School of Medicine, St. Louis, Missouri ABSTRACT Homologous sequences usually have the same,
Grundlagen der Bioinformatik, SS 08, D. Huson, May 2,
 Grundlagen der Bioinformatik, SS 08, D. Huson, May 2, 2008 39 5 Blast This lecture is based on the following, which are all recommended reading: R. Merkl, S. Waack: Bioinformatik Interaktiv. Chapter 11.4-11.7
Grundlagen der Bioinformatik, SS 08, D. Huson, May 2, 2008 39 5 Blast This lecture is based on the following, which are all recommended reading: R. Merkl, S. Waack: Bioinformatik Interaktiv. Chapter 11.4-11.7
Giri Narasimhan. CAP 5510: Introduction to Bioinformatics CGS 5166: Bioinformatics Tools. Evaluation. Course Homepage.
 CAP 5510: Introduction to Bioinformatics CGS 5166: Bioinformatics Tools Giri Narasimhan ECS 389; Phone: x3748 giri@cis.fiu.edu www.cis.fiu.edu/~giri/teach/bioinfs06.html 1/12/06 CAP5510/CGS5166 1 Evaluation
CAP 5510: Introduction to Bioinformatics CGS 5166: Bioinformatics Tools Giri Narasimhan ECS 389; Phone: x3748 giri@cis.fiu.edu www.cis.fiu.edu/~giri/teach/bioinfs06.html 1/12/06 CAP5510/CGS5166 1 Evaluation
Implications of Structural Genomics Target Selection Strategies: Pfam5000, Whole Genome, and Random Approaches
 PROTEINS: Structure, Function, and Bioinformatics 58:166 179 (2005) Implications of Structural Genomics Target Selection Strategies: Pfam5000, Whole Genome, and Random Approaches John-Marc Chandonia 1
PROTEINS: Structure, Function, and Bioinformatics 58:166 179 (2005) Implications of Structural Genomics Target Selection Strategies: Pfam5000, Whole Genome, and Random Approaches John-Marc Chandonia 1
Bioinformatics tools for phylogeny and visualization. Yanbin Yin
 Bioinformatics tools for phylogeny and visualization Yanbin Yin 1 Homework assignment 5 1. Take the MAFFT alignment http://cys.bios.niu.edu/yyin/teach/pbb/purdue.cellwall.list.lignin.f a.aln as input and
Bioinformatics tools for phylogeny and visualization Yanbin Yin 1 Homework assignment 5 1. Take the MAFFT alignment http://cys.bios.niu.edu/yyin/teach/pbb/purdue.cellwall.list.lignin.f a.aln as input and
1. Protein Data Bank (PDB) 1. Protein Data Bank (PDB)
 1. Protein Data Bank (PDB)") Protein structure databases; visualization; and classifications 1. Introduction to Protein Data Bank (PDB) 2. Free graphic software for 3D structure visualization 3. Hierarchical classification of protein
Protein structure databases; visualization; and classifications 1. Introduction to Protein Data Bank (PDB) 2. Free graphic software for 3D structure visualization 3. Hierarchical classification of protein
Carson Andorf 1,3, Adrian Silvescu 1,3, Drena Dobbs 2,3,4, Vasant Honavar 1,3,4. University, Ames, Iowa, 50010, USA. Ames, Iowa, 50010, USA
 Learning Classifiers for Assigning Protein Sequences to Gene Ontology Functional Families: Combining of Function Annotation Using Sequence Homology With that Based on Amino Acid k-gram Composition Yields
Learning Classifiers for Assigning Protein Sequences to Gene Ontology Functional Families: Combining of Function Annotation Using Sequence Homology With that Based on Amino Acid k-gram Composition Yields
ROUGH SET PROTEIN CLASSIFIER
 ROUGH SET PROTEIN CLASSIFIER RAMADEVI YELLASIRI 1, C.R.RAO 2 1 Dept. of CSE, Chaitanya Bharathi Institute of Technology, Hyderabad, INDIA. 2 DCIS, School of MCIS, University of Hyderabad, Hyderabad, INDIA.
ROUGH SET PROTEIN CLASSIFIER RAMADEVI YELLASIRI 1, C.R.RAO 2 1 Dept. of CSE, Chaitanya Bharathi Institute of Technology, Hyderabad, INDIA. 2 DCIS, School of MCIS, University of Hyderabad, Hyderabad, INDIA.
BMD645. Integration of Omics
 BMD645 Integration of Omics Shu-Jen Chen, Chang Gung University Dec. 11, 2009 1 Traditional Biology vs. Systems Biology Traditional biology : Single genes or proteins Systems biology: Simultaneously study
BMD645 Integration of Omics Shu-Jen Chen, Chang Gung University Dec. 11, 2009 1 Traditional Biology vs. Systems Biology Traditional biology : Single genes or proteins Systems biology: Simultaneously study
In-Depth Assessment of Local Sequence Alignment
 2012 International Conference on Environment Science and Engieering IPCBEE vol.3 2(2012) (2012)IACSIT Press, Singapoore In-Depth Assessment of Local Sequence Alignment Atoosa Ghahremani and Mahmood A.
2012 International Conference on Environment Science and Engieering IPCBEE vol.3 2(2012) (2012)IACSIT Press, Singapoore In-Depth Assessment of Local Sequence Alignment Atoosa Ghahremani and Mahmood A.
ATLAS of Biochemistry
 ATLAS of Biochemistry USER GUIDE http://lcsb-databases.epfl.ch/atlas/ CONTENT 1 2 3 GET STARTED Create your user account NAVIGATE Curated KEGG reactions ATLAS reactions Pathways Maps USE IT! Fill a gap
ATLAS of Biochemistry USER GUIDE http://lcsb-databases.epfl.ch/atlas/ CONTENT 1 2 3 GET STARTED Create your user account NAVIGATE Curated KEGG reactions ATLAS reactions Pathways Maps USE IT! Fill a gap
Tiffany Samaroo MB&B 452a December 8, Take Home Final. Topic 1
 Tiffany Samaroo MB&B 452a December 8, 2003 Take Home Final Topic 1 Prior to 1970, protein and DNA sequence alignment was limited to visual comparison. This was a very tedious process; even proteins with
Tiffany Samaroo MB&B 452a December 8, 2003 Take Home Final Topic 1 Prior to 1970, protein and DNA sequence alignment was limited to visual comparison. This was a very tedious process; even proteins with
Introduction to Bioinformatics Online Course: IBT
 Introduction to Bioinformatics Online Course: IBT Multiple Sequence Alignment Building Multiple Sequence Alignment Lec1 Building a Multiple Sequence Alignment Learning Outcomes 1- Understanding Why multiple
Introduction to Bioinformatics Online Course: IBT Multiple Sequence Alignment Building Multiple Sequence Alignment Lec1 Building a Multiple Sequence Alignment Learning Outcomes 1- Understanding Why multiple
Development of a Cell Signaling Networks Database. Division of Chem-Bio Informatics, National Institute of Health Sciences,
 Development of a Cell Signaling Networks Database TAKAKO IGARASHI and TSUGUCHIKA KAMINUMA Division of Chem-Bio Informatics, National Institute of Health Sciences, 1-18-1 Kami-Yoga, Setagaya-ku, Tokyo 158,
Development of a Cell Signaling Networks Database TAKAKO IGARASHI and TSUGUCHIKA KAMINUMA Division of Chem-Bio Informatics, National Institute of Health Sciences, 1-18-1 Kami-Yoga, Setagaya-ku, Tokyo 158,
Clustering. Genome 559: Introduction to Statistical and Computational Genomics Elhanan Borenstein. Some slides adapted from Jacques van Helden
 Clustering Genome 559: Introduction to Statistical and Computational Genomics Elhanan Borenstein Some slides adapted from Jacques van Helden Gene expression profiling A quick review Which molecular processes/functions
Clustering Genome 559: Introduction to Statistical and Computational Genomics Elhanan Borenstein Some slides adapted from Jacques van Helden Gene expression profiling A quick review Which molecular processes/functions
Application of Topology to Complex Object Identification. Eliseo CLEMENTINI University of L Aquila
 Application of Topology to Complex Object Identification Eliseo CLEMENTINI University of L Aquila Agenda Recognition of complex objects in ortophotos Some use cases Complex objects definition An ontology
Application of Topology to Complex Object Identification Eliseo CLEMENTINI University of L Aquila Agenda Recognition of complex objects in ortophotos Some use cases Complex objects definition An ontology
Integrating Ontological Prior Knowledge into Relational Learning
 Stefan Reckow reckow@mpipsykl.mpg.de Max Planck Institute of Psychiatry, Proteomics and Biomarkers, 80804 Munich, Germany Volker Tresp volker.tresp@siemens.com Siemens AG, Corporate Research & Technology,
Stefan Reckow reckow@mpipsykl.mpg.de Max Planck Institute of Psychiatry, Proteomics and Biomarkers, 80804 Munich, Germany Volker Tresp volker.tresp@siemens.com Siemens AG, Corporate Research & Technology,
Bayesian Hierarchical Classification. Seminar on Predicting Structured Data Jukka Kohonen
 Bayesian Hierarchical Classification Seminar on Predicting Structured Data Jukka Kohonen 17.4.2008 Overview Intro: The task of hierarchical gene annotation Approach I: SVM/Bayes hybrid Barutcuoglu et al:
Bayesian Hierarchical Classification Seminar on Predicting Structured Data Jukka Kohonen 17.4.2008 Overview Intro: The task of hierarchical gene annotation Approach I: SVM/Bayes hybrid Barutcuoglu et al: