Octa-Acid Binding Affinities with QM Methods
|
|
|
- Jeffery Owens
- 5 years ago
- Views:
Transcription
 6 5 4 3 2 1 0 0 1 2 3 4 5 6 Time (ns) 7 8 9 10 1. Intro 2. DFT-D3 3. CCSD(T) 4.")
1 Octa-Acid Binding Affinities with QM Methods Ulf Ryde Theoretical Chemistry Lund University Sweden 8 H B 7 Distance (Å) Time (ns) Intro 2. DFT-D3 3. CCSD(T) 4. MD snaps
2 1. Introduction 2. DFT Optimisation 3. DLPNO-CCSD(T) 4. MD snapshots
3 Results from SAMPL4 FEP with MM RESP or AM1-BCC charges QM/MM-FEP with NBB or ssea charge 1 DFT on optimised structures LCCSD(T0)-PMISP on DFT structures Varying results MAD RESP 4 AM1-BCC 4 QM-FEP 26 DFT opt 6 LCCSD(T0) 24 R tr
4 SAMPL5 Gusts more dissimilar Problems for FEP Net charge 1 or +1 not for DFT continuum solvation MAD RESP 4 AM1-BCC 4 QM-FEP 26 DFT opt 6 LCCSD(T0) 24
5 Philosophy Ligands not proper for FEP Dissimilar ligands Varying net charge In SAMPL4 DFT-opt gave MAD 6 9 kj/mol Might be good enough if FEP does not work Improve the DFT approach with gained experience
6 Improvements Minimise effect of flexibility Reduce effects of negative charge Try MD sampling Improve CCSD(T) with DLPNO-CCSD(T) (domain-based local pair natural orbital) no fractionation needed
7 DFT-opt in SAMPL4 Three approaches Opt in vacuum Vac Cos Opt in COSMO continuum COSMO + 4 water Vac Cos Wat Cons MAD MADtr R t Wat
8 Flexibility of host from MD Extensive fluctuation 8Å Breathing motion Rapid dynamics No difference between guests in average 8 Distances HB CA 7 Distance (Å) Time (ns) HB Bz 11.7 MeBz 11.7 EtBz 11.8 pclbz 11.6 mclbz 11.6 Hx 11.7 MeHx 11.7 Pen 11.7 Hep 11.8 CC
9 Problem for optimised structures
10 Movement of Propionate Groups Extensive dynamics with 3 minima All 8 torsions change on a time-scale of ns 1 D11 All 3 minima visited, but not for all propionates D21 D31 D41 D12 D22 D23 D24 Dihedral angle (degree) Time (ns)
11 Problem also for optimised structures Less for vacuum optimisations
12 Attempt 1 Start from C4 symmetric host Keep all structures as symmetric and similar as possible Use vacuum optimisation Vac Cos Wat Cons MAD MADtr R t

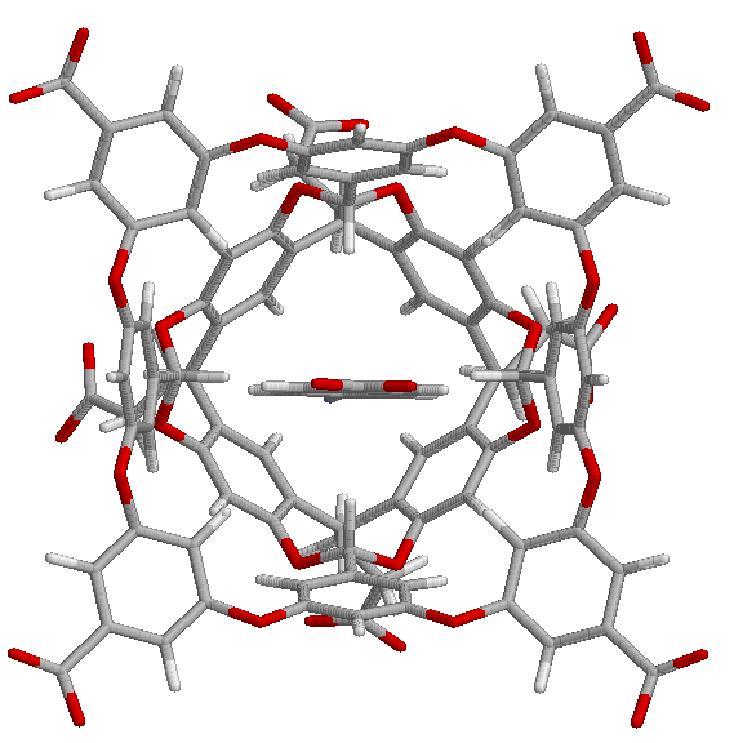

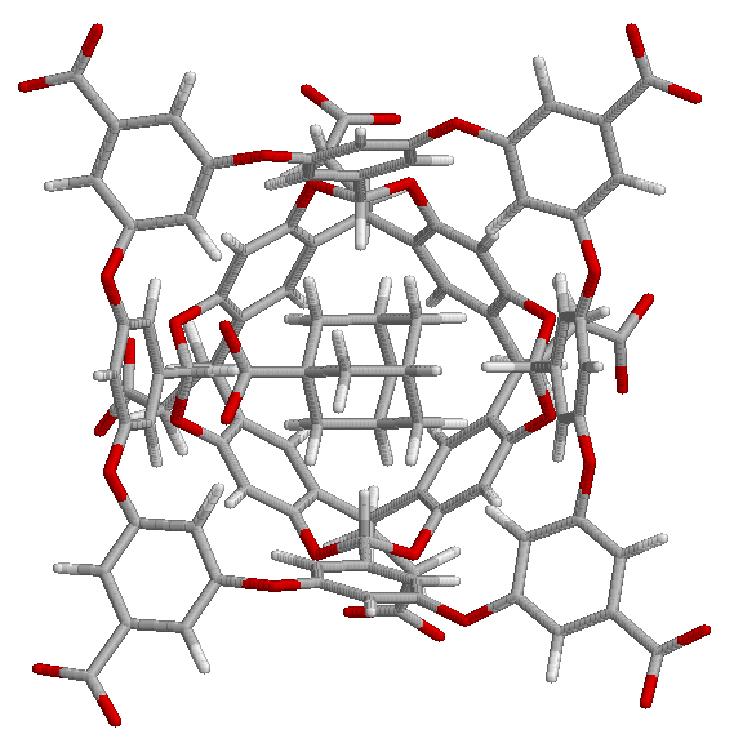

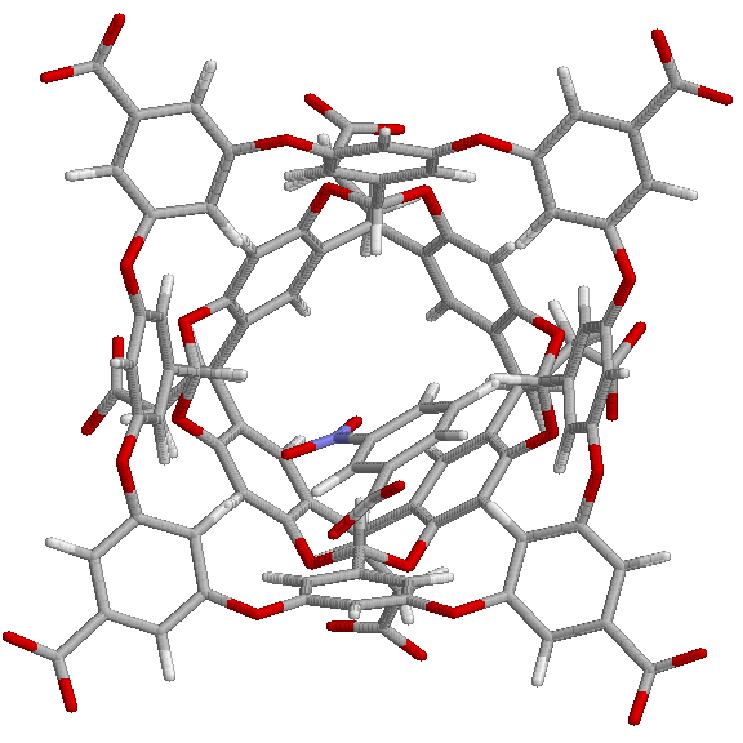
13 Partly Successful G3 G5
14 Partly Successful G3 G5 G3 and G5 (with NMe3+) groups still distorted
15 Attempt 2 Remove all 4 propionate and 4 bezoate groups Reduces solvation energy from 6600 to 300 kj/mol Reduced flexibility Coulombic (ε = 80) correction for +/ series (~23 kj/mol)
 Identical results for 5 perturbations 12 10 8 6 4 2 0 0 2 4 6 8 Exp.")
16 Minimal effect at MM level The differences come from propionate groups MAD R2 tr 2 kj/mol difference for 3 perturbations Nearly same performance compared to Exp. OA NOA 3.6 ± ± ± ± ± ±0.00 HOA 3.9± ± ± OA NOA HOA Calc. (kj/mol) Identical results for 5 perturbations Exp. (kj/mol)
17 1. Introduction 2. DFT Optimisation 3. DLPNO-CCSD(T) 4. MD snapshots
18 Method Approach suggested by Grimme Chem Eur J 18(12)9955 Gbind Optimise structures with TPSS-D3/def2-SV(P) = EDFT Single-point TPSS/def2-QZVP' No counter-poise correction + Edisp + Gsolv + Gfreq DFT-D3 Becke Johnson damping and 3rd-order terms COSMO-RS solvation energy from BP/TZVP in vacuum and in COSMO(ε= ) ZPE, entropy & thermal corrections from HF-3c optimisation & frequency calculation Rigid-rotor harmonic-oscillator ideal-gas approximation low-lying modes with free-rotor approximation
19 Rigid Interaction Energies Results strongly stabilised when using rigid interaction energies i.e. with the geometry of the complex Ebind = E (HG@HG) E(H@HG) E(G@HG) Relaxation energy of guests 0 8 kj/mol for G2, G4 G6 G1 & G kj/mol Deteriorates the results Reduces the effect of host flexibility
20 Four sets Fully charged hosts ( atoms) Neutralised hosts DLPNO-CCSD(T) calculations on DFT structures neutralised hosts MD snapshots for neutralised host
21 DFT Results Gbind = EDFT Charged host Large variation in DFT binding energy 1062 to +973 kj/mol Compensated by solvation energy 906 to Edisp + Gsolv + Gfreq Neutral host Less variation in DFT energy 72 to +30 kj/mol Also solvation energy kj/mol Sum shows small correlation with Exp. (0 0.4)
22 DFT Results Gbind = EDFT Dispersion energy 165 to 94 kj/mol Somewhat more variation with charged host + Some correlation with Exp. R = Edisp + Gsolv + Gfreq Entropy and thermal corrections kj/mol No correlation with Exp.
23 Total Energies Gbind Poor results = EDFT All binding energies kj/mol too positive Calc. (kj/mol) 80 R2 = MADtr = kj/mol OAH NOAH OAM NOAM G1 + Edisp + Gsolv + Gfreq G2 G G Exp. (kj/mol) OAH OAMe NOAH NOAMe MADtr R
24 Sometimes Strange Structures G1 G3 G5 G6 G1 carboxylate inside host G3 & G5 NMe3 partly inside G6 also too deep



25 Structure Problems G4 More distorted than charged hosts All distorted, except G4
26 OAMe structures G1 G4 does not bind G1 carboxylate still inside host


27 OAMe structures G1 G2 G6 G1 & G6 less distorted G2 more distorted
28 OAH vs. OAMe Differences G2 upright in OAMe G3 & G5 NMe3 more inside in OAH G6 more inside in OAH
29 1. Introduction 2. DFT structures 3. DLPNO-CCSD(T) 4. MD snapshots
30 DLPNO-CCSD(T) Calculations Gbind = EDFT + Tried to improve the DFT-D3 results with DLPNO-CCSD(T) calculations 1 Neese et al. J Chem Theory Comput 2011, 7 (2011) 33 Tight PNO thresholds Full complex ( atoms) Edisp CBS extrapolation with def2-svp and def2-tzvp TZ QZ on way + Gsolv + Gfreq Counter-poise corrections Br in G4 treated by ZORA CCSD(T) replaced the TPSS/def2-QZVP'+DFT-D3 energies Gbind = ECCSD(T) + Gsolv + Gfreq
31 DLPNO-CCSD(T) Results Quite similar to DFT results 7 to +11 kj/mol difference MAD = 5 kj/mol Much less than in SAMPL4 LCCSD(T)+PMISP kj/mol Charged host? Calc. (kj/mol) 100 NOAH NOAM CCH CCMe Exp. (kj/mol)
32 DLPNO-CCSD(T) Results Reproduce Exp equally poorly MADtr = kj/mol R2 = Same DFT structures NOAH NOAMe CCH CCMe MADtr R Calc. (kj/mol) 100 NOAH NOAM CCH CCMe Exp. (kj/mol)
33 1. Introduction 2. DFT structures 3. DLPNO-CCSD(T) 4. MD snapshots
34 Structures from MD Gbind = EDFT Run 10 ns MD simulation of each complex with MM + Edisp + Gsolv + Gfreq Took 10 snapshots Optimised with HF-3c TPSS/def2-QZVP' DFT-D3 BJ 3rd-order dispersion COSMO-RS solvation energy (BP/TZVP) HF-3c ZPE, entropy & thermal corrections (same as DFT-opt)
35 Variation of binding free energies Restricted variation for most guests; 3 15 kj/mol Standard Error = kj/mol Sometimes a one outlier G6 large variation (28 52 kj/mol; SE 3 5 kj/mol) G4 bimodal in OAH Binding Energy (kj/mol) G1 G2 G3 G4 G5 G6 OAM G1 G2 G3 G4 G5 G6
 G3 much more")

36 Structures different (NOAH) G3 much more outside G5 much more outside G6 sometimes other orientation
 G1 less")


37 Structures different (NOAMe) G1 less inside G3 & G5 rather similar G4 forced inside in MD and stayed there Distorted host
38 Results R2 much worse 0.0 to OAH OAMe partly owing to forced binding of G4 (host distortion E missing) Calc. (kj/mol) MAD better kj/mol G3 & G5 too strong G1 too weak Exp. (kj/mol) NOAH NOAMe MADtr R MDH MDMe
39 Conclusions MD Promising approach More black-box Gives a precision of 1 ( 5) kj/mol Trying better methods for optimisation

40 Improved Optimisation Methods MD snapshot Vacuum opt. COSMO opt. COSMO + 4 Water
41 Conclusions Poor results Partly owing to the use of vacuum structures Possibly additional un-recovered problems (unexperienced PhD) Flexibility a major problem Optimisation time-consuming (~1 month) Need much nursing MD approach may solve some problems


42 Worth Investigating Alternative QM/MM FEP with MM reference potential Latest results with SAMPL4 data QM systems of atoms NOA and PM6-DH2X QM calculations are needed for convergence to 1 kj/mol DFT optimisation ~200 QM calculations 1/4000!






43 Acknowledgements Octav Caldararu Martin Olsson Dr. Christoph Riplinger Prof. Frank Neese Swedish Research Council Lunarc HPC2N
44
45 Submitted Data We submitted three sets of data 1. Charged host with DFT 2. Neutral host with DFT 3. Neutral host with DLPNO-CCSD(T)//DFT OAH, not rigid energies (missing at submission time) With guest relaxation energies (suboptimal) Error in isolated G2 energy MD not finished in time No Coulombic correction

46 MM-MD structures G1 G1 carboxylate actually inside host


47 MM-MD structures OAMe G1 G2 G1 carboxylate more exposed G2 more up-right
48 LCCSD(T) PMISP Tried to improve the DFT-D3 results with Local CCSD(T) calculations Hampel & Werner, J Chem Phys 104 (1996) 6286 Employing the PMISP approach Polarised Multipolar Interactions with Supermolecular Pairs Söderhjelm & Ryde, J Phys Chem A 113 (2009) 617 Based on the Cos structures solvation energies, and thermal corrections Replaced the TPSS/def2-QZVP'/TZVP+DFT-D3 energies with LCCSD(T) energies

49 LCCSD(T) calculations Together with Prof. Ricardo Mata, Göttingen cc-pvtz basis set Extrapolated to basis-set limit at conventional MP2 level with aug-cc-pvtz and aug-cc-pvqz basis sets and n 3 scheme Full counter-poise corrections Density fitting Pipek Mezey localisation
50 PMISP Etot = Eele + Eind + Eother < Eother = Σ cj(eqm(bai) Eele(BAi) Eind(BAi) MM (NEMO) electrostatics and induction QM calculations on Guest (B) and Host fragments (Aj) (to get multipole expansion and polarisabilities) and all BAi pairs (to get dispersion, repulsion, penetration, CT,... = other )
51 PMISP details Multipoles up to octupoles and anisotropic polarisabilities in atomic centres and all bond midpoints Calculated at the MP2/cc-pVTZ level Interaction energies between guest and host calculated for guest+fragment at the LCCSD(T)/CBS level Many-body effects treated at the MM (NEMO) level with the multipoles and polarisabilities
52 Fractionation Host fractionated according to the MFCC approach Molecular Fractionation with Conjugate Caps Adapted to the complicated host 24 fragments 32 conjugate fragments 4 doubly-conjugate fragments
53 Results CCSD(T) correction rather large and negative 27 (Hx) to 54 (mclbz) kj/mol Bz MeBz EtBz pclbz mclbz Hx MeHx Pen Hep All quality measures significantly worse Constant shift + exaggerate differences Submitted absolute affinities with one outlier Cos CC corr Cos MAD 9 MADtr 8 R2 0.7 t 0.7 t PI 0.9 RMSD 10 MSD -5 slope 2 intercept 21 CC Calculated (kj/mol) Experimental (kj/mol) -15
54 Conclusions MM FEP acceptable results Not much better than for proteins Best MAD = 6.0 kj/mol for 91 transformations in 10 proteins No difference between RESP & BCC charges DFT-FEP has severe convergence problems Poor Optimised DFT intermediate performance Intermediate Severe problems with flexibility Hard to improve LCCSD(T) possible for ligand binding with PMISP Worst But poor results why?




55 Structures different (NOAH) G1 deeper G2 similar G3 much more outside G4 similar G5 much more outside G6 other orientation
")


56 Structures different (NOAMe) G1 less inside G2 similar G3 rather similar G4 forced inside G5 similar G6 similar
Free-energy perturbation and quantum mechanical study of SAMPL4 octa-acid hostguest binding energies.
 Free-energy perturbation and quantum mechanical study of SAMPL4 octa-acid hostguest binding energies. Mikulskis, Paulius; Cioloboc, Daniela; Andrejić, Milica; Khare, Sakshi; Brorsson, Joakim; Genheden,
Free-energy perturbation and quantum mechanical study of SAMPL4 octa-acid hostguest binding energies. Mikulskis, Paulius; Cioloboc, Daniela; Andrejić, Milica; Khare, Sakshi; Brorsson, Joakim; Genheden,
Binding free energies in the SAMPL5 octa-acid host guest challenge calculated with DFT-D3 and CCSD(T)
") J Comput Aided Mol Des (2017) 31:87 106 DOI 10.1007/s10822-016-9957-5 Binding free energies in the SAMPL5 octa-acid host guest challenge calculated with DFT-D3 and CCSD(T) Octav Caldararu 1 Martin A. Olsson
J Comput Aided Mol Des (2017) 31:87 106 DOI 10.1007/s10822-016-9957-5 Binding free energies in the SAMPL5 octa-acid host guest challenge calculated with DFT-D3 and CCSD(T) Octav Caldararu 1 Martin A. Olsson
Supporting Information: Predicting the Ionic Product of Water
 Supporting Information: Predicting the Ionic Product of Water Eva Perlt 1,+, Michael von Domaros 1,+, Barbara Kirchner 1, Ralf Ludwig 2, and Frank Weinhold 3,* 1 Mulliken Center for Theoretical Chemistry,
Supporting Information: Predicting the Ionic Product of Water Eva Perlt 1,+, Michael von Domaros 1,+, Barbara Kirchner 1, Ralf Ludwig 2, and Frank Weinhold 3,* 1 Mulliken Center for Theoretical Chemistry,
Quantum Chemistry for Supramolecular Complexes
 Quantum Chemistry for Supramolecular Complexes Stefan Grimme Mulliken Center for Theoretical Chemistry, University of Bonn COSMO Symposium, March 2015 Supramolecular chemistry: chemistry beyond the covalent
Quantum Chemistry for Supramolecular Complexes Stefan Grimme Mulliken Center for Theoretical Chemistry, University of Bonn COSMO Symposium, March 2015 Supramolecular chemistry: chemistry beyond the covalent
Advanced in silico drug design
 Advanced in silico drug design RNDr. Martin Lepšík, Ph.D. Lecture: Advanced scoring Palacky University, Olomouc 2016 1 Outline 1. Scoring Definition, Types 2. Physics-based Scoring: Master Equation Terms
Advanced in silico drug design RNDr. Martin Lepšík, Ph.D. Lecture: Advanced scoring Palacky University, Olomouc 2016 1 Outline 1. Scoring Definition, Types 2. Physics-based Scoring: Master Equation Terms
Non-covalent force fields computed ab initio
 Non-covalent force fields computed ab initio Supermolecule calculations Symmetry-adapted perturbation theory (SAPT) Supermolecule calculations Requirements: E = E AB E A E B. Include electron correlation,
Non-covalent force fields computed ab initio Supermolecule calculations Symmetry-adapted perturbation theory (SAPT) Supermolecule calculations Requirements: E = E AB E A E B. Include electron correlation,
High-level Quantum Chemistry Methods and Benchmark Datasets for Molecules
 High-level Quantum Chemistry Methods and Benchmark Datasets for Molecules Markus Schneider Fritz Haber Institute of the MPS, Berlin, Germany École Polytechnique Fédérale de Lausanne, Switzerland دانشگاه
High-level Quantum Chemistry Methods and Benchmark Datasets for Molecules Markus Schneider Fritz Haber Institute of the MPS, Berlin, Germany École Polytechnique Fédérale de Lausanne, Switzerland دانشگاه
Interaction energy of DNA base pairs and amino acid pairs: DFT, DFTB and WFT calculations
 Interaction energy of DNA base pairs and amino acid pairs: DFT, DFTB and WFT calculations Tomáš Kubař, Petr Jurečka, Jirka Černý, Honza Řezáč, Michal Otyepka, Haydée Valdés and Pavel Hobza* Institute of
Interaction energy of DNA base pairs and amino acid pairs: DFT, DFTB and WFT calculations Tomáš Kubař, Petr Jurečka, Jirka Černý, Honza Řezáč, Michal Otyepka, Haydée Valdés and Pavel Hobza* Institute of
Comparison of the Efficiency of the LIE and MM/GBSA Methods to Calculate LigandBinding Energies
 Comparison of the Efficiency of the LIE and MM/GBSA Methods to Calculate LigandBinding Energies Genheden, Samuel; Ryde, Ulf Published in: Journal of Chemical Theory and Computation DOI: 10.1021/ct200163c
Comparison of the Efficiency of the LIE and MM/GBSA Methods to Calculate LigandBinding Energies Genheden, Samuel; Ryde, Ulf Published in: Journal of Chemical Theory and Computation DOI: 10.1021/ct200163c
Prediction of spectroscopic parameters for bio-organic and bio-inorganic intermediates in complex systems
 Prediction of spectroscopic parameters for bio-organic and bio-inorganic intermediates in complex systems Erik Donovan Hedegård Department of Physics, Chemistry and Pharmacy University of Southern Denmark
Prediction of spectroscopic parameters for bio-organic and bio-inorganic intermediates in complex systems Erik Donovan Hedegård Department of Physics, Chemistry and Pharmacy University of Southern Denmark
Jack Simons, Henry Eyring Scientist and Professor Chemistry Department University of Utah
 1. Born-Oppenheimer approx.- energy surfaces 2. Mean-field (Hartree-Fock) theory- orbitals 3. Pros and cons of HF- RHF, UHF 4. Beyond HF- why? 5. First, one usually does HF-how? 6. Basis sets and notations
1. Born-Oppenheimer approx.- energy surfaces 2. Mean-field (Hartree-Fock) theory- orbitals 3. Pros and cons of HF- RHF, UHF 4. Beyond HF- why? 5. First, one usually does HF-how? 6. Basis sets and notations
Introduction to Electronic Structure Theory
 CSC/PRACE Spring School in Computational Chemistry 2017 Introduction to Electronic Structure Theory Mikael Johansson http://www.iki.fi/~mpjohans Objective: To get familiarised with the, subjectively chosen,
CSC/PRACE Spring School in Computational Chemistry 2017 Introduction to Electronic Structure Theory Mikael Johansson http://www.iki.fi/~mpjohans Objective: To get familiarised with the, subjectively chosen,
Calculation of Ligand Dissociation Energies in Large Transition-Metal Complexes arxiv: v2 [physics.chem-ph] 20 Mar 2018
![Calculation of Ligand Dissociation Energies in Large Transition-Metal Complexes arxiv: v2 [physics.chem-ph] 20 Mar 2018](/thumbs/90/103453741.jpg "Calculation of Ligand Dissociation Energies in Large Transition-Metal Complexes arxiv: v2 [physics.chem-ph] 20 Mar 2018") Calculation of Ligand Dissociation Energies in Large Transition-Metal Complexes arxiv:1801.06584v2 [physics.chem-ph] 20 Mar 2018 Tamara Husch, Leon Freitag, and Markus Reiher ETH Zürich, Laboratorium für
Calculation of Ligand Dissociation Energies in Large Transition-Metal Complexes arxiv:1801.06584v2 [physics.chem-ph] 20 Mar 2018 Tamara Husch, Leon Freitag, and Markus Reiher ETH Zürich, Laboratorium für
Macrocycle Conformational Sampling by DFT-
 Supporting Information Macrocycle Conformational Sampling by DFT- D3/COSMO-RS Methodology. Ondrej Gutten, Daniel Bím, Jan Řezáč, Lubomír Rulíšek* The Institute of Organic Chemistry and Biochemistry of
Supporting Information Macrocycle Conformational Sampling by DFT- D3/COSMO-RS Methodology. Ondrej Gutten, Daniel Bím, Jan Řezáč, Lubomír Rulíšek* The Institute of Organic Chemistry and Biochemistry of
Binding affinities by alchemical perturbation using QM/MM with a large QM system and polarizable MM model.
 Binding affinities by alchemical perturbation using QM/MM with a large QM system and polarizable MM model. Genheden, Samuel; Ryde, Ulf; Söderhjelm, Pär Published in: Journal of Computational Chemistry
Binding affinities by alchemical perturbation using QM/MM with a large QM system and polarizable MM model. Genheden, Samuel; Ryde, Ulf; Söderhjelm, Pär Published in: Journal of Computational Chemistry
Large Density-Functional and Basis-Set Effects for the DMSO Reductase Catalyzed Oxo-Transfer Reaction
 Large Density-Functional and Basis-Set Effects for the DMSO Reductase Catalyzed Oxo-Transfer Reaction Li, Jilai; Mata, Ricardo A.; Ryde, Ulf Published in: Journal of Chemical Theory and Computation DOI:
Large Density-Functional and Basis-Set Effects for the DMSO Reductase Catalyzed Oxo-Transfer Reaction Li, Jilai; Mata, Ricardo A.; Ryde, Ulf Published in: Journal of Chemical Theory and Computation DOI:
Tailoring the Properties of Quadruplex Nucleobases for Biological and Nanomaterial Applications
 Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 2014 Supporting Information for: Tailoring the Properties of Quadruplex Nucleobases
Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 2014 Supporting Information for: Tailoring the Properties of Quadruplex Nucleobases
How are hydrogen bonds modified by metal binding?
 How are hydrogen bonds modified by metal binding? Husberg, Charlotte; Ryde, Ulf Published in: Journal of Biological Irganic Chemistry DOI:.7/s775-3-996-3 Link to publication Citation for published version
How are hydrogen bonds modified by metal binding? Husberg, Charlotte; Ryde, Ulf Published in: Journal of Biological Irganic Chemistry DOI:.7/s775-3-996-3 Link to publication Citation for published version
QUANTUM CHEMISTRY PROJECT 3: PARTS B AND C
 Chemistry 460 Fall 2017 Dr. Jean M. Standard November 6, 2017 QUANTUM CHEMISTRY PROJECT 3: PARTS B AND C PART B: POTENTIAL CURVE, SPECTROSCOPIC CONSTANTS, AND DISSOCIATION ENERGY OF DIATOMIC HYDROGEN (20
Chemistry 460 Fall 2017 Dr. Jean M. Standard November 6, 2017 QUANTUM CHEMISTRY PROJECT 3: PARTS B AND C PART B: POTENTIAL CURVE, SPECTROSCOPIC CONSTANTS, AND DISSOCIATION ENERGY OF DIATOMIC HYDROGEN (20
T. Helgaker, Department of Chemistry, University of Oslo, Norway. T. Ruden, University of Oslo, Norway. W. Klopper, University of Karlsruhe, Germany
 1 The a priori calculation of molecular properties to chemical accuarcy T. Helgaker, Department of Chemistry, University of Oslo, Norway T. Ruden, University of Oslo, Norway W. Klopper, University of Karlsruhe,
1 The a priori calculation of molecular properties to chemical accuarcy T. Helgaker, Department of Chemistry, University of Oslo, Norway T. Ruden, University of Oslo, Norway W. Klopper, University of Karlsruhe,
platinum and iridium complexes
 SUPPORTING INFORMATION A relativistic DFT methodology for calculating the structures and NMR chemical shifts of octahedral platinum and iridium complexes Jan Vícha, Michael Patzschke, Radek Marek Figure
SUPPORTING INFORMATION A relativistic DFT methodology for calculating the structures and NMR chemical shifts of octahedral platinum and iridium complexes Jan Vícha, Michael Patzschke, Radek Marek Figure
Effect of geometry optimisations on QM-cluster and QM/MM studies of reaction energies in proteins
 Effect of geometry optimisations on QM-cluster and QM/MM studies of reaction energies in proteins Sumner, Sophie; Söderhjelm, Pär; Ryde, Ulf Published in: Journal of Chemical Theory and Computation DOI:
Effect of geometry optimisations on QM-cluster and QM/MM studies of reaction energies in proteins Sumner, Sophie; Söderhjelm, Pär; Ryde, Ulf Published in: Journal of Chemical Theory and Computation DOI:
This is a simple input file for the calculation of NMR chemical shieldings for a given molecule using the B3LYP functional and def2-tzvpp basis set:
 Computing NMR parameters using ORCA This practical comes with a short lecture on the basics of the computation of NMR parameters using standard electronic structure theory methods. By now you should have
Computing NMR parameters using ORCA This practical comes with a short lecture on the basics of the computation of NMR parameters using standard electronic structure theory methods. By now you should have
Alanine: Then There Was Water
 Chemistry Publications Chemistry 6-2009 Alanine: Then There Was Water Jonathan M. Mullin Iowa State University Mark S. Gordon Iowa State University, mgordon@iastate.edu Follow this and additional works
Chemistry Publications Chemistry 6-2009 Alanine: Then There Was Water Jonathan M. Mullin Iowa State University Mark S. Gordon Iowa State University, mgordon@iastate.edu Follow this and additional works
Force Fields for Classical Molecular Dynamics simulations of Biomolecules. Emad Tajkhorshid
 Force Fields for Classical Molecular Dynamics simulations of Biomolecules Emad Tajkhorshid Theoretical and Computational Biophysics Group, Beckman Institute Departments of Biochemistry and Pharmacology,
Force Fields for Classical Molecular Dynamics simulations of Biomolecules Emad Tajkhorshid Theoretical and Computational Biophysics Group, Beckman Institute Departments of Biochemistry and Pharmacology,
3rd Advanced in silico Drug Design KFC/ADD Molecular mechanics intro Karel Berka, Ph.D. Martin Lepšík, Ph.D. Pavel Polishchuk, Ph.D.
 3rd Advanced in silico Drug Design KFC/ADD Molecular mechanics intro Karel Berka, Ph.D. Martin Lepšík, Ph.D. Pavel Polishchuk, Ph.D. Thierry Langer, Ph.D. Jana Vrbková, Ph.D. UP Olomouc, 23.1.-26.1. 2018
3rd Advanced in silico Drug Design KFC/ADD Molecular mechanics intro Karel Berka, Ph.D. Martin Lepšík, Ph.D. Pavel Polishchuk, Ph.D. Thierry Langer, Ph.D. Jana Vrbková, Ph.D. UP Olomouc, 23.1.-26.1. 2018
Introduction to DFTB. Marcus Elstner. July 28, 2006
 Introduction to DFTB Marcus Elstner July 28, 2006 I. Non-selfconsistent solution of the KS equations DFT can treat up to 100 atoms in routine applications, sometimes even more and about several ps in MD
Introduction to DFTB Marcus Elstner July 28, 2006 I. Non-selfconsistent solution of the KS equations DFT can treat up to 100 atoms in routine applications, sometimes even more and about several ps in MD
Computational Studies of the Photoreceptor Rhodopsin. Scott E. Feller Wabash College
 Computational Studies of the Photoreceptor Rhodopsin Scott E. Feller Wabash College Rhodopsin Photocycle Dark-adapted Rhodopsin hn Isomerize retinal Photorhodopsin ~200 fs Bathorhodopsin Meta-II ms timescale
Computational Studies of the Photoreceptor Rhodopsin Scott E. Feller Wabash College Rhodopsin Photocycle Dark-adapted Rhodopsin hn Isomerize retinal Photorhodopsin ~200 fs Bathorhodopsin Meta-II ms timescale
Noncovalent interactions in biochemistry
 Noncovalent interactions in biochemistry Kevin E. Riley 1 and Pavel Hobza 2,3 Noncovalent interactions are known to play a key role in biochemistry. The knowledge of stabilization (relative) energies and
Noncovalent interactions in biochemistry Kevin E. Riley 1 and Pavel Hobza 2,3 Noncovalent interactions are known to play a key role in biochemistry. The knowledge of stabilization (relative) energies and
Multi-scale approaches in description and design of enzymes
 Multi-scale approaches in description and design of enzymes Anastassia Alexandrova and Manuel Sparta UCLA & CNSI Catalysis: it is all about the barrier The inside-out protocol: Big Aim: development of
Multi-scale approaches in description and design of enzymes Anastassia Alexandrova and Manuel Sparta UCLA & CNSI Catalysis: it is all about the barrier The inside-out protocol: Big Aim: development of
List of Figures Page Figure No. Figure Caption No. Figure 1.1.
 List of Figures Figure No. Figure Caption Page No. Figure 1.1. Cation- interactions and their modulations. 4 Figure 1.2. Three conformations of benzene dimer, S is not a minimum on the potential energy
List of Figures Figure No. Figure Caption Page No. Figure 1.1. Cation- interactions and their modulations. 4 Figure 1.2. Three conformations of benzene dimer, S is not a minimum on the potential energy
Electronic quantum effect on hydrogen bond geometry in. water dimer
 Electronic quantum effect on hydrogen bond geometry in water dimer Danhui Li 1,2, Zhiyuan Zhang 1,2 Wanrun Jiang 1,2 Depeng Zhang 1,2 Yu Zhu 1,2 and Zhigang Wang 1,2* 1 Institute of Atomic and Molecular
Electronic quantum effect on hydrogen bond geometry in water dimer Danhui Li 1,2, Zhiyuan Zhang 1,2 Wanrun Jiang 1,2 Depeng Zhang 1,2 Yu Zhu 1,2 and Zhigang Wang 1,2* 1 Institute of Atomic and Molecular
4 Post-Hartree Fock Methods: MPn and Configuration Interaction
 4 Post-Hartree Fock Methods: MPn and Configuration Interaction In the limit of a complete basis, the Hartree-Fock (HF) energy in the complete basis set limit (ECBS HF ) yields an upper boundary to the
4 Post-Hartree Fock Methods: MPn and Configuration Interaction In the limit of a complete basis, the Hartree-Fock (HF) energy in the complete basis set limit (ECBS HF ) yields an upper boundary to the
Fragmentation methods
 Fragmentation methods Scaling of QM Methods HF, DFT scale as N 4 MP2 scales as N 5 CC methods scale as N 7 What if we could freeze the value of N regardless of the size of the system? Then each method
Fragmentation methods Scaling of QM Methods HF, DFT scale as N 4 MP2 scales as N 5 CC methods scale as N 7 What if we could freeze the value of N regardless of the size of the system? Then each method
Solution of the Electronic Schrödinger Equation. Using Basis Sets to Solve the Electronic Schrödinger Equation with Electron Correlation
 Solution of the Electronic Schrödinger Equation Using Basis Sets to Solve the Electronic Schrödinger Equation with Electron Correlation Errors in HF Predictions: Binding Energies D e (kcal/mol) HF Expt
Solution of the Electronic Schrödinger Equation Using Basis Sets to Solve the Electronic Schrödinger Equation with Electron Correlation Errors in HF Predictions: Binding Energies D e (kcal/mol) HF Expt
Towards gas-phase accuracy for condensed phase problems
 Towards gas-phase accuracy for condensed phase problems Fred Manby Centre for Computational Chemistry, School of Chemistry University of Bristol STC 2006: Quantum Chemistry Methods and Applications Erkner,
Towards gas-phase accuracy for condensed phase problems Fred Manby Centre for Computational Chemistry, School of Chemistry University of Bristol STC 2006: Quantum Chemistry Methods and Applications Erkner,
Basis Set for Molecular Orbital Theory
 Basis Set for Molecular Orbital Theory! Different Types of Basis Functions! Different Types of Atom Center Basis Functions! Classifications of Gaussian Basis Sets! Pseudopotentials! Molecular Properties
Basis Set for Molecular Orbital Theory! Different Types of Basis Functions! Different Types of Atom Center Basis Functions! Classifications of Gaussian Basis Sets! Pseudopotentials! Molecular Properties
Electronic excitations in conjugated. Many-Body Green's Functions. Behnaz Bagheri Varnousfaderani. Behnaz Bagheri Varnousfaderani
 Technische Universiteit Eindhoven University of Technology Electronic excitations in conjugated polymers Electronic from excitations QM/MM simulations in conjugated pol based from on Many-Body QM/MM simulations
Technische Universiteit Eindhoven University of Technology Electronic excitations in conjugated polymers Electronic from excitations QM/MM simulations in conjugated pol based from on Many-Body QM/MM simulations
Force Fields for MD simulations
 Force Fields for MD simulations Topology/parameter files Where do the numbers an MD code uses come from? ow to make topology files for ligands, cofactors, special amino acids, ow to obtain/develop missing
Force Fields for MD simulations Topology/parameter files Where do the numbers an MD code uses come from? ow to make topology files for ligands, cofactors, special amino acids, ow to obtain/develop missing
Electronic structure theory: Fundamentals to frontiers. VI. Analysis and more.
 Electronic structure theory: Fundamentals to frontiers. VI. Analysis and more. MARTIN HEAD-GORDON Department of Chemistry, University of California, Berkeley, and, Chemical Sciences Division, Lawrence
Electronic structure theory: Fundamentals to frontiers. VI. Analysis and more. MARTIN HEAD-GORDON Department of Chemistry, University of California, Berkeley, and, Chemical Sciences Division, Lawrence
Computational Chemistry I
 Computational Chemistry I Text book Cramer: Essentials of Quantum Chemistry, Wiley (2 ed.) Chapter 3. Post Hartree-Fock methods (Cramer: chapter 7) There are many ways to improve the HF method. Most of
Computational Chemistry I Text book Cramer: Essentials of Quantum Chemistry, Wiley (2 ed.) Chapter 3. Post Hartree-Fock methods (Cramer: chapter 7) There are many ways to improve the HF method. Most of
Importing ab-initio theory into DFT: Some applications of the Lieb variation principle
 Importing ab-initio theory into DFT: Some applications of the Lieb variation principle Trygve Helgaker, Andy Teale, and Sonia Coriani Centre for Theoretical and Computational Chemistry (CTCC), Department
Importing ab-initio theory into DFT: Some applications of the Lieb variation principle Trygve Helgaker, Andy Teale, and Sonia Coriani Centre for Theoretical and Computational Chemistry (CTCC), Department
Structures and infrared spectra of fluoride hydrogen sulfide clusters from ab initio calculations: F -(H 2 S) n, n = 1 5w
 n, n = 1 5w") RESEARCH PAPER Structures and infrared spectra of fluoride hydrogen sulfide clusters from ab initio calculations: F -(H 2 S) n, n = 1 5w D. A. Wild* and T. Lenzer PCCP www.rsc.org/pccp MPI fu r biophysikalische
RESEARCH PAPER Structures and infrared spectra of fluoride hydrogen sulfide clusters from ab initio calculations: F -(H 2 S) n, n = 1 5w D. A. Wild* and T. Lenzer PCCP www.rsc.org/pccp MPI fu r biophysikalische
Specific Ion Solvtion in Ethylene Carbonate and Propylene Carbonate
 Specific Ion Solvtion in Ethylene Carbonate and Propylene Carbonate A. Arslanargin, A. Powers, S. Rick, T. Pollard, T. Beck Univ Cincinnati Chemistry Support: NSF, OSC TSRC 2016 November 2, 2016 A. Arslanargin,
Specific Ion Solvtion in Ethylene Carbonate and Propylene Carbonate A. Arslanargin, A. Powers, S. Rick, T. Pollard, T. Beck Univ Cincinnati Chemistry Support: NSF, OSC TSRC 2016 November 2, 2016 A. Arslanargin,
Supporting Information
 Supporting Information Ionic Liquid Designed for PEDOT:PSS Conductivity Enhancement Ambroise de Izarra, a,b,1 Seongjin Park, a,1 Jinhee Lee, a Yves Lansac, b,c, * Yun Hee Jang a, * a Department of Energy
Supporting Information Ionic Liquid Designed for PEDOT:PSS Conductivity Enhancement Ambroise de Izarra, a,b,1 Seongjin Park, a,1 Jinhee Lee, a Yves Lansac, b,c, * Yun Hee Jang a, * a Department of Energy
Accurate van der Waals interactions from ground state electron density
 Accurate van der Waals interactions from ground state electron density Alexandre Tkatchenko Theory Department, Fritz Haber Institut der MPG Berlin, Germany tkatchen@fhi berlin.mpg.de Haber Institute EXCITCM09,
Accurate van der Waals interactions from ground state electron density Alexandre Tkatchenko Theory Department, Fritz Haber Institut der MPG Berlin, Germany tkatchen@fhi berlin.mpg.de Haber Institute EXCITCM09,
Supplementary information
 Matthias Heger, Tina Scharge, and Martin A. Suhm Institute of Physical Chemistry, Georg-August-Universität, Tammannstraße 6, 37077 Göttingen, Germany. E-mail: msuhm@gwdg.de Current address: Gesellschaft
Matthias Heger, Tina Scharge, and Martin A. Suhm Institute of Physical Chemistry, Georg-August-Universität, Tammannstraße 6, 37077 Göttingen, Germany. E-mail: msuhm@gwdg.de Current address: Gesellschaft
Jack Simons, Henry Eyring Scientist and Professor Chemistry Department University of Utah
 1. Born-Oppenheimer approx.- energy surfaces 2. Mean-field (Hartree-Fock) theory- orbitals 3. Pros and cons of HF- RHF, UHF 4. Beyond HF- why? 5. First, one usually does HF-how? 6. Basis sets and notations
1. Born-Oppenheimer approx.- energy surfaces 2. Mean-field (Hartree-Fock) theory- orbitals 3. Pros and cons of HF- RHF, UHF 4. Beyond HF- why? 5. First, one usually does HF-how? 6. Basis sets and notations
CE 530 Molecular Simulation
 1 CE 530 Molecular Simulation Lecture 14 Molecular Models David A. Kofke Department of Chemical Engineering SUNY Buffalo kofke@eng.buffalo.edu 2 Review Monte Carlo ensemble averaging, no dynamics easy
1 CE 530 Molecular Simulation Lecture 14 Molecular Models David A. Kofke Department of Chemical Engineering SUNY Buffalo kofke@eng.buffalo.edu 2 Review Monte Carlo ensemble averaging, no dynamics easy
Lec20 Fri 3mar17
 564-17 Lec20 Fri 3mar17 [PDF]GAUSSIAN 09W TUTORIAL www.molcalx.com.cn/wp-content/uploads/2015/01/gaussian09w_tutorial.pdf by A Tomberg - Cited by 8 - Related articles GAUSSIAN 09W TUTORIAL. AN INTRODUCTION
564-17 Lec20 Fri 3mar17 [PDF]GAUSSIAN 09W TUTORIAL www.molcalx.com.cn/wp-content/uploads/2015/01/gaussian09w_tutorial.pdf by A Tomberg - Cited by 8 - Related articles GAUSSIAN 09W TUTORIAL. AN INTRODUCTION
Tools for QM studies of large systems
 Tools for QM studies of large systems Automated, hessian-free saddle point search & characterization QM/MM implementation for zeolites Shaama Mallikarjun Sharada Advisors: Prof. Alexis T Bell, Prof. Martin
Tools for QM studies of large systems Automated, hessian-free saddle point search & characterization QM/MM implementation for zeolites Shaama Mallikarjun Sharada Advisors: Prof. Alexis T Bell, Prof. Martin
MM-PBSA Validation Study. Trent E. Balius Department of Applied Mathematics and Statistics AMS
 MM-PBSA Validation Study Trent. Balius Department of Applied Mathematics and Statistics AMS 535 11-26-2008 Overview MM-PBSA Introduction MD ensembles one snap-shots relaxed structures nrichment Computational
MM-PBSA Validation Study Trent. Balius Department of Applied Mathematics and Statistics AMS 535 11-26-2008 Overview MM-PBSA Introduction MD ensembles one snap-shots relaxed structures nrichment Computational
Computational Methods. Chem 561
 Computational Methods Chem 561 Lecture Outline 1. Ab initio methods a) HF SCF b) Post-HF methods 2. Density Functional Theory 3. Semiempirical methods 4. Molecular Mechanics Computational Chemistry " Computational
Computational Methods Chem 561 Lecture Outline 1. Ab initio methods a) HF SCF b) Post-HF methods 2. Density Functional Theory 3. Semiempirical methods 4. Molecular Mechanics Computational Chemistry " Computational
Molecular dynamics simulation of Aquaporin-1. 4 nm
 Molecular dynamics simulation of Aquaporin-1 4 nm Molecular Dynamics Simulations Schrödinger equation i~@ t (r, R) =H (r, R) Born-Oppenheimer approximation H e e(r; R) =E e (R) e(r; R) Nucleic motion described
Molecular dynamics simulation of Aquaporin-1 4 nm Molecular Dynamics Simulations Schrödinger equation i~@ t (r, R) =H (r, R) Born-Oppenheimer approximation H e e(r; R) =E e (R) e(r; R) Nucleic motion described
Q-Chem 4.0: Expanding the Frontiers. Jing Kong Q-Chem Inc. Pittsburgh, PA
 Q-Chem 4.0: Expanding the Frontiers Jing Kong Q-Chem Inc. Pittsburgh, PA Q-Chem: Profile Q-Chem is a high performance quantum chemistry program; Contributed by best quantum chemists from 40 universities
Q-Chem 4.0: Expanding the Frontiers Jing Kong Q-Chem Inc. Pittsburgh, PA Q-Chem: Profile Q-Chem is a high performance quantum chemistry program; Contributed by best quantum chemists from 40 universities
Towards a force field based on density fitting
 THE JOURNAL OF CHEMICAL PHYSICS 124, 104101 2006 Towards a force field based on density fitting Jean-Philip Piquemal a and G. Andrés Cisneros Laboratory of Structural Biology, National Institute of Environmental
THE JOURNAL OF CHEMICAL PHYSICS 124, 104101 2006 Towards a force field based on density fitting Jean-Philip Piquemal a and G. Andrés Cisneros Laboratory of Structural Biology, National Institute of Environmental
Supporting Information Computational Part
 Supporting Information Computational Part Ruthenium-Catalyzed Alkyne trans-hydrometalation: Mechanistic Insights and Preparative Implications Dragoş Adrian Roşca, Karin Radkowski, Larry M. Wolf, Minal
Supporting Information Computational Part Ruthenium-Catalyzed Alkyne trans-hydrometalation: Mechanistic Insights and Preparative Implications Dragoş Adrian Roşca, Karin Radkowski, Larry M. Wolf, Minal
Competing, Coverage-Dependent Decomposition Pathways for C 2 H y Species on Nickel (111)
") 20028 J. Phys. Chem. C 2010, 114, 20028 20041 Competing, Coverage-Dependent Decomposition Pathways for C 2 H y Species on Nickel (111) Jonathan E. Mueller, Adri C. T. van Duin, and William A. Goddard III*,
20028 J. Phys. Chem. C 2010, 114, 20028 20041 Competing, Coverage-Dependent Decomposition Pathways for C 2 H y Species on Nickel (111) Jonathan E. Mueller, Adri C. T. van Duin, and William A. Goddard III*,
D3RGC2: free energy scoring by alchemical free energy implementation in SOMD
 D3RGC2: free energy scoring by alchemical free energy implementation in SOMD Julien Michel EaStCHEM School of Chemistry, University of Edinburgh, United Kingdom D3R webinar 27/03/17 Acknowledgements: group
D3RGC2: free energy scoring by alchemical free energy implementation in SOMD Julien Michel EaStCHEM School of Chemistry, University of Edinburgh, United Kingdom D3R webinar 27/03/17 Acknowledgements: group
Lecture 11: Potential Energy Functions
 Lecture 11: Potential Energy Functions Dr. Ronald M. Levy ronlevy@temple.edu Originally contributed by Lauren Wickstrom (2011) Microscopic/Macroscopic Connection The connection between microscopic interactions
Lecture 11: Potential Energy Functions Dr. Ronald M. Levy ronlevy@temple.edu Originally contributed by Lauren Wickstrom (2011) Microscopic/Macroscopic Connection The connection between microscopic interactions
Supporting Information Computational Part
 Supporting Information Computational Part The Cinchona Primary Amine-Catalyzed Asymmetric Epoxidation and Hydroperoxidation of, -Unsaturated Carbonyl Compounds with Hydrogen Peroxide Olga Lifchits, Manuel
Supporting Information Computational Part The Cinchona Primary Amine-Catalyzed Asymmetric Epoxidation and Hydroperoxidation of, -Unsaturated Carbonyl Compounds with Hydrogen Peroxide Olga Lifchits, Manuel
BUDE. A General Purpose Molecular Docking Program Using OpenCL. Richard B Sessions
 BUDE A General Purpose Molecular Docking Program Using OpenCL Richard B Sessions 1 The molecular docking problem receptor ligand Proteins typically O(1000) atoms Ligands typically O(100) atoms predicted
BUDE A General Purpose Molecular Docking Program Using OpenCL Richard B Sessions 1 The molecular docking problem receptor ligand Proteins typically O(1000) atoms Ligands typically O(100) atoms predicted
CHEM6085: Density Functional Theory Lecture 10
 CHEM6085: Density Functional Theory Lecture 10 1) Spin-polarised calculations 2) Geometry optimisation C.-K. Skylaris 1 Unpaired electrons So far we have developed Kohn-Sham DFT for the case of paired
CHEM6085: Density Functional Theory Lecture 10 1) Spin-polarised calculations 2) Geometry optimisation C.-K. Skylaris 1 Unpaired electrons So far we have developed Kohn-Sham DFT for the case of paired
General Physical Chemistry II
 General Physical Chemistry II Lecture 13 Aleksey Kocherzhenko October 16, 2014" Last time " The Hückel method" Ø Used to study π systems of conjugated molecules" Ø π orbitals are treated separately from
General Physical Chemistry II Lecture 13 Aleksey Kocherzhenko October 16, 2014" Last time " The Hückel method" Ø Used to study π systems of conjugated molecules" Ø π orbitals are treated separately from
Practical Advice for Quantum Chemistry Computations. C. David Sherrill School of Chemistry and Biochemistry Georgia Institute of Technology
 Practical Advice for Quantum Chemistry Computations C. David Sherrill School of Chemistry and Biochemistry Georgia Institute of Technology Choice of Basis Set STO-3G is too small 6-31G* or 6-31G** 6 probably
Practical Advice for Quantum Chemistry Computations C. David Sherrill School of Chemistry and Biochemistry Georgia Institute of Technology Choice of Basis Set STO-3G is too small 6-31G* or 6-31G** 6 probably
Hyeyoung Shin a, Tod A. Pascal ab, William A. Goddard III abc*, and Hyungjun Kim a* Korea
 The Scaled Effective Solvent Method for Predicting the Equilibrium Ensemble of Structures with Analysis of Thermodynamic Properties of Amorphous Polyethylene Glycol-Water Mixtures Hyeyoung Shin a, Tod
The Scaled Effective Solvent Method for Predicting the Equilibrium Ensemble of Structures with Analysis of Thermodynamic Properties of Amorphous Polyethylene Glycol-Water Mixtures Hyeyoung Shin a, Tod
Lecture: P1_Wk1_L1 IntraMolecular Interactions. Ron Reifenberger Birck Nanotechnology Center Purdue University 2012
 Lecture: IntraMolecular Interactions Distinguish between IntraMolecular (within a molecule) and InterMolecular (between molecules) Ron Reifenberger Birck Nanotechnology Center Purdue University 2012 1
Lecture: IntraMolecular Interactions Distinguish between IntraMolecular (within a molecule) and InterMolecular (between molecules) Ron Reifenberger Birck Nanotechnology Center Purdue University 2012 1
Uptake of OH radical to aqueous aerosol: a computational study
 Uptake of OH radical to aqueous aerosol: a computational study Grigory Andreev Karpov Institute of Physical Chemistry 10 Vorontsovo pole, Moscow, 105064, Russia Institute of Physical Chemistry and Electrochemistry
Uptake of OH radical to aqueous aerosol: a computational study Grigory Andreev Karpov Institute of Physical Chemistry 10 Vorontsovo pole, Moscow, 105064, Russia Institute of Physical Chemistry and Electrochemistry
Force Fields for Classical Molecular Dynamics simulations of Biomolecules. Emad Tajkhorshid
 Force Fields for Classical Molecular Dynamics simulations of Biomolecules Emad Tajkhorshid Beckman Institute Departments of Biochemistry Center for Biophysics and Computational Biology University of Illinois
Force Fields for Classical Molecular Dynamics simulations of Biomolecules Emad Tajkhorshid Beckman Institute Departments of Biochemistry Center for Biophysics and Computational Biology University of Illinois
On the selection of domains and orbital pairs in local correlation treatments
 On the selection of domains and orbital pairs in local correlation treatments Hans-Joachim Werner and Klaus Pflüger Institut für Theoretische Chemie, Universität Stuttgart, Pfaffenwaldring 55, D-70569
On the selection of domains and orbital pairs in local correlation treatments Hans-Joachim Werner and Klaus Pflüger Institut für Theoretische Chemie, Universität Stuttgart, Pfaffenwaldring 55, D-70569
Ab initio calculations for potential energy surfaces. D. Talbi GRAAL- Montpellier
 Ab initio calculations for potential energy surfaces D. Talbi GRAAL- Montpellier A theoretical study of a reaction is a two step process I-Electronic calculations : techniques of quantum chemistry potential
Ab initio calculations for potential energy surfaces D. Talbi GRAAL- Montpellier A theoretical study of a reaction is a two step process I-Electronic calculations : techniques of quantum chemistry potential
ONETEP PB/SA: Application to G-Quadruplex DNA Stability. Danny Cole
 ONETEP PB/SA: Application to G-Quadruplex DNA Stability Danny Cole Introduction Historical overview of structure and free energy calculation of complex molecules using molecular mechanics and continuum
ONETEP PB/SA: Application to G-Quadruplex DNA Stability Danny Cole Introduction Historical overview of structure and free energy calculation of complex molecules using molecular mechanics and continuum
Lecture 5: More about one- Final words about the Hartree-Fock theory. First step above it by the Møller-Plesset perturbation theory.
 Lecture 5: More about one- determinant wave functions Final words about the Hartree-Fock theory. First step above it by the Møller-Plesset perturbation theory. Items from Lecture 4 Could the Koopmans theorem
Lecture 5: More about one- determinant wave functions Final words about the Hartree-Fock theory. First step above it by the Møller-Plesset perturbation theory. Items from Lecture 4 Could the Koopmans theorem
Charge-Transfer and Dispersion Energies in Water Clusters
 II.26 Charge-Transfer and Dispersion Energies in Water Clusters Suehiro Iwata 1,2, Pradipta Bandyopadhyay 3, Sotiris S. Xantheas 4 1)Toyota Physical and Chemical Research Institute (2008-2012, fellow)
II.26 Charge-Transfer and Dispersion Energies in Water Clusters Suehiro Iwata 1,2, Pradipta Bandyopadhyay 3, Sotiris S. Xantheas 4 1)Toyota Physical and Chemical Research Institute (2008-2012, fellow)
This semester. Books
 Models mostly proteins from detailed to more abstract models Some simulation methods This semester Books None necessary for my group and Prof Rarey Molecular Modelling: Principles and Applications Leach,
Models mostly proteins from detailed to more abstract models Some simulation methods This semester Books None necessary for my group and Prof Rarey Molecular Modelling: Principles and Applications Leach,
Gilles Frison, Gilles Ohanessian. To cite this version: HAL Id: hal
 A comparative study of semiempirical, ab initio, and DFT methods in evaluating metal-ligand bond strength, proton affinity, and interactions between first and second shell ligands in Zn-biomimetic complexes
A comparative study of semiempirical, ab initio, and DFT methods in evaluating metal-ligand bond strength, proton affinity, and interactions between first and second shell ligands in Zn-biomimetic complexes
The Molecular Dynamics Method
 The Molecular Dynamics Method Thermal motion of a lipid bilayer Water permeation through channels Selective sugar transport Potential Energy (hyper)surface What is Force? Energy U(x) F = d dx U(x) Conformation
The Molecular Dynamics Method Thermal motion of a lipid bilayer Water permeation through channels Selective sugar transport Potential Energy (hyper)surface What is Force? Energy U(x) F = d dx U(x) Conformation
Characteristics of the interaction in azulene (H 2 X) n=1,2 (X=O,S) clusters.
 n=1,2 (X=O,S) clusters.") Characteristics of the interaction in azulene (H 2 X) n=1,2 (X=O,S) clusters. Enrique M. Cabaleiro-Lago (a), Ángeles Peña-Gallego (b), Jesús Rodríguez-Otero (b), M. Merced Montero-Campillo (b) (a) Departamento
Characteristics of the interaction in azulene (H 2 X) n=1,2 (X=O,S) clusters. Enrique M. Cabaleiro-Lago (a), Ángeles Peña-Gallego (b), Jesús Rodríguez-Otero (b), M. Merced Montero-Campillo (b) (a) Departamento
The Potential Energy Surface
 The Potential Energy Surface In this section we will explore the information that can be obtained by solving the Schrödinger equation for a molecule, or series of molecules. Of course, the accuracy of
The Potential Energy Surface In this section we will explore the information that can be obtained by solving the Schrödinger equation for a molecule, or series of molecules. Of course, the accuracy of
Ligand affinities estimated by quantum chemical calculations
 Ligand affinities estimated by quantum chemical calculations Söderhjelm, Pär; Kongsted, Jacob; Ryde, Ulf Published in: Journal of Chemical Theory and Computation DOI: 10.1021/ct9006986 2010 Link to publication
Ligand affinities estimated by quantum chemical calculations Söderhjelm, Pär; Kongsted, Jacob; Ryde, Ulf Published in: Journal of Chemical Theory and Computation DOI: 10.1021/ct9006986 2010 Link to publication
An introduction to Molecular Dynamics. EMBO, June 2016
 An introduction to Molecular Dynamics EMBO, June 2016 What is MD? everything that living things do can be understood in terms of the jiggling and wiggling of atoms. The Feynman Lectures in Physics vol.
An introduction to Molecular Dynamics EMBO, June 2016 What is MD? everything that living things do can be understood in terms of the jiggling and wiggling of atoms. The Feynman Lectures in Physics vol.
PROTEIN-PROTEIN DOCKING REFINEMENT USING RESTRAINT MOLECULAR DYNAMICS SIMULATIONS
 TASKQUARTERLYvol.20,No4,2016,pp.353 360 PROTEIN-PROTEIN DOCKING REFINEMENT USING RESTRAINT MOLECULAR DYNAMICS SIMULATIONS MARTIN ZACHARIAS Physics Department T38, Technical University of Munich James-Franck-Str.
TASKQUARTERLYvol.20,No4,2016,pp.353 360 PROTEIN-PROTEIN DOCKING REFINEMENT USING RESTRAINT MOLECULAR DYNAMICS SIMULATIONS MARTIN ZACHARIAS Physics Department T38, Technical University of Munich James-Franck-Str.
PH 548 Atomistic Simulation Techniques
 PH 548 Atomistic Simulation Techniques Lectures: Lab: 2-0-2-6 Tuesday 12-1 PM Thursday 12-1 PM Monday 2-5 PM P. K. Padmanabhan PH548: Two Excellent Books How to do it? 1 2 F 12 F 23 3 F i = F ij j i F
PH 548 Atomistic Simulation Techniques Lectures: Lab: 2-0-2-6 Tuesday 12-1 PM Thursday 12-1 PM Monday 2-5 PM P. K. Padmanabhan PH548: Two Excellent Books How to do it? 1 2 F 12 F 23 3 F i = F ij j i F
Example questions for Molecular modelling (Level 4) Dr. Adrian Mulholland
 Dr. Adrian Mulholland") Example questions for Molecular modelling (Level 4) Dr. Adrian Mulholland 1) Question. Two methods which are widely used for the optimization of molecular geometies are the Steepest descents and Newton-Raphson
Example questions for Molecular modelling (Level 4) Dr. Adrian Mulholland 1) Question. Two methods which are widely used for the optimization of molecular geometies are the Steepest descents and Newton-Raphson
The Potential Energy Surface (PES) And the Basic Force Field Chem 4021/8021 Video II.iii
 And the Basic Force Field Chem 4021/8021 Video II.iii") The Potential Energy Surface (PES) And the Basic Force Field Chem 4021/8021 Video II.iii Fundamental Points About Which to Be Thinking It s clear the PES is useful, so how can I construct it for an arbitrary
The Potential Energy Surface (PES) And the Basic Force Field Chem 4021/8021 Video II.iii Fundamental Points About Which to Be Thinking It s clear the PES is useful, so how can I construct it for an arbitrary
Electron Correlation Methods
 Electron Correlation Methods HF method: electron-electron interaction is replaced by an average interaction E HF c = E 0 E HF E 0 exact ground state energy E HF HF energy for a given basis set HF E c
Electron Correlation Methods HF method: electron-electron interaction is replaced by an average interaction E HF c = E 0 E HF E 0 exact ground state energy E HF HF energy for a given basis set HF E c
COSMO-RS Theory. The Basics
 Theory The Basics From µ to properties Property µ 1 µ 2 activity coefficient vapor pressure Infinite dilution Gas phase Pure compound Pure bulk compound Partition coefficient Phase 1 Phase 2 Liquid-liquid
Theory The Basics From µ to properties Property µ 1 µ 2 activity coefficient vapor pressure Infinite dilution Gas phase Pure compound Pure bulk compound Partition coefficient Phase 1 Phase 2 Liquid-liquid
Mechanism of Cu/Pd-Catalyzed Decarboxylative Cross-Couplings: A DFT Investigation
 Mechanism of Cu/Pd-Catalyzed Decarboxylative Cross-Couplings: A DFT Investigation Andreas Fromm, a Christoph van Wüllen, b Dagmar Hackenberger a and Lukas J. Gooßen* a a Fachbereich Chemie Organische Chemie
Mechanism of Cu/Pd-Catalyzed Decarboxylative Cross-Couplings: A DFT Investigation Andreas Fromm, a Christoph van Wüllen, b Dagmar Hackenberger a and Lukas J. Gooßen* a a Fachbereich Chemie Organische Chemie
Electric properties of molecules
 Electric properties of molecules For a molecule in a uniform electric fielde the Hamiltonian has the form: Ĥ(E) = Ĥ + E ˆµ x where we assume that the field is directed along the x axis and ˆµ x is the
Electric properties of molecules For a molecule in a uniform electric fielde the Hamiltonian has the form: Ĥ(E) = Ĥ + E ˆµ x where we assume that the field is directed along the x axis and ˆµ x is the
Biochemistry,530:,, Introduc5on,to,Structural,Biology, Autumn,Quarter,2015,
 Biochemistry,530:,, Introduc5on,to,Structural,Biology, Autumn,Quarter,2015, Course,Informa5on, BIOC%530% GraduateAlevel,discussion,of,the,structure,,func5on,,and,chemistry,of,proteins,and, nucleic,acids,,control,of,enzyma5c,reac5ons.,please,see,the,course,syllabus,and,
Biochemistry,530:,, Introduc5on,to,Structural,Biology, Autumn,Quarter,2015, Course,Informa5on, BIOC%530% GraduateAlevel,discussion,of,the,structure,,func5on,,and,chemistry,of,proteins,and, nucleic,acids,,control,of,enzyma5c,reac5ons.,please,see,the,course,syllabus,and,
Accounting for Solvation in Quantum Chemistry. Comp chem spring school 2014 CSC, Finland
 Accounting for Solvation in Quantum Chemistry Comp chem spring school 2014 CSC, Finland In solution or in a vacuum? Solvent description is important when: Polar solvent: electrostatic stabilization Solvent
Accounting for Solvation in Quantum Chemistry Comp chem spring school 2014 CSC, Finland In solution or in a vacuum? Solvent description is important when: Polar solvent: electrostatic stabilization Solvent
Structure and Dynamics of Chitin Nanofibrils in Aqueous Environment Revealed by Molecular Dynamics Simulations
 Electronic Supplementary Material (ESI) for RSC Advances. This journal is The Royal Society of Chemistry 2016 Structure and Dynamics of Chitin Nanofibrils in Aqueous Environment Revealed by Molecular Dynamics
Electronic Supplementary Material (ESI) for RSC Advances. This journal is The Royal Society of Chemistry 2016 Structure and Dynamics of Chitin Nanofibrils in Aqueous Environment Revealed by Molecular Dynamics
Comparison of frozen-density embedding and discrete reaction field solvent models for molecular propertiesw
 PAPER www.rsc.org/pccp Physical Chemistry Chemical Physics Comparison of frozen-density embedding and discrete reaction field solvent models for molecular propertiesw Christoph R. Jacob,* a Johannes Neugebauer,
PAPER www.rsc.org/pccp Physical Chemistry Chemical Physics Comparison of frozen-density embedding and discrete reaction field solvent models for molecular propertiesw Christoph R. Jacob,* a Johannes Neugebauer,
What Makes for a Good Catalytic Cycle? A Theoretical Study of the. SPhos Ligand in the Suzuki-Miyaura Reaction.
 This journal is (c) The Royal Society of Chemistry 2011 Supplementary Information for: What Makes for a Good Catalytic Cycle? A Theoretical Study of the SPhos Ligand in the Suzuki-Miyaura Reaction. Sebastian
This journal is (c) The Royal Society of Chemistry 2011 Supplementary Information for: What Makes for a Good Catalytic Cycle? A Theoretical Study of the SPhos Ligand in the Suzuki-Miyaura Reaction. Sebastian
AN INTRODUCTION TO QUANTUM CHEMISTRY. Mark S. Gordon Iowa State University
 AN INTRODUCTION TO QUANTUM CHEMISTRY Mark S. Gordon Iowa State University 1 OUTLINE Theoretical Background in Quantum Chemistry Overview of GAMESS Program Applications 2 QUANTUM CHEMISTRY In principle,
AN INTRODUCTION TO QUANTUM CHEMISTRY Mark S. Gordon Iowa State University 1 OUTLINE Theoretical Background in Quantum Chemistry Overview of GAMESS Program Applications 2 QUANTUM CHEMISTRY In principle,
METHODS FOR TREATING SOLVENT EFFECTS AND INTERMOLECULAR FORCES. Mark S. Gordon Iowa State University Ames Laboratory
 METHODS FOR TREATING SOLVENT EFFECTS AND INTERMOLECULAR FORCES Mark S. Gordon Iowa State University Ames Laboratory OUTLINE Solvation Methods Explicit vs. implicit methods Explicit Methods TIP3P, TIP4P
METHODS FOR TREATING SOLVENT EFFECTS AND INTERMOLECULAR FORCES Mark S. Gordon Iowa State University Ames Laboratory OUTLINE Solvation Methods Explicit vs. implicit methods Explicit Methods TIP3P, TIP4P
Exercise 1: Structure and dipole moment of a small molecule
 Introduction to computational chemistry Exercise 1: Structure and dipole moment of a small molecule Vesa Hänninen 1 Introduction In this exercise the equilibrium structure and the dipole moment of a small
Introduction to computational chemistry Exercise 1: Structure and dipole moment of a small molecule Vesa Hänninen 1 Introduction In this exercise the equilibrium structure and the dipole moment of a small
4 th Advanced in silico Drug Design KFC/ADD Molecular Modelling Intro. Karel Berka, Ph.D.
 4 th Advanced in silico Drug Design KFC/ADD Molecular Modelling Intro Karel Berka, Ph.D. UP Olomouc, 21.1.-25.1. 2019 Motto A theory is something nobody believes, except the person who made it An experiment
4 th Advanced in silico Drug Design KFC/ADD Molecular Modelling Intro Karel Berka, Ph.D. UP Olomouc, 21.1.-25.1. 2019 Motto A theory is something nobody believes, except the person who made it An experiment
Exploring the Sequence Dependent Structure and Dynamics of DNA with Molecular Dynamics Simulation
 Exploring the Sequence Dependent Structure and Dynamics of DNA with Molecular Dynamics Simulation Sarah Harris School of Physics and Astronomy University of Leeds Introduction Calculations of the charge
Exploring the Sequence Dependent Structure and Dynamics of DNA with Molecular Dynamics Simulation Sarah Harris School of Physics and Astronomy University of Leeds Introduction Calculations of the charge
A. MP2 - Inclusion of counterpoise in the optimisation step
 A. MP2 - Inclusion of counterpoise in the optimisation step Figure S1. Top and side views of the M_FS_SF_A and M_FS_SF_R IP-dimer structures computed at the MP2 level with (orange) and without (blue) counterpoise
A. MP2 - Inclusion of counterpoise in the optimisation step Figure S1. Top and side views of the M_FS_SF_A and M_FS_SF_R IP-dimer structures computed at the MP2 level with (orange) and without (blue) counterpoise