A Method for Aligning RNA Secondary Structures
|
|
|
- Clyde Manning
- 5 years ago
- Views:
Transcription
1 Method for ligning RN Secondary Structures Jason T. L. Wang New Jersey Institute of Technology J Liu, JTL Wang, J Hu and B Tian, BM Bioinformatics,
2 Outline Introduction Structural alignment of RN (preliminaries, RSmatch algorithm, software) Experiments (RN motif detection) Multiple structural alignment (RMulti) ombining RSmatch with RNView onclusion and future work 2

3 Molecule building blocks Protein building blocks: 20 types of amino acid RN building blocks: Purine: denine, uanine Pyrimidine: ytosine, racil 3
4 RN structure elements RN sequence folds to form secondary/tertiary structure Majority of base connections involve two bases Watson-rick: or Non-canonical: or Basic structure elements of RN 4
5 Definition of structural components iven an RN sequence: : r 1 r 2 r 3 r n Two types of structural components [1] : Single bases (blue) Bonded base pairs (red) [1] Zuker, M. (1989) Science 5
6 Secondary structure constraint (1) Prohibited! No common base can be shared by any two pairs [2]. Bad: is shared by two pairs: - and - (a) OOD (b) BD [2] Hofacker, I.L. (2003) NR 6
7 Secondary structure constraint (2) hairpin Prohibited! hairpin element must have at least 3 bases on the loop part [3]. Bad: only two bases ( and ) present in the loop (a) OOD (b) BD [3] Zuker, M. (1991) NR 7
8 Secondary structure constraint (3) Pseudoknots are not included [4] (a) BD (b) OOD (nested structure) (c) OOD (branching) Prohibited! [4] Mathews, D.H. (1999) JMB 8
9 RN secondary structure representation schemes a. Bond annotation [5] b. rc representation [6] c. Tree representation [7] d. Nested parenthesis representation [8] [5] Shapiro, B. (1990) BIOS [6] Zhang, K. (1999) PM [7] Ma, B. (2002) TS [8] Hofacker, I.L. (2002) JMB 9
10 Outline Introduction Structural alignment of RN (preliminaries, RSmatch algorithm, software) Experiments (RN motif detection) Multiple structural alignment (RMulti) ombining RSmatch with RNView onclusion and future work 10
11 Extended circle model circle 5 circle 4 circle 3 circle 2 circle 1 circle 7 circle 0 circle 6 circle 8 ircle model [9] : circle 0:,,,,, circle 1:,,, circle 7:,,,, circle 8:,,,,,, Sequential order between components: > > -> > -> - [9] Liu, J. (2005) BM Bioinformatics 11
12 Hierarchical organization circles are organized in a tree-like hierarchy circle 5 circle 4 circle 3 circle 2 circle 1 circle 7 circle 0 circle 6 circle 8 circle 3 circle 4 circle 5 circle 0 circle 1 circle 2 circle 6 circle 7 circle 8 12
13 Hierarchical relationship between two structural components (1) the same circle: e.g. each pair from,,, -, -,, - (2) descendant/ancestor circles: e.g. pair (, -) (3) cousin circles: e.g. pairs (, ), (-, -) and (, -) (1) (2) (3) circle 13
14 Partial structure induced by a structural component parent structure child structure 14
15 Structural alignment rules (1) 1 precedes 2 iff B 1 precedes B 2 where 1, 2, B 1,B 2 are structural components. 15
16 Structural alignment rules (2) RN 1 RN 2 (a) (a) Same loop relationship preserved: 1 is in the same loop as 2 iff B 1 is in the same loop as B 2 (b) ncestor/descendant relationship preserved: 1 is ancestor of 2 iff B 1 is ancestor of B 2 (b) (c) ousin relationship preserved: 1 is cousin of 2 iff B 1 is cousin of B 2 (c) 16
17 Example alignment First RN..((...(((...)))((.(...))).)).. Second RN..((..((...))(((...))).)).. ll structural alignment rules must be satisfied for a valid alignment In addition, a single base can not be aligned with a base pair lignment Result..((...(((...)))((.(.....))).)) ((.. ((... ))(( (...))).)).. 17
18 Dynamic programming algorithm: overview First structure Second structure DP scoring table The best alignment between partial structures of and - 18
19 ase 1 19
20 ase 2 20
21 ase 3 21
22 ase
23 ase
24 Example of matching score function Score function of matching two equal-length structural components: i.e. 1, if both a and b are single bases and a = g( a, b ) = 2, if both a and b are base pairs and a = b 0, otherwise ap penalty equals 0 Extending g to the whole set of matched component pairs, our goal is to maximize f(r 1, R 2 ) f ( R, R2 ) = g(, 1 a i bi i ) b 24
25 ell type 1 : single base vs. single base?..(...)....(...). ()..(...) (...). (B)..(...) (...). ()..(...) (...). 25
26 ell type 2: base pair vs. single base? first score second score?? 26
27 ell type 2: base pair vs. single base (first score) (...)?...(...). (...) (...). (... ) (...). 27
28 ell type 2: base pair vs. single base (second score)..(...)?...(...). ()..(...) (...). (B).. (...) (...). ().. (...) (...). 28
29 ell type 3: base pair vs. base pair..(...)?...(...) () (B) ()?? (b1)?? (b2) 29
30 ell type 3: base pair vs. base pair (first score) (...)? (...) () (B) () (...) (...) (... ) (...) (...) -- (... ) 30
31 ell type 3: base pair vs. base pair (2 nd & 3 rd score)..(...)? (...) (...)?...(...) (... ) (...) (...) (...) (...) (...) 31
32 ell type 3: base pair vs. base pair (final score)? ()..(...)..(...) --...(...)...(...) (B) ().. (...) (...)..(...) (...) (D).. (...) (...)..(...) (...) 32
33 nalysis of algorithm Time and space complexity Each score is calculated only once. Time is bounded by the number of score calculations needed to fill up the table. Each base pair will contribute to two or four score calculations. Single bases: N s ; base pairs: N p Total number of score calculations: N s2 +4N s N p +4N 2 p =O(N 2 ) N 2 s score calculations are contributed by two single bases 4N s N p score calculations are contributed by one single base and one base pair 4N p2 score calculations are contributed by two base pairs 33

34 Software RSmatch 34
35 Outline Introduction Structural alignment of RN (preliminaries, RSmatch algorithm, software) Experiments (RN motif detection) Multiple structural alignment (RMulti) ombining RSmatch with RNView onclusion and future work 35
36 Motif example: detection/instantiation Motif structure is known IB ambiguity symbols: N: W: H: not 36
37 ap Penalty Example motif structure subject structure 37
38 Position independent scoring matrices Two scoring matrices ap penalty: -3 for each single base, -6 for each base pair, involved in the gap 38
39 Motifs used in the experiments (a) HSL3 (b) IRE HSL3 has a typical stem loop structure with two flanking tails IRE has specific stem-loop structure for gene regulation related to cell iron metabolism Wildcard n is allowed to match with 0 or 1 nucleotide IB code: M:, T/; Y:, T/; H: not ; R:, ; W:, T; 39
40 Experiments Performance measurements: sensitivity (recall) and specificity (precision) 19,986 human RefSeq mrn sequences were obtained from NBI; 39,972 TR regions were extracted Each TR sequence was chopped and folded into secondary structures using Vienna RN package, yielding ~575,000 structures ompare RSmatch with PatSearch [10] [10] Pesole. (2000) Bioinformatics 40
41 hop and fold TR sequences TR ORF TR ORF ORF: Open Reading Frame 41
42 Detecting HSL3 motif PatSearch: specificity (98.2%), sensitivity (87.1%). Several histone genes (i.e. NM_003542, NM_003548) were found by RSmatch, but not by PatSearch. 42
43 Detecting IRE motif se PatSearch to search 39,972 TR sequences for IRE motif and get 27 hit structures belonging to 18 TR sequences The 18 TR sequences were chopped and folded into 1,196 structures ompare RSmatch, Rsearch [11] and stemloc [12]. well-known IRE-containing structure (NM_000032) was used as the query (it does not have wildcard or ambiguity symbols since Rsearch and stemloc cannot handle them) [11] Klein, R.J. (2003) BM Bioinformatics [12] Holms, I. (2002) PSB 43

44 Experimental results for IRE motif 44
45 Dealing with complex structures 45
46 Outline Introduction Structural alignment of RN (preliminaries, RSmatch algorithm, software) Experiments (RN motif detection) Multiple structural alignment (RMulti) ombining RSmatch with RNView onclusion and future work 46
47 Extension to multiple structural alignment search small database YES expand best alignment score (best alignment) < δ OR non-expandable NO pairwise match profile expand seed alignment seed alignment 47
48 Example expand expand 48

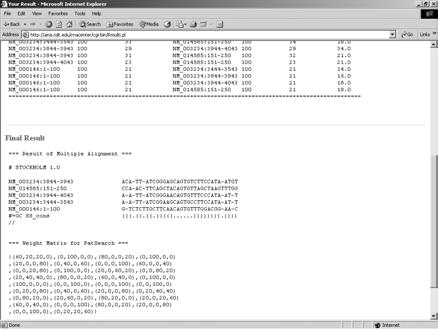
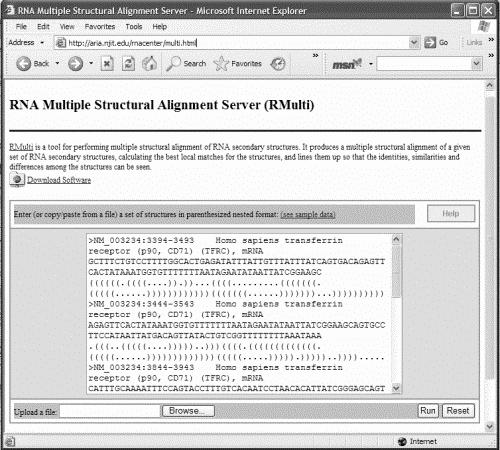
49 RMulti Webserver 49
50 Outline Introduction Structural alignment of RN (preliminaries, RSmatch algorithm, software) Experiments (RN motif detection) Multiple structural alignment (Rmulti) ombining RSmatch with RNView onclusion and future work 50
51 51
52 52
53 Outline Introduction Structural alignment of RN (preliminaries, RSmatch algorithm, software) Experiments (RN motif detection) Multiple structural alignment (RMulti) ombining RSmatch with RNView onclusion and future work 53
54 onclusion n efficient algorithm RSmatch to align and analyze RN secondary structures multiple structural alignment tool RMulti visualization tool combining RSmatch with RNView 54
55 Future Work Extending RSmatch to handle pseudoknots Large-scale genome-wide motif mining Indexing very large RN structure databases Improved multiple structural alignment of RN sequences RN classification and clustering RN-RN interactions and protein-rn interactions 55
56 56
proteins are the basic building blocks and active players in the cell, and
 12 RN Secondary Structure Sources for this lecture: R. Durbin, S. Eddy,. Krogh und. Mitchison, Biological sequence analysis, ambridge, 1998 J. Setubal & J. Meidanis, Introduction to computational molecular
12 RN Secondary Structure Sources for this lecture: R. Durbin, S. Eddy,. Krogh und. Mitchison, Biological sequence analysis, ambridge, 1998 J. Setubal & J. Meidanis, Introduction to computational molecular
Conserved RNA Structures. Ivo L. Hofacker. Institut for Theoretical Chemistry, University Vienna.
 onserved RN Structures Ivo L. Hofacker Institut for Theoretical hemistry, University Vienna http://www.tbi.univie.ac.at/~ivo/ Bled, January 2002 Energy Directed Folding Predict structures from sequence
onserved RN Structures Ivo L. Hofacker Institut for Theoretical hemistry, University Vienna http://www.tbi.univie.ac.at/~ivo/ Bled, January 2002 Energy Directed Folding Predict structures from sequence
Structure-Based Comparison of Biomolecules
 Structure-Based Comparison of Biomolecules Benedikt Christoph Wolters Seminar Bioinformatics Algorithms RWTH AACHEN 07/17/2015 Outline 1 Introduction and Motivation Protein Structure Hierarchy Protein
Structure-Based Comparison of Biomolecules Benedikt Christoph Wolters Seminar Bioinformatics Algorithms RWTH AACHEN 07/17/2015 Outline 1 Introduction and Motivation Protein Structure Hierarchy Protein
RNA Abstract Shape Analysis
 ourse: iegerich RN bstract nalysis omplete shape iegerich enter of Biotechnology Bielefeld niversity robert@techfak.ni-bielefeld.de ourse on omputational RN Biology, Tübingen, March 2006 iegerich ourse:
ourse: iegerich RN bstract nalysis omplete shape iegerich enter of Biotechnology Bielefeld niversity robert@techfak.ni-bielefeld.de ourse on omputational RN Biology, Tübingen, March 2006 iegerich ourse:
In Genomes, Two Types of Genes
 In Genomes, Two Types of Genes Protein-coding: [Start codon] [codon 1] [codon 2] [ ] [Stop codon] + DNA codons translated to amino acids to form a protein Non-coding RNAs (NcRNAs) No consistent patterns
In Genomes, Two Types of Genes Protein-coding: [Start codon] [codon 1] [codon 2] [ ] [Stop codon] + DNA codons translated to amino acids to form a protein Non-coding RNAs (NcRNAs) No consistent patterns
EVALUATION OF RNA SECONDARY STRUCTURE MOTIFS USING REGRESSION ANALYSIS
 EVLTION OF RN SEONDRY STRTRE MOTIFS SIN RERESSION NLYSIS Mohammad nwar School of Information Technology and Engineering, niversity of Ottawa e-mail: manwar@site.uottawa.ca bstract Recent experimental evidences
EVLTION OF RN SEONDRY STRTRE MOTIFS SIN RERESSION NLYSIS Mohammad nwar School of Information Technology and Engineering, niversity of Ottawa e-mail: manwar@site.uottawa.ca bstract Recent experimental evidences
Homology Modeling. Roberto Lins EPFL - summer semester 2005
 Homology Modeling Roberto Lins EPFL - summer semester 2005 Disclaimer: course material is mainly taken from: P.E. Bourne & H Weissig, Structural Bioinformatics; C.A. Orengo, D.T. Jones & J.M. Thornton,
Homology Modeling Roberto Lins EPFL - summer semester 2005 Disclaimer: course material is mainly taken from: P.E. Bourne & H Weissig, Structural Bioinformatics; C.A. Orengo, D.T. Jones & J.M. Thornton,
THEORY. Based on sequence Length According to the length of sequence being compared it is of following two types
 Exp 11- THEORY Sequence Alignment is a process of aligning two sequences to achieve maximum levels of identity between them. This help to derive functional, structural and evolutionary relationships between
Exp 11- THEORY Sequence Alignment is a process of aligning two sequences to achieve maximum levels of identity between them. This help to derive functional, structural and evolutionary relationships between
Multiple Sequence Alignment
 Multiple equence lignment Four ami Khuri Dept of omputer cience an José tate University Multiple equence lignment v Progressive lignment v Guide Tree v lustalw v Toffee v Muscle v MFFT * 20 * 0 * 60 *
Multiple equence lignment Four ami Khuri Dept of omputer cience an José tate University Multiple equence lignment v Progressive lignment v Guide Tree v lustalw v Toffee v Muscle v MFFT * 20 * 0 * 60 *
98 Algorithms in Bioinformatics I, WS 06, ZBIT, D. Huson, December 6, 2006
 98 Algorithms in Bioinformatics I, WS 06, ZBIT, D. Huson, December 6, 2006 8.3.1 Simple energy minimization Maximizing the number of base pairs as described above does not lead to good structure predictions.
98 Algorithms in Bioinformatics I, WS 06, ZBIT, D. Huson, December 6, 2006 8.3.1 Simple energy minimization Maximizing the number of base pairs as described above does not lead to good structure predictions.
Algorithms in Bioinformatics FOUR Pairwise Sequence Alignment. Pairwise Sequence Alignment. Convention: DNA Sequences 5. Sequence Alignment
 Algorithms in Bioinformatics FOUR Sami Khuri Department of Computer Science San José State University Pairwise Sequence Alignment Homology Similarity Global string alignment Local string alignment Dot
Algorithms in Bioinformatics FOUR Sami Khuri Department of Computer Science San José State University Pairwise Sequence Alignment Homology Similarity Global string alignment Local string alignment Dot
Copyright (c) 2007 IEEE. Personal use of this material is permitted. However, permission to use this material for any other purposes must be obtained
 2007 IEEE. Personal use of this material is permitted. However, permission to use this material for any other purposes must be obtained") Copyright (c) 2007 IEEE. Personal use of this material is permitted. However, permission to use this material for any other purposes must be obtained from the IEEE by sending an email to pubs-permissions@ieee.org.
Copyright (c) 2007 IEEE. Personal use of this material is permitted. However, permission to use this material for any other purposes must be obtained from the IEEE by sending an email to pubs-permissions@ieee.org.
Bio nformatics. Lecture 23. Saad Mneimneh
 Bio nformatics Lecture 23 Protein folding The goal is to determine the three-dimensional structure of a protein based on its amino acid sequence Assumption: amino acid sequence completely and uniquely
Bio nformatics Lecture 23 Protein folding The goal is to determine the three-dimensional structure of a protein based on its amino acid sequence Assumption: amino acid sequence completely and uniquely
Chapter 5. Proteomics and the analysis of protein sequence Ⅱ
 Proteomics Chapter 5. Proteomics and the analysis of protein sequence Ⅱ 1 Pairwise similarity searching (1) Figure 5.5: manual alignment One of the amino acids in the top sequence has no equivalent and
Proteomics Chapter 5. Proteomics and the analysis of protein sequence Ⅱ 1 Pairwise similarity searching (1) Figure 5.5: manual alignment One of the amino acids in the top sequence has no equivalent and
Sequence alignment methods. Pairwise alignment. The universe of biological sequence analysis
 he universe of biological sequence analysis Word/pattern recognition- Identification of restriction enzyme cleavage sites Sequence alignment methods PstI he universe of biological sequence analysis - prediction
he universe of biological sequence analysis Word/pattern recognition- Identification of restriction enzyme cleavage sites Sequence alignment methods PstI he universe of biological sequence analysis - prediction
Statistical Machine Learning Methods for Biomedical Informatics II. Hidden Markov Model for Biological Sequences
 Statistical Machine Learning Methods for Biomedical Informatics II. Hidden Markov Model for Biological Sequences Jianlin Cheng, PhD William and Nancy Thompson Missouri Distinguished Professor Department
Statistical Machine Learning Methods for Biomedical Informatics II. Hidden Markov Model for Biological Sequences Jianlin Cheng, PhD William and Nancy Thompson Missouri Distinguished Professor Department
Sequence analysis and comparison
 The aim with sequence identification: Sequence analysis and comparison Marjolein Thunnissen Lund September 2012 Is there any known protein sequence that is homologous to mine? Are there any other species
The aim with sequence identification: Sequence analysis and comparison Marjolein Thunnissen Lund September 2012 Is there any known protein sequence that is homologous to mine? Are there any other species
A Structure-Based Flexible Search Method for Motifs in RNA
 JOURNAL OF COMPUTATIONAL BIOLOGY Volume 14, Number 7, 2007 Mary Ann Liebert, Inc. Pp. 908 926 DOI: 10.1089/cmb.2007.0061 A Structure-Based Flexible Search Method for Motifs in RNA ISANA VEKSLER-LUBLINSKY,
JOURNAL OF COMPUTATIONAL BIOLOGY Volume 14, Number 7, 2007 Mary Ann Liebert, Inc. Pp. 908 926 DOI: 10.1089/cmb.2007.0061 A Structure-Based Flexible Search Method for Motifs in RNA ISANA VEKSLER-LUBLINSKY,
Genomics and bioinformatics summary. Finding genes -- computer searches
 Genomics and bioinformatics summary 1. Gene finding: computer searches, cdnas, ESTs, 2. Microarrays 3. Use BLAST to find homologous sequences 4. Multiple sequence alignments (MSAs) 5. Trees quantify sequence
Genomics and bioinformatics summary 1. Gene finding: computer searches, cdnas, ESTs, 2. Microarrays 3. Use BLAST to find homologous sequences 4. Multiple sequence alignments (MSAs) 5. Trees quantify sequence
Combinatorial approaches to RNA folding Part I: Basics
 Combinatorial approaches to RNA folding Part I: Basics Matthew Macauley Department of Mathematical Sciences Clemson University http://www.math.clemson.edu/~macaule/ Math 4500, Spring 2015 M. Macauley (Clemson)
Combinatorial approaches to RNA folding Part I: Basics Matthew Macauley Department of Mathematical Sciences Clemson University http://www.math.clemson.edu/~macaule/ Math 4500, Spring 2015 M. Macauley (Clemson)
Searching for Noncoding RNA
 Searching for Noncoding RN Larry Ruzzo omputer Science & Engineering enome Sciences niversity of Washington http://www.cs.washington.edu/homes/ruzzo Bio 2006, Seattle, 8/4/2006 1 Outline Noncoding RN Why
Searching for Noncoding RN Larry Ruzzo omputer Science & Engineering enome Sciences niversity of Washington http://www.cs.washington.edu/homes/ruzzo Bio 2006, Seattle, 8/4/2006 1 Outline Noncoding RN Why
Comparative Bioinformatics Midterm II Fall 2004
 Comparative Bioinformatics Midterm II Fall 2004 Objective Answer, part I: For each of the following, select the single best answer or completion of the phrase. (3 points each) 1. Deinococcus radiodurans
Comparative Bioinformatics Midterm II Fall 2004 Objective Answer, part I: For each of the following, select the single best answer or completion of the phrase. (3 points each) 1. Deinococcus radiodurans
HMM applications. Applications of HMMs. Gene finding with HMMs. Using the gene finder
 HMM applications Applications of HMMs Gene finding Pairwise alignment (pair HMMs) Characterizing protein families (profile HMMs) Predicting membrane proteins, and membrane protein topology Gene finding
HMM applications Applications of HMMs Gene finding Pairwise alignment (pair HMMs) Characterizing protein families (profile HMMs) Predicting membrane proteins, and membrane protein topology Gene finding
Mitochondrial Genome Annotation
 Protein Genes 1,2 1 Institute of Bioinformatics University of Leipzig 2 Department of Bioinformatics Lebanese University TBI Bled 2015 Outline Introduction Mitochondrial DNA Problem Tools Training Annotation
Protein Genes 1,2 1 Institute of Bioinformatics University of Leipzig 2 Department of Bioinformatics Lebanese University TBI Bled 2015 Outline Introduction Mitochondrial DNA Problem Tools Training Annotation
5. MULTIPLE SEQUENCE ALIGNMENT BIOINFORMATICS COURSE MTAT
 5. MULTIPLE SEQUENCE ALIGNMENT BIOINFORMATICS COURSE MTAT.03.239 03.10.2012 ALIGNMENT Alignment is the task of locating equivalent regions of two or more sequences to maximize their similarity. Homology:
5. MULTIPLE SEQUENCE ALIGNMENT BIOINFORMATICS COURSE MTAT.03.239 03.10.2012 ALIGNMENT Alignment is the task of locating equivalent regions of two or more sequences to maximize their similarity. Homology:
Sequence Alignments. Dynamic programming approaches, scoring, and significance. Lucy Skrabanek ICB, WMC January 31, 2013
 Sequence Alignments Dynamic programming approaches, scoring, and significance Lucy Skrabanek ICB, WMC January 31, 213 Sequence alignment Compare two (or more) sequences to: Find regions of conservation
Sequence Alignments Dynamic programming approaches, scoring, and significance Lucy Skrabanek ICB, WMC January 31, 213 Sequence alignment Compare two (or more) sequences to: Find regions of conservation
Sara C. Madeira. Universidade da Beira Interior. (Thanks to Ana Teresa Freitas, IST for useful resources on this subject)
") Bioinformática Sequence Alignment Pairwise Sequence Alignment Universidade da Beira Interior (Thanks to Ana Teresa Freitas, IST for useful resources on this subject) 1 16/3/29 & 23/3/29 27/4/29 Outline
Bioinformática Sequence Alignment Pairwise Sequence Alignment Universidade da Beira Interior (Thanks to Ana Teresa Freitas, IST for useful resources on this subject) 1 16/3/29 & 23/3/29 27/4/29 Outline
COMP 598 Advanced Computational Biology Methods & Research. Introduction. Jérôme Waldispühl School of Computer Science McGill University
 COMP 598 Advanced Computational Biology Methods & Research Introduction Jérôme Waldispühl School of Computer Science McGill University General informations (1) Office hours: by appointment Office: TR3018
COMP 598 Advanced Computational Biology Methods & Research Introduction Jérôme Waldispühl School of Computer Science McGill University General informations (1) Office hours: by appointment Office: TR3018
Statistical Machine Learning Methods for Bioinformatics II. Hidden Markov Model for Biological Sequences
 Statistical Machine Learning Methods for Bioinformatics II. Hidden Markov Model for Biological Sequences Jianlin Cheng, PhD Department of Computer Science University of Missouri 2008 Free for Academic
Statistical Machine Learning Methods for Bioinformatics II. Hidden Markov Model for Biological Sequences Jianlin Cheng, PhD Department of Computer Science University of Missouri 2008 Free for Academic
BLAST. Varieties of BLAST
 BLAST Basic Local Alignment Search Tool (1990) Altschul, Gish, Miller, Myers, & Lipman Uses short-cuts or heuristics to improve search speed Like speed-reading, does not examine every nucleotide of database
BLAST Basic Local Alignment Search Tool (1990) Altschul, Gish, Miller, Myers, & Lipman Uses short-cuts or heuristics to improve search speed Like speed-reading, does not examine every nucleotide of database
Protein folding. α-helix. Lecture 21. An α-helix is a simple helix having on average 10 residues (3 turns of the helix)
") Computat onal Biology Lecture 21 Protein folding The goal is to determine the three-dimensional structure of a protein based on its amino acid sequence Assumption: amino acid sequence completely and uniquely
Computat onal Biology Lecture 21 Protein folding The goal is to determine the three-dimensional structure of a protein based on its amino acid sequence Assumption: amino acid sequence completely and uniquely
CMPS 6630: Introduction to Computational Biology and Bioinformatics. Structure Comparison
 CMPS 6630: Introduction to Computational Biology and Bioinformatics Structure Comparison Protein Structure Comparison Motivation Understand sequence and structure variability Understand Domain architecture
CMPS 6630: Introduction to Computational Biology and Bioinformatics Structure Comparison Protein Structure Comparison Motivation Understand sequence and structure variability Understand Domain architecture
Computational Approaches for determination of Most Probable RNA Secondary Structure Using Different Thermodynamics Parameters
 Computational Approaches for determination of Most Probable RNA Secondary Structure Using Different Thermodynamics Parameters 1 Binod Kumar, Assistant Professor, Computer Sc. Dept, ISTAR, Vallabh Vidyanagar,
Computational Approaches for determination of Most Probable RNA Secondary Structure Using Different Thermodynamics Parameters 1 Binod Kumar, Assistant Professor, Computer Sc. Dept, ISTAR, Vallabh Vidyanagar,
Week 10: Homology Modelling (II) - HHpred
 - HHpred") Week 10: Homology Modelling (II) - HHpred Course: Tools for Structural Biology Fabian Glaser BKU - Technion 1 2 Identify and align related structures by sequence methods is not an easy task All comparative
Week 10: Homology Modelling (II) - HHpred Course: Tools for Structural Biology Fabian Glaser BKU - Technion 1 2 Identify and align related structures by sequence methods is not an easy task All comparative
RNA secondary structure prediction. Farhat Habib
 RNA secondary structure prediction Farhat Habib RNA RNA is similar to DNA chemically. It is usually only a single strand. T(hyamine) is replaced by U(racil) Some forms of RNA can form secondary structures
RNA secondary structure prediction Farhat Habib RNA RNA is similar to DNA chemically. It is usually only a single strand. T(hyamine) is replaced by U(racil) Some forms of RNA can form secondary structures
Sequence Alignment Techniques and Their Uses
 Sequence Alignment Techniques and Their Uses Sarah Fiorentino Since rapid sequencing technology and whole genomes sequencing, the amount of sequence information has grown exponentially. With all of this
Sequence Alignment Techniques and Their Uses Sarah Fiorentino Since rapid sequencing technology and whole genomes sequencing, the amount of sequence information has grown exponentially. With all of this
Algorithms in Bioinformatics
 Algorithms in Bioinformatics Sami Khuri Department of omputer Science San José State University San José, alifornia, USA khuri@cs.sjsu.edu www.cs.sjsu.edu/faculty/khuri Pairwise Sequence Alignment Homology
Algorithms in Bioinformatics Sami Khuri Department of omputer Science San José State University San José, alifornia, USA khuri@cs.sjsu.edu www.cs.sjsu.edu/faculty/khuri Pairwise Sequence Alignment Homology
Page 1. Evolutionary Trees. Why build evolutionary tree? Outline
 Page Evolutionary Trees Russ. ltman MI S 7 Outline. Why build evolutionary trees?. istance-based vs. character-based methods. istance-based: Ultrametric Trees dditive Trees. haracter-based: Perfect phylogeny
Page Evolutionary Trees Russ. ltman MI S 7 Outline. Why build evolutionary trees?. istance-based vs. character-based methods. istance-based: Ultrametric Trees dditive Trees. haracter-based: Perfect phylogeny
SUPPLEMENTARY INFORMATION
 Supplementary information S1 (box). Supplementary Methods description. Prokaryotic Genome Database Archaeal and bacterial genome sequences were downloaded from the NCBI FTP site (ftp://ftp.ncbi.nlm.nih.gov/genomes/all/)
Supplementary information S1 (box). Supplementary Methods description. Prokaryotic Genome Database Archaeal and bacterial genome sequences were downloaded from the NCBI FTP site (ftp://ftp.ncbi.nlm.nih.gov/genomes/all/)
Multiple Alignment. Slides revised and adapted to Bioinformática IST Ana Teresa Freitas
 n Introduction to Bioinformatics lgorithms Multiple lignment Slides revised and adapted to Bioinformática IS 2005 na eresa Freitas n Introduction to Bioinformatics lgorithms Outline Dynamic Programming
n Introduction to Bioinformatics lgorithms Multiple lignment Slides revised and adapted to Bioinformática IS 2005 na eresa Freitas n Introduction to Bioinformatics lgorithms Outline Dynamic Programming
A Novel Statistical Model for the Secondary Structure of RNA
 ISBN 978-1-8466-93-3 Proceedings of the 5th International ongress on Mathematical Biology (IMB11) Vol. 3 Nanjing, P. R. hina, June 3-5, 11 Novel Statistical Model for the Secondary Structure of RN Liu
ISBN 978-1-8466-93-3 Proceedings of the 5th International ongress on Mathematical Biology (IMB11) Vol. 3 Nanjing, P. R. hina, June 3-5, 11 Novel Statistical Model for the Secondary Structure of RN Liu
RNA-Strukturvorhersage Strukturelle Bioinformatik WS16/17
 RNA-Strukturvorhersage Strukturelle Bioinformatik WS16/17 Dr. Stefan Simm, 01.11.2016 simm@bio.uni-frankfurt.de RNA secondary structures a. hairpin loop b. stem c. bulge loop d. interior loop e. multi
RNA-Strukturvorhersage Strukturelle Bioinformatik WS16/17 Dr. Stefan Simm, 01.11.2016 simm@bio.uni-frankfurt.de RNA secondary structures a. hairpin loop b. stem c. bulge loop d. interior loop e. multi
Basic Local Alignment Search Tool
 Basic Local Alignment Search Tool Alignments used to uncover homologies between sequences combined with phylogenetic studies o can determine orthologous and paralogous relationships Local Alignment uses
Basic Local Alignment Search Tool Alignments used to uncover homologies between sequences combined with phylogenetic studies o can determine orthologous and paralogous relationships Local Alignment uses
Protein Structures. Sequences of amino acid residues 20 different amino acids. Quaternary. Primary. Tertiary. Secondary. 10/8/2002 Lecture 12 1
 Protein Structures Sequences of amino acid residues 20 different amino acids Primary Secondary Tertiary Quaternary 10/8/2002 Lecture 12 1 Angles φ and ψ in the polypeptide chain 10/8/2002 Lecture 12 2
Protein Structures Sequences of amino acid residues 20 different amino acids Primary Secondary Tertiary Quaternary 10/8/2002 Lecture 12 1 Angles φ and ψ in the polypeptide chain 10/8/2002 Lecture 12 2
Protein Threading. BMI/CS 776 Colin Dewey Spring 2015
 Protein Threading BMI/CS 776 www.biostat.wisc.edu/bmi776/ Colin Dewey cdewey@biostat.wisc.edu Spring 2015 Goals for Lecture the key concepts to understand are the following the threading prediction task
Protein Threading BMI/CS 776 www.biostat.wisc.edu/bmi776/ Colin Dewey cdewey@biostat.wisc.edu Spring 2015 Goals for Lecture the key concepts to understand are the following the threading prediction task
Sequence Alignment: A General Overview. COMP Fall 2010 Luay Nakhleh, Rice University
 Sequence Alignment: A General Overview COMP 571 - Fall 2010 Luay Nakhleh, Rice University Life through Evolution All living organisms are related to each other through evolution This means: any pair of
Sequence Alignment: A General Overview COMP 571 - Fall 2010 Luay Nakhleh, Rice University Life through Evolution All living organisms are related to each other through evolution This means: any pair of
Overview Multiple Sequence Alignment
 Overview Multiple Sequence Alignment Inge Jonassen Bioinformatics group Dept. of Informatics, UoB Inge.Jonassen@ii.uib.no Definition/examples Use of alignments The alignment problem scoring alignments
Overview Multiple Sequence Alignment Inge Jonassen Bioinformatics group Dept. of Informatics, UoB Inge.Jonassen@ii.uib.no Definition/examples Use of alignments The alignment problem scoring alignments
Domain-based computational approaches to understand the molecular basis of diseases
 Domain-based computational approaches to understand the molecular basis of diseases Dr. Maricel G. Kann Assistant Professor Dept of Biological Sciences UMBC http://bioinf.umbc.edu Research at Kann s Lab.
Domain-based computational approaches to understand the molecular basis of diseases Dr. Maricel G. Kann Assistant Professor Dept of Biological Sciences UMBC http://bioinf.umbc.edu Research at Kann s Lab.
Pairwise & Multiple sequence alignments
 Pairwise & Multiple sequence alignments Urmila Kulkarni-Kale Bioinformatics Centre 411 007 urmila@bioinfo.ernet.in Basis for Sequence comparison Theory of evolution: gene sequences have evolved/derived
Pairwise & Multiple sequence alignments Urmila Kulkarni-Kale Bioinformatics Centre 411 007 urmila@bioinfo.ernet.in Basis for Sequence comparison Theory of evolution: gene sequences have evolved/derived
A phylogenetic view on RNA structure evolution
 3 2 9 4 7 3 24 23 22 8 phylogenetic view on RN structure evolution 9 26 6 52 7 5 6 37 57 45 5 84 63 86 77 65 3 74 7 79 8 33 9 97 96 89 47 87 62 32 34 42 73 43 44 4 76 58 75 78 93 39 54 82 99 28 95 52 46
3 2 9 4 7 3 24 23 22 8 phylogenetic view on RN structure evolution 9 26 6 52 7 5 6 37 57 45 5 84 63 86 77 65 3 74 7 79 8 33 9 97 96 89 47 87 62 32 34 42 73 43 44 4 76 58 75 78 93 39 54 82 99 28 95 52 46
An Introduction to Bioinformatics Algorithms Hidden Markov Models
 Hidden Markov Models Outline 1. CG-Islands 2. The Fair Bet Casino 3. Hidden Markov Model 4. Decoding Algorithm 5. Forward-Backward Algorithm 6. Profile HMMs 7. HMM Parameter Estimation 8. Viterbi Training
Hidden Markov Models Outline 1. CG-Islands 2. The Fair Bet Casino 3. Hidden Markov Model 4. Decoding Algorithm 5. Forward-Backward Algorithm 6. Profile HMMs 7. HMM Parameter Estimation 8. Viterbi Training
Local Alignment of RNA Sequences with Arbitrary Scoring Schemes
 Local Alignment of RNA Sequences with Arbitrary Scoring Schemes Rolf Backofen 1, Danny Hermelin 2, ad M. Landau 2,3, and Oren Weimann 4 1 Institute of omputer Science, Albert-Ludwigs niversität Freiburg,
Local Alignment of RNA Sequences with Arbitrary Scoring Schemes Rolf Backofen 1, Danny Hermelin 2, ad M. Landau 2,3, and Oren Weimann 4 1 Institute of omputer Science, Albert-Ludwigs niversität Freiburg,
13 Comparative RNA analysis
 13 Comparative RNA analysis Sources for this lecture: R. Durbin, S. Eddy, A. Krogh und G. Mitchison, Biological sequence analysis, Cambridge, 1998 D.W. Mount. Bioinformatics: Sequences and Genome analysis,
13 Comparative RNA analysis Sources for this lecture: R. Durbin, S. Eddy, A. Krogh und G. Mitchison, Biological sequence analysis, Cambridge, 1998 D.W. Mount. Bioinformatics: Sequences and Genome analysis,
Background: comparative genomics. Sequence similarity. Homologs. Similarity vs homology (2) Similarity vs homology. Sequence Alignment (chapter 6)
 Similarity vs homology. Sequence Alignment (chapter 6)") Sequence lignment (chapter ) he biological problem lobal alignment Local alignment Multiple alignment Background: comparative genomics Basic question in biology: what properties are shared among organisms?
Sequence lignment (chapter ) he biological problem lobal alignment Local alignment Multiple alignment Background: comparative genomics Basic question in biology: what properties are shared among organisms?
Genome Annotation. Bioinformatics and Computational Biology. Genome sequencing Assembly. Gene prediction. Protein targeting.
 Genome Annotation Bioinformatics and Computational Biology Genome Annotation Frank Oliver Glöckner 1 Genome Analysis Roadmap Genome sequencing Assembly Gene prediction Protein targeting trna prediction
Genome Annotation Bioinformatics and Computational Biology Genome Annotation Frank Oliver Glöckner 1 Genome Analysis Roadmap Genome sequencing Assembly Gene prediction Protein targeting trna prediction
Bioinformatics. Dept. of Computational Biology & Bioinformatics
 Bioinformatics Dept. of Computational Biology & Bioinformatics 3 Bioinformatics - play with sequences & structures Dept. of Computational Biology & Bioinformatics 4 ORGANIZATION OF LIFE ROLE OF BIOINFORMATICS
Bioinformatics Dept. of Computational Biology & Bioinformatics 3 Bioinformatics - play with sequences & structures Dept. of Computational Biology & Bioinformatics 4 ORGANIZATION OF LIFE ROLE OF BIOINFORMATICS
Position-specific scoring matrices (PSSM)
") Regulatory Sequence nalysis Position-specific scoring matrices (PSSM) Jacques van Helden Jacques.van-Helden@univ-amu.fr Université d ix-marseille, France Technological dvances for Genomics and Clinics
Regulatory Sequence nalysis Position-specific scoring matrices (PSSM) Jacques van Helden Jacques.van-Helden@univ-amu.fr Université d ix-marseille, France Technological dvances for Genomics and Clinics
Motivating the need for optimal sequence alignments...
 1 Motivating the need for optimal sequence alignments... 2 3 Note that this actually combines two objectives of optimal sequence alignments: (i) use the score of the alignment o infer homology; (ii) use
1 Motivating the need for optimal sequence alignments... 2 3 Note that this actually combines two objectives of optimal sequence alignments: (i) use the score of the alignment o infer homology; (ii) use
Comparative Network Analysis
 Comparative Network Analysis BMI/CS 776 www.biostat.wisc.edu/bmi776/ Spring 2016 Anthony Gitter gitter@biostat.wisc.edu These slides, excluding third-party material, are licensed under CC BY-NC 4.0 by
Comparative Network Analysis BMI/CS 776 www.biostat.wisc.edu/bmi776/ Spring 2016 Anthony Gitter gitter@biostat.wisc.edu These slides, excluding third-party material, are licensed under CC BY-NC 4.0 by
Molecular Modeling. Prediction of Protein 3D Structure from Sequence. Vimalkumar Velayudhan. May 21, 2007
 Molecular Modeling Prediction of Protein 3D Structure from Sequence Vimalkumar Velayudhan Jain Institute of Vocational and Advanced Studies May 21, 2007 Vimalkumar Velayudhan Molecular Modeling 1/23 Outline
Molecular Modeling Prediction of Protein 3D Structure from Sequence Vimalkumar Velayudhan Jain Institute of Vocational and Advanced Studies May 21, 2007 Vimalkumar Velayudhan Molecular Modeling 1/23 Outline
Bioinformatics. Proteins II. - Pattern, Profile, & Structure Database Searching. Robert Latek, Ph.D. Bioinformatics, Biocomputing
 Bioinformatics Proteins II. - Pattern, Profile, & Structure Database Searching Robert Latek, Ph.D. Bioinformatics, Biocomputing WIBR Bioinformatics Course, Whitehead Institute, 2002 1 Proteins I.-III.
Bioinformatics Proteins II. - Pattern, Profile, & Structure Database Searching Robert Latek, Ph.D. Bioinformatics, Biocomputing WIBR Bioinformatics Course, Whitehead Institute, 2002 1 Proteins I.-III.
Genome 559 Wi RNA Function, Search, Discovery
 Genome 559 Wi 2009 RN Function, Search, Discovery The Message Cells make lots of RN noncoding RN Functionally important, functionally diverse Structurally complex New tools required alignment, discovery,
Genome 559 Wi 2009 RN Function, Search, Discovery The Message Cells make lots of RN noncoding RN Functionally important, functionally diverse Structurally complex New tools required alignment, discovery,
Sequence Alignment (chapter 6)
") Sequence lignment (chapter 6) he biological problem lobal alignment Local alignment Multiple alignment Introduction to bioinformatics, utumn 6 Background: comparative genomics Basic question in biology:
Sequence lignment (chapter 6) he biological problem lobal alignment Local alignment Multiple alignment Introduction to bioinformatics, utumn 6 Background: comparative genomics Basic question in biology:
Computational approaches for RNA energy parameter estimation
 omputational approaches for RNA energy parameter estimation by Mirela Ştefania Andronescu M.Sc., The University of British olumbia, 2003 B.Sc., Bucharest Academy of Economic Studies, 1999 A THESIS SUBMITTED
omputational approaches for RNA energy parameter estimation by Mirela Ştefania Andronescu M.Sc., The University of British olumbia, 2003 B.Sc., Bucharest Academy of Economic Studies, 1999 A THESIS SUBMITTED
STRUCTURAL BIOINFORMATICS I. Fall 2015
 STRUCTURAL BIOINFORMATICS I Fall 2015 Info Course Number - Classification: Biology 5411 Class Schedule: Monday 5:30-7:50 PM, SERC Room 456 (4 th floor) Instructors: Vincenzo Carnevale - SERC, Room 704C;
STRUCTURAL BIOINFORMATICS I Fall 2015 Info Course Number - Classification: Biology 5411 Class Schedule: Monday 5:30-7:50 PM, SERC Room 456 (4 th floor) Instructors: Vincenzo Carnevale - SERC, Room 704C;
Protein Bioinformatics. Rickard Sandberg Dept. of Cell and Molecular Biology Karolinska Institutet sandberg.cmb.ki.
 Protein Bioinformatics Rickard Sandberg Dept. of Cell and Molecular Biology Karolinska Institutet rickard.sandberg@ki.se sandberg.cmb.ki.se Outline Protein features motifs patterns profiles signals 2 Protein
Protein Bioinformatics Rickard Sandberg Dept. of Cell and Molecular Biology Karolinska Institutet rickard.sandberg@ki.se sandberg.cmb.ki.se Outline Protein features motifs patterns profiles signals 2 Protein
Tools and Algorithms in Bioinformatics
 Tools and Algorithms in Bioinformatics GCBA815, Fall 2013 Week3: Blast Algorithm, theory and practice Babu Guda, Ph.D. Department of Genetics, Cell Biology & Anatomy Bioinformatics and Systems Biology
Tools and Algorithms in Bioinformatics GCBA815, Fall 2013 Week3: Blast Algorithm, theory and practice Babu Guda, Ph.D. Department of Genetics, Cell Biology & Anatomy Bioinformatics and Systems Biology
Lecture 14: Multiple Sequence Alignment (Gene Finding, Conserved Elements) Scribe: John Ekins
 Scribe: John Ekins") Lecture 14: Multiple Sequence Alignment (Gene Finding, Conserved Elements) 2 19 2015 Scribe: John Ekins Multiple Sequence Alignment Given N sequences x 1, x 2,, x N : Insert gaps in each of the sequences
Lecture 14: Multiple Sequence Alignment (Gene Finding, Conserved Elements) 2 19 2015 Scribe: John Ekins Multiple Sequence Alignment Given N sequences x 1, x 2,, x N : Insert gaps in each of the sequences
2MHR. Protein structure classification is important because it organizes the protein structure universe that is independent of sequence similarity.
 Protein structure classification is important because it organizes the protein structure universe that is independent of sequence similarity. A global picture of the protein universe will help us to understand
Protein structure classification is important because it organizes the protein structure universe that is independent of sequence similarity. A global picture of the protein universe will help us to understand
Bioinformatics and BLAST
 Bioinformatics and BLAST Overview Recap of last time Similarity discussion Algorithms: Needleman-Wunsch Smith-Waterman BLAST Implementation issues and current research Recap from Last Time Genome consists
Bioinformatics and BLAST Overview Recap of last time Similarity discussion Algorithms: Needleman-Wunsch Smith-Waterman BLAST Implementation issues and current research Recap from Last Time Genome consists
Detecting local deviations. Optimisation and applications to RNA-gene searching.
 Detecting Local Deviations Detecting local deviations. Optimisation and applications to R-gene searching. iels Richard ansen niversity of openhagen Department of pplied Mathematics and Statistics p. 1/20
Detecting Local Deviations Detecting local deviations. Optimisation and applications to R-gene searching. iels Richard ansen niversity of openhagen Department of pplied Mathematics and Statistics p. 1/20
The Double Helix. CSE 417: Algorithms and Computational Complexity! The Central Dogma of Molecular Biology! DNA! RNA! Protein! Protein!
 The Double Helix SE 417: lgorithms and omputational omplexity! Winter 29! W. L. Ruzzo! Dynamic Programming, II" RN Folding! http://www.rcsb.org/pdb/explore.do?structureid=1t! Los lamos Science The entral
The Double Helix SE 417: lgorithms and omputational omplexity! Winter 29! W. L. Ruzzo! Dynamic Programming, II" RN Folding! http://www.rcsb.org/pdb/explore.do?structureid=1t! Los lamos Science The entral
Bioinformatics. Scoring Matrices. David Gilbert Bioinformatics Research Centre
 Bioinformatics Scoring Matrices David Gilbert Bioinformatics Research Centre www.brc.dcs.gla.ac.uk Department of Computing Science, University of Glasgow Learning Objectives To explain the requirement
Bioinformatics Scoring Matrices David Gilbert Bioinformatics Research Centre www.brc.dcs.gla.ac.uk Department of Computing Science, University of Glasgow Learning Objectives To explain the requirement
Homology Modeling (Comparative Structure Modeling) GBCB 5874: Problem Solving in GBCB
 GBCB 5874: Problem Solving in GBCB") Homology Modeling (Comparative Structure Modeling) Aims of Structural Genomics High-throughput 3D structure determination and analysis To determine or predict the 3D structures of all the proteins encoded
Homology Modeling (Comparative Structure Modeling) Aims of Structural Genomics High-throughput 3D structure determination and analysis To determine or predict the 3D structures of all the proteins encoded
Cluster Analysis of Gene Expression Microarray Data. BIOL 495S/ CS 490B/ MATH 490B/ STAT 490B Introduction to Bioinformatics April 8, 2002
 Cluster Analysis of Gene Expression Microarray Data BIOL 495S/ CS 490B/ MATH 490B/ STAT 490B Introduction to Bioinformatics April 8, 2002 1 Data representations Data are relative measurements log 2 ( red
Cluster Analysis of Gene Expression Microarray Data BIOL 495S/ CS 490B/ MATH 490B/ STAT 490B Introduction to Bioinformatics April 8, 2002 1 Data representations Data are relative measurements log 2 ( red
Searching genomes for non-coding RNA using FastR
 Searching genomes for non-coding RNA using FastR Shaojie Zhang Brian Haas Eleazar Eskin Vineet Bafna Keywords: non-coding RNA, database search, filtration, riboswitch, bacterial genome. Address for correspondence:
Searching genomes for non-coding RNA using FastR Shaojie Zhang Brian Haas Eleazar Eskin Vineet Bafna Keywords: non-coding RNA, database search, filtration, riboswitch, bacterial genome. Address for correspondence:
SA-REPC - Sequence Alignment with a Regular Expression Path Constraint
 SA-REPC - Sequence Alignment with a Regular Expression Path Constraint Nimrod Milo Tamar Pinhas Michal Ziv-Ukelson Ben-Gurion University of the Negev, Be er Sheva, Israel Graduate Seminar, BGU 2010 Milo,
SA-REPC - Sequence Alignment with a Regular Expression Path Constraint Nimrod Milo Tamar Pinhas Michal Ziv-Ukelson Ben-Gurion University of the Negev, Be er Sheva, Israel Graduate Seminar, BGU 2010 Milo,
Predicting RNA Secondary Structure
 7.91 / 7.36 / BE.490 Lecture #6 Mar. 11, 2004 Predicting RNA Secondary Structure Chris Burge Review of Markov Models & DNA Evolution CpG Island HMM The Viterbi Algorithm Real World HMMs Markov Models for
7.91 / 7.36 / BE.490 Lecture #6 Mar. 11, 2004 Predicting RNA Secondary Structure Chris Burge Review of Markov Models & DNA Evolution CpG Island HMM The Viterbi Algorithm Real World HMMs Markov Models for
Algorithms in Bioinformatics
 Algorithms in Bioinformatics Sami Khuri Department of Computer Science San José State University San José, California, USA khuri@cs.sjsu.edu www.cs.sjsu.edu/faculty/khuri RNA Structure Prediction Secondary
Algorithms in Bioinformatics Sami Khuri Department of Computer Science San José State University San José, California, USA khuri@cs.sjsu.edu www.cs.sjsu.edu/faculty/khuri RNA Structure Prediction Secondary
USING BLAST TO IDENTIFY PROTEINS THAT ARE EVOLUTIONARILY RELATED ACROSS SPECIES
 USING BLAST TO IDENTIFY PROTEINS THAT ARE EVOLUTIONARILY RELATED ACROSS SPECIES HOW CAN BIOINFORMATICS BE USED AS A TOOL TO DETERMINE EVOLUTIONARY RELATIONSHPS AND TO BETTER UNDERSTAND PROTEIN HERITAGE?
USING BLAST TO IDENTIFY PROTEINS THAT ARE EVOLUTIONARILY RELATED ACROSS SPECIES HOW CAN BIOINFORMATICS BE USED AS A TOOL TO DETERMINE EVOLUTIONARY RELATIONSHPS AND TO BETTER UNDERSTAND PROTEIN HERITAGE?
Grundlagen der Bioinformatik Summer semester Lecturer: Prof. Daniel Huson
 Grundlagen der Bioinformatik, SS 10, D. Huson, April 12, 2010 1 1 Introduction Grundlagen der Bioinformatik Summer semester 2010 Lecturer: Prof. Daniel Huson Office hours: Thursdays 17-18h (Sand 14, C310a)
Grundlagen der Bioinformatik, SS 10, D. Huson, April 12, 2010 1 1 Introduction Grundlagen der Bioinformatik Summer semester 2010 Lecturer: Prof. Daniel Huson Office hours: Thursdays 17-18h (Sand 14, C310a)
An Introduction to Sequence Similarity ( Homology ) Searching
 Searching") An Introduction to Sequence Similarity ( Homology ) Searching Gary D. Stormo 1 UNIT 3.1 1 Washington University, School of Medicine, St. Louis, Missouri ABSTRACT Homologous sequences usually have the same,
An Introduction to Sequence Similarity ( Homology ) Searching Gary D. Stormo 1 UNIT 3.1 1 Washington University, School of Medicine, St. Louis, Missouri ABSTRACT Homologous sequences usually have the same,
Protein Structure Prediction II Lecturer: Serafim Batzoglou Scribe: Samy Hamdouche
 Protein Structure Prediction II Lecturer: Serafim Batzoglou Scribe: Samy Hamdouche The molecular structure of a protein can be broken down hierarchically. The primary structure of a protein is simply its
Protein Structure Prediction II Lecturer: Serafim Batzoglou Scribe: Samy Hamdouche The molecular structure of a protein can be broken down hierarchically. The primary structure of a protein is simply its
Study and Implementation of Various Techniques Involved in DNA and Protein Sequence Analysis
 Study and Implementation of Various Techniques Involved in DNA and Protein Sequence Analysis Kumud Joseph Kujur, Sumit Pal Singh, O.P. Vyas, Ruchir Bhatia, Varun Singh* Indian Institute of Information
Study and Implementation of Various Techniques Involved in DNA and Protein Sequence Analysis Kumud Joseph Kujur, Sumit Pal Singh, O.P. Vyas, Ruchir Bhatia, Varun Singh* Indian Institute of Information
Statistical Machine Learning Methods for Bioinformatics IV. Neural Network & Deep Learning Applications in Bioinformatics
 Statistical Machine Learning Methods for Bioinformatics IV. Neural Network & Deep Learning Applications in Bioinformatics Jianlin Cheng, PhD Department of Computer Science University of Missouri, Columbia
Statistical Machine Learning Methods for Bioinformatics IV. Neural Network & Deep Learning Applications in Bioinformatics Jianlin Cheng, PhD Department of Computer Science University of Missouri, Columbia
Large-Scale Genomic Surveys
 Bioinformatics Subtopics Fold Recognition Secondary Structure Prediction Docking & Drug Design Protein Geometry Protein Flexibility Homology Modeling Sequence Alignment Structure Classification Gene Prediction
Bioinformatics Subtopics Fold Recognition Secondary Structure Prediction Docking & Drug Design Protein Geometry Protein Flexibility Homology Modeling Sequence Alignment Structure Classification Gene Prediction
Hidden Markov Models
 Hidden Markov Models Outline 1. CG-Islands 2. The Fair Bet Casino 3. Hidden Markov Model 4. Decoding Algorithm 5. Forward-Backward Algorithm 6. Profile HMMs 7. HMM Parameter Estimation 8. Viterbi Training
Hidden Markov Models Outline 1. CG-Islands 2. The Fair Bet Casino 3. Hidden Markov Model 4. Decoding Algorithm 5. Forward-Backward Algorithm 6. Profile HMMs 7. HMM Parameter Estimation 8. Viterbi Training
Sequence analysis and Genomics
 Sequence analysis and Genomics October 12 th November 23 rd 2 PM 5 PM Prof. Peter Stadler Dr. Katja Nowick Katja: group leader TFome and Transcriptome Evolution Bioinformatics group Paul-Flechsig-Institute
Sequence analysis and Genomics October 12 th November 23 rd 2 PM 5 PM Prof. Peter Stadler Dr. Katja Nowick Katja: group leader TFome and Transcriptome Evolution Bioinformatics group Paul-Flechsig-Institute
RNA and Protein Structure Prediction
 RNA and Protein Structure Prediction Bioinformatics: Issues and Algorithms CSE 308-408 Spring 2007 Lecture 18-1- Outline Multi-Dimensional Nature of Life RNA Secondary Structure Prediction Protein Structure
RNA and Protein Structure Prediction Bioinformatics: Issues and Algorithms CSE 308-408 Spring 2007 Lecture 18-1- Outline Multi-Dimensional Nature of Life RNA Secondary Structure Prediction Protein Structure
EECS730: Introduction to Bioinformatics
 EECS730: Introduction to Bioinformatics Lecture 07: profile Hidden Markov Model http://bibiserv.techfak.uni-bielefeld.de/sadr2/databasesearch/hmmer/profilehmm.gif Slides adapted from Dr. Shaojie Zhang
EECS730: Introduction to Bioinformatics Lecture 07: profile Hidden Markov Model http://bibiserv.techfak.uni-bielefeld.de/sadr2/databasesearch/hmmer/profilehmm.gif Slides adapted from Dr. Shaojie Zhang
10-810: Advanced Algorithms and Models for Computational Biology. microrna and Whole Genome Comparison
 10-810: Advanced Algorithms and Models for Computational Biology microrna and Whole Genome Comparison Central Dogma: 90s Transcription factors DNA transcription mrna translation Proteins Central Dogma:
10-810: Advanced Algorithms and Models for Computational Biology microrna and Whole Genome Comparison Central Dogma: 90s Transcription factors DNA transcription mrna translation Proteins Central Dogma:
Massachusetts Institute of Technology Computational Evolutionary Biology, Fall, 2005 Notes for November 7: Molecular evolution
 Massachusetts Institute of Technology 6.877 Computational Evolutionary Biology, Fall, 2005 Notes for November 7: Molecular evolution 1. Rates of amino acid replacement The initial motivation for the neutral
Massachusetts Institute of Technology 6.877 Computational Evolutionary Biology, Fall, 2005 Notes for November 7: Molecular evolution 1. Rates of amino acid replacement The initial motivation for the neutral
Computational Design of New and Recombinant Selenoproteins
 Computational Design of ew and Recombinant Selenoproteins Rolf Backofen and Friedrich-Schiller-University Jena Institute of Computer Science Chair for Bioinformatics 1 Computational Design of ew and Recombinant
Computational Design of ew and Recombinant Selenoproteins Rolf Backofen and Friedrich-Schiller-University Jena Institute of Computer Science Chair for Bioinformatics 1 Computational Design of ew and Recombinant
DNA/RNA Structure Prediction
 C E N T R E F O R I N T E G R A T I V E B I O I N F O R M A T I C S V U Master Course DNA/Protein Structurefunction Analysis and Prediction Lecture 12 DNA/RNA Structure Prediction Epigenectics Epigenomics:
C E N T R E F O R I N T E G R A T I V E B I O I N F O R M A T I C S V U Master Course DNA/Protein Structurefunction Analysis and Prediction Lecture 12 DNA/RNA Structure Prediction Epigenectics Epigenomics:
EBI web resources II: Ensembl and InterPro
 EBI web resources II: Ensembl and InterPro Yanbin Yin http://www.ebi.ac.uk/training/online/course/ 1 Homework 3 Go to http://www.ebi.ac.uk/interpro/training.htmland finish the second online training course
EBI web resources II: Ensembl and InterPro Yanbin Yin http://www.ebi.ac.uk/training/online/course/ 1 Homework 3 Go to http://www.ebi.ac.uk/interpro/training.htmland finish the second online training course
HMMs and biological sequence analysis
 HMMs and biological sequence analysis Hidden Markov Model A Markov chain is a sequence of random variables X 1, X 2, X 3,... That has the property that the value of the current state depends only on the
HMMs and biological sequence analysis Hidden Markov Model A Markov chain is a sequence of random variables X 1, X 2, X 3,... That has the property that the value of the current state depends only on the
Introduction to Comparative Protein Modeling. Chapter 4 Part I
 Introduction to Comparative Protein Modeling Chapter 4 Part I 1 Information on Proteins Each modeling study depends on the quality of the known experimental data. Basis of the model Search in the literature
Introduction to Comparative Protein Modeling Chapter 4 Part I 1 Information on Proteins Each modeling study depends on the quality of the known experimental data. Basis of the model Search in the literature
Computational Molecular Biology (
 Computational Molecular Biology (http://cmgm cmgm.stanford.edu/biochem218/) Biochemistry 218/Medical Information Sciences 231 Douglas L. Brutlag, Lee Kozar Jimmy Huang, Josh Silverman Lecture Syllabus
Computational Molecular Biology (http://cmgm cmgm.stanford.edu/biochem218/) Biochemistry 218/Medical Information Sciences 231 Douglas L. Brutlag, Lee Kozar Jimmy Huang, Josh Silverman Lecture Syllabus
Hairpin Database: Why and How?
 Hairpin Database: Why and How? Clark Jeffries Research Professor Renaissance Computing Institute and School of Pharmacy University of North Carolina at Chapel Hill, United States Why should a database
Hairpin Database: Why and How? Clark Jeffries Research Professor Renaissance Computing Institute and School of Pharmacy University of North Carolina at Chapel Hill, United States Why should a database
Lecture 4: Evolutionary Models and Substitution Matrices (PAM and BLOSUM)
") Bioinformatics II Probability and Statistics Universität Zürich and ETH Zürich Spring Semester 2009 Lecture 4: Evolutionary Models and Substitution Matrices (PAM and BLOSUM) Dr Fraser Daly adapted from
Bioinformatics II Probability and Statistics Universität Zürich and ETH Zürich Spring Semester 2009 Lecture 4: Evolutionary Models and Substitution Matrices (PAM and BLOSUM) Dr Fraser Daly adapted from