Igor A. Abrikosov Theoretical Physics, Department of Physics, Chemistry and Biology, Linköping University, Sweden
|
|
|
- Earl York
- 6 years ago
- Views:
Transcription
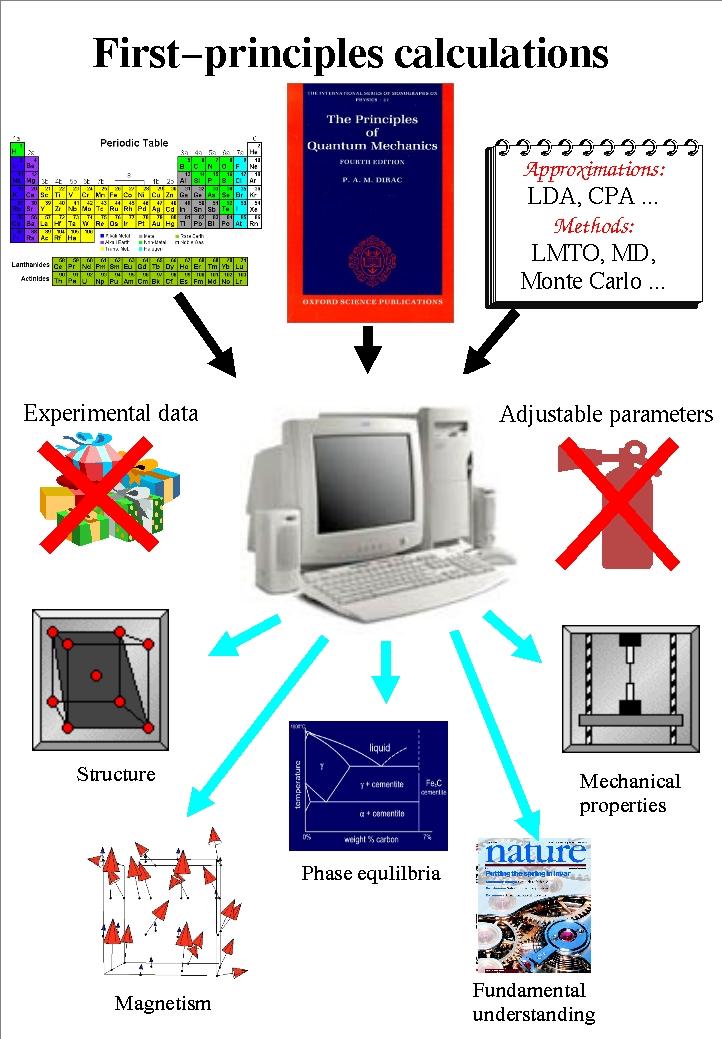
1 Ab initio simulations of materials properties: from fundamental theory towards materials design Igor A. Abrikosov Theoretical Physics Department of Physics Chemistry and Biology Linköping University Sweden


2 Theory and Modeling Theoretical Physics Computational Physics Bioinformatics Computational Biology Theoretical Biology Igor Abrikosov Sven Stafström Bengt Persson Jesper Tegner Bo Ebenman








3 Igor Abrikosov Bo Sernelius Theoretical Physics Igor Abrikosov Professor Ab initio electronic structure theory Materials simulations Mesoscopic physics semiconductor structures in the quantum regime transport and chaos Understanding of fundamental molecular interactions Dynamical simulations of metallic heterostructures Energy localization in descrete systems Non-linear dynamics of anharmonic lattices Karl-Fredrik Berggren Magnus Johansson Sergei Simak Leonid Pourovskii Irina Yakimenko Ferenc Tasnadi Peter Münger











4 Students and Postdoctoral Fellows (totally 46 PhDs from Theoretical Physics): Hans Lind Marcus Ekholm Olga Vekilova Tobias Marten Postdoc Olle Hellman Peter Steneteg Prof. Eyvaz Isaev ALVA lector Björn Alling Assistant Professor Dr. Arkady Mikhaylushkin ALVA lectro








5 Prof. Igor A. Abrikosov Head of Theory and Modelling Division IFM Linköping University Sweden Electronic Theory of Materials Properties Basic research Applied Studies Methodological developments Properties vs external parameters Theory of magnetism (INVA effect) Materials for nuclear Hard coatings waste disposal Environmentally friendly materials Magnetism for nanotechnology


6 Linköping KTH
7 DECISION Your application has been granted in the manner described below HPCN: Akka 30 x 000 core hours/month NSC: Neolith 30 x 000 core hours/month Kappa 0 x 000 core hours/month UPPMAX: Kalkyl 0 x 000 core hours/month LUNAC: Platon 0 x 000 core hours/month C3SE: Beda 0 x 000 core hours/month PDC: Ferlin 60 x 000 core hours/month Lindgren 500 x 000 core hours/month
 method that works for metals Conventional band structure methods (VASP Quantum Espresso) DMFT implementation in FLAPW Multiscale")
8 INALLOY toolkit Coherent potential approximation: KK-ASA basis set Exact Muffin-Tin Orbitals (EMTO) basis set Screened Impurity Model for charge fluctuations Model treatment of the local lattice relaxations Locally self-consistent Green s function method an O(N) method that works for metals Conventional band structure methods (VASP Quantum Espresso) DMFT implementation in FLAPW Multiscale modeling based on Hamiltonians with parameters determined ab initio
9 Density Functional Theory (DFT) ) ( ) ( ) ( M M M N N N N N N r r r V N i i m H r r r el many E r r r H σ σ σ σ σ σ σ σ σ + = = Ψ = Ψ h = + + = + = = occ r v v v r eff v r eff v m eff H r el one r eff H XC EXT H ψ ψ σ ρ ρ ρ ρ σ ρ σ ρ σ ψ ε σ ψ * )... ( ] [ ] [ ] [ )]... ( [ )]... ( [ )... ( )... ( M M M M M h
 W.")
10 Density Functional Theory (DFT) W. Kohn
11 A. V. uban and I. A. Abrikosov ep. Prog. Phys (008). 990s: LDAGGA LDA+U 000+: DMFT hybrid etc VASP WienkCASTEP ABINIT KK EMTO etc. + V KS (x... N m I ) φ i (x... N e I ) = ε i φ i (x... N I ) h Supercell CPA O(N) AIMD etc. It is NOT a trivial task to run ab initio software!



12 CoSn S. I. Simak U. Häußermann I. A. Abrikosov et al. Phys. ev. Lett (997)


13 From T. S. Duffy Nature (004) ecent seismological model of the Earth
 Structural complexity: pure")
14 From Vocadlo et al. Phys. Earth Planet. Sci. 7 3 (000) Structural complexity: pure Fe
; L. Vocadlo et al.")
 ΔG fcc-hcp ~ -(3-5) mev. fcc? A. Mikhailushkin et al. Phys. ev.")
15 Structural complexity: pure Fe A. B. Belonoshko. Ahuja and B. Johansson Nature (003); L. Vocadlo et al. Nature (003): ΔG bcc-hcp ~ mev A. Mikhailushkin et al. Phys. ev. Lett (007) ΔG fcc-hcp ~ -(3-5) mev. fcc? A. Mikhailushkin et al. Phys. ev. B (009) From Vocadlo et al. Phys. Earth Planet. Sci. 7 3 (000)
 φ i (x... N e I ) = ε i φ i (x.")
16 Ab initio molecular dynamics simulations of Fe-rich FeNi alloy with the bcc crystal structure simulated with an SQS. P=300 GPa T=6000 K + V KS (x... N m I ) φ i (x... N e I ) = ε i φ i (x... N I ) h
17 4000 Melt Shen et al. 004 Lin et al. 00 Boehler 993 HP-bcc 3000 Temperature K 000 fcc 000 bcc hcp L. Dubrovinsky... I. A. Abrikosov Science (007) Pressure GPa
18 A. V. uban and I. A. Abrikosov ep. Prog. Phys (008). 990s: LDAGGA LDA+U 000+: DMFT hybrid etc VASP WienkCASTEP ABINIT KK EMTO etc. + V KS (x... N m I ) φ i (x... N e I ) = ε i φ i (x... N I ) h Supercell CPA O(N) AIMD etc. It is NOT a trivial task to run ab initio software!
19 [ ev. Mod. Phys (996)]
20 DMFT calculations: FLAPW implementation [M. Aichhorn et al. PB (009)] continuous-time quantum Monte Carlo impurity solver Wannier-like functions for the Fe-3d shell U varied in the range from.0 to 3. ev J=0.9 ev

21 V = 67.5 a.u. 3 /at DMFT LDA
22 V = 70 a.u. 3 /at

23 V = 67.5 a.u. 3 /at
24 V = 65 a.u. 3 /at

25 V = 6.5 a.u. 3 /at

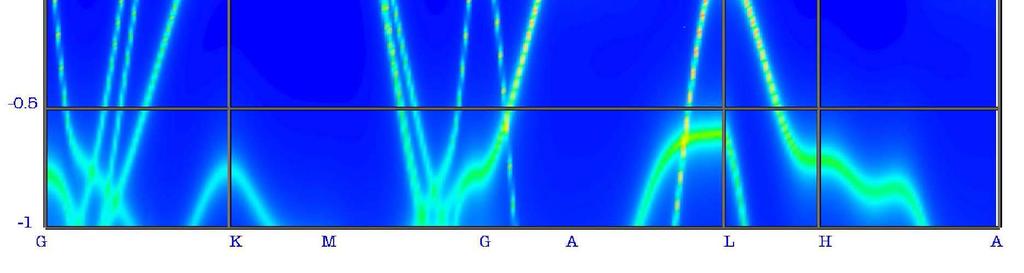
26 V = 60 a.u. 3 /at
27 Optimization of ionic conductivity in doped ceria CeO is known to be good solid elictrolyte when it is doped with cations of lower valence than the host cation. Attractive electrolyte for solid-oxide fuel cells It is important to optimize the ionic conductivity in order to decrease the operation temperature
28 Activation energy for diffusion E a determines the ionic conductivity (σ): E = a σ σ 0 T exp kbt E = E + a ass E m D. A. Andersson S. I. Simak N. V. Skorodumova I. A. Abrikosov and B. Johansson PNAS (006)
.")
29 Confirmed experimentally: S. Omar E. D. Wachsman Activation energy for diffusion and J. C. Nino Appl. Phys. Lett (007). E a determines the ionic conductivity (σ): E = a σ σ 0 T exp kbt E = E + a ass E m
30
31 Dramatic advances in theory and algorithms In Computational Materials Science the amplification from intellect (new theory algorithms and insights) is at least equal to the speed -up from hardware E0 E9 E8 E7 relative performance Moore s law computer speed David Landau: UGA elative performance increase of Ising model simulations ( ) compared the normalized speed of the computers ( ) the simulations were executed on. The dashed line is a schematic of the increase in peak performance of the fastest supercomputers since 97.
Igor A. Abrikosov Department of Physics, Chemistry, and Biology (IFM), Linköping University, Sweden
, Linköping University, Sweden") Correlation between electronic structure, magnetism and physical properties of Fe-Cr alloys: ab initio modeling Igor A. Abrikosov Department of Physics, Chemistry, and Biology (IFM), Linköping University,
Correlation between electronic structure, magnetism and physical properties of Fe-Cr alloys: ab initio modeling Igor A. Abrikosov Department of Physics, Chemistry, and Biology (IFM), Linköping University,
MAGNETIC EFFECTS IN SIMULATIONS OF STRUCTURAL TRANSFORMATIONS BELOW AND ABOVE THE MAGNETIC TRANSITION TEMPERATURE:
 MAGNETIC EFFECTS IN SIMULATIONS OF STRUCTURAL TRANSFORMATIONS BELOW AND ABOVE THE MAGNETIC TRANSITION TEMPERATURE: Navigatig i space o atoms ad spis I. A. Abrikosov Departmet o Physics, Chemistry ad Biology
MAGNETIC EFFECTS IN SIMULATIONS OF STRUCTURAL TRANSFORMATIONS BELOW AND ABOVE THE MAGNETIC TRANSITION TEMPERATURE: Navigatig i space o atoms ad spis I. A. Abrikosov Departmet o Physics, Chemistry ad Biology
An introduction to the dynamical mean-field theory. L. V. Pourovskii
 An introduction to the dynamical mean-field theory L. V. Pourovskii Nordita school on Photon-Matter interaction, Stockholm, 06.10.2016 OUTLINE The standard density-functional-theory (DFT) framework An
An introduction to the dynamical mean-field theory L. V. Pourovskii Nordita school on Photon-Matter interaction, Stockholm, 06.10.2016 OUTLINE The standard density-functional-theory (DFT) framework An
arxiv: v1 [cond-mat.mtrl-sci] 19 Mar 2014
![arxiv: v1 [cond-mat.mtrl-sci] 19 Mar 2014](/thumbs/71/66018354.jpg "arxiv: v1 [cond-mat.mtrl-sci] 19 Mar 2014") Vibrational free energy and phase stability of paramagnetic and antiferromagnetic CrN from ab-initio molecular dynamics arxiv:1403.4766v1 [cond-mat.mtrl-sci] 19 Mar 2014 Nina Shulumba, 1 Björn Alling,
Vibrational free energy and phase stability of paramagnetic and antiferromagnetic CrN from ab-initio molecular dynamics arxiv:1403.4766v1 [cond-mat.mtrl-sci] 19 Mar 2014 Nina Shulumba, 1 Björn Alling,
Ab initio Berechungen für Datenbanken
 J Ab initio Berechungen für Datenbanken Jörg Neugebauer University of Paderborn Lehrstuhl Computational Materials Science Computational Materials Science Group CMS Group Scaling Problem in Modeling length
J Ab initio Berechungen für Datenbanken Jörg Neugebauer University of Paderborn Lehrstuhl Computational Materials Science Computational Materials Science Group CMS Group Scaling Problem in Modeling length
Introduction to Density Functional Theory
 1 Introduction to Density Functional Theory 21 February 2011; V172 P.Ravindran, FME-course on Ab initio Modelling of solar cell Materials 21 February 2011 Introduction to DFT 2 3 4 Ab initio Computational
1 Introduction to Density Functional Theory 21 February 2011; V172 P.Ravindran, FME-course on Ab initio Modelling of solar cell Materials 21 February 2011 Introduction to DFT 2 3 4 Ab initio Computational
André Schleife Department of Materials Science and Engineering
 André Schleife Department of Materials Science and Engineering Yesterday you (should have) learned this: http://upload.wikimedia.org/wikipedia/commons/e/ea/ Simple_Harmonic_Motion_Orbit.gif 1. deterministic
André Schleife Department of Materials Science and Engineering Yesterday you (should have) learned this: http://upload.wikimedia.org/wikipedia/commons/e/ea/ Simple_Harmonic_Motion_Orbit.gif 1. deterministic
Modified Becke-Johnson (mbj) exchange potential
 exchange potential") Modified Becke-Johnson (mbj) exchange potential Hideyuki Jippo Fujitsu Laboratories LTD. 2015.12.21-22 OpenMX developer s meeting @ Kobe Overview: mbj potential The semilocal exchange potential adding
Modified Becke-Johnson (mbj) exchange potential Hideyuki Jippo Fujitsu Laboratories LTD. 2015.12.21-22 OpenMX developer s meeting @ Kobe Overview: mbj potential The semilocal exchange potential adding
Complementary approaches to high T- high p crystal structure stability and melting!
 Complementary approaches to high T- high p crystal structure stability and melting! Dario ALFÈ Department of Earth Sciences & Department of Physics and Astronomy, Thomas Young Centre@UCL & London Centre
Complementary approaches to high T- high p crystal structure stability and melting! Dario ALFÈ Department of Earth Sciences & Department of Physics and Astronomy, Thomas Young Centre@UCL & London Centre
Spectral Density Functional Theory
 Spectral Density Functional Theory Sergej Savrasov Financial support NSF US DOE LANL Collaborators and Content Constructing New Functionals to Access Energetics and Spectra of Correlated Solids Phonons
Spectral Density Functional Theory Sergej Savrasov Financial support NSF US DOE LANL Collaborators and Content Constructing New Functionals to Access Energetics and Spectra of Correlated Solids Phonons
Is there a future for quantum chemistry on supercomputers? Jürg Hutter Physical-Chemistry Institute, University of Zurich
 Is there a future for quantum chemistry on supercomputers? Jürg Hutter Physical-Chemistry Institute, University of Zurich Chemistry Chemistry is the science of atomic matter, especially its chemical reactions,
Is there a future for quantum chemistry on supercomputers? Jürg Hutter Physical-Chemistry Institute, University of Zurich Chemistry Chemistry is the science of atomic matter, especially its chemical reactions,
Fundamentals and applications of Density Functional Theory Astrid Marthinsen PhD candidate, Department of Materials Science and Engineering
 Fundamentals and applications of Density Functional Theory Astrid Marthinsen PhD candidate, Department of Materials Science and Engineering Outline PART 1: Fundamentals of Density functional theory (DFT)
Fundamentals and applications of Density Functional Theory Astrid Marthinsen PhD candidate, Department of Materials Science and Engineering Outline PART 1: Fundamentals of Density functional theory (DFT)
Effect of magnetic disorder and strong electron correlations on the thermodynamics of CrN
 Effect of magnetic disorder and strong electron correlations on the thermodynamics of CrN Björn Alling, Tobias Marten and Igor Abrikosov Linköping University Post Print N.B.: When citing this work, cite
Effect of magnetic disorder and strong electron correlations on the thermodynamics of CrN Björn Alling, Tobias Marten and Igor Abrikosov Linköping University Post Print N.B.: When citing this work, cite
Outline. Introduction: graphene. Adsorption on graphene: - Chemisorption - Physisorption. Summary
 Outline Introduction: graphene Adsorption on graphene: - Chemisorption - Physisorption Summary 1 Electronic band structure: Electronic properties K Γ M v F = 10 6 ms -1 = c/300 massless Dirac particles!
Outline Introduction: graphene Adsorption on graphene: - Chemisorption - Physisorption Summary 1 Electronic band structure: Electronic properties K Γ M v F = 10 6 ms -1 = c/300 massless Dirac particles!
Linköping University Post Print. Mixing and decomposition thermodynamics of c-ti1-xalxn from first-principles calculations
 Linköping University Post Print Mixing and decomposition thermodynamics of c-ti1-xalxn from first-principles calculations Björn Alling, A. V. Ruban, A. Karimi, O. E. Peil, Sergey Simak, Lars Hultman and
Linköping University Post Print Mixing and decomposition thermodynamics of c-ti1-xalxn from first-principles calculations Björn Alling, A. V. Ruban, A. Karimi, O. E. Peil, Sergey Simak, Lars Hultman and
Linköping University Post Print. First principle calculations of core-level binding energy and Auger kinetic energy shifts in metallic solids
 Linköping University Post Print First principle calculations of core-level binding energy and Auger kinetic energy shifts in metallic solids Weine Olovsson, Tobias Marten, Erik Holmstrom, Borje Johansson
Linköping University Post Print First principle calculations of core-level binding energy and Auger kinetic energy shifts in metallic solids Weine Olovsson, Tobias Marten, Erik Holmstrom, Borje Johansson
Band calculations: Theory and Applications
 Band calculations: Theory and Applications Lecture 2: Different approximations for the exchange-correlation correlation functional in DFT Local density approximation () Generalized gradient approximation
Band calculations: Theory and Applications Lecture 2: Different approximations for the exchange-correlation correlation functional in DFT Local density approximation () Generalized gradient approximation
Dynamical Mean-Field Theory for Correlated Electron Materials Dieter Vollhardt
 Center for Electronic Correlations and Magnetism University of Augsburg Dynamical Mean-Field Theory for Correlated Electron Materials Dieter Vollhardt XXIII Latin American Symposium on Solid State Physics
Center for Electronic Correlations and Magnetism University of Augsburg Dynamical Mean-Field Theory for Correlated Electron Materials Dieter Vollhardt XXIII Latin American Symposium on Solid State Physics
Linköping University Post Print. Origin of the Anomalous Piezoelectric Response in Wurtzite Sc x Al 1-x N Alloys
 Linköping University Post Print Origin of the Anomalous Piezoelectric Response in Wurtzite Sc x Al 1-x N Alloys Ferenc Tasnadi, Björn Alling, Carina Höglund, Gunilla Wingqvist, Jens Birch, Lars Hultman
Linköping University Post Print Origin of the Anomalous Piezoelectric Response in Wurtzite Sc x Al 1-x N Alloys Ferenc Tasnadi, Björn Alling, Carina Höglund, Gunilla Wingqvist, Jens Birch, Lars Hultman
A Green Function Method for Large Scale Electronic Structure Calculations. Rudolf Zeller
 A Green Function Method for Large Scale Electronic Structure Calculations Rudolf Zeller Institute for Advanced Simulation, Forschungszentrum Jülich Electronic structure calculations (density functional
A Green Function Method for Large Scale Electronic Structure Calculations Rudolf Zeller Institute for Advanced Simulation, Forschungszentrum Jülich Electronic structure calculations (density functional
Point defect interactions and structural stability of compounds.
 Point defect interactions and structural stability of compounds. VITALY BAYKOV Doctoral Thesis School of Industrial Engineering and Management Department of Material Science and Engineering Royal Institute
Point defect interactions and structural stability of compounds. VITALY BAYKOV Doctoral Thesis School of Industrial Engineering and Management Department of Material Science and Engineering Royal Institute
Electrochemistry project, Chemistry Department, November Ab-initio Molecular Dynamics Simulation
 Electrochemistry project, Chemistry Department, November 2006 Ab-initio Molecular Dynamics Simulation Outline Introduction Ab-initio concepts Total energy concepts Adsorption energy calculation Project
Electrochemistry project, Chemistry Department, November 2006 Ab-initio Molecular Dynamics Simulation Outline Introduction Ab-initio concepts Total energy concepts Adsorption energy calculation Project
Electronic structure of solids: basic concepts and methods
 Electronic structure of solids: basic concepts and methods Ondřej Šipr II. NEVF 514 Surface Physics Winter Term 2016-2017 Troja, 21st October 2016 Outline A bit of formal mathematics for the beginning
Electronic structure of solids: basic concepts and methods Ondřej Šipr II. NEVF 514 Surface Physics Winter Term 2016-2017 Troja, 21st October 2016 Outline A bit of formal mathematics for the beginning
Defects in TiO 2 Crystals
 , March 13-15, 2013, Hong Kong Defects in TiO 2 Crystals Richard Rivera, Arvids Stashans 1 Abstract-TiO 2 crystals, anatase and rutile, have been studied using Density Functional Theory (DFT) and the Generalized
, March 13-15, 2013, Hong Kong Defects in TiO 2 Crystals Richard Rivera, Arvids Stashans 1 Abstract-TiO 2 crystals, anatase and rutile, have been studied using Density Functional Theory (DFT) and the Generalized
Determination of dopant of ceria system by density functional theory
 J Mater Sci (2007) 42:7461 7466 DOI 10.1007/s10853-006-1486-5 Determination of dopant of ceria system by density functional theory K. Muthukkumaran Æ Roshan Bokalawela Æ Tom Mathews Æ S. Selladurai Received:
J Mater Sci (2007) 42:7461 7466 DOI 10.1007/s10853-006-1486-5 Determination of dopant of ceria system by density functional theory K. Muthukkumaran Æ Roshan Bokalawela Æ Tom Mathews Æ S. Selladurai Received:
2. TranSIESTA 1. SIESTA. DFT In a Nutshell. Introduction to SIESTA. Boundary Conditions: Open systems. Greens functions and charge density
 1. SIESTA DFT In a Nutshell Introduction to SIESTA Atomic Orbitals Capabilities Resources 2. TranSIESTA Transport in the Nanoscale - motivation Boundary Conditions: Open systems Greens functions and charge
1. SIESTA DFT In a Nutshell Introduction to SIESTA Atomic Orbitals Capabilities Resources 2. TranSIESTA Transport in the Nanoscale - motivation Boundary Conditions: Open systems Greens functions and charge
Impact of magnetism upon chemical interactions in Fe alloys
 Impact of magnetism upon chemical interactions in Fe alloys A.V. Ruban KTH, Stockholm 2009 M. Ohr, 1985 (Pettifor) C. Wolverton, 2005 Multi-scale engineering design FEM CAD-CAM Engineering design Density
Impact of magnetism upon chemical interactions in Fe alloys A.V. Ruban KTH, Stockholm 2009 M. Ohr, 1985 (Pettifor) C. Wolverton, 2005 Multi-scale engineering design FEM CAD-CAM Engineering design Density
All-Electron Full-Potential Calculations at O(ASA) Speed A Fata Morgana?
 Speed A Fata Morgana?") All-Electron Full-Potential Calculations at O(ASA) Speed A Fata Morgana? SFB 484, Teilprojekt D6 October 5, 2007 Outline 1 2 3 Outline 1 2 3 Outline 1 2 3 Outline 1 2 3 Back in the 1930 s... John C. Slater
All-Electron Full-Potential Calculations at O(ASA) Speed A Fata Morgana? SFB 484, Teilprojekt D6 October 5, 2007 Outline 1 2 3 Outline 1 2 3 Outline 1 2 3 Outline 1 2 3 Back in the 1930 s... John C. Slater
Phonon wavefunctions and electron phonon interactions in semiconductors
 Phonon wavefunctions and electron phonon interactions in semiconductors Bartomeu Monserrat bm418@cam.ac.uk University of Cambridge Quantum Monte Carlo in the Apuan Alps VII QMC in the Apuan Alps VII Bartomeu
Phonon wavefunctions and electron phonon interactions in semiconductors Bartomeu Monserrat bm418@cam.ac.uk University of Cambridge Quantum Monte Carlo in the Apuan Alps VII QMC in the Apuan Alps VII Bartomeu
arxiv: v1 [cond-mat.mtrl-sci] 21 Feb 2009
![arxiv: v1 [cond-mat.mtrl-sci] 21 Feb 2009](/thumbs/74/70570791.jpg "arxiv: v1 [cond-mat.mtrl-sci] 21 Feb 2009") The f-electron challenge: localized and itinerant states in lanthanide oxides united by GW@LDA+U arxiv:0902.3697v1 [cond-mat.mtrl-sci] 21 Feb 2009 Hong Jiang, 1 Ricardo I. Gomez-Abal, 1 Patrick Rinke,
The f-electron challenge: localized and itinerant states in lanthanide oxides united by GW@LDA+U arxiv:0902.3697v1 [cond-mat.mtrl-sci] 21 Feb 2009 Hong Jiang, 1 Ricardo I. Gomez-Abal, 1 Patrick Rinke,
Binding energy of 2D materials using Quantum Monte Carlo
 Quantum Monte Carlo in the Apuan Alps IX International Workshop, 26th July to 2nd August 2014 The Apuan Alps Centre for Physics @ TTI, Vallico Sotto, Tuscany, Italy Binding energy of 2D materials using
Quantum Monte Carlo in the Apuan Alps IX International Workshop, 26th July to 2nd August 2014 The Apuan Alps Centre for Physics @ TTI, Vallico Sotto, Tuscany, Italy Binding energy of 2D materials using
Strained Silicon, Electronic Band Structure and Related Issues.
 Strained Silicon, Electronic Band Structure and Related Issues. D. Rideau, F. Gilibert, M. Minondo, C. Tavernier and H. Jaouen STMicroelectronics,, Device Modeling 850 rue Jean Monnet, BP 16, F-38926 Crolles
Strained Silicon, Electronic Band Structure and Related Issues. D. Rideau, F. Gilibert, M. Minondo, C. Tavernier and H. Jaouen STMicroelectronics,, Device Modeling 850 rue Jean Monnet, BP 16, F-38926 Crolles
6. Computational Design of Energy-related Materials
 6. Computational Design of Energy-related Materials Contents 6.1 Atomistic Simulation Methods for Energy Materials 6.2 ab initio design of photovoltaic materials 6.3 Solid Ion Conductors for Fuel Cells
6. Computational Design of Energy-related Materials Contents 6.1 Atomistic Simulation Methods for Energy Materials 6.2 ab initio design of photovoltaic materials 6.3 Solid Ion Conductors for Fuel Cells
Re-evaluating CeO 2 Expansion Upon Reduction: Non-counterpoised Forces, Not Ionic Radius Effects, are the Cause
 Re-evaluating CeO 2 Expansion Upon Reduction: Non-counterpoised Forces, Not Ionic Radius Effects, are the Cause Christopher L. Muhich, a* a ETH Zurich, Department of Mechanical and Process Engineering,
Re-evaluating CeO 2 Expansion Upon Reduction: Non-counterpoised Forces, Not Ionic Radius Effects, are the Cause Christopher L. Muhich, a* a ETH Zurich, Department of Mechanical and Process Engineering,
Temperature-dependence of magnetism of free Fe clusters
 Temperature-dependence of magnetism of free Fe clusters O. Šipr 1, S. Bornemann 2, J. Minár 2, S. Polesya 2, H. Ebert 2 1 Institute of Physics, Academy of Sciences CR, Prague, Czech Republic 2 Universität
Temperature-dependence of magnetism of free Fe clusters O. Šipr 1, S. Bornemann 2, J. Minár 2, S. Polesya 2, H. Ebert 2 1 Institute of Physics, Academy of Sciences CR, Prague, Czech Republic 2 Universität
Double exchange in double perovskites: Ferromagnetism and Antiferromagnetism
 Double exchange in double perovskites: Ferromagnetism and Antiferromagnetism Prabuddha Sanyal University of Hyderabad with H. Das, T. Saha Dasgupta, P. Majumdar, S. Ray, D.D. Sarma H. Das, P. Sanyal, D.D.
Double exchange in double perovskites: Ferromagnetism and Antiferromagnetism Prabuddha Sanyal University of Hyderabad with H. Das, T. Saha Dasgupta, P. Majumdar, S. Ray, D.D. Sarma H. Das, P. Sanyal, D.D.
Prof. John M. Wills (1), and Ann E Mattsson (2) (1) Head of Theoretical Division, Los Alamos National Laboratiry, Los Alamos, USA,
, and Ann E Mattsson (2) (1) Head of Theoretical Division, Los Alamos National Laboratiry, Los Alamos, USA,") Abstracts for the WORKSHOP in Electronic Structure Theory organized by Linköping Linnaeus Initiative for Novel Functional Materials, Theory and Modeling Platform Prof. Ann E. Mattson, Sandia National Laboratories,
Abstracts for the WORKSHOP in Electronic Structure Theory organized by Linköping Linnaeus Initiative for Novel Functional Materials, Theory and Modeling Platform Prof. Ann E. Mattson, Sandia National Laboratories,
All-Electron Full-Potential Calculations at O(ASA) Speed A Fata Morgana?
 Speed A Fata Morgana?") All-Electron Full-Potential Calculations at O(ASA) Speed A Fata Morgana? Center for Electronic Correlations and Magnetism Institute for Physics, University of Augsburg February 4, 2008 Outline 1 2 3 Outline
All-Electron Full-Potential Calculations at O(ASA) Speed A Fata Morgana? Center for Electronic Correlations and Magnetism Institute for Physics, University of Augsburg February 4, 2008 Outline 1 2 3 Outline
Spin crossovers in the Earth mantle. Spin crossovers in the Earth mantle
 Spin crossovers in the Earth mantle Spin crossovers in the Earth mantle Renata M. Wentzcovitch Dept. of Chemical Engineering and Materials Science Minnesota Supercomputing Institute Collaborators Han Hsu
Spin crossovers in the Earth mantle Spin crossovers in the Earth mantle Renata M. Wentzcovitch Dept. of Chemical Engineering and Materials Science Minnesota Supercomputing Institute Collaborators Han Hsu
Density-functional theory of superconductivity
 Density-functional theory of superconductivity E. K. U. Gross MPI for Microstructure Physics Halle http://users.physi.fu-berlin.de/~ag-gross CO-WORKERS: HALLE A. Sanna C. Bersier A. Linscheid H. Glawe
Density-functional theory of superconductivity E. K. U. Gross MPI for Microstructure Physics Halle http://users.physi.fu-berlin.de/~ag-gross CO-WORKERS: HALLE A. Sanna C. Bersier A. Linscheid H. Glawe
NiO - hole doping and bandstructure of charge transfer insulator
 NiO - hole doping and bandstructure of charge transfer insulator Jan Kuneš Institute for Physics, Uni. Augsburg Collaboration: V. I. Anisimov S. L. Skornyakov A. V. Lukoyanov D. Vollhardt Outline NiO -
NiO - hole doping and bandstructure of charge transfer insulator Jan Kuneš Institute for Physics, Uni. Augsburg Collaboration: V. I. Anisimov S. L. Skornyakov A. V. Lukoyanov D. Vollhardt Outline NiO -
Melting curve and Hugoniot of molybdenum up to 400 GPa by ab initio simulations
 Melting curve and Hugoniot of molybdenum up to 4 GPa by ab initio simulations C. Cazorla 1,2, M. J. Gillan 1,2, S. Taioli 3 and D. Alfè 1,2,3 1 London Centre for Nanotechnology, UCL, London WC1H OAH, U.K.
Melting curve and Hugoniot of molybdenum up to 4 GPa by ab initio simulations C. Cazorla 1,2, M. J. Gillan 1,2, S. Taioli 3 and D. Alfè 1,2,3 1 London Centre for Nanotechnology, UCL, London WC1H OAH, U.K.
Electronic Structure Theory for Periodic Systems: The Concepts. Christian Ratsch
 Electronic Structure Theory for Periodic Systems: The Concepts Christian Ratsch Institute for Pure and Applied Mathematics and Department of Mathematics, UCLA Motivation There are 10 20 atoms in 1 mm 3
Electronic Structure Theory for Periodic Systems: The Concepts Christian Ratsch Institute for Pure and Applied Mathematics and Department of Mathematics, UCLA Motivation There are 10 20 atoms in 1 mm 3
Multi-Scale Modeling from First Principles
 m mm Multi-Scale Modeling from First Principles μm nm m mm μm nm space space Predictive modeling and simulations must address all time and Continuum Equations, densityfunctional space scales Rate Equations
m mm Multi-Scale Modeling from First Principles μm nm m mm μm nm space space Predictive modeling and simulations must address all time and Continuum Equations, densityfunctional space scales Rate Equations
First principle calculations of plutonium and plutonium compounds: part 1
 First principle calculations of plutonium and plutonium compounds: part 1 A. B. Shick Institute of Physics ASCR, Prague, CZ Outline: u Lecture 1: Methods of Correlated band theory DFT and DFT+U u Lecture
First principle calculations of plutonium and plutonium compounds: part 1 A. B. Shick Institute of Physics ASCR, Prague, CZ Outline: u Lecture 1: Methods of Correlated band theory DFT and DFT+U u Lecture
Kevin Driver 1 Shuai Zhang 1 Burkhard Militzer 1 R. E. Cohen 2.
 Quantum Monte Carlo Simulations of a Single Iron Impurity in MgO Kevin Driver 1 Shuai Zhang 1 Burkhard Militzer 1 R. E. Cohen 2 1 Department of Earth & Planetary Science University of California, Berkeley
Quantum Monte Carlo Simulations of a Single Iron Impurity in MgO Kevin Driver 1 Shuai Zhang 1 Burkhard Militzer 1 R. E. Cohen 2 1 Department of Earth & Planetary Science University of California, Berkeley
Defects and diffusion in metal oxides: Challenges for first-principles modelling
 Defects and diffusion in metal oxides: Challenges for first-principles modelling Karsten Albe, FG Materialmodellierung, TU Darmstadt Johan Pohl, Peter Agoston, Paul Erhart, Manuel Diehm FUNDING: ICTP Workshop
Defects and diffusion in metal oxides: Challenges for first-principles modelling Karsten Albe, FG Materialmodellierung, TU Darmstadt Johan Pohl, Peter Agoston, Paul Erhart, Manuel Diehm FUNDING: ICTP Workshop
Tuning magnetic anisotropy, Kondo screening and Dzyaloshinskii-Moriya interaction in pairs of Fe adatoms
 Tuning magnetic anisotropy, Kondo screening and Dzyaloshinskii-Moriya interaction in pairs of Fe adatoms Department of Physics, Hamburg University, Hamburg, Germany SPICE Workshop, Mainz Outline Tune magnetic
Tuning magnetic anisotropy, Kondo screening and Dzyaloshinskii-Moriya interaction in pairs of Fe adatoms Department of Physics, Hamburg University, Hamburg, Germany SPICE Workshop, Mainz Outline Tune magnetic
Simulating charge transfer in (organic) solar cells
 solar cells") Simulating charge transfer in (organic) solar cells July 24, 2013 Organic solar cells 1 c Fraunhofer ISE Organic solar cells Step 1: Microscopic structure Organic solar cells 1 c Fraunhofer ISE Organic
Simulating charge transfer in (organic) solar cells July 24, 2013 Organic solar cells 1 c Fraunhofer ISE Organic solar cells Step 1: Microscopic structure Organic solar cells 1 c Fraunhofer ISE Organic
Basics of density-functional theory and fast guide to actual calculations Matthias Scheffler
 Basics of density-functional theory and fast guide to actual calculations Matthias Scheffler http://www.fhi-berlin.mpg.de/th/th.html I. From the many-particle problem to the Kohn-Sham functional II. From
Basics of density-functional theory and fast guide to actual calculations Matthias Scheffler http://www.fhi-berlin.mpg.de/th/th.html I. From the many-particle problem to the Kohn-Sham functional II. From
Density Functional Theory (DFT)
") Density Functional Theory (DFT) An Introduction by A.I. Al-Sharif Irbid, Aug, 2 nd, 2009 Density Functional Theory Revolutionized our approach to the electronic structure of atoms, molecules and solid
Density Functional Theory (DFT) An Introduction by A.I. Al-Sharif Irbid, Aug, 2 nd, 2009 Density Functional Theory Revolutionized our approach to the electronic structure of atoms, molecules and solid
Key concepts in Density Functional Theory (II)
") Kohn-Sham scheme and band structures European Theoretical Spectroscopy Facility (ETSF) CNRS - Laboratoire des Solides Irradiés Ecole Polytechnique, Palaiseau - France Present Address: LPMCN Université
Kohn-Sham scheme and band structures European Theoretical Spectroscopy Facility (ETSF) CNRS - Laboratoire des Solides Irradiés Ecole Polytechnique, Palaiseau - France Present Address: LPMCN Université
Electronic correlations in models and materials. Jan Kuneš
 Electronic correlations in models and materials Jan Kuneš Outline Dynamical-mean field theory Implementation (impurity problem) Single-band Hubbard model MnO under pressure moment collapse metal-insulator
Electronic correlations in models and materials Jan Kuneš Outline Dynamical-mean field theory Implementation (impurity problem) Single-band Hubbard model MnO under pressure moment collapse metal-insulator
Solid State Theory: Band Structure Methods
 Solid State Theory: Band Structure Methods Lilia Boeri Wed., 11:00-12:30 HS P3 (PH02112) http://itp.tugraz.at/lv/boeri/ele/ Who am I? Assistant Professor, Institute for Theoretical and Computational Physics,
Solid State Theory: Band Structure Methods Lilia Boeri Wed., 11:00-12:30 HS P3 (PH02112) http://itp.tugraz.at/lv/boeri/ele/ Who am I? Assistant Professor, Institute for Theoretical and Computational Physics,
Supporting Information
 Supporting Information The Origin of Active Oxygen in a Ternary CuO x /Co 3 O 4 -CeO Catalyst for CO Oxidation Zhigang Liu, *, Zili Wu, *, Xihong Peng, ++ Andrew Binder, Songhai Chai, Sheng Dai *,, School
Supporting Information The Origin of Active Oxygen in a Ternary CuO x /Co 3 O 4 -CeO Catalyst for CO Oxidation Zhigang Liu, *, Zili Wu, *, Xihong Peng, ++ Andrew Binder, Songhai Chai, Sheng Dai *,, School
Density Functional Theory. Martin Lüders Daresbury Laboratory
 Density Functional Theory Martin Lüders Daresbury Laboratory Ab initio Calculations Hamiltonian: (without external fields, non-relativistic) impossible to solve exactly!! Electrons Nuclei Electron-Nuclei
Density Functional Theory Martin Lüders Daresbury Laboratory Ab initio Calculations Hamiltonian: (without external fields, non-relativistic) impossible to solve exactly!! Electrons Nuclei Electron-Nuclei
Dept of Mechanical Engineering MIT Nanoengineering group
 1 Dept of Mechanical Engineering MIT Nanoengineering group » Recap of HK theorems and KS equations» The physical meaning of the XC energy» Solution of a one-particle Schroedinger equation» Pseudo Potentials»
1 Dept of Mechanical Engineering MIT Nanoengineering group » Recap of HK theorems and KS equations» The physical meaning of the XC energy» Solution of a one-particle Schroedinger equation» Pseudo Potentials»
Molecular Mechanics: The Ab Initio Foundation
 Molecular Mechanics: The Ab Initio Foundation Ju Li GEM4 Summer School 2006 Cell and Molecular Mechanics in BioMedicine August 7 18, 2006, MIT, Cambridge, MA, USA 2 Outline Why are electrons quantum? Born-Oppenheimer
Molecular Mechanics: The Ab Initio Foundation Ju Li GEM4 Summer School 2006 Cell and Molecular Mechanics in BioMedicine August 7 18, 2006, MIT, Cambridge, MA, USA 2 Outline Why are electrons quantum? Born-Oppenheimer
Classification of Solids, Fermi Level and Conductivity in Metals Dr. Anurag Srivastava
 Classification of Solids, Fermi Level and Conductivity in Metals Dr. Anurag Srivastava Web address: http://tiiciiitm.com/profanurag Email: profanurag@gmail.com Visit me: Room-110, Block-E, IIITM Campus
Classification of Solids, Fermi Level and Conductivity in Metals Dr. Anurag Srivastava Web address: http://tiiciiitm.com/profanurag Email: profanurag@gmail.com Visit me: Room-110, Block-E, IIITM Campus
Is the homogeneous electron gas homogeneous?
 Is the homogeneous electron gas homogeneous? Electron gas (jellium): simplest way to view a metal homogeneous and normal Hartree-Fock: simplest method for many-electron systems a single Slater determinant
Is the homogeneous electron gas homogeneous? Electron gas (jellium): simplest way to view a metal homogeneous and normal Hartree-Fock: simplest method for many-electron systems a single Slater determinant
Interstitial Mn in (Ga,Mn)As: Hybridization with Conduction Band and Electron Mediated Exchange Coupling
As: Hybridization with Conduction Band and Electron Mediated Exchange Coupling") Vol. 112 (2007) ACTA PHYSICA POLONICA A No. 2 Proceedings of the XXXVI International School of Semiconducting Compounds, Jaszowiec 2007 Interstitial Mn in (Ga,Mn)As: Hybridization with Conduction Band
Vol. 112 (2007) ACTA PHYSICA POLONICA A No. 2 Proceedings of the XXXVI International School of Semiconducting Compounds, Jaszowiec 2007 Interstitial Mn in (Ga,Mn)As: Hybridization with Conduction Band
Calculation of thermal expansion coefficient of Fe3Al with the addition of transition metal elements Abstract Introduction Methodology
 Calculation of thermal expansion coefficient of Fe Al with the addition of transition metal elements Tatiana Seletskaia, Leonid Muratov, Bernard Cooper, West Virginia University, Physics Departement, Hodges
Calculation of thermal expansion coefficient of Fe Al with the addition of transition metal elements Tatiana Seletskaia, Leonid Muratov, Bernard Cooper, West Virginia University, Physics Departement, Hodges
Order- N Green's Function Technique for Local Environment Effects in Alloys
 Downloaded from orbit.dtu.dk on: Dec 16, 2018 Order- N Green's Function Technique for Local Environment Effects in Alloys Abrikosov, I. A.; Niklasson, A. M. N.; Simak, S. I.; Johansson, B.; Ruban, Andrei;
Downloaded from orbit.dtu.dk on: Dec 16, 2018 Order- N Green's Function Technique for Local Environment Effects in Alloys Abrikosov, I. A.; Niklasson, A. M. N.; Simak, S. I.; Johansson, B.; Ruban, Andrei;
Computational Materials Physics
 Computational Materials Physics narrated by Hans L. Skriver Center for Atomic-scale Materials Physics CAMP-DTU Lyngby thanks to L. Vitos P. Söderlind S.I. Simak A. Ruban N. Rosengård J. Nørskov A. Niklasson
Computational Materials Physics narrated by Hans L. Skriver Center for Atomic-scale Materials Physics CAMP-DTU Lyngby thanks to L. Vitos P. Söderlind S.I. Simak A. Ruban N. Rosengård J. Nørskov A. Niklasson
Ab Initio Calculation of Exchange Interactions, Adiabatic Spin-Waves, and Curie Temperature of Itinerant Ferromagnets
 Ab Initio Calculation of Exchange Interactions, Adiabatic Spin-Waves, and Curie Temperature of Itinerant Ferromagnets Patrick Bruno Max-Planck-Institut für Mikrostrukturphysik, Halle, Germany People involved:
Ab Initio Calculation of Exchange Interactions, Adiabatic Spin-Waves, and Curie Temperature of Itinerant Ferromagnets Patrick Bruno Max-Planck-Institut für Mikrostrukturphysik, Halle, Germany People involved:
Linköping University Post Print. Dynamic stability of palladium hydride: An ab initio study
 Linköping University Post Print Dynamic stability of palladium hydride: An ab initio study L E Isaeva, D I Bazhanov, Eyvas Isaev, S V Eremeev, S E Kulkova and Igor Abrikosov N.B.: When citing this work,
Linköping University Post Print Dynamic stability of palladium hydride: An ab initio study L E Isaeva, D I Bazhanov, Eyvas Isaev, S V Eremeev, S E Kulkova and Igor Abrikosov N.B.: When citing this work,
Improved Electronic Structure and Optical Properties of sp-hybridized Semiconductors Using LDA+U SIC
 286 Brazilian Journal of Physics, vol. 36, no. 2A, June, 2006 Improved Electronic Structure and Optical Properties of sp-hybridized Semiconductors Using LDA+U SIC Clas Persson and Susanne Mirbt Department
286 Brazilian Journal of Physics, vol. 36, no. 2A, June, 2006 Improved Electronic Structure and Optical Properties of sp-hybridized Semiconductors Using LDA+U SIC Clas Persson and Susanne Mirbt Department
All electron optimized effective potential method for solids
 All electron optimized effective potential method for solids Institut für Theoretische Physik Freie Universität Berlin, Germany and Fritz Haber Institute of the Max Planck Society, Berlin, Germany. 22
All electron optimized effective potential method for solids Institut für Theoretische Physik Freie Universität Berlin, Germany and Fritz Haber Institute of the Max Planck Society, Berlin, Germany. 22
GEM4 Summer School OpenCourseWare
 GEM4 Summer School OpenCourseWare http://gem4.educommons.net/ http://www.gem4.org/ Lecture: Molecular Mechanics by Ju Li. Given August 9, 2006 during the GEM4 session at MIT in Cambridge, MA. Please use
GEM4 Summer School OpenCourseWare http://gem4.educommons.net/ http://www.gem4.org/ Lecture: Molecular Mechanics by Ju Li. Given August 9, 2006 during the GEM4 session at MIT in Cambridge, MA. Please use
Transition of Iron Ions from High-Spin to Low-Spin State and Pressure-Induced Insulator Metal Transition in Hematite Fe 2 O 3
 ISSN 63-776, Journal of Experimental and Theoretical Physics, 7, Vol. 5, No. 5, pp. 35. Pleiades Publishing, Inc., 7. Original Russian Text A.V. Kozhevnikov, A.V. Lukoyanov, V.I. Anisimov, M.A. Korotin,
ISSN 63-776, Journal of Experimental and Theoretical Physics, 7, Vol. 5, No. 5, pp. 35. Pleiades Publishing, Inc., 7. Original Russian Text A.V. Kozhevnikov, A.V. Lukoyanov, V.I. Anisimov, M.A. Korotin,
*Supported by NSF Grants DMR and DMR and WFU s Center for Energy, Environment, and Sustainability.
 First Principles Modeling of Electrolye Materials in All-Solid-State Batteries* N. A. W. Holzwarth** Department of Physics Wake Forest University, Winston-Salem, NC, USA, 27109 *Supported by NSF Grants
First Principles Modeling of Electrolye Materials in All-Solid-State Batteries* N. A. W. Holzwarth** Department of Physics Wake Forest University, Winston-Salem, NC, USA, 27109 *Supported by NSF Grants
The Exact Muffin-Tin Orbitals method and applications
 HUNGARIAN ACADEMY OF SCIENCES DOCTORAL DISSERTATION The Exact Muffin-Tin Orbitals method and applications Levente Vitos Ph.D. Research Institute for Solid State Physics and Optics Budapest February 2,
HUNGARIAN ACADEMY OF SCIENCES DOCTORAL DISSERTATION The Exact Muffin-Tin Orbitals method and applications Levente Vitos Ph.D. Research Institute for Solid State Physics and Optics Budapest February 2,
SnO 2 Physical and Chemical Properties due to the Impurity Doping
 , March 13-15, 2013, Hong Kong SnO 2 Physical and Chemical Properties due to the Impurity Doping Richard Rivera, Freddy Marcillo, Washington Chamba, Patricio Puchaicela, Arvids Stashans Abstract First-principles
, March 13-15, 2013, Hong Kong SnO 2 Physical and Chemical Properties due to the Impurity Doping Richard Rivera, Freddy Marcillo, Washington Chamba, Patricio Puchaicela, Arvids Stashans Abstract First-principles
Pseudopotentials for hybrid density functionals and SCAN
 Pseudopotentials for hybrid density functionals and SCAN Jing Yang, Liang Z. Tan, Julian Gebhardt, and Andrew M. Rappe Department of Chemistry University of Pennsylvania Why do we need pseudopotentials?
Pseudopotentials for hybrid density functionals and SCAN Jing Yang, Liang Z. Tan, Julian Gebhardt, and Andrew M. Rappe Department of Chemistry University of Pennsylvania Why do we need pseudopotentials?
Local lattice relaxations in random metallic alloys: Effective tetrahedron model and supercell approach
 PHYSICAL REVIEW B 67, 214302 2003 Local lattice relaxations in random metallic alloys: Effective tetrahedron model and supercell approach A. V. Ruban, 1 S. I. Simak, 2 S. Shallcross, 2 and H. L. Skriver
PHYSICAL REVIEW B 67, 214302 2003 Local lattice relaxations in random metallic alloys: Effective tetrahedron model and supercell approach A. V. Ruban, 1 S. I. Simak, 2 S. Shallcross, 2 and H. L. Skriver
Electronic band structure, sx-lda, Hybrid DFT, LDA+U and all that. Keith Refson STFC Rutherford Appleton Laboratory
 Electronic band structure, sx-lda, Hybrid DFT, LDA+U and all that Keith Refson STFC Rutherford Appleton Laboratory LDA/GGA DFT is good but... Naive LDA/GGA calculation severely underestimates band-gaps.
Electronic band structure, sx-lda, Hybrid DFT, LDA+U and all that Keith Refson STFC Rutherford Appleton Laboratory LDA/GGA DFT is good but... Naive LDA/GGA calculation severely underestimates band-gaps.
SMARTMET project: Towards breaking the inverse ductility-strength relation
 SMARTMET project: Towards breaking the inverse ductility-strength relation B. Grabowski, C. Tasan SMARTMET ERC advanced grant 3.8 Mio Euro for 5 years (Raabe/Neugebauer) Adaptive Structural Materials group
SMARTMET project: Towards breaking the inverse ductility-strength relation B. Grabowski, C. Tasan SMARTMET ERC advanced grant 3.8 Mio Euro for 5 years (Raabe/Neugebauer) Adaptive Structural Materials group
Non-collinear OEP for solids: SDFT vs CSDFT
 Non-collinear OEP for solids: SDFT vs Sangeeta Sharma 1,2, J. K. Dewhurst 3 S. Pittalis 2, S. Kurth 2, S. Shallcross 4 and E. K. U. Gross 2 1. Fritz-Haber nstitut of the Max Planck Society, Berlin, Germany
Non-collinear OEP for solids: SDFT vs Sangeeta Sharma 1,2, J. K. Dewhurst 3 S. Pittalis 2, S. Kurth 2, S. Shallcross 4 and E. K. U. Gross 2 1. Fritz-Haber nstitut of the Max Planck Society, Berlin, Germany
Observation of a robust zero-energy bound state in iron-based superconductor Fe(Te,Se)
") Materials and Methods: SUPPLEMENTARY INFORMATION Observation of a robust zero-energy bound state in iron-based superconductor Fe(Te,Se) All the crystals, with nominal composition FeTe0.5Se0.5, used in
Materials and Methods: SUPPLEMENTARY INFORMATION Observation of a robust zero-energy bound state in iron-based superconductor Fe(Te,Se) All the crystals, with nominal composition FeTe0.5Se0.5, used in
arxiv: v1 [cond-mat.mtrl-sci] 14 Apr 2010
![arxiv: v1 [cond-mat.mtrl-sci] 14 Apr 2010](/thumbs/72/66735818.jpg "arxiv: v1 [cond-mat.mtrl-sci] 14 Apr 2010") APS/123-QED Theory of the ferromagnetism in Ti 1 x Cr x N solid solutions B. Alling Department of Physics, Chemistry and Biology (IFM), arxiv:1004.2337v1 [cond-mat.mtrl-sci] 14 Apr 2010 Linköping University,
APS/123-QED Theory of the ferromagnetism in Ti 1 x Cr x N solid solutions B. Alling Department of Physics, Chemistry and Biology (IFM), arxiv:1004.2337v1 [cond-mat.mtrl-sci] 14 Apr 2010 Linköping University,
Spins and spin-orbit coupling in semiconductors, metals, and nanostructures
 B. Halperin Spin lecture 1 Spins and spin-orbit coupling in semiconductors, metals, and nanostructures Behavior of non-equilibrium spin populations. Spin relaxation and spin transport. How does one produce
B. Halperin Spin lecture 1 Spins and spin-orbit coupling in semiconductors, metals, and nanostructures Behavior of non-equilibrium spin populations. Spin relaxation and spin transport. How does one produce
The LDA+U method: a primer and implementation within SIESTA
 The LDA+U method: a primer and implementation within SIESTA Daniel Sánchez-Portal Thanks to Javier Junquera, Sampsa Riikonen and Eduardo Anglada Source of the failure of LDA to describe Mott insulators
The LDA+U method: a primer and implementation within SIESTA Daniel Sánchez-Portal Thanks to Javier Junquera, Sampsa Riikonen and Eduardo Anglada Source of the failure of LDA to describe Mott insulators
Computational strongly correlated materials R. Torsten Clay Physics & Astronomy
 Computational strongly correlated materials R. Torsten Clay Physics & Astronomy Current/recent students Saurabh Dayal (current PhD student) Wasanthi De Silva (new grad student 212) Jeong-Pil Song (finished
Computational strongly correlated materials R. Torsten Clay Physics & Astronomy Current/recent students Saurabh Dayal (current PhD student) Wasanthi De Silva (new grad student 212) Jeong-Pil Song (finished
Serendipitous. *Supported by NSF Grants DMR and DMR and WFU s Center for Energy, Environment, and Sustainability.
 Serendipitous ^ Design and synthesis of a crystalline LiPON electrolyte* N. A. W. Holzwarth** Department of Physics Wake Forest University, Winston-Salem, NC, USA, 27109 *Supported by NSF Grants DMR-0705239
Serendipitous ^ Design and synthesis of a crystalline LiPON electrolyte* N. A. W. Holzwarth** Department of Physics Wake Forest University, Winston-Salem, NC, USA, 27109 *Supported by NSF Grants DMR-0705239
Quantum anomalous Hall states on decorated magnetic surfaces
 Quantum anomalous Hall states on decorated magnetic surfaces David Vanderbilt Rutgers University Kevin Garrity & D.V. Phys. Rev. Lett.110, 116802 (2013) Recently: Topological insulators (TR-invariant)
Quantum anomalous Hall states on decorated magnetic surfaces David Vanderbilt Rutgers University Kevin Garrity & D.V. Phys. Rev. Lett.110, 116802 (2013) Recently: Topological insulators (TR-invariant)
Research Projects. Dr Martin Paul Vaughan. Research Background
 Research Projects Dr Martin Paul Vaughan Research Background Research Background Transport theory Scattering in highly mismatched alloys Density functional calculations First principles approach to alloy
Research Projects Dr Martin Paul Vaughan Research Background Research Background Transport theory Scattering in highly mismatched alloys Density functional calculations First principles approach to alloy
From Atoms to Materials: Predictive Theory and Simulations
 From Atoms to Materials: Predictive Theory and Simulations Week 3 Lecture 4 Potentials for metals and semiconductors Ale Strachan strachan@purdue.edu School of Materials Engineering & Birck anotechnology
From Atoms to Materials: Predictive Theory and Simulations Week 3 Lecture 4 Potentials for metals and semiconductors Ale Strachan strachan@purdue.edu School of Materials Engineering & Birck anotechnology
BONDING AND ELECTRONIC STRUCTURE OF MINERALS
 BONDING AND ELECTRONIC STRUCTURE OF MINERALS RONALD E. COHEN Carnegie Institution of Washington Geophysical Laboratory and Center for High Pressure Research 5251 Broad Branch Rd., N.W. Washington, D.C.
BONDING AND ELECTRONIC STRUCTURE OF MINERALS RONALD E. COHEN Carnegie Institution of Washington Geophysical Laboratory and Center for High Pressure Research 5251 Broad Branch Rd., N.W. Washington, D.C.
Molybdenum Sulfide based electronics. Gotthard Seifert Physikalische Chemie,Technische Universität Dresden, Germany
 start Molybdenum Sulfide based electronics Gotthard Seifert Physikalische Chemie,Technische Universität Dresden, Germany Early studies 1960-ies R. Fivaz, E. Mooser Mobility of Charge Carriers in semiconducting
start Molybdenum Sulfide based electronics Gotthard Seifert Physikalische Chemie,Technische Universität Dresden, Germany Early studies 1960-ies R. Fivaz, E. Mooser Mobility of Charge Carriers in semiconducting
Basics of DFT applications to solids and surfaces
 Basics of DFT applications to solids and surfaces Peter Kratzer Physics Department, University Duisburg-Essen, Duisburg, Germany E-mail: Peter.Kratzer@uni-duisburg-essen.de Periodicity in real space and
Basics of DFT applications to solids and surfaces Peter Kratzer Physics Department, University Duisburg-Essen, Duisburg, Germany E-mail: Peter.Kratzer@uni-duisburg-essen.de Periodicity in real space and
The Plane-Wave Pseudopotential Method
 Hands-on Workshop on Density Functional Theory and Beyond: Computational Materials Science for Real Materials Trieste, August 6-15, 2013 The Plane-Wave Pseudopotential Method Ralph Gebauer ICTP, Trieste
Hands-on Workshop on Density Functional Theory and Beyond: Computational Materials Science for Real Materials Trieste, August 6-15, 2013 The Plane-Wave Pseudopotential Method Ralph Gebauer ICTP, Trieste
Introduction to First-Principles Method
 Joint ICTP/CAS/IAEA School & Workshop on Plasma-Materials Interaction in Fusion Devices, July 18-22, 2016, Hefei Introduction to First-Principles Method by Guang-Hong LU ( 吕广宏 ) Beihang University Computer
Joint ICTP/CAS/IAEA School & Workshop on Plasma-Materials Interaction in Fusion Devices, July 18-22, 2016, Hefei Introduction to First-Principles Method by Guang-Hong LU ( 吕广宏 ) Beihang University Computer
Many-body effects in iron pnictides and chalcogenides
 Many-body effects in iron pnictides and chalcogenides separability of non-local and dynamical correlation effects Jan M. Tomczak Vienna University of Technology jan.tomczak@tuwien.ac.at Emergent Quantum
Many-body effects in iron pnictides and chalcogenides separability of non-local and dynamical correlation effects Jan M. Tomczak Vienna University of Technology jan.tomczak@tuwien.ac.at Emergent Quantum
Ionic Bonding. Example: Atomic Radius: Na (r = 0.192nm) Cl (r = 0.099nm) Ionic Radius : Na (r = 0.095nm) Cl (r = 0.181nm)
 Cl (r = 0.099nm) Ionic Radius : Na (r = 0.095nm) Cl (r = 0.181nm)") Ionic Bonding Ion: an atom or molecule that gains or loses electrons (acquires an electrical charge). Atoms form cations (+charge), when they lose electrons, or anions (- charge), when they gain electrons.
Ionic Bonding Ion: an atom or molecule that gains or loses electrons (acquires an electrical charge). Atoms form cations (+charge), when they lose electrons, or anions (- charge), when they gain electrons.
From Quantum Mechanics to Materials Design
 The Basics of Density Functional Theory Volker Eyert Center for Electronic Correlations and Magnetism Institute of Physics, University of Augsburg December 03, 2010 Outline Formalism 1 Formalism Definitions
The Basics of Density Functional Theory Volker Eyert Center for Electronic Correlations and Magnetism Institute of Physics, University of Augsburg December 03, 2010 Outline Formalism 1 Formalism Definitions
Electronic Structure of Crystalline Solids
 Electronic Structure of Crystalline Solids Computing the electronic structure of electrons in solid materials (insulators, conductors, semiconductors, superconductors) is in general a very difficult problem
Electronic Structure of Crystalline Solids Computing the electronic structure of electrons in solid materials (insulators, conductors, semiconductors, superconductors) is in general a very difficult problem
The electronic structure of materials 2 - DFT
 Quantum mechanics 2 - Lecture 9 December 19, 2012 1 Density functional theory (DFT) 2 Literature Contents 1 Density functional theory (DFT) 2 Literature Historical background The beginnings: L. de Broglie
Quantum mechanics 2 - Lecture 9 December 19, 2012 1 Density functional theory (DFT) 2 Literature Contents 1 Density functional theory (DFT) 2 Literature Historical background The beginnings: L. de Broglie
DEFECTS IN 2D MATERIALS: HOW WE TAUGHT ELECTRONIC SCREENING TO MACHINES
 DEFECTS IN 2D MATERIALS: HOW WE TAUGHT ELECTRONIC SCREENING TO MACHINES Johannes Lischner Imperial College London LISCHNER GROUP AT IMPERIAL COLLEGE LONDON Theory and simulation of materials: focus on
DEFECTS IN 2D MATERIALS: HOW WE TAUGHT ELECTRONIC SCREENING TO MACHINES Johannes Lischner Imperial College London LISCHNER GROUP AT IMPERIAL COLLEGE LONDON Theory and simulation of materials: focus on
Magnetism in transition metal oxides by post-dft methods
 Magnetism in transition metal oxides by post-dft methods Cesare Franchini Faculty of Physics & Center for Computational Materials Science University of Vienna, Austria Workshop on Magnetism in Complex
Magnetism in transition metal oxides by post-dft methods Cesare Franchini Faculty of Physics & Center for Computational Materials Science University of Vienna, Austria Workshop on Magnetism in Complex
Spin effects (spin polarized systems, spin-orbit ) G. Zérah CEA-DAM Ile de France Bruyères-le-Châtel
 G. Zérah CEA-DAM Ile de France Bruyères-le-Châtel") Spin effects (spin polarized systems, spin-orbit ) G. Zérah CEA-DAM Ile de France 91680 Bruyères-le-Châtel 1 Macroscopic magnetization A crystal can be found in different magnetization states. The direction
Spin effects (spin polarized systems, spin-orbit ) G. Zérah CEA-DAM Ile de France 91680 Bruyères-le-Châtel 1 Macroscopic magnetization A crystal can be found in different magnetization states. The direction