Novel Monte Carlo Methods for Protein Structure Modeling. Jinfeng Zhang Department of Statistics Harvard University
|
|
|
- Tracy Cook
- 5 years ago
- Views:
Transcription
1 Novel Monte Carlo Methods for Protein Structure Modeling Jinfeng Zhang Department of Statistics Harvard University




2 Introduction Machines of life Proteins play crucial roles in virtually all biological processes. Myosin Rhodopsin Hemoglobin Pepsin From Protein Data Bank (PDB) 2


3 Introduction From Sequence to Structure to Function The function of a protein is governed by its three-dimensional structure. Structure is determined by sequence. Genome Sequencing Projects Function 3
4 Structural Genomics The next step beyond the human genome project X-ray, NMR At least one structure in each protein family (~8000) Computational prediction More than 3,000,000 proteins 4
5 Outline Markov Chain Monte Carlo (CMC) method for protein structure prediction Sequential Monte Carlo (SMC) method for characterizing ensemble of protein conformations Summary & Discussions 5
 = e Z( T ) i E( x )/ k i B T")
6 Protein Folding p( x) E( x)/ kbt e = Z( T ) = e Z( T ) i E( x )/ k i B T 6

7 Simplified Protein Folding Model HP Model Protein sequence Hydrophobic (H) and polar (P) residues e.g. HHHPHHPHHHHPHHP. Protein conformation Self-avoiding chain on a 2D square or 3D cubic lattice. Energy E = - n, where n is number of hydrophobic contacts. The assumption The native conformation is the one with minimum energy. Finding the conformation with minimum energy NP complete problem. 7
8 A Sequence of 64 Residues HHHHHHHHHHHHPHPHPPHHPPHHPPHPPHHP PHHPPHPPHHPPHHPPHPHPHHHHHHHHHHHH E = -42 8
9 HP Model and Lattice Polymers Lattice models. A Brief Review Polymer science since 1950s HP model, 1985 by Dill. Simple with basic features of protein folding. Studying protein thermodynamics, folding principle, designability, protein evolution Studies on folding HP sequences HZ, CHCC, CI, GA, SA, GTS, EMC, GMC, GSA, PERM, SISPER, ACO, EES, npermis, npermh. Unsolved problem for sequences with medium length. 9
 = = π ( y) q( y, x) min 1, π ( x) q( x, y) min { [ E( x) E( y)]/ k T } B 1, e")
10 Exploring the Energy Landscape Metropolis-Hastings Algorithm a( x, Start with a conformation x. Draw y from q(x,y) ) = q(y x). Accept the new conformation with probability y) = = π ( y) q( y, x) min 1, π ( x) q( x, y) min { [ E( x) E( y)]/ k T } B 1, e 10
11 Moves on Lattice Pivot move Local moves End move Corner move Crankshaft move 11
12 Fragment Re-growth via Energyguided Sequential Sampling (FRESS) 1. Sample a l from L min to L max. For example, p(l) U[L min = 2, L max = 12], and l = Select and delete a random fragment of length l. 3. Sample the fragment sequentially. p j exp( E j / T ) 4. Accept or reject the newly sampled conformation using Metropolis criterion: p = min{1, e t+ ( E 1 E )/ T t } p = min{1, w w t t+ 1 e ( E t+ 1 Et )/ T } 5. Simulated Annealing. 12
13 FRESS Movie 13
14 2D Sequences Seq. code L Sequence 2D50 50 HHPHPHPHPHHHHPHPPPHPPPHPPPPHPPPHPPPHPHHHHPHPHPHPHH 2D D D D100a 100 2D100b 100 PPHHHPHHHHHHHHPPPHHHHHHHHHHPHPPPHHHHHHHHHHHHPPP PHHHHHHPHHPHP HHHHHHHHHHHHPHPHPPHHPPHHPPHPPHHPPHHPPHPPHHPPHHPPH PHPHHHHHHHHHHHH HHHHPPPPHHHHHHHHHHHHPPPPPPHHHHHHHHHHHHPPPHHHHHH HHHHHHPPPHHHHHHHHHHHHPPPHPPHHPPHHPPHPH PPPPPPHPHHPPPPPHHHPHHHHHPHHPPPPHHPPHHPHHHHHPHHHHH HHHHHPHHPHHHHHHHPPPPPPPPPPPHHHHHHHPPHPHHHPPPPPPHP HH PPPHHPPHHHHPPHHHPHHPHHPHHHHPPPPPPPPHHHHHHPPHHHHHH PPPPPPPPPHPHHPHHHHHHHHHHHPPHHHPHHPHPPHPHHHPPPPPPHH H 14
15 Comparison on Folding Long HP Sequences 2D seq. EMC SISPER GSA EES npermis FRESS 2D NA -21 NA -21 2D D64 2D85 2D100a 2D100b -39 NA NA NA
16 Minimum Energy Conformations of 2D64 16
17 Minimum Energy Conformations of 2D100 17
18 3D Sequences Seq. code L Sequence 3D D D D D D D PHPHHHPHHHPPHHPHPHHPHHHPHPHPHHPPHHHPPHPHPPPPHP PHPPHHPPHPPH PHHPHHPHHHPPHPHPPHPHPPHHHPHHPHHPPHHPHHPHHHPPH PHPPHPHPPHHHPHHPHHP PHPHHPHHPHPPHHHPPPHPHHPHHPHPPHHHPPPHPHHPHHPHPP HHHPPPHPHHPHHPHPPHHHP PHPHHPHHPHPPHHPPHPPHPPHPPHPPHPPHHPPHHHPPHHHPPH HHPPHHHPPHPHHPHHPHPPHPPHPPHHPPHPPHPPHHPPHP PPHHPPPPPHHPPHHPHPPHPPPPPPPHPPPHHPHHPPPPPPHPPHPH PPHPPPPPHHHPPPPHHPHHPPPPPHHPPPPHHHHPHPPPPPPPPHHH HHPPHPP PPPHHHPHPPPPHPPPPPHHPPPPHHPPHHPPPPHPPPPHPPHPPHHP PPHHPHPHHHPPPPHHHPPPPPPHHPPHPPHPHPPHPPPPPPPHPPHH HPPPPHPPPHHHHHPPPPHHPHPHPHPH HPPPPPHPPPPHPHHPHHPPPPHPHHHPPPPHPHPHHHHPPPPPPPPP PPHPPHPPPHPHHPPPHHPPHPPHPHPHPPPPPPPPHPPPHHHHHHPP PHHPPHHHPPPHHPHHHHHPPPPPPPPPHPPPPHPHPPPP 18
19 Comparison on 3D HP Sequences 3D seq. CI (1996) npermis (2003) npermh (2005) FRESS (2006) 3D (0.19*) -44 (1.10) -44 (0.09) 3D64 NA -56 (0.45) -56 (0.47) -56 (0.53) 3D67 NA -56 (1.10) -56 (0.33) -56 (1.41) 3D88 NA -69 (NA) -69 (0.45) -72 (5.03) 3D (3.12) -55 (0.25) -57 (4.47) 3D (12.3) -71 (1.19) -75 (280) 3D (110) NA -83 (350) * Times are in CPU hours. CPU: npermis 667MHz, npermh 1.84 GHz, FRESS 1.4 GHz. For 3D124, -74 was found in 4.8 hours, and for 3D136, -82 was found in 6.4 hours. 19
20 Minimum Energy Conformations Sequence 3D88: PHPHHPHHPHPPHHPPHPPHPPHPPHPPHPPHHPPHHHPPHHHP PHHHPPHHHPPHPHHPHHPHPPHPPHPPHHPPHPPHPPHHPPHP, E = -72, previous minimum energy is
21 Minimum Energy Conformations Sequence 3D103, E = -57, previous minimum energy is -55. Sequence 3D124:, E = -75, previous minimum energy is -71. c 21
22 Minimum Energy Conformations Sequence 3D136: HPPPPPHPPPPHPHHPHHPPPPHPHHHPPPPHPHPHHHHPPPP PPPPPPPHPPHPPPHPHHPPPHHPPHPPHPHPHPPPPPPPPHPPPHHHHHHPPPH HPPHHHPPPHHPHHHHHPPPPPPPPPHPPPPHPHPPPP, E = -83, previous minimum energy is
23 Characterizing Ensemble Protein Conformations by Sequential Monte Carlo Method 23
24 X-ray Structures 24


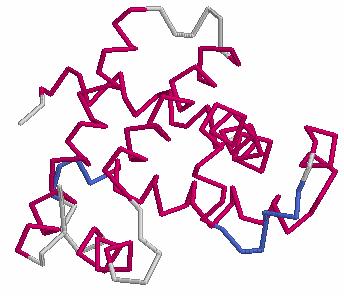

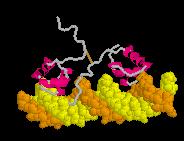
25 Proteins are Dynamic Molecules 25

26 YJ. Huang and GT. Montelione, Nature, 438, (2005),
27 N Furnham, T Blundell, M DePristo, Nature Structure & Molecular Biology, (2006) 13: A more suitable representation of a macro-molecular crystal structure would be an ensemble of models. The range of structures in the ensemble would be considered by any user of the structural information. 27
, PLoS Comp Biol, 2(12): e168. 28")
28 Characterizing Ensemble Conformations Backbone Ensemble of structures with different backbone conformations. J. Zhang et. al. (2007), Proteins, 66: Side-chain Ensemble of structures with the same backbone but different side-chain conformations. J. Zhang & JS Liu (2006), PLoS Comp Biol, 2(12): e

29 Ensemble of Side-chain Conformations 29
30 Ensemble of Side-chain Conformations Number of side chain conformations, N sc. Side chain conformational entropy. S sc = k B ln(n sc ) Protein stability. G = H-T S 30
31 Side-chain Modeling All heavy atoms are explicitly modeled. Side-chain flexibility Rotamer library by D. Richardson Excluded volume effect A pair of atoms i and j are considered to be a hard clash if r ij < a ( r0 ( i) + r0 ( j)) r ij : distance; r 0 (i) and r 0 (j) : van der Waals radii of the two atoms; a : scaling coefficient. 31
32 Sequential Monte Carlo (SMC) S n = (r 1,, r j,, r n ), r j R j = {1,, M j }. SMC: sample a side-chain (r) one at a time and fix the residues that are already sampled. S n Ω n h ( S n ) For each sample i, there is an associated weight, w (i). At step t, a residue, r t, is picked, and a rotamer, k, is sampled from a given distribution with probability p k. Update weight by w w / ( i) = t+1 h( S = 1 m n S Ω m n n i= 1 ) ( i) t p k w ( i) n h( S ( i) n ) 32
33 Performance S sc k B a 2ovo 3ebx Enumeration SMC Length Standard deviation b 2erl (40 aa, 48.1) 4rnt (104 aa, 109.3) 1fi2a (201 aa, 250.0) 1uzba (516 aa, 672.5) Sample size The total number of self-avoiding side-chain conformations for the fragment of 3ebx, residue 1-17, is 396,325,923, , SMC estimate is with a sample size of

34 Sequential Sampling of Side Chains 34
35 Effect of SCE on Protein Stability Native & Decoy Structures Decoy R Us database 35
36 Native & Decoys Structures 1ctf Native S sc k B Contact Number S sc can differ by more than 20 in k B unit, which corresponds to kcal/mol of free energy at 300K. The stability of a protein is around -5 to -20 kcal/mol. 36
37 Incorporating SCE in Energy Function G = H H : Residue contact potential. G = H - T S sc H : Residue contact potential. S sc : Side-chain entropy. T = 1. 37
38 ΔH vs. ΔH - ΔS sc Protein ID ΔH ΔH - ΔS sc Protein ID ΔH ΔH - ΔS sc 1ctf (A, 630)* 6 1 1beo (D, 2000) r69 (A, 675) ctf (D, 2000) sn3 (A, 660) dkt-A (D, 2000) cro (A, 674) fca (D, 2000) icb (A, 653) nkl (D, 2000) pti (A, 687) pgb (D, 1572) rxn (A, 677) b0n-B (E, 497) fc2 (B, 500) 7 5 1ctf (E, 497) hdd-C (B, 500) dtk (E, 215) 1 1 2cro (B, 500) fc2 (E, 500) icb (B, 500) 1 1 1igd (E, 500) bl0 (C, 971) shf-A (E, 437) 2 2 1eh2 (C, 2413) cro (E, 500) 1 1 1jwe (C, 1407) ovo (E, 347) 19 2 smd3 (C, 1200) pti (E, 343) 1 1 * A: 4state_reduced, B: fisa, C: fisa_casp3, D: lattice_ssfit, E: lmds. 38
39 Protein Interactions and SCE 39
40 Native & Decoy Structure of Protein Complexes 1spb 1brc S sc k B Native Contact Number S sc k B Native Contact Number 40



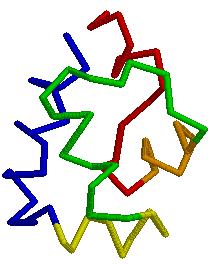
41 X-ray & NMR Structures Protein in crystal Protein in solution 41
42 SCE vs. R g of X-ray and NMR Structures 23 proteins with both X-ray and NMR structures Average S sc k B S sc = Ssc, X ray Ssc, NMR R g = Rg, X ray Rg, NMR Average R g 42
43 SCE in Protein Design 1bth 2ptc S sc S sc Contact Number Contact Number S sc = S sc,complex -S sc,protein_1 -S sc,protein_2 43
44 Summary Sampling method: a new MC method for protein structure prediction Global optimization and sampling. Simple and effective. Characterizing ensemble conformations SMC for estimating entropy and free energy. SCE is important for protein folding and structure modeling. 44
45 Acknowledgement Prof. Jun Liu Prof. Jie Liang Prof. Rong Chen Prof. Sam Kou Department of Statistics Harvard University Bioengineering Department University of Illinois at Chicago Department of Information and Decision Science University of Illinois at Chicago Department of Statistics Harvard University NIH, NSF for financial support! 45
46 Future Work Extend FRESS to real protein simulation. Apply FRESS to other optimization and sampling problems. Apply side-chain modeling to protein structure prediction, protein interaction, and protein design. SMC for other statistical and computational problems. 46
47 10 Benchmark Sequences of Length 48 s48a HPHHPPHHHHPHHHPPHHPPHPHHHPHPHHPPHHPPPHPPPPPPPPHH -32 s48b HHHHPHHPHHHHHPPHPPHHPPHPPPPPPHPPHPPPHPPHHPPHHHPH -34 s48c PHPHHPHHHHHHPPHPHPPHPHHPHPHPPPHPPHHPPHHPPHPHPPHP -34 s48d PHPHHPPHPHHHPPHHPHHPPPHHHHHPPHPHHPHPHPPPPHPPHPHP -33 s48e PPHPPPHPHHHHPPHHHHPHHPHHHPPHPHPHPPHPPPPPPHHPHHPH -32 s48f HHHPPPHHPHPHHPHHPHHPHPPPPPPPHPHPPHPPPHPPHHHHHHPH -32 s48g PHPPPPHPHHHPHPHHHHPHHPHHPPPHPHPPPHHHPPHHPPHHPPPH -32 s48h PHHPHHHPHHHHPPHHHPPPPPPHPHHPPHHPHPPPHHPHPHPHHPPP -31 s48i PHPHPPPPHPHPHPPHPHHHHHHPPHHHPHPPHPHHPPHPHHHPPPPH -34 s48j PHHPPPPPPHHPPPHHHPHPPHPHHPPHPPHPPHHPPHHHHHHHPPHH -33 Yue, K., et al. Proc. Natl. Acad. Sci. U.S.A. 92:325,
48 Comparison on Benchmark Sequences Method # Seq Avg. Time* FRESS LM npermh ACO CG MA * CPU time in minutes. CPUs: FRESS 1.4 GHz, npermh 1.84 GHz, ACO 2.4 GHz. 48
49 The Key Ingredients of FRESS Method # Seq Avg. Time Optimal NE L max = L = Optimal condition: starting temperature T h = 3.5, minimum temperature T l = 0.1, temperature decreases by 0.98 geometrically; L min = 2, L max = 12, with moves at each temperature. 49

50 The Strategy of FRESS 50
51 3D Sequences 3D seq. CI npermis npermh FRESS (2006) E* 3D (0.19*) -44 (1.10) -44 (0.09) -47 3D64 3D67 3D88 3D103 3D124 3D136 NA NA NA (0.45) -56 (1.10) -69 (NA) -54 (3.12) -71 (12.3) -80 (110) -56 (0.47) -56 (0.33) -69 (0.45) -55 (0.25) -71 (1.19) NA -56 (0.53) -56 (1.41) -72 (5.03) -57 (4.47) -75 (280) -83 (350)
52 Large Scale Study of SCE S sc k B a α = α = Length S sc,buried S sc b α = α = Length J Zhang, JS Liu (2006) PLoS Comp Biol, 2(12): e


53 Protein Interactions and SCE 53
Protein Structure Analysis with Sequential Monte Carlo Method. Jinfeng Zhang Computational Biology Lab Department of Statistics Harvard University
 Protein Structure Analysis with Sequential Monte Carlo Method Jinfeng Zhang Computational Biology Lab Department of Statistics Harvard University Introduction Structure Function & Interaction Protein structure
Protein Structure Analysis with Sequential Monte Carlo Method Jinfeng Zhang Computational Biology Lab Department of Statistics Harvard University Introduction Structure Function & Interaction Protein structure
Biopolymer structure simulation and optimization via fragment regrowth Monte Carlo
 THE JOURNAL OF CHEMICAL PHYSICS 126, 225101 2007 Biopolymer structure simulation and optimization via fragment regrowth Monte Carlo Jinfeng Zhang, S. C. Kou, and Jun S. Liu a Department of Statistics,
THE JOURNAL OF CHEMICAL PHYSICS 126, 225101 2007 Biopolymer structure simulation and optimization via fragment regrowth Monte Carlo Jinfeng Zhang, S. C. Kou, and Jun S. Liu a Department of Statistics,
Importance of chirality and reduced flexibility of protein side chains: A study with square and tetrahedral lattice models
 JOURNAL OF CHEMICAL PHYSICS VOLUME 121, NUMBER 1 1 JULY 2004 Importance of chirality and reduced flexibility of protein side chains: A study with square and tetrahedral lattice models Jinfeng Zhang Department
JOURNAL OF CHEMICAL PHYSICS VOLUME 121, NUMBER 1 1 JULY 2004 Importance of chirality and reduced flexibility of protein side chains: A study with square and tetrahedral lattice models Jinfeng Zhang Department
Supporting Online Material for
 www.sciencemag.org/cgi/content/full/309/5742/1868/dc1 Supporting Online Material for Toward High-Resolution de Novo Structure Prediction for Small Proteins Philip Bradley, Kira M. S. Misura, David Baker*
www.sciencemag.org/cgi/content/full/309/5742/1868/dc1 Supporting Online Material for Toward High-Resolution de Novo Structure Prediction for Small Proteins Philip Bradley, Kira M. S. Misura, David Baker*
3D HP Protein Folding Problem using Ant Algorithm
 3D HP Protein Folding Problem using Ant Algorithm Fidanova S. Institute of Parallel Processing BAS 25A Acad. G. Bonchev Str., 1113 Sofia, Bulgaria Phone: +359 2 979 66 42 E-mail: stefka@parallel.bas.bg
3D HP Protein Folding Problem using Ant Algorithm Fidanova S. Institute of Parallel Processing BAS 25A Acad. G. Bonchev Str., 1113 Sofia, Bulgaria Phone: +359 2 979 66 42 E-mail: stefka@parallel.bas.bg
Monte Carlo Simulations of Protein Folding using Lattice Models
 Monte Carlo Simulations of Protein Folding using Lattice Models Ryan Cheng 1,2 and Kenneth Jordan 1,3 1 Bioengineering and Bioinformatics Summer Institute, Department of Computational Biology, University
Monte Carlo Simulations of Protein Folding using Lattice Models Ryan Cheng 1,2 and Kenneth Jordan 1,3 1 Bioengineering and Bioinformatics Summer Institute, Department of Computational Biology, University
Template Free Protein Structure Modeling Jianlin Cheng, PhD
 Template Free Protein Structure Modeling Jianlin Cheng, PhD Associate Professor Computer Science Department Informatics Institute University of Missouri, Columbia 2013 Protein Energy Landscape & Free Sampling
Template Free Protein Structure Modeling Jianlin Cheng, PhD Associate Professor Computer Science Department Informatics Institute University of Missouri, Columbia 2013 Protein Energy Landscape & Free Sampling
Template Free Protein Structure Modeling Jianlin Cheng, PhD
 Template Free Protein Structure Modeling Jianlin Cheng, PhD Professor Department of EECS Informatics Institute University of Missouri, Columbia 2018 Protein Energy Landscape & Free Sampling http://pubs.acs.org/subscribe/archive/mdd/v03/i09/html/willis.html
Template Free Protein Structure Modeling Jianlin Cheng, PhD Professor Department of EECS Informatics Institute University of Missouri, Columbia 2018 Protein Energy Landscape & Free Sampling http://pubs.acs.org/subscribe/archive/mdd/v03/i09/html/willis.html
Outline. The ensemble folding kinetics of protein G from an all-atom Monte Carlo simulation. Unfolded Folded. What is protein folding?
 The ensemble folding kinetics of protein G from an all-atom Monte Carlo simulation By Jun Shimada and Eugine Shaknovich Bill Hawse Dr. Bahar Elisa Sandvik and Mehrdad Safavian Outline Background on protein
The ensemble folding kinetics of protein G from an all-atom Monte Carlo simulation By Jun Shimada and Eugine Shaknovich Bill Hawse Dr. Bahar Elisa Sandvik and Mehrdad Safavian Outline Background on protein
Absolute Entropy of a 2D Lattice Model for a Denatured Protein
 Absolute Entropy of a 2D Lattice Model for a Denatured Protein Jason Funt 1,4 and Hagai Meirovitch 2,3 1 Bioengineering and Bioinformatics Summer Institute 2 Center for Computational Biology and Bioinformatics,
Absolute Entropy of a 2D Lattice Model for a Denatured Protein Jason Funt 1,4 and Hagai Meirovitch 2,3 1 Bioengineering and Bioinformatics Summer Institute 2 Center for Computational Biology and Bioinformatics,
Monte Carlo Sampling of Near-Native Structures of Proteins With Applications
 66:61 68 (2007) Monte Carlo Sampling of Near-Native Structures of Proteins With Applications Jinfeng Zhang, 1 Ming Lin, 2 Rong Chen, 2,3,4 * Jie Liang, 3 * and Jun S. Liu 1 * 1 Department of Statistics,
66:61 68 (2007) Monte Carlo Sampling of Near-Native Structures of Proteins With Applications Jinfeng Zhang, 1 Ming Lin, 2 Rong Chen, 2,3,4 * Jie Liang, 3 * and Jun S. Liu 1 * 1 Department of Statistics,
Extraction of an Effective Pairwise Potential for Amino Acids
 Graduate Theses and Dissertations Graduate College 2011 Extraction of an Effective Pairwise Potential for Amino Acids Jie Luo Iowa State University Follow this and additional works at: http://lib.dr.iastate.edu/etd
Graduate Theses and Dissertations Graduate College 2011 Extraction of an Effective Pairwise Potential for Amino Acids Jie Luo Iowa State University Follow this and additional works at: http://lib.dr.iastate.edu/etd
Distance-Dependent, Pair Potential for Protein Folding: Results From Linear Optimization
 PROTEINS: Structure, Function, and Genetics 41:40 46 (2000) Distance-Dependent, Pair Potential for Protein Folding: Results From Linear Optimization Dror Tobi 1 and Ron Elber 1,2 * 1 Department of Biological
PROTEINS: Structure, Function, and Genetics 41:40 46 (2000) Distance-Dependent, Pair Potential for Protein Folding: Results From Linear Optimization Dror Tobi 1 and Ron Elber 1,2 * 1 Department of Biological
Clustering of low-energy conformations near the native structures of small proteins
 Proc. Natl. Acad. Sci. USA Vol. 95, pp. 11158 11162, September 1998 Biophysics Clustering of low-energy conformations near the native structures of small proteins DAVID SHORTLE*, KIM T. SIMONS, AND DAVID
Proc. Natl. Acad. Sci. USA Vol. 95, pp. 11158 11162, September 1998 Biophysics Clustering of low-energy conformations near the native structures of small proteins DAVID SHORTLE*, KIM T. SIMONS, AND DAVID
Protein Structure Prediction, Engineering & Design CHEM 430
 Protein Structure Prediction, Engineering & Design CHEM 430 Eero Saarinen The free energy surface of a protein Protein Structure Prediction & Design Full Protein Structure from Sequence - High Alignment
Protein Structure Prediction, Engineering & Design CHEM 430 Eero Saarinen The free energy surface of a protein Protein Structure Prediction & Design Full Protein Structure from Sequence - High Alignment
A new combination of replica exchange Monte Carlo and histogram analysis for protein folding and thermodynamics
 JOURNAL OF CHEMICAL PHYSICS VOLUME 115, NUMBER 3 15 JULY 2001 A new combination of replica exchange Monte Carlo and histogram analysis for protein folding and thermodynamics Dominik Gront Department of
JOURNAL OF CHEMICAL PHYSICS VOLUME 115, NUMBER 3 15 JULY 2001 A new combination of replica exchange Monte Carlo and histogram analysis for protein folding and thermodynamics Dominik Gront Department of
Computer Simulation of Peptide Adsorption
 Computer Simulation of Peptide Adsorption M P Allen Department of Physics University of Warwick Leipzig, 28 November 2013 1 Peptides Leipzig, 28 November 2013 Outline Lattice Peptide Monte Carlo 1 Lattice
Computer Simulation of Peptide Adsorption M P Allen Department of Physics University of Warwick Leipzig, 28 November 2013 1 Peptides Leipzig, 28 November 2013 Outline Lattice Peptide Monte Carlo 1 Lattice
Discrimination of Near-Native Protein Structures From Misfolded Models by Empirical Free Energy Functions
 PROTEINS: Structure, Function, and Genetics 41:518 534 (2000) Discrimination of Near-Native Protein Structures From Misfolded Models by Empirical Free Energy Functions David W. Gatchell, Sheldon Dennis,
PROTEINS: Structure, Function, and Genetics 41:518 534 (2000) Discrimination of Near-Native Protein Structures From Misfolded Models by Empirical Free Energy Functions David W. Gatchell, Sheldon Dennis,
Many proteins spontaneously refold into native form in vitro with high fidelity and high speed.
 Macromolecular Processes 20. Protein Folding Composed of 50 500 amino acids linked in 1D sequence by the polypeptide backbone The amino acid physical and chemical properties of the 20 amino acids dictate
Macromolecular Processes 20. Protein Folding Composed of 50 500 amino acids linked in 1D sequence by the polypeptide backbone The amino acid physical and chemical properties of the 20 amino acids dictate
Can a continuum solvent model reproduce the free energy landscape of a β-hairpin folding in water?
 Can a continuum solvent model reproduce the free energy landscape of a β-hairpin folding in water? Ruhong Zhou 1 and Bruce J. Berne 2 1 IBM Thomas J. Watson Research Center; and 2 Department of Chemistry,
Can a continuum solvent model reproduce the free energy landscape of a β-hairpin folding in water? Ruhong Zhou 1 and Bruce J. Berne 2 1 IBM Thomas J. Watson Research Center; and 2 Department of Chemistry,
Frontiers in Physics 27-29, Sept Self Avoiding Growth Walks and Protein Folding
 Frontiers in Physics 27-29, Sept. 2012 Self Avoiding Growth Walks and Protein Folding K P N Murthy, K Manasa, and K V K Srinath School of Physics, School of Life Sciences University of Hyderabad September
Frontiers in Physics 27-29, Sept. 2012 Self Avoiding Growth Walks and Protein Folding K P N Murthy, K Manasa, and K V K Srinath School of Physics, School of Life Sciences University of Hyderabad September
Lecture 18 Generalized Belief Propagation and Free Energy Approximations
 Lecture 18, Generalized Belief Propagation and Free Energy Approximations 1 Lecture 18 Generalized Belief Propagation and Free Energy Approximations In this lecture we talked about graphical models and
Lecture 18, Generalized Belief Propagation and Free Energy Approximations 1 Lecture 18 Generalized Belief Propagation and Free Energy Approximations In this lecture we talked about graphical models and
arxiv:cond-mat/ v1 [cond-mat.soft] 16 Nov 2002
![arxiv:cond-mat/ v1 [cond-mat.soft] 16 Nov 2002](/thumbs/91/105680653.jpg "arxiv:cond-mat/ v1 [cond-mat.soft] 16 Nov 2002") Dependence of folding rates on protein length Mai Suan Li 1, D. K. Klimov 2 and D. Thirumalai 2 1 Institute of Physics, Polish Academy of Sciences, Al. Lotnikow 32/46, 02-668 Warsaw, Poland 2 Institute
Dependence of folding rates on protein length Mai Suan Li 1, D. K. Klimov 2 and D. Thirumalai 2 1 Institute of Physics, Polish Academy of Sciences, Al. Lotnikow 32/46, 02-668 Warsaw, Poland 2 Institute
Proteins are not rigid structures: Protein dynamics, conformational variability, and thermodynamic stability
 Proteins are not rigid structures: Protein dynamics, conformational variability, and thermodynamic stability Dr. Andrew Lee UNC School of Pharmacy (Div. Chemical Biology and Medicinal Chemistry) UNC Med
Proteins are not rigid structures: Protein dynamics, conformational variability, and thermodynamic stability Dr. Andrew Lee UNC School of Pharmacy (Div. Chemical Biology and Medicinal Chemistry) UNC Med
Polypeptide Folding Using Monte Carlo Sampling, Concerted Rotation, and Continuum Solvation
 Polypeptide Folding Using Monte Carlo Sampling, Concerted Rotation, and Continuum Solvation Jakob P. Ulmschneider and William L. Jorgensen J.A.C.S. 2004, 126, 1849-1857 Presented by Laura L. Thomas and
Polypeptide Folding Using Monte Carlo Sampling, Concerted Rotation, and Continuum Solvation Jakob P. Ulmschneider and William L. Jorgensen J.A.C.S. 2004, 126, 1849-1857 Presented by Laura L. Thomas and
Algorithm for protein folding problem in 3D lattice HP model
 Algorithm for protein folding problem in 3D lattice HP model METODI TRAYKOV Department of Electrical Engineering, Electronics and Automatics University Center for Advanced Bioinformatics Research South-West
Algorithm for protein folding problem in 3D lattice HP model METODI TRAYKOV Department of Electrical Engineering, Electronics and Automatics University Center for Advanced Bioinformatics Research South-West
April, The energy functions include:
 REDUX A collection of Python scripts for torsion angle Monte Carlo protein molecular simulations and analysis The program is based on unified residue peptide model and is designed for more efficient exploration
REDUX A collection of Python scripts for torsion angle Monte Carlo protein molecular simulations and analysis The program is based on unified residue peptide model and is designed for more efficient exploration
Design of a Novel Globular Protein Fold with Atomic-Level Accuracy
 Design of a Novel Globular Protein Fold with Atomic-Level Accuracy Brian Kuhlman, Gautam Dantas, Gregory C. Ireton, Gabriele Varani, Barry L. Stoddard, David Baker Presented by Kate Stafford 4 May 05 Protein
Design of a Novel Globular Protein Fold with Atomic-Level Accuracy Brian Kuhlman, Gautam Dantas, Gregory C. Ireton, Gabriele Varani, Barry L. Stoddard, David Baker Presented by Kate Stafford 4 May 05 Protein
Monte Carlo (MC) Simulation Methods. Elisa Fadda
 Simulation Methods. Elisa Fadda") Monte Carlo (MC) Simulation Methods Elisa Fadda 1011-CH328, Molecular Modelling & Drug Design 2011 Experimental Observables A system observable is a property of the system state. The system state i is
Monte Carlo (MC) Simulation Methods Elisa Fadda 1011-CH328, Molecular Modelling & Drug Design 2011 Experimental Observables A system observable is a property of the system state. The system state i is
Statistical geometry of packing defects of lattice chain polymer from enumeration and sequential Monte Carlo method
 JOURNAL OF CHEMICAL PHYSICS VOLUME 117, NUMBER 7 15 AUGUST 2002 Statistical geometry of packing defects of lattice chain polymer from enumeration and sequential Monte Carlo method Jie Liang a) and Jinfeng
JOURNAL OF CHEMICAL PHYSICS VOLUME 117, NUMBER 7 15 AUGUST 2002 Statistical geometry of packing defects of lattice chain polymer from enumeration and sequential Monte Carlo method Jie Liang a) and Jinfeng
Molecular Modeling. Prediction of Protein 3D Structure from Sequence. Vimalkumar Velayudhan. May 21, 2007
 Molecular Modeling Prediction of Protein 3D Structure from Sequence Vimalkumar Velayudhan Jain Institute of Vocational and Advanced Studies May 21, 2007 Vimalkumar Velayudhan Molecular Modeling 1/23 Outline
Molecular Modeling Prediction of Protein 3D Structure from Sequence Vimalkumar Velayudhan Jain Institute of Vocational and Advanced Studies May 21, 2007 Vimalkumar Velayudhan Molecular Modeling 1/23 Outline
Protein Structure Prediction
 Protein Structure Prediction Michael Feig MMTSB/CTBP 2006 Summer Workshop From Sequence to Structure SEALGDTIVKNA Ab initio Structure Prediction Protocol Amino Acid Sequence Conformational Sampling to
Protein Structure Prediction Michael Feig MMTSB/CTBP 2006 Summer Workshop From Sequence to Structure SEALGDTIVKNA Ab initio Structure Prediction Protocol Amino Acid Sequence Conformational Sampling to
Lecture 34 Protein Unfolding Thermodynamics
 Physical Principles in Biology Biology 3550 Fall 2018 Lecture 34 Protein Unfolding Thermodynamics Wednesday, 21 November c David P. Goldenberg University of Utah goldenberg@biology.utah.edu Clicker Question
Physical Principles in Biology Biology 3550 Fall 2018 Lecture 34 Protein Unfolding Thermodynamics Wednesday, 21 November c David P. Goldenberg University of Utah goldenberg@biology.utah.edu Clicker Question
A Quasi-physical Algorithm for the Structure Optimization Off-lattice Protein Model
 Method A Quasi-physical Algorithm for the Structure Optimization Off-lattice Protein Model Jing-Fa Liul>'* and Wen-Qi Huang' in an School of Computer Science and Technology, Huazhong University of Science
Method A Quasi-physical Algorithm for the Structure Optimization Off-lattice Protein Model Jing-Fa Liul>'* and Wen-Qi Huang' in an School of Computer Science and Technology, Huazhong University of Science
Protein Folding Prof. Eugene Shakhnovich
 Protein Folding Eugene Shakhnovich Department of Chemistry and Chemical Biology Harvard University 1 Proteins are folded on various scales As of now we know hundreds of thousands of sequences (Swissprot)
Protein Folding Eugene Shakhnovich Department of Chemistry and Chemical Biology Harvard University 1 Proteins are folded on various scales As of now we know hundreds of thousands of sequences (Swissprot)
Docking. GBCB 5874: Problem Solving in GBCB
 Docking Benzamidine Docking to Trypsin Relationship to Drug Design Ligand-based design QSAR Pharmacophore modeling Can be done without 3-D structure of protein Receptor/Structure-based design Molecular
Docking Benzamidine Docking to Trypsin Relationship to Drug Design Ligand-based design QSAR Pharmacophore modeling Can be done without 3-D structure of protein Receptor/Structure-based design Molecular
Alpha-helical Topology and Tertiary Structure Prediction of Globular Proteins Scott R. McAllister Christodoulos A. Floudas Princeton University
 Alpha-helical Topology and Tertiary Structure Prediction of Globular Proteins Scott R. McAllister Christodoulos A. Floudas Princeton University Department of Chemical Engineering Program of Applied and
Alpha-helical Topology and Tertiary Structure Prediction of Globular Proteins Scott R. McAllister Christodoulos A. Floudas Princeton University Department of Chemical Engineering Program of Applied and
Timothy Chan. Bojana Jankovic. Viet Le. Igor Naverniouk. March 15, 2004
 Comparative Study of Hydrophobic-Polar and Miyazawa-Jernigan Energy Functions in Protein Folding on a Cubic Lattice Using Pruned-Enriched Rosenbluth Monte Carlo Algorithm Timothy Chan University of British
Comparative Study of Hydrophobic-Polar and Miyazawa-Jernigan Energy Functions in Protein Folding on a Cubic Lattice Using Pruned-Enriched Rosenbluth Monte Carlo Algorithm Timothy Chan University of British
Long Range Moves for High Density Polymer Simulations
 arxiv:cond-mat/9610116v1 [cond-mat.soft] 15 Oct 1996 Long Range Moves for High Density Polymer Simulations J.M.Deutsch University of California, Santa Cruz, U.S.A. Abstract Monte Carlo simulations of proteins
arxiv:cond-mat/9610116v1 [cond-mat.soft] 15 Oct 1996 Long Range Moves for High Density Polymer Simulations J.M.Deutsch University of California, Santa Cruz, U.S.A. Abstract Monte Carlo simulations of proteins
ICCP Project 2 - Advanced Monte Carlo Methods Choose one of the three options below
 ICCP Project 2 - Advanced Monte Carlo Methods Choose one of the three options below Introduction In statistical physics Monte Carlo methods are considered to have started in the Manhattan project (1940
ICCP Project 2 - Advanced Monte Carlo Methods Choose one of the three options below Introduction In statistical physics Monte Carlo methods are considered to have started in the Manhattan project (1940
CMPS 6630: Introduction to Computational Biology and Bioinformatics. Tertiary Structure Prediction
 CMPS 6630: Introduction to Computational Biology and Bioinformatics Tertiary Structure Prediction Tertiary Structure Prediction Why Should Tertiary Structure Prediction Be Possible? Molecules obey the
CMPS 6630: Introduction to Computational Biology and Bioinformatics Tertiary Structure Prediction Tertiary Structure Prediction Why Should Tertiary Structure Prediction Be Possible? Molecules obey the
Crystal Structure Prediction using CRYSTALG program
 Crystal Structure Prediction using CRYSTALG program Yelena Arnautova Baker Laboratory of Chemistry and Chemical Biology, Cornell University Problem of crystal structure prediction: - theoretical importance
Crystal Structure Prediction using CRYSTALG program Yelena Arnautova Baker Laboratory of Chemistry and Chemical Biology, Cornell University Problem of crystal structure prediction: - theoretical importance
Effect of Sequences on the Shape of Protein Energy Landscapes Yue Li Department of Computer Science Florida State University Tallahassee, FL 32306
 Effect of Sequences on the Shape of Protein Energy Landscapes Yue Li Department of Computer Science Florida State University Tallahassee, FL 32306 yli@cs.fsu.edu Gary Tyson Department of Computer Science
Effect of Sequences on the Shape of Protein Energy Landscapes Yue Li Department of Computer Science Florida State University Tallahassee, FL 32306 yli@cs.fsu.edu Gary Tyson Department of Computer Science
F. Piazza Center for Molecular Biophysics and University of Orléans, France. Selected topic in Physical Biology. Lecture 1
 Zhou Pei-Yuan Centre for Applied Mathematics, Tsinghua University November 2013 F. Piazza Center for Molecular Biophysics and University of Orléans, France Selected topic in Physical Biology Lecture 1
Zhou Pei-Yuan Centre for Applied Mathematics, Tsinghua University November 2013 F. Piazza Center for Molecular Biophysics and University of Orléans, France Selected topic in Physical Biology Lecture 1
Structural Bioinformatics (C3210) Molecular Docking
 Molecular Docking") Structural Bioinformatics (C3210) Molecular Docking Molecular Recognition, Molecular Docking Molecular recognition is the ability of biomolecules to recognize other biomolecules and selectively interact
Structural Bioinformatics (C3210) Molecular Docking Molecular Recognition, Molecular Docking Molecular recognition is the ability of biomolecules to recognize other biomolecules and selectively interact
Homework 9: Protein Folding & Simulated Annealing : Programming for Scientists Due: Thursday, April 14, 2016 at 11:59 PM
 Homework 9: Protein Folding & Simulated Annealing 02-201: Programming for Scientists Due: Thursday, April 14, 2016 at 11:59 PM 1. Set up We re back to Go for this assignment. 1. Inside of your src directory,
Homework 9: Protein Folding & Simulated Annealing 02-201: Programming for Scientists Due: Thursday, April 14, 2016 at 11:59 PM 1. Set up We re back to Go for this assignment. 1. Inside of your src directory,
Protein structure prediction. CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror
 Protein structure prediction CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror 1 Outline Why predict protein structure? Can we use (pure) physics-based methods? Knowledge-based methods Two major
Protein structure prediction CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror 1 Outline Why predict protein structure? Can we use (pure) physics-based methods? Knowledge-based methods Two major
Kd = koff/kon = [R][L]/[RL]
![Kd = koff/kon = [R][L]/[RL]](/thumbs/96/127564193.jpg "Kd = koff/kon = [R][L]/[RL]") Taller de docking y cribado virtual: Uso de herramientas computacionales en el diseño de fármacos Docking program GLIDE El programa de docking GLIDE Sonsoles Martín-Santamaría Shrödinger is a scientific
Taller de docking y cribado virtual: Uso de herramientas computacionales en el diseño de fármacos Docking program GLIDE El programa de docking GLIDE Sonsoles Martín-Santamaría Shrödinger is a scientific
Protein structure forces, and folding
 Harvard-MIT Division of Health Sciences and Technology HST.508: Quantitative Genomics, Fall 2005 Instructors: Leonid Mirny, Robert Berwick, Alvin Kho, Isaac Kohane Protein structure forces, and folding
Harvard-MIT Division of Health Sciences and Technology HST.508: Quantitative Genomics, Fall 2005 Instructors: Leonid Mirny, Robert Berwick, Alvin Kho, Isaac Kohane Protein structure forces, and folding
Molecular Interactions F14NMI. Lecture 4: worked answers to practice questions
 Molecular Interactions F14NMI Lecture 4: worked answers to practice questions http://comp.chem.nottingham.ac.uk/teaching/f14nmi jonathan.hirst@nottingham.ac.uk (1) (a) Describe the Monte Carlo algorithm
Molecular Interactions F14NMI Lecture 4: worked answers to practice questions http://comp.chem.nottingham.ac.uk/teaching/f14nmi jonathan.hirst@nottingham.ac.uk (1) (a) Describe the Monte Carlo algorithm
The sequences of naturally occurring proteins are defined by
 Protein topology and stability define the space of allowed sequences Patrice Koehl* and Michael Levitt Department of Structural Biology, Fairchild Building, D109, Stanford University, Stanford, CA 94305
Protein topology and stability define the space of allowed sequences Patrice Koehl* and Michael Levitt Department of Structural Biology, Fairchild Building, D109, Stanford University, Stanford, CA 94305
Protein Structures. 11/19/2002 Lecture 24 1
 Protein Structures 11/19/2002 Lecture 24 1 All 3 figures are cartoons of an amino acid residue. 11/19/2002 Lecture 24 2 Peptide bonds in chains of residues 11/19/2002 Lecture 24 3 Angles φ and ψ in the
Protein Structures 11/19/2002 Lecture 24 1 All 3 figures are cartoons of an amino acid residue. 11/19/2002 Lecture 24 2 Peptide bonds in chains of residues 11/19/2002 Lecture 24 3 Angles φ and ψ in the
3.320 Lecture 18 (4/12/05)
") 3.320 Lecture 18 (4/12/05) Monte Carlo Simulation II and free energies Figure by MIT OCW. General Statistical Mechanics References D. Chandler, Introduction to Modern Statistical Mechanics D.A. McQuarrie,
3.320 Lecture 18 (4/12/05) Monte Carlo Simulation II and free energies Figure by MIT OCW. General Statistical Mechanics References D. Chandler, Introduction to Modern Statistical Mechanics D.A. McQuarrie,
Context of the project...3. What is protein design?...3. I The algorithms...3 A Dead-end elimination procedure...4. B Monte-Carlo simulation...
 Laidebeure Stéphane Context of the project...3 What is protein design?...3 I The algorithms...3 A Dead-end elimination procedure...4 B Monte-Carlo simulation...5 II The model...6 A The molecular model...6
Laidebeure Stéphane Context of the project...3 What is protein design?...3 I The algorithms...3 A Dead-end elimination procedure...4 B Monte-Carlo simulation...5 II The model...6 A The molecular model...6
Why Proteins Fold. How Proteins Fold? e - ΔG/kT. Protein Folding, Nonbonding Forces, and Free Energy
 Why Proteins Fold Proteins are the action superheroes of the body. As enzymes, they make reactions go a million times faster. As versatile transport vehicles, they carry oxygen and antibodies to fight
Why Proteins Fold Proteins are the action superheroes of the body. As enzymes, they make reactions go a million times faster. As versatile transport vehicles, they carry oxygen and antibodies to fight
CMPS 3110: Bioinformatics. Tertiary Structure Prediction
 CMPS 3110: Bioinformatics Tertiary Structure Prediction Tertiary Structure Prediction Why Should Tertiary Structure Prediction Be Possible? Molecules obey the laws of physics! Conformation space is finite
CMPS 3110: Bioinformatics Tertiary Structure Prediction Tertiary Structure Prediction Why Should Tertiary Structure Prediction Be Possible? Molecules obey the laws of physics! Conformation space is finite
DETECTING NATIVE PROTEIN FOLDS AMONG LARGE DECOY SETS WITH THE OPLS ALL-ATOM POTENTIAL AND THE SURFACE GENERALIZED BORN SOLVENT MODEL
 Computational Methods for Protein Folding: Advances in Chemical Physics, Volume 12. Edited by Richard A. Friesner. Series Editors: I. Prigogine and Stuart A. Rice. Copyright # 22 John Wiley & Sons, Inc.
Computational Methods for Protein Folding: Advances in Chemical Physics, Volume 12. Edited by Richard A. Friesner. Series Editors: I. Prigogine and Stuart A. Rice. Copyright # 22 John Wiley & Sons, Inc.
Free energy, electrostatics, and the hydrophobic effect
 Protein Physics 2016 Lecture 3, January 26 Free energy, electrostatics, and the hydrophobic effect Magnus Andersson magnus.andersson@scilifelab.se Theoretical & Computational Biophysics Recap Protein structure
Protein Physics 2016 Lecture 3, January 26 Free energy, electrostatics, and the hydrophobic effect Magnus Andersson magnus.andersson@scilifelab.se Theoretical & Computational Biophysics Recap Protein structure
Other Cells. Hormones. Viruses. Toxins. Cell. Bacteria
 Other Cells Hormones Viruses Toxins Cell Bacteria ΔH < 0 reaction is exothermic, tells us nothing about the spontaneity of the reaction Δ H > 0 reaction is endothermic, tells us nothing about the spontaneity
Other Cells Hormones Viruses Toxins Cell Bacteria ΔH < 0 reaction is exothermic, tells us nothing about the spontaneity of the reaction Δ H > 0 reaction is endothermic, tells us nothing about the spontaneity
Generating folded protein structures with a lattice chain growth algorithm
 JOURAL OF CHEMICAL PHYSICS VOLUME 113, UMBER 13 1 OCTOBER 2000 Generating folded protein structures with a lattice chain growth algorithm Hin Hark Gan a) Department of Chemistry and Courant Institute of
JOURAL OF CHEMICAL PHYSICS VOLUME 113, UMBER 13 1 OCTOBER 2000 Generating folded protein structures with a lattice chain growth algorithm Hin Hark Gan a) Department of Chemistry and Courant Institute of
MCB100A/Chem130 MidTerm Exam 2 April 4, 2013
 MCBA/Chem Miderm Exam 2 April 4, 2 Name Student ID rue/false (2 points each).. he Boltzmann constant, k b sets the energy scale for observing energy microstates 2. Atoms with favorable electronic configurations
MCBA/Chem Miderm Exam 2 April 4, 2 Name Student ID rue/false (2 points each).. he Boltzmann constant, k b sets the energy scale for observing energy microstates 2. Atoms with favorable electronic configurations
Presenter: She Zhang
 Presenter: She Zhang Introduction Dr. David Baker Introduction Why design proteins de novo? It is not clear how non-covalent interactions favor one specific native structure over many other non-native
Presenter: She Zhang Introduction Dr. David Baker Introduction Why design proteins de novo? It is not clear how non-covalent interactions favor one specific native structure over many other non-native
arxiv:physics/ v1 [physics.bio-ph] 7 Mar 2002
![arxiv:physics/ v1 [physics.bio-ph] 7 Mar 2002](/thumbs/74/69872546.jpg "arxiv:physics/ v1 [physics.bio-ph] 7 Mar 2002") Statistical Geometry of Packing Defects of Lattice Chain Polymer from Enumeration and Sequential Monte Carlo Method arxiv:physics/0203015v1 [physics.bio-ph] 7 Mar 2002 Jie Liang 1, Jinfeng Zhang 1, Rong
Statistical Geometry of Packing Defects of Lattice Chain Polymer from Enumeration and Sequential Monte Carlo Method arxiv:physics/0203015v1 [physics.bio-ph] 7 Mar 2002 Jie Liang 1, Jinfeng Zhang 1, Rong
There are self-avoiding walks of steps on Z 3
 There are 7 10 26 018 276 self-avoiding walks of 38 797 311 steps on Z 3 Nathan Clisby MASCOS, The University of Melbourne Institut für Theoretische Physik Universität Leipzig November 9, 2012 1 / 37 Outline
There are 7 10 26 018 276 self-avoiding walks of 38 797 311 steps on Z 3 Nathan Clisby MASCOS, The University of Melbourne Institut für Theoretische Physik Universität Leipzig November 9, 2012 1 / 37 Outline
Local Interactions Dominate Folding in a Simple Protein Model
 J. Mol. Biol. (1996) 259, 988 994 Local Interactions Dominate Folding in a Simple Protein Model Ron Unger 1,2 * and John Moult 2 1 Department of Life Sciences Bar-Ilan University Ramat-Gan, 52900, Israel
J. Mol. Biol. (1996) 259, 988 994 Local Interactions Dominate Folding in a Simple Protein Model Ron Unger 1,2 * and John Moult 2 1 Department of Life Sciences Bar-Ilan University Ramat-Gan, 52900, Israel
Biology Chemistry & Physics of Biomolecules. Examination #1. Proteins Module. September 29, Answer Key
 Biology 5357 Chemistry & Physics of Biomolecules Examination #1 Proteins Module September 29, 2017 Answer Key Question 1 (A) (5 points) Structure (b) is more common, as it contains the shorter connection
Biology 5357 Chemistry & Physics of Biomolecules Examination #1 Proteins Module September 29, 2017 Answer Key Question 1 (A) (5 points) Structure (b) is more common, as it contains the shorter connection
Protein Docking by Exploiting Multi-dimensional Energy Funnels
 Protein Docking by Exploiting Multi-dimensional Energy Funnels Ioannis Ch. Paschalidis 1 Yang Shen 1 Pirooz Vakili 1 Sandor Vajda 2 1 Center for Information and Systems Engineering and Department of Manufacturing
Protein Docking by Exploiting Multi-dimensional Energy Funnels Ioannis Ch. Paschalidis 1 Yang Shen 1 Pirooz Vakili 1 Sandor Vajda 2 1 Center for Information and Systems Engineering and Department of Manufacturing
Scaling Law for the Radius of Gyration of Proteins and Its Dependence on Hydrophobicity
 Scaling Law for the Radius of Gyration of Proteins and Its Dependence on Hydrophobicity LIU HONG, JINZHI LEI Zhou Pei-Yuan Center for Applied Mathematics, Tsinghua University, Beijing, People s Republic
Scaling Law for the Radius of Gyration of Proteins and Its Dependence on Hydrophobicity LIU HONG, JINZHI LEI Zhou Pei-Yuan Center for Applied Mathematics, Tsinghua University, Beijing, People s Republic
EE512 Graphical Models Fall 2009
 EE512 Graphical Models Fall 2009 Prof. Jeff Bilmes University of Washington, Seattle Department of Electrical Engineering Fall Quarter, 2009 http://ssli.ee.washington.edu/~bilmes/ee512fa09 Lecture 3 -
EE512 Graphical Models Fall 2009 Prof. Jeff Bilmes University of Washington, Seattle Department of Electrical Engineering Fall Quarter, 2009 http://ssli.ee.washington.edu/~bilmes/ee512fa09 Lecture 3 -
Computer simulation of polypeptides in a confinement
 J Mol Model (27) 13:327 333 DOI 1.17/s894-6-147-6 ORIGINAL PAPER Computer simulation of polypeptides in a confinement Andrzej Sikorski & Piotr Romiszowski Received: 3 November 25 / Accepted: 27 June 26
J Mol Model (27) 13:327 333 DOI 1.17/s894-6-147-6 ORIGINAL PAPER Computer simulation of polypeptides in a confinement Andrzej Sikorski & Piotr Romiszowski Received: 3 November 25 / Accepted: 27 June 26
Folding of small proteins using a single continuous potential
 JOURNAL OF CHEMICAL PHYSICS VOLUME 120, NUMBER 17 1 MAY 2004 Folding of small proteins using a single continuous potential Seung-Yeon Kim School of Computational Sciences, Korea Institute for Advanced
JOURNAL OF CHEMICAL PHYSICS VOLUME 120, NUMBER 17 1 MAY 2004 Folding of small proteins using a single continuous potential Seung-Yeon Kim School of Computational Sciences, Korea Institute for Advanced
Modular Bayesian uncertainty assessment for Structural Health Monitoring
 uncertainty assessment for Structural Health Monitoring Warwick Centre for Predictive Modelling André Jesus a.jesus@warwick.ac.uk June 26, 2017 Thesis advisor: Irwanda Laory & Peter Brommer Structural
uncertainty assessment for Structural Health Monitoring Warwick Centre for Predictive Modelling André Jesus a.jesus@warwick.ac.uk June 26, 2017 Thesis advisor: Irwanda Laory & Peter Brommer Structural
Ж У Р Н А Л С Т Р У К Т У Р Н О Й Х И М И И Том 50, 5 Сентябрь октябрь С
 Ж У Р Н А Л С Т Р У К Т У Р Н О Й Х И М И И 2009. Том 50, 5 Сентябрь октябрь С. 873 877 UDK 539.27 STRUCTURAL STUDIES OF L-SERYL-L-HISTIDINE DIPEPTIDE BY MEANS OF MOLECULAR MODELING, DFT AND 1 H NMR SPECTROSCOPY
Ж У Р Н А Л С Т Р У К Т У Р Н О Й Х И М И И 2009. Том 50, 5 Сентябрь октябрь С. 873 877 UDK 539.27 STRUCTURAL STUDIES OF L-SERYL-L-HISTIDINE DIPEPTIDE BY MEANS OF MOLECULAR MODELING, DFT AND 1 H NMR SPECTROSCOPY
A rule of seven in Watson-Crick base-pairing of mismatched sequences
 A rule of seven in Watson-Crick base-pairing of mismatched sequences Ibrahim I. Cisse 1,3, Hajin Kim 1,2, Taekjip Ha 1,2 1 Department of Physics and Center for the Physics of Living Cells, University of
A rule of seven in Watson-Crick base-pairing of mismatched sequences Ibrahim I. Cisse 1,3, Hajin Kim 1,2, Taekjip Ha 1,2 1 Department of Physics and Center for the Physics of Living Cells, University of
Protein structure prediction. CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror
 Protein structure prediction CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror 1 Outline Why predict protein structure? Can we use (pure) physics-based methods? Knowledge-based methods Two major
Protein structure prediction CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror 1 Outline Why predict protein structure? Can we use (pure) physics-based methods? Knowledge-based methods Two major
Programme Last week s quiz results + Summary Fold recognition Break Exercise: Modelling remote homologues
 Programme 8.00-8.20 Last week s quiz results + Summary 8.20-9.00 Fold recognition 9.00-9.15 Break 9.15-11.20 Exercise: Modelling remote homologues 11.20-11.40 Summary & discussion 11.40-12.00 Quiz 1 Feedback
Programme 8.00-8.20 Last week s quiz results + Summary 8.20-9.00 Fold recognition 9.00-9.15 Break 9.15-11.20 Exercise: Modelling remote homologues 11.20-11.40 Summary & discussion 11.40-12.00 Quiz 1 Feedback
Protein Structure Prediction II Lecturer: Serafim Batzoglou Scribe: Samy Hamdouche
 Protein Structure Prediction II Lecturer: Serafim Batzoglou Scribe: Samy Hamdouche The molecular structure of a protein can be broken down hierarchically. The primary structure of a protein is simply its
Protein Structure Prediction II Lecturer: Serafim Batzoglou Scribe: Samy Hamdouche The molecular structure of a protein can be broken down hierarchically. The primary structure of a protein is simply its
Introduction to Comparative Protein Modeling. Chapter 4 Part I
 Introduction to Comparative Protein Modeling Chapter 4 Part I 1 Information on Proteins Each modeling study depends on the quality of the known experimental data. Basis of the model Search in the literature
Introduction to Comparative Protein Modeling Chapter 4 Part I 1 Information on Proteins Each modeling study depends on the quality of the known experimental data. Basis of the model Search in the literature
Protein Dynamics. The space-filling structures of myoglobin and hemoglobin show that there are no pathways for O 2 to reach the heme iron.
 Protein Dynamics The space-filling structures of myoglobin and hemoglobin show that there are no pathways for O 2 to reach the heme iron. Below is myoglobin hydrated with 350 water molecules. Only a small
Protein Dynamics The space-filling structures of myoglobin and hemoglobin show that there are no pathways for O 2 to reach the heme iron. Below is myoglobin hydrated with 350 water molecules. Only a small
Application of the Markov State Model to Molecular Dynamics of Biological Molecules. Speaker: Xun Sang-Ni Supervisor: Prof. Wu Dr.
 Application of the Markov State Model to Molecular Dynamics of Biological Molecules Speaker: Xun Sang-Ni Supervisor: Prof. Wu Dr. Jiang Introduction Conformational changes of proteins : essential part
Application of the Markov State Model to Molecular Dynamics of Biological Molecules Speaker: Xun Sang-Ni Supervisor: Prof. Wu Dr. Jiang Introduction Conformational changes of proteins : essential part
Molecular dynamics simulation. CS/CME/BioE/Biophys/BMI 279 Oct. 5 and 10, 2017 Ron Dror
 Molecular dynamics simulation CS/CME/BioE/Biophys/BMI 279 Oct. 5 and 10, 2017 Ron Dror 1 Outline Molecular dynamics (MD): The basic idea Equations of motion Key properties of MD simulations Sample applications
Molecular dynamics simulation CS/CME/BioE/Biophys/BMI 279 Oct. 5 and 10, 2017 Ron Dror 1 Outline Molecular dynamics (MD): The basic idea Equations of motion Key properties of MD simulations Sample applications
Monte Carlo simulations of polyalanine using a reduced model and statistics-based interaction potentials
 THE JOURNAL OF CHEMICAL PHYSICS 122, 024904 2005 Monte Carlo simulations of polyalanine using a reduced model and statistics-based interaction potentials Alan E. van Giessen and John E. Straub Department
THE JOURNAL OF CHEMICAL PHYSICS 122, 024904 2005 Monte Carlo simulations of polyalanine using a reduced model and statistics-based interaction potentials Alan E. van Giessen and John E. Straub Department
October 2016 v1 12/10/2015 Page 1 of 10
 State Section S s Effective October 1, 2016 Overview The tables list the Section S items that will be active on records with a target date on or after October 1, 2016. The active on each item subset code
State Section S s Effective October 1, 2016 Overview The tables list the Section S items that will be active on records with a target date on or after October 1, 2016. The active on each item subset code
THE TANGO ALGORITHM: SECONDARY STRUCTURE PROPENSITIES, STATISTICAL MECHANICS APPROXIMATION
 THE TANGO ALGORITHM: SECONDARY STRUCTURE PROPENSITIES, STATISTICAL MECHANICS APPROXIMATION AND CALIBRATION Calculation of turn and beta intrinsic propensities. A statistical analysis of a protein structure
THE TANGO ALGORITHM: SECONDARY STRUCTURE PROPENSITIES, STATISTICAL MECHANICS APPROXIMATION AND CALIBRATION Calculation of turn and beta intrinsic propensities. A statistical analysis of a protein structure
arxiv:cond-mat/ v1 2 Feb 94
 cond-mat/9402010 Properties and Origins of Protein Secondary Structure Nicholas D. Socci (1), William S. Bialek (2), and José Nelson Onuchic (1) (1) Department of Physics, University of California at San
cond-mat/9402010 Properties and Origins of Protein Secondary Structure Nicholas D. Socci (1), William S. Bialek (2), and José Nelson Onuchic (1) (1) Department of Physics, University of California at San
Monte Carlo simulation of proteins through a random walk in energy space
 JOURNAL OF CHEMICAL PHYSICS VOLUME 116, NUMBER 16 22 APRIL 2002 Monte Carlo simulation of proteins through a random walk in energy space Nitin Rathore and Juan J. de Pablo a) Department of Chemical Engineering,
JOURNAL OF CHEMICAL PHYSICS VOLUME 116, NUMBER 16 22 APRIL 2002 Monte Carlo simulation of proteins through a random walk in energy space Nitin Rathore and Juan J. de Pablo a) Department of Chemical Engineering,
Contact map guided ab initio structure prediction
 Contact map guided ab initio structure prediction S M Golam Mortuza Postdoctoral Research Fellow I-TASSER Workshop 2017 North Carolina A&T State University, Greensboro, NC Outline Ab initio structure prediction:
Contact map guided ab initio structure prediction S M Golam Mortuza Postdoctoral Research Fellow I-TASSER Workshop 2017 North Carolina A&T State University, Greensboro, NC Outline Ab initio structure prediction:
Ab-initio protein structure prediction
 Ab-initio protein structure prediction Jaroslaw Pillardy Computational Biology Service Unit Cornell Theory Center, Cornell University Ithaca, NY USA Methods for predicting protein structure 1. Homology
Ab-initio protein structure prediction Jaroslaw Pillardy Computational Biology Service Unit Cornell Theory Center, Cornell University Ithaca, NY USA Methods for predicting protein structure 1. Homology
Abstract. Introduction
 In silico protein design: the implementation of Dead-End Elimination algorithm CS 273 Spring 2005: Project Report Tyrone Anderson 2, Yu Bai1 3, and Caroline E. Moore-Kochlacs 2 1 Biophysics program, 2
In silico protein design: the implementation of Dead-End Elimination algorithm CS 273 Spring 2005: Project Report Tyrone Anderson 2, Yu Bai1 3, and Caroline E. Moore-Kochlacs 2 1 Biophysics program, 2
Lecture 2 and 3: Review of forces (ctd.) and elementary statistical mechanics. Contributions to protein stability
 and elementary statistical mechanics. Contributions to protein stability") Lecture 2 and 3: Review of forces (ctd.) and elementary statistical mechanics. Contributions to protein stability Part I. Review of forces Covalent bonds Non-covalent Interactions: Van der Waals Interactions
Lecture 2 and 3: Review of forces (ctd.) and elementary statistical mechanics. Contributions to protein stability Part I. Review of forces Covalent bonds Non-covalent Interactions: Van der Waals Interactions
Computer Modeling of Protein Folding: Conformational and Energetic Analysis of Reduced and Detailed Protein Models
 Cust. Ref. No. PEW 62/94 [SGML] J. Mol. Biol. (1995) 247, 995 1012 : Conformational and Energetic Analysis of Reduced and Detailed Protein Models Alessandro Monge, Elizabeth J. P. Lathrop, John R. Gunn
Cust. Ref. No. PEW 62/94 [SGML] J. Mol. Biol. (1995) 247, 995 1012 : Conformational and Energetic Analysis of Reduced and Detailed Protein Models Alessandro Monge, Elizabeth J. P. Lathrop, John R. Gunn
Monte Carlo Simulations of the Hyaluronan-Aggrecan Complex in the Pericellular Matrix
 Monte Carlo Simulations of the Hyaluronan-Aggrecan Complex in the Pericellular Matrix Marcel Hellmann BIOMS Group Cellular Biophysics, Deutsches Krebsforschungszentrum (DKFZ) Theoretical Biophysics Group,
Monte Carlo Simulations of the Hyaluronan-Aggrecan Complex in the Pericellular Matrix Marcel Hellmann BIOMS Group Cellular Biophysics, Deutsches Krebsforschungszentrum (DKFZ) Theoretical Biophysics Group,
Simulating Folding of Helical Proteins with Coarse Grained Models
 366 Progress of Theoretical Physics Supplement No. 138, 2000 Simulating Folding of Helical Proteins with Coarse Grained Models Shoji Takada Department of Chemistry, Kobe University, Kobe 657-8501, Japan
366 Progress of Theoretical Physics Supplement No. 138, 2000 Simulating Folding of Helical Proteins with Coarse Grained Models Shoji Takada Department of Chemistry, Kobe University, Kobe 657-8501, Japan
Wang-Landau sampling for Quantum Monte Carlo. Stefan Wessel Institut für Theoretische Physik III Universität Stuttgart
 Wang-Landau sampling for Quantum Monte Carlo Stefan Wessel Institut für Theoretische Physik III Universität Stuttgart Overview Classical Monte Carlo First order phase transitions Classical Wang-Landau
Wang-Landau sampling for Quantum Monte Carlo Stefan Wessel Institut für Theoretische Physik III Universität Stuttgart Overview Classical Monte Carlo First order phase transitions Classical Wang-Landau
Steering on-surface polymerization with metal-directed template
 Supporting Information Steering on-surface polymerization with metal-directed template Tao Lin 1, Xue Song Shang 2, Jinne Adisoejoso 1, Pei Nian Liu 2,*, Nian Lin 1,* 1. Synthesis Compounds 1 and 2 were
Supporting Information Steering on-surface polymerization with metal-directed template Tao Lin 1, Xue Song Shang 2, Jinne Adisoejoso 1, Pei Nian Liu 2,*, Nian Lin 1,* 1. Synthesis Compounds 1 and 2 were
Markov Chain Monte Carlo Lecture 1
 What are Monte Carlo Methods? The subject of Monte Carlo methods can be viewed as a branch of experimental mathematics in which one uses random numbers to conduct experiments. Typically the experiments
What are Monte Carlo Methods? The subject of Monte Carlo methods can be viewed as a branch of experimental mathematics in which one uses random numbers to conduct experiments. Typically the experiments
Protein Structure Prediction
 Page 1 Protein Structure Prediction Russ B. Altman BMI 214 CS 274 Protein Folding is different from structure prediction --Folding is concerned with the process of taking the 3D shape, usually based on
Page 1 Protein Structure Prediction Russ B. Altman BMI 214 CS 274 Protein Folding is different from structure prediction --Folding is concerned with the process of taking the 3D shape, usually based on
Conformational Sampling in Template-Free Protein Loop Structure Modeling: An Overview
 # of Loops, http://dx.doi.org/10.5936/csbj.201302003 CSBJ Conformational Sampling in Template-Free Protein Loop Structure Modeling: An Overview Yaohang Li a,* Abstract: Accurately modeling protein loops
# of Loops, http://dx.doi.org/10.5936/csbj.201302003 CSBJ Conformational Sampling in Template-Free Protein Loop Structure Modeling: An Overview Yaohang Li a,* Abstract: Accurately modeling protein loops
BIOC : Homework 1 Due 10/10
 Contact information: Name: Student # BIOC530 2012: Homework 1 Due 10/10 Department Email address The following problems are based on David Baker s lectures of forces and protein folding. When numerical
Contact information: Name: Student # BIOC530 2012: Homework 1 Due 10/10 Department Email address The following problems are based on David Baker s lectures of forces and protein folding. When numerical
Protein folding. Today s Outline
 Protein folding Today s Outline Review of previous sessions Thermodynamics of folding and unfolding Determinants of folding Techniques for measuring folding The folding process The folding problem: Prediction
Protein folding Today s Outline Review of previous sessions Thermodynamics of folding and unfolding Determinants of folding Techniques for measuring folding The folding process The folding problem: Prediction