Text Algorithms (4AP) Biological measures of similarity. Jaak Vilo 2008 fall. MTAT Text Algorithms. Complete genomes
|
|
|
- Leon Sherman
- 5 years ago
- Views:
Transcription

1 Text Algorithms (4AP) Biological measures of similarity Jaak Vilo 2008 fall Jaak Vilo MTAT Text Algorithms 1 Complete genomes Lecture 1, Tuesday April 1, 2003 Serafim Batzoglou 1
2 Evolution Lecture 1, Tuesday April 1, 2003 Serafim Batzoglou W. Gilbert NATURE 349: For 15 years, the DNA databases have grown by 60 per cent a year, a factor of ten every five years. The human genome project will continue and accelerate this rate of increase. Thus I expect that sequence data for all of the model organisms and half of the total knowledge of the human organism will be available in five to seven years, and all of it by the end of the decade.... To use this flood of knowledge, which will pour across the computer networks of the world, biologists not only must become computer literate, but also change their approach to the problem of understanding life. 2


 Affine gap")
3 Technology Review, Oct 6, Dot plot Methods Needleman Wunsch (global alignment) Affine gap penalties Smith Waterman (lovcal alignment) Affine gap penalties BLAST BLAT, 3
 Brian Golding,")
 Dot plot: black if")
4 Dot plots Lecture notes on Dot plot (Benjamin Corum) Brian Golding, McMaster Univ. (course outline) Dot plot: black if ai=bi, white otherwise 4
5 5 matches 8 matches 5


6 8 positions, 1 mismatch Priit Adler: 10,2 Priit Adler: 6
7 Evolution at the DNA level C ACGGTGCAGTCACCA ACGTTGCAGTCCACCA SEQUENCE EDITS Lecture 1, Tuesday April 1, 2003 REARRANGEMENTS Evolutionary Rates next generation OK OK OK X X Lecture 1, Tuesday April 1, 2003 Still OK? 7
8 Sequence conservation implies function Interleukin region in human and mouse Lecture 1, Tuesday April 1, 2003 Sequence Alignment AGGCTATCACCTGACCTCCAGGCCGATGCCC TAGCTATCACGACCGCGGTCGATTTGCCCGAC -AGGCTATCACCTGACCTCCAGGCCGA--TGCCC--- TAG-CTATCAC--GACCGC--GGTCGATTTGCCCGAC Definition Given two strings x = x 1 x 2...x M, y = y 1 y 2 y N, an alignment is an assignment of gaps to positions 0,, N in x, and 0,, N in y, so as to line up each sequence with either a letter, or a gap in the other sequence letter in one Lecture 1, Tuesday April 1,
9 What is a good alignment? Alignment: The best way to match the letters of one sequence with those of the other How do we define best? Alignment: A hypothesis that the two sequences come from a common ancestor through sequence edits Parsimonious explanation: Find the minimum number of edits that transform one sequence into the other Lecture 1, Tuesday April 1, 2003 Serafim Batzoglou Scoring Function Sequence edits: Mutations ti Insertions Deletions Scoring Function: Match: +m Mismatch: -s Gap: -d AGGCCTC AGGACTC AGGGCCTC AGG.CTC Score F = (# matches) m - (# mismatches) s (#gaps) d Lecture 1, Tuesday April 1,
10 How do we compute the best alignment? AGTGCCCTGGAACCCTGACGGTGGGTCACAAAACTTCTGGA AGTGACCTGGGAAGACCCTGACCCTGGG GTCACAAAACTC Too many possible alignments: O( 2 M+N ) Lecture 1, Tuesday April 1, 2003 Three Main Steps 1. Assign similarity scores A numerical value (score) is assigned to every cell in the array depending on similarity/dissimilarity il it i il it Similarity scores may be simple, or related to chemical similarities or frequency of observed substitutions 2. Score paths through array For each cell want to know the maximum possible score for an alignment ending at that point Cumulative score by adding in a path through the matrix Searches subrow and subcolumn for the highest score Gap penalty independent d of the length of the gap The best match is the pathway with the highest score Maximum match = largest number of amino acids of one protein that can be matched with those of another protein while allowing for all possible deletions 3. Construct an alignment 10
11 1. Values are assigned to each matching pair of residues based upon a similarity matrix. Here the matrix is the simplest: match = 1 mismatch = 0 More complex similarity matrices, such as PAM or BLOSUM, may be used. Assign Relative Values 2. Rlti Relative values are then assigned dto each cell of the array, based upon the maximum scoring pathway through the array. * Search subrow and subcolumn to assign the score for each cell. Each cell is assigned the highest possible score based upon it's own score + the highest score in the diagonal preceeding it. * Gaps are allowed and gap penalties may be assigned. In this example there are no gap penalties. 11

12 Pathfinding 3 Alignments producing a maximum match are constructed 3. Alignments producing a maximum match are constructed. * The maximum match score will always be in the outermost row or column. * A maximum alignment can be constructed by following the maximum scores in each row backwards from the outermost maximum score. * The two equal maximal alignments resulting from this example are displayed below the array. * These alignments are global every nucleotide or amino acid must be included in the alignment. This algorithm therefore misses many motif or domain similarities. 12
: G(i, j): score of alignment x 1 x i to y 1 y j if x i aligns to y j score if x i, or y j, aligns to a gap Lecture 2, Thursday April 3, 2003")
13 affine gaps : 1 longer is more likely than (n) = d + (n 1) e gap gap open extend To compute optimal alignment, many small gaps (n) At position i,j, need to remember best score if gap is open best score if gap is not open d e F(i, j): G(i, j): score of alignment x 1 x i to y 1 y j if x i aligns to y j score if x i, or y j, aligns to a gap Lecture 2, Thursday April 3,
14 Scoring the gaps more accurately Current model: (n) Gap of length incurs penalty n n d However, gaps usually occur in bunches Convex gap penalty function: (n) ( ) (n): for all n, (n + 1) (n) (n) (n 1) Lecture 2, Thursday April 3, 2003 Needleman Wunsch with affine gaps Initialization: F(i, 0) = d + (i 1) e F(0, j) = d + (j 1) e Iteration: F(i, j) = max F(i 1, j 1) + s(x i, y j ) G(i 1, j 1) + s(x i, y j ) Termination: G(i, j) = max same F(i 1, j) d F(i, (,j 1) d G(i, j 1) e G(i 1, j) e Lecture 2, Thursday April 3,
15 Needleman, S. B., Wunsch, C. D., J. Mol. Biol. (1970) 48: (from Aoife McLysaght and NCBI workshop slides) General algorithm for sequence comparison Fundamental principle to calculate the alignment score S(i,j) you only need to enumerate and score all ways in which one aligned pair can be added to a shorter alignment to produce an alignment of the first i residues of seq1 and the first j residues of seq2 (dynamic programming) Allpossible pairs are represented by a two dimensional array, and all possible comparisons are represented by pathways through this array Global alignments... i.e. every residue of the two sequences has to participate therefore will not detect motif or active site homology alone Global vs local alignment Global alignment methods give the optimal end to end alignment between two sequences. However, homologous sequences may share similarity in a sub region of the sequence. In trying to choose the optimal end to end alignment, global alignments can fail to pick up local homology. Local alignment methods will return alignments with high scoring regional homology. 15
16 Global vs local alignment Needleman and Wunsch ("Global" Alignment) Usesa global alignmentalgorithm algorithm, meaningthat: a. the full sequence must be aligned. b. gaps at the end of the sequence are penalized as much as internal gaps. Smith Waterman ("Local" Alignment) Uses a local alignment algorithm, meaning that: a. it searches for both full and partial sequence matches. b. gaps at the end of the sequence essentially have a penalty of zero. The local alignment problem Given two strings x = x 1 x M, y = y 1 y N Find substrings x, y whose similarity (optimal global alignment value) is maximum e.g. x = aaaacccccgggg y = cccgggaaccaacc Lecture 2, Thursday April 3,
 = F(i, 0) = 0 0 Iteration: F(i, j) = max F(i 1, j) d F(i, j 1) d F(i 1, j 1) + s(x i, y j ) Lecture 2, Thursday April")
17 Why local alignment Genes are shuffled between genomes Portions of proteins (domains) are often conserved Lecture 2, Thursday April 3, 2003 The Smith Waterman algorithm Idea: Ignore badly aligning regions Modifications to Needleman Wunsch: Initialization: F(0, j) = F(i, 0) = 0 0 Iteration: F(i, j) = max F(i 1, j) d F(i, j 1) d F(i 1, j 1) + s(x i, y j ) Lecture 2, Thursday April 3,
18 The Smith Waterman algorithm Termination: 1. If we want the best local alignment F OPT = max i,j F(i, j) 2. If we want all local alignments scoring > t Lecture 2, Thursday April 3, 2003 For all i, j find F(i, j) > t, and trace back General gap dynamic programming Initialization: same Iteration: F(i 1, j 1) + s(x i, y j ) F(i, j) = max max k=0 i 1 F(k,j) (i k) max k=0 j 1 F(i,k) (j k) Termination: same Running Time: O(N 2 M) Space: O(NM) (assume N>M) Lecture 2, Thursday April 3,
19 Smith, T. F., Waterman, M. S., J. Mol. Biol. (1981) 147: Based on N W Algorithm Instead of looking at each sequence in its entirety, this compares segments of all possible lengths and chooses whichever optimise the similarity measure (local alignments) Comparing two sequences A (=a 1 a 2 a 3... a n ) and B (=b 1 b 2 b 3... b m ) H ij =max{h i 1, j 1 +s(a i,b j ), max{h i k,j W k }, max{h i, j l W l }, 0} H ij is the maximum similarity of two segments ending in a i and b j respectively. Including the alternative that the similarity score be zero in the expression for H ij allows the local alignment to restart at any pair of aligned residues. In addition, it makes the calculation much more sensitive to the precise match and mismatch scores and gap penalties. Smith Waterman Uses a local alignment algorithm, meaning that: a. it searches for both full and partial sequence matches. b. gaps at the end of the sequence essentially have a penalty of zero. Assigns a score to each pair of bases Uses similarity scores only Uses positive scores for related residues Uses negative scores for substitutions and gaps Initializes edges of the matrix with zeros As the scores are summed in the matrix, ti any score bl below 0 is recorded ddas 0. Begins the trace back at the maximum value found anywhere in the matrix Continues until the score falls to 0. 19
20 Scoring metrics are of two types: Distance (cost) Scores The distance between identical residues must be 0. The distance, or cost, for aligning two non identical residues must be greater than 0 (i.e. negative distances are not allowed). Similarity Scores Identical or similar residues have positive scores Dissimilar residues can have 0 or even negative scores 20
 (* where k = number of")
21 Smith Waterman example Penalties Assigning values in the S W 2 dimensional array requires the use of gap penalties. In this example scores are derived from a simple similarity matrix plus a gap initiation and gap extension penalty as follows: Match = + 1 MisMatch = 1/3 Gap = (1 + 1/3k*) (* where k = number of residues included in gap) All cell values start at zero and are not allowed to fall below zero (so a new ( alignment path can begin at any point). Assigns value to cell based upon value of cell plus the highest value in subrow, subcolumn or direct diagonal (as shown) when gap penalties are taken into account. 21
22 Relative values in adjacent cells of a path can rise, stay the same or fll fall. Alignments can begin and end anywhere in the sequence, not just the final row or column. The value in any cell is the highest score for an alignment of any length ending at that cell. * The alignment for the path shown in red in the 2-dimensional array to the right is displayed below. 22


23 * The alignment for the path shown in red in the 2-dimensional array to the right is displayed below. The alignment for the path shown in red in the 2-dimensional i array to the right is displayed below. 23
24 Alignment Scores: The higher the score... the better the alignment. Alignment Scores and Search Algorithms (Baylor College of Medicine) A Substitution Score is chosen for each aligned pair of letters. The set of these scores is called the substitution matrix. The matrix scores highly identical matches of bases, and also gives 'better' scores to alignments of non identical bases that are similiar in some way, and a 'worse' score to pairs that are very dissimilar. This system can preserve alignments based on function by identifying, for example, sections of the two sequences that are similarly hydrophobic, even if not identical in sequence. A gap scoring system (affine gap costs) is used to chose scores for 'gaps'. A gap is one or more adjacent nulls in one sequence aligned with letters in the other sequence. Ideally, the gap scoring system charges a large initial penality for the existence of a gap, and smaller penalitiesfor each individualresidue. Thistakes intoaccount thateach mutationalevent can insert or delete multiple residues at a time the bulk of the gap cost penalty is for the existence of the mutation itself, not the length. The alignment score is the sum of the scores specified for each of the aligned pairs of letters, and letters with nulls, in the alignment. The higher the alignment score, the better the alignment. Vaata eestikeelseid referaate: Leopold Parts Mark Tehver 24
25 Smith Waterman algorithm finds best matching substrings between two sequences It uses simple dynamic programming, except the longer gaps can be supported with affine penalty scores. Maximum is taken between 1) comparison scores for last characters plus score without the last characters 2) max for all prefixes of first sequence plus the gap cost 3) max for all prefixes of second sequence plus the gap cost The used parameters: GAP_PENALTY penalty to start a gap GAP_EXTENSION penalty to extend an existing gap MATCH_PENALTY match score MISMATCH_PENALTY mismatch score SwTable(String A, String B): table[i][0] = 0, 0 <= j <= length(b) table[0][i] = 0, 0 <= i <= length(a) for(i = 1; i <= length(a); ++i) for(j = 1; j <= length(b); ++j) maxrow = -INFINITY; maxcolumn = -INFINITY; // find the best score from column maxcolumn = max(maxcolumn, table[i - k][j] - gapscore(k)); 0 < k < i // find the best score from row maxrow = max(maxcolumn, table[i][j - k] - gapscore(k)); 0 < k < j table[i][j] = max( // new value is a max of 4: table[i-1][j-1] + letterscore(ai, Bj), maxrow, maxcolumn, 0 // no negative values ); gapscore(k) = GAP_PENALTY + GAP_EXTENSION*k letterscore(c, d) = MATCH_PENALTY, if c = d MISMATCH_PENALTY, if c!= d 25
26 Gap penalties Gaps were rather freely permitted in the overly simple implementations of the Needleman Wunsch and Smith Waterman algorithms shown above. What would happen if you feel that gaps should be rarer events in your particular protein? It is possible to assign a different weight to gaps. This can be done by subtracting from the score, some predetermined value every time a gap is required. In this case you can define a weight as W = a + bk, where k is the length of the gap. Hence you can control whether many short gaps occur or whether long gaps occur but more infrequently. Deletions do occur but when they occur it is seldom many small, short deletions but rather fewer and longer deletions. How do you choose a gap penalty? Unfortunately, there is little knowledge to help here. Most of the tests done so far depend on an empirical basis designed to achieve some end. For example, Smith and Fitch have derived (by exhaustive search) gap penalties that will best align distantly related haemoglobin genes. But there is no guarantee that these values would work well for the protein or (worse) the nucleotide sequence that you are interested in. Typical values are 0.5 < a < < b < 1.0 b < a but there is nothing special about these values other than the fact that they seem to work well for some of the common comparisons. Note that in general a > b. This corresponds with biological knowledge of how gaps are generated it is easier to generate one gap of two residues rather than two gaps of one residue since the former can be created by a single mutational event. Substitution rates Weights for different mismatches should be permitted. A transition is more likely than a transversion; a Ile Val more likely than Ile Arg change. If the two sequences have no obvious relationship at their right and left ends, then end gaps should not be penalized. Unless two sequences are known to be homologous over their entire length, a local alignment is preferable to a global alignment. An optimal alignment is by no means necessarily statistically significant. One must make some estimate of the probability that a given alignment is due to chance. An alignment demonstrates similarity, not necessarily, homology. Homology is an evolutionary inference based on examination of the similarity and its biological meaning. Sequence similarity may result from homology but it may also result from chance, convergence or analogy. 26
![PAM: [Taken from the NCBI Glossary PAM definition]. Percent Accepted Mutation. A unit introduced by Dayhoff et al.](/docs-images/96/126596819/images/27-0.jpg "to quantify the amount of evolutionary change in a protein sequence. 1.")
 substitution matrix is a look up table in which scores for each amino acid substitution have been calculated based on the")
27 PAM: [Taken from the NCBI Glossary PAM definition]. Percent Accepted Mutation. A unit introduced by Dayhoff et al. to quantify the amount of evolutionary change in a protein sequence. 1.0 PAM unit, is the amount of evolution which will change, on average, 1% of amino acids in a protein sequence. A PAM(x) substitution matrix is a look up table in which scores for each amino acid substitution have been calculated based on the frequency of that substitution in closely related proteins that have experienced a certain amount (x) of evolutionary divergence. 27


28 BLOSUM Blocks Substitution Matrix. A substitution matrix in which scores for each position are derived from observations of the frequencies of substitutions in blocks of local alignments in related proteins. Each matrix is tailored to a particular evolutionary distance. In the BLOSUM62 matrix, for example, the alignment from which scores were derived was created using sequences sharing no more than 62% identity. Sequences more identical than 62% are represented by a single sequence in the alignment so as to avoid overweighting closely related family members. (Henikoff and Henikoff) 28
29 Homology Orthology / orthologous the same gene in different species Paralogy / Paralogous Similar gene in the same organism E.g. gene duplication Paralogous genes can evolve independently and acquire new different functions Or become redundant, and disappear 29
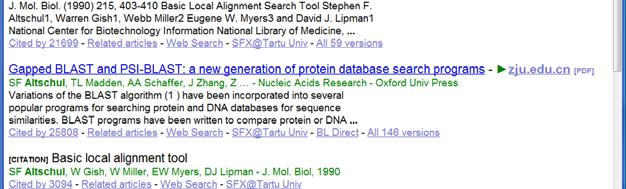
30 BLAST Basic Local Alignment Search Tool. (Altschul et al. J. Mol. Biol. 215(3):403 10, 10, 1990) A sequence comparison algorithm optimized for speed used to search sequence databases for optimal local alignments to a query. The initial search is done for a word of length "W" that scores at least "T" when compared to the query using a substitution matrix. Word hits are then extended in either direction in an attempt to generate an alignment with a score exceeding the threshold of "S". The "T" parameter dictates the speed and sensitivity of the search. 30
31 Indexing-based local alignment (BLAST- Basic Local Alignment Search Tool) Main idea: query 1. Construct a dictionary of all the words in the query 2. Initiate a local alignment for each word match between query and DB DB Running Time: O(MN) However, orders of magnitude faster than Smith-Waterman CS262 Lecture 3, Win06, Batzoglou Indexing-based local alignment Dictionary: All words of length k (~10) Alignment initiated between words of alignment score T (typically T = k) query Alignment: Ungapped extensions until score below statistical ti ti threshold h Output: All local alignments with score > statistical threshold CS262 Lecture 3, Win06, Batzoglou scan DB query 31
32 Indexing-based local alignment Extensions Example: k = 4 The matching word GGTC initiates an alignment Extension to the left and right with no gaps until alignment falls < C below best so far Output: GTAAGGTCC GTTAGGTCC CS262 Lecture 3, Win06, Batzoglou C C C T T C C T G G A T T G C G A A C G A A G T A A G G T C C A G T Indexing-based local alignment Extensions Gapped extensions G A A C G A A G T A A G G T C C A G T Extensions with gaps in a band around anchor Output: GTAAGGTCCAGT GTTAGGTC-AGT CS262 Lecture 3, Win06, Batzoglou C T G A T C C T G G A T T G C 32

 short words (k = 7) Speed Sens.")
33 Indexing-based local alignment Extensions Gapped extensions until threshold G A A C G A A G T A A G G T C C A G T Extensions with gaps until score < C below best score so far Output: GTAAGGTCCAGT GTTAGGTC-AGT CS262 Lecture 3, Win06, Batzoglou C T G A T C C T G G A T T G C Sensitivity-Speed Tradeoff X% Sensitivity long words (k = 15) short words (k = 7) Speed Sens. Speed CS262 Lecture 3, Win06, Batzoglou Kent WJ, Genome Research



34 Sensitivity-Speed Tradeoff Methods to improve sensitivity/speed 1. Using pairs of words ATAACGGACGACTGATTACACTGATTCTTAC GGCACGGACCAGTGACTACTCTGATTCCCAG 2. Using inexact words ATAACGGACGACTGATTACACTGATTCTTAC 3. Patterns non consecutive positions CS262 Lecture 3, Win06, Batzoglou GGCGCCGACGAGTGATTACACAGATTGCCAG TTTGATTACACAGAT T G TT CAC G Measured improvement CS262 Lecture 3, Win06, Batzoglou Kent WJ, Genome Research
35 Non-consecutive words Patterns Patterns increase the likelihood of at least one match within a long conserved region Consecutive Positions Non-Consecutive Positions 6 common 7 common 5 common 3 common On a 100-long 70% conserved region: Consecutive Non-consecutive Expected # hits: Prob[at least one hit]: CS262 Lecture 3, Win06, Batzoglou Advantage of Patterns 11 positions 11 positions 10 positions CS262 Lecture 3, Win06, Batzoglou 35
 for homology between two regions Find: best set of K patterns")
36 Multiple patterns TTTGATTACACAGAT T G TT CAC G T G T C CAG TTGATT A G How long does it take to search the query? K patterns Takes K times longer to scan Patterns can complement one another Computational problem: Given: a model (prob distribution) for homology between two regions Find: best set of K patterns that maximizes Prob(at least one match) Buhler et al. RECOMB 2003 CS262 Lecture 3, Win06, Batzoglou Sun & Buhler RECOMB 2004 Variants of BLAST NCBI BLAST: search the universe MEGABLAST: Optimized to align very similar sequences Works best when k = 4i 16 Linear gap penalty WU-BLAST: (Wash U BLAST) Very good optimizations Good set of features & command line arguments BLAT Faster, less sensitive than BLAST Good for aligning huge numbers of queries CHAOS Uses inexact k-mers, sensitive PatternHunter p p p p Uses patterns instead of k-mers BlastZ Uses patterns, good for finding genes Typhon Uses multiple alignments to improve sensitivity/speed tradeoff CS262 Lecture 3, Win06, Batzoglou 36
37 Example Query: gattacaccccgattacaccccgattaca (29 letters) [2 mins] Database: All GenBank+EMBL+DDBJ+PDB sequences (but no EST, STS, GSS, or phase 0, 1 or 2 HTGS sequences) 1,726,556 sequences; 8,074,398,388 total t letters >gi gb AC Oryza sativa chromosome 3 BAC OSJNBa0087C10 genomic sequence, complete sequence Length = Score = 34.2 bits (17), Expect = 4.5 Identities = 20/21 (95%) Strand = Plus / Plus Query: 4 tacaccccgattacaccccga 24 Sbjct: tacacccagattacaccccga Score = 34.2 bits (17), Expect = 4.5 Identities = 20/21 (95%) Strand = Plus / Plus Query: 4 tacaccccgattacaccccga 24 Sbjct: tacacccagattacaccccga >gi gb AC Oryza sativa chromosome 3 BAC OSJNBa0052F07 genomic sequence, complete sequence Length = Score = 34.2 bits (17), Expect = 4.5 Identities = 20/21 (95%) Strand = Plus / Plus Query: 4 tacaccccgattacaccccga 24 Sbjct: 3891 tacacccagattacaccccga 3911 CS262 Lecture 3, Win06, Batzoglou Example Query: Human atoh enhancer, 179 letters [1.5 min] Result: 57 blast hits 1. gi gb AF AF Homo sapiens ATOH1 enhanc e gi gb AC Mus musculus Strain C57BL6/J ch e gi gb AF AF Mus musculus Atoh1 enhanc e gi gb AF Gallus gallus CATH1 (CATH1) gene e gi emb AL Zebrafish DNA sequence from clo e gi gb AC Oryza sativa chromosome 10 BAC O gi ref NM_ Mus musculus suppressor of Ty gi gb BC Mus musculus, Similar to suppres gi gb AF AF Mus musculus Atoh1 enhancer sequence Length = 1517 Score = 256 bits (129), Expect = 9e-66 Identities = 167/177 (94%), Gaps = 2/177 (1%) Strand = Plus / Plus Query: 3 tgacaatagagggtctggcagaggctcctggccgcggtgcggagcgtctggagcggagca 62 Sbjct: 1144 tgacaatagaggggctggcagaggctcctggccccggtgcggagcgtctggagcggagca 1203 Query: 63 cgcgctgtcagctggtgagcgcactctcctttcaggcagctccccggggagctgtgcggc 122 Sbjct: 1204 cgcgctgtcagctggtgagcgcactc-gctttcaggccgctccccggggagctgagcggc 1262 Query: 123 cacatttaacaccatcatcacccctccccggcctcctcaacctcggcctcctcctcg 179 Sbjct: 1263 cacatttaacaccgtcgtca-ccctccccggcctcctcaacatcggcctcctcctcg 1318 CS262 Lecture 3, Win06, Batzoglou 37
38 Examples: >sp P48432 SOX2_MOUSE Transcription factor SOX 2 OS=Mus musculus GN=Sox2 PE=1 SV=2 MYNMMETELKPPGPQQASGGGGGGGNATAAATGGNQKNSPDRVKRPMNAFMVWSRGQRRK MAQENPKMHNSEISKRLGAEWKLLSETEKRPFIDEAKRLRALHMKEHPDYKYRPRRKTKT LMKKDKYTLPGGLLAPGGNSMASGVGVGAGLGGGLNQRMDSYAHMNGWSNGSYSMMQEQL GYPQHPGLNAHGAAQMQPMHRYVVSALQYNSMTSSQTYMNGSPTYSMSYSQQGTPGMALG SMGSVVKSEASSSPPVVTSSSHSRAPCQAGDLRDMISMYLPGAEVPEPAAPSRLHMAQHY QSGPVPGTAKYGTLPLSHM ctattaactt gttcaaaaaa gtatcaggag ttgtcaaggc agagaagaga gtgtttgcaa aaagggaaaa gtactttgct gcctctttaa gactagggct gggagaaaga agaggagaga gaaagaaagg agagaagttt ggagcccgag gcttaagcct ttccaaaaac taatcacaac bin/hgblat?command=start Lessons learned? New searching technique: Filter based on partial hits Refine the search on the candidate hits Statistical significance Probability of a good looking hit increases when DB grows 38



39 CS262 Lecture 3, Win06, Batzoglou Multiple alignment: find the alignment for tens, if not hundreds d or thousands of sequences Very importamt for actual biological interpretation, say describing protein families through the evolution. 39
40 Relevant Citations Needleman, S.B. and Wunsch, C.D. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J Mol Biol. 48(3): (1970).PubMed: Smith, T.F. and Waterman, M.S. Identification of common molecular subsequences. J Mol Biol. 147(1):195 7 (1981). PubMed: Pearson, W.R. and Lipman, D.J. Improved tools for biological sequence comparison. Proc Natl Acad Sci U S A. 85(8): (1988). PubMed: Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 215(3): (1990). PubMed: Evolution sets restrictions on possible mutations One needs to use substitution cost matrices that somehow reflect the evolutionary distance Links Distance Measures, edited by B.Golding, Feb 1996 web (Itshack Pe`er) Sequence alignment algorithms (Benjamin Corum) And some of them with local copies: Amino acids distances Amino acids distances PAM matrices (Margaret Dayhoff) PAM units PAM matrices Amino acids distances 40



41 41




42 42
EECS730: Introduction to Bioinformatics
 EECS730: Introduction to Bioinformatics Lecture 05: Index-based alignment algorithms Slides adapted from Dr. Shaojie Zhang (University of Central Florida) Real applications of alignment Database search
EECS730: Introduction to Bioinformatics Lecture 05: Index-based alignment algorithms Slides adapted from Dr. Shaojie Zhang (University of Central Florida) Real applications of alignment Database search
Evolution. CT Amemiya et al. Nature 496, (2013) doi: /nature12027
 doi: /nature12027") Sequence Alignment Evolution CT Amemiya et al. Nature 496, 311-316 (2013) doi:10.1038/nature12027 Evolutionary Rates next generation OK OK OK X X Still OK? Sequence conservation implies function Alignment
Sequence Alignment Evolution CT Amemiya et al. Nature 496, 311-316 (2013) doi:10.1038/nature12027 Evolutionary Rates next generation OK OK OK X X Still OK? Sequence conservation implies function Alignment
Sequence Database Search Techniques I: Blast and PatternHunter tools
 Sequence Database Search Techniques I: Blast and PatternHunter tools Zhang Louxin National University of Singapore Outline. Database search 2. BLAST (and filtration technique) 3. PatternHunter (empowered
Sequence Database Search Techniques I: Blast and PatternHunter tools Zhang Louxin National University of Singapore Outline. Database search 2. BLAST (and filtration technique) 3. PatternHunter (empowered
In-Depth Assessment of Local Sequence Alignment
 2012 International Conference on Environment Science and Engieering IPCBEE vol.3 2(2012) (2012)IACSIT Press, Singapoore In-Depth Assessment of Local Sequence Alignment Atoosa Ghahremani and Mahmood A.
2012 International Conference on Environment Science and Engieering IPCBEE vol.3 2(2012) (2012)IACSIT Press, Singapoore In-Depth Assessment of Local Sequence Alignment Atoosa Ghahremani and Mahmood A.
Linear-Space Alignment
 Linear-Space Alignment Subsequences and Substrings Definition A string x is a substring of a string x, if x = ux v for some prefix string u and suffix string v (similarly, x = x i x j, for some 1 i j x
Linear-Space Alignment Subsequences and Substrings Definition A string x is a substring of a string x, if x = ux v for some prefix string u and suffix string v (similarly, x = x i x j, for some 1 i j x
Bioinformatics (GLOBEX, Summer 2015) Pairwise sequence alignment
 Pairwise sequence alignment") Bioinformatics (GLOBEX, Summer 2015) Pairwise sequence alignment Substitution score matrices, PAM, BLOSUM Needleman-Wunsch algorithm (Global) Smith-Waterman algorithm (Local) BLAST (local, heuristic) E-value
Bioinformatics (GLOBEX, Summer 2015) Pairwise sequence alignment Substitution score matrices, PAM, BLOSUM Needleman-Wunsch algorithm (Global) Smith-Waterman algorithm (Local) BLAST (local, heuristic) E-value
Single alignment: Substitution Matrix. 16 march 2017
 Single alignment: Substitution Matrix 16 march 2017 BLOSUM Matrix BLOSUM Matrix [2] (Blocks Amino Acid Substitution Matrices ) It is based on the amino acids substitutions observed in ~2000 conserved block
Single alignment: Substitution Matrix 16 march 2017 BLOSUM Matrix BLOSUM Matrix [2] (Blocks Amino Acid Substitution Matrices ) It is based on the amino acids substitutions observed in ~2000 conserved block
3. SEQUENCE ANALYSIS BIOINFORMATICS COURSE MTAT
 3. SEQUENCE ANALYSIS BIOINFORMATICS COURSE MTAT.03.239 25.09.2012 SEQUENCE ANALYSIS IS IMPORTANT FOR... Prediction of function Gene finding the process of identifying the regions of genomic DNA that encode
3. SEQUENCE ANALYSIS BIOINFORMATICS COURSE MTAT.03.239 25.09.2012 SEQUENCE ANALYSIS IS IMPORTANT FOR... Prediction of function Gene finding the process of identifying the regions of genomic DNA that encode
CISC 889 Bioinformatics (Spring 2004) Sequence pairwise alignment (I)
 Sequence pairwise alignment (I)") CISC 889 Bioinformatics (Spring 2004) Sequence pairwise alignment (I) Contents Alignment algorithms Needleman-Wunsch (global alignment) Smith-Waterman (local alignment) Heuristic algorithms FASTA BLAST
CISC 889 Bioinformatics (Spring 2004) Sequence pairwise alignment (I) Contents Alignment algorithms Needleman-Wunsch (global alignment) Smith-Waterman (local alignment) Heuristic algorithms FASTA BLAST
Quantifying sequence similarity
 Quantifying sequence similarity Bas E. Dutilh Systems Biology: Bioinformatic Data Analysis Utrecht University, February 16 th 2016 After this lecture, you can define homology, similarity, and identity
Quantifying sequence similarity Bas E. Dutilh Systems Biology: Bioinformatic Data Analysis Utrecht University, February 16 th 2016 After this lecture, you can define homology, similarity, and identity
Practical considerations of working with sequencing data
 Practical considerations of working with sequencing data File Types Fastq ->aligner -> reference(genome) coordinates Coordinate files SAM/BAM most complete, contains all of the info in fastq and more!
Practical considerations of working with sequencing data File Types Fastq ->aligner -> reference(genome) coordinates Coordinate files SAM/BAM most complete, contains all of the info in fastq and more!
Algorithms in Bioinformatics FOUR Pairwise Sequence Alignment. Pairwise Sequence Alignment. Convention: DNA Sequences 5. Sequence Alignment
 Algorithms in Bioinformatics FOUR Sami Khuri Department of Computer Science San José State University Pairwise Sequence Alignment Homology Similarity Global string alignment Local string alignment Dot
Algorithms in Bioinformatics FOUR Sami Khuri Department of Computer Science San José State University Pairwise Sequence Alignment Homology Similarity Global string alignment Local string alignment Dot
Algorithms in Bioinformatics
 Algorithms in Bioinformatics Sami Khuri Department of omputer Science San José State University San José, alifornia, USA khuri@cs.sjsu.edu www.cs.sjsu.edu/faculty/khuri Pairwise Sequence Alignment Homology
Algorithms in Bioinformatics Sami Khuri Department of omputer Science San José State University San José, alifornia, USA khuri@cs.sjsu.edu www.cs.sjsu.edu/faculty/khuri Pairwise Sequence Alignment Homology
Pairwise & Multiple sequence alignments
 Pairwise & Multiple sequence alignments Urmila Kulkarni-Kale Bioinformatics Centre 411 007 urmila@bioinfo.ernet.in Basis for Sequence comparison Theory of evolution: gene sequences have evolved/derived
Pairwise & Multiple sequence alignments Urmila Kulkarni-Kale Bioinformatics Centre 411 007 urmila@bioinfo.ernet.in Basis for Sequence comparison Theory of evolution: gene sequences have evolved/derived
An Introduction to Sequence Similarity ( Homology ) Searching
 Searching") An Introduction to Sequence Similarity ( Homology ) Searching Gary D. Stormo 1 UNIT 3.1 1 Washington University, School of Medicine, St. Louis, Missouri ABSTRACT Homologous sequences usually have the same,
An Introduction to Sequence Similarity ( Homology ) Searching Gary D. Stormo 1 UNIT 3.1 1 Washington University, School of Medicine, St. Louis, Missouri ABSTRACT Homologous sequences usually have the same,
Sequence analysis and Genomics
 Sequence analysis and Genomics October 12 th November 23 rd 2 PM 5 PM Prof. Peter Stadler Dr. Katja Nowick Katja: group leader TFome and Transcriptome Evolution Bioinformatics group Paul-Flechsig-Institute
Sequence analysis and Genomics October 12 th November 23 rd 2 PM 5 PM Prof. Peter Stadler Dr. Katja Nowick Katja: group leader TFome and Transcriptome Evolution Bioinformatics group Paul-Flechsig-Institute
Moreover, the circular logic
 Moreover, the circular logic How do we know what is the right distance without a good alignment? And how do we construct a good alignment without knowing what substitutions were made previously? ATGCGT--GCAAGT
Moreover, the circular logic How do we know what is the right distance without a good alignment? And how do we construct a good alignment without knowing what substitutions were made previously? ATGCGT--GCAAGT
Biochemistry 324 Bioinformatics. Pairwise sequence alignment
 Biochemistry 324 Bioinformatics Pairwise sequence alignment How do we compare genes/proteins? When we have sequenced a genome, we try and identify the function of unknown genes by finding a similar gene
Biochemistry 324 Bioinformatics Pairwise sequence alignment How do we compare genes/proteins? When we have sequenced a genome, we try and identify the function of unknown genes by finding a similar gene
Sara C. Madeira. Universidade da Beira Interior. (Thanks to Ana Teresa Freitas, IST for useful resources on this subject)
") Bioinformática Sequence Alignment Pairwise Sequence Alignment Universidade da Beira Interior (Thanks to Ana Teresa Freitas, IST for useful resources on this subject) 1 16/3/29 & 23/3/29 27/4/29 Outline
Bioinformática Sequence Alignment Pairwise Sequence Alignment Universidade da Beira Interior (Thanks to Ana Teresa Freitas, IST for useful resources on this subject) 1 16/3/29 & 23/3/29 27/4/29 Outline
Pairwise sequence alignment
 Department of Evolutionary Biology Example Alignment between very similar human alpha- and beta globins: GSAQVKGHGKKVADALTNAVAHVDDMPNALSALSDLHAHKL G+ +VK+HGKKV A+++++AH+D++ +++++LS+LH KL GNPKVKAHGKKVLGAFSDGLAHLDNLKGTFATLSELHCDKL
Department of Evolutionary Biology Example Alignment between very similar human alpha- and beta globins: GSAQVKGHGKKVADALTNAVAHVDDMPNALSALSDLHAHKL G+ +VK+HGKKV A+++++AH+D++ +++++LS+LH KL GNPKVKAHGKKVLGAFSDGLAHLDNLKGTFATLSELHCDKL
Pairwise Alignment. Guan-Shieng Huang. Dept. of CSIE, NCNU. Pairwise Alignment p.1/55
 Pairwise Alignment Guan-Shieng Huang shieng@ncnu.edu.tw Dept. of CSIE, NCNU Pairwise Alignment p.1/55 Approach 1. Problem definition 2. Computational method (algorithms) 3. Complexity and performance Pairwise
Pairwise Alignment Guan-Shieng Huang shieng@ncnu.edu.tw Dept. of CSIE, NCNU Pairwise Alignment p.1/55 Approach 1. Problem definition 2. Computational method (algorithms) 3. Complexity and performance Pairwise
Computational Biology
 Computational Biology Lecture 6 31 October 2004 1 Overview Scoring matrices (Thanks to Shannon McWeeney) BLAST algorithm Start sequence alignment 2 1 What is a homologous sequence? A homologous sequence,
Computational Biology Lecture 6 31 October 2004 1 Overview Scoring matrices (Thanks to Shannon McWeeney) BLAST algorithm Start sequence alignment 2 1 What is a homologous sequence? A homologous sequence,
Introduction to Bioinformatics
 Introduction to Bioinformatics Jianlin Cheng, PhD Department of Computer Science Informatics Institute 2011 Topics Introduction Biological Sequence Alignment and Database Search Analysis of gene expression
Introduction to Bioinformatics Jianlin Cheng, PhD Department of Computer Science Informatics Institute 2011 Topics Introduction Biological Sequence Alignment and Database Search Analysis of gene expression
THEORY. Based on sequence Length According to the length of sequence being compared it is of following two types
 Exp 11- THEORY Sequence Alignment is a process of aligning two sequences to achieve maximum levels of identity between them. This help to derive functional, structural and evolutionary relationships between
Exp 11- THEORY Sequence Alignment is a process of aligning two sequences to achieve maximum levels of identity between them. This help to derive functional, structural and evolutionary relationships between
Sequence Comparison. mouse human
 Sequence Comparison Sequence Comparison mouse human Why Compare Sequences? The first fact of biological sequence analysis In biomolecular sequences (DNA, RNA, or amino acid sequences), high sequence similarity
Sequence Comparison Sequence Comparison mouse human Why Compare Sequences? The first fact of biological sequence analysis In biomolecular sequences (DNA, RNA, or amino acid sequences), high sequence similarity
Tiffany Samaroo MB&B 452a December 8, Take Home Final. Topic 1
 Tiffany Samaroo MB&B 452a December 8, 2003 Take Home Final Topic 1 Prior to 1970, protein and DNA sequence alignment was limited to visual comparison. This was a very tedious process; even proteins with
Tiffany Samaroo MB&B 452a December 8, 2003 Take Home Final Topic 1 Prior to 1970, protein and DNA sequence alignment was limited to visual comparison. This was a very tedious process; even proteins with
Introduction to sequence alignment. Local alignment the Smith-Waterman algorithm
 Lecture 2, 12/3/2003: Introduction to sequence alignment The Needleman-Wunsch algorithm for global sequence alignment: description and properties Local alignment the Smith-Waterman algorithm 1 Computational
Lecture 2, 12/3/2003: Introduction to sequence alignment The Needleman-Wunsch algorithm for global sequence alignment: description and properties Local alignment the Smith-Waterman algorithm 1 Computational
Large-Scale Genomic Surveys
 Bioinformatics Subtopics Fold Recognition Secondary Structure Prediction Docking & Drug Design Protein Geometry Protein Flexibility Homology Modeling Sequence Alignment Structure Classification Gene Prediction
Bioinformatics Subtopics Fold Recognition Secondary Structure Prediction Docking & Drug Design Protein Geometry Protein Flexibility Homology Modeling Sequence Alignment Structure Classification Gene Prediction
Sequence Alignments. Dynamic programming approaches, scoring, and significance. Lucy Skrabanek ICB, WMC January 31, 2013
 Sequence Alignments Dynamic programming approaches, scoring, and significance Lucy Skrabanek ICB, WMC January 31, 213 Sequence alignment Compare two (or more) sequences to: Find regions of conservation
Sequence Alignments Dynamic programming approaches, scoring, and significance Lucy Skrabanek ICB, WMC January 31, 213 Sequence alignment Compare two (or more) sequences to: Find regions of conservation
Sequence Alignment Techniques and Their Uses
 Sequence Alignment Techniques and Their Uses Sarah Fiorentino Since rapid sequencing technology and whole genomes sequencing, the amount of sequence information has grown exponentially. With all of this
Sequence Alignment Techniques and Their Uses Sarah Fiorentino Since rapid sequencing technology and whole genomes sequencing, the amount of sequence information has grown exponentially. With all of this
Bioinformatics and BLAST
 Bioinformatics and BLAST Overview Recap of last time Similarity discussion Algorithms: Needleman-Wunsch Smith-Waterman BLAST Implementation issues and current research Recap from Last Time Genome consists
Bioinformatics and BLAST Overview Recap of last time Similarity discussion Algorithms: Needleman-Wunsch Smith-Waterman BLAST Implementation issues and current research Recap from Last Time Genome consists
Lecture 2: Pairwise Alignment. CG Ron Shamir
 Lecture 2: Pairwise Alignment 1 Main source 2 Why compare sequences? Human hexosaminidase A vs Mouse hexosaminidase A 3 www.mathworks.com/.../jan04/bio_genome.html Sequence Alignment עימוד רצפים The problem:
Lecture 2: Pairwise Alignment 1 Main source 2 Why compare sequences? Human hexosaminidase A vs Mouse hexosaminidase A 3 www.mathworks.com/.../jan04/bio_genome.html Sequence Alignment עימוד רצפים The problem:
Local Alignment Statistics
 Local Alignment Statistics Stephen Altschul National Center for Biotechnology Information National Library of Medicine National Institutes of Health Bethesda, MD Central Issues in Biological Sequence Comparison
Local Alignment Statistics Stephen Altschul National Center for Biotechnology Information National Library of Medicine National Institutes of Health Bethesda, MD Central Issues in Biological Sequence Comparison
Welcome to CS262: Computational Genomics
 Instructor: Welcome to CS262: Computational Genomics Serafim Batzoglou TA: Paul Chen email: cs262-win2015-staff@lists.stanford.edu Tuesdays & Thursdays 12:50-2:05pm Clark S361 http://cs262.stanford.edu
Instructor: Welcome to CS262: Computational Genomics Serafim Batzoglou TA: Paul Chen email: cs262-win2015-staff@lists.stanford.edu Tuesdays & Thursdays 12:50-2:05pm Clark S361 http://cs262.stanford.edu
Bioinformatics for Biologists
 Bioinformatics for Biologists Sequence Analysis: Part I. Pairwise alignment and database searching Fran Lewitter, Ph.D. Head, Biocomputing Whitehead Institute Bioinformatics Definitions The use of computational
Bioinformatics for Biologists Sequence Analysis: Part I. Pairwise alignment and database searching Fran Lewitter, Ph.D. Head, Biocomputing Whitehead Institute Bioinformatics Definitions The use of computational
Sequence analysis and comparison
 The aim with sequence identification: Sequence analysis and comparison Marjolein Thunnissen Lund September 2012 Is there any known protein sequence that is homologous to mine? Are there any other species
The aim with sequence identification: Sequence analysis and comparison Marjolein Thunnissen Lund September 2012 Is there any known protein sequence that is homologous to mine? Are there any other species
Lecture 5,6 Local sequence alignment
 Lecture 5,6 Local sequence alignment Chapter 6 in Jones and Pevzner Fall 2018 September 4,6, 2018 Evolution as a tool for biological insight Nothing in biology makes sense except in the light of evolution
Lecture 5,6 Local sequence alignment Chapter 6 in Jones and Pevzner Fall 2018 September 4,6, 2018 Evolution as a tool for biological insight Nothing in biology makes sense except in the light of evolution
Grundlagen der Bioinformatik, SS 08, D. Huson, May 2,
 Grundlagen der Bioinformatik, SS 08, D. Huson, May 2, 2008 39 5 Blast This lecture is based on the following, which are all recommended reading: R. Merkl, S. Waack: Bioinformatik Interaktiv. Chapter 11.4-11.7
Grundlagen der Bioinformatik, SS 08, D. Huson, May 2, 2008 39 5 Blast This lecture is based on the following, which are all recommended reading: R. Merkl, S. Waack: Bioinformatik Interaktiv. Chapter 11.4-11.7
CONCEPT OF SEQUENCE COMPARISON. Natapol Pornputtapong 18 January 2018
 CONCEPT OF SEQUENCE COMPARISON Natapol Pornputtapong 18 January 2018 SEQUENCE ANALYSIS - A ROSETTA STONE OF LIFE Sequence analysis is the process of subjecting a DNA, RNA or peptide sequence to any of
CONCEPT OF SEQUENCE COMPARISON Natapol Pornputtapong 18 January 2018 SEQUENCE ANALYSIS - A ROSETTA STONE OF LIFE Sequence analysis is the process of subjecting a DNA, RNA or peptide sequence to any of
Tools and Algorithms in Bioinformatics
 Tools and Algorithms in Bioinformatics GCBA815, Fall 2013 Week3: Blast Algorithm, theory and practice Babu Guda, Ph.D. Department of Genetics, Cell Biology & Anatomy Bioinformatics and Systems Biology
Tools and Algorithms in Bioinformatics GCBA815, Fall 2013 Week3: Blast Algorithm, theory and practice Babu Guda, Ph.D. Department of Genetics, Cell Biology & Anatomy Bioinformatics and Systems Biology
Alignment & BLAST. By: Hadi Mozafari KUMS
 Alignment & BLAST By: Hadi Mozafari KUMS SIMILARITY - ALIGNMENT Comparison of primary DNA or protein sequences to other primary or secondary sequences Expecting that the function of the similar sequence
Alignment & BLAST By: Hadi Mozafari KUMS SIMILARITY - ALIGNMENT Comparison of primary DNA or protein sequences to other primary or secondary sequences Expecting that the function of the similar sequence
Sequence Alignment (chapter 6)
") Sequence lignment (chapter 6) he biological problem lobal alignment Local alignment Multiple alignment Introduction to bioinformatics, utumn 6 Background: comparative genomics Basic question in biology:
Sequence lignment (chapter 6) he biological problem lobal alignment Local alignment Multiple alignment Introduction to bioinformatics, utumn 6 Background: comparative genomics Basic question in biology:
Sequence Alignment: A General Overview. COMP Fall 2010 Luay Nakhleh, Rice University
 Sequence Alignment: A General Overview COMP 571 - Fall 2010 Luay Nakhleh, Rice University Life through Evolution All living organisms are related to each other through evolution This means: any pair of
Sequence Alignment: A General Overview COMP 571 - Fall 2010 Luay Nakhleh, Rice University Life through Evolution All living organisms are related to each other through evolution This means: any pair of
Bio nformatics. Lecture 3. Saad Mneimneh
 Bio nformatics Lecture 3 Sequencing As before, DNA is cut into small ( 0.4KB) fragments and a clone library is formed. Biological experiments allow to read a certain number of these short fragments per
Bio nformatics Lecture 3 Sequencing As before, DNA is cut into small ( 0.4KB) fragments and a clone library is formed. Biological experiments allow to read a certain number of these short fragments per
1.5 Sequence alignment
 1.5 Sequence alignment The dramatic increase in the number of sequenced genomes and proteomes has lead to development of various bioinformatic methods and algorithms for extracting information (data mining)
1.5 Sequence alignment The dramatic increase in the number of sequenced genomes and proteomes has lead to development of various bioinformatic methods and algorithms for extracting information (data mining)
First generation sequencing and pairwise alignment (High-tech, not high throughput) Analysis of Biological Sequences
 Analysis of Biological Sequences") First generation sequencing and pairwise alignment (High-tech, not high throughput) Analysis of Biological Sequences 140.638 where do sequences come from? DNA is not hard to extract (getting DNA from a
First generation sequencing and pairwise alignment (High-tech, not high throughput) Analysis of Biological Sequences 140.638 where do sequences come from? DNA is not hard to extract (getting DNA from a
Minimum Edit Distance. Defini'on of Minimum Edit Distance
 Minimum Edit Distance Defini'on of Minimum Edit Distance How similar are two strings? Spell correc'on The user typed graffe Which is closest? graf gra@ grail giraffe Computa'onal Biology Align two sequences
Minimum Edit Distance Defini'on of Minimum Edit Distance How similar are two strings? Spell correc'on The user typed graffe Which is closest? graf gra@ grail giraffe Computa'onal Biology Align two sequences
Module: Sequence Alignment Theory and Applications Session: Introduction to Searching and Sequence Alignment
 Module: Sequence Alignment Theory and Applications Session: Introduction to Searching and Sequence Alignment Introduction to Bioinformatics online course : IBT Jonathan Kayondo Learning Objectives Understand
Module: Sequence Alignment Theory and Applications Session: Introduction to Searching and Sequence Alignment Introduction to Bioinformatics online course : IBT Jonathan Kayondo Learning Objectives Understand
Pairwise sequence alignments
 Pairwise sequence alignments Volker Flegel VI, October 2003 Page 1 Outline Introduction Definitions Biological context of pairwise alignments Computing of pairwise alignments Some programs VI, October
Pairwise sequence alignments Volker Flegel VI, October 2003 Page 1 Outline Introduction Definitions Biological context of pairwise alignments Computing of pairwise alignments Some programs VI, October
BLAST. Varieties of BLAST
 BLAST Basic Local Alignment Search Tool (1990) Altschul, Gish, Miller, Myers, & Lipman Uses short-cuts or heuristics to improve search speed Like speed-reading, does not examine every nucleotide of database
BLAST Basic Local Alignment Search Tool (1990) Altschul, Gish, Miller, Myers, & Lipman Uses short-cuts or heuristics to improve search speed Like speed-reading, does not examine every nucleotide of database
Lecture 1, 31/10/2001: Introduction to sequence alignment. The Needleman-Wunsch algorithm for global sequence alignment: description and properties
 Lecture 1, 31/10/2001: Introduction to sequence alignment The Needleman-Wunsch algorithm for global sequence alignment: description and properties 1 Computational sequence-analysis The major goal of computational
Lecture 1, 31/10/2001: Introduction to sequence alignment The Needleman-Wunsch algorithm for global sequence alignment: description and properties 1 Computational sequence-analysis The major goal of computational
Alignment principles and homology searching using (PSI-)BLAST. Jaap Heringa Centre for Integrative Bioinformatics VU (IBIVU)
BLAST. Jaap Heringa Centre for Integrative Bioinformatics VU (IBIVU)") Alignment principles and homology searching using (PSI-)BLAST Jaap Heringa Centre for Integrative Bioinformatics VU (IBIVU) http://ibivu.cs.vu.nl Bioinformatics Nothing in Biology makes sense except in
Alignment principles and homology searching using (PSI-)BLAST Jaap Heringa Centre for Integrative Bioinformatics VU (IBIVU) http://ibivu.cs.vu.nl Bioinformatics Nothing in Biology makes sense except in
BLAST Database Searching. BME 110: CompBio Tools Todd Lowe April 8, 2010
 BLAST Database Searching BME 110: CompBio Tools Todd Lowe April 8, 2010 Admin Reading: Read chapter 7, and the NCBI Blast Guide and tutorial http://www.ncbi.nlm.nih.gov/blast/why.shtml Read Chapter 8 for
BLAST Database Searching BME 110: CompBio Tools Todd Lowe April 8, 2010 Admin Reading: Read chapter 7, and the NCBI Blast Guide and tutorial http://www.ncbi.nlm.nih.gov/blast/why.shtml Read Chapter 8 for
Pairwise sequence alignments. Vassilios Ioannidis (From Volker Flegel )
") Pairwise sequence alignments Vassilios Ioannidis (From Volker Flegel ) Outline Introduction Definitions Biological context of pairwise alignments Computing of pairwise alignments Some programs Importance
Pairwise sequence alignments Vassilios Ioannidis (From Volker Flegel ) Outline Introduction Definitions Biological context of pairwise alignments Computing of pairwise alignments Some programs Importance
Lecture 4: Evolutionary Models and Substitution Matrices (PAM and BLOSUM)
") Bioinformatics II Probability and Statistics Universität Zürich and ETH Zürich Spring Semester 2009 Lecture 4: Evolutionary Models and Substitution Matrices (PAM and BLOSUM) Dr Fraser Daly adapted from
Bioinformatics II Probability and Statistics Universität Zürich and ETH Zürich Spring Semester 2009 Lecture 4: Evolutionary Models and Substitution Matrices (PAM and BLOSUM) Dr Fraser Daly adapted from
CSE : Computational Issues in Molecular Biology. Lecture 6. Spring 2004
 CSE 397-497: Computational Issues in Molecular Biology Lecture 6 Spring 2004-1 - Topics for today Based on premise that algorithms we've studied are too slow: Faster method for global comparison when sequences
CSE 397-497: Computational Issues in Molecular Biology Lecture 6 Spring 2004-1 - Topics for today Based on premise that algorithms we've studied are too slow: Faster method for global comparison when sequences
Basic Local Alignment Search Tool
 Basic Local Alignment Search Tool Alignments used to uncover homologies between sequences combined with phylogenetic studies o can determine orthologous and paralogous relationships Local Alignment uses
Basic Local Alignment Search Tool Alignments used to uncover homologies between sequences combined with phylogenetic studies o can determine orthologous and paralogous relationships Local Alignment uses
Lecture 2, 5/12/2001: Local alignment the Smith-Waterman algorithm. Alignment scoring schemes and theory: substitution matrices and gap models
 Lecture 2, 5/12/2001: Local alignment the Smith-Waterman algorithm Alignment scoring schemes and theory: substitution matrices and gap models 1 Local sequence alignments Local sequence alignments are necessary
Lecture 2, 5/12/2001: Local alignment the Smith-Waterman algorithm Alignment scoring schemes and theory: substitution matrices and gap models 1 Local sequence alignments Local sequence alignments are necessary
Tools and Algorithms in Bioinformatics
 Tools and Algorithms in Bioinformatics GCBA815, Fall 2015 Week-4 BLAST Algorithm Continued Multiple Sequence Alignment Babu Guda, Ph.D. Department of Genetics, Cell Biology & Anatomy Bioinformatics and
Tools and Algorithms in Bioinformatics GCBA815, Fall 2015 Week-4 BLAST Algorithm Continued Multiple Sequence Alignment Babu Guda, Ph.D. Department of Genetics, Cell Biology & Anatomy Bioinformatics and
Handling Rearrangements in DNA Sequence Alignment
 Handling Rearrangements in DNA Sequence Alignment Maneesh Bhand 12/5/10 1 Introduction Sequence alignment is one of the core problems of bioinformatics, with a broad range of applications such as genome
Handling Rearrangements in DNA Sequence Alignment Maneesh Bhand 12/5/10 1 Introduction Sequence alignment is one of the core problems of bioinformatics, with a broad range of applications such as genome
L3: Blast: Keyword match basics
 L3: Blast: Keyword match basics Fa05 CSE 182 Silly Quiz TRUE or FALSE: In New York City at any moment, there are 2 people (not bald) with exactly the same number of hairs! Assignment 1 is online Due 10/6
L3: Blast: Keyword match basics Fa05 CSE 182 Silly Quiz TRUE or FALSE: In New York City at any moment, there are 2 people (not bald) with exactly the same number of hairs! Assignment 1 is online Due 10/6
Motivating the need for optimal sequence alignments...
 1 Motivating the need for optimal sequence alignments... 2 3 Note that this actually combines two objectives of optimal sequence alignments: (i) use the score of the alignment o infer homology; (ii) use
1 Motivating the need for optimal sequence alignments... 2 3 Note that this actually combines two objectives of optimal sequence alignments: (i) use the score of the alignment o infer homology; (ii) use
Homology Modeling. Roberto Lins EPFL - summer semester 2005
 Homology Modeling Roberto Lins EPFL - summer semester 2005 Disclaimer: course material is mainly taken from: P.E. Bourne & H Weissig, Structural Bioinformatics; C.A. Orengo, D.T. Jones & J.M. Thornton,
Homology Modeling Roberto Lins EPFL - summer semester 2005 Disclaimer: course material is mainly taken from: P.E. Bourne & H Weissig, Structural Bioinformatics; C.A. Orengo, D.T. Jones & J.M. Thornton,
20 Grundlagen der Bioinformatik, SS 08, D. Huson, May 27, Global and local alignment of two sequences using dynamic programming
 20 Grundlagen der Bioinformatik, SS 08, D. Huson, May 27, 2008 4 Pairwise alignment We will discuss: 1. Strings 2. Dot matrix method for comparing sequences 3. Edit distance 4. Global and local alignment
20 Grundlagen der Bioinformatik, SS 08, D. Huson, May 27, 2008 4 Pairwise alignment We will discuss: 1. Strings 2. Dot matrix method for comparing sequences 3. Edit distance 4. Global and local alignment
C E N T R. Introduction to bioinformatics 2007 E B I O I N F O R M A T I C S V U F O R I N T. Lecture 5 G R A T I V. Pair-wise Sequence Alignment
 C E N T R E F O R I N T E G R A T I V E B I O I N F O R M A T I C S V U Introduction to bioinformatics 2007 Lecture 5 Pair-wise Sequence Alignment Bioinformatics Nothing in Biology makes sense except in
C E N T R E F O R I N T E G R A T I V E B I O I N F O R M A T I C S V U Introduction to bioinformatics 2007 Lecture 5 Pair-wise Sequence Alignment Bioinformatics Nothing in Biology makes sense except in
String Matching Problem
 String Matching Problem Pattern P Text T Set of Locations L 9/2/23 CAP/CGS 5991: Lecture 2 Computer Science Fundamentals Specify an input-output description of the problem. Design a conceptual algorithm
String Matching Problem Pattern P Text T Set of Locations L 9/2/23 CAP/CGS 5991: Lecture 2 Computer Science Fundamentals Specify an input-output description of the problem. Design a conceptual algorithm
Collected Works of Charles Dickens
 Collected Works of Charles Dickens A Random Dickens Quote If there were no bad people, there would be no good lawyers. Original Sentence It was a dark and stormy night; the night was dark except at sunny
Collected Works of Charles Dickens A Random Dickens Quote If there were no bad people, there would be no good lawyers. Original Sentence It was a dark and stormy night; the night was dark except at sunny
MATHEMATICAL MODELS - Vol. III - Mathematical Modeling and the Human Genome - Hilary S. Booth MATHEMATICAL MODELING AND THE HUMAN GENOME
 MATHEMATICAL MODELING AND THE HUMAN GENOME Hilary S. Booth Australian National University, Australia Keywords: Human genome, DNA, bioinformatics, sequence analysis, evolution. Contents 1. Introduction:
MATHEMATICAL MODELING AND THE HUMAN GENOME Hilary S. Booth Australian National University, Australia Keywords: Human genome, DNA, bioinformatics, sequence analysis, evolution. Contents 1. Introduction:
BIO 285/CSCI 285/MATH 285 Bioinformatics Programming Lecture 8 Pairwise Sequence Alignment 2 And Python Function Instructor: Lei Qian Fisk University
 BIO 285/CSCI 285/MATH 285 Bioinformatics Programming Lecture 8 Pairwise Sequence Alignment 2 And Python Function Instructor: Lei Qian Fisk University Measures of Sequence Similarity Alignment with dot
BIO 285/CSCI 285/MATH 285 Bioinformatics Programming Lecture 8 Pairwise Sequence Alignment 2 And Python Function Instructor: Lei Qian Fisk University Measures of Sequence Similarity Alignment with dot
Introduction to Bioinformatics
 Introduction to Bioinformatics Lecture : p he biological problem p lobal alignment p Local alignment p Multiple alignment 6 Background: comparative genomics p Basic question in biology: what properties
Introduction to Bioinformatics Lecture : p he biological problem p lobal alignment p Local alignment p Multiple alignment 6 Background: comparative genomics p Basic question in biology: what properties
EECS730: Introduction to Bioinformatics
 EECS730: Introduction to Bioinformatics Lecture 03: Edit distance and sequence alignment Slides adapted from Dr. Shaojie Zhang (University of Central Florida) KUMC visit How many of you would like to attend
EECS730: Introduction to Bioinformatics Lecture 03: Edit distance and sequence alignment Slides adapted from Dr. Shaojie Zhang (University of Central Florida) KUMC visit How many of you would like to attend
Background: comparative genomics. Sequence similarity. Homologs. Similarity vs homology (2) Similarity vs homology. Sequence Alignment (chapter 6)
 Similarity vs homology. Sequence Alignment (chapter 6)") Sequence lignment (chapter ) he biological problem lobal alignment Local alignment Multiple alignment Background: comparative genomics Basic question in biology: what properties are shared among organisms?
Sequence lignment (chapter ) he biological problem lobal alignment Local alignment Multiple alignment Background: comparative genomics Basic question in biology: what properties are shared among organisms?
Scoring Matrices. Shifra Ben-Dor Irit Orr
 Scoring Matrices Shifra Ben-Dor Irit Orr Scoring matrices Sequence alignment and database searching programs compare sequences to each other as a series of characters. All algorithms (programs) for comparison
Scoring Matrices Shifra Ben-Dor Irit Orr Scoring matrices Sequence alignment and database searching programs compare sequences to each other as a series of characters. All algorithms (programs) for comparison
Bioinformatics. Scoring Matrices. David Gilbert Bioinformatics Research Centre
 Bioinformatics Scoring Matrices David Gilbert Bioinformatics Research Centre www.brc.dcs.gla.ac.uk Department of Computing Science, University of Glasgow Learning Objectives To explain the requirement
Bioinformatics Scoring Matrices David Gilbert Bioinformatics Research Centre www.brc.dcs.gla.ac.uk Department of Computing Science, University of Glasgow Learning Objectives To explain the requirement
Practical Bioinformatics
 5/2/2017 Dictionaries d i c t i o n a r y = { A : T, T : A, G : C, C : G } d i c t i o n a r y [ G ] d i c t i o n a r y [ N ] = N d i c t i o n a r y. h a s k e y ( C ) Dictionaries g e n e t i c C o
5/2/2017 Dictionaries d i c t i o n a r y = { A : T, T : A, G : C, C : G } d i c t i o n a r y [ G ] d i c t i o n a r y [ N ] = N d i c t i o n a r y. h a s k e y ( C ) Dictionaries g e n e t i c C o
Whole Genome Alignments and Synteny Maps
 Whole Genome Alignments and Synteny Maps IINTRODUCTION It was not until closely related organism genomes have been sequenced that people start to think about aligning genomes and chromosomes instead of
Whole Genome Alignments and Synteny Maps IINTRODUCTION It was not until closely related organism genomes have been sequenced that people start to think about aligning genomes and chromosomes instead of
Advanced topics in bioinformatics
 Feinberg Graduate School of the Weizmann Institute of Science Advanced topics in bioinformatics Shmuel Pietrokovski & Eitan Rubin Spring 2003 Course WWW site: http://bioinformatics.weizmann.ac.il/courses/atib
Feinberg Graduate School of the Weizmann Institute of Science Advanced topics in bioinformatics Shmuel Pietrokovski & Eitan Rubin Spring 2003 Course WWW site: http://bioinformatics.weizmann.ac.il/courses/atib
InDel 3-5. InDel 8-9. InDel 3-5. InDel 8-9. InDel InDel 8-9
 Lecture 5 Alignment I. Introduction. For sequence data, the process of generating an alignment establishes positional homologies; that is, alignment provides the identification of homologous phylogenetic
Lecture 5 Alignment I. Introduction. For sequence data, the process of generating an alignment establishes positional homologies; that is, alignment provides the identification of homologous phylogenetic
BIOINFORMATICS: An Introduction
 BIOINFORMATICS: An Introduction What is Bioinformatics? The term was first coined in 1988 by Dr. Hwa Lim The original definition was : a collective term for data compilation, organisation, analysis and
BIOINFORMATICS: An Introduction What is Bioinformatics? The term was first coined in 1988 by Dr. Hwa Lim The original definition was : a collective term for data compilation, organisation, analysis and
Introduction to protein alignments
 Introduction to protein alignments Comparative Analysis of Proteins Experimental evidence from one or more proteins can be used to infer function of related protein(s). Gene A Gene X Protein A compare
Introduction to protein alignments Comparative Analysis of Proteins Experimental evidence from one or more proteins can be used to infer function of related protein(s). Gene A Gene X Protein A compare
Similarity searching summary (2)
") Similarity searching / sequence alignment summary Biol4230 Thurs, February 22, 2016 Bill Pearson wrp@virginia.edu 4-2818 Pinn 6-057 What have we covered? Homology excess similiarity but no excess similarity
Similarity searching / sequence alignment summary Biol4230 Thurs, February 22, 2016 Bill Pearson wrp@virginia.edu 4-2818 Pinn 6-057 What have we covered? Homology excess similiarity but no excess similarity
Chapter 5. Proteomics and the analysis of protein sequence Ⅱ
 Proteomics Chapter 5. Proteomics and the analysis of protein sequence Ⅱ 1 Pairwise similarity searching (1) Figure 5.5: manual alignment One of the amino acids in the top sequence has no equivalent and
Proteomics Chapter 5. Proteomics and the analysis of protein sequence Ⅱ 1 Pairwise similarity searching (1) Figure 5.5: manual alignment One of the amino acids in the top sequence has no equivalent and
Bioinformatics for Computer Scientists (Part 2 Sequence Alignment) Sepp Hochreiter
 Sepp Hochreiter") Bioinformatics for Computer Scientists (Part 2 Sequence Alignment) Institute of Bioinformatics Johannes Kepler University, Linz, Austria Sequence Alignment 2. Sequence Alignment Sequence Alignment 2.1
Bioinformatics for Computer Scientists (Part 2 Sequence Alignment) Institute of Bioinformatics Johannes Kepler University, Linz, Austria Sequence Alignment 2. Sequence Alignment Sequence Alignment 2.1
Similarity or Identity? When are molecules similar?
 Similarity or Identity? When are molecules similar? Mapping Identity A -> A T -> T G -> G C -> C or Leu -> Leu Pro -> Pro Arg -> Arg Phe -> Phe etc If we map similarity using identity, how similar are
Similarity or Identity? When are molecules similar? Mapping Identity A -> A T -> T G -> G C -> C or Leu -> Leu Pro -> Pro Arg -> Arg Phe -> Phe etc If we map similarity using identity, how similar are
Sequence Analysis '17 -- lecture 7
 Sequence Analysis '17 -- lecture 7 Significance E-values How significant is that? Please give me a number for......how likely the data would not have been the result of chance,......as opposed to......a
Sequence Analysis '17 -- lecture 7 Significance E-values How significant is that? Please give me a number for......how likely the data would not have been the result of chance,......as opposed to......a
Fundamentals of database searching
 Fundamentals of database searching Aligning novel sequences with previously characterized genes or proteins provides important insights into their common attributes and evolutionary origins. The principles
Fundamentals of database searching Aligning novel sequences with previously characterized genes or proteins provides important insights into their common attributes and evolutionary origins. The principles
BLAST: Target frequencies and information content Dannie Durand
 Computational Genomics and Molecular Biology, Fall 2016 1 BLAST: Target frequencies and information content Dannie Durand BLAST has two components: a fast heuristic for searching for similar sequences
Computational Genomics and Molecular Biology, Fall 2016 1 BLAST: Target frequencies and information content Dannie Durand BLAST has two components: a fast heuristic for searching for similar sequences
Substitution matrices
 Introduction to Bioinformatics Substitution matrices Jacques van Helden Jacques.van-Helden@univ-amu.fr Université d Aix-Marseille, France Lab. Technological Advances for Genomics and Clinics (TAGC, INSERM
Introduction to Bioinformatics Substitution matrices Jacques van Helden Jacques.van-Helden@univ-amu.fr Université d Aix-Marseille, France Lab. Technological Advances for Genomics and Clinics (TAGC, INSERM
Sequence Analysis 17: lecture 5. Substitution matrices Multiple sequence alignment
 Sequence Analysis 17: lecture 5 Substitution matrices Multiple sequence alignment Substitution matrices Used to score aligned positions, usually of amino acids. Expressed as the log-likelihood ratio of
Sequence Analysis 17: lecture 5 Substitution matrices Multiple sequence alignment Substitution matrices Used to score aligned positions, usually of amino acids. Expressed as the log-likelihood ratio of
Chapter 7: Rapid alignment methods: FASTA and BLAST
 Chapter 7: Rapid alignment methods: FASTA and BLAST The biological problem Search strategies FASTA BLAST Introduction to bioinformatics, Autumn 2007 117 BLAST: Basic Local Alignment Search Tool BLAST (Altschul
Chapter 7: Rapid alignment methods: FASTA and BLAST The biological problem Search strategies FASTA BLAST Introduction to bioinformatics, Autumn 2007 117 BLAST: Basic Local Alignment Search Tool BLAST (Altschul
BLAST: Basic Local Alignment Search Tool
 .. CSC 448 Bioinformatics Algorithms Alexander Dekhtyar.. (Rapid) Local Sequence Alignment BLAST BLAST: Basic Local Alignment Search Tool BLAST is a family of rapid approximate local alignment algorithms[2].
.. CSC 448 Bioinformatics Algorithms Alexander Dekhtyar.. (Rapid) Local Sequence Alignment BLAST BLAST: Basic Local Alignment Search Tool BLAST is a family of rapid approximate local alignment algorithms[2].
Local Alignment: Smith-Waterman algorithm
 Local Alignment: Smith-Waterman algorithm Example: a shared common domain of two protein sequences; extended sections of genomic DNA sequence. Sensitive to detect similarity in highly diverged sequences.
Local Alignment: Smith-Waterman algorithm Example: a shared common domain of two protein sequences; extended sections of genomic DNA sequence. Sensitive to detect similarity in highly diverged sequences.
Exercise 5. Sequence Profiles & BLAST
 Exercise 5 Sequence Profiles & BLAST 1 Substitution Matrix (BLOSUM62) Likelihood to substitute one amino acid with another Figure taken from https://en.wikipedia.org/wiki/blosum 2 Substitution Matrix (BLOSUM62)
Exercise 5 Sequence Profiles & BLAST 1 Substitution Matrix (BLOSUM62) Likelihood to substitute one amino acid with another Figure taken from https://en.wikipedia.org/wiki/blosum 2 Substitution Matrix (BLOSUM62)
Statistical Distributions of Optimal Global Alignment Scores of Random Protein Sequences
 BMC Bioinformatics This Provisional PDF corresponds to the article as it appeared upon acceptance. The fully-formatted PDF version will become available shortly after the date of publication, from the
BMC Bioinformatics This Provisional PDF corresponds to the article as it appeared upon acceptance. The fully-formatted PDF version will become available shortly after the date of publication, from the
Lecture 4: Evolutionary models and substitution matrices (PAM and BLOSUM).
.") 1 Bioinformatics: In-depth PROBABILITY & STATISTICS Spring Semester 2011 University of Zürich and ETH Zürich Lecture 4: Evolutionary models and substitution matrices (PAM and BLOSUM). Dr. Stefanie Muff
1 Bioinformatics: In-depth PROBABILITY & STATISTICS Spring Semester 2011 University of Zürich and ETH Zürich Lecture 4: Evolutionary models and substitution matrices (PAM and BLOSUM). Dr. Stefanie Muff
A profile-based protein sequence alignment algorithm for a domain clustering database
 A profile-based protein sequence alignment algorithm for a domain clustering database Lin Xu,2 Fa Zhang and Zhiyong Liu 3, Key Laboratory of Computer System and architecture, the Institute of Computing
A profile-based protein sequence alignment algorithm for a domain clustering database Lin Xu,2 Fa Zhang and Zhiyong Liu 3, Key Laboratory of Computer System and architecture, the Institute of Computing
Giri Narasimhan. CAP 5510: Introduction to Bioinformatics. ECS 254; Phone: x3748
 CAP 5510: Introduction to Bioinformatics Giri Narasimhan ECS 254; Phone: x3748 giri@cis.fiu.edu www.cis.fiu.edu/~giri/teach/bioinfs07.html 1/23/07 CAP5510 1 Genomic Databases Entrez Portal at National
CAP 5510: Introduction to Bioinformatics Giri Narasimhan ECS 254; Phone: x3748 giri@cis.fiu.edu www.cis.fiu.edu/~giri/teach/bioinfs07.html 1/23/07 CAP5510 1 Genomic Databases Entrez Portal at National
Introduction to Sequence Alignment. Manpreet S. Katari
 Introduction to Sequence Alignment Manpreet S. Katari 1 Outline 1. Global vs. local approaches to aligning sequences 1. Dot Plots 2. BLAST 1. Dynamic Programming 3. Hash Tables 1. BLAT 4. BWT (Burrow Wheeler
Introduction to Sequence Alignment Manpreet S. Katari 1 Outline 1. Global vs. local approaches to aligning sequences 1. Dot Plots 2. BLAST 1. Dynamic Programming 3. Hash Tables 1. BLAT 4. BWT (Burrow Wheeler
Pairwise alignment. 2.1 Introduction GSAQVKGHGKKVADALTNAVAHVDDMPNALSALSD----LHAHKL
 2 Pairwise alignment 2.1 Introduction The most basic sequence analysis task is to ask if two sequences are related. This is usually done by first aligning the sequences (or parts of them) and then deciding
2 Pairwise alignment 2.1 Introduction The most basic sequence analysis task is to ask if two sequences are related. This is usually done by first aligning the sequences (or parts of them) and then deciding
Genomics and bioinformatics summary. Finding genes -- computer searches
 Genomics and bioinformatics summary 1. Gene finding: computer searches, cdnas, ESTs, 2. Microarrays 3. Use BLAST to find homologous sequences 4. Multiple sequence alignments (MSAs) 5. Trees quantify sequence
Genomics and bioinformatics summary 1. Gene finding: computer searches, cdnas, ESTs, 2. Microarrays 3. Use BLAST to find homologous sequences 4. Multiple sequence alignments (MSAs) 5. Trees quantify sequence