Communication: An accurate full fifteen dimensional permutationally invariant. Jun Li, 1,* and Hua Guo 2,* China. New Mexico 87131, United States
|
|
|
- Augustine Butler
- 5 years ago
- Views:
Transcription
1 Submitted to J. Chem. Phys., 11/7/2015, revised, 11/28/2015 Communication: An accurate full fifteen dimensional permutationally invariant potential energy surface for the OH + CH 4 H 2 O + CH 3 reaction Jun Li, 1,* and Hua Guo 2,* 1 School of Chemistry and Chemical Engineering, Chongqing University, Chongqing , China 2 Department of Chemistry and Chemical Biology, University of New Mexico, Albuquerque, New Mexico 87131, United States *: corresponding authors, s: jli15@cqu.edu.edu (JL), hguo@unm.edu (HG) 1
2 Abstract A globally accurate full-dimensional potential energy surface (PES) for the OH + CH 4 H 2 O + CH 3 reaction is developed using the permutation invariant polynomial-neural network (PIP-NN) approach based on ~135,000 points at the level of UCCSD(T)-F12a/AVTZ. The total root mean square fitting error is only 3.9 mev or 0.09 kcal/mol. This PES is shown to reproduce energies, geometries, and harmonic frequencies of stationary points along the reaction path. Kinetic and dynamical calculations on the PES indicated a good agreement with available experimental data. 2
3 An important pre-requisite for studying dynamics of chemical reactions is an accurate representation of the Born-Oppenheimer potential energy surfaces (PESs). Despite the tremendous advances in electronic structure theory, the analytical representation of multi-dimensional global PESs based on large number of ab initio points has only recently become a reality for reactive systems beyond three atoms. 1-3 Indeed, several analytical functional forms with the necessary permutation invariance have been proposed to represent the complex topography of the PES. The two main forms are the permutation invariant polynomials (PIP) 2 and neural network (NN) methods. 4, 5 Our recent work combining the two approaches led to the so-called PIP-NN method, 6, 7 which has been demonstrated to be both accurate and efficient in representing global PESs for reactive systems up to six atoms In this Communication, we report a globally accurate PIP-NN PES for a seven-atom reactive system based on ~135,000 high-level ab initio points, which spans unprecedented fifteen degrees of freedom. The hydrogen abstraction reaction involving the simplest hydrocarbon, methane, by the hydroxyl radical, i.e., OH + CH 4 H 2 O + CH 3 (ΔH = kcal/mol), is of great importance in atmospheric chemistry because of its role in removing the greenhouse gas methane, which in turn provides a main sink for atmospheric OH radicals. This reaction is also relevant in combustion at high temperatures. For these reasons, its thermal rate coefficients have been measured using different techniques over a wide range ( K) of temperatures (see Ref. 12 and references therein). In addition, the state-to-state dynamics of the OH/OD + CH 4 /CD 4 /CHD 3 reactions has been investigated experimentally by Liu and co-workers In addition, the vibrational spectroscopy and decay dynamics of the CH 4 OH 3
4 16, 17 complex in the entrance channel have been studied by Lester and co-workers. Much theoretical work has also been devoted to the reaction barrier and rate coefficients at various levels of theory for this prototypical bimolecular reaction with seven atoms However, a complete understanding of the reaction dynamics requires a full-dimensional global PES. The first full-dimensional PES for the title reaction was constructed by Espinosa-Garcia and co-workers using a molecular mechanics functional form based on both theoretical and experimental information. 25 Several kinetics and dynamics calculations have since been performed on this so-called PES-2000, and the agreement with available experimental data has generally been good. A refined PES, PES-2014, was later constructed by fitting exclusively to ab initio data, using essentially the same approach. 31 Additional studies have been performed on the new PES to provide insight into the kinetics and dynamics of this reaction Despite their immense value as the first full-dimensional global PESs, their limitations are also apparent. Due to the simple analytical forms and the limited number of ab initio points used to determine the small set of parameters, these PESs are not expected to be accurate outside the region near the minimum energy path (MEP). 31 Even for configurations along the MEP, it has been pointed out that the saddle points in these PESs are significantly different from that determined at a higher level of theory. 30 In addition, only the permutation symmetry of the four hydrogens in CH 4 was taken into account. To better understand the dynamics of this important reaction, a PES that is uniformly accurate in all relevant configurations is highly desired. To this end, we report a new globally accurate PES based on 135,000 ab initio points at the explicitly correlated coupled cluster singles, doubles, and perturbative triples level with the augmented correlation consistent 4
5 polarized valence triple-zeta basis set (UCCSD(T)-F12a/AVTZ). 34, 35 These points were fit using the PIP-NN approach, 6, 7 with a total root-mean square error (RMSE) of only 3.9 mev. The construction of the PES consists of two steps: sampling of the data points in the relevant configuration space and fitting them with an analytic function. To this end, a small set of data points around the vicinity of the MEP was first selected by running direct dynamics trajectories at a low theory level. Then, UCCSD(T)-F12a/AVTZ calculations were performed at these selected points, and a primitive PES was constructed. Trajectories with various initial conditions were then dispatched on this primitive PES to further explore the configuration space and to generate new points. Points were added to the data set if they are not too close to those already in the existing data set and a new PES is subsequently generated. This procedure was repeated until convergence. Finally, a total of ~135,000 points were calculated and fit using the PIP-NN method. 6, 7 They correspond to points because of the hydrogen (5!=120 fold) permutation symmetries. The permutation symmetry of the system can be enforced by symmetry functions as the input layer of the NN and the permutation symmetry of all five hydrogens is taken into account. In PIP-NN, the PIPs are symmetrized monomials of Morse-like variables of the internuclear distances ( p exp( r ) with =1.0 Å -1 ): 36 ij ij 7 PIP( ) ˆ lij r S p (1) ij i j ij where Ŝ is the symmetrization operator and l the order. The PIP-NN PES can be formally expressed as V f (PIP( r )) (2) NN ij All 1331 PIPs up to the fifth order were used in the input layer for this A 5 BC type molecule 5
6 and the NN architecture was selected to be of 4 and 100 neurons for the two hidden layers, resulting in 5929 parameters. To avoid overfitting, the data are randomly divided into the training (90%), validating (5%) and testing (5%) sets. The target function is defined as the RMSE of the PES points from their ab initio counterparts. Further details of the PIP-NN fitting can be found in our recent publications The final PES is the average of three best PIP-NN fits, which have RMSEs for the training/validating/testing sets and maximum deviation of 4.8/6.4/5.6/187.4, 4.2/5.0/6.8/225.7, and 4.4/6.1/6.3/212.1 mev, respectively. The RMSE of the final PES is 3.9 mev with a maximum deviation mev. The PES can be obtained from the authors upon request. Figure 1 presents the contour plot of the PIP-NN PES along the two reactive coordinates, r OH and r CH, with other internal coordinates fixed at saddle point. It is clearly seen that this reaction has a reactant-like barrier, much earlier than that on the PES-2014.{Espinosa-Garcia, 2015 #9133} The reaction energy of kcal/mol is in reasonably good agreement with the latest theoretical values of -13.5, 20 and kcal/mol. 31 In Figure 2, geometric parameters of the stationary points along MEP, namely the reactants (OH + CH 4 ), transition state (TS), products (H 2 O + CH 3 ), as well as van der Waals complexes in the entrance (R-vdW) and exit (P-vdW) channels are displayed. The general agreement with previous theoretical results 18, 21-23, 30 is quite good except for TS and R-vdW (vide infra). The energies and geometries of the stationary points are all reproduced well by the PIP-NN PES, as illustrated in Figure 2. The differences in energy are less than 0.02 kcal/mol. The geometries of the reactants, products, and TS agree well with the ab initio ones, and the deviations of the bond lengths and angles are less than Å and 0.4º, respectively. The agreement for the 6
7 geometries of R- and P-vdW is not as good because of the floppy nature of these complexes. Similarly, the harmonic frequencies, which are collected in Supplementary Information (SI), 37 of the rigid modes are reproduced better than those of the floppy ones. In early work, both eclipsed and staggered geometries were found for TS at relative low levels of theory. 19 Later, the staggered conformation was identified as a second-order saddle point, 21 and the eclipsed conformation was thus considered as the TS, confirmed subsequently at the MCG3/3 23 and MP2/6-31G(d,p) levels. 31 However, a staggered and slightly non-linear TS is found at the UCCSD(T)-F12a/AVTZ level, as shown in Figure 2. Our TS geometry is quite different from that in PES-2014: which overestimates the breaking C-H bond by ~0.1 Å and the forming O-H bond by ~0.04 Å. The classical barrier of 6.29 kcal/mol obtained here can be compared to the literature values of 6.7, , , 24 and 6.4 kcal/mol. 31 The van der Waals complexes in the entrance and exit channels are not present in PES-2000, but included in PES At the level of UCCSD(T)-F12a/AVTZ, we did not find the o.4 kcal/mol deep H 3 CH OH well present in PES-2014, 31 but identified a C 3v H 4 C HO well (Figure 2), which is 1.22 kcal/mol lower than the reactant asymptote. This is consistent with the recent theoretical work. 16, 38 The P-vdW complex, on the other hand, was found in an H 3 C HOH configuration, stabilized by 1.84 kcal/mol with respect to the product asymptote, consistent with previous theoretical reports. 21, 31 It is interesting that some small imaginary frequencies persist and are hard to eliminate at the ab initio level, as shown in SI, due apparently to the floppy nature of the system. The thermal rate coefficients of the title reaction were computed on the PIP-NN PES 7
8 with the canonical variational transition-state theory (CVTST) 39 using POLYRATE. 40 Motions orthogonal to the reaction path were treated using quantum mechanical vibrational partition functions under the harmonic approximation, except for the torsional mode of the OH group with respect to CH 3 group. The hindered rotor model was used to account for the anharmonicity in that mode. 23 Quantum effects in the reaction coordinate were included by using the micro-canonical optimized multidimensional tunneling (μomt) approach, 39 in which, at each total energy, the larger of the small-curvature (SCT) and large-curvature (LCT) tunneling probabilities was taken as the best estimate. The rotational partition functions were calculated classically. The symmetry factor of 12 and the OH spin-orbit splitting (140.0 cm -1 ), were taken into account in the reactant electronic partition function. As shown in Figure 3(a), the calculated rate coefficients on the current PIP-NN PES and PES-2014{Espinosa-Garcia, 2015 #9133} are both in good agreement with experimental data, 12 reproducing the tunneling facilitated non-arrhenius feature at low temperatures. As discussed by Ellingson et al.,{ellingson, 2007 #7618} the treatment of the torsional problem and the coordinate system have a significant effect on the kinetics, and they remain open questions. We plan to perform a systematic study on the kinetics of the title reaction, including the kinetic isotope effects, in the near future. The kinetics of the reaction is mostly sensitive to the TS region of the PES. To further test the PIP-NN PES in a wider range of configurations, quasi-classical trajectory calculations were performed using VENUS 41 for the experimentally studied OH + CD 4 HOD + CD 3 reaction at collision energies ranging from 4 to 20 kcal/mol. At each collision energy, 10 5 or trajectories were calculated starting from the ro-vibrational ground states of OH and 8
9 CD 4, and the maximal impact parameter (b max ) was determined using small batches of trajectories with trial values. The trajectories were initiated with a reactant separation of 8.0 Å and terminated when products or reactants reached a separation of 8.0 Å. Other scattering parameters including the spatial orientation of the initial reactants, vibrational phases, and impact parameter were determined according to the Monte Carlo approach as implemented in VENUS. 41 The gradient of the PES is obtained numerically by a central-difference algorithm. The propagation time step was 0.05 fs. Almost all trajectories conserved energy to within a chosen criteria (10-4 kcal/mol), which testifies the smoothness of the PES. Figure 3(b) presents the reaction cross sections, compared to the experiment 14 and recent calculations. 31 Since only the relative cross sections were reported experimentally, the theoretical QCT values on the PIP-NN PES and PES-2014 are both normalized at the collision energy of 16 kcal/mol. Overall, they agree well with each other, and the PIP-NN PES appears slightly better than PES-2014 in reproducing the experiment. To summarize, we have developed a full fifteen-dimensional highly accurate global PES for the OH + CH 4 H 2 O + CH 3 reaction based on ~135,000 ab initio points calculated at the level of UCCSD(T)-F12a/AVTZ. Full permutation symmetry is enforced, which not only reduces computational costs, but also provides correct description of the PES, particularly at high symmetry regions. The preliminary kinetic and dynamical calculations on this PES are very promising. It is our expectation that this PES will provide a reliable platform for dynamical studies of this important bimolecular reaction in the future. Acknowledgments: This work was supported by the Hundred-Talent Foundation of Chongqing University (project no to JL) and National Natural Science 9
10 Foundation of China ( to JL), and by US Department of Energy (DE-FG02-05ER15694 to HG). References: 1. M. A. Collins, Theo. Chem. Acc. 108, 313 (2002). 2. J. M. Bowman, G. Czakó and B. Fu, Phys. Chem. Chem. Phys. 13, 8094 (2011). 3. J. Li, B. Jiang, H. Song, J. Ma, B. Zhao, R. Dawes and H. Guo, J. Phys. Chem. A 119, 4667 (2015). 4. L. M. Raff, R. Komanduri, M. Hagan and S. T. S. Bukkapatnam, Neural Networks in Chemical Reaction Dynamics. (Oxford University Press, Oxford, 2012). 5. J. Chen, X. Xu and D. H. Zhang, J. Chem. Phys. 138, (2013). 6. B. Jiang and H. Guo, J. Chem. Phys. 139, (2013). 7. J. Li, B. Jiang and H. Guo, J. Chem. Phys. 139, (2013). 8. A. Li and H. Guo, J. Chem. Phys. 140, (2014). 9. J. Li, J. Chen, D. H. Zhang and H. Guo, J. Chem. Phys. 140, (2014). 10. J. Li and H. Guo, Phys. Chem. Chem. Phys. 16, 6753 (2014). 11. J. Li, J. Chen, Z. Zhao, D. Xie, D. H. Zhang and H. Guo, J. Chem. Phys. 142, (2015). 12. N. K. Srinivasan, M. C. Su, J. W. Sutherland and J. V. Michael, J. Phys. Chem. A 109, 1857 (2005). 13. B. Zhang, W. C. Shiu, J. J. Lin and K. Liu, J. Chem. Phys. 122, (2005). 14. B. Zhang, W. C. Shiu and K. Liu, J. Phys. Chem. A 109, 8983 (2005). 15. B. Zhang, W. C. Shiu and K. Liu, J. Phys. Chem. A 109, 8989 (2005). 16. M. D. Wheeler, M. Tsiouris, M. I. Lester and G. Lendvay, J. Chem. Phys. 112, 6590 (2000). 17. M. Tsiouris, M. D. Wheeler and M. I. Lester, J. Chem. Phys. 114, 187 (2001). 18. K. D. Dobbs, D. A. Dixon and A. Komornicki, J. Chem. Phys. 98, 8852 (1993). 19. V. S. Melissas and D. G. Truhlar, J. Chem. Phys. 99, 1013 (1993). 20. B. J. Lynch, P. L. Fast, M. Harris and D. G. Truhlar, J. Phys. Chem. A 104, 4811 (2000). 21. L. Masgrau, A. Gonzalez-Lafont and J. M. Lluch, J. Chem. Phys. 114, 2154 (2001). 22. S. R. Sellevag, G. Nyman and C. J. Nielsen, J. Phys. Chem. A 110, 141 (2006). 23. B. A. Ellingson, J. Pu, H. Lin, Y. Zhao and D. G. Truhlar, J. Phys. Chem. A 111, (2007). 24. A. Karton, A. Tarnopolsky, J.-F. Lamere, G. C. Schatz and J. M. L. Martin, J. Phys. Chem. A 112, (2008). 25. J. Espinosa-García and J. C. Corchado, J. Chem. Phys. 112, 5731 (2000). 26. H.-G. Yu, J. Chem. Phys. 114, 2967 (2001). 27. W. Wang and Y. Zhao, J. Chem. Phys. 137, (2012). 28. J. W. Allen, W. H. Green, Y. Li, H. Guo and Y. V. Suleimanov, J. Chem. Phys. 138, (2013). 29. H. Song, S.-Y. Lee, M. Yang and Y. Lu, J. Chem. Phys. 139, (2013). 30. H. Song, J. Li, B. Jiang, M. Yang, Y. Lu and H. Guo, J. Chem. Phys. 140, (2014). 31. J. Espinosa-Garcia and J. C. Corchado, Theo. Chem. Acc. 134, 1 (2015). 32. J. Espinosa-Garcia and J. C. Corchado, J. Phys. Chem. B ASAP (2015). 33. Y. V. Suleimanov and J. Espinosa-Garcia, J. Phys. Chem. B ASAP (2015). 34. T. B. Adler, G. Knizia and H.-J. Werner, J. Chem. Phys. 127, (2007). 35. G. Knizia, T. B. Adler and H.-J. Werner, J. Chem. Phys. 130, (2009). 36. Z. Xie and J. M. Bowman, J. Chem. Theo. Comp. 6, 26 (2010). 10
11 37. See supplementary material at for additional results. 38. H. Basch and S. Hoz, J. Phys. Chem. A 101, 4416 (1997). 39. D. G. Truhlar, A. D. Issacson and B. C. Garrett, in Theory of Chemical Reaction Dynamics, edited by M. Bear (CRC, Boca Raton, 1985), pp J. C. Corchado, Y.-Y. Chuang, P. L. Fast, W.-P. Hu, Y.-P. Liu, G. C. Lynch, K. A. Nguyen, C. F. Jackels, A. Fernandez Ramos, B. A. Ellingson, B. J. Lynch, J. Zheng, V. S. Melissas, J. Villà, I. Rossi, E. L. Coitiño, J. Pu, T. V. Albu, R. Steckler, B. C. Garrett, A. D. Isaacson and D. G. Truhlar, (University of Minnesota, Minneapolis, 2007). 41. X. Hu, W. L. Hase and T. Pirraglia, J. Comp. Chem. 12, 1014 (1991). 11
12 Figures Captions Figure 1. Contour plot for the OH + CH 4 H 2 O + CH 3 reaction along the two reactive coordinates, r OH and r CH, (in Å) with other internal coordinates fixed at the transition state. The energies are in kcal/mol relative to the reactant asymptote. The comparison of the minimum energy path on the PIP-NN PES and by the ab initio calculation is also shown in the inset. Figure 2. Schematic illustration of the reaction pathway for the reaction OH + CH 4 H 2 O + CH 3. All energies are in kcal/mol relative to the OH + CH 4 asymptote. The geometries (distances in Å and angels in deg.) of the stationary points are also shown. The first value (in black) is obtained on the present PIP-NN PES and the second (in red) is determined at the level of UCCSD(T)-F12a/AVTZ. The third entry in blue corresponds to those on PES The geometries of TS on PES-2014 are also included (the third entry in blue) for comparison. The numbers are not meant to indicate the significant figures, but are used here to compare with other theoretical values. Figure 3. (a) Comparison of thermal rate coefficients for the title reaction OH + CH 4 H 2 O + CH 3. (b) Relative reaction cross section as a function of the collision energy (E c ) for the reaction OH + CD 4 HOD + CD 3. The experimental values (only the ground state CD 3 was considered) are adopted from Ref. 14, and the QCT results on the PES-2014 are adopted from Ref
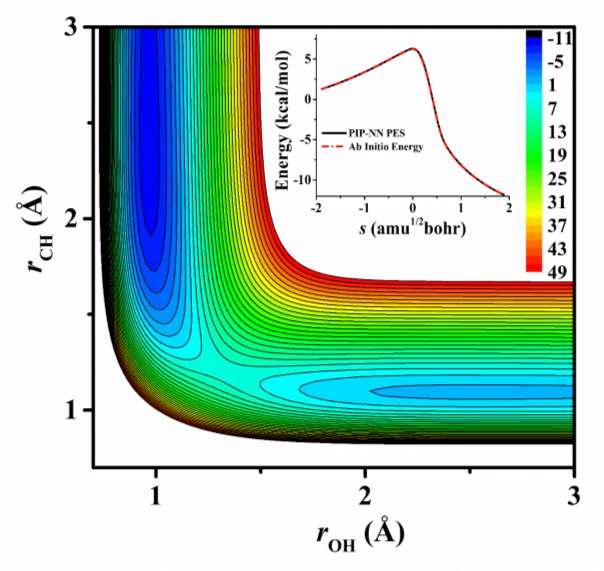
13 Fig. 1 13
14 Fig. 2 14
15 Fig. 3 15
16
17
18
Equivalence between Symmetric and Antisymmetric Stretching Modes of NH 3 in
 Submitted to JCP, 9/8/2016 Equivalence between Symmetric and Antisymmetric Stretching Modes of NH 3 in Promoting H + NH 3 H 2 + NH 2 Reaction Hongwei Song, 1,* Minghui Yang, 1 and Hua Guo 2 1 Key Laboratory
Submitted to JCP, 9/8/2016 Equivalence between Symmetric and Antisymmetric Stretching Modes of NH 3 in Promoting H + NH 3 H 2 + NH 2 Reaction Hongwei Song, 1,* Minghui Yang, 1 and Hua Guo 2 1 Key Laboratory
A Combined Experimental-Theoretical Study of the OH + CO H + CO 2. Reaction Dynamics
 A Combined Experimental-Theoretical Study of the OH + CO H + CO 2 Reaction Dynamics Adriana Caracciolo, 1,# Dandan Lu, 2,# Nadia Balucani, 1 Gianmarco Vanuzzo, 1 Domenico Stranges, 3 Xingan Wang, 4 Jun
A Combined Experimental-Theoretical Study of the OH + CO H + CO 2 Reaction Dynamics Adriana Caracciolo, 1,# Dandan Lu, 2,# Nadia Balucani, 1 Gianmarco Vanuzzo, 1 Domenico Stranges, 3 Xingan Wang, 4 Jun
Ring Polymer Molecular Dynamics Calculations of Thermal Rate Constants for the O([superscript 3]P) + CH[subscript 4] OH + CH[subscript 3] Reaction:
![Ring Polymer Molecular Dynamics Calculations of Thermal Rate Constants for the O([superscript 3]P) + CH[subscript 4] OH + CH[subscript 3] Reaction:](/thumbs/89/99065962.jpg "Ring Polymer Molecular Dynamics Calculations of Thermal Rate Constants for the O([superscript 3]P) + CH[subscript 4] OH + CH[subscript 3] Reaction:") Ring Polymer Molecular Dynamics Calculations of Thermal Rate Constants for the O([superscript 3]P) + CH[subscript 4] OH + CH[subscript 3] Reaction: The MIT Faculty has made this article openly available.
Ring Polymer Molecular Dynamics Calculations of Thermal Rate Constants for the O([superscript 3]P) + CH[subscript 4] OH + CH[subscript 3] Reaction: The MIT Faculty has made this article openly available.
OH CH 3 D H 2 O CH 2 D and HDO CH 3, R2
 JOURNAL OF CHEMICAL PHYSICS VOLUME 114, NUMBER 5 1 FEBRUARY 2001 The reactions CH n D 4Àn OH\P and CH 4 OD\CH 3 HOD as a test of current direct dynamics computational methods to determine variational transition-state
JOURNAL OF CHEMICAL PHYSICS VOLUME 114, NUMBER 5 1 FEBRUARY 2001 The reactions CH n D 4Àn OH\P and CH 4 OD\CH 3 HOD as a test of current direct dynamics computational methods to determine variational transition-state
Manual for SS-QRRK utility code
 Manual for SS-QRRK utility code SS-QRRK: A Program for System-Specific Quantum Rice-Ramsperger-Kassel Theory Junwei Lucas Bao and Donald G. Truhlar Department of Chemistry Chemical Theory Center, and Supercomputing
Manual for SS-QRRK utility code SS-QRRK: A Program for System-Specific Quantum Rice-Ramsperger-Kassel Theory Junwei Lucas Bao and Donald G. Truhlar Department of Chemistry Chemical Theory Center, and Supercomputing
Stereodynamics of the O( 3 P) with H 2 (D 2 ) (ν = 0, j = 0) reaction
 with H 2 (D 2 ) (ν = 0, j = 0) reaction") Stereodynamics of the O( 3 P) with H 2 (D 2 ) (ν = 0, j = 0) reaction Liu Yu-Fang( ), He Xiao-Hu( ), Shi De-Heng( ), and Sun Jin-Feng( ) Department of Physics, Henan Normal University, Xinxiang 453007,
Stereodynamics of the O( 3 P) with H 2 (D 2 ) (ν = 0, j = 0) reaction Liu Yu-Fang( ), He Xiao-Hu( ), Shi De-Heng( ), and Sun Jin-Feng( ) Department of Physics, Henan Normal University, Xinxiang 453007,
Citation. As Published Publisher. Version
 Rate coefficients and kinetic isotope effects of the X + CH[subscript 4] CH[subscript 3] + HX (X = H, D, Mu) reactions from ring polymer molecular dynamics The MIT Faculty has made this article openly
Rate coefficients and kinetic isotope effects of the X + CH[subscript 4] CH[subscript 3] + HX (X = H, D, Mu) reactions from ring polymer molecular dynamics The MIT Faculty has made this article openly
Manual for SS-QRRK utility code
 Manual for SS-QRRK utility code SS-QRRK: A Program for System-Specific Quantum Rice-Ramsperger-Kassel Theory Junwei Lucas Bao and Donald G. Truhlar Department of Chemistry Chemical Theory Center, and Supercomputing
Manual for SS-QRRK utility code SS-QRRK: A Program for System-Specific Quantum Rice-Ramsperger-Kassel Theory Junwei Lucas Bao and Donald G. Truhlar Department of Chemistry Chemical Theory Center, and Supercomputing
Hongwei Song and Hua Guo * Department of Chemistry and Chemical Biology, University of New Mexico, Albuquerque, New Mexico, 87131, USA
 Submitted to JCP, 10/21/2014 Effects of reactant rotational excitations on + N H + NH 3 reactivity Hongwei Song and Hua Guo * Department of Chemistry and Chemical Biology, University of New Mexico, Albuquerque,
Submitted to JCP, 10/21/2014 Effects of reactant rotational excitations on + N H + NH 3 reactivity Hongwei Song and Hua Guo * Department of Chemistry and Chemical Biology, University of New Mexico, Albuquerque,
Direct ab initio dynamics studies of N H 2^NH H reaction
 JOURNAL OF CHEMICAL PHYSICS VOLUME 113, NUMBER 15 15 OCTOBER 2000 Direct ab initio dynamics studies of N H 2^NH H reaction Shaowen Zhang and Thanh N. Truong a) Henry Eyring Center for Theoretical Chemistry,
JOURNAL OF CHEMICAL PHYSICS VOLUME 113, NUMBER 15 15 OCTOBER 2000 Direct ab initio dynamics studies of N H 2^NH H reaction Shaowen Zhang and Thanh N. Truong a) Henry Eyring Center for Theoretical Chemistry,
Variational Transition State Theory Calculations of Thermal Rate Coefficients for the O( 3 P ) + HCl Reaction
 + HCl Reaction") Variational Transition State Theory Calculations of Thermal Rate Coefficients for the O( 3 P ) + HCl Reaction Thomas C. Allison (a), B. Ramachandran (b), Jörg Senekowitsch (c), Donald G. Truhlar (a), and
Variational Transition State Theory Calculations of Thermal Rate Coefficients for the O( 3 P ) + HCl Reaction Thomas C. Allison (a), B. Ramachandran (b), Jörg Senekowitsch (c), Donald G. Truhlar (a), and
Resonances in Chemical Reactions : Theory and Experiment. Toshiyuki Takayanagi Saitama University Department of Chemistry
 Resonances in Chemical Reactions : Theory and Experiment Toshiyuki Takayanagi Saitama University Department of Chemistry What is Chemical Reaction? Collision process between molecules (atoms) containing
Resonances in Chemical Reactions : Theory and Experiment Toshiyuki Takayanagi Saitama University Department of Chemistry What is Chemical Reaction? Collision process between molecules (atoms) containing
PCCP PAPER. Tracking the energy flow in the hydrogen exchange reaction OH + H 2 O - H 2 O+OH. I. Introduction. Yongfa Zhu, Jun Li * d a
 PCCP PAPER Cite this: Phys. Chem. Chem. Phys., 2018, 20, 12543 Tracking the energy flow in the hydrogen exchange reaction OH + H 2 O - H 2 O+OH Yongfa Zhu, ab Leilei Ping, ac Mengna Bai, d Yang Liu, d
PCCP PAPER Cite this: Phys. Chem. Chem. Phys., 2018, 20, 12543 Tracking the energy flow in the hydrogen exchange reaction OH + H 2 O - H 2 O+OH Yongfa Zhu, ab Leilei Ping, ac Mengna Bai, d Yang Liu, d
Manual for SS-QRRK utility code
 Manual for SS-QRRK utility code SS-QRRK: A Program for System-Specific Quantum Rice-Ramsperger-Kassel Theory SS-QRRK Version: 2018.12.22 Junwei Lucas Bao and Donald G. Truhlar Department of Chemistry Chemical
Manual for SS-QRRK utility code SS-QRRK: A Program for System-Specific Quantum Rice-Ramsperger-Kassel Theory SS-QRRK Version: 2018.12.22 Junwei Lucas Bao and Donald G. Truhlar Department of Chemistry Chemical
Roaming and Spectroscopy
 Roaming and Spectroscopy D. Townsend, S. A. Lahankar, S. K. Lee, S. D. Chambreau, A. G. Suits, X. Zhang, and J. Rheinecker, L. B. Harding, J. M. Bowman, Science. 306, 1158 (2004). J. M. Bowman, X. Zhang,
Roaming and Spectroscopy D. Townsend, S. A. Lahankar, S. K. Lee, S. D. Chambreau, A. G. Suits, X. Zhang, and J. Rheinecker, L. B. Harding, J. M. Bowman, Science. 306, 1158 (2004). J. M. Bowman, X. Zhang,
Laser-Cooled Be + Ions
 Supporting Information Optical Control of Reactions between Water and Laser-Cooled Be + Ions Tiangang Yang 1,*, Anyang Li 2,*, Gary K. Chen 1, Changjian Xie 3, Arthur G. Suits 4, Wesley C. Campbell 1,
Supporting Information Optical Control of Reactions between Water and Laser-Cooled Be + Ions Tiangang Yang 1,*, Anyang Li 2,*, Gary K. Chen 1, Changjian Xie 3, Arthur G. Suits 4, Wesley C. Campbell 1,
Example questions for Molecular modelling (Level 4) Dr. Adrian Mulholland
 Dr. Adrian Mulholland") Example questions for Molecular modelling (Level 4) Dr. Adrian Mulholland 1) Question. Two methods which are widely used for the optimization of molecular geometies are the Steepest descents and Newton-Raphson
Example questions for Molecular modelling (Level 4) Dr. Adrian Mulholland 1) Question. Two methods which are widely used for the optimization of molecular geometies are the Steepest descents and Newton-Raphson
Accurate ab initio potential energy surface, thermochemistry, and dynamics of the Cl(2P, 2P3/2) + CH4 HCl + CH3 and H + CH3Cl reactions
 + CH4 HCl + CH3 and H + CH3Cl reactions") Accurate ab initio potential energy surface, thermochemistry, and dynamics of the Cl(2P, 2P3/2) + CH4 HCl + CH3 and H + CH3Cl reactions Gábor Czakó and Joel M. Bowman Citation: J. Chem. Phys. 136, 044307
Accurate ab initio potential energy surface, thermochemistry, and dynamics of the Cl(2P, 2P3/2) + CH4 HCl + CH3 and H + CH3Cl reactions Gábor Czakó and Joel M. Bowman Citation: J. Chem. Phys. 136, 044307
A VTST Study of the H + O 3 and O + HO 2 Reactions Using a Six-dimensional DMBE Potential Energy Surface for Ground State HO 3
 J. Phys. Chem. A 2002, 106, 4077-4083 4077 A VTST Study of the H + O 3 and O + HO 2 Reactions Using a Six-dimensional DMBE Potential Energy Surface for Ground State HO 3 A. Fernández-Ramos and A. J. C.
J. Phys. Chem. A 2002, 106, 4077-4083 4077 A VTST Study of the H + O 3 and O + HO 2 Reactions Using a Six-dimensional DMBE Potential Energy Surface for Ground State HO 3 A. Fernández-Ramos and A. J. C.
Tests of Potential Energy Surfaces for H + CH 4 CH 3 + H 2 : Deuterium and Muonium Kinetic Isotope Effects for the Forward and Reverse Reaction
 Revised: July 12, 2002 Tests of Potential Energy Surfaces for H + CH 4 CH 3 + H 2 : Deuterium and Muonium Kinetic Isotope Effects for the Forward and Reverse Reaction Jingzhi Pu and Donald G. Truhlar Department
Revised: July 12, 2002 Tests of Potential Energy Surfaces for H + CH 4 CH 3 + H 2 : Deuterium and Muonium Kinetic Isotope Effects for the Forward and Reverse Reaction Jingzhi Pu and Donald G. Truhlar Department
and Technology of China, Hefei, P.R. China and
 Electronic Supplementary Material (ESI) for Chemical Science. This journal is The Royal Society of Chemistry 2015 Supplementary Information A seven-dimensional quantum dynamics study of the dissociative
Electronic Supplementary Material (ESI) for Chemical Science. This journal is The Royal Society of Chemistry 2015 Supplementary Information A seven-dimensional quantum dynamics study of the dissociative
Theoretical study of the OH-initiated atmospheric oxidation mechanism. of perfluoro methyl vinyl ether, CF 3 OCF=CF 2
 Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 2015 Theoretical study of the OH-initiated atmospheric oxidation mechanism of perfluoro
Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 2015 Theoretical study of the OH-initiated atmospheric oxidation mechanism of perfluoro
Seven dimensional quantum dynamics study of the H 2 +NH 2 \H+NH 3 reaction
 THE OURNAL OF CHEMICAL PHYSICS 17, 184308 007 Seven dimensional quantum dynamics study of the H +NH \H+NH 3 reaction Minghui Yang a State Key Laboratory of Magnetic Resonance and Atomic and Molecular Physics,
THE OURNAL OF CHEMICAL PHYSICS 17, 184308 007 Seven dimensional quantum dynamics study of the H +NH \H+NH 3 reaction Minghui Yang a State Key Laboratory of Magnetic Resonance and Atomic and Molecular Physics,
Chemical Science EDGE ARTICLE
 Chemical Science View Online Dynamic Article Links C < Cite this: Chem. Sci., 2011, 2, 2199 www.rsc.org/chemicalscience Multi-structural variational transition state theory. Kinetics of the 1,4-hydrogen
Chemical Science View Online Dynamic Article Links C < Cite this: Chem. Sci., 2011, 2, 2199 www.rsc.org/chemicalscience Multi-structural variational transition state theory. Kinetics of the 1,4-hydrogen
Supporting Information. I. A refined two-state diabatic potential matrix
 Signatures of a Conical Intersection in Adiabatic Dissociation on the Ground Electronic State Changjian Xie, Christopher L. Malbon, # David R. Yarkony, #,* Daiqian Xie,,%,* and Hua Guo,* Department of
Signatures of a Conical Intersection in Adiabatic Dissociation on the Ground Electronic State Changjian Xie, Christopher L. Malbon, # David R. Yarkony, #,* Daiqian Xie,,%,* and Hua Guo,* Department of
Optimizing the Performance of the Multiconfiguration Molecular Mechanics Method
 Article Subscriber access provided by University of Minnesota Libraries Optimizing the Performance of the Multiconfiguration Molecular Mechanics Method Oksana Tishchenko, and Donald G. Truhlar J. Phys.
Article Subscriber access provided by University of Minnesota Libraries Optimizing the Performance of the Multiconfiguration Molecular Mechanics Method Oksana Tishchenko, and Donald G. Truhlar J. Phys.
prepared for publication in Computer Physics Communications original manuscript October 8, 1997 revised November 4, 1997
 prepared for publication in Computer Physics Communications original manuscript October 8, 1997 revised November 4, 1997 ABCRATE: A Program for the Calculation of Atom-Diatom Reaction Rates Bruce C. GARRETT
prepared for publication in Computer Physics Communications original manuscript October 8, 1997 revised November 4, 1997 ABCRATE: A Program for the Calculation of Atom-Diatom Reaction Rates Bruce C. GARRETT
Multipath Variational Transition State Theory: Rate Constant of the 1,4-Hydrogen Shift Isomerization of the 2-Cyclohexylethyl Radical
 This is an open access article published under an ACS AuthorChoice License, which permits copying and redistribution of the article or any adaptations for non-commercial purposes. pubs.acs.org/jpca Multipath
This is an open access article published under an ACS AuthorChoice License, which permits copying and redistribution of the article or any adaptations for non-commercial purposes. pubs.acs.org/jpca Multipath
Theoretical Gas Phase Chemical Kinetics. Stephen J. Klippenstein
 Theoretical Gas Phase Chemical Kinetics Stephen J. Klippenstein Goal Contribute to Improving the Accuracy of Mechanisms Theoretical Kinetics Predictions for Key Reactions Guided by Modeling Efforts Butanol
Theoretical Gas Phase Chemical Kinetics Stephen J. Klippenstein Goal Contribute to Improving the Accuracy of Mechanisms Theoretical Kinetics Predictions for Key Reactions Guided by Modeling Efforts Butanol
New Reaction Classes in the Kinetic Modeling of Low Temperature Oxidation of n-alkanes
 Supplemental Material for paper New Reaction Classes in the Kinetic Modeling of Low Temperature Oxidation of n-alkanes Eliseo Ranzi, Carlo Cavallotti, Alberto Cuoci, Alessio Frassoldati, Matteo Pelucchi,
Supplemental Material for paper New Reaction Classes in the Kinetic Modeling of Low Temperature Oxidation of n-alkanes Eliseo Ranzi, Carlo Cavallotti, Alberto Cuoci, Alessio Frassoldati, Matteo Pelucchi,
Vibrationally Mediated Bond Selective Dissociative Chemisorption of HOD on Cu(111) Supporting Information
 Supporting Information") Submitted to Chem. Sci. 8/30/202 Vibrationally Mediated Bond Selective Dissociative Chemisorption of HOD on Cu() Supporting Information Bin Jiang,,2 Daiqian Xie,,a) and Hua Guo 2,a) Institute of Theoretical
Submitted to Chem. Sci. 8/30/202 Vibrationally Mediated Bond Selective Dissociative Chemisorption of HOD on Cu() Supporting Information Bin Jiang,,2 Daiqian Xie,,a) and Hua Guo 2,a) Institute of Theoretical
Accurate Computed Rate Coefficients for the Hydrogen Atom Abstraction Reactions from Methanol and n-butanol by the Hydroperoxyl Radical.
 Accurate Computed Rate Coefficients for the Hydrogen Atom Abstraction Reactions from Methanol and n-butanol by the Hydroperoxyl Radical John Alecu Second Annual CEFRC Conference August 17, 2011 Acknowledgments
Accurate Computed Rate Coefficients for the Hydrogen Atom Abstraction Reactions from Methanol and n-butanol by the Hydroperoxyl Radical John Alecu Second Annual CEFRC Conference August 17, 2011 Acknowledgments
Non-Equilibrium Reaction Rates in Hydrogen Combustion
 25 th ICDERS August 2 7, 25 Leeds, UK Non-Equilibrium Reaction Rates in Hydrogen Combustion Stephen J. Voelkel, Venkat Raman 2, Philip Varghese The University of Texas at Austin, Austin, TX 7872, USA 2
25 th ICDERS August 2 7, 25 Leeds, UK Non-Equilibrium Reaction Rates in Hydrogen Combustion Stephen J. Voelkel, Venkat Raman 2, Philip Varghese The University of Texas at Austin, Austin, TX 7872, USA 2
A Theoretical Study of Oxidation of Phenoxy and Benzyl Radicals by HO 2
 A Theoretical Study of xidation of Phenoxy and Benzyl adicals by 2 S. Skokov, A. Kazakov, and F. L. Dryer Department of Mechanical and Aerospace Engineering Princeton University, Princeton, NJ 08544 We
A Theoretical Study of xidation of Phenoxy and Benzyl adicals by 2 S. Skokov, A. Kazakov, and F. L. Dryer Department of Mechanical and Aerospace Engineering Princeton University, Princeton, NJ 08544 We
A Reactant-Coordinate-Based Wave Packet Method for Full-dimensional State-to-State
 Submitted to J. Chem. Phys. //06 A Reactant-Coordinate-Based Wave Packet Method for Full-dimensional State-to-State Quantum Dynamics of Tetra-Atomic Reactions: Application to Both the Abstraction and Exchange
Submitted to J. Chem. Phys. //06 A Reactant-Coordinate-Based Wave Packet Method for Full-dimensional State-to-State Quantum Dynamics of Tetra-Atomic Reactions: Application to Both the Abstraction and Exchange
Inclusion of Machine Learning Kernel Ridge Regression. Potential Energy Surfaces in On-the-Fly Nonadiabatic
 Supporting information for Inclusion of Machine Learning Kernel Ridge Regression Potential Energy Surfaces in On-the-Fly Nonadiabatic Molecular Dynamics Simulation Deping Hu, Yu Xie, Xusong Li, Lingyue
Supporting information for Inclusion of Machine Learning Kernel Ridge Regression Potential Energy Surfaces in On-the-Fly Nonadiabatic Molecular Dynamics Simulation Deping Hu, Yu Xie, Xusong Li, Lingyue
Annual Report Combustion Energy Frontier Research Center Princeton, NJ Sept. 23, Truhlar group University of Minnesota
 Annual Report Combustion Energy Frontier Research Center Princeton, NJ Sept. 23, 2010 Truhlar group University of Minnesota John Steven Ewa Xuefei Tao Jingjing Alecu Mielke Papajak Xu Yu Zheng Practical
Annual Report Combustion Energy Frontier Research Center Princeton, NJ Sept. 23, 2010 Truhlar group University of Minnesota John Steven Ewa Xuefei Tao Jingjing Alecu Mielke Papajak Xu Yu Zheng Practical
Theoretical comparative study on hydrogen storage of BC 3 and carbon nanotubes
 J. At. Mol. Sci. doi: 10.4208/jams.121011.011412a Vol. 3, No. 4, pp. 367-374 November 2012 Theoretical comparative study on hydrogen storage of BC 3 and carbon nanotubes Xiu-Ying Liu a,, Li-Ying Zhang
J. At. Mol. Sci. doi: 10.4208/jams.121011.011412a Vol. 3, No. 4, pp. 367-374 November 2012 Theoretical comparative study on hydrogen storage of BC 3 and carbon nanotubes Xiu-Ying Liu a,, Li-Ying Zhang
Figure 1: Transition State, Saddle Point, Reaction Pathway
 Computational Chemistry Workshops West Ridge Research Building-UAF Campus 9:00am-4:00pm, Room 009 Electronic Structure - July 19-21, 2016 Molecular Dynamics - July 26-28, 2016 Potential Energy Surfaces
Computational Chemistry Workshops West Ridge Research Building-UAF Campus 9:00am-4:00pm, Room 009 Electronic Structure - July 19-21, 2016 Molecular Dynamics - July 26-28, 2016 Potential Energy Surfaces
D. De Fazio, T. V. Tscherbul 2, S. Cavalli 3, and V. Aquilanti 3
 D. De Fazio, T. V. Tscherbul, S. Cavalli 3, and V. Aquilanti 3 1 Istituto di Struttura della Materia C.N.R., 00016 Roma, Italy Department of Chemistry, University of Toronto, M5S 3H6, Canada 3 Dipartimento
D. De Fazio, T. V. Tscherbul, S. Cavalli 3, and V. Aquilanti 3 1 Istituto di Struttura della Materia C.N.R., 00016 Roma, Italy Department of Chemistry, University of Toronto, M5S 3H6, Canada 3 Dipartimento
Prediction of Absolute Rate Constants for the Reactions of NH 2 with Alkanes from ab Initio G2M/TST Calculations
 2088 J. Phys. Chem. A 1999, 103, 2088-2096 Prediction of Absolute Rate Constants for the Reactions of NH 2 with Alkanes from ab Initio G2M/TST Calculations A. M. Mebel, and M. C. Lin*, Institute of Atomic
2088 J. Phys. Chem. A 1999, 103, 2088-2096 Prediction of Absolute Rate Constants for the Reactions of NH 2 with Alkanes from ab Initio G2M/TST Calculations A. M. Mebel, and M. C. Lin*, Institute of Atomic
Direct dynamics for QM? Fitting strategies Coordinates The curse of dimensionality Permutational invariance Direct fitting on n-mode grids
 Lecture II Ab initio Potential Energy Surfaces (and Dipole Moments) in High Dimensionality Direct dynamics for QM? Fitting strategies Coordinates The curse of dimensionality Permutational invariance Direct
Lecture II Ab initio Potential Energy Surfaces (and Dipole Moments) in High Dimensionality Direct dynamics for QM? Fitting strategies Coordinates The curse of dimensionality Permutational invariance Direct
Supporting information for Polymer interactions with Reduced Graphene Oxide: Van der Waals binding energies of Benzene on defected Graphene
 Supporting information for Polymer interactions with Reduced Graphene Oxide: Van der Waals binding energies of Benzene on defected Graphene Mohamed Hassan, Michael Walter *,,, and Michael Moseler, Freiburg
Supporting information for Polymer interactions with Reduced Graphene Oxide: Van der Waals binding energies of Benzene on defected Graphene Mohamed Hassan, Michael Walter *,,, and Michael Moseler, Freiburg
Reduced Dimensionality Quantum Reaction Dynamics of OH + CH 4 H 2 O + CH 3
 Reduced Dimensionality Quantum Reaction Dynamics of OH + CH 4 H 2 O + CH 3 Liesbeth M. C. Janssen January 5, 2008 Supervisors: Dr. S. T. Banks and Prof. dr. D. C. Clary Physical and Theoretical Chemistry
Reduced Dimensionality Quantum Reaction Dynamics of OH + CH 4 H 2 O + CH 3 Liesbeth M. C. Janssen January 5, 2008 Supervisors: Dr. S. T. Banks and Prof. dr. D. C. Clary Physical and Theoretical Chemistry
Isotope effect on the stereodynamics for the collision reaction H+LiF(v = 0, j = 0) HF+Li
 HF+Li") Isotope effect on the stereodynamics for the collision reaction H+LiF(v = 0, j = 0) HF+Li Yue Xian-Fang( 岳现房 ) Department of Physics and Information Engineering, Jining University, Qufu 273155, China (Received
Isotope effect on the stereodynamics for the collision reaction H+LiF(v = 0, j = 0) HF+Li Yue Xian-Fang( 岳现房 ) Department of Physics and Information Engineering, Jining University, Qufu 273155, China (Received
Supporting information for: Mechanisms and Dynamics of Protonation and. Lithiation of Ferrocene
 Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 215 Supporting information for: Mechanisms and Dynamics of Protonation and Lithiation
Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 215 Supporting information for: Mechanisms and Dynamics of Protonation and Lithiation
DFT Study on the Reaction of Molybdenum with Acetaldehyde in Gas Phase
 Asian Journal of Chemistry; Vol. 25, No. 1 (2013), 89-94 http://dx.doi.org/10.14233/ajchem.2013.12753 DFT Study on the Reaction of Molybdenum with Acetaldehyde in Gas Phase YONG WANG 1,2 and GUO-LIANG
Asian Journal of Chemistry; Vol. 25, No. 1 (2013), 89-94 http://dx.doi.org/10.14233/ajchem.2013.12753 DFT Study on the Reaction of Molybdenum with Acetaldehyde in Gas Phase YONG WANG 1,2 and GUO-LIANG
In: European Combustion Meeting (szerk.) Proceedings of the European Combustion Meeting Konferencia helye, ideje: Budapest, Magyarország,
 Proceedings of the European Combustion Meeting Konferencia helye, ideje: Budapest, Magyarország,") In: European Combustion Meeting (szerk.) Proceedings of the European Combustion Meeting Konferencia helye, ideje: Budapest, Magyarország, 2015.03.30-2015.04.02. Budapest: Magyar Égéstudományi Bizottság,
In: European Combustion Meeting (szerk.) Proceedings of the European Combustion Meeting Konferencia helye, ideje: Budapest, Magyarország, 2015.03.30-2015.04.02. Budapest: Magyar Égéstudományi Bizottság,
A DFT study on the mechanism of the gas phase reaction of niobium with acetaldehyde
 Indian Journal of Chemistry Vol. 51A, November 2012, pp. 1553-1560 A DFT study on the mechanism of the gas phase reaction of niobium with acetaldehyde Yong Wang a, b & Gui-hua Chen a, * a School of Pharmaceutical
Indian Journal of Chemistry Vol. 51A, November 2012, pp. 1553-1560 A DFT study on the mechanism of the gas phase reaction of niobium with acetaldehyde Yong Wang a, b & Gui-hua Chen a, * a School of Pharmaceutical
T6.2 Molecular Mechanics
 T6.2 Molecular Mechanics We have seen that Benson group additivities are capable of giving heats of formation of molecules with accuracies comparable to those of the best ab initio procedures. However,
T6.2 Molecular Mechanics We have seen that Benson group additivities are capable of giving heats of formation of molecules with accuracies comparable to those of the best ab initio procedures. However,
Multi-Configuration Molecular Mechanics Based on Combined Quantum Mechanical and Molecular Mechanical Calculations
 July 18, 2006 JCTC Final Author Version Multi-Configuration Molecular Mechanics Based on Combined Quantum Mechanical and Molecular Mechanical Calculations Hai Lin,*,, Yan Zhao, Oksana Tishchenko, and Donald
July 18, 2006 JCTC Final Author Version Multi-Configuration Molecular Mechanics Based on Combined Quantum Mechanical and Molecular Mechanical Calculations Hai Lin,*,, Yan Zhao, Oksana Tishchenko, and Donald
Quantum Rate Coefficients and Kinetic Isotope Effect for the Reaction Cl + CH[subscript 4] HCl + CH[subscript 3] from Ring Polymer Molecular Dynamics
![Quantum Rate Coefficients and Kinetic Isotope Effect for the Reaction Cl + CH[subscript 4] HCl + CH[subscript 3] from Ring Polymer Molecular Dynamics](/thumbs/83/87268093.jpg "Quantum Rate Coefficients and Kinetic Isotope Effect for the Reaction Cl + CH[subscript 4] HCl + CH[subscript 3] from Ring Polymer Molecular Dynamics") Quantum Rate Coefficients and Kinetic Isotope Effect for the Reaction Cl + CH[subscript 4] HCl + CH[subscript 3] from Ring Polymer Molecular Dynamics The MIT Faculty has made this article openly available.
Quantum Rate Coefficients and Kinetic Isotope Effect for the Reaction Cl + CH[subscript 4] HCl + CH[subscript 3] from Ring Polymer Molecular Dynamics The MIT Faculty has made this article openly available.
Computational Chemistry. An Introduction to Molecular Dynamic Simulations
 Computational Chemistry An Introduction to Molecular Dynamic Simulations Computational chemistry simulates chemical structures and reactions numerically, based in full or in part on the fundamental laws
Computational Chemistry An Introduction to Molecular Dynamic Simulations Computational chemistry simulates chemical structures and reactions numerically, based in full or in part on the fundamental laws
Electron detachment process in collisions of negative hydrogen ions with hydrogen molecules
 Journal of Physics: Conference Series PAPER OPEN ACCESS Electron detachment process in collisions of negative hydrogen ions with hydrogen molecules To cite this article: O V Aleksandrovich et al 1 J. Phys.:
Journal of Physics: Conference Series PAPER OPEN ACCESS Electron detachment process in collisions of negative hydrogen ions with hydrogen molecules To cite this article: O V Aleksandrovich et al 1 J. Phys.:
Throwing Light on Reaction Dynamics: H + HBr
 Throwing Light on Reaction Dynamics: H + HBr The thermal reaction of hydrogen gas (H 2 ) and bromine gas (Br 2 ) to form hydrogen bromide vapor (HBr) is a classic reaction: 22+2rHBrHBææÆ Energetics (thermodynamics)
Throwing Light on Reaction Dynamics: H + HBr The thermal reaction of hydrogen gas (H 2 ) and bromine gas (Br 2 ) to form hydrogen bromide vapor (HBr) is a classic reaction: 22+2rHBrHBææÆ Energetics (thermodynamics)
MO Calculation for a Diatomic Molecule. /4 0 ) i=1 j>i (1/r ij )
 i=1 j>i (1/r ij )") MO Calculation for a Diatomic Molecule Introduction The properties of any molecular system can in principle be found by looking at the solutions to the corresponding time independent Schrodinger equation
MO Calculation for a Diatomic Molecule Introduction The properties of any molecular system can in principle be found by looking at the solutions to the corresponding time independent Schrodinger equation
Topic 4 Thermodynamics
 Topic 4 Thermodynamics Thermodynamics We need thermodynamic data to: Determine the heat release in a combustion process (need enthalpies and heat capacities) Calculate the equilibrium constant for a reaction
Topic 4 Thermodynamics Thermodynamics We need thermodynamic data to: Determine the heat release in a combustion process (need enthalpies and heat capacities) Calculate the equilibrium constant for a reaction
Computational Methods. Chem 561
 Computational Methods Chem 561 Lecture Outline 1. Ab initio methods a) HF SCF b) Post-HF methods 2. Density Functional Theory 3. Semiempirical methods 4. Molecular Mechanics Computational Chemistry " Computational
Computational Methods Chem 561 Lecture Outline 1. Ab initio methods a) HF SCF b) Post-HF methods 2. Density Functional Theory 3. Semiempirical methods 4. Molecular Mechanics Computational Chemistry " Computational
Molecular body vs. molecular skeleton Ideal surface 1, 29
 Subject Index Acid catalysis 244, 245, 247, 249, 252 Bonds-pairs 177, 178, 184, 185, 190, 191, Activation 207, 208 202-204 Activation barrier 160, 233, 242, 249 Bottleneck 291, 299, 307, 323-325, 336,
Subject Index Acid catalysis 244, 245, 247, 249, 252 Bonds-pairs 177, 178, 184, 185, 190, 191, Activation 207, 208 202-204 Activation barrier 160, 233, 242, 249 Bottleneck 291, 299, 307, 323-325, 336,
Machine learning the Born-Oppenheimer potential energy surface: from molecules to materials. Gábor Csányi Engineering Laboratory
 Machine learning the Born-Oppenheimer potential energy surface: from molecules to materials Gábor Csányi Engineering Laboratory Interatomic potentials for molecular dynamics Transferability biomolecular
Machine learning the Born-Oppenheimer potential energy surface: from molecules to materials Gábor Csányi Engineering Laboratory Interatomic potentials for molecular dynamics Transferability biomolecular
Semi-Empirical MO Methods
 Semi-Empirical MO Methods the high cost of ab initio MO calculations is largely due to the many integrals that need to be calculated (esp. two electron integrals) semi-empirical MO methods start with the
Semi-Empirical MO Methods the high cost of ab initio MO calculations is largely due to the many integrals that need to be calculated (esp. two electron integrals) semi-empirical MO methods start with the
Titus V. Albu PROFESSIONAL EXPERIENCE
 Oct 23, 2018 Titus V. Albu Associate Professor, Chemistry University of Tennessee at Chattanooga Phone: (423) 425-4143 615 Mc Callie Avenue, Box 2252 Fax: (423) 425-5234 Chattanooga, TN 37403 E-mail: titus-albu@utc.edu
Oct 23, 2018 Titus V. Albu Associate Professor, Chemistry University of Tennessee at Chattanooga Phone: (423) 425-4143 615 Mc Callie Avenue, Box 2252 Fax: (423) 425-5234 Chattanooga, TN 37403 E-mail: titus-albu@utc.edu
Physical Chemistry Chemical Physics
 Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 2018 Physical Chemistry Chemical Physics Supporting Information Thermochemistry of the
Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 2018 Physical Chemistry Chemical Physics Supporting Information Thermochemistry of the
Chemical Reaction Rate Coefficients from Ring Polymer Molecular Dynamics: Theory and Practical Applications
 Chemical Reaction Rate Coefficients from Ring Polymer Molecular Dynamics: Theory and Practical Applications Yury V. Suleimanov* Computation-based Science and Technology Research Center, Cyprus Institute,
Chemical Reaction Rate Coefficients from Ring Polymer Molecular Dynamics: Theory and Practical Applications Yury V. Suleimanov* Computation-based Science and Technology Research Center, Cyprus Institute,
A Two Transition State Model for Radical-Molecule Reactions: Applications to Isomeric Branching in the OH-Isoprene Reaction
 5582 J. Phys. Chem. A 2007, 111, 5582-5592 A Two Transition State Model for Radical-Molecule Reactions: Applications to Isomeric Branching in the OH-Isoprene Reaction Erin E. Greenwald, Simon W. North,*,
5582 J. Phys. Chem. A 2007, 111, 5582-5592 A Two Transition State Model for Radical-Molecule Reactions: Applications to Isomeric Branching in the OH-Isoprene Reaction Erin E. Greenwald, Simon W. North,*,
Ab initio calculations of F-H-Br system with linear geometry
 Current Chemistry Letters 5 (016) 1 6 Contents lists available atgrowingscience Current Chemistry Letters homepage: www.growingscience.com/ccl Ab initio calculations of F-H-Br system with linear geometry
Current Chemistry Letters 5 (016) 1 6 Contents lists available atgrowingscience Current Chemistry Letters homepage: www.growingscience.com/ccl Ab initio calculations of F-H-Br system with linear geometry
Theoretical investigation of mechanism for the gas-phase reaction of OH radical and ethane
 J. At. Mol. Sci. doi: 10.4208/jams.122810.011811a Vol. 2, No. 3, pp. 225-233 August 2011 Theoretical investigation of mechanism for the gas-phase reaction of OH radical and ethane Xiao-Ping Hu a, Bing-Xing
J. At. Mol. Sci. doi: 10.4208/jams.122810.011811a Vol. 2, No. 3, pp. 225-233 August 2011 Theoretical investigation of mechanism for the gas-phase reaction of OH radical and ethane Xiao-Ping Hu a, Bing-Xing
A Two Transition State Model for Radical-Molecule Reactions: A Case Study of the Addition of OH to C 2 H 4
 J. Phys. Chem. A 2005, 109, 6031-6044 6031 A Two Transition State Model for Radical-Molecule Reactions: A Case Study of the Addition of OH to C 2 H 4 Erin E. Greenwald and Simon W. North Department of
J. Phys. Chem. A 2005, 109, 6031-6044 6031 A Two Transition State Model for Radical-Molecule Reactions: A Case Study of the Addition of OH to C 2 H 4 Erin E. Greenwald and Simon W. North Department of
This paper is part of the following report: UNCLASSIFIED
 UNCLASSIFIED Defense Technical Information Center Compilation Part Notice ADP023624 TITLE: Ignition Kinetics in Fuels Oxidation DISTRIBUTION: Approved for public release, distribution unlimited This paper
UNCLASSIFIED Defense Technical Information Center Compilation Part Notice ADP023624 TITLE: Ignition Kinetics in Fuels Oxidation DISTRIBUTION: Approved for public release, distribution unlimited This paper
Homework Problem Set 5 Solutions. E e + H corr (a.u.) a.) Determine the bond dissociation enthalpy of ethane in kcal/mol (1 a.u. = kcal/mol).
 a.) Determine the bond dissociation enthalpy of ethane in kcal/mol (1 a.u. = kcal/mol).") Chemistry 380.37 Dr. Jean M. Standard Homework Problem Set 5 Solutions 1. Given below are the sum of electronic and thermal enthalpies, E e + H corr, from Hartree-Fock calculations using a 6-31G(d) basis
Chemistry 380.37 Dr. Jean M. Standard Homework Problem Set 5 Solutions 1. Given below are the sum of electronic and thermal enthalpies, E e + H corr, from Hartree-Fock calculations using a 6-31G(d) basis
Uptake of OH radical to aqueous aerosol: a computational study
 Uptake of OH radical to aqueous aerosol: a computational study Grigory Andreev Karpov Institute of Physical Chemistry 10 Vorontsovo pole, Moscow, 105064, Russia Institute of Physical Chemistry and Electrochemistry
Uptake of OH radical to aqueous aerosol: a computational study Grigory Andreev Karpov Institute of Physical Chemistry 10 Vorontsovo pole, Moscow, 105064, Russia Institute of Physical Chemistry and Electrochemistry
Reaction class transition state theory: Hydrogen abstraction reactions by hydrogen atoms as test cases
 JOURNAL OF CHEMICAL PHYSICS VOLUME 113, NUMBER 12 22 SEPTEMBER 2000 Reaction class transition state theory: Hydrogen abstraction reactions by hydrogen atoms as test cases Thanh N. Truong Henry Eyring Center
JOURNAL OF CHEMICAL PHYSICS VOLUME 113, NUMBER 12 22 SEPTEMBER 2000 Reaction class transition state theory: Hydrogen abstraction reactions by hydrogen atoms as test cases Thanh N. Truong Henry Eyring Center
Quasi-classical trajectory study of the stereodynamics of a Ne+H + 2 NeH+ +H reaction
 Quasi-classical trajectory study of the stereodynamics of a Ne+H + 2 NeH+ +H reaction Ge Mei-Hua( ) and Zheng Yu-Jun( ) School of Physics, Shandong University, Jinan 250100, China (Received 19 February
Quasi-classical trajectory study of the stereodynamics of a Ne+H + 2 NeH+ +H reaction Ge Mei-Hua( ) and Zheng Yu-Jun( ) School of Physics, Shandong University, Jinan 250100, China (Received 19 February
Supporting Information for Atmospheric Hydroxyl Radical Source: Reaction of Triplet SO 2 and Water
 Supporting Information for Atmospheric Hydroxyl Radical Source: Reaction of Triplet SO 2 and Water Authors: Jay A. Kroll 1,2,#, Benjamin N. Frandsen 3,#, Henrik G. Kjaergaard 3,*, and Veronica Vaida 1,2,*
Supporting Information for Atmospheric Hydroxyl Radical Source: Reaction of Triplet SO 2 and Water Authors: Jay A. Kroll 1,2,#, Benjamin N. Frandsen 3,#, Henrik G. Kjaergaard 3,*, and Veronica Vaida 1,2,*
Rate Constant for the NH 3 NO 2. HONO Reaction: Comparison of Kinetically Modeled and Predicted Results
 Rate Constant for the NH 3 HONO Reaction: Comparison of Kinetically Modeled and Predicted Results A. GRANT THAXTON, C.-C. HSU, M. C. LIN Department of Chemistry, Emory University, Atlanta, Georgia 30322
Rate Constant for the NH 3 HONO Reaction: Comparison of Kinetically Modeled and Predicted Results A. GRANT THAXTON, C.-C. HSU, M. C. LIN Department of Chemistry, Emory University, Atlanta, Georgia 30322
ARTICLES. Normal-mode analysis without the Hessian: A driven molecular-dynamics approach
 JOURNAL OF CHEMICAL PHYSICS VOLUME 119, NUMBER 2 8 JULY 2003 ARTICLES Normal-mode analysis without the Hessian: A driven molecular-dynamics approach Joel M. Bowman, a) Xiubin Zhang, and Alex Brown Cherry
JOURNAL OF CHEMICAL PHYSICS VOLUME 119, NUMBER 2 8 JULY 2003 ARTICLES Normal-mode analysis without the Hessian: A driven molecular-dynamics approach Joel M. Bowman, a) Xiubin Zhang, and Alex Brown Cherry
JOURNAL OF CHEMICAL PHYSICS VOLUME 120, NUMBER 7 15 FEBRUARY 2004
 JOURAL OF CHEMICAL HYSICS VOLUME 20, UMBER 7 5 FEBRUARY 2004 ath integral calculation of thermal rate constants within the quantum instanton approximation: Application to the H CH 4 \H 2 CH 3 hydrogen
JOURAL OF CHEMICAL HYSICS VOLUME 20, UMBER 7 5 FEBRUARY 2004 ath integral calculation of thermal rate constants within the quantum instanton approximation: Application to the H CH 4 \H 2 CH 3 hydrogen
Density Functional Theory Study on Mechanism of Forming Spiro-Geheterocyclic Ring Compound from Me 2 Ge=Ge: and Acetaldehyde
 CHINESE JURNAL F CHEMICAL PHYSICS VLUME 26, NUMBER 1 FEBRUARY 27, 2013 ARTICLE Density Functional Theory Study on Mechanism of Forming Spiro-Geheterocyclic Ring Compound from Me 2 Ge=Ge: and Acetaldehyde
CHINESE JURNAL F CHEMICAL PHYSICS VLUME 26, NUMBER 1 FEBRUARY 27, 2013 ARTICLE Density Functional Theory Study on Mechanism of Forming Spiro-Geheterocyclic Ring Compound from Me 2 Ge=Ge: and Acetaldehyde
QUANTUM MECHANICS. Franz Schwabl. Translated by Ronald Kates. ff Springer
 Franz Schwabl QUANTUM MECHANICS Translated by Ronald Kates Second Revised Edition With 122Figures, 16Tables, Numerous Worked Examples, and 126 Problems ff Springer Contents 1. Historical and Experimental
Franz Schwabl QUANTUM MECHANICS Translated by Ronald Kates Second Revised Edition With 122Figures, 16Tables, Numerous Worked Examples, and 126 Problems ff Springer Contents 1. Historical and Experimental
Effect of isotope substitution on the stereodynamics for O+H(D)Br OH(D)+Br reactions. 1 Introduction
Br OH(D)+Br reactions. 1 Introduction") J. At. Mol. Sci. doi: 10.4208/jams.052411.070811a Vol. 3, No. 2, pp. 114-121 May 2012 Effect of isotope substitution on the stereodynamics for O+H(D)Br OH(D)+Br reactions Hong Li, Bin Zheng, Ji-Qing Yin,
J. At. Mol. Sci. doi: 10.4208/jams.052411.070811a Vol. 3, No. 2, pp. 114-121 May 2012 Effect of isotope substitution on the stereodynamics for O+H(D)Br OH(D)+Br reactions Hong Li, Bin Zheng, Ji-Qing Yin,
Title. Author(s)Tachikawa, Hiroto. CitationThe Journal of Chemical Physics, 125(133119) Issue Date Doc URL. Rights. Type.
Tachikawa, Hiroto. CitationThe Journal of Chemical Physics, 125(133119) Issue Date Doc URL. Rights. Type.") Title Direct ab initio molecular dynamics study on a micro Author(s)Tachikawa, Hiroto CitationThe Journal of Chemical Physics, 125(133119) Issue Date 2006-10-07 Doc URL http://hdl.handle.net/2115/14882
Title Direct ab initio molecular dynamics study on a micro Author(s)Tachikawa, Hiroto CitationThe Journal of Chemical Physics, 125(133119) Issue Date 2006-10-07 Doc URL http://hdl.handle.net/2115/14882
Chemistry 334 Part 2: Computational Quantum Chemistry
 Chemistry 334 Part 2: Computational Quantum Chemistry 1. Definition Louis Scudiero, Ben Shepler and Kirk Peterson Washington State University January 2006 Computational chemistry is an area of theoretical
Chemistry 334 Part 2: Computational Quantum Chemistry 1. Definition Louis Scudiero, Ben Shepler and Kirk Peterson Washington State University January 2006 Computational chemistry is an area of theoretical
Quantum chemistry and vibrational spectra
 Chapter 3 Quantum chemistry and vibrational spectra This chapter presents the quantum chemical results for the systems studied in this work, FHF (Section 3.) and OHF (Section 3.3). These triatomic anions
Chapter 3 Quantum chemistry and vibrational spectra This chapter presents the quantum chemical results for the systems studied in this work, FHF (Section 3.) and OHF (Section 3.3). These triatomic anions
Ab initio interpolated potential energy surface and classical reaction dynamics for HCO + +H, HOC + +H, and deuterated analogues
 THE JOURNAL OF CHEMICAL PHYSICS 124, 124318 2006 Ab initio interpolated potential energy surface and classical reaction dynamics for HCO + +H, HOC + +H, and deuterated analogues Gloria E. Moyano, Seth
THE JOURNAL OF CHEMICAL PHYSICS 124, 124318 2006 Ab initio interpolated potential energy surface and classical reaction dynamics for HCO + +H, HOC + +H, and deuterated analogues Gloria E. Moyano, Seth
Introduction to Theories of Chemical Reactions. Graduate Course Seminar Beate Flemmig FHI
 Introduction to Theories of Chemical Reactions Graduate Course Seminar Beate Flemmig FHI I. Overview What kind of reactions? gas phase / surface unimolecular / bimolecular thermal / photochemical What
Introduction to Theories of Chemical Reactions Graduate Course Seminar Beate Flemmig FHI I. Overview What kind of reactions? gas phase / surface unimolecular / bimolecular thermal / photochemical What
Effects of Vibrational Excitation on the F + H2O HF + OH Reaction: Dissociative Photodetachment of Overtone Excited [F H OH]ˉ
![Effects of Vibrational Excitation on the F + H2O HF + OH Reaction: Dissociative Photodetachment of Overtone Excited [F H OH]ˉ](/thumbs/82/85526767.jpg "Effects of Vibrational Excitation on the F + H2O HF + OH Reaction: Dissociative Photodetachment of Overtone Excited [F H OH]ˉ") Electronic Supplementary Material (ESI) for Chemical Science. This journal is The Royal Society of Chemistry 2017 Effects of Vibrational Excitation on the F + H2O HF + OH Reaction: Dissociative Photodetachment
Electronic Supplementary Material (ESI) for Chemical Science. This journal is The Royal Society of Chemistry 2017 Effects of Vibrational Excitation on the F + H2O HF + OH Reaction: Dissociative Photodetachment
Supporting Information
 Supporting Information Conflict in the Mechanism and Kinetics of the Barrierless Reaction between SH and NO 2 Radicals Ramanpreet Kaur and Vikas * Quantum Chemistry Group, Department of Chemistry & Centre
Supporting Information Conflict in the Mechanism and Kinetics of the Barrierless Reaction between SH and NO 2 Radicals Ramanpreet Kaur and Vikas * Quantum Chemistry Group, Department of Chemistry & Centre
Transition states and reaction paths
 Transition states and reaction paths Lab 4 Theoretical background Transition state A transition structure is the molecular configuration that separates reactants and products. In a system with a single
Transition states and reaction paths Lab 4 Theoretical background Transition state A transition structure is the molecular configuration that separates reactants and products. In a system with a single
Spin-orbit effect in the energy pooling reaction
 THE JOURNAL OF CHEMICAL PHYSICS 126, 124304 2007 Spin-orbit effect in the energy pooling reaction O 2 a 1 +O 2 a 1 \O 2 b 1 +O 2 X 3 Rui-Feng Lu and Pei-Yu Zhang Academy of Sciences, Dalian 116023, China
THE JOURNAL OF CHEMICAL PHYSICS 126, 124304 2007 Spin-orbit effect in the energy pooling reaction O 2 a 1 +O 2 a 1 \O 2 b 1 +O 2 X 3 Rui-Feng Lu and Pei-Yu Zhang Academy of Sciences, Dalian 116023, China
CH Stretching Excitation Promotes its Cleavage in. Collision Energies
 Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 2017 Electronic supplementary information for CH Stretching Excitation Promotes its
Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 2017 Electronic supplementary information for CH Stretching Excitation Promotes its
Stress Test for Quantum Dynamics Approximations: Deep Tunneling in the Muonium Exchange Reaction D + HMu DMu + H
 Stress Test for Quantum Dynamics Approximations: Deep Tunneling in the Muonium Exchange Reaction D + HMu DMu + H The MIT Faculty has made this article openly available. Please share how this access benefits
Stress Test for Quantum Dynamics Approximations: Deep Tunneling in the Muonium Exchange Reaction D + HMu DMu + H The MIT Faculty has made this article openly available. Please share how this access benefits
VIRT&L-COMM
 CHEMICAL REACTIONS LEARNING OBJECTS A. Moriconi, S. Pasqua, G. Squarta, LABEL, University of Perugia, IT A. Laganà, Department of Chemistry, Biology and Biotechnologies, University of Perugia, IT Introduction
CHEMICAL REACTIONS LEARNING OBJECTS A. Moriconi, S. Pasqua, G. Squarta, LABEL, University of Perugia, IT A. Laganà, Department of Chemistry, Biology and Biotechnologies, University of Perugia, IT Introduction
Chem 442 Review for Exam 2. Exact separation of the Hamiltonian of a hydrogenic atom into center-of-mass (3D) and relative (3D) components.
 and relative (3D) components.") Chem 44 Review for Exam Hydrogenic atoms: The Coulomb energy between two point charges Ze and e: V r Ze r Exact separation of the Hamiltonian of a hydrogenic atom into center-of-mass (3D) and relative
Chem 44 Review for Exam Hydrogenic atoms: The Coulomb energy between two point charges Ze and e: V r Ze r Exact separation of the Hamiltonian of a hydrogenic atom into center-of-mass (3D) and relative
Theoretical Study of the Pyrolysis of Methyltrichlorosilane in the Gas Phase. 3. Reaction Rate Constant Calculations
 Chemical and Biological Engineering Publications Chemical and Biological Engineering 2010 Theoretical Study of the Pyrolysis of Methyltrichlorosilane in the Gas Phase. 3. Reaction Rate Constant Calculations
Chemical and Biological Engineering Publications Chemical and Biological Engineering 2010 Theoretical Study of the Pyrolysis of Methyltrichlorosilane in the Gas Phase. 3. Reaction Rate Constant Calculations
Mechanisms of H- and OH-assisted CO activation as well as C-C coupling on the flat Co(0001) surface Revisited
 surface Revisited") Electronic Supplementary Material (ESI) for Catalysis Science & Technology. This journal is The Royal Society of Chemistry 2016 Mechanisms of H- and OH-assisted CO activation as well as C-C coupling on
Electronic Supplementary Material (ESI) for Catalysis Science & Technology. This journal is The Royal Society of Chemistry 2016 Mechanisms of H- and OH-assisted CO activation as well as C-C coupling on
Brazilian Journal of Physics, vol. 36, no. 3A, September,
 Brazilian Journal of Physics, vol. 36, no. 3A, September, 2006 725 Effects of Molecular Rovibrational States and Surface Topologies for Molecule-Surface Interaction: Chemisorption Dynamics of D 2 Collision
Brazilian Journal of Physics, vol. 36, no. 3A, September, 2006 725 Effects of Molecular Rovibrational States and Surface Topologies for Molecule-Surface Interaction: Chemisorption Dynamics of D 2 Collision
Semi-Classical Theory for Non-separable Systems :
 Semi-Classical Theory for Non-separable Systems : Construction of Good Action-Angle Variables for Reaction Rate Constants* BY WILLIAM H. MILLER-^ Department of Chemistry, and Materials and Molecular Research
Semi-Classical Theory for Non-separable Systems : Construction of Good Action-Angle Variables for Reaction Rate Constants* BY WILLIAM H. MILLER-^ Department of Chemistry, and Materials and Molecular Research
Free Energy Simulation Methods
 Free Energy Simulation Methods Free energy simulation methods Many methods have been developed to compute (relative) free energies on the basis of statistical mechanics Free energy perturbation Thermodynamic
Free Energy Simulation Methods Free energy simulation methods Many methods have been developed to compute (relative) free energies on the basis of statistical mechanics Free energy perturbation Thermodynamic
Chemistry 881 Lecture Topics Fall 2001
 Chemistry 881 Lecture Topics Fall 2001 Texts PHYSICAL CHEMISTRY A Molecular Approach McQuarrie and Simon MATHEMATICS for PHYSICAL CHEMISTRY, Mortimer i. Mathematics Review (M, Chapters 1,2,3 & 4; M&S,
Chemistry 881 Lecture Topics Fall 2001 Texts PHYSICAL CHEMISTRY A Molecular Approach McQuarrie and Simon MATHEMATICS for PHYSICAL CHEMISTRY, Mortimer i. Mathematics Review (M, Chapters 1,2,3 & 4; M&S,
Chemical reactions occur when one reactant collides with another
 HF(v 3) forward scattering in the F H 2 reaction: Shape resonance and slow-down mechanism Xingan Wang*, Wenrui Dong*, Minghui Qiu, Zefeng Ren*, Li Che*, Dongxu Dai*, Xiuyan Wang*, Xueming Yang*, Zhigang
HF(v 3) forward scattering in the F H 2 reaction: Shape resonance and slow-down mechanism Xingan Wang*, Wenrui Dong*, Minghui Qiu, Zefeng Ren*, Li Che*, Dongxu Dai*, Xiuyan Wang*, Xueming Yang*, Zhigang