|
|
|
- Horatio Booker
- 5 years ago
- Views:
Transcription

1
2 Table of Contents Preface Getting Started Introduction Drawing Molecules Making Selections Tools Draw Tool Navigate Tool Bond-Centric Manipulate Tool Manipulate Tool Selection Tool Auto-Rotate Tool Auto-Optimize Tool Measure Tool Align Tool Menus File Menu Edit Menu View Menu Build Menu Select Menu Extension Menu Building Molecules Importing Molecules by Name Importing from the Protein Data Bank (PDB) Building a Peptide Building DNA or RNA Building Carbon Nanotubes Insert Molecular Fragments Building with SMILES
3 Building Materials Building a Supercell Making a Crystal Surface Slab Building a Polymer Unit Cell Perceiving Crystall Symmetry Reducing Crystals to a Primitive Unit Cell Scaling Crystal Cell Volume Building Molecule-Surface Interactions Optimizing Geometry Introduction to Molecular Mechanics Finding Conformers of Molecules Geometry Constraints Display Types Different Display Styles Coloring Part of a Molecules Tutorials Using QTAIM (Atoms in Molecules) Analysis Viewing Vibrations Viewing Vibrational Spectra Calculations Viewing Molecular Orbitals Viewing Electrostatic Potential Maps Naming a Molecule Extensions ABINIT Input Generator LAMMPS Input
4 Preface layout: single title: Avogadro: Molecular Editor and Visualization Avogadro: Molecular Editor and Visualization Avogadro is a free, open source molecular editor and visualization tool, designed for use on Mac, Windows, and Linux in computational chemistry, molecular modeling, bioinformatics, materials science, and related areas. It offers flexible high quality rendering and a powerful plugin architecture. More about Avogadro, including development details and downloads can be found at Thanks This book would not be possible without the help and effort of many people, including Avogadro developers, translators, and users world-wide. Funding for the Avogadro manual was provided by the University of Pittsburgh Department of Chemistry. -Taylor Cornell and Geoffrey Hutchison Summer 2015 Pittsburgh, Pennsylvania 4

5 Introduction Introduction Avogadro is a "molecular editor," designed to be easy to use to construct and view molecules and materials in 3D. It runs on Windows, Linux, and Mac. This manual was largely made on a Mac, but the interface should be very similar on any computer. When you initially open Avogadro you will be presented with a screen such as the one shown below. Opening a File The first thing you will probably want to do is open a file and navigate around the molecule. To do this click on the File menu and select Open.... 5


6 Introduction You can then look through the files on your disk and find an appropriate chemical file. Thanks to Open Babel, a large number of file types are supported including CML, XYZ, SDF, Mol2, PDB etc. Several example molecules are supplied with Avogadro. The screen shot above shows the ethanol.cml file opened up and displayed using the default Ball and Stick display type. Notice that when a new file is opened Avogadro switches from the Draw Tool to the Navigate Tool, which allows you to view the molecule without editing it. Navigation You can zoom in/out using the scroll wheel on your mouse or holding down the middle mouse button and moving the mouse cursor up/down. You can rotate the view by holding down the left mouse button and moving the mouse cursor. You can also translate the view by holding down the right mouse button and moving the mouse cursor. 6
7 Introduction Note: if your mouse only has one or two buttons you can also use the modifier keys (shift and control) along with the left mouse button to perform actions where you would normally use the middle or right mouse buttons respectively. 7
8 Drawing Molecules Drawing Molecules Molecules are built and edited with the draw tool. Left clicking on the black display will allow you to begin your journey into molecule creation. A left click will generate a carbon atom. A right click will delete the atom. Left clicking the initial atom and dragging your mouse will generate a bond to another carbon atom. 8
9 Drawing Molecules Avogadro uses carbon as the default element. A different element can be selected through the "Element" drop down menu. Typing the atomic symbol (e.g., "A-s" for Arsenic) is a shortcut for changing the selected element. Let's say you wanted to create water. You can either type in "O", or select "Oxygen (8)" from the drop down menu, and then click on the black display. Left clicking on an atom that has already been generated will also change the element. In this case, clicking on the initial carbon atom changed it into an oxygen atom. 9
10 Drawing Molecules If the "Adjust Hydrogens" box is checked, hydrogen atoms in the molecule will be automatically adjusted to satisfy valency (as shown above). Bond order is changed through the "Bond Order" drop down menu, or by typing the numbers "1", "2", or "3". Bonds are added by left clicking on a bond that has already been created. Right clicking on a bond deletes the bond, and the atom it's bonded to. 10
11 Drawing Molecules Creating Carbon Dioxide: Begin drawing the "O-C-O" structure. After the structure is drawn, all you need to do is left click on the bonds. Left clicking on the bonds will create a double bond (shown below). 11
12 Drawing Molecules 12
13 Drawing Molecules Once you've created your molecule, you can optimize it's geometry through the extensions menu. Selecting the "Extensions" menu, and clicking "Optimize Geometry" will provide your molecule with proper bond lengths and angles. You now know the basics of drawing a molecule in Avogadro! 13
14 Making Selections Making Selections The selection tool is also a useful feature to master when beginning to learn Avogadro. Generally, the selection tool allows for the individual selection of atoms, bonds, or fragments. There are three types of selection modes: "Atom/Bond", "Residue", and "Molecule". The "Atom/Bond" selection mode provides you with the ability to select a single atom within a molecule. This is achieved by left clicking the atom. Press and holding the "shift" key allows for the selection of multiple atoms. Right clicking on the black display will clear the selection made. 14
15 Making Selections The "Residue" selection mode selects an entire residue within a molecule. A residue is selected by clicking on a single atom within the residue. 15
16 Making Selections The "Molecule" selection mode selects the entire molecule by clicking on an atom. Double clicking an atom in the molecule will also select the entire molecule. 16
17 Making Selections Clicking and dragging your cursor is another way molecules, or fragments of molecules can be selected. 17
18 Making Selections More information on selections can be found in the "Tools" section. 18
19 Draw Tool The Draw Tool 1. Creating a Molecule Molecules can be built and edited with the draw tool. 2. You can begin creating a molecule by left clicking on the black display. This will generate a carbon atom. Right clicking on the atom will delete it. 19
20 Draw Tool 3. Left clicking and dragging the mouse will generate a bond to another carbon atom. 4. Avogadro uses carbon as the default element. A different element can be selected through the "Element" drop down menu. Typing the atomic symbol (e.g., "A-s" for Arsenic) is a shortcut for changing the selected element. 20
21 Draw Tool 5. Let's say you wanted to create water. You can either type in "O", or select "Oxygen (8)" from the drop down menu, and then click on the black display. Left clicking on an atom that has already been generated will also change the element. In this case, clicking on the initial carbon atom changed it into an oxygen atom. 6. If the "Adjust Hydrogens" box is checked, hydrogen atoms in the molecule are automatically adjusted to satisfy valency (as shown above). 21
22 Draw Tool 7. Bond order is changed through the "Bond Order" drop down menu, or by typing the numbers "1", "2", or "3". Bonds are added by left clicking on a bond that has already been created. Right clicking on a bond deletes the bond, and the atom it's bonded to. 8. Creating Carbon Dioxide: Begin drawing the "O-C-O" structure. After the structure is drawn, all you need to do is left click on the bonds. Left clicking on the bonds will create a double bond (shown below). 22
23 Draw Tool 23
24 Draw Tool 9. Once you've created your molecule, you can optimize it's geometry through the extensions menu. Selecting the "Extensions" menu, and clicking "Optimize Geometry" will provide your molecule with proper bond lengths and angles. 24


25 Navigate Tool The Navigate Tool The navigation tool is used to pan, rotate, and scale the view of a molecule. If the "Display visual cues" box is checked, yellow arrows will display what type of navigation is taking place. Clicking anywhere on the black display and dragging the mouse will tilt, and rotate the entire molecule (as shown below). 25
26 Navigate Tool A molecule can also be rotated about an atom by clicking the atom and dragging the mouse. Depicted below, acetone is being rotated about its initial carbon. Right clicking allows you to change the molecules location on the display. Double clicking the molecule will reset the molecule's view. 26
27 Navigate Tool Using the middle scroll bar on a mouse will allow you to zoom in and out. 27


28 Bond-Centric Manipulate Tool The Bond-Centric Manipulate Tool The Bond-Centric Manipulate tool alters angles, bonds, and torsions of a molecule. Starting with the basics: To begin using the bond centric manipulate tool, click on a bond within your molecule. Clicking on a bond and dragging your cursor allows you to adjust the plane. If the "Show Angles" box is checked, the angles from the selected bond to all adjacent bonds are displayed. If the "Snap-to Bonds" box is checked you'll notice that the plane changes from yellow 28
29 Bond-Centric Manipulate Tool to blue as it's rotated. A yellow plane indicates that an adjacent bond is in line with the plane. If the plane depicted is blue, the plane is not in line with any adjacent bonds. "Snap-to Threshold" determines how many degrees away a plane has to be to snap to an adjacent bond plane. For example, the "Snap-to Threshold" shown below is 10 degrees. Therefore if the plane being rotated comes within 10 degrees of an adjacent bond, it will snap to the adjacent bond's plane. Changing the Snap-to Threshold to 90 degrees is a quick trick for the rotating plane to only snap to adjacent bond planes. Adjusting Bonds and Torsion Angles Once a plane is selected the atoms on either end of the plane can be manipulated, by left clicking on the atom and dragging. The displayed angles will automatically adjust, and the selected bond will not change in length. 29
30 Bond-Centric Manipulate Tool Left clicking on a substituent, or in this case a hydrogen bonded to one of the selected carbon atoms will allow you to adjust the torsion angle. 30
31 Bond-Centric Manipulate Tool 31
 into the desired positon.")
32 Manipulate Tool The Manipulate Tool The manipulate tool allows you to move atoms and selected fragments. The manipulate tool is automatically selected when inserting a fragment. This allows the fragment to be rotated into position. The "Translate by:" section allows you to easily translate the length of a bond (to a fragment or atom) into the desired positon. For a translation to take place, enter in the desired translation and then click "Apply". 32
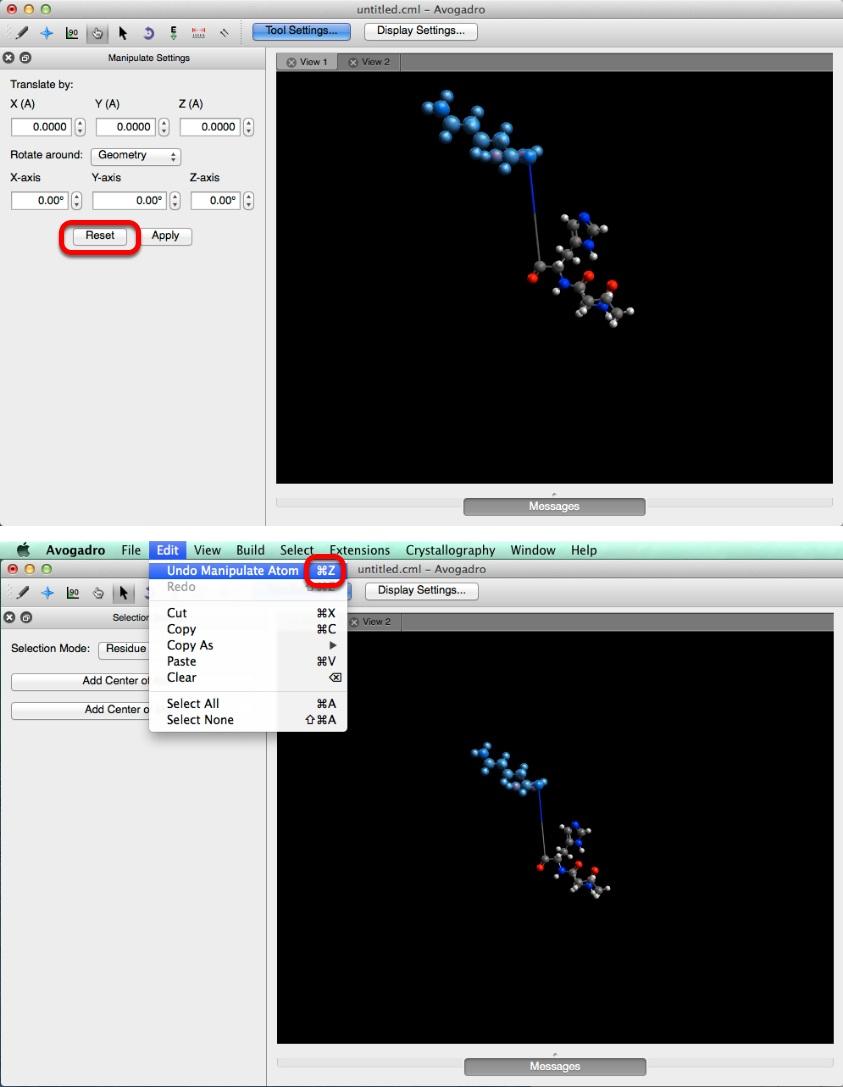
33 Manipulate Tool The "Rotate around:" section allows you to rotate your selection around the current geometry, or the origin. After choosing how many degrees to rotate your selection click "Apply". Clicking "Reset" will reset all of the information in the translation, and rotation boxes. This does not reset the molecule, if you want to undo your adjustments go to the "Edit" menu in the top bar and select "Undo Manipulate Atom". 33
34 Manipulate Tool 34
35 Selection Tool The Selection Tool The selection tool allows the indiviual selection of atoms, bonds, or fragments. There are three types of selection modes: "Atom/Bond", "Residue", and "Molecule". The "Atom/Bond" selection mode provides you with the ability to select a single atom within a molecule. This is achieved by left clicking the atom. Pressing down the "Shift" button on your keyboard allows the selection of multiple atoms. Right clicking on the black display will clear the selection made. 35
36 Selection Tool The "Residue" selection mode will select an entire residue within the molecule, by clicking on a single atom in the residue. The "molecule" selection mode will select the entire molecule by clicking on an atom. Double clicking the molecule will also select the entire molecule. 36
37 Selection Tool Molecules can also be selected by clicking and dragging your cursor across the display. 37
38 Selection Tool Clicking on "Add Center of Atoms" will display a black ball where the center of the molecule is found. Clicking on the "Add Center of Mass" will display a black ball where the center of mass of the molecule is found. 38
39 Selection Tool 39

40 Auto-Rotate Tool The Auto-Rotate Tool The auto rotate tool allows for the continuous rotation of a molecule. To rotate the molecule, adjust the x, y, and z rotation scales as desired. Then select "Start". You can also click and drag from an atom to rotate the molecule in a specific direction. 40
41 Auto-Rotate Tool Select "Stop" to end the molecule's rotation. Clicking "Reset" will zero out the rotation scales and stop the rotation. 41
42 Auto-Rotate Tool 42


43 Auto-Optimize Tool The Auto-Optimize Tool The Auto Optimize tool continuously optimizes molecular geometry through molecular mechanics. This tool provides an interactive interface, allowing you to manipulate a molecule while it's molecular geometry is being optimized. Force Fields & Algorithms The Auto-Optimization settings provide several force field options. The default force field in Avogadro is UFF (Universal Force Field). UFF can generally reproduce the most structural features across the periodic table. However, depending on the molecule being optimized, the other force fields may be better suited to optimize the molecular parameters. The force field options are shown below. For more information on force fields refer to the optimizing geometry section of this lab manual. 43


44 Auto-Optimize Tool The default setting for "Steps per Update" is 4. This number can be preferentially increased or decreased. If you have a slower computer consider decreasing this number. Avogadro also has the ability to apply specific algorithms dependent on your need. Steepest descent is the default algorithm, and has the most fluid and interactive system. Fixing & Ignoring Selections If necessary, atoms can be fixed into place before optimization so that they don't move. This is done by going to the "Extensions" menu, holding your cursor over "Molecular Mechanics" and then selecting "Fix Selected Atoms". 44

45 Auto-Optimize Tool Clicking on "Start" will allow you to manipulate the molecule by left clicking on an atom and dragging your cursor. Notice that the fixed atoms don't bend with the manipulation of the molecule. 45
46 Auto-Optimize Tool If the "Fixed atoms are movable" box is checked, the bonds between atoms can be manipulated into a new configuration. Manipulating a bond using this feature will lock that bond into place, and display the number of constraints cast on that molecule. Below is an exaggerated version of a bond manipulation using this feature. Selecting the "Extensions" drop down menu, holding your cursor over the "Molecular Mechanics" option, and choosing "Ignore Selection" will allow you to ignore a selection of atoms. Avogadro registers an ignored selection as if the selection doesn't exist. Therefore optimization only takes place in correspondence to atoms that aren't ignored. Atoms that have been ignored can still be adjusted if the "Ignored atoms are movable" box is checked, however they will not be optimized. Molecules will reoptimize until de=0 or "Stop" is clicked. 46
47 Auto-Optimize Tool 47
48 Measure Tool The Measure Tool The measure tool determines bond lengths, angles, and dihedrals. The measure tool allows you to select and assess up to four atoms. As you click on atoms Avogadro will automatically calculate the distances between atoms in a respective order. For example, the distance between atom 1 and 2 is Å (displayed below). 48
49 Measure Tool Avogadro will also determine the angle between atoms, if at least three atoms have been selected. The second atom is used as the vertex. If four atoms are selected, a dihedral angle is determined. 49
50 Measure Tool Right clicking the display will reset the atoms previously selected. 50
51 Measure Tool 51

52 Align Tool The Align Tool The align tool rotates, and translates a molecule(s) into a specific reference frame. An alignment axis can be chosen from the "Axis" drop down menu. Typing "x", "y", or "z" is a shortcut for changing the alignment axis. The align tool can be used to align everything in the frame, or a specific molecule with "Molecule" option in the "Align" drop down menu. As shown below, two atoms will be used as reference points to align your selection. 52
53 Align Tool Clicking "Align" will then reposition the molecule/frame with the new alignment. The alignment axes are displayed in the bottom left corner. The red axis is designated as the X axis, green is designated as the Y axis, and blue is designated as the z axis. 53
54 Align Tool 54

55 File Menu The File Menu The file menu provides the standard abilities of creating a new file, opening & closing documents, as well as saving documents. It also yields the capability to import files from various databases. New The "New" selection will open a new file in Avogadro. Open After selecting "Open", a file that has previously been saved is accessible through the pop up browser. Open Recent "Open Recent" displays a list of documents recently launched. Close "Close" dismisses the window currently open. Save "Save" will maintain your progress. 55


56 File Menu Save As... "Save As..." allows you to save progress without overwriting the original file. Revert to Saved "Revert to Saved" will revert any changes made to the previously saved file. Import "Import" will open chemical files stored in a database. Export "Export" will make files created in Avogadro suitable for other programs. 56
57 File Menu 57

58 Edit Menu The Edit Menu The edit menu administers basic file revisions. Undo "Undo" will negate the last change to the document. Redo "Redo" will recover the last change to the document. Cut "Cut" will remove and copy a selection. Copy "Copy" will create a duplicate of the entire molecule or a selection, and place it on a clipboard. Copy As 58
59 Edit Menu "Copy As" provides text representations of the molecules present in the viewing screen. For example, selecting "Copy As" and "SMILES", renders "C(=O)(C)C.O.O" as the output for the viewing screen below. This selection can then be pasted in a text document for external projects. Paste "Paste" recalls the last data copied onto the clipboard. Clear "Clear" removes all chemical structures from the viewing window. Select All "Select All" highlights everything in the screen (this feature can also be found under the "Select" menu). 59
60 Edit Menu Select None "Select None" will dismiss everything in the display (this feature is also found under the "Select" menu). 60

61 View Menu The View Menu The view menu gives the user the ability to add, and adjust the display views currently in use. New View "New View" creates a new, blank viewing window. Duplicate View By selecting "Duplicate View" from the drop down bar, a duplicate of the current view will be created. Any changes made to the display window will automatically update in all of the views. 61
62 View Menu Detach View The "Detach View" selection will display the current view in a new window. 62

63 View Menu Close View "Close View" deletes the display that's open. A view can also be closed by clicking the x on the left of the view tab. 63

64 View Menu Center "Center" will align the molecule(s) to the middle of the viewing screen. Align View to Axes "Align View to Axes" adjusts the display view to be in the x, y plane with the positive z-axis pointing toward you. Full Screen 64

65 View Menu "Full Screen" expands the window to fill the computer screen. Reset Display Types "Reset Display Types" will deselect all display types checked, and revert back to the default "Ball and Stick" display type. Set Background Color... The "Set Background Color..." feature will allow you to change the background color of the viewing window. 65
 view of a molecule in space.")
66 View Menu Projection There are two types of projection features (prospective, and orthographic projection), the default projection is perspective projection. Perspective projection provides a more realistic (3D) view of a molecule in space. Orthographic projection provides, and adjusts the molecule into a planar (2D) view, where all like atoms are adjusted to stay the same size. The projection views are most evident when drawing molecules. Display Axes "Display Axes" will render an axes display in the lower left hand corner. 66
67 View Menu Debug Information "Debug Information" provides additional information about the view, and what's currently taking place on your screen. 67
68 View Menu Use Quick Render Quick render adjusts the 3D molecular image in the viewing screen to achieve a faster image rendering on slow computers. On most modern (2012 or later) computers, this is not necessary. All Molecules in File... "All Molecules in File..." allows you to look at all of the molecules that have previously been created and embedded into one file. From the dialog box that pops up, you can select and edit a molecule by clicking on the molecule's title. 68

69 View Menu Crystal View Options... "Crystal View Options..." when selected will open the toolbar shown below. This toolbar allows you to edit the Miller indices, and the Unit Cell for any crystal structure. 69
70 View Menu Properties The "Properties" selection will provides you with molecule, atom, bond, angle, torsion, and conformer properties. These settings display general compository information about the molecule and atoms present. 70

71 View Menu For example, clicking on "Molecule Properties" will display general molecular information. 71

72 View Menu 72

73 Build Menu The Build Menu The build menu helps to ease the process of constructing molecules. Cartesian Editor... "Cartesian Editor..." when selected provides you with the capability to manually adjust bond lengths. The dialog box for the cartesian editor is displayed below. 1. Sort by... 73

74 Build Menu The sort by drop down menu will rearrange the data in the dialog box for your convenience. Sort by can arrange the data by element, or by location of the atom. All of the data for sorting by X, Y, and Z coordinates will start reading at the atom to the furthest left in the molecule, and continue until it reaches the atom at the right most point. 2. Unit of Measure Avogadro provides three units of measure to adjust bond lengths, Angstroms, Bohrs, and Fractional coordinates. A unit cell must be defined to use fractional coordinates. 74

75 Build Menu 3. Editing & Modifying Data Editing the data is as simple as clicking on the number you wish to edit, and typing in a new coordinate. After clicking "Apply" and returning back to the Avogadro display screen, you should notice that the atom has changed position. The displayed data can also be modified according to your personal preference, or for the use of additional plugins. 75

76 Build Menu Change H to Methyl "Change H to Methyl" will replace any Hydrogens present in the display window with methyl groups. Depicted below is acetone with all of its hydrogens replaced by methyl groups. 76
77 Build Menu Add Hydrogens "Add Hydrogens" will satisfy the valency of the atoms present with hydrogens. 77
 acidic hydrogens to ionizable groups in peptides, according to the desired ph.")
78 Build Menu Add Hydrogens for ph... "Add Hydrogens for ph..." will create a dialog box (displayed below) that allows you to adjust the ph of the molelcular environment. This feature will add (or subtract) acidic hydrogens to ionizable groups in peptides, according to the desired ph. Remove Hydrogens "Remove Hyrogens" will delete all hydrogens in the display screen. 78

79 Build Menu Insert "Insert" provides a faster, simpler way of building molecules. A depicted below, you can insert DNA/RNA, a Fragment, a Peptide, and can also insert a molecule based on SMILES text. 79
80 Build Menu Invert Chirality "Invert Chirality" will reverse (invert) the initial chirality to the opposite R/S configuration. 80

81 Build Menu 81
82 Build Menu Super Cell Builder... Information on the Super Cell Builder can be found in "Building and Editing Crystals and Materials" section. Nanotube Builder... Information on building Carbon Nanotubes in Avogadro can be found in the "Building Molecules" section. 82
.")
83 Select Menu The Select Menu The select menu makes chemical alterations more efficient through various modes of selection. Select All "Select All" highlights everything in the display (this feature can also be found under the "Edit" menu). Select None "Select None" will dismiss everything in the display (this feature is also found under the "Edit" menu). Invert Selection "Invert Selection" reverses the selection made. The first image displayed below is the orginal selection, and the second image demonstrates the inverted selection. 83
84 Select Menu 84
85 Select Menu Select SMARTS... SMARTS (SMiles ARbitrary Target Specification) is a more general chemical language extension of SMILES. "Select SMARTS..." allows you to use this chemical language to select various atoms, or groups of atoms within the molecule. For example, typing "a" into the dialog box and clicking ok will select all atoms with aromaticity. More information can be found at the Daylight SMARTS webpage. Select by Element... "Select by Element..." generates a periodic table pop up screen that allows you to select an element throughout the viewing display. 85
 to select an alanine residue.")
86 Select Menu Select by Residue... "Select by Residue..." generates a pop up screen that allows you to select residues with specified names. For example, typing in "ALA" (this feature is case sensitive) to select an alanine residue. This feauture only works with residues that were made using the peptide builder. 86
87 Select Menu Select Solvent This feature will select "HOH" residues in PDB (protein data bank) files. Add Name Selection... "Add Name Selection..." allows you to add a new selection to the current data base, which you can then recall at your convenience. 87

88 Extension Menu The Extensions Menu The Extensions Menu is a catalog of computational plugins equipped with Avogadro. These plugins can interact with molecules, generate input file dialogs for quantum codes, and create molecule property dialogs. Animation Selecting "Animation" will open the animate trajectory dialog box shown below. From here you can load a file, view and edit the animation, as well as save the file in a PC compatible format. 88


89 Extension Menu Optimize Geometry "Optimize Geometry" provides a quick, realistic rendition of a molecule using molecular mechanics. Molecular Mechanics "Molecular Mechanics" allows you to edit the geometry optimization of a molecule, so that it best suits your purposes. 89

.")
90 Extension Menu Setup Force Field... A dialog box will open when "Setup Force Field..." is selected. This dialog box provides you with the ability to choose the type of force field, and algorithm that can best optimize your molecular parameters, and preferences. Calculate Energy "Calculate Energy" determines the amount of energy per the amount of material (kj/mol), and displays this number in a pop up dialog box. Conformer Search "Conformer Search" is a way to easily search for conformers within a molecule (dialog box shown below). A more detailed outline on how to perform a conformer search is found in the "Optimizing Geometry" section of this manual. Avogadro only renders staggered conformations, and does not calculate ring conformers. 90
.")

91 Extension Menu Constraints "Constraints" is a way to ensure atom stability in various selections (dialog box depicted below). The constraints that can be applied to a molecule include Ignore Atom, Fix Atom, Fix Atom X, Fix Atom Y, Fix Atom Z, Distance, Angle, and Torsion Angle. A detailed outline on how to use the constraints feature is found in the "Optimizing Geometry" section of this manual. 91
92 Extension Menu Ignore Selection "Ignore Selection" allows you to select a specific part of a molecule to omit during a geometry optimization. Fix Selected Atoms "Fix Selected Atoms" also allows you to set a certain part of a molecule to fix during optimization. Avogadro Extensions--Plugins Avogadro provides you with the ability to interface your molecules with other dialog based plugins. These extensions interact with a molecule to provide further molecular information, and additional computation abilities. These plugins include but aren't limited to GAMESS, Abinit, Dalton, GAMESS-UK, Gaussian, MOLPRO, MOPAC, NWChem, PSI4, Q-Chem, and LAMMPS. General "How To" for Plugins Avogadro (as you will see below) can be used to display molecular orbitals, QTAIM, spectra, as well as create surfaces. However, many of these features can not be used to their full potential without first running one of the plugins listed in the section above. Gaussian is one of the most common plugins used, due to it's wide range of basis sets/functions. Running Gaussian After selecting "Gaussian" from the Extensions menu, the dialog box depicted below will appear. You can edit the dialog box and add specific keywords to utilize these features in Avogadro. For example, typing "freq" in the dialog box will compute force constants and vibrational frequencies. More information on keywords for Gaussian can be found at the Gaussian website ( Then clicking generate will let you save the file to your computer, so you can run the file in external software. 92

93 Extension Menu Once the file has been run through the external software, you will have a.g03 or.g09 file that will open the keyword selection in a toolbar on the right hand side of the screen. "Freq" will open the vibrations toolbar shown below. 93

94 Extension Menu Molecular Orbitals The "Molecular Orbitals" selection will display the molecular orbitals for orbitals with full status bars. The quality of the orbitals can be adjusted an reconfigured if need be. This feature only works by running gaussian extension files (.fchk,.g03,.g09, etc.). 94
95 Extension Menu QTAIM (Quantum Theory of Atoms in Molecules) QTAIM displays the implicit bonding that is theorized to take place between the hydrogens of organic crystals (the implicit bonding is conveyed through dots). This display type is utilized by importing a.wfn file from the "QTAIM", "Molecular Graph" selection under the "Extensions" menu. Selecting "Molecular Graph with Lone Pairs" or "Atomic Charge" will provide concurrent information about the molecule. More information can be found on this process in the Tutorial section of this manual. 95
96 Extension Menu Spectra... Clicking on "Spectra..." will create a spectra visualization of a.g03, or.g09 file that has been run with the keyword "freq". A spectral visualization can also be created through the vibrations toolbar by selecting "Show Spectra...". 96


97 Extension Menu Create Surfaces... "Create Surfaces..." allows you to view the Van der Waals, Electrostatic Potential, Electron Density, and Molecular Orbital Surfaces. The surface type options for viewing depend on what type of calculations have previously been run on the molecule. The type of file you open/create allows for more or less surface viewing options (generally discussed under Avogadro Extensions--Plugins). This feature also allows you to edit the color, resolution, and iso value to further enhance your surface. 97


98 Importing Molecules by Name Importing Molecules by Name Select the "File" menu. Then select "Import", and "Fetch by chemical name..." A dialog box will pop up (depicted below), where you can type in any chemical name. Avogadro will import the molecule into the viewing screen after you click "OK". 98

99 Importing Molecules by Name It may take a few seconds or even a minute to download the molecule online. Avogadro uses the NIH "Chemical Resolver" to convert the name into a molecular structure. 99


100 Importing from the Protein Data Bank (PDB) Importing from the Protein Data Bank (PDB) You can read PDB files that you download yourself from or access the PDB code yourself. Importing Directly Older versions of Avogadro have a bug with direct access to the PDB (since the website has moved) but using v 1.2.0, you can again use File > Import > Fetch from PDB... to download proteins. A dialog will come up and allow you to enter a PDB code (e.g., 1CRN for crambin) There it is... the PDB data direct from the website. 100
\" format of the file.")
101 Importing from the Protein Data Bank (PDB) Reading a Downloaded PDB file If the direct import doesn't work, you can also use the website yourself. Go to and select a protein, then download the "PDB File (text)" format of the file. From your downloads folder you can either drag and drop the file onto an Avogadro display screen, or drag and drop the file over the Avogadro application icon. Dragging and dropping the file will open the PDB import with a unit cell (Avogadro displays unit cells for all PDB imports). 101
102 Importing from the Protein Data Bank (PDB) If the unit cell is unwanted, you can remove the unit cell under the "Crystallography" menu by selecting "Remove Unit Cell". 102

103 Importing from the Protein Data Bank (PDB) 103
104 Importing from the Protein Data Bank (PDB) 104


105 Building a Peptide Building a Peptide A walkthrough on how to create a custom peptide model in Avogadro. Select the Build menu. 105


106 Building a Peptide Bring up the peptide builder window. You can select amino acids to insert into the new peptide. As you click on particular amino acids, they will be added to the sequence on the right. The peptide will build up as a sequence, starting from the N terminus. Of course you can also type the residues directly or paste from an online database. 106



107 Building a Peptide You can pick the secondary structure Click to insert the sequence into the main window. The new oligopeptide will be selected automatically, and the manipulate tool will allow you to translate and rotate the chain into the position you want. 107

108 Building a Peptide You may wish to re-center the view, since the new peptide may be large. 108

 and the rest of the window will update")
109 Building DNA or RNA Building DNA/RNA Avogadro now has a builder for nucleic acid sequences and this walk-through will show you how to use it. The DNA/RNA builder is under the "Build" menu and "Insert" submenu. Select either DNA or RNA (1) and the rest of the window will update accordingly. 109
.")
, you can insert either single-stranded or double-stranded DNA.")
110 Building DNA or RNA You can also control the number of bases per turn as shown (with defaults for A-DNA, B- DNA, Z-DNA, or RNA). You can enter the sequence either by clicking the buttons, or by typing the one-letter codes directly. For DNA sequences (as shown here), you can insert either single-stranded or double-stranded DNA. You may wish to re-center the view or align the view to axes to see the whole molecule. 110

111 Building DNA or RNA There we go -- the well-known DNA double-helix! 111
 Under the Build menu, there s a new option for the nanotube builder.")

112 Building Carbon Nanotubes Building Carbon Nanotubes Avogadro 1.1 includes a new nanotube builder, based on the well-known TubeGen code and website from the Doren group at U. Delaware. ( Under the Build menu, there s a new option for the nanotube builder. At the moment only single-walled nanotubes (SWNT) can be built in one step, although it s easy to generate several nested tubes for multi-walled (MWNT) as shown here. The builder will show up at the bottom of the Avogadro window. You can set the n,m indexes to determine the type of nanotube (1) the length of the tube (2), in Angstrom, bohr, picometers, nanometers, or periodic unit cells (e.g., if you wish to do a calculation with periodic boundar conditions), and how to terminate the nanotube (3). NOTE: determining double bonds can be time-consuming on large nanotubes. 112



113 Building Carbon Nanotubes The nanotube will be generated aligned along the z-axis, so you may want to re-center the view. Here, we've added a 6,6 nanotube after inserting our 4,4 nanotube. We'll need to re-center the tube to produce a more accurate double-walled system. Here, we use the manual translation options, new in Avogadro 1.1, to nudge the 6,6 nanotube in the XY plane to properly center around the 4,4 nanotube. 113


114 Building Carbon Nanotubes Here we ve nudged the 6,6 tube into an approximately correct position. We ll now use Avogadro s built-in force fields and the Auto-Optimize tool to relax the structure. We (1) select the Auto-Optimize tool to allow interactive minimization of the nanotubes, and (2) select the MMFF94 force field. Other forcefields would also likely work well. Finally (3) start the optimization. 114

115 Building Carbon Nanotubes After a few steps, you can see a nicely relaxed double-walled nanotube. You could repeat the process as desired. 115


116 Insert Molecular Fragments Insert Fragments Avogadro includes over 300 common molecules and molecular fragments to make building larger structures easy. Under the "Build" menu, hold your cursor over "Insert", and then select "Fragment...". A database of fragments will then pop up (shown below). You can filter the selection if need be. After you've made your fragment selection, click "Insert". 116

117 Insert Molecular Fragments The fragment will be inserted and the Manipulate tool will be selected so you can move the new fragment around the window. 117
118 Insert Molecular Fragments 118


119 Building with SMILES Building with SMILES SMILES (Simplified molecular-input line-entry system) allows you to build molecules through a string of text. If you have a SMILES string (e.g., copied from a paper or website) or prefer to enter one for a complicated molecule, Avogadro will build a 3D geometry from the SMILES. Under the "Build" menu, hold your cursor over "Insert", and select "SMILES...". Enter your SMILES fragment, and select "OK". There it is.. 119
120 Building with SMILES 120

121 Building a Supercell Super Cell Builder Once a crystal surface has been built, the Super Cell Builder can expand atoms within a space group, replicate the unit cell, and perform simple bonding. When "Super Cell Builder..." is selected under the "Build" menu, the dialog box below pops up. This dialog box will allow you to replicate a unit cell that has already been created (if need be, a unit cell can be created by selecting "Add Unit Cell" under the "Crystallography" menu). Creating a Surface One way supercell can be utilized is by creating a surface. Below is an elemental unit cell comprised of silver. This cell was imported through the "File" menu, under "Import", "Crystal...". When the dialog box appears follow the procedure displayed below. 121

122 Building a Supercell A unit cell can then be replicated to make a slab or a surface. For this example, the parameters were edited as shown in the image below. After editing the parameters, clicking "Generate Cell" will expand your surface. 122

123 Building a Supercell A surface can then be modified by introducing impurities. Here, copper impurities were added to the silver surface. This file can now be exported to another program to determine, through calculations, how the impurities will impact the surface. 123
124 Building a Supercell 124


125 Making a Crystal Surface Slab Building a Crystal Surface (Slab) Build up a crystal surface, e.g., Pt <111> for a defined Miller Plane. Import the appropriate crystal structure. Either open a CIF file with the crystal structure needed, or import one from the built-in Avogadro crystal library. The tutorial will assume you import a structure from the Avogadro library. Choose File > Import > Crystal to bring up the library. Either browse through the crystals, or type a filter -- by element or name. Click "Insert" to import the selected structure. 125
. To build a specified surface (e.g., Ag <121>) choose Crystallography > Build > Slab.")

126 Making a Crystal Surface Slab Importing a crystal will show the asymmetric unit cell (e.g., one atom for Silver here). To build a specified surface (e.g., Ag <121>) choose Crystallography > Build > Slab... to bring up the slab builder settings. Future crystal builders (e.g., nanoparticles, supercells) will also appear in this menu. 126
127 Making a Crystal Surface Slab Specify the indices of the Miller plane desired (for hexagonal unit cells, all 4 indices will appear), and choose the dimensions in either distances or repeating cells of the resulting surface. The generated surface is aligned in the XY plane, and a specified thickness will be cleaved in the z-axis below the XY plane. This feature allows easy alignment between a new surface and a molecule for surface interaction calculations. Click "Build" to start the surface generation. After clicking "Build," Avogadro will generate a large supercell, align, rotate, and cleave the designated surface. This may take some time, depending on the size of the crystal cell. Here translucent van der Waals spheres are used to illustrate the corrugation of the Ag <121> surface. The resulting surface is a 2x2 supercell, with a large spacing (40 Å) in the z-axis. 127


128 Building a Polymer Unit Cell Building a Polymer Unit Cell A walk-through on creating a unit cell (of a polymer) using Avogadro and the Align tool. This specific example uses Gaussian, but translation vectors for other programs can be performed similarly. Build out the molecule for the unit cell. Notice that while the repeat unit here is 2 rings, we have built 3 rings. This way, we will properly model the bond which spans two unit cells. Optimize the geometry of the molecule. 128




129 Building a Polymer Unit Cell Switch to the Align tool to translate and orient the unit cell coordinates. Make sure to open the Tool Settings window, which will allow you to work with the Align tool. First click on the "start" atom of the polythiophene. This atom will be translated to the origin (0, 0, 0). Then click on the corresponding atom in the "next" unit cell. The distance between these two atoms will define one axis in the unit cell. In the "Align Settings" window, define an axis for the unit cell. Then click the Align button. This will change the coordinate set to have atom #1 at the origin, and atom #2 (from the step above) projected onto the x-axis. 129


 and the \"end\" hydrogen atoms.")
130 Building a Polymer Unit Cell Open the Cartesian Editor window to verify the results of the Align operation. Notice that atom #1 is at the origin, and atom #11 is projected onto the X-axis. The size of the unit cell is 7.806Å -- the distance between atom #1 and atom #11. Now delete "extra" atoms which should not be included in the unit cell calculations. This includes the third ring (including atom 11) and the "end" hydrogen atoms. For example, you can use the select tool and drag over the atoms to be deleted to pick them. 130



131 Building a Polymer Unit Cell Once selected, you can use the "Clear" menu command to delete the atoms. If you wish to submit the unit cell to Gaussian, pick the Gaussian input extension. Set options as you desire. Make sure to add a "TV " line at the bottom of the preview text. This will enable the unit cell calculation by setting the translation vector for the unit cell. 131

132 Perceiving Crystall Symmetry Crystal Symmetry Perception Calculation results often specify all atoms and translation vectors, but not the space group. Here we see how to perceive the space group from a set of crystallographic coordinates. Open a Crystal File Here we open an example VASP calculation by opening the POSCAR file. 132



133 Perceiving Crystall Symmetry This example is triclinic, looking for Li / H structures. Note that VASP files do not specify a space group, so it is reported as "Unknown." We can either set the spacegroup manually, or here, perceive the space group, using the open source spglib code. 133
134 Perceiving Crystall Symmetry We need to set the tolerance, since some atoms may be slightly out of place in Cartesian coordinates. Our example VASP file isn't very interesting -- the space group is P1. Here's another example, where the space group is P

135 Reducing Crystals to a Primitive Unit Cell Reducing Crystals to Primitive Unit Cells Some simulations use "supercells" -- larger periodic boundary systems than the primitive unit cell. Here is a walk-through on reducing a large supercell to the primitive unit cell. Open or import the file with the supercell -- here, CaCO3. Note that the space group is unknown, since the file came from VASP, which does not specify a space group with the coordinates. After perceiving the space group, we see correctly that the system is R -3 c. Now we can reduce the supercell to a primitive cell of CaCO3. 135
.")
136 Reducing Crystals to a Primitive Unit Cell Avogadro provides two algorithms for reducing the unit cell to a primitive or Niggli cell. Here, pick "Primitive." Note that the volume of this supercell was over 4,000 Å3. You will need to set a tolerance for the Cartesian coordinates (here, in Å). After reduction, note that the space group is retained, the lattice is properly Rhombohedral, and the unit cell volume is 36 times smaller. 136
, we see the normal unit cell and lattice information.")

137 Scaling Crystal Cell Volume Scaling Crystal Volumes Avogadro 1.1 allows you to adjust the volume or spacing of a unit cell. After creating or opening the crystal (here ice), we see the normal unit cell and lattice information. We will now adjust the cell volume. Before we scale volume, we can either choose to preseve Cartesian coordinates (which will add empty space to the edges of the unit cell) or preserve fractional coordinates (which will symmetrically scale the entire unit cell). This walk-through will show both. 137


 around the outside of the unit cell boundaries, when preserving")
138 Scaling Crystal Cell Volume First we'll scale the cell while preserving Cartesian coordinates. The units of the volume are determined by your settings (here Å). We adjust the volume from the original Å3, and click "OK." Here, we've greatly exaggerated the volume, to show the empty space (arrows) around the outside of the unit cell boundaries, when preserving Cartesian coordinates. The space group has also changed (to C 1 m 1). 138

 are")
139 Scaling Crystal Cell Volume If you preserve fractional coordinates, you can scale the unit cell symmetrically. Note that while the volume is significantly expanded, the space group (and fractional coordinates) are retained. 139

140 Building Molecule-Surface Interactions Molecule-Surface Interactions Beyond building a crystal surface, new features in Avogadro make it easy to consider molecule-surface interactions. The lesson picks up at the end of the "Building a Crystal Surface" lesson. Start with a generated Crystal Surface Generate the desired crystal surface. Avogadro will center the surface cell, aligned in the XY plane, with slab atoms defined below Z = 0. The Slab Builder also leaves a large space along the z-axis to allow insertion of molecules for surface interaction calculations. You can control this padding as indicated above. New Window: Create our Molecule 140



141 Building Molecule-Surface Interactions In a new window, draw the desired molecule, or open a file. Here we consider ethanol. We will use the "Align Tool" to allow us to rotate and align the molecule with the OH group at the origin, and the molecule aligned along the z-axis. We will click on the terminal H atom (which will be translated to the origin) followed by the carbon atom (which will define the z-axis of the molecule). 141




142 Building Molecule-Surface Interactions After defining the atoms (they will show colored spheres and numbers once selected), click on the "Align" button to translate and rotate the molecule. You may wish to alter the current camera view. Choosing View > Align View to Axes will reset the view to project the z-axis of the molecule to point towards you. Perfect! Now we can copy our ethanol to the surface document. After copying, we can switch to our surface. 142

143 Building Molecule-Surface Interactions Now we'll paste in the ethanol molecule. Note that the ethanol is now embedded in the surface, centered as desired. The Manipulate tool has been selected, allow us to translate the molecule as needed. 143
.")

144 Building Molecule-Surface Interactions New in version 1.1 is an option to specify the exact amount to translate or rotate the selection (i.e., the molecule we just pasted). Here, we've specified that we want to move the molecule +2.5Å along the z-axis, above the surface, and then we click "Apply" to complete. We could also rotate around the z-axis if the positioning isn't as desired. Here we have translated the ethanol 2.5 Å above the Ag <121> surface and are ready to submit for a calculation. 144
145 Introduction to Molecular Mechanics Molecular Mechanics & Force Fields Avogadro comes equipped with multiple different force fields. Below is general information regarding the force fields to help you select the best optimization method. UFF UFF (Universal Force Field) is capable of reproducing the most structural feature across the periodic table. This force field can optimize the geometry for all elements, and does well with inorganic materials, and organometallic materials. MMFF94(s) MMFF94 & MMFF94s (designed by Merck), is particularly good with organic compounds. MMFF94 has specifically been parameterized for alkanes, alkenes, alcohols, phenols, ethers, aldehydes, ketones, ketals, acetals, hemiketals, hemiacetals, amines, amides, peptide analogs, ureas, imides, carboxylic acids, esters, carboxylate anions, ammonium cations, thiols, mercaptans, disulfides, halides (chlorides and fluorides), imines, iminium cations, amine N-oxides, hydroxylamines, hydroxamic acids, amidines, guanidines, amidinium cations, guanidinium cations, imidazolium cations, aromatic hydrocarbons, and heteroaromatic compounds. MMFF94 and MMFF94s use the same functional form to calculate the potential energy. They only differ in the Torsion and Out-Of-Plane bending parameters used. The 's' in MMFF94s stands for static and this set of parameters is more suited for tasks where the output is static. These force fields also add electrostatic charges, and hydrogen bonds (displayed below). 145
146 Introduction to Molecular Mechanics GAFF GAFF (General AMBER Force Field) is often used for optimizing the geometries of drugs. AMBER (Assisted Model Building with Energy Refinement) is a common protein force field. GAFF has specifically been parameterized for organic molecules made of C, N, O, H, S, P, F, Cl, Br, and I. 146

147 Finding Conformers of Molecules Conformers How to search for low-energy conformations Start with your molecule of interest Optimize the geometry Before finding a low-energy conformer, we should make sure the current geometry is at least a near-optimal geometry. Perform an "Optimize Geometry" for a quick clean-up. 147


148 Finding Conformers of Molecules Now we will want to change the force field and/or the number of optimization steps. Change the force field options You may want to change the force field used. MMFF94 is a good default for organic-like molecules. If the molecule contains metals or is otherwise unusual, UFF is a better choice. Next, change the number of geometry optimization steps for each conformer tested, it will be optimized. Switch to a small number like 20 or 50 for a quick clean-up to prevent "clashing" atoms when bonds are rotated. Set the conformer search options 148

 If you pick a Weighted Search, set the number of conformers to test (e.g., 200).")
149 Finding Conformers of Molecules Pick a search method and start the search 1) Pick a search method. Systematic searches are exhaustive, but will always find the global minimum. A Weighted rotor search or Genetic Algorithm search are preferred. 2) If you pick a Weighted Search, set the number of conformers to test (e.g., 200). 3) If you pick the Genetic Algorithm search, set the options, including the Energy scoring method. 4) Click "OK" to start the search. 149
150 Finding Conformers of Molecules 150

151 Geometry Constraints Constraints & Optimizations Avogadro allows for the optimization of an object, with respect to a variable(s). Below is one example of how constraints can be applied while optimizing a molecule. Constraints Constraints can be applied to fix or ignore a specific selection of atoms in a molecule, as well as to fix distances, and angles. For additional information review the "Auto-Optimization Tool" and "The Extensions Menu" sections of this manual. Example Let's say we have a diene, that we want to fix in a cisoid conformation before optimizing the geometry of the rest of the molecule (example shown below). After drawing your molecule, check the label display type, and click the wrench to the right of the name. Select the "Atom number" label form and close the dialog box. 151
152 Geometry Constraints This will label all of the atom indices. Before we can apply the constraint, we'll need to figure out the distance between atom 3 & 6. Following the procedure displayed below, select the measure tool, and then choose the two atoms that you want to apply a constraint to. This will output a distance in angstroms at the bottom of the screen. 152
153 Geometry Constraints From here go to the "Extensions" menu, and under "Molecular Mechanics", select "Constraints...". Then choose "Distance", as the type of constraint, enter in the length (3.092 Å), and the atom indices. Select "Add", and then click "OK", to add the constraint and close the dialog box. 153

154 Geometry Constraints Optimizations Optimizations can then be applied to work around the constraint. For more information about force fields refer to the "Molecular Mechanics and Force Fields" section of this manual. Example cont. Now (post addition of constraint), Avogadro will selectively keep the cisoid conformation while concurrently adjusting the parameters of the other atoms in the molecule. 154
155 Geometry Constraints 155
156 Different Display Styles Different Display Types in Avogadro Avogadro comes equipped with various display types to aid in molecular interpretation. The default engine plugins include Axes, Ball and Stick, Cartoon, Dipole, Force, Hydrogen Bond, Label, Polygon, QTAIM, Ribbon, Ring, Simple Wireframe, Stick, Surface, Van der Waals Spheres, and Wireframe. Locating Display Types The different display types can be accessed by clicking "Display Settings..." in the top middle of the open Avogadro window. The display types toolbar will be added above the "Tool Settings..." toolbar that is currently opened. All of the plugins featured below can be used in conjunction with one another. Clicking on the wrench located on the right of some display types will allow various adjustments to be made to the display. For example, you can choose to edit the opacity of the Van der Waals Spheres feature so that you can still view the ball and stick model underneath. 156

157 Different Display Styles Axes Clicking on the Axes plugin will provide the cartesian axes of the molecule from the origin. Note that the red, green, and blue arrows represent the x, y, and z axes respectively. 157
158 Different Display Styles Ball and Stick Ball and Stick is the default plugin when Avogadro is opened. This plugin provides the standard ball and stick representation of a molecule. 158
159 Different Display Styles Cartoon The cartoon feature only applies to secondary biological structures (α helix and β sheet). Below is the cartoon for hemoglobin. 159
160 Different Display Styles Dipole The Dipole plugin will display an overall net dipole if one is present. 160
161 Different Display Styles Force The Force plugin displays green arrows on atoms (as shown below), to qualitatively demonstrate the forces being applied to the atoms. 161
162 Different Display Styles Hydrogen Bond The Hydrogen Bond plugin demonstrates implicit hydrogen bonding that can occur between atoms. 162
163 Different Display Styles Label The label plugin numbers and labels all atoms present in a molecule. 163
164 Different Display Styles Polygon The Polygon feature takes metallic centers with three or more atoms bonded to them, and draws a polygon around them. 164
165 Different Display Styles QTAIM (Quantum Theory of Atoms in Molecules) QTAIM displays the implicit bonding that is theorized to take place between the hydrogens of organic crystals (the implicit bonding is conveyed through dots). This display type is utilized by importing a.wfn file from the "QTAIM", "Molecular Graph" selection under the "Extensions" menu. More information can be found on this process in the Tutorial section of this manual. 165
166 Different Display Styles Ribbon Similar to the Cartoon plugin, the Ribbon plugin conveys secondary biological structures as a simple ribbon rendering. 166
167 Different Display Styles Ring This feature distinguishes rings with different colors dependent on their size. As shown below a six-membered ring is purple, and five membered ring is blue, etc. 167
168 Different Display Styles Simple Wireframe This feature provides a basic wireframe display of a molecule. 168
169 Different Display Styles Stick Stick is another molecular visual display type, that renders a stick representation of a molecule. 169
170 Different Display Styles Surfaces Once a surface has been created (Extensions Menu -> Create Surfaces...), the Surface display type can be used. This display type allows adjustments to the orbital, opacity, rendering, style, and color. 170
171 Different Display Styles Van der Waals Spheres The Van der Waals plugin provides the classic sphere rendered Van der Waals image. 171
172 Different Display Styles Wireframe Unlike Simple Wireframe, Wireframe draws atoms and bond order into the molecule. 172
173 Different Display Styles 173
174 Coloring Part of a Molecules Coloring Part of a Molecule Coloring various parts of a molecule can provide a more visually stimulating way to display qualitative information. Hemoglobin Below hemoglobin is depicted in its ball and stick form (file imported from PDB). Select by Residue We can select specific residues in the molecule through the "Select" menu. 174
175 Coloring Part of a Molecules After typing in the residue name (this feature is case sensitive), and clicking "OK", adjustments can be made to emphasize the selection. 175

176 Coloring Part of a Molecules After making a selection, click the wrench next to the display type you're choosing to edit (in this instance the Ball and Stick display). When the dialog box pops up select "Objects", and then click the blue table button in the bottom right hand corner. This feature adjusts what was initally considered an object (Hemoglobin), and edits the selection so that the display type will only encompass the heme residues. 176
177 Coloring Part of a Molecules From there you can edit other settings of the display type by clicking on "Settings" or "Colors". 177
178 Coloring Part of a Molecules Select by Element Coloring parts of a molecule doesn't have to end after a single adjustment. Say we want to stress that iron is at the center of the hem residue. We can go to the "Select" menu, "Select by Element...", and choose to select all of the iron atoms in the molecule. 178

179 Coloring Part of a Molecules After the selection has been made, with the same procedure as before, we can choose a display type and edit the selection so that it only encompasses the iron atoms. 179

180 Coloring Part of a Molecules Then adjustments can be made to the settings. 180
181 Coloring Part of a Molecules The final product is a more intuitive view of qualitative information about hemoglobin. 181

182 Using QTAIM (Atoms in Molecules) Analysis Using Atoms-In-Molecules (Bader) Analysis: QTAIM and WFN Avogadro now includes support for the QTAIM analysis developed by Prof. Richard Bader and his group. This technique allows Avogadro to determine bond connections and bond orders directly from the quantum mechanical electron density. Start with a WFN File Most quantum chemistry software allows creating a WFN format file from the output of a calculation. We can read this file in through the QTAIM Extension for visualization and analysis. In the future, other formats will also be supported, including deriving the data directly from checkpoint files. 182
")


183 Using QTAIM (Atoms in Molecules) Analysis Here we've picked a WFN file of interest (HCO2) using the Molecular Graph + Lone Pairs command. Avogadro's QTAIM support will read in the atoms, determine bonds and bond orders using AIM analysis and then continue with further analysis. 183

184 Using QTAIM (Atoms in Molecules) Analysis Initially, you may not see anything different. QTAIM includes a separate Display Type for viewing the results of the AIM analysis. Here, we've enabled the QTAIM display, which will show lone pairs, bond critical points, etc. You may also want to turn off the Ball and Stick model to see all AIM annotations. Here, we've also turned off the classical ball and stick display type to leave only the QTAIM view of HCO2. 184
 After opening an output file that has been run with the keyword \"freq\", the \"Vibrations\" toolbar will automatically open. This toolbar allows you to view the various vibrations of a molecule.")
185 Viewing Vibrations Viewing Vibrations This feature allows visualizing vibrations from a "frequency" calculation with quantum chemistry codes (e.g., Gaussian, Q-Chem, etc.) After opening an output file that has been run with the keyword "freq", the "Vibrations" toolbar will automatically open. This toolbar allows you to view the various vibrations of a molecule. Selecting a frequency, and clicking "Start Animation" will begin a vibration. 185
186 Viewing Vibrations Avogadro then provides you with the ability to edit the vibration amplitude, it allows you to display the force vectors on the atoms present, and it allows you to adjust the animation speed by the frequency. 186
187 Viewing Vibrations 187
188 Viewing Vibrational Spectra Calculations Viewing Spectral Properties This tutorial covers plotting vibrational spectra, but other types of spectra are possible with the output files from quantum chemical programs After opening an output file that has been run with the keyword "freq", the "Vibrations" toolbar will automatically open. Show Spectra... Clicking on "Show Spectra..." in the Vibrations toolbar, or selecting "Spectra..." under the "Extensions" menu will open a spectra visualization of the molecule (displayed below). 188

189 Viewing Vibrational Spectra Calculations Selecting "Advanced>>" in the spectra visualization will allow you to change the visual settings, as well as the spectral settings, and export the image. The "Advanced>>" selection provides a multitude of general edits to use at your convenience. For example, you can change % transmittance, to % absorbance (under Infrared Spectra Settings), and change the scheme from dark to publication (Appearance Settings). 189

190 Viewing Vibrational Spectra Calculations 190
191 Viewing Molecular Orbitals Viewing Molecular Orbitals This feature requires a "checkpoint" or "formatted checkpoint" from quantum chemistry codes When the output file is opened, if a matching checkpoint file is found, it automatically opens the Orbitals toolbar. All potential molecular orbitals will have full status bars (you may need to scroll down considerably to find the potential orbitals). Clicking in the row of an orbital, with a full status bar, will create a quick low quality rendition of the orbital. 191
192 Viewing Molecular Orbitals A higher orbital quality can be selected and applied if desired. This is done by selecting a new image quality from the drop down menu, and clicking "render". These renditions can take a moment to load. 192

193 Viewing Molecular Orbitals Configure Selecting "Configure" allows you to adjust the default parameters for the orbital toolbar. Once the parameters have been adjusted, click "Recalculate All" before closing the dialog box. "Recalculate All" reevaluates, and updates all of the parameters. 193
194 Viewing Electrostatic Potential Maps Viewing Electrostatic Potential Maps The electrostatic potential maps help to visualize charge distribution, and other charge related properties of molecules. Overall, let's say you want to determine visually if a specific proton has more or less electron density. First, you'll want to begin with your molecule of choice (shown below is trifluoracetic acid). Then under the "Extensions" menu, select "Create Surfaces...". 194

195 Viewing Electrostatic Potential Maps A dialog box will pop up providing you with various surface options. Under "Color By:" select "Electrostatic Potential", and then click "Calculate". After Avogadro calculates the surface select "Close". 195
, and where the least electron density resides (deep blue areas).")
196 Viewing Electrostatic Potential Maps An electrostatic surface has now been created. From this surface, you can interpret where the most electron density resides (in the more red areas), and where the least electron density resides (deep blue areas). You can further determine, and compare the acidity of various protons, and how surrounding atoms impact the overall electron density. 196

197 Viewing Electrostatic Potential Maps This example was taken from "Exploring the Acidity of Organic Molecules with Avogadro" written by Tamika Madison. Changing Surface Settings The opacity, rendition, and colors of the surface can the be changed by clicking the wrench next to the "Surfaces" display type. 197

198 Viewing Electrostatic Potential Maps 198

 while the server returns the name, (unknown) if the molecule is not")
199 Naming a Molecule Naming a molecule with PubChem Avogadro 1.1 and later includes support for naming compounds using the NIH Chemical Resolver system and the PubChem database. Open up the Molecule Properties window, under the View menu. The IUPAC name will initially show as (pending) while the server returns the name, (unknown) if the molecule is not found in the resolver, or (unavailable) if your network connection is down or the resolver service is otherwise unreachable. 199

200 Naming a Molecule As you modify the molecule (e.g., adding a Cl atom), the name will update automatically with the other properties. 200

201 ABINIT Input Generator ABINIT Generator Avogadro has a new interface to the open source solid-state code ABINIT, provided by one of the ABINIT developers, Prof. Matthieu Verstraete Start with a crystal structure here a clay mineral from the new Avogadro crystal library. 201

 generated as you change")
202 ABINIT Input Generator Open up the Abinit input generator under the Extensions menu Most common options for Abinit are available here, with a text preview (highlighted in red) generated as you change options above. 202
 pick the type of geometry optimization (if any).")
 if Abinit is available locally on your")
203 ABINIT Input Generator The input generator allows you to (1) pick the type of coordinates: real-space Cartesians or reduced coordinates and (2) pick the type of geometry optimization (if any). Another important option is setting the occupation scheme (1) and SCF options (2) and (3). Finally, when finished, you can click the Compute button (not pictured) if Abinit is available locally on your computer, or Generate to save the input file for submission to a remote queue. 203

204 ABINIT Input Generator Save the input file and you re finished! 204
205 LAMMPS Input LAMMPS input for water Prepare initial geometry The independently developed Packmol extension can be used to generate a box of water molecules. Open the LAMMPS input dialog 205


206 LAMMPS Input Prepare simulation parameters 1. Choose the number of repeating units of the input coordintes in x, y and z directions 2. Choose the water potential. The current version supports SPC and SPC/E model potentials 3. Choose the name of LAMMPS formatted coordinates. The name will be used in a later 206

207 LAMMPS Input step when the lmpdat file is created. 4. Choose the total number of MD steps. 5. Choose the file name of the XYZ formatted trajectory file. Generate the LAMMPS parameters file 1. Click the Generate button 2. Choose a file name 3. Click save 4. Close the input generator dialog Generate the LAMMPS Coordintes file 207

208 LAMMPS Input 1. Select "Save As" from the file menu 2. Input the "water.lmpdat" file name from above 3. Select "All files" 4. Save the LAMMPS formatted coordinates file Run LAMMPS 208

209 LAMMPS Input After 2700 time steps, the temperature is begining to stabilize. 209
Exercises for Windows
 Exercises for Windows CAChe User Interface for Windows Select tool Application window Document window (workspace) Style bar Tool palette Select entire molecule Select Similar Group Select Atom tool Rotate
Exercises for Windows CAChe User Interface for Windows Select tool Application window Document window (workspace) Style bar Tool palette Select entire molecule Select Similar Group Select Atom tool Rotate
NMR Predictor. Introduction
 NMR Predictor This manual gives a walk-through on how to use the NMR Predictor: Introduction NMR Predictor QuickHelp NMR Predictor Overview Chemical features GUI features Usage Menu system File menu Edit
NMR Predictor This manual gives a walk-through on how to use the NMR Predictor: Introduction NMR Predictor QuickHelp NMR Predictor Overview Chemical features GUI features Usage Menu system File menu Edit
General Chemistry Lab Molecular Modeling
 PURPOSE The objectives of this experiment are PROCEDURE General Chemistry Lab Molecular Modeling To learn how to use molecular modeling software, a commonly used tool in chemical research and industry.
PURPOSE The objectives of this experiment are PROCEDURE General Chemistry Lab Molecular Modeling To learn how to use molecular modeling software, a commonly used tool in chemical research and industry.
Application Note. U. Heat of Formation of Ethyl Alcohol and Dimethyl Ether. Introduction
 Application Note U. Introduction The molecular builder (Molecular Builder) is part of the MEDEA standard suite of building tools. This tutorial provides an overview of the Molecular Builder s basic functionality.
Application Note U. Introduction The molecular builder (Molecular Builder) is part of the MEDEA standard suite of building tools. This tutorial provides an overview of the Molecular Builder s basic functionality.
Introduction Molecular Structure Script Console External resources Advanced topics. JMol tutorial. Giovanni Morelli.
 Gen 19th, 2017 1 2 Create and edit Display and view Mesurament and labelling Surface and Orbitals 3 4 from Database Protein Enzyme Crystal Structure and Unit Cell 5 Symmetry Animation General information
Gen 19th, 2017 1 2 Create and edit Display and view Mesurament and labelling Surface and Orbitals 3 4 from Database Protein Enzyme Crystal Structure and Unit Cell 5 Symmetry Animation General information
Molecular Modeling and Conformational Analysis with PC Spartan
 Molecular Modeling and Conformational Analysis with PC Spartan Introduction Molecular modeling can be done in a variety of ways, from using simple hand-held models to doing sophisticated calculations on
Molecular Modeling and Conformational Analysis with PC Spartan Introduction Molecular modeling can be done in a variety of ways, from using simple hand-held models to doing sophisticated calculations on
Tutorial. Getting started. Sample to Insight. March 31, 2016
 Getting started March 31, 2016 Sample to Insight CLC bio, a QIAGEN Company Silkeborgvej 2 Prismet 8000 Aarhus C Denmark Telephone: +45 70 22 32 44 www.clcbio.com support-clcbio@qiagen.com Getting started
Getting started March 31, 2016 Sample to Insight CLC bio, a QIAGEN Company Silkeborgvej 2 Prismet 8000 Aarhus C Denmark Telephone: +45 70 22 32 44 www.clcbio.com support-clcbio@qiagen.com Getting started
Assignment 1: Molecular Mechanics (PART 1 25 points)
") Chemistry 380.37 Fall 2015 Dr. Jean M. Standard August 19, 2015 Assignment 1: Molecular Mechanics (PART 1 25 points) In this assignment, you will perform some molecular mechanics calculations using the
Chemistry 380.37 Fall 2015 Dr. Jean M. Standard August 19, 2015 Assignment 1: Molecular Mechanics (PART 1 25 points) In this assignment, you will perform some molecular mechanics calculations using the
Chemistry 14CL. Worksheet for the Molecular Modeling Workshop. (Revised FULL Version 2012 J.W. Pang) (Modified A. A. Russell)
 (Modified A. A. Russell)") Chemistry 14CL Worksheet for the Molecular Modeling Workshop (Revised FULL Version 2012 J.W. Pang) (Modified A. A. Russell) Structure of the Molecular Modeling Assignment The molecular modeling assignment
Chemistry 14CL Worksheet for the Molecular Modeling Workshop (Revised FULL Version 2012 J.W. Pang) (Modified A. A. Russell) Structure of the Molecular Modeling Assignment The molecular modeling assignment
ISIS/Draw "Quick Start"
 ISIS/Draw "Quick Start" Click to print, or click Drawing Molecules * Basic Strategy 5.1 * Drawing Structures with Template tools and template pages 5.2 * Drawing bonds and chains 5.3 * Drawing atoms 5.4
ISIS/Draw "Quick Start" Click to print, or click Drawing Molecules * Basic Strategy 5.1 * Drawing Structures with Template tools and template pages 5.2 * Drawing bonds and chains 5.3 * Drawing atoms 5.4
Chem 253. Tutorial for Materials Studio
 Chem 253 Tutorial for Materials Studio This tutorial is designed to introduce Materials Studio 7.0, which is a program used for modeling and simulating materials for predicting and rationalizing structure
Chem 253 Tutorial for Materials Studio This tutorial is designed to introduce Materials Studio 7.0, which is a program used for modeling and simulating materials for predicting and rationalizing structure
Molecular Visualization. Introduction
 Molecular Visualization Jeffry D. Madura Department of Chemistry & Biochemistry Center for Computational Sciences Duquesne University Introduction Assessments of change, dynamics, and cause and effect
Molecular Visualization Jeffry D. Madura Department of Chemistry & Biochemistry Center for Computational Sciences Duquesne University Introduction Assessments of change, dynamics, and cause and effect
SeeSAR 7.1 Beginners Guide. June 2017
 SeeSAR 7.1 Beginners Guide June 2017 Part 1: Basics 1 Type a pdb code and press return or Load your own protein or already existing project, or Just load molecules To begin, let s type 2zff and download
SeeSAR 7.1 Beginners Guide June 2017 Part 1: Basics 1 Type a pdb code and press return or Load your own protein or already existing project, or Just load molecules To begin, let s type 2zff and download
PQSMol. User s Manual
 PQSMol User s Manual c Parallel Quantum Solutions, 2006 www.pqs-chem.com Conventions used in this manual Filenames, commands typed in at the command prompt are written in typewriter font: pqsview aspirin.out
PQSMol User s Manual c Parallel Quantum Solutions, 2006 www.pqs-chem.com Conventions used in this manual Filenames, commands typed in at the command prompt are written in typewriter font: pqsview aspirin.out
Let s continue our discussion on the interaction between Fe(III) and 6,7-dihydroxynaphthalene-2- sulfonate.
 and 6,7-dihydroxynaphthalene-2- sulfonate.") Chemistry 5995(133)-8990(013) Bioinorganic Chemistry: The Good, the Bad, and the Potential of Metals Assignment 2- Aqueous Speciation, Magnetism, Redox, UV-Vis Spectroscopy, and Pymol Let s continue our
Chemistry 5995(133)-8990(013) Bioinorganic Chemistry: The Good, the Bad, and the Potential of Metals Assignment 2- Aqueous Speciation, Magnetism, Redox, UV-Vis Spectroscopy, and Pymol Let s continue our
Preparing a PDB File
 Figure 1: Schematic view of the ligand-binding domain from the vitamin D receptor (PDB file 1IE9). The crystallographic waters are shown as small spheres and the bound ligand is shown as a CPK model. HO
Figure 1: Schematic view of the ligand-binding domain from the vitamin D receptor (PDB file 1IE9). The crystallographic waters are shown as small spheres and the bound ligand is shown as a CPK model. HO
Literature values: ΔH f, gas = % error Source: ΔH f, solid = % error. For comparison, your experimental value was ΔH f = phase:
 1 Molecular Calculations Lab: Some guideline given at the bottom of page 3. 1. Use the semi-empirical AM1 method to calculate ΔH f for the compound you used in the heat of combustion experiment. Be sure
1 Molecular Calculations Lab: Some guideline given at the bottom of page 3. 1. Use the semi-empirical AM1 method to calculate ΔH f for the compound you used in the heat of combustion experiment. Be sure
Comparing whole genomes
 BioNumerics Tutorial: Comparing whole genomes 1 Aim The Chromosome Comparison window in BioNumerics has been designed for large-scale comparison of sequences of unlimited length. In this tutorial you will
BioNumerics Tutorial: Comparing whole genomes 1 Aim The Chromosome Comparison window in BioNumerics has been designed for large-scale comparison of sequences of unlimited length. In this tutorial you will
3D - Structure Graphics Capabilities with PDF-4 Database Products
 3D - Structure Graphics Capabilities with PDF-4 Database Products Atomic Structure Information in the PDF-4 Databases ICDD s PDF-4 databases contain atomic structure information for a significant number
3D - Structure Graphics Capabilities with PDF-4 Database Products Atomic Structure Information in the PDF-4 Databases ICDD s PDF-4 databases contain atomic structure information for a significant number
1 Introduction to Computational Chemistry (Spartan)
") 1 Introduction to Computational Chemistry (Spartan) Start Spartan by clicking Start / Programs / Spartan Then click File / New Exercise 1 Study of H-X-H Bond Angles (Suitable for general chemistry) Structure
1 Introduction to Computational Chemistry (Spartan) Start Spartan by clicking Start / Programs / Spartan Then click File / New Exercise 1 Study of H-X-H Bond Angles (Suitable for general chemistry) Structure
Ligand Scout Tutorials
 Ligand Scout Tutorials Step : Creating a pharmacophore from a protein-ligand complex. Type ke6 in the upper right area of the screen and press the button Download *+. The protein will be downloaded and
Ligand Scout Tutorials Step : Creating a pharmacophore from a protein-ligand complex. Type ke6 in the upper right area of the screen and press the button Download *+. The protein will be downloaded and
Introduction to Spark
 1 As you become familiar or continue to explore the Cresset technology and software applications, we encourage you to look through the user manual. This is accessible from the Help menu. However, don t
1 As you become familiar or continue to explore the Cresset technology and software applications, we encourage you to look through the user manual. This is accessible from the Help menu. However, don t
Space Objects. Section. When you finish this section, you should understand the following:
 GOLDMC02_132283433X 8/24/06 2:21 PM Page 97 Section 2 Space Objects When you finish this section, you should understand the following: How to create a 2D Space Object and label it with a Space Tag. How
GOLDMC02_132283433X 8/24/06 2:21 PM Page 97 Section 2 Space Objects When you finish this section, you should understand the following: How to create a 2D Space Object and label it with a Space Tag. How
Mnova Software for Analyzing Reaction Monitoring NMR Spectra
 Mnova Software for Analyzing Reaction Monitoring NMR Spectra Version 10 Chen Peng, PhD, VP of Business Development, US & China Mestrelab Research SL San Diego, CA, USA chen.peng@mestrelab.com 858.736.4563
Mnova Software for Analyzing Reaction Monitoring NMR Spectra Version 10 Chen Peng, PhD, VP of Business Development, US & China Mestrelab Research SL San Diego, CA, USA chen.peng@mestrelab.com 858.736.4563
Silica surface - Materials Studio tutorial. CREATING SiO 2 SURFACE
 Silica surface - Materials Studio tutorial CREATING SiO 2 SURFACE Our goal surface of SiO2 6.948 Ǻ Import structure The XRD experiment gives us such parameters as: lattice parameters, symmetry group and
Silica surface - Materials Studio tutorial CREATING SiO 2 SURFACE Our goal surface of SiO2 6.948 Ǻ Import structure The XRD experiment gives us such parameters as: lattice parameters, symmetry group and
Dock Ligands from a 2D Molecule Sketch
 Dock Ligands from a 2D Molecule Sketch March 31, 2016 Sample to Insight CLC bio, a QIAGEN Company Silkeborgvej 2 Prismet 8000 Aarhus C Denmark Telephone: +45 70 22 32 44 www.clcbio.com support-clcbio@qiagen.com
Dock Ligands from a 2D Molecule Sketch March 31, 2016 Sample to Insight CLC bio, a QIAGEN Company Silkeborgvej 2 Prismet 8000 Aarhus C Denmark Telephone: +45 70 22 32 44 www.clcbio.com support-clcbio@qiagen.com
Assignment 1: Molecular Mechanics (PART 2 25 points)
") Chemistry 380.37 Fall 2015 Dr. Jean M. Standard September 2, 2015 Assignment 1: Molecular Mechanics (PART 2 25 points) In this assignment, you will perform some additional molecular mechanics calculations
Chemistry 380.37 Fall 2015 Dr. Jean M. Standard September 2, 2015 Assignment 1: Molecular Mechanics (PART 2 25 points) In this assignment, you will perform some additional molecular mechanics calculations
Calculating NMR Chemical Shifts for beta-ionone O
 Calculating NMR Chemical Shifts for beta-ionone O Molecular orbital calculations can be used to get good estimates for chemical shifts. In this exercise we will calculate the chemical shifts for beta-ionone.
Calculating NMR Chemical Shifts for beta-ionone O Molecular orbital calculations can be used to get good estimates for chemical shifts. In this exercise we will calculate the chemical shifts for beta-ionone.
Pymol Practial Guide
 Pymol Practial Guide Pymol is a powerful visualizor very convenient to work with protein molecules. Its interface may seem complex at first, but you will see that with a little practice is simple and powerful
Pymol Practial Guide Pymol is a powerful visualizor very convenient to work with protein molecules. Its interface may seem complex at first, but you will see that with a little practice is simple and powerful
Introduc)on to IQmol: Part I.!!! Shirin Faraji, Ilya Kaliman, and Anna Krylov
on to IQmol: Part I.!!! Shirin Faraji, Ilya Kaliman, and Anna Krylov") Introduc)on to IQmol: Part I!!! Shirin Faraji, Ilya Kaliman, and Anna Krylov! 1 Resources! Written by Dr. Andrew Gilbert Keep yourself up to date with IQmol website: http://iqmol.org! IQmol Youtube channel:
Introduc)on to IQmol: Part I!!! Shirin Faraji, Ilya Kaliman, and Anna Krylov! 1 Resources! Written by Dr. Andrew Gilbert Keep yourself up to date with IQmol website: http://iqmol.org! IQmol Youtube channel:
Version 1.2 October 2017 CSD v5.39
 Mogul Geometry Check Table of Contents Introduction... 2 Example 1. Using Mogul to assess intramolecular geometry... 3 Example 2. Using Mogul to explain activity data... 5 Conclusions... 8 Further Exercises...
Mogul Geometry Check Table of Contents Introduction... 2 Example 1. Using Mogul to assess intramolecular geometry... 3 Example 2. Using Mogul to explain activity data... 5 Conclusions... 8 Further Exercises...
Physical Chemistry Analyzing a Crystal Structure and the Diffraction Pattern Virginia B. Pett The College of Wooster
 Physical Chemistry Analyzing a Crystal Structure and the Diffraction Pattern Virginia B. Pett The College of Wooster L. W. Haynes and his Senior Independent Study students conducted the 2 + 2 photo addition
Physical Chemistry Analyzing a Crystal Structure and the Diffraction Pattern Virginia B. Pett The College of Wooster L. W. Haynes and his Senior Independent Study students conducted the 2 + 2 photo addition
Protein Bioinformatics Computer lab #1 Friday, April 11, 2008 Sean Prigge and Ingo Ruczinski
 Protein Bioinformatics 260.655 Computer lab #1 Friday, April 11, 2008 Sean Prigge and Ingo Ruczinski Goals: Approx. Time [1] Use the Protein Data Bank PDB website. 10 minutes [2] Use the WebMol Viewer.
Protein Bioinformatics 260.655 Computer lab #1 Friday, April 11, 2008 Sean Prigge and Ingo Ruczinski Goals: Approx. Time [1] Use the Protein Data Bank PDB website. 10 minutes [2] Use the WebMol Viewer.
Calculating Bond Enthalpies of the Hydrides
 Proposed Exercise for the General Chemistry Section of the Teaching with Cache Workbook: Calculating Bond Enthalpies of the Hydrides Contributed by James Foresman, Rachel Fogle, and Jeremy Beck, York College
Proposed Exercise for the General Chemistry Section of the Teaching with Cache Workbook: Calculating Bond Enthalpies of the Hydrides Contributed by James Foresman, Rachel Fogle, and Jeremy Beck, York College
Examples of Protein Modeling. Protein Modeling. Primary Structure. Protein Structure Description. Protein Sequence Sources. Importing Sequences to MOE
 Examples of Protein Modeling Protein Modeling Visualization Examination of an experimental structure to gain insight about a research question Dynamics To examine the dynamics of protein structures To
Examples of Protein Modeling Protein Modeling Visualization Examination of an experimental structure to gain insight about a research question Dynamics To examine the dynamics of protein structures To
Computer simulation of radioactive decay
 Computer simulation of radioactive decay y now you should have worked your way through the introduction to Maple, as well as the introduction to data analysis using Excel Now we will explore radioactive
Computer simulation of radioactive decay y now you should have worked your way through the introduction to Maple, as well as the introduction to data analysis using Excel Now we will explore radioactive
LAB 2 - ONE DIMENSIONAL MOTION
 Name Date Partners L02-1 LAB 2 - ONE DIMENSIONAL MOTION OBJECTIVES Slow and steady wins the race. Aesop s fable: The Hare and the Tortoise To learn how to use a motion detector and gain more familiarity
Name Date Partners L02-1 LAB 2 - ONE DIMENSIONAL MOTION OBJECTIVES Slow and steady wins the race. Aesop s fable: The Hare and the Tortoise To learn how to use a motion detector and gain more familiarity
Tutorial: Structural Analysis of a Protein-Protein Complex
 Molecular Modeling Section (MMS) Department of Pharmaceutical and Pharmacological Sciences University of Padova Via Marzolo 5-35131 Padova (IT) @contact: stefano.moro@unipd.it Tutorial: Structural Analysis
Molecular Modeling Section (MMS) Department of Pharmaceutical and Pharmacological Sciences University of Padova Via Marzolo 5-35131 Padova (IT) @contact: stefano.moro@unipd.it Tutorial: Structural Analysis
Zeeman Effect Physics 481
 Zeeman Effect Introduction You are familiar with Atomic Spectra, especially the H- atom energy spectrum. Atoms emit or absorb energies in packets, or quanta which are photons. The orbital motion of electrons
Zeeman Effect Introduction You are familiar with Atomic Spectra, especially the H- atom energy spectrum. Atoms emit or absorb energies in packets, or quanta which are photons. The orbital motion of electrons
Table of Contents. Table of Contents Converting lattices: Rhombohedral to hexagonal and back
 Table of Contents Table of Contents Converting lattices: Rhombohedral to hexagonal and back Conversion between hp and hr representations Converting hp supercell to hr primitive cell Crystal classifications
Table of Contents Table of Contents Converting lattices: Rhombohedral to hexagonal and back Conversion between hp and hr representations Converting hp supercell to hr primitive cell Crystal classifications
Computational Chemistry Lab Module: Conformational Analysis of Alkanes
 Introduction Computational Chemistry Lab Module: Conformational Analysis of Alkanes In this experiment, we will use CAChe software package to model the conformations of butane, 2-methylbutane, and substituted
Introduction Computational Chemistry Lab Module: Conformational Analysis of Alkanes In this experiment, we will use CAChe software package to model the conformations of butane, 2-methylbutane, and substituted
POC via CHEMnetBASE for Identifying Unknowns
 Table of Contents A red arrow was used to identify where buttons and functions are located in CHEMnetBASE. Figure Description Page Entering the Properties of Organic Compounds (POC) Database 1 Swain Home
Table of Contents A red arrow was used to identify where buttons and functions are located in CHEMnetBASE. Figure Description Page Entering the Properties of Organic Compounds (POC) Database 1 Swain Home
Maestro 8.5. Tutorial. Schrödinger Press
 Maestro Tutorial Maestro 8.5 Tutorial Schrödinger Press Maestro Tutorial Copyright 2008 Schrödinger, LLC. All rights reserved. While care has been taken in the preparation of this publication, Schrödinger
Maestro Tutorial Maestro 8.5 Tutorial Schrödinger Press Maestro Tutorial Copyright 2008 Schrödinger, LLC. All rights reserved. While care has been taken in the preparation of this publication, Schrödinger
Learning to Use Scigress Wagner, Eugene P. (revised May 15, 2018)
") Learning to Use Scigress Wagner, Eugene P. (revised May 15, 2018) Abstract Students are introduced to basic features of Scigress by building molecules and performing calculations on them using semi-empirical
Learning to Use Scigress Wagner, Eugene P. (revised May 15, 2018) Abstract Students are introduced to basic features of Scigress by building molecules and performing calculations on them using semi-empirical
Assignment 2: Conformation Searching (50 points)
") Chemistry 380.37 Fall 2015 Dr. Jean M. Standard September 16, 2015 Assignment 2: Conformation Searching (50 points) In this assignment, you will use the Spartan software package to investigate some conformation
Chemistry 380.37 Fall 2015 Dr. Jean M. Standard September 16, 2015 Assignment 2: Conformation Searching (50 points) In this assignment, you will use the Spartan software package to investigate some conformation
How to Make or Plot a Graph or Chart in Excel
 This is a complete video tutorial on How to Make or Plot a Graph or Chart in Excel. To make complex chart like Gantt Chart, you have know the basic principles of making a chart. Though I have used Excel
This is a complete video tutorial on How to Make or Plot a Graph or Chart in Excel. To make complex chart like Gantt Chart, you have know the basic principles of making a chart. Though I have used Excel
ICM-Chemist-Pro How-To Guide. Version 3.6-1h Last Updated 12/29/2009
 ICM-Chemist-Pro How-To Guide Version 3.6-1h Last Updated 12/29/2009 ICM-Chemist-Pro ICM 3D LIGAND EDITOR: SETUP 1. Read in a ligand molecule or PDB file. How to setup the ligand in the ICM 3D Ligand Editor.
ICM-Chemist-Pro How-To Guide Version 3.6-1h Last Updated 12/29/2009 ICM-Chemist-Pro ICM 3D LIGAND EDITOR: SETUP 1. Read in a ligand molecule or PDB file. How to setup the ligand in the ICM 3D Ligand Editor.
ICM-Chemist How-To Guide. Version 3.6-1g Last Updated 12/01/2009
 ICM-Chemist How-To Guide Version 3.6-1g Last Updated 12/01/2009 ICM-Chemist HOW TO IMPORT, SKETCH AND EDIT CHEMICALS How to access the ICM Molecular Editor. 1. Click here 2. Start sketching How to sketch
ICM-Chemist How-To Guide Version 3.6-1g Last Updated 12/01/2009 ICM-Chemist HOW TO IMPORT, SKETCH AND EDIT CHEMICALS How to access the ICM Molecular Editor. 1. Click here 2. Start sketching How to sketch
POC via CHEMnetBASE for Identifying Unknowns
 Table of Contents A red arrow is used to identify where buttons and functions are located in CHEMnetBASE. Figure Description Page Entering the Properties of Organic Compounds (POC) Database 1 CHEMnetBASE
Table of Contents A red arrow is used to identify where buttons and functions are located in CHEMnetBASE. Figure Description Page Entering the Properties of Organic Compounds (POC) Database 1 CHEMnetBASE
Jaguar DFT Optimizations and Transition State Searches
 Jaguar DFT Optimizations and Transition State Searches Density Functional Theory (DFT) is a quantum mechanical (QM) method that gives results superior to Hartree Fock (HF) in less computational time. A
Jaguar DFT Optimizations and Transition State Searches Density Functional Theory (DFT) is a quantum mechanical (QM) method that gives results superior to Hartree Fock (HF) in less computational time. A
Chem 310, Organic Chemistry Lab Molecular Modeling Using Macromodel
 Chem 310, Organic Chemistry Lab Molecular Modeling Using Macromodel This is a molecular modeling experiment, and should be written up in your lab notebook just as if it were a normal "wet-chemistry" experiment.
Chem 310, Organic Chemistry Lab Molecular Modeling Using Macromodel This is a molecular modeling experiment, and should be written up in your lab notebook just as if it were a normal "wet-chemistry" experiment.
User Guide (Windows and Macintosh)
") User Guide (Windows and Macintosh) WAVEFUNCTION Wavefunction, Inc. 18401 Von Karman Avenue, Suite 370 Irvine, CA 92612 U.S.A. www.wavefun.com Wavefunction, Inc., Japan Branch Office 3-5-2, Kouji-machi,
User Guide (Windows and Macintosh) WAVEFUNCTION Wavefunction, Inc. 18401 Von Karman Avenue, Suite 370 Irvine, CA 92612 U.S.A. www.wavefun.com Wavefunction, Inc., Japan Branch Office 3-5-2, Kouji-machi,
BUILDING BASICS WITH HYPERCHEM LITE
 BUILDING BASICS WITH HYPERCHEM LITE LAB MOD1.COMP From Gannon University SIM INTRODUCTION A chemical bond is a link between atoms resulting from the mutual attraction of their nuclei for electrons. There
BUILDING BASICS WITH HYPERCHEM LITE LAB MOD1.COMP From Gannon University SIM INTRODUCTION A chemical bond is a link between atoms resulting from the mutual attraction of their nuclei for electrons. There
Assignment #0 Using Stellarium
 Name: Class: Date: Assignment #0 Using Stellarium The purpose of this exercise is to familiarize yourself with the Stellarium program and its many capabilities and features. Stellarium is a visually beautiful
Name: Class: Date: Assignment #0 Using Stellarium The purpose of this exercise is to familiarize yourself with the Stellarium program and its many capabilities and features. Stellarium is a visually beautiful
1 An Experimental and Computational Investigation of the Dehydration of 2-Butanol
 1 An Experimental and Computational Investigation of the Dehydration of 2-Butanol Summary. 2-Butanol will be dehydrated to a mixture of 1-butene and cis- and trans-2-butene using the method described in
1 An Experimental and Computational Investigation of the Dehydration of 2-Butanol Summary. 2-Butanol will be dehydrated to a mixture of 1-butene and cis- and trans-2-butene using the method described in
IFM Chemistry Computational Chemistry 2010, 7.5 hp LAB2. Computer laboratory exercise 1 (LAB2): Quantum chemical calculations
: Quantum chemical calculations") Computer laboratory exercise 1 (LAB2): Quantum chemical calculations Introduction: The objective of the second computer laboratory exercise is to get acquainted with a program for performing quantum chemical
Computer laboratory exercise 1 (LAB2): Quantum chemical calculations Introduction: The objective of the second computer laboratory exercise is to get acquainted with a program for performing quantum chemical
Building Inflation Tables and CER Libraries
 Building Inflation Tables and CER Libraries January 2007 Presented by James K. Johnson Tecolote Research, Inc. Copyright Tecolote Research, Inc. September 2006 Abstract Building Inflation Tables and CER
Building Inflation Tables and CER Libraries January 2007 Presented by James K. Johnson Tecolote Research, Inc. Copyright Tecolote Research, Inc. September 2006 Abstract Building Inflation Tables and CER
CHEMDRAW ULTRA ITEC107 - Introduction to Computing for Pharmacy. ITEC107 - Introduction to Computing for Pharmacy 1
 CHEMDRAW ULTRA 12.0 ITEC107 - Introduction to Computing for Pharmacy 1 Objectives Basic drawing skills with ChemDraw Bonds, captions, hotkeys, chains, arrows Checking and cleaning up structures Chemical
CHEMDRAW ULTRA 12.0 ITEC107 - Introduction to Computing for Pharmacy 1 Objectives Basic drawing skills with ChemDraw Bonds, captions, hotkeys, chains, arrows Checking and cleaning up structures Chemical
How many states. Record high temperature
 Record high temperature How many states Class Midpoint Label 94.5 99.5 94.5-99.5 0 97 99.5 104.5 99.5-104.5 2 102 102 104.5 109.5 104.5-109.5 8 107 107 109.5 114.5 109.5-114.5 18 112 112 114.5 119.5 114.5-119.5
Record high temperature How many states Class Midpoint Label 94.5 99.5 94.5-99.5 0 97 99.5 104.5 99.5-104.5 2 102 102 104.5 109.5 104.5-109.5 8 107 107 109.5 114.5 109.5-114.5 18 112 112 114.5 119.5 114.5-119.5
Viewing and Analyzing Proteins, Ligands and their Complexes 2
 2 Viewing and Analyzing Proteins, Ligands and their Complexes 2 Overview Viewing the accessible surface Analyzing the properties of proteins containing thousands of atoms is best accomplished by representing
2 Viewing and Analyzing Proteins, Ligands and their Complexes 2 Overview Viewing the accessible surface Analyzing the properties of proteins containing thousands of atoms is best accomplished by representing
Physical Chemistry Final Take Home Fall 2003
 Physical Chemistry Final Take Home Fall 2003 Do one of the following questions. These projects are worth 30 points (i.e. equivalent to about two problems on the final). Each of the computational problems
Physical Chemistry Final Take Home Fall 2003 Do one of the following questions. These projects are worth 30 points (i.e. equivalent to about two problems on the final). Each of the computational problems
Searching Substances in Reaxys
 Searching Substances in Reaxys Learning Objectives Understand that substances in Reaxys have different sources (e.g., Reaxys, PubChem) and can be found in Document, Reaction and Substance Records Recognize
Searching Substances in Reaxys Learning Objectives Understand that substances in Reaxys have different sources (e.g., Reaxys, PubChem) and can be found in Document, Reaction and Substance Records Recognize
The CSC Interface to Sky in Google Earth
 The CSC Interface to Sky in Google Earth CSC Threads The CSC Interface to Sky in Google Earth 1 Table of Contents The CSC Interface to Sky in Google Earth - CSC Introduction How to access CSC data with
The CSC Interface to Sky in Google Earth CSC Threads The CSC Interface to Sky in Google Earth 1 Table of Contents The CSC Interface to Sky in Google Earth - CSC Introduction How to access CSC data with
Creating a Pharmacophore Query from a Reference Molecule & Scaffold Hopping in CSD-CrossMiner
 Table of Contents Creating a Pharmacophore Query from a Reference Molecule & Scaffold Hopping in CSD-CrossMiner Introduction... 2 CSD-CrossMiner Terminology... 2 Overview of CSD-CrossMiner... 3 Features
Table of Contents Creating a Pharmacophore Query from a Reference Molecule & Scaffold Hopping in CSD-CrossMiner Introduction... 2 CSD-CrossMiner Terminology... 2 Overview of CSD-CrossMiner... 3 Features
v Prerequisite Tutorials GSSHA WMS Basics Watershed Delineation using DEMs and 2D Grid Generation Time minutes
 v. 10.1 WMS 10.1 Tutorial GSSHA WMS Basics Creating Feature Objects and Mapping Attributes to the 2D Grid Populate hydrologic parameters in a GSSHA model using land use and soil data Objectives This tutorial
v. 10.1 WMS 10.1 Tutorial GSSHA WMS Basics Creating Feature Objects and Mapping Attributes to the 2D Grid Populate hydrologic parameters in a GSSHA model using land use and soil data Objectives This tutorial
GIS Workshop UCLS_Fall Forum 2014 Sowmya Selvarajan, PhD TABLE OF CONTENTS
 TABLE OF CONTENTS TITLE PAGE NO. 1. ArcGIS Basics I 2 a. Open and Save a Map Document 2 b. Work with Map Layers 2 c. Navigate in a Map Document 4 d. Measure Distances 4 2. ArcGIS Basics II 5 a. Work with
TABLE OF CONTENTS TITLE PAGE NO. 1. ArcGIS Basics I 2 a. Open and Save a Map Document 2 b. Work with Map Layers 2 c. Navigate in a Map Document 4 d. Measure Distances 4 2. ArcGIS Basics II 5 a. Work with
Login -the operator screen should be in view when you first sit down at the spectrometer console:
 Lab #2 1D 1 H Double Resonance (Selective Decoupling) operation of the 400 MHz instrument using automated sample insertion (robot) and automated locking and shimming collection of 1D 1 H spectra retrieving
Lab #2 1D 1 H Double Resonance (Selective Decoupling) operation of the 400 MHz instrument using automated sample insertion (robot) and automated locking and shimming collection of 1D 1 H spectra retrieving
PDF-4+ Tools and Searches
 PDF-4+ Tools and Searches PDF-4+ 2018 The PDF-4+ 2018 database is powered by our integrated search display software. PDF-4+ 2018 boasts 72 search selections coupled with 125 display fields resulting in
PDF-4+ Tools and Searches PDF-4+ 2018 The PDF-4+ 2018 database is powered by our integrated search display software. PDF-4+ 2018 boasts 72 search selections coupled with 125 display fields resulting in
PDF-4+ Tools and Searches
 PDF-4+ Tools and Searches PDF-4+ 2019 The PDF-4+ 2019 database is powered by our integrated search display software. PDF-4+ 2019 boasts 74 search selections coupled with 126 display fields resulting in
PDF-4+ Tools and Searches PDF-4+ 2019 The PDF-4+ 2019 database is powered by our integrated search display software. PDF-4+ 2019 boasts 74 search selections coupled with 126 display fields resulting in
Using Microsoft Excel
 Using Microsoft Excel Objective: Students will gain familiarity with using Excel to record data, display data properly, use built-in formulae to do calculations, and plot and fit data with linear functions.
Using Microsoft Excel Objective: Students will gain familiarity with using Excel to record data, display data properly, use built-in formulae to do calculations, and plot and fit data with linear functions.
Create Satellite Image, Draw Maps
 Create Satellite Image, Draw Maps 1. The goal Using Google Earth, we want to create and import a background file into our Adviser program. From there, we will be creating paddock boundaries. The accuracy
Create Satellite Image, Draw Maps 1. The goal Using Google Earth, we want to create and import a background file into our Adviser program. From there, we will be creating paddock boundaries. The accuracy
ON SITE SYSTEMS Chemical Safety Assistant
 ON SITE SYSTEMS Chemical Safety Assistant CS ASSISTANT WEB USERS MANUAL On Site Systems 23 N. Gore Ave. Suite 200 St. Louis, MO 63119 Phone 314-963-9934 Fax 314-963-9281 Table of Contents INTRODUCTION
ON SITE SYSTEMS Chemical Safety Assistant CS ASSISTANT WEB USERS MANUAL On Site Systems 23 N. Gore Ave. Suite 200 St. Louis, MO 63119 Phone 314-963-9934 Fax 314-963-9281 Table of Contents INTRODUCTION
Performing a Pharmacophore Search using CSD-CrossMiner
 Table of Contents Introduction... 2 CSD-CrossMiner Terminology... 2 Overview of CSD-CrossMiner... 3 Searching with a Pharmacophore... 4 Performing a Pharmacophore Search using CSD-CrossMiner Version 2.0
Table of Contents Introduction... 2 CSD-CrossMiner Terminology... 2 Overview of CSD-CrossMiner... 3 Searching with a Pharmacophore... 4 Performing a Pharmacophore Search using CSD-CrossMiner Version 2.0
2G1/3G4 GIS TUTORIAL >>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>
 > University of Michigan >Taubman College of Architecture > ARCH 552, Perimeter @ Work Out [T]here, Fall 2009 >September 24, 2009 2G1/3G4 GIS TUTORIAL >>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>
> University of Michigan >Taubman College of Architecture > ARCH 552, Perimeter @ Work Out [T]here, Fall 2009 >September 24, 2009 2G1/3G4 GIS TUTORIAL >>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>
PDF-2 Tools and Searches
 PDF-2 Tools and Searches PDF-2 2019 The PDF-2 2019 database is powered by our integrated search display software. PDF-2 2019 boasts 69 search selections coupled with 53 display fields resulting in a nearly
PDF-2 Tools and Searches PDF-2 2019 The PDF-2 2019 database is powered by our integrated search display software. PDF-2 2019 boasts 69 search selections coupled with 53 display fields resulting in a nearly
Data Structures & Database Queries in GIS
 Data Structures & Database Queries in GIS Objective In this lab we will show you how to use ArcGIS for analysis of digital elevation models (DEM s), in relationship to Rocky Mountain bighorn sheep (Ovis
Data Structures & Database Queries in GIS Objective In this lab we will show you how to use ArcGIS for analysis of digital elevation models (DEM s), in relationship to Rocky Mountain bighorn sheep (Ovis
(THIS IS AN OPTIONAL BUT WORTHWHILE EXERCISE)
") PART 2: Analysis in ArcGIS (THIS IS AN OPTIONAL BUT WORTHWHILE EXERCISE) Step 1: Start ArcCatalog and open a geodatabase If you have a shortcut icon for ArcCatalog on your desktop, double-click it to start
PART 2: Analysis in ArcGIS (THIS IS AN OPTIONAL BUT WORTHWHILE EXERCISE) Step 1: Start ArcCatalog and open a geodatabase If you have a shortcut icon for ArcCatalog on your desktop, double-click it to start
Molecular modeling with InsightII
 Molecular modeling with InsightII Yuk Sham Computational Biology/Biochemistry Consultant Phone: (612) 624 7427 (Walter Library) Phone: (612) 624 0783 (VWL) Email: shamy@msi.umn.edu How to run InsightII
Molecular modeling with InsightII Yuk Sham Computational Biology/Biochemistry Consultant Phone: (612) 624 7427 (Walter Library) Phone: (612) 624 0783 (VWL) Email: shamy@msi.umn.edu How to run InsightII
Athena Visual Software, Inc. 1
 Athena Visual Studio Visual Kinetics Tutorial VisualKinetics is an integrated tool within the Athena Visual Studio software environment, which allows scientists and engineers to simulate the dynamic behavior
Athena Visual Studio Visual Kinetics Tutorial VisualKinetics is an integrated tool within the Athena Visual Studio software environment, which allows scientists and engineers to simulate the dynamic behavior
Introduction to Structure Preparation and Visualization
 Introduction to Structure Preparation and Visualization Created with: Release 2018-4 Prerequisites: Release 2018-2 or higher Access to the internet Categories: Molecular Visualization, Structure-Based
Introduction to Structure Preparation and Visualization Created with: Release 2018-4 Prerequisites: Release 2018-2 or higher Access to the internet Categories: Molecular Visualization, Structure-Based
Project 2. Chemistry of Transient Species in Planetary Atmospheres: Exploring the Potential Energy Surfaces of CH 2 S
 Chemistry 362 Spring 2018 Dr. Jean M. Standard March 21, 2018 Project 2. Chemistry of Transient Species in Planetary Atmospheres: Exploring the Potential Energy Surfaces of CH 2 S In this project, you
Chemistry 362 Spring 2018 Dr. Jean M. Standard March 21, 2018 Project 2. Chemistry of Transient Species in Planetary Atmospheres: Exploring the Potential Energy Surfaces of CH 2 S In this project, you
Chapter: 22. Visualization: Making INPUT File and Processing of Output Results
 Chapter: 22 Visualization: Making INPUT File and Processing of Output Results Keywords: visualization, input and output structure, molecular orbital, electron density. In the previous chapters, we have
Chapter: 22 Visualization: Making INPUT File and Processing of Output Results Keywords: visualization, input and output structure, molecular orbital, electron density. In the previous chapters, we have
Lewis Structures and Molecular Shapes
 Lewis Structures and Molecular Shapes Rules for Writing Lewis Structures 1. Determine the correct skeleton structure (connectivity of atoms). Usually, the most electronegative atoms go around the edges
Lewis Structures and Molecular Shapes Rules for Writing Lewis Structures 1. Determine the correct skeleton structure (connectivity of atoms). Usually, the most electronegative atoms go around the edges
Tutorial I: IQ MOL and Basic DFT and MP2 Calculations 1 / 30
 Tutorial I: IQ MOL and Basic DFT and MP2 Calculations Q-Chem User Workshop, Denver March 21, 2015 1 / 30 2 / 30 Introduction to IQMOL DFT and MP2 Calculations 3 / 30 IQMOL and Q-CHEM IQMOL is an open-source
Tutorial I: IQ MOL and Basic DFT and MP2 Calculations Q-Chem User Workshop, Denver March 21, 2015 1 / 30 2 / 30 Introduction to IQMOL DFT and MP2 Calculations 3 / 30 IQMOL and Q-CHEM IQMOL is an open-source
Lab 1 Uniform Motion - Graphing and Analyzing Motion
 Lab 1 Uniform Motion - Graphing and Analyzing Motion Objectives: < To observe the distance-time relation for motion at constant velocity. < To make a straight line fit to the distance-time data. < To interpret
Lab 1 Uniform Motion - Graphing and Analyzing Motion Objectives: < To observe the distance-time relation for motion at constant velocity. < To make a straight line fit to the distance-time data. < To interpret
BOND LENGTH WITH HYPERCHEM LITE
 BOND LENGTH WITH HYPERCHEM LITE LAB MOD2.COMP From Gannon University SIM INTRODUCTION The electron cloud surrounding the nucleus of the atom determines the size of the atom. Since this distance is somewhat
BOND LENGTH WITH HYPERCHEM LITE LAB MOD2.COMP From Gannon University SIM INTRODUCTION The electron cloud surrounding the nucleus of the atom determines the size of the atom. Since this distance is somewhat
Introduction to Hartree-Fock calculations in Spartan
 EE5 in 2008 Hannes Jónsson Introduction to Hartree-Fock calculations in Spartan In this exercise, you will get to use state of the art software for carrying out calculations of wavefunctions for molecues,
EE5 in 2008 Hannes Jónsson Introduction to Hartree-Fock calculations in Spartan In this exercise, you will get to use state of the art software for carrying out calculations of wavefunctions for molecues,
Getting Started with Spartan 3rd Edition
 Getting Started with Spartan 3rd Edition WAVEFUNCTION Wavefunction, Inc. 18401 Von Karman Avenue, Suite 370 Irvine, CA 92612 U.S.A. www.wavefun.com Wavefunction, Inc., Japan Branch Office Level 14 ibiya
Getting Started with Spartan 3rd Edition WAVEFUNCTION Wavefunction, Inc. 18401 Von Karman Avenue, Suite 370 Irvine, CA 92612 U.S.A. www.wavefun.com Wavefunction, Inc., Japan Branch Office Level 14 ibiya
Lewis Structures and Molecular Shapes
 Lewis Structures and Molecular Shapes Rules for Writing Lewis Structures 1. Determine the correct skeleton structure (connectivity of atoms). Usually, the most electronegative atoms go around the edges
Lewis Structures and Molecular Shapes Rules for Writing Lewis Structures 1. Determine the correct skeleton structure (connectivity of atoms). Usually, the most electronegative atoms go around the edges
Introduction to ArcMap
 Introduction to ArcMap ArcMap ArcMap is a Map-centric GUI tool used to perform map-based tasks Mapping Create maps by working geographically and interactively Display and present Export or print Publish
Introduction to ArcMap ArcMap ArcMap is a Map-centric GUI tool used to perform map-based tasks Mapping Create maps by working geographically and interactively Display and present Export or print Publish
Polar alignment in 5 steps based on the Sánchez Valente method
 1 Polar alignment in 5 steps based on the Sánchez Valente method Compared to the drift alignment method, this one, allows you to easily achieve a perfect polar alignment in just one step. By "perfect polar
1 Polar alignment in 5 steps based on the Sánchez Valente method Compared to the drift alignment method, this one, allows you to easily achieve a perfect polar alignment in just one step. By "perfect polar
Motion II. Goals and Introduction
 Motion II Goals and Introduction As you have probably already seen in lecture or homework, and if you ve performed the experiment Motion I, it is important to develop a strong understanding of how to model
Motion II Goals and Introduction As you have probably already seen in lecture or homework, and if you ve performed the experiment Motion I, it is important to develop a strong understanding of how to model
Moving into the information age: From records to Google Earth
 Moving into the information age: From records to Google Earth David R. R. Smith Psychology, School of Life Sciences, University of Hull e-mail: davidsmith.butterflies@gmail.com Introduction Many of us
Moving into the information age: From records to Google Earth David R. R. Smith Psychology, School of Life Sciences, University of Hull e-mail: davidsmith.butterflies@gmail.com Introduction Many of us
Synteny Portal Documentation
 Synteny Portal Documentation Synteny Portal is a web application portal for visualizing, browsing, searching and building synteny blocks. Synteny Portal provides four main web applications: SynCircos,
Synteny Portal Documentation Synteny Portal is a web application portal for visualizing, browsing, searching and building synteny blocks. Synteny Portal provides four main web applications: SynCircos,
Task 1: Start ArcMap and add the county boundary data from your downloaded dataset to the data frame.
 Exercise 6 Coordinate Systems and Map Projections The following steps describe the general process that you will follow to complete the exercise. Specific steps will be provided later in the step-by-step
Exercise 6 Coordinate Systems and Map Projections The following steps describe the general process that you will follow to complete the exercise. Specific steps will be provided later in the step-by-step
Electric Fields and Equipotentials
 OBJECTIVE Electric Fields and Equipotentials To study and describe the two-dimensional electric field. To map the location of the equipotential surfaces around charged electrodes. To study the relationship
OBJECTIVE Electric Fields and Equipotentials To study and describe the two-dimensional electric field. To map the location of the equipotential surfaces around charged electrodes. To study the relationship
You w i ll f ol l ow these st eps : Before opening files, the S c e n e panel is active.
 You w i ll f ol l ow these st eps : A. O pen a n i m a g e s t a c k. B. Tr a c e t h e d e n d r i t e w i t h t h e user-guided m ode. C. D e t e c t t h e s p i n e s a u t o m a t i c a l l y. D. C
You w i ll f ol l ow these st eps : A. O pen a n i m a g e s t a c k. B. Tr a c e t h e d e n d r i t e w i t h t h e user-guided m ode. C. D e t e c t t h e s p i n e s a u t o m a t i c a l l y. D. C
Avogadro with YAeHMOP Manual
 Avogadro with YAeHMOP Manual Patrick Avery, Herbert Ludowieg, Jochen Autschbach, and Eva Zurek Department of Chemistry University at Buffalo Buffalo, NY 14260-3000, USA September 11, 2017 Contents 1 Installation
Avogadro with YAeHMOP Manual Patrick Avery, Herbert Ludowieg, Jochen Autschbach, and Eva Zurek Department of Chemistry University at Buffalo Buffalo, NY 14260-3000, USA September 11, 2017 Contents 1 Installation
Multiphysics Modeling
 11 Multiphysics Modeling This chapter covers the use of FEMLAB for multiphysics modeling and coupled-field analyses. It first describes the various ways of building multiphysics models. Then a step-by-step
11 Multiphysics Modeling This chapter covers the use of FEMLAB for multiphysics modeling and coupled-field analyses. It first describes the various ways of building multiphysics models. Then a step-by-step
Jack Smith. Center for Environmental, Geotechnical and Applied Science. Marshall University
 Jack Smith Center for Environmental, Geotechnical and Applied Science Marshall University -- Division of Science and Research WV Higher Education Policy Commission WVU HPC Summer Institute June 20, 2014
Jack Smith Center for Environmental, Geotechnical and Applied Science Marshall University -- Division of Science and Research WV Higher Education Policy Commission WVU HPC Summer Institute June 20, 2014