Optimisation: From theory to better prediction
|
|
|
- Abel Henderson
- 6 years ago
- Views:
Transcription

1 Optimisation: From theory to better prediction Claire Adjiman Claire Adjiman 23 May 2013 Christodoulos A. Floudas Memorial Symposium 6 May 2017
 provides certainty safety and risk: finding the worst-case solution to minimise/control risk thermodynamics: local minima are just wrong model")
2 Why look for the global solution? Deterministic global optimisation (DGO) provides certainty safety and risk: finding the worst-case solution to minimise/control risk thermodynamics: local minima are just wrong model parameterisation/identification: gaining a correct physical understanding DGO can provide enhanced performance (technical or economic) why settle for second (or third, or fourth) best?
 g( x) 0 h( x) = 0 x X f(x) Nonconvexity f(x) Convex relaxations x 2 x x 2 x x 1 x 1")
3 Deterministic global optimisation: Basic concepts Department of Chemical Engineering min x s. t. f ( x) g( x) 0 h( x) = 0 x X f(x) Nonconvexity f(x) Convex relaxations x 2 x x 2 x x 1 x 1
4 Branch-and-Bound First iteration

5 Branch-and-Bound First iteration Second iteration
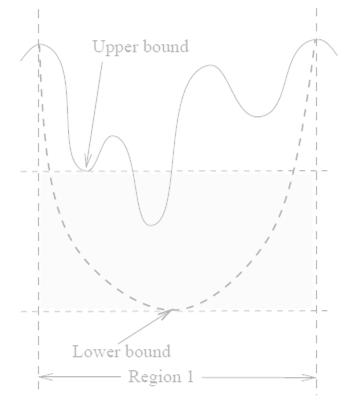

6 Branch-and-Bound First iteration Second iteration


7 The ααbb underestimator / algorithm ααbb underestimator for a CC 2 general function ff(xx): nn ff xx = ff xx + αα ii (x ii LL x ii )(x ii UU x ii ) ii=1 Properties of ff xx for αα ii 0 ff xx ff xx, xx [xx L, xx U ] ff xx is convex xx [xx L, xx U ] ff x = sin 5x + x 2, x XX = 2,2 iff αα max 0, 1 2 λλ mmmmmm where λλ mmmmmm is the minimum eigenvalue of HH xx = 2 ff(xx), xx XX Maranas, Floudas, 1992, The Journal of Chemical Physics, 97, Androulakis, Maranas, Floudas, 1995, Journal of Global Optimization, 7, 337. Adjiman, Dallwig, Floudas, Neumaier, 1998, Computers & Chemical Engineering, 22, 1137.



8 αbb algorithm > 1600 citations Snapshot of a PhD with Chris Department of Chemical Engineering Paper co-authors: I.P. Androulakis S. Dallwig C. D. Maranas A. Neumaier C.A. Schweiger






9 The ability to construct convex underestimators enables the solution of more and more problems Nonlinear and mixed-integer nonlinear problems Dynamic problems Papamichail Bounding trajectories Kazazakis New underestimators Bilevel problems Kleniati, Paulavicius Branch-and-Sandwich algorithm

10 Optimisation for better prediction of solid form

11 Polymorphism Molecules can pack in different forms Abbott Laboratories believed that the HIV drug Ritanovir Form I was stable, but Form II appeared unexpectedly in the manufacturing process Form II is more stable and less soluble: bioavailability of the drug changed Ritanovir had to be recalled at a cost of several hundred million dollars

12 Predicting the unpredictable There are many mysteries of nature that we have not solved. Hurricanes, for example, continue to occur and often cause massive devastation. Meteorologists cannot predict months in advance when and with what velocity a hurricane will strike a specific community. Polymorphism is a parallel phenomenon. We know that it will probably happen. But not why or when. Unfortunately, there is nothing that we can do today to prevent a hurricane from striking any community or polymorphism from striking any drug. Dr. Eugene Sun, Abbott Laboratories, 1998, at press conference on ritonavir crisis, when a more stable polymorph of this HIV drug appeared in manufacture




13 Predicting the unpredictable 4/11 1/8 1/11 1/6


 ( T, P) G U latt = U U intra + P V + U rep/disp inter T")
14 Ab initio crystal structure prediction: A large and expensive nonlinear optimisation problem Unit cell is determined by: β lattice lengths a, b and c lattice angles α, β and γ positions of all atoms within unit cell rˆ, = 1,..,, = 1,.., ji i N j Z c ˆr ji,z α γ b ˆr ji,y min a, b, c, α, β, γ, r min a, b, c, α, β, γ, r ij ij ( T, P) ( T, P) G U latt = U U intra + P V + U rep/disp inter T S + U All low-energy local minima, including the global minimum ˆr ji,x ele inter a
15 Crystal structure prediction methodology Department of Chemical Engineering isolated-molecule quantum mechanical calculations empirical models for dispersion/repulsion vdw U, ii, ' 1,.., N = ii s CrystalPredictor 1-4 global search for low-energy structures with limited molecular flexibility and simple models molecular connectivity conformational analysis intramolecular energies & intermolecular electrostatic potentials as functions of molecular conformation polymorphs charge density 1 Karamertzanis & Pantelides J. Comp. Chem., 2005, 26, Karamertzanis & Pantelides Mol. Phys., 2007, 105, Habgood, Sugden, Kazantsev, Adjiman, Pantelides, JCTC, 2015, 11, Sugden, Adjiman, Pantelides, Acta Cryst B, 2016, B72, 864






16 Crystal structure prediction methodology Department of Chemical Engineering isolated-molecule quantum mechanical calculations empirical models for dispersion/repulsion vdw U, ii, ' 1,.., N = ii s Lowest-energy structures only CrystalPredictor 1-4 CrystalOptimizer 5 global search for low-energy structures with limited molecular flexibility and simple models local minimization of lattice energy with extensive molecular flexibility and more accurate models molecular connectivity conformational analysis intramolecular energies & intermolecular electrostatic potentials as functions of molecular conformation polymorphs charge density 5 Kazantsev, Karamertzanis, Adjiman, & Pantelides, JCTC, 2011, 7, 1998.
 + 45 40 35 30 25 20 15 Intermolecular Energy, U inter 195 190 185 180 175 170 165-151.7695-154.5903-148.9487-154.5903-146.1278-140.4862-151.")
17 Molecular flexibility Intramolecular energy U intra O1-C1-C2-C3 ( o ) O1-C1-C2-C3 ( o ) H1-O1-C1-C2 ( o ) Intermolecular Energy, U inter H1-O1-C1-C2 ( o ) H1 O1 Successful prediction depends on correct balance between inter- and intramolecular forces C1 C3 C2 O1-C1-C2-C3 ( o ) Lattice energy, U inter + U intra H1-O1-C1-C2 ( o ) Accounting for flexibility comes at a huge computational expense
 260 240 220 200 180 160 140 120")

18 Achieving high accuracy at lower cost: Adaptive Local Approximate Models (LAMs) Carry out QM calculations at specific points T2 (º) T1 (º) Use an adaptive grid: o High density of LAMs in chemically interesting areas o A large search space covered with fewer LAMs



19 Accuracy of adaptive LAMs Intramolecular energy for ROY molecule Exact ab initio surface (5 o scan) Uniform coarse grid (40 o spacing) Adaptive grid

 -232-237 -242-247 Experimental Rank=1 rmsd 20 =0.")

20 Target XXV 6 th blind test of crystal structure prediction Overlays of experimental & predicted structures U latt (kj/mol) Experimental Rank=1 rmsd 20 =0.317Å rmsd 20 = Å rmsd 1 = Å including H atoms rmsd 1 =0.035 Å Not including H atoms rmsd 1 = Å including H atoms rmsd 1 = Å Not including H atoms ρ(g/cm 3 )
 -185-190 -195 Only 3643 non-uniform")

21 Target XXVI 6 th blind test CrystalPredictor Department of Chemical Engineering U latt (kj/mol) Only 3643 non-uniform LAMS needed A 70% reduction on the 11,858 LAMs that an equivalent uniform grid would require Density (g/cm 3 )


22 -180 Target XXVI 6 th blind test Final results: CrystalOptimiser Department of Chemical Engineering U latt (kj/mol) RMSD 20 : Å RMSD 1 : Å Density (g/cm 3 )
 (5 th")
 Axitinib 5 (Pfizer)")
 1 Bhardwaj R.M., et al.")
, Int. J. Pharm.; Bardwell et al.")
, Cryst. Growth Des.; 4 Price L.S., et al.")
 Chem Eng Sci; 6 Habgood et al.")
23 Applications of crystal structure prediction Olanzapine 1 Molecule XX 2 (Eli Lilly) (5 th blind test) GSK269984B 3 (GlaxoSmithKline) Tazofelone 4 (Eli Lilly) Axitinib 5 (Pfizer) Target XXV 6 (6 th blind test) BMS (Bristol Myers Squibb) 1 Bhardwaj R.M., et al.,(2013), Cryst. Growth Des.; 2 Kazantsev A.V., et al., (2011), Int. J. Pharm.; Bardwell et al., (2011), Acta Cryst. B; 3 Ismail S.Z., et al., (2013), Cryst. Growth Des.; 4 Price L.S., et al., (2014), J. Mol. Struct. 5 Vasileiadis et al. (2015) Chem Eng Sci; 6 Habgood et al.(2015) JCTC; 7 Sugden et al. (2016) Acta Cryst B
24 Concluding remarks Optimisation is ubiquitous Nonlinear, mixed-integer, large-scale, bilevel, dynamic problems A key tool to predict behaviour/properties and to design better systems Advances are made through better theories, algorithms, and implementations but also via applications: problem formulation, tailored algorithms
25 Thank you Department of Chemical Engineering Students and postdocs Collaborators Ioannis Papamichail Andrei Kazantsev Manolis Vasileiadis Christina-Anna Gatsiou Nikolaos Kazazakis Panos Karamertzanis Polyxeni Kleniati Matthew Habgood Isaac Sugden Remigijus Paulavicius While at Princeton: Ioannis Androulakis Costas Maranas Carl Schweiger Arnold Neumaier (Vienna) Stefan Dallwig (Vienna) Costas Pantelides Sally Price (UCL) EPSRC And above all Thank you, Chris
Prediction of the crystal structures of axitinib, a polymorphic pharmaceutical molecule
 Prediction of the crystal structures of axitinib, a polymorphic pharmaceutical molecule Manolis Vasileiadis, Constantinos C. Pantelides, Claire S. Adjiman 1 Centre for Process Systems Engineering Department
Prediction of the crystal structures of axitinib, a polymorphic pharmaceutical molecule Manolis Vasileiadis, Constantinos C. Pantelides, Claire S. Adjiman 1 Centre for Process Systems Engineering Department
Improving the accuracy of lattice energy calculations in crystal structure prediction using experimental data
 Improving the accuracy of lattice energy calculations in crystal structure prediction using experimental data Christina-Anna Gatsiou A thesis submitted for the Doctor of Philosophy degree of Imperial College
Improving the accuracy of lattice energy calculations in crystal structure prediction using experimental data Christina-Anna Gatsiou A thesis submitted for the Doctor of Philosophy degree of Imperial College
Computational Crystal Energy Landscapes as an aid to polymorph screening
 Control and Prediction of the rganic Solid State A Basic Technology project of the Research Councils UK Computational Crystal Energy Landscapes as an aid to polymorph screening Sarah (Sally) L Price Department
Control and Prediction of the rganic Solid State A Basic Technology project of the Research Councils UK Computational Crystal Energy Landscapes as an aid to polymorph screening Sarah (Sally) L Price Department
Deterministic Global Optimization Algorithm and Nonlinear Dynamics
 Deterministic Global Optimization Algorithm and Nonlinear Dynamics C. S. Adjiman and I. Papamichail Centre for Process Systems Engineering Department of Chemical Engineering and Chemical Technology Imperial
Deterministic Global Optimization Algorithm and Nonlinear Dynamics C. S. Adjiman and I. Papamichail Centre for Process Systems Engineering Department of Chemical Engineering and Chemical Technology Imperial
Alpha-helical Topology and Tertiary Structure Prediction of Globular Proteins Scott R. McAllister Christodoulos A. Floudas Princeton University
 Alpha-helical Topology and Tertiary Structure Prediction of Globular Proteins Scott R. McAllister Christodoulos A. Floudas Princeton University Department of Chemical Engineering Program of Applied and
Alpha-helical Topology and Tertiary Structure Prediction of Globular Proteins Scott R. McAllister Christodoulos A. Floudas Princeton University Department of Chemical Engineering Program of Applied and
Many-body electronic structure theory: Is Quantum Monte-Carlo affordable for molecular crystals?
 Many-body electronic structure theory: Is Quantum Monte-Carlo affordable for molecular crystals? Gerit Brandenburg 23 rd of November 2017 CHEMISTRY DEPARTMENT, UNIVERSITY OF TURIN,
Many-body electronic structure theory: Is Quantum Monte-Carlo affordable for molecular crystals? Gerit Brandenburg 23 rd of November 2017 CHEMISTRY DEPARTMENT, UNIVERSITY OF TURIN,
Screening for cocrystals of succinic acid and 4-aminobenzoic acid. Supplementary Information
 Screening for cocrystals of succinic acid and 4-aminobenzoic acid Nizar Issa, Sarah A. Barnett, Sharmarke Mohamed, Doris E. Braun, Royston C. B. Copley, Derek A. Tocher, Sarah L Price* Supplementary Information
Screening for cocrystals of succinic acid and 4-aminobenzoic acid Nizar Issa, Sarah A. Barnett, Sharmarke Mohamed, Doris E. Braun, Royston C. B. Copley, Derek A. Tocher, Sarah L Price* Supplementary Information
Implementation of an αbb-type underestimator in the SGO-algorithm
 Implementation of an αbb-type underestimator in the SGO-algorithm Process Design & Systems Engineering November 3, 2010 Refining without branching Could the αbb underestimator be used without an explicit
Implementation of an αbb-type underestimator in the SGO-algorithm Process Design & Systems Engineering November 3, 2010 Refining without branching Could the αbb underestimator be used without an explicit
Unit 2: Problem Classification and Difficulty in Optimization
 Unit 2: Problem Classification and Difficulty in Optimization Learning goals Unit 2 I. What is the subject area of multiobjective decision analysis and multiobjective optimization; How does it relate to
Unit 2: Problem Classification and Difficulty in Optimization Learning goals Unit 2 I. What is the subject area of multiobjective decision analysis and multiobjective optimization; How does it relate to
Supporting Information
 Supporting Information Three Polymorphic Forms of Ciprofloxacin Maleate: Formation Pathways, Crystal Structures, Calculations and Thermodynamic Stability Aspects Artem O. Surov a, Andrei V. Churakov b,
Supporting Information Three Polymorphic Forms of Ciprofloxacin Maleate: Formation Pathways, Crystal Structures, Calculations and Thermodynamic Stability Aspects Artem O. Surov a, Andrei V. Churakov b,
A global optimization method, αbb, for general twice-differentiable constrained NLPs I. Theoretical advances
 Computers Chem. Engng Vol. 22, No. 9, pp. 1137 1158, 1998 Copyright 1998 Elsevier Science Ltd All rights reserved. Printed in Great Britain PII: S0098-1354(98)00027-1 0098 1354/98 $19.00#0.00 A global
Computers Chem. Engng Vol. 22, No. 9, pp. 1137 1158, 1998 Copyright 1998 Elsevier Science Ltd All rights reserved. Printed in Great Britain PII: S0098-1354(98)00027-1 0098 1354/98 $19.00#0.00 A global
Crystal Structure Prediction using CRYSTALG program
 Crystal Structure Prediction using CRYSTALG program Yelena Arnautova Baker Laboratory of Chemistry and Chemical Biology, Cornell University Problem of crystal structure prediction: - theoretical importance
Crystal Structure Prediction using CRYSTALG program Yelena Arnautova Baker Laboratory of Chemistry and Chemical Biology, Cornell University Problem of crystal structure prediction: - theoretical importance
Crystal Structure Prediction A Decade of Blind Tests. Frank Leusen
 Crystal Structure Prediction A Decade of Blind Tests Frank Leusen Outline Crystal structure prediction Blind tests in crystal structure prediction 1999, 2001 and 2004 editions The 2007 edition of the blind
Crystal Structure Prediction A Decade of Blind Tests Frank Leusen Outline Crystal structure prediction Blind tests in crystal structure prediction 1999, 2001 and 2004 editions The 2007 edition of the blind
In Memoriam: Christodoulos Achilleus Floudas
 In Memoriam: Christodoulos Achilleus Floudas Ruth Misener Department of Computing, Imperial College London South Kensington SW7 2AZ, UK r.misener@imperial.ac.uk September 30, 2016 The Centre for Process
In Memoriam: Christodoulos Achilleus Floudas Ruth Misener Department of Computing, Imperial College London South Kensington SW7 2AZ, UK r.misener@imperial.ac.uk September 30, 2016 The Centre for Process
Density Functional Theory Including van der Waals Forces:
 Density Functional Theory Including van der Waals Forces: Semi-classical correction provides versatile electronic structure tool Gerit Brandenburg 22 nd of Feb. 2017 57 TH SANIBEL
Density Functional Theory Including van der Waals Forces: Semi-classical correction provides versatile electronic structure tool Gerit Brandenburg 22 nd of Feb. 2017 57 TH SANIBEL
Calculation of the free energy of crystalline solids
 Imperial College London Calculation of the free energy of crystalline solids Manolis Vasileiadis A thesis submitted for the Doctor of Philosophy degree of Imperial College London and for the Diploma of
Imperial College London Calculation of the free energy of crystalline solids Manolis Vasileiadis A thesis submitted for the Doctor of Philosophy degree of Imperial College London and for the Diploma of
Department of Chemistry, University College London, 20 Gordon Street, London WC1H 0AJ, UK. gerit.
 Cite this: Faraday Discuss., 2018,211, 275 PAPER Crystal structure prediction of flexible pharmaceutical-like molecules: density functional tight-binding as an intermediate optimisation method and for
Cite this: Faraday Discuss., 2018,211, 275 PAPER Crystal structure prediction of flexible pharmaceutical-like molecules: density functional tight-binding as an intermediate optimisation method and for
1 Introduction A large proportion of all optimization problems arising in an industrial or scientic context are characterized by the presence of nonco
 A Global Optimization Method, BB, for General Twice-Dierentiable Constrained NLPs I. Theoretical Advances C.S. Adjiman, S. Dallwig 1, C.A. Floudas 2 and A. Neumaier 1 Department of Chemical Engineering,
A Global Optimization Method, BB, for General Twice-Dierentiable Constrained NLPs I. Theoretical Advances C.S. Adjiman, S. Dallwig 1, C.A. Floudas 2 and A. Neumaier 1 Department of Chemical Engineering,
Structure and interactions in benzamide molecular crystals
 Structure and interactions in benzamide molecular crystals Philipp Ectors 1, Dominique Ectors 2, Dirk Zahn 1 1) Lehrstuhl für Theoretische Chemie/Computer-Chemie-Centrum Friedrich-Alexander- Universität
Structure and interactions in benzamide molecular crystals Philipp Ectors 1, Dominique Ectors 2, Dirk Zahn 1 1) Lehrstuhl für Theoretische Chemie/Computer-Chemie-Centrum Friedrich-Alexander- Universität
Solving Global Optimization Problems by Interval Computation
 Solving Global Optimization Problems by Interval Computation Milan Hladík Department of Applied Mathematics, Faculty of Mathematics and Physics, Charles University in Prague, Czech Republic, http://kam.mff.cuni.cz/~hladik/
Solving Global Optimization Problems by Interval Computation Milan Hladík Department of Applied Mathematics, Faculty of Mathematics and Physics, Charles University in Prague, Czech Republic, http://kam.mff.cuni.cz/~hladik/
Selected Publications through 2007 Christodoulos A. Floudas
 Selected Publications through 2007 Christodoulos A. Floudas For a complete listing of downloadable publications, click here. 2007 A New Robust Optimization Approach for Scheduling under Uncertainty: II.
Selected Publications through 2007 Christodoulos A. Floudas For a complete listing of downloadable publications, click here. 2007 A New Robust Optimization Approach for Scheduling under Uncertainty: II.
Brief Review of Statistical Mechanics
 Brief Review of Statistical Mechanics Introduction Statistical mechanics: a branch of physics which studies macroscopic systems from a microscopic or molecular point of view (McQuarrie,1976) Also see (Hill,1986;
Brief Review of Statistical Mechanics Introduction Statistical mechanics: a branch of physics which studies macroscopic systems from a microscopic or molecular point of view (McQuarrie,1976) Also see (Hill,1986;
MOLECULAR DYNAMICS SIMULATIONS OF HMX CRYSTAL POLYMORPHS USING A FLEXIBLE MOLECULE FORCE FIELD
 MOLECULAR DYNAMICS SIMULATIONS OF HMX CRYSTAL POLYMORPHS USING A FLEXIBLE MOLECULE FORCE FIELD Dmitry Bedrov 1, Grant D. Smith 1, and Thomas D. Sewell 2 1 Department of Materials Science & Engineering,
MOLECULAR DYNAMICS SIMULATIONS OF HMX CRYSTAL POLYMORPHS USING A FLEXIBLE MOLECULE FORCE FIELD Dmitry Bedrov 1, Grant D. Smith 1, and Thomas D. Sewell 2 1 Department of Materials Science & Engineering,
Global Optimization by Interval Analysis
 Global Optimization by Interval Analysis Milan Hladík Department of Applied Mathematics, Faculty of Mathematics and Physics, Charles University in Prague, Czech Republic, http://kam.mff.cuni.cz/~hladik/
Global Optimization by Interval Analysis Milan Hladík Department of Applied Mathematics, Faculty of Mathematics and Physics, Charles University in Prague, Czech Republic, http://kam.mff.cuni.cz/~hladik/
Pharmaceutical Process & Product Development: What can Process Systems Engineering contribute?
 fff THE ADVANCED PROCESS MODELLING COMPANY Pharmaceutical Process & Product Development: What can Process Systems Engineering contribute? Costas Pantelides FIPSE 30 August 2012 Outline 1. Systems Engineering
fff THE ADVANCED PROCESS MODELLING COMPANY Pharmaceutical Process & Product Development: What can Process Systems Engineering contribute? Costas Pantelides FIPSE 30 August 2012 Outline 1. Systems Engineering
Data-Driven Global Optimization
 Data-Driven Global Optimization Fani Boukouvala & Chris Kieslich Christodoulos A Floudas Memorial Symposium Princeton University May 6 th 2017 A story of global optimization & more September 17 th 2014
Data-Driven Global Optimization Fani Boukouvala & Chris Kieslich Christodoulos A Floudas Memorial Symposium Princeton University May 6 th 2017 A story of global optimization & more September 17 th 2014
CE 530 Molecular Simulation
 1 CE 530 Molecular Simulation Lecture 14 Molecular Models David A. Kofke Department of Chemical Engineering SUNY Buffalo kofke@eng.buffalo.edu 2 Review Monte Carlo ensemble averaging, no dynamics easy
1 CE 530 Molecular Simulation Lecture 14 Molecular Models David A. Kofke Department of Chemical Engineering SUNY Buffalo kofke@eng.buffalo.edu 2 Review Monte Carlo ensemble averaging, no dynamics easy
Lecture 3. Linear Regression
 Lecture 3. Linear Regression COMP90051 Statistical Machine Learning Semester 2, 2017 Lecturer: Andrey Kan Copyright: University of Melbourne Weeks 2 to 8 inclusive Lecturer: Andrey Kan MS: Moscow, PhD:
Lecture 3. Linear Regression COMP90051 Statistical Machine Learning Semester 2, 2017 Lecturer: Andrey Kan Copyright: University of Melbourne Weeks 2 to 8 inclusive Lecturer: Andrey Kan MS: Moscow, PhD:
Fondamenti di Chimica Farmaceutica. Computer Chemistry in Drug Research: Introduction
 Fondamenti di Chimica Farmaceutica Computer Chemistry in Drug Research: Introduction Introduction Introduction Introduction Computer Chemistry in Drug Design Drug Discovery: Target identification Lead
Fondamenti di Chimica Farmaceutica Computer Chemistry in Drug Research: Introduction Introduction Introduction Introduction Computer Chemistry in Drug Design Drug Discovery: Target identification Lead
Protein Docking by Exploiting Multi-dimensional Energy Funnels
 Protein Docking by Exploiting Multi-dimensional Energy Funnels Ioannis Ch. Paschalidis 1 Yang Shen 1 Pirooz Vakili 1 Sandor Vajda 2 1 Center for Information and Systems Engineering and Department of Manufacturing
Protein Docking by Exploiting Multi-dimensional Energy Funnels Ioannis Ch. Paschalidis 1 Yang Shen 1 Pirooz Vakili 1 Sandor Vajda 2 1 Center for Information and Systems Engineering and Department of Manufacturing
Heuristics and Upper Bounds for a Pooling Problem with Cubic Constraints
 Heuristics and Upper Bounds for a Pooling Problem with Cubic Constraints Matthew J. Real, Shabbir Ahmed, Helder Inàcio and Kevin Norwood School of Chemical & Biomolecular Engineering 311 Ferst Drive, N.W.
Heuristics and Upper Bounds for a Pooling Problem with Cubic Constraints Matthew J. Real, Shabbir Ahmed, Helder Inàcio and Kevin Norwood School of Chemical & Biomolecular Engineering 311 Ferst Drive, N.W.
FORCE FIELDS AND CRYSTAL STRUCTURE PREDICTION
 FORCE FIELDS AND CRYSTAL STRUCTURE PREDICTION Bouke P. van Eijck Utrecht University (Retired) Department of Crystal and Structural Chemistry Padualaan 8, 3584 CH Utrecht, The Netherlands January 9, 2017
FORCE FIELDS AND CRYSTAL STRUCTURE PREDICTION Bouke P. van Eijck Utrecht University (Retired) Department of Crystal and Structural Chemistry Padualaan 8, 3584 CH Utrecht, The Netherlands January 9, 2017
Why polymorphism? An Evaluation using Experimental Charge Densities Analysis
 Why polymorphism? An Evaluation using Experimental Charge Densities Analysis T. N. Guru Row Solid State and Structural Chemistry Unit Indian Institute of Science Bangalore 560012 INDIA Email: ssctng@sscu.iisc.ernet.in
Why polymorphism? An Evaluation using Experimental Charge Densities Analysis T. N. Guru Row Solid State and Structural Chemistry Unit Indian Institute of Science Bangalore 560012 INDIA Email: ssctng@sscu.iisc.ernet.in
Chem 263 Winter 2018 Problem Set #2 Due: February 16
 Chem 263 Winter 2018 Problem Set #2 Due: February 16 1. Use size considerations to predict the crystal structures of PbF2, CoF2, and BeF2. Do your predictions agree with the actual structures of these
Chem 263 Winter 2018 Problem Set #2 Due: February 16 1. Use size considerations to predict the crystal structures of PbF2, CoF2, and BeF2. Do your predictions agree with the actual structures of these
Ab initio crystal structure determination of two polymorphic modifications of a local anesthetic agent, Tetracaine Hydrochloride
 1 / 30 Ab initio crystal structure determination of two polymorphic modifications of a local anesthetic agent, Tetracaine Hydrochloride Robert J. Papoular Leon Brillouin Laboratory papou@llb.saclay.cea.fr
1 / 30 Ab initio crystal structure determination of two polymorphic modifications of a local anesthetic agent, Tetracaine Hydrochloride Robert J. Papoular Leon Brillouin Laboratory papou@llb.saclay.cea.fr
2.4 Error Analysis for Iterative Methods
 2.4 Error Analysis for Iterative Methods 1 Definition 2.7. Order of Convergence Suppose {pp nn } nn=0 is a sequence that converges to pp with pp nn pp for all nn. If positive constants λλ and αα exist
2.4 Error Analysis for Iterative Methods 1 Definition 2.7. Order of Convergence Suppose {pp nn } nn=0 is a sequence that converges to pp with pp nn pp for all nn. If positive constants λλ and αα exist
Mixed Integer Non Linear Programming
 Mixed Integer Non Linear Programming Claudia D Ambrosio CNRS Research Scientist CNRS & LIX, École Polytechnique MPRO PMA 2016-2017 Outline What is a MINLP? Dealing with nonconvexities Global Optimization
Mixed Integer Non Linear Programming Claudia D Ambrosio CNRS Research Scientist CNRS & LIX, École Polytechnique MPRO PMA 2016-2017 Outline What is a MINLP? Dealing with nonconvexities Global Optimization
Current approaches to predicting molecular organic crystal structures
 Crystallography Reviews ISSN: 0889-311X (Print) 1476-3508 (Online) Journal homepage: http://www.tandfonline.com/loi/gcry20 Current approaches to predicting molecular organic crystal structures Graeme M.
Crystallography Reviews ISSN: 0889-311X (Print) 1476-3508 (Online) Journal homepage: http://www.tandfonline.com/loi/gcry20 Current approaches to predicting molecular organic crystal structures Graeme M.
Reliable and practical computational description of molecular crystal polymorphs
 MATERIALS SCIENCE Copyright 9 The Authors, some Reliable and practical computational description of molecular crystal polymorphs Johannes Hoja, Hsin-Yu Ko, Marcus A. Neumann, Roberto Car, Robert A. DiStasio
MATERIALS SCIENCE Copyright 9 The Authors, some Reliable and practical computational description of molecular crystal polymorphs Johannes Hoja, Hsin-Yu Ko, Marcus A. Neumann, Roberto Car, Robert A. DiStasio
The potential of computed crystal energy landscapes to aid solid form development
 The potential of computed crystal energy landscapes to aid solid form development Sarah L. Price, Department of Chemistry, University College London, 0 Gordon Street, London WC1H 0AJ, UK Susan M. Reutzel-Edens,
The potential of computed crystal energy landscapes to aid solid form development Sarah L. Price, Department of Chemistry, University College London, 0 Gordon Street, London WC1H 0AJ, UK Susan M. Reutzel-Edens,
MULTIOBJECTIVE OPTIMIZATION CONSIDERING ECONOMICS AND ENVIRONMENTAL IMPACT
 MULTIOBJECTIVE OPTIMIZATION CONSIDERING ECONOMICS AND ENVIRONMENTAL IMPACT Young-il Lim, Pascal Floquet, Xavier Joulia* Laboratoire de Génie Chimique (LGC, UMR-CNRS 5503) INPT-ENSIGC, 8 chemin de la loge,
MULTIOBJECTIVE OPTIMIZATION CONSIDERING ECONOMICS AND ENVIRONMENTAL IMPACT Young-il Lim, Pascal Floquet, Xavier Joulia* Laboratoire de Génie Chimique (LGC, UMR-CNRS 5503) INPT-ENSIGC, 8 chemin de la loge,
Online generation via offline selection - Low dimensional linear cuts from QP SDP relaxation -
 Online generation via offline selection - Low dimensional linear cuts from QP SDP relaxation - Radu Baltean-Lugojan Ruth Misener Computational Optimisation Group Department of Computing Pierre Bonami Andrea
Online generation via offline selection - Low dimensional linear cuts from QP SDP relaxation - Radu Baltean-Lugojan Ruth Misener Computational Optimisation Group Department of Computing Pierre Bonami Andrea
A Decomposition-based MINLP Solution Method Using Piecewise Linear Relaxations
 A Decomposition-based MINLP Solution Method Using Piecewise Linear Relaxations Pradeep K. Polisetty and Edward P. Gatzke 1 Department of Chemical Engineering University of South Carolina Columbia, SC 29208
A Decomposition-based MINLP Solution Method Using Piecewise Linear Relaxations Pradeep K. Polisetty and Edward P. Gatzke 1 Department of Chemical Engineering University of South Carolina Columbia, SC 29208
Deterministic Global Optimization of Nonlinear Dynamic Systems
 Deterministic Global Optimization of Nonlinear Dynamic Systems Youdong Lin and Mark A. Stadtherr Department of Chemical and Biomolecular Engineering University of Notre Dame, Notre Dame, IN 46556, USA
Deterministic Global Optimization of Nonlinear Dynamic Systems Youdong Lin and Mark A. Stadtherr Department of Chemical and Biomolecular Engineering University of Notre Dame, Notre Dame, IN 46556, USA
From Dynamics to Thermodynamics using Molecular Simulation
 From Dynamics to Thermodynamics using Molecular Simulation David van der Spoel Computational Chemistry Physical models to describe molecules Software to evaluate models and do predictions - GROMACS Model
From Dynamics to Thermodynamics using Molecular Simulation David van der Spoel Computational Chemistry Physical models to describe molecules Software to evaluate models and do predictions - GROMACS Model
On the Design of Optimal Solvent Mixtures using Generalised Disjunctive Programming
 Ian David Lockhart Bogle and Michael Fairweather (Editors), Proceedings of the 22nd European Symposium on Computer Aided Process Engineering, 17-20 June 2012, London. 2012 Elsevier B.V. All rights reserved.
Ian David Lockhart Bogle and Michael Fairweather (Editors), Proceedings of the 22nd European Symposium on Computer Aided Process Engineering, 17-20 June 2012, London. 2012 Elsevier B.V. All rights reserved.
Supplementary Information
 Electronic Supplementary Material (ESI) for Catalysis Science & Technology. This journal is The Royal Society of Chemistry 2015 Supplementary Information Insights into the Synergistic Role of Metal-Lattice
Electronic Supplementary Material (ESI) for Catalysis Science & Technology. This journal is The Royal Society of Chemistry 2015 Supplementary Information Insights into the Synergistic Role of Metal-Lattice
SUPPLEMENTARY INFORMATION
 Computer-aided molecular design of solvents for accelerated reaction kinetics Heiko Struebing, Zara Ganase, Panagiotis G. Karamertzanis, Eirini Siougkrou, Peter Haycock, Patrick Piccione, Alan Armstrong,
Computer-aided molecular design of solvents for accelerated reaction kinetics Heiko Struebing, Zara Ganase, Panagiotis G. Karamertzanis, Eirini Siougkrou, Peter Haycock, Patrick Piccione, Alan Armstrong,
Liquid-Liquid Equilibria of C8H18 O3 and H2O by Molecular Dynamics
 Liquid-Liquid Equilibria of C8H18 O3 and H2O by Molecular Dynamics Thermodynamics and Energy Technology, University of Paderborn Computational Chemical Engineering (CoChE) Fraunhofer SCAI 1 2 2 V r = K
Liquid-Liquid Equilibria of C8H18 O3 and H2O by Molecular Dynamics Thermodynamics and Energy Technology, University of Paderborn Computational Chemical Engineering (CoChE) Fraunhofer SCAI 1 2 2 V r = K
Supplementary Information
 Supplementary Information Supplementary Figure 1: Electronic Kohn-Sham potential profile of a charged monolayer MoTe 2 calculated using PBE-DFT. Plotted is the averaged electronic Kohn- Sham potential
Supplementary Information Supplementary Figure 1: Electronic Kohn-Sham potential profile of a charged monolayer MoTe 2 calculated using PBE-DFT. Plotted is the averaged electronic Kohn- Sham potential
CHEM-UA 652: Thermodynamics and Kinetics
 1 CHEM-UA 652: Thermodynamics and Kinetics Notes for Lecture 11 I. PHYSICAL AND CHEMICAL RELEVANCE OF FREE ENERGY In this section, we will consider some examples showing the significance of free energies.
1 CHEM-UA 652: Thermodynamics and Kinetics Notes for Lecture 11 I. PHYSICAL AND CHEMICAL RELEVANCE OF FREE ENERGY In this section, we will consider some examples showing the significance of free energies.
Intermolecular Forces in Density Functional Theory
 Intermolecular Forces in Density Functional Theory Problems of DFT Peter Pulay at WATOC2005: There are 3 problems with DFT 1. Accuracy does not converge 2. Spin states of open shell systems often incorrect
Intermolecular Forces in Density Functional Theory Problems of DFT Peter Pulay at WATOC2005: There are 3 problems with DFT 1. Accuracy does not converge 2. Spin states of open shell systems often incorrect
Selectivity in the initial C-H bond cleavage of n-butane on PdO(101)
") Supporting Information for Selectivity in the initial C-H bond cleavage of n-butane on PdO(101) Can Hakanoglu (a), Feng Zhang (a), Abbin Antony (a), Aravind Asthagiri (b) and Jason F. Weaver (a) * (a)
Supporting Information for Selectivity in the initial C-H bond cleavage of n-butane on PdO(101) Can Hakanoglu (a), Feng Zhang (a), Abbin Antony (a), Aravind Asthagiri (b) and Jason F. Weaver (a) * (a)
A Molecular Dynamics Simulation of a Homogeneous Organic-Inorganic Hybrid Silica Membrane
 A Molecular Dynamics Simulation of a Homogeneous Organic-Inorganic Hybrid Silica Membrane Supplementary Information: Simulation Procedure and Physical Property Analysis Simulation Procedure The molecular
A Molecular Dynamics Simulation of a Homogeneous Organic-Inorganic Hybrid Silica Membrane Supplementary Information: Simulation Procedure and Physical Property Analysis Simulation Procedure The molecular
Methods for van der Waals Interactions
 Methods for van der Waals Interactions Alexandre Tkatchenko Theory Department, Fritz Haber Institut der MPG Berlin, Germany tkatchen@fhi berlin.mpg.de Haber Institute FHI DFT and Beyond Workshop, Jul.
Methods for van der Waals Interactions Alexandre Tkatchenko Theory Department, Fritz Haber Institut der MPG Berlin, Germany tkatchen@fhi berlin.mpg.de Haber Institute FHI DFT and Beyond Workshop, Jul.
Generation of crystal structures using known crystal structures as analogues
 Supporting information Volume 72 (2016) Supporting information for article: Generation of crystal structures using known crystal structures as analogues Jason C. Cole, Colin R. Groom, Murray G. Read, Ilenia
Supporting information Volume 72 (2016) Supporting information for article: Generation of crystal structures using known crystal structures as analogues Jason C. Cole, Colin R. Groom, Murray G. Read, Ilenia
The statement, Many people think that polymorphism and
 pubs.acs.org/crystal Terms of Use Complex Polymorphic System of Gallic Acid Five Monohydrates, Three Anhydrates, and over 20 Solvates Doris E. Braun,*,, Rajni M. Bhardwaj, Alastair J. Florence, Derek A.
pubs.acs.org/crystal Terms of Use Complex Polymorphic System of Gallic Acid Five Monohydrates, Three Anhydrates, and over 20 Solvates Doris E. Braun,*,, Rajni M. Bhardwaj, Alastair J. Florence, Derek A.
Approximate Second Order Algorithms. Seo Taek Kong, Nithin Tangellamudi, Zhikai Guo
 Approximate Second Order Algorithms Seo Taek Kong, Nithin Tangellamudi, Zhikai Guo Why Second Order Algorithms? Invariant under affine transformations e.g. stretching a function preserves the convergence
Approximate Second Order Algorithms Seo Taek Kong, Nithin Tangellamudi, Zhikai Guo Why Second Order Algorithms? Invariant under affine transformations e.g. stretching a function preserves the convergence
Analysis of the simulation
 Analysis of the simulation Marcus Elstner and Tomáš Kubař January 7, 2014 Thermodynamic properties time averages of thermodynamic quantites correspond to ensemble averages (ergodic theorem) some quantities
Analysis of the simulation Marcus Elstner and Tomáš Kubař January 7, 2014 Thermodynamic properties time averages of thermodynamic quantites correspond to ensemble averages (ergodic theorem) some quantities
Thermodynamic stability and transformation of pharmaceutical polymorphs*
 Pure Appl. Chem., Vol. 77, No. 3, pp. 581 591, 2005. DOI: 10.1351/pac200577030581 2005 IUPAC Thermodynamic stability and transformation of pharmaceutical polymorphs* Mitsutaka Kitamura Department of Mechanical
Pure Appl. Chem., Vol. 77, No. 3, pp. 581 591, 2005. DOI: 10.1351/pac200577030581 2005 IUPAC Thermodynamic stability and transformation of pharmaceutical polymorphs* Mitsutaka Kitamura Department of Mechanical
BUDE. A General Purpose Molecular Docking Program Using OpenCL. Richard B Sessions
 BUDE A General Purpose Molecular Docking Program Using OpenCL Richard B Sessions 1 The molecular docking problem receptor ligand Proteins typically O(1000) atoms Ligands typically O(100) atoms predicted
BUDE A General Purpose Molecular Docking Program Using OpenCL Richard B Sessions 1 The molecular docking problem receptor ligand Proteins typically O(1000) atoms Ligands typically O(100) atoms predicted
Robust Parameter Estimation in Nonlinear Dynamic Process Models
 European Symposium on Computer Arded Aided Process Engineering 15 L. Puigjaner and A. Espuña (Editors) 2005 Elsevier Science B.V. All rights reserved. Robust Parameter Estimation in Nonlinear Dynamic Process
European Symposium on Computer Arded Aided Process Engineering 15 L. Puigjaner and A. Espuña (Editors) 2005 Elsevier Science B.V. All rights reserved. Robust Parameter Estimation in Nonlinear Dynamic Process
User Guide for LeDock
 User Guide for LeDock Hongtao Zhao, PhD Email: htzhao@lephar.com Website: www.lephar.com Copyright 2017 Hongtao Zhao. All rights reserved. Introduction LeDock is flexible small-molecule docking software,
User Guide for LeDock Hongtao Zhao, PhD Email: htzhao@lephar.com Website: www.lephar.com Copyright 2017 Hongtao Zhao. All rights reserved. Introduction LeDock is flexible small-molecule docking software,
Applications: Molecular crystals Graphite MgO(001)/CO MIL-53(Al) 2
/CO MIL-53(Al) 2") Bartolomeo Civalleri Voice: Loredana Valenzano B3LYP augmented with an empirical dispersion term (B3LYP-D*) as applied to solids Università di Torino Dipartimento di Chimica IFM & NIS Torino - MSSC2009-10/09/09
Bartolomeo Civalleri Voice: Loredana Valenzano B3LYP augmented with an empirical dispersion term (B3LYP-D*) as applied to solids Università di Torino Dipartimento di Chimica IFM & NIS Torino - MSSC2009-10/09/09
A Stochastic-Oriented NLP Relaxation for Integer Programming
 A Stochastic-Oriented NLP Relaxation for Integer Programming John Birge University of Chicago (With Mihai Anitescu (ANL/U of C), Cosmin Petra (ANL)) Motivation: The control of energy systems, particularly
A Stochastic-Oriented NLP Relaxation for Integer Programming John Birge University of Chicago (With Mihai Anitescu (ANL/U of C), Cosmin Petra (ANL)) Motivation: The control of energy systems, particularly
DENSITY FUNCTIONAL THEORY FOR NON-THEORISTS JOHN P. PERDEW DEPARTMENTS OF PHYSICS AND CHEMISTRY TEMPLE UNIVERSITY
 DENSITY FUNCTIONAL THEORY FOR NON-THEORISTS JOHN P. PERDEW DEPARTMENTS OF PHYSICS AND CHEMISTRY TEMPLE UNIVERSITY A TUTORIAL FOR PHYSICAL SCIENTISTS WHO MAY OR MAY NOT HATE EQUATIONS AND PROOFS REFERENCES
DENSITY FUNCTIONAL THEORY FOR NON-THEORISTS JOHN P. PERDEW DEPARTMENTS OF PHYSICS AND CHEMISTRY TEMPLE UNIVERSITY A TUTORIAL FOR PHYSICAL SCIENTISTS WHO MAY OR MAY NOT HATE EQUATIONS AND PROOFS REFERENCES
CO Adsorption Site Preference on Platinum: Charge Is the Essence
 Supporting Information CO Adsorption Site Preference on Platinum: Charge Is the Essence G.T. Kasun Kalhara Gunasooriya, and Mark Saeys *, Laboratory for Chemical Technology, Ghent University, Technologiepark
Supporting Information CO Adsorption Site Preference on Platinum: Charge Is the Essence G.T. Kasun Kalhara Gunasooriya, and Mark Saeys *, Laboratory for Chemical Technology, Ghent University, Technologiepark
Periodic DFT Study of Molecular Crystals
 , March 13-15, 2013, Hong Kong Periodic DFT Study of Molecular Crystals Richard Rivera, Soraya Jácome, Darwin Castillo, Arvids Stashans 1 Abstract Two molecular crystals have been studied using the first-principles
, March 13-15, 2013, Hong Kong Periodic DFT Study of Molecular Crystals Richard Rivera, Soraya Jácome, Darwin Castillo, Arvids Stashans 1 Abstract Two molecular crystals have been studied using the first-principles
Packing_Similarity_Dendrogram.py
 Packing_Similarity_Dendrogram.py Summary: This command-line script is designed to compare the packing of a set of input structures of a molecule (polymorphs, co-crystals, solvates, and hydrates). An all-to-all
Packing_Similarity_Dendrogram.py Summary: This command-line script is designed to compare the packing of a set of input structures of a molecule (polymorphs, co-crystals, solvates, and hydrates). An all-to-all
Deterministic Global Optimization for Dynamic Systems Using Interval Analysis
 Deterministic Global Optimization for Dynamic Systems Using Interval Analysis Youdong Lin and Mark A. Stadtherr Department of Chemical and Biomolecular Engineering University of Notre Dame, Notre Dame,
Deterministic Global Optimization for Dynamic Systems Using Interval Analysis Youdong Lin and Mark A. Stadtherr Department of Chemical and Biomolecular Engineering University of Notre Dame, Notre Dame,
Worst case analysis of mechanical structures by interval methods
 Worst case analysis of mechanical structures by interval methods Arnold Neumaier (University of Vienna, Austria) (joint work with Andrzej Pownuk) Safety Safety studies in structural engineering are supposed
Worst case analysis of mechanical structures by interval methods Arnold Neumaier (University of Vienna, Austria) (joint work with Andrzej Pownuk) Safety Safety studies in structural engineering are supposed
Valid Inequalities and Convex Hulls for Multilinear Functions
 Electronic Notes in Discrete Mathematics 36 (2010) 805 812 www.elsevier.com/locate/endm Valid Inequalities and Convex Hulls for Multilinear Functions Pietro Belotti Department of Industrial and Systems
Electronic Notes in Discrete Mathematics 36 (2010) 805 812 www.elsevier.com/locate/endm Valid Inequalities and Convex Hulls for Multilinear Functions Pietro Belotti Department of Industrial and Systems
Static and lattice vibrational energy differences between polymorphs
 Static and lattice vibrational energy differences between polymorphs Jonas Nyman a and Graeme M. Day a Received Xth XXXXXXXXXX 2XX, Accepted Xth XXXXXXXXX 2XX First published on the web Xth XXXXXXXXXX
Static and lattice vibrational energy differences between polymorphs Jonas Nyman a and Graeme M. Day a Received Xth XXXXXXXXXX 2XX, Accepted Xth XXXXXXXXX 2XX First published on the web Xth XXXXXXXXXX
Research Article Polymorphs of Tolfenamic Acids: Stability Analysis Using Cluster Method
 Physics Research International Volume 2016, Article ID 3537842, 5 pages http://dx.doi.org/10.1155/2016/3537842 Research Article Polymorphs of Tolfenamic Acids: Stability Analysis Using Cluster Method Lee
Physics Research International Volume 2016, Article ID 3537842, 5 pages http://dx.doi.org/10.1155/2016/3537842 Research Article Polymorphs of Tolfenamic Acids: Stability Analysis Using Cluster Method Lee
McCormick Relaxations: Convergence Rate and Extension to Multivariate Outer Function
 Introduction Relaxations Rate Multivariate Relaxations Conclusions McCormick Relaxations: Convergence Rate and Extension to Multivariate Outer Function Alexander Mitsos Systemverfahrenstecnik Aachener
Introduction Relaxations Rate Multivariate Relaxations Conclusions McCormick Relaxations: Convergence Rate and Extension to Multivariate Outer Function Alexander Mitsos Systemverfahrenstecnik Aachener
(3) E LJ (r ab ) = 4ɛ r ab
 E LJ (r ab ) = 4ɛ r ab") Hybrid Ensembles for Improved Force Matching Lee-Ping Wang and Troy Van Voorhis Department of Chemistry, Massachusetts Institute of Technology 77 Massachusetts Ave. Cambridge, MA 2139 USA Dated: 24 April
Hybrid Ensembles for Improved Force Matching Lee-Ping Wang and Troy Van Voorhis Department of Chemistry, Massachusetts Institute of Technology 77 Massachusetts Ave. Cambridge, MA 2139 USA Dated: 24 April
Temperature and pressure dependence of the Raman frequency shifts in anthracene
 Indian Journal of Pure & Applied Physics Vol. 54, August 2016, pp. 489-494 Temperature and pressure dependence of the Raman frequency shifts in anthracene H Özdemir & H Yurtseven* Department of Physics,
Indian Journal of Pure & Applied Physics Vol. 54, August 2016, pp. 489-494 Temperature and pressure dependence of the Raman frequency shifts in anthracene H Özdemir & H Yurtseven* Department of Physics,
Report on Atomistic Modeling of Bonding in Carbon-Based Nanostructures
 Report on Atomistic Modeling of Bonding in Carbon-Based Nanostructures Timothy Stillings Department of Physics, Astronomy and Materials Science Missouri State University Advisor: Ridwan Sakidja Abstract
Report on Atomistic Modeling of Bonding in Carbon-Based Nanostructures Timothy Stillings Department of Physics, Astronomy and Materials Science Missouri State University Advisor: Ridwan Sakidja Abstract
Polymorphism. 6 June 2012 Lecture 4 RUB
 Polymorphism 1 Polymorphism It is the phenomenon in which the same chemical substance exhibits different internal crystal packing arrangements. Polymorphism is an exclusively solid state phenomenon. Polymorphs
Polymorphism 1 Polymorphism It is the phenomenon in which the same chemical substance exhibits different internal crystal packing arrangements. Polymorphism is an exclusively solid state phenomenon. Polymorphs
ν + = e2 qq φ = 36.9,andθ = Pankratov [4] obtained ν 0 = ν + ν = e2 qq φ = 34.1, and θ = Boogaarts et al. [5]
![ν + = e2 qq φ = 36.9,andθ = Pankratov [4] obtained ν 0 = ν + ν = e2 qq φ = 34.1, and θ = Boogaarts et al. [5]](/thumbs/73/68415777.jpg "ν + = e2 qq φ = 36.9,andθ = Pankratov [4] obtained ν 0 = ν + ν = e2 qq φ = 34.1, and θ = Boogaarts et al. [5]") 14 N NQR Study of Diphenylamine Janez Seliger a,b and Veselko Žagar a a Jozef Stefan Institute, Jamova 39, 1000 Ljubljana, Slovenia b University of Ljubljana, Faculty of Mathematics and Physics, Department
14 N NQR Study of Diphenylamine Janez Seliger a,b and Veselko Žagar a a Jozef Stefan Institute, Jamova 39, 1000 Ljubljana, Slovenia b University of Ljubljana, Faculty of Mathematics and Physics, Department
Schrodinger ebootcamp #3, Summer EXPLORING METHODS FOR CONFORMER SEARCHING Jas Bhachoo, Senior Applications Scientist
 Schrodinger ebootcamp #3, Summer 2016 EXPLORING METHODS FOR CONFORMER SEARCHING Jas Bhachoo, Senior Applications Scientist Numerous applications Generating conformations MM Agenda http://www.schrodinger.com/macromodel
Schrodinger ebootcamp #3, Summer 2016 EXPLORING METHODS FOR CONFORMER SEARCHING Jas Bhachoo, Senior Applications Scientist Numerous applications Generating conformations MM Agenda http://www.schrodinger.com/macromodel
Fit point-wise ab initio calculation potential energies to a multi-dimension Morse/Long-Range model
 Fit point-wise ab initio calculation potential energies to a multi-dimension Morse/Long-Range model Yu Zhai, Hui Li Institute of Theoretical Chemistry, Jilin University, Changchun, Jilin, China Robert
Fit point-wise ab initio calculation potential energies to a multi-dimension Morse/Long-Range model Yu Zhai, Hui Li Institute of Theoretical Chemistry, Jilin University, Changchun, Jilin, China Robert
Fig. 3.1? Hard core potential
 6 Hard Sphere Gas The interactions between the atoms or molecules of a real gas comprise a strong repulsion at short distances and a weak attraction at long distances Both of these are important in determining
6 Hard Sphere Gas The interactions between the atoms or molecules of a real gas comprise a strong repulsion at short distances and a weak attraction at long distances Both of these are important in determining
Chapter 2 Experimental sources of intermolecular potentials
 Chapter 2 Experimental sources of intermolecular potentials 2.1 Overview thermodynamical properties: heat of vaporization (Trouton s rule) crystal structures ionic crystals rare gas solids physico-chemical
Chapter 2 Experimental sources of intermolecular potentials 2.1 Overview thermodynamical properties: heat of vaporization (Trouton s rule) crystal structures ionic crystals rare gas solids physico-chemical
Quantum Mechanics. An essential theory to understand properties of matter and light. Chemical Electronic Magnetic Thermal Optical Etc.
 Quantum Mechanics An essential theory to understand properties of matter and light. Chemical Electronic Magnetic Thermal Optical Etc. Fall 2018 Prof. Sergio B. Mendes 1 CHAPTER 3 Experimental Basis of
Quantum Mechanics An essential theory to understand properties of matter and light. Chemical Electronic Magnetic Thermal Optical Etc. Fall 2018 Prof. Sergio B. Mendes 1 CHAPTER 3 Experimental Basis of
Softwares for Molecular Docking. Lokesh P. Tripathi NCBS 17 December 2007
 Softwares for Molecular Docking Lokesh P. Tripathi NCBS 17 December 2007 Molecular Docking Attempt to predict structures of an intermolecular complex between two or more molecules Receptor-ligand (or drug)
Softwares for Molecular Docking Lokesh P. Tripathi NCBS 17 December 2007 Molecular Docking Attempt to predict structures of an intermolecular complex between two or more molecules Receptor-ligand (or drug)
Protein Structure Determination
 Protein Structure Determination Given a protein sequence, determine its 3D structure 1 MIKLGIVMDP IANINIKKDS SFAMLLEAQR RGYELHYMEM GDLYLINGEA 51 RAHTRTLNVK QNYEEWFSFV GEQDLPLADL DVILMRKDPP FDTEFIYATY 101
Protein Structure Determination Given a protein sequence, determine its 3D structure 1 MIKLGIVMDP IANINIKKDS SFAMLLEAQR RGYELHYMEM GDLYLINGEA 51 RAHTRTLNVK QNYEEWFSFV GEQDLPLADL DVILMRKDPP FDTEFIYATY 101
Exact Computation of Global Minima of a Nonconvex Portfolio Optimization Problem
 Frontiers In Global Optimization, pp. 1-2 C. A. Floudas and P. M. Pardalos, Editors c 2003 Kluwer Academic Publishers Exact Computation of Global Minima of a Nonconvex Portfolio Optimization Problem Josef
Frontiers In Global Optimization, pp. 1-2 C. A. Floudas and P. M. Pardalos, Editors c 2003 Kluwer Academic Publishers Exact Computation of Global Minima of a Nonconvex Portfolio Optimization Problem Josef
What is Classical Molecular Dynamics?
 What is Classical Molecular Dynamics? Simulation of explicit particles (atoms, ions,... ) Particles interact via relatively simple analytical potential functions Newton s equations of motion are integrated
What is Classical Molecular Dynamics? Simulation of explicit particles (atoms, ions,... ) Particles interact via relatively simple analytical potential functions Newton s equations of motion are integrated
An interval-matrix branch-and-bound algorithm for bounding eigenvalues
 Optimization Methods and Software ISSN: 1055-6788 (Print) 1029-4937 (Online) Journal homepage: https://www.tandfonline.com/loi/goms20 An interval-matrix branch-and-bound algorithm for bounding eigenvalues
Optimization Methods and Software ISSN: 1055-6788 (Print) 1029-4937 (Online) Journal homepage: https://www.tandfonline.com/loi/goms20 An interval-matrix branch-and-bound algorithm for bounding eigenvalues
74 these states cannot be reliably obtained from experiments. In addition, the barriers between the local minima can also not be obtained reliably fro
 73 Chapter 5 Development of Adiabatic Force Field for Polyvinyl Chloride (PVC) and Chlorinated PVC (CPVC) 5.1 Introduction Chlorinated polyvinyl chloride has become an important specialty polymer due to
73 Chapter 5 Development of Adiabatic Force Field for Polyvinyl Chloride (PVC) and Chlorinated PVC (CPVC) 5.1 Introduction Chlorinated polyvinyl chloride has become an important specialty polymer due to
Non-Convex Optimization. CS6787 Lecture 7 Fall 2017
 Non-Convex Optimization CS6787 Lecture 7 Fall 2017 First some words about grading I sent out a bunch of grades on the course management system Everyone should have all their grades in Not including paper
Non-Convex Optimization CS6787 Lecture 7 Fall 2017 First some words about grading I sent out a bunch of grades on the course management system Everyone should have all their grades in Not including paper
Pose and affinity prediction by ICM in D3R GC3. Max Totrov Molsoft
 Pose and affinity prediction by ICM in D3R GC3 Max Totrov Molsoft Pose prediction method: ICM-dock ICM-dock: - pre-sampling of ligand conformers - multiple trajectory Monte-Carlo with gradient minimization
Pose and affinity prediction by ICM in D3R GC3 Max Totrov Molsoft Pose prediction method: ICM-dock ICM-dock: - pre-sampling of ligand conformers - multiple trajectory Monte-Carlo with gradient minimization
Why study protein dynamics?
 Why study protein dynamics? Protein flexibility is crucial for function. One average structure is not enough. Proteins constantly sample configurational space. Transport - binding and moving molecules
Why study protein dynamics? Protein flexibility is crucial for function. One average structure is not enough. Proteins constantly sample configurational space. Transport - binding and moving molecules
Thermodynamic Integration with Enhanced Sampling (TIES)
") Thermodynamic Integration with Enhanced Sampling (TIES) A. P. Bhati, S. Wan, D. W. Wright and P. V. Coveney agastya.bhati.14@ucl.ac.uk Centre for Computational Science Department of Chemistry University
Thermodynamic Integration with Enhanced Sampling (TIES) A. P. Bhati, S. Wan, D. W. Wright and P. V. Coveney agastya.bhati.14@ucl.ac.uk Centre for Computational Science Department of Chemistry University
André Schleife Department of Materials Science and Engineering
 André Schleife Department of Materials Science and Engineering Yesterday you (should have) learned this: http://upload.wikimedia.org/wikipedia/commons/e/ea/ Simple_Harmonic_Motion_Orbit.gif 1. deterministic
André Schleife Department of Materials Science and Engineering Yesterday you (should have) learned this: http://upload.wikimedia.org/wikipedia/commons/e/ea/ Simple_Harmonic_Motion_Orbit.gif 1. deterministic
AB INITIO MOLECULAR-DYNAMICS SIMULATIONS OF ADSORPTION OF DYE MOLECULES AT SURFACES
 AB INITIO MOLECULAR-DYNAMICS SIMULATIONS OF ADSORPTION OF DYE MOLECULES AT SURFACES M. SUGIHARA, H. MEYER, AND P. ENTEL Theoretische Tieftemperaturphysik, Universität Duisburg, 47048 Duisburg, Germany
AB INITIO MOLECULAR-DYNAMICS SIMULATIONS OF ADSORPTION OF DYE MOLECULES AT SURFACES M. SUGIHARA, H. MEYER, AND P. ENTEL Theoretische Tieftemperaturphysik, Universität Duisburg, 47048 Duisburg, Germany
Michael W. Mahoney Department of Physics, Yale University, New Haven, Connecticut 06520
 JOURNAL OF CHEMICAL PHYSICS VOLUME 115, NUMBER 23 15 DECEMBER 2001 Quantum, intramolecular flexibility, and polarizability effects on the reproduction of the density anomaly of liquid water by simple potential
JOURNAL OF CHEMICAL PHYSICS VOLUME 115, NUMBER 23 15 DECEMBER 2001 Quantum, intramolecular flexibility, and polarizability effects on the reproduction of the density anomaly of liquid water by simple potential
V(φ) CH 3 CH 2 CH 2 CH 3. High energy states. Low energy states. Views along the C2-C3 bond
 CH 3 CH 2 CH 2 CH 3. High energy states. Low energy states. Views along the C2-C3 bond") Example V(φ): Rotational conformations of n-butane C 3 C C C 3 Potential energy of a n-butane molecule as a function of the angle φ of bond rotation. V(φ) Potential energy/kj mol -1 0 15 10 5 eclipse gauche
Example V(φ): Rotational conformations of n-butane C 3 C C C 3 Potential energy of a n-butane molecule as a function of the angle φ of bond rotation. V(φ) Potential energy/kj mol -1 0 15 10 5 eclipse gauche
My Career in the Pharmaceutical Industry
 SCI Career Options Seminar My Career in the Pharmaceutical Industry Simon Yates, AstraZeneca University of Sheffield 27 th November 2013 1998-2002 - Degree MChem. Undergraduate at University of York Final
SCI Career Options Seminar My Career in the Pharmaceutical Industry Simon Yates, AstraZeneca University of Sheffield 27 th November 2013 1998-2002 - Degree MChem. Undergraduate at University of York Final