Supporting Information: Highly Sampled Tetranucleotide and Tetraloop Motifs Enable Evaluation of Common RNA Force Fields
|
|
|
- Emil Gardner
- 5 years ago
- Views:
Transcription
1 Supporting Information: Highly Sampled Tetranucleotide and Tetraloop Motifs Enable Evaluation of Common RNA Force Fields Christina Bergonzo, Niel M. Henriksen,, Daniel R. Roe, Thomas E. Cheatham III * Department of Medicinal Chemistry, College of Pharmacy, University of Utah, Salt Lake City, Utah 84112, United States Current Address: Skaggs School of Pharmacy and Pharmaceutical Sciences, University of California San Diego, 9500 Gilman Drive, La Jolla, California , United States Contents Supporting Figure 1: Loop region overlay of the final trajectory frames from the Stable-15 and Unstable-5 subsets on the NMR structure Supporting Table 1: Detailed cluster analysis for GACC Supporting Figure 2: Representative GACC structures Supporting Table 2: Detailed cluster analysis for CCCC Supporting Figure 3: Representative CCCC structures... 7 Supporting Table 3: Detailed NOE analysis for CCCC C36 top cluster trajectory Supporting Figure 4: Overlap of representative structure for most populated clusters in Charmm K ensembles of GACC and CCCC tetranucleotides Supporting Figure 5: C36 backbone dihedral angle distributions for all structures in the top cluster for GACC and CCCC tetranucleotide Supporting Table 4: Hbond analysis for ff12 intercalated cluster trajectories Supporting Figure 6: Overlap of intercalated representative structures from GACC and CCCC Supporting Figure 7a: Cluster correlation analysis for UUCG M-REMD simulations Supporting Figure 7b: Cluster correlation analysis for UUCG M-REMD simulations Supporting Figure 8: Close up of Cluster 2 from combined cluster analysis at 277 K coordinating a K+ ion Supporting Figure 9: RMSD to ladder reference
2 Supporting Figure 10: Unrestrained MD simulations starting from ladder representative structure do not transition back to a helix in 500 ns Supporting Figure 11: Combined clustering results for UUCG tetraloop simulations at 300 K Supporting Figure 12: Cluster population vs. Time and cluster population correlation of unrestrained M-REMD UUCG folding simulations Supporting Text: Anton Simulation Details Supporting Table 5: amd parameters used in M-REMD simulations of GACC with ff99 + χ Yil Supporting CPPTRAJ Script 1: Clustering command for analysis of GACC tetranucleotide simulations Supporting CPPTRAJ Script 2: Clustering command for analysis of CCCC tetranucleotide simulations Supporting CPPTRAJ Script 3. UUCG Combined Cluster Analysis Supporting File: Frcmod.ff99CG force modification file for building ff99 + Chen-Garcia LJ and dihedral modifications Supporting File: Parmed.py script for modifying off-diagonal LJ terms for ff99 + Chen-Garcia Supporting File: Frcmod.vdW force modification file for building ff12 + vdw all modifications Supporting File: Parmed.py script for modifying O4 atom from ff12 + vdw all to default ff12, generating ff12 + vdw bb modifications
 and Unstable-5 (right) subsets on the NMR structure (model 1 PDB: 2KOC). Loop region consists of residues 5-10.")
3 Supporting Figure 1: Loop region overlay of the final trajectory frames from the Stable-15 and Unstable-5 subsets on the NMR structure. Loop region overlay of the final trajectory frames from the Stable-15 (left) and Unstable-5 (right) subsets on the NMR structure (model 1 PDB: 2KOC). Loop region consists of residues The simulation structures are colored black. The NMR structure is magenta. Overlay was performed by RMS fitting to the NMR structure using residues 5 and 10 3
4 Supporting Table 1: Detailed cluster analysis for GACC. ff12+ ff12+ ff12+ ff12+ ff99+ ff99+ ff99+x ff99+ ff12 ff12 vdwal vdwal vdwb vdwb C36 C36 CG CG yil run Xyil 277 K Replicas run 1 run 2 l run 1 l run 2 b run1 b run2 run 1 run 2 run 1 run 2 1 run 2 Experimental Structures A-form-major A-form-minor Alternate NMR Major & Minor Conformations A-form-major-1-syn General A-form major A-form-minor-1-syn A-form-minor-4-syn General A-form minor Conformations Classified as Intercalated Intercalated-anti Intercalated-2-anti-extruded Intercalated-syn Intercalated-syn-3-extruded Intercalated-syn-2-extruded Pre-intercalated General Intercalated Conformations Classified as Inverted Inverted-anti Inverted-anti_ Inverted-syn Inverted-syn-conf Conformations Classified as Base Paired 1_3-basepair _4-basepair _4-basepair-conf General Basepaired Conformations Classified as "Other" 1_3-stack _3-2_4-stack _4-stack (1_4-2_3) extruded extruded _hbond Backbone hbonds General hbonds

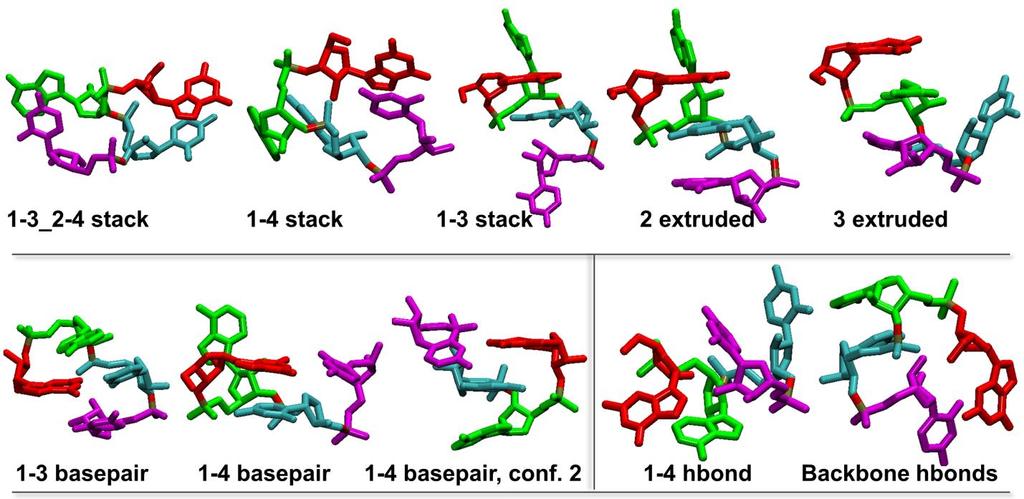
5 Supporting Figure 2: Representative GACC structures. 5
6 Supporting Table 2: Detailed cluster analysis for CCCC. RMSD to A-form (Å) ff12 run 1 ff12 run 2 ff12+ vdwall run 1 ff12+ vdwall run 2 ff12+ ff12+ ff99+ vdwbb vdwbb CG run1 run2 run 1 ff99+ CG run 2 C36 run 1 C36 run 2 Representative StructureCluster Intercalated 2-extruded % 86.0% 79.3% 80.7% 82.7% 83.8% 85.3% 87.0% 2.8% 3.0% A-form hbonded % 3.4% 5.5% 5.8% 6.7% 6.5% 1.1% 1.1% 58.4% 58.2% A-form rotated % 3.1% 3.7% 3.5% 3.8% 3.9% 1.2% 1.0% 3.6% 3.4% Inverted % 0.2% 0.6% 0.7% 0.6% 0.5% 0.3% 0.2% 3.6% 3.2% 2-3-4_stack % 0.0% 0.0% 0.0% 0.0% 0.0% 0.0% 0.0% 1.9% 2.4% 1-3_stack % 0.3% 0.6% 0.3% 0.2% 0.2% 0.9% 0.8% 0.0% 0.0% 1-3_2-4_stack % 0.3% 1.2% 0.3% 0.3% 0.2% 0.2% 0.1% 0.0% 0.0% 1-3_2-4_stack, 1 syn % 0.2% 0.7% 0.6% 0.2% 0.1% 0.5% 0.5% 0.0% 0.0% Above: dbscan minpoints 25 epsilon 0.9 rms :1@O2,H5,C1',P,:2@O2,H5,C1',P,:3@O2,H5,C1',P,:4@O2,H5,C1',P Intercalated % 53.7% 39.8% 43.1% 52.6% 52.1% 20.3% 20.5% 1.3% 1.5% Intercalated 2-extruded % 28.8% 34.2% 32.3% 26.7% 28.1% 59.8% 61.1% 0.0% 0.0% A-form hbonded % 0.6% 1.0% 0.8% 0.8% 0.8% 0.2% 0.3% 44.7% 43.6% Intercalated 4-dangle % 1.1% 1.7% 1.7% 1.4% 1.5% 0.7% 0.8% 0.3% 0.2% A-form hbonded - rotate % 0.7% 1.0% 1.4% 1.5% 1.7% 0.1% 0.1% 0.1% 0.1% Pre-inverted/2-3-4_stack % 0.0% 0.0% 0.0% 0.0% 0.0% 0.0% 0.0% 1.7% 1.6% Above: dbscan minpoints 25 epsilon 0.5 rms :1@O2,H5,C1',P,:2@O2,H5,C1',P,:3@O2,H5,C1',P,:4@O2,H5,C1',P Intercalated 2-extruded % 87.6% 81.5% 83.1% 83.9% 85.0% 88.3% 90.1% 5.8% 5.9% A-form hbonded % 4.8% 7.2% 7.0% 7.9% 7.9% 1.5% 1.6% 62.5% 62.1% A-form 4 dangle % 2.4% 3.1% 3.2% 2.7% 2.8% 2.7% 2.2% 9.9% 10.1% A-form/NMR % 2.3% 2.7% 2.6% 2.3% 2.0% 1.6% 1.3% 8.7% 8.2% Pre-inverted/2-3-4_stack % 0.9% 1.4% 1.4% 1.1% 0.9% 1.1% 1.1% 11.4% 11.4% 1-2-4_stack_3-extruded % 0.9% 1.1% 1.0% 0.8% 0.7% 1.6% 1.0% 1.0% 1.1% H-bonded % 0.4% 0.9% 0.6% 0.6% 0.3% 2.0% 1.7% 0.7% 1.2% 1-3_2-4_stack, 1 syn % 0.7% 2.3% 1.2% 0.7% 0.4% 1.3% 1.1% 0.2% 0.1% Above: kmeans clusters 8 rms :1@O2,H5,C1',P,:2@O2,H5,C1',P,:3@O2,H5,C1',P,:4@O2,H5,C1',P 6


7 Supporting Figure 3: Representative CCCC structures. 7
8 Supporting Table 3: Detailed NOE analysis for CCCC C36 top cluster trajectory. Analysis performed using Cpptraj to calculate distances with r6 averaging. Average values which violate NMR bounds are shaded red. All values are in units of Å. NOE NMR value NMR lower NMR upper C36 Run 1 C36 Run 2 C36 Avg C36 StDev :1@H2'-:2@H1' :1@H2'-:2@H5' :1@H5-:2@H :1@H6-:1@H1' :1@H6-:1@H2' :1@H6-:2@H :2@H5-:1@H2' :2@H5-:3@H :2@H6-:1@H2' :2@H6-:2@H1' :2@H6-:2@H5'' :3@H3'-:4@H2' :3@H5-:4@H :3@H6-:3@H2' :3@H6-:3@H3' :3@H6-:3@H5'' :3@H6-:4@H2' :3@H6-:4@H :4@H1'-:3@H2' :4@H5-:3@H2' :4@H5-:3@H3' :4@H6-:3@H2' :4@H6-:3@H3' :4@H6-:4@H2' :4@H6-:4@H3' :4@H6-:4@H4' :4@H6-:4@H5''

9 Supporting Figure 4: Overlap of representative structure for most populated clusters in Charmm K ensembles of GACC and CCCC tetranucleotides. 9

10 Supporting Figure 5: C36 backbone dihedral angle distributions for all structures in the top cluster for GACC and CCCC tetranucleotide. Values reported in the literature for each base are denoted by grey lines and labeled α-ζ. These are the expected values for the ensembles. The histogram peaks denote the actual dihedral angles adopted in the top cluster for C36. Though hard to see, two runs per tetranucleotide are shown as solid and dashed lines this is perhaps most noticeable in GACC residue G1, zeta backbone dihedral. 10

11 Supporting Table 4: Hbond analysis for ff12 intercalated cluster trajectories. Equivalent hydrogen bonds are aligned between CCCC (left) and GACC (right). Italicized red hydrogen bonds are less than 1% occupied, but were included if occupied at > 1% in the other system. CCCC Run 1 ff12, cluster 0, >1% occupancy GACC Run 1 ff12, cluster 2, >1% occupancy Acceptor DonorH Fraction AvgDist AvgAng Acceptor DonorH Fraction AvgDist AvgAng C_3@OP2 C5_1@HO5' C_3@OP2 G5_1@HO5' C_3@OP2 C3_4@HO3' C_3@OP2 C3_4@HO3' C_3@OP1 C_2@HO2' C_3@OP1 A_2@HO2' C3_4@OP1 C5_1@H C3_4@OP1 C5_1@H C_2@OP1 C_3@H A_2@OP1 C_3@H C_2@OP1 C_3@H A_2@OP1 C_3@H C_3@OP1 C3_4@HO3' C_3@OP1 C3_4@HO3' C_3@OP2 C3_4@HO2' C_3@OP2 C3_4@HO2' C_2@OP1 C5_1@HO2' A_2@OP1 G5_1@HO2' C_2@O3' C5_1@HO5' A_2@O3' G5_1@HO5' C3_4@O2 C5_1@HO5' C3_4@O2 G5_1@HO5' C3_4@OP1 C_3@HO2' C3_4@OP1 C_3@HO2' C3_4@O2' C5_1@HO5' C3_4@O2' G5_1@HO5' C_3@OP1 C3_4@HO2' C_3@OP1 C3_4@HO2' C5_1@O5' C3_4@HO3' G5_1@O5' C3_4@HO3' C_3@O5' C3_4@HO3' C_3@O5' C3_4@HO3' C_3@O4' C_2@HO2' C_3@O4' A_2@HO2' C_3@O5' C_2@HO2' C_3@O5' A_2@HO2' C_3@O5' C5_1@HO5' C_3@O5' G5_1@HO5' C5_1@O5' C3_4@HO2' G5_1@O5' C3_4@HO2' Supporting Figure 6: Overlap of intercalated representative structures from GACC and CCCC. RMSD of GACC to CCCC is 0.36 Å (mass weighted backbone heavy atoms only). 11
12 Supporting Figure 7a: Cluster correlation analysis for UUCG M- REMD simulations. Last 1 μs of each 277 K and 300 K replica are used. Two independent M-REMD simulations were performed for each force field and their cluster populations show convergence between runs. Top: AMBER ff12; Bottom: AMBER ff12 + vdw all. 12
13 Supporting Figure 7b: Cluster correlation analysis for UUCG M- REMD simulations. Last 1 μs of each 277 K and 300 K replica are used. Two independent M-REMD simulations were performed for each force field and their cluster populations show convergence between runs. Top: AMBER ff12 + vdw bb ; Bottom: AMBER ff99 + Chen-Garcia. 13

14 Supporting Figure 8: Close up of Cluster 2 from combined cluster analysis at 277 K coordinating a K+ ion. 14
15 Supporting Figure 9: RMSD to ladder reference. Average RMSD to a ladder-like stem reference structure of the last 1 μs of 277 K replicas for AMBER ff12, AMBER ff12 + vdw all, AMBER ff12 + vdw bb, and ff99 + Chen-Garcia systems. Reference structure is top cluster of 277 K combined cluster analysis, shown in main text Figure 4. Error bars represent standard deviation between two independent runs. 15
 to the ladder reference structure at 277 K (left) and at 300 K (right).")
16 Supporting Figure 10: Unrestrained MD simulations starting from ladder representative structure do not transition back to a helix in 500 ns. Top row: RMSD of the stem (:1-3,8-10) to the ladder reference structure at 277 K (left) and at 300 K (right). Bottom Row: RMSD of the stem to the NMR reference structure at 277 K (left) and at 300 K (right). It is important to note that two of the 300 K simulations move away from the ladder-like reference, but they do not move towards the NMR reference. 16

17 Supporting Figure 11: Combined clustering results for UUCG tetraloop simulations at 300 K. Cluster number is shown on the x-axis and percentage of each simulation s 300 K ensemble is shown on the y-axis. Representative structures are shown below the bar graph. The native structure is found in the 31 st cluster, accounting for 0.02% of structures in the ff12 ensemble, 0.025% of structures in the ff12 + vdw all ensemble, 0.07% of structures in the ff12 + vdw bb ensemble and 0.97% of structures in ff99 + Chen-Garcia. 17
18 Supporting Figure 12: Cluster population vs. Time and cluster population correlation of unrestrained M-REMD UUCG folding simulations. Top: Cluster population vs. Time of unrestrained M-REMD UUCG folding simulations from folded and unfolded starting structures. Bottom: Cluster Population correlation between the last 500 ns of unrestrained M-REMD simulations from folded and unfolded do not sample similar structure populations and remain unconverged. 18
19 Supporting Text: Anton Simulation Details Production dynamics were performed on Anton using versions and Conversion of the Amber parameter and topology files into Anton appropriate formats was enabled by the supplied amber_topnrst2cms.py script on the computer anton.psc.edu with Desmond (Bowers et al. 2006) to create the needed *.cms file. A bug in the amber_topnrst2cms.py script erroneously assigned zero mass to C5 atoms when converting from Amber topologies, so the files generated were hand-edited to fix the mass, and further checked to make sure the resulting *.cms file contained the correct Amber ff99 + parmbsc0 force field parameters. The Anton guess_chem, refinesigma, and subboxer programs were used to set up inputs for Anton and a series of anton_run commands performed to do the MD simulations. For the Anton runs, constant 300 K temperature and 1 bar pressure with weak coupling using a coupling time tau of 10.0, a maximum and minimum velocity scaling of 1.2 and 0.85, and a maximum and minimum expansion per step of 1.1 and 0.95 and kappa of 4.5x10-5 were imposed. The integration time step used was set to 2 fs and max_strain was set to 0.15 performing RESPA on the long-range non-bonded interactions every third step.(tuckerman et al. 1992; Procacci et al. 1996) Bowers KJ, Chow E, Xu H, Dror RO, Eastwood MP, Gregersen BA, Klepeis JL, Kolossváry I, Moraes MA, Sacerdoti FD, et al Scalable algorithms for molecular dynamics simulations on commodity clusters. In Proceedings of the ACM/IEEE Conference on Supercomputing, pp , Tampa. Procacci P, Darden T, Marchi M A very fast molecular dynamics method to simulate biomolecular systems with realistic electrostatic interactions. J Phys Chem 100: Tuckerman M, Berne BJ, Martyna GJ Reversible multiple time scale molecular dynamics. J Chem Phys 97:
20 Supporting Table 5: amd parameters used in M-REMD simulations of GACC with ff99 + χyil. Hamiltonian Ethreshold (kcal/mol) α (kcal/mol) 1 No Boost No Boost
21 Supporting CPPTRAJ Script 1: Clustering command for analysis of GACC tetranucleotide simulations. parm../../nowat.gacc.parm7 trajin../nowat.nc.0 #Set sieve so there are 10,000 frames during 1st pass cluster dbscan minpoints 25 epsilon 0.9 \ rms :1@N2,O6,C1',P,:2@H2,N6,C1',P,:3@O2,H5,C1',P,:4@O2,H5,C1',P \ sievetoframe sieve 3 out cvt.dat summary summary.dat \ repout rep repfmt pdb cpopvtime cpop.agr normframe Supporting CPPTRAJ Script 2: Clustering command for analysis of CCCC tetranucleotide simulations. parm../nowat.cccc.topo trajin../nowat.277k.cccc.nc cluster dbscan minpoints 25 epsilon 0.5 \ rms :1@O2,H5,C1',P,:2@O2,H5,C1',P,:3@O2,H5,C1',P,:4@O2,H5,C1',P \ sievetoframe sieve 10 out cvt.0.5.dat summary summary.0.5.dat \ clusterout cluster.nc clusterfmt cdf repout rep.new repfmt pdb \ cpopvtime cpop.0.5.agr normframe Supporting CPPTRAJ Script 3. UUCG Combined Cluster Analysis parm../nowat.uucgrest.parm7 trajin../trn-rest/2us/nowat.2us.300k.nc trajin../hmr-rest1/2us/nowat.2us.300k.nc trajin../ff12-run1/nowat.2.5us.300k.nc trajin../ff12-run2/nowat.2.5us.300k.nc trajin../ff99-chengar-run1/2us/nowat.cg.2us.300k.nc trajin../ff99-chengar-run2/nowat.2us.300k.nc trajin../ff12dacbb-run1/nowat.300k.nc trajin../ ff12dacbb-run2/nowat.300k.2us.nc cluster dbscan minpoints 25 epsilon 1.5 rms mass :1-10 sievetoframe \ sieve 60 summaryhalf split300.dat \ splitframe ,200000,300000,400000, \ singlerepout singlerep300.mdcrd singlerepfmt crd \ summary Replica0.summary.300.dat out cvt.300.dat 21
22 Supporting File: Frcmod.ff99CG force modification file for building ff99 + Chen-Garcia LJ and dihedral modifications. glycosdic bond torsion modifications to parm99 from Chen and Garcia (PNAS, 2013, 110, 16820) MASS CA CB CK CM CQ CP CS N* NA N NC NB O DIHE OS-CT-N*-CK OS-CT-N*-CM OS-CT-N*-CM NONB CA CB CK CM CQ CP CS N* NA N NC NB O Supporting File: Parmed.py script for modifying off-diagonal LJ terms for ff99 + Chen-Garcia. changeljpair parmout ff99chengarcia.parm7 go 22
23 Supporting File: Frcmod.vdW force modification file for building ff12 + vdwall modifications. Steinbrecher-Latzer-Case Phosphate Oxygen parameters (JCTC, 8, , 2012) MASS O OS NONB O OS Supporting File: Parmed.py script for modifying O4 atom from ff12 + vdwall to default ff12, generating ff12 + vdwbb modifications. radius epsilon parmout ff12vdwbb.parm7 go 23
SANDRO BOTTARO, PAVEL BANÁŠ, JIŘÍ ŠPONER, AND GIOVANNI BUSSI
 FREE ENERGY LANDSCAPE OF GAGA AND UUCG RNA TETRALOOPS. SANDRO BOTTARO, PAVEL BANÁŠ, JIŘÍ ŠPONER, AND GIOVANNI BUSSI Contents 1. Supplementary Text 1 2 Sample PLUMED input file. 2. Supplementary Figure
FREE ENERGY LANDSCAPE OF GAGA AND UUCG RNA TETRALOOPS. SANDRO BOTTARO, PAVEL BANÁŠ, JIŘÍ ŠPONER, AND GIOVANNI BUSSI Contents 1. Supplementary Text 1 2 Sample PLUMED input file. 2. Supplementary Figure
Assessing the current state of AMBER force field modifications for DNA.
 Assessing the current state of AMBER force field modifications for DNA. Supporting Information. Table S1 Average structural parameters for the DDD system with the ε/ζ OL1 +χ OL4 force field modification.
Assessing the current state of AMBER force field modifications for DNA. Supporting Information. Table S1 Average structural parameters for the DDD system with the ε/ζ OL1 +χ OL4 force field modification.
Replica Exchange with Solute Scaling: A More Efficient Version of Replica Exchange with Solute Tempering (REST2)
") pubs.acs.org/jpcb Replica Exchange with Solute Scaling: A More Efficient Version of Replica Exchange with Solute Tempering (REST2) Lingle Wang, Richard A. Friesner, and B. J. Berne* Department of Chemistry,
pubs.acs.org/jpcb Replica Exchange with Solute Scaling: A More Efficient Version of Replica Exchange with Solute Tempering (REST2) Lingle Wang, Richard A. Friesner, and B. J. Berne* Department of Chemistry,
Enhancing Amber for Use on the Blue Waters High- Performance Computing Resource
 Enhancing Amber for Use on the Blue Waters High- Performance Computing Resource Daniel R. Roe a, Jason M. Swails b, Romelia Salomon-Ferrer c, Adrian E. Roitberg b, and Thomas E. Cheatham III a * a. Department
Enhancing Amber for Use on the Blue Waters High- Performance Computing Resource Daniel R. Roe a, Jason M. Swails b, Romelia Salomon-Ferrer c, Adrian E. Roitberg b, and Thomas E. Cheatham III a * a. Department
Routine access to millisecond timescale events with accelerated molecular dynamics
 Routine access to millisecond timescale events with accelerated molecular dynamics Levi C.T. Pierce, Romelia Salomon-Ferrer, Cesar Augusto F. de Oliveira #, J. Andrew McCammon #, Ross C. Walker * SUPPORTING
Routine access to millisecond timescale events with accelerated molecular dynamics Levi C.T. Pierce, Romelia Salomon-Ferrer, Cesar Augusto F. de Oliveira #, J. Andrew McCammon #, Ross C. Walker * SUPPORTING
Multidimensional Replica Exchange Molecular Dynamics Yields a Converged Ensemble of an RNA Tetranucleotide
 pubs.acs.org/jctc Terms of Use Multidimensional Replica Exchange Molecular Dynamics Yields a Converged Ensemble of an RNA Tetranucleotide Christina Bergonzo, Niel M. Henriksen, Daniel R. Roe, Jason M.
pubs.acs.org/jctc Terms of Use Multidimensional Replica Exchange Molecular Dynamics Yields a Converged Ensemble of an RNA Tetranucleotide Christina Bergonzo, Niel M. Henriksen, Daniel R. Roe, Jason M.
Supporting Information Quantitative characterization of the binding and unbinding of millimolar drug fragments with molecular dynamics simulations
 Supporting Information Quantitative characterization of the binding and unbinding of millimolar drug fragments with molecular dynamics simulations Simulation Details All binding simulations were run on
Supporting Information Quantitative characterization of the binding and unbinding of millimolar drug fragments with molecular dynamics simulations Simulation Details All binding simulations were run on
Supporting Material for. Microscopic origin of gating current fluctuations in a potassium channel voltage sensor
 Supporting Material for Microscopic origin of gating current fluctuations in a potassium channel voltage sensor J. Alfredo Freites, * Eric V. Schow, * Stephen H. White, and Douglas J. Tobias * * Department
Supporting Material for Microscopic origin of gating current fluctuations in a potassium channel voltage sensor J. Alfredo Freites, * Eric V. Schow, * Stephen H. White, and Douglas J. Tobias * * Department
Supplementary Information
 Supplementary Information Resveratrol Serves as a Protein-Substrate Interaction Stabilizer in Human SIRT1 Activation Xuben Hou,, David Rooklin, Hao Fang *,,, Yingkai Zhang Department of Medicinal Chemistry
Supplementary Information Resveratrol Serves as a Protein-Substrate Interaction Stabilizer in Human SIRT1 Activation Xuben Hou,, David Rooklin, Hao Fang *,,, Yingkai Zhang Department of Medicinal Chemistry
Time-dependence of key H-bond/electrostatic interaction distances in the sirna5-hago2 complexes... Page S14
 Supporting Information Probing the Binding Interactions between Chemically Modified sirnas and Human Argonaute 2 Using Microsecond Molecular Dynamics Simulations S. Harikrishna* and P. I. Pradeepkumar*
Supporting Information Probing the Binding Interactions between Chemically Modified sirnas and Human Argonaute 2 Using Microsecond Molecular Dynamics Simulations S. Harikrishna* and P. I. Pradeepkumar*
Dihedral Angles. Homayoun Valafar. Department of Computer Science and Engineering, USC 02/03/10 CSCE 769
 Dihedral Angles Homayoun Valafar Department of Computer Science and Engineering, USC The precise definition of a dihedral or torsion angle can be found in spatial geometry Angle between to planes Dihedral
Dihedral Angles Homayoun Valafar Department of Computer Science and Engineering, USC The precise definition of a dihedral or torsion angle can be found in spatial geometry Angle between to planes Dihedral
Supplementary Figures. Measuring Similarity Between Dynamic Ensembles of Biomolecules
 Supplementary Figures Measuring Similarity Between Dynamic Ensembles of Biomolecules Shan Yang, Loïc Salmon 2, and Hashim M. Al-Hashimi 3*. Department of Chemistry, University of Michigan, Ann Arbor, MI,
Supplementary Figures Measuring Similarity Between Dynamic Ensembles of Biomolecules Shan Yang, Loïc Salmon 2, and Hashim M. Al-Hashimi 3*. Department of Chemistry, University of Michigan, Ann Arbor, MI,
SUPPLEMENTAL MATERIAL
 SUPPLEMENTAL MATERIAL Systematic Coarse-Grained Modeling of Complexation between Small Interfering RNA and Polycations Zonghui Wei 1 and Erik Luijten 1,2,3,4,a) 1 Graduate Program in Applied Physics, Northwestern
SUPPLEMENTAL MATERIAL Systematic Coarse-Grained Modeling of Complexation between Small Interfering RNA and Polycations Zonghui Wei 1 and Erik Luijten 1,2,3,4,a) 1 Graduate Program in Applied Physics, Northwestern
Thomas E. Cheatham III
 NSF OCI-1036208: PRAC Hierarchical molecular dynamics sampling for assessing pathways and free energies of RNA catalysis, ligand binding, and conformational change. NEIS-P2 update, May 2013 Thomas E. Cheatham
NSF OCI-1036208: PRAC Hierarchical molecular dynamics sampling for assessing pathways and free energies of RNA catalysis, ligand binding, and conformational change. NEIS-P2 update, May 2013 Thomas E. Cheatham
Supporting Protocol This protocol describes the construction and the force-field parameters of the non-standard residue for the Ag + -site using CNS
 Supporting Protocol This protocol describes the construction and the force-field parameters of the non-standard residue for the Ag + -site using CNS CNS input file generatemetal.inp: remarks file generate/generatemetal.inp
Supporting Protocol This protocol describes the construction and the force-field parameters of the non-standard residue for the Ag + -site using CNS CNS input file generatemetal.inp: remarks file generate/generatemetal.inp
April, The energy functions include:
 REDUX A collection of Python scripts for torsion angle Monte Carlo protein molecular simulations and analysis The program is based on unified residue peptide model and is designed for more efficient exploration
REDUX A collection of Python scripts for torsion angle Monte Carlo protein molecular simulations and analysis The program is based on unified residue peptide model and is designed for more efficient exploration
2008 Biowerkzeug Ltd.
 2008 Biowerkzeug Ltd. 1 Contents Summary...3 1 Simulation...4 1.1 Setup...4 1.2 Output...4 2 Settings...5 3 Analysis...9 3.1 Setup...9 3.2 Input options...9 3.3 Descriptions...10 Please note that we cannot
2008 Biowerkzeug Ltd. 1 Contents Summary...3 1 Simulation...4 1.1 Setup...4 1.2 Output...4 2 Settings...5 3 Analysis...9 3.1 Setup...9 3.2 Input options...9 3.3 Descriptions...10 Please note that we cannot
Electronic Supplementary Information
 Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 28 Electronic Supplementary Information ph-dependent Cooperativity and Existence of
Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 28 Electronic Supplementary Information ph-dependent Cooperativity and Existence of
Nature Structural & Molecular Biology: doi: /nsmb Supplementary Figure 1
 Supplementary Figure 1 Resonance assignment and NMR spectra for hairpin and duplex A 6 constructs. (a) 2D HSQC spectra of hairpin construct (hp-a 6 -RNA) with labeled assignments. (b) 2D HSQC or SOFAST-HMQC
Supplementary Figure 1 Resonance assignment and NMR spectra for hairpin and duplex A 6 constructs. (a) 2D HSQC spectra of hairpin construct (hp-a 6 -RNA) with labeled assignments. (b) 2D HSQC or SOFAST-HMQC
Developing Monovalent Ion Parameters for the Optimal Point Charge (OPC) Water Model. John Dood Hope College
 Water Model. John Dood Hope College") Developing Monovalent Ion Parameters for the Optimal Point Charge (OPC) Water Model John Dood Hope College What are MD simulations? Model and predict the structure and dynamics of large macromolecules.
Developing Monovalent Ion Parameters for the Optimal Point Charge (OPC) Water Model John Dood Hope College What are MD simulations? Model and predict the structure and dynamics of large macromolecules.
Experimental and Computational Mutagenesis to Investigate the. Positioning of a General Base within an Enzyme Active Site
 Experimental and Computational Mutagenesis to Investigate the Positioning of a General Base within an Enzyme Active Site Jason P. Schwans, Philip Hanoian, Benjamin J. Lengerich, Fanny Sunden, Ana Gonzalez
Experimental and Computational Mutagenesis to Investigate the Positioning of a General Base within an Enzyme Active Site Jason P. Schwans, Philip Hanoian, Benjamin J. Lengerich, Fanny Sunden, Ana Gonzalez
Christine M. Isborn, Andreas W. Götz, Matthew A. Clark, Ross C. Walker, and Todd J. Martínez
 Supporting Information for Electronic Absorption Spectra from MM and ab initio QM/MM Molecular Dynamics: Environmental Effects on the Absorption Spectrum of Photoactive Yellow Protein Christine M. Isborn,
Supporting Information for Electronic Absorption Spectra from MM and ab initio QM/MM Molecular Dynamics: Environmental Effects on the Absorption Spectrum of Photoactive Yellow Protein Christine M. Isborn,
L718Q mutant EGFR escapes covalent inhibition by stabilizing. a non-reactive conformation of the lung cancer drug. osimertinib
 Electronic Supplementary Material (ESI) for Chemical Science. This journal is The Royal Society of Chemistry 2018 Electronic Supplementary Information (ESI) for L718Q mutant EGFR escapes covalent inhibition
Electronic Supplementary Material (ESI) for Chemical Science. This journal is The Royal Society of Chemistry 2018 Electronic Supplementary Information (ESI) for L718Q mutant EGFR escapes covalent inhibition
Solutions to Assignment #4 Getting Started with HyperChem
 Solutions to Assignment #4 Getting Started with HyperChem 1. This first exercise is meant to familiarize you with the different methods for visualizing molecules available in HyperChem. (a) Create a molecule
Solutions to Assignment #4 Getting Started with HyperChem 1. This first exercise is meant to familiarize you with the different methods for visualizing molecules available in HyperChem. (a) Create a molecule
Limitations of temperature replica exchange (T-REMD) for protein folding simulations
 for protein folding simulations") Limitations of temperature replica exchange (T-REMD) for protein folding simulations Jed W. Pitera, William C. Swope IBM Research pitera@us.ibm.com Anomalies in protein folding kinetic thermodynamic 322K
Limitations of temperature replica exchange (T-REMD) for protein folding simulations Jed W. Pitera, William C. Swope IBM Research pitera@us.ibm.com Anomalies in protein folding kinetic thermodynamic 322K
HIV-1 TAR RNA Spontaneously Undergoes Relevant Apo-to-Holo Conformational Transitions in Molecular Dynamics and Constrained Geometrical Simulations
 J. Chem. Inf. Model. 2010, 50, 1489 1501 1489 HIV-1 TAR RNA Spontaneously Undergoes Relevant Apo-to-Holo Conformational Transitions in Molecular Dynamics and Constrained Geometrical Simulations Simone
J. Chem. Inf. Model. 2010, 50, 1489 1501 1489 HIV-1 TAR RNA Spontaneously Undergoes Relevant Apo-to-Holo Conformational Transitions in Molecular Dynamics and Constrained Geometrical Simulations Simone
Supporting Information
 Supporting Information Constant ph molecular dynamics reveals ph-modulated binding of two small-molecule BACE1 inhibitors Christopher R. Ellis 1,, Cheng-Chieh Tsai 1,, Xinjun Hou 2, and Jana Shen 1, 1
Supporting Information Constant ph molecular dynamics reveals ph-modulated binding of two small-molecule BACE1 inhibitors Christopher R. Ellis 1,, Cheng-Chieh Tsai 1,, Xinjun Hou 2, and Jana Shen 1, 1
Unfolding CspB by means of biased molecular dynamics
 Chapter 4 Unfolding CspB by means of biased molecular dynamics 4.1 Introduction Understanding the mechanism of protein folding has been a major challenge for the last twenty years, as pointed out in the
Chapter 4 Unfolding CspB by means of biased molecular dynamics 4.1 Introduction Understanding the mechanism of protein folding has been a major challenge for the last twenty years, as pointed out in the
Structural Insights from Molecular Dynamics. Simulations of Tryptophan 7-Halogenase and
 Supporting Information Structural Insights from Molecular Dynamics Simulations of Tryptophan 7-Halogenase and Tryptophan 5-halogenase Jon Ainsley 1, Adrian J. Mulholland 2, Gary W. Black 1, Olivier Sparagano
Supporting Information Structural Insights from Molecular Dynamics Simulations of Tryptophan 7-Halogenase and Tryptophan 5-halogenase Jon Ainsley 1, Adrian J. Mulholland 2, Gary W. Black 1, Olivier Sparagano
All-atom Molecular Mechanics. Trent E. Balius AMS 535 / CHE /27/2010
 All-atom Molecular Mechanics Trent E. Balius AMS 535 / CHE 535 09/27/2010 Outline Molecular models Molecular mechanics Force Fields Potential energy function functional form parameters and parameterization
All-atom Molecular Mechanics Trent E. Balius AMS 535 / CHE 535 09/27/2010 Outline Molecular models Molecular mechanics Force Fields Potential energy function functional form parameters and parameterization
The Molecular Dynamics Method
 H-bond energy (kcal/mol) - 4.0 The Molecular Dynamics Method Fibronectin III_1, a mechanical protein that glues cells together in wound healing and in preventing tumor metastasis 0 ATPase, a molecular
H-bond energy (kcal/mol) - 4.0 The Molecular Dynamics Method Fibronectin III_1, a mechanical protein that glues cells together in wound healing and in preventing tumor metastasis 0 ATPase, a molecular
Supplemental Information
 Chemistry & Biology, Volume22 Supplemental Information Overcoming Chemical, Biological, and Computational Challenges in the Development of Inhibitors Targeting Protein-Protein Interactions Luca Laraia,
Chemistry & Biology, Volume22 Supplemental Information Overcoming Chemical, Biological, and Computational Challenges in the Development of Inhibitors Targeting Protein-Protein Interactions Luca Laraia,
NMR, X-ray Diffraction, Protein Structure, and RasMol
 NMR, X-ray Diffraction, Protein Structure, and RasMol Introduction So far we have been mostly concerned with the proteins themselves. The techniques (NMR or X-ray diffraction) used to determine a structure
NMR, X-ray Diffraction, Protein Structure, and RasMol Introduction So far we have been mostly concerned with the proteins themselves. The techniques (NMR or X-ray diffraction) used to determine a structure
CHEM 498Q / CHEM 630Q: Molecular Modeling of Proteins TUTORIAL #3a: Empirical force fields
 Department of Chemistry and Biochemistry, Concordia University! page 1 of 6 CHEM 498Q / CHEM 630Q: Molecular Modeling of Proteins TUTORIAL #3a: Empirical force fields INTRODUCTION The goal of this tutorial
Department of Chemistry and Biochemistry, Concordia University! page 1 of 6 CHEM 498Q / CHEM 630Q: Molecular Modeling of Proteins TUTORIAL #3a: Empirical force fields INTRODUCTION The goal of this tutorial
ψ j φ j+1 Motif position
 1.0 % Occupancy 0.8 0.6 0.4 0.2 0.0 ψ i-1 φ i ψ j φ j+1 ψ k-1 Motif position φ Supplementary Figure 1 Residue profiles for the positions in the β-sheet motif. Residue types as percentages for the six positions
1.0 % Occupancy 0.8 0.6 0.4 0.2 0.0 ψ i-1 φ i ψ j φ j+1 ψ k-1 Motif position φ Supplementary Figure 1 Residue profiles for the positions in the β-sheet motif. Residue types as percentages for the six positions
Long-timescale molecular dynamics (MD) simulations have
 simulations have") RNA force field with accuracy comparable to state-ofthe-art protein force fields Dazhi Tan a,1, Stefano Piana a,2, Robert M. Dirks a,3, and David E. Shaw a,b,2 PNAS PLUS a D. E. Shaw Research, New York,
RNA force field with accuracy comparable to state-ofthe-art protein force fields Dazhi Tan a,1, Stefano Piana a,2, Robert M. Dirks a,3, and David E. Shaw a,b,2 PNAS PLUS a D. E. Shaw Research, New York,
Figure 1. Molecules geometries of 5021 and Each neutral group in CHARMM topology was grouped in dash circle.
 Project I Chemistry 8021, Spring 2005/2/23 This document was turned in by a student as a homework paper. 1. Methods First, the cartesian coordinates of 5021 and 8021 molecules (Fig. 1) are generated, in
Project I Chemistry 8021, Spring 2005/2/23 This document was turned in by a student as a homework paper. 1. Methods First, the cartesian coordinates of 5021 and 8021 molecules (Fig. 1) are generated, in
Molecular Mechanics. I. Quantum mechanical treatment of molecular systems
 Molecular Mechanics I. Quantum mechanical treatment of molecular systems The first principle approach for describing the properties of molecules, including proteins, involves quantum mechanics. For example,
Molecular Mechanics I. Quantum mechanical treatment of molecular systems The first principle approach for describing the properties of molecules, including proteins, involves quantum mechanics. For example,
Can a continuum solvent model reproduce the free energy landscape of a β-hairpin folding in water?
 Can a continuum solvent model reproduce the free energy landscape of a β-hairpin folding in water? Ruhong Zhou 1 and Bruce J. Berne 2 1 IBM Thomas J. Watson Research Center; and 2 Department of Chemistry,
Can a continuum solvent model reproduce the free energy landscape of a β-hairpin folding in water? Ruhong Zhou 1 and Bruce J. Berne 2 1 IBM Thomas J. Watson Research Center; and 2 Department of Chemistry,
Performance of Molecular Mechanics Force Fields for RNA Simulations: Stability of UUCG and GNRA Hairpins
 3836 J. Chem. Theory Comput. 2010, 6, 3836 3849 Performance of Molecular Mechanics Force Fields for RNA Simulations: Stability of UUCG and GNRA Hairpins Pavel Banáš,, Daniel Hollas, Marie Zgarbová, Petr
3836 J. Chem. Theory Comput. 2010, 6, 3836 3849 Performance of Molecular Mechanics Force Fields for RNA Simulations: Stability of UUCG and GNRA Hairpins Pavel Banáš,, Daniel Hollas, Marie Zgarbová, Petr
Supplementary Materials for
 advances.sciencemag.org/cgi/content/full/4/1/eaau413/dc1 Supplementary Materials for Structure and dynamics conspire in the evolution of affinity between intrinsically disordered proteins Per Jemth*, Elin
advances.sciencemag.org/cgi/content/full/4/1/eaau413/dc1 Supplementary Materials for Structure and dynamics conspire in the evolution of affinity between intrinsically disordered proteins Per Jemth*, Elin
Supporting Information Comparative Molecular Dynamics Studies of Human DNA Polymerase η
 Supporting Information Comparative Molecular Dynamics Studies of Human DNA Polymerase η Melek N. Ucisik and Sharon Hammes-Schiffer* Department of Chemistry, University of Illinois at Urbana-Champaign,
Supporting Information Comparative Molecular Dynamics Studies of Human DNA Polymerase η Melek N. Ucisik and Sharon Hammes-Schiffer* Department of Chemistry, University of Illinois at Urbana-Champaign,
What makes a good graphene-binding peptide? Adsorption of amino acids and peptides at aqueous graphene interfaces: Electronic Supplementary
 Electronic Supplementary Material (ESI) for Journal of Materials Chemistry B. This journal is The Royal Society of Chemistry 21 What makes a good graphene-binding peptide? Adsorption of amino acids and
Electronic Supplementary Material (ESI) for Journal of Materials Chemistry B. This journal is The Royal Society of Chemistry 21 What makes a good graphene-binding peptide? Adsorption of amino acids and
Molecular dynamics re-refinement of two different small RNA loop structures using the original NMR data suggest a common structure
 J Biomol NMR (2012) 53:321 339 DOI 10.1007/s10858-012-9642-5 ARTICLE Molecular dynamics re-refinement of two different small RNA loop structures using the original NMR data suggest a common structure Niel
J Biomol NMR (2012) 53:321 339 DOI 10.1007/s10858-012-9642-5 ARTICLE Molecular dynamics re-refinement of two different small RNA loop structures using the original NMR data suggest a common structure Niel
ONETEP PB/SA: Application to G-Quadruplex DNA Stability. Danny Cole
 ONETEP PB/SA: Application to G-Quadruplex DNA Stability Danny Cole Introduction Historical overview of structure and free energy calculation of complex molecules using molecular mechanics and continuum
ONETEP PB/SA: Application to G-Quadruplex DNA Stability Danny Cole Introduction Historical overview of structure and free energy calculation of complex molecules using molecular mechanics and continuum
LysinebasedTrypsinActSite. A computer application for modeling Chymotrypsin
 LysinebasedTrypsinActSite A computer application for modeling Chymotrypsin Version.2 May 2006 LysTAS A computer application for modeling chymotrypsin Version.2 May 2006 Table of Contents Page. Introduction
LysinebasedTrypsinActSite A computer application for modeling Chymotrypsin Version.2 May 2006 LysTAS A computer application for modeling chymotrypsin Version.2 May 2006 Table of Contents Page. Introduction
Structural and mechanistic insight into the substrate. binding from the conformational dynamics in apo. and substrate-bound DapE enzyme
 Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 215 Structural and mechanistic insight into the substrate binding from the conformational
Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 215 Structural and mechanistic insight into the substrate binding from the conformational
Challenges in generation of conformational ensembles for peptides and small proteins
 Challenges in generation of conformational ensembles for peptides and small proteins Carlos Simmerling Stony Brook University What could (and does) go wrong? 1. Sampling: difficult to obtain converged
Challenges in generation of conformational ensembles for peptides and small proteins Carlos Simmerling Stony Brook University What could (and does) go wrong? 1. Sampling: difficult to obtain converged
Simulating Folding of Helical Proteins with Coarse Grained Models
 366 Progress of Theoretical Physics Supplement No. 138, 2000 Simulating Folding of Helical Proteins with Coarse Grained Models Shoji Takada Department of Chemistry, Kobe University, Kobe 657-8501, Japan
366 Progress of Theoretical Physics Supplement No. 138, 2000 Simulating Folding of Helical Proteins with Coarse Grained Models Shoji Takada Department of Chemistry, Kobe University, Kobe 657-8501, Japan
Force Fields for Classical Molecular Dynamics simulations of Biomolecules. Emad Tajkhorshid
 Force Fields for Classical Molecular Dynamics simulations of Biomolecules Emad Tajkhorshid Theoretical and Computational Biophysics Group, Beckman Institute Departments of Biochemistry and Pharmacology,
Force Fields for Classical Molecular Dynamics simulations of Biomolecules Emad Tajkhorshid Theoretical and Computational Biophysics Group, Beckman Institute Departments of Biochemistry and Pharmacology,
Free Energy Landscape of Protein Folding in Water: Explicit vs. Implicit Solvent
 PROTEINS: Structure, Function, and Genetics 53:148 161 (2003) Free Energy Landscape of Protein Folding in Water: Explicit vs. Implicit Solvent Ruhong Zhou* IBM T.J. Watson Research Center, Yorktown Heights,
PROTEINS: Structure, Function, and Genetics 53:148 161 (2003) Free Energy Landscape of Protein Folding in Water: Explicit vs. Implicit Solvent Ruhong Zhou* IBM T.J. Watson Research Center, Yorktown Heights,
Structural fidelity and NMR relaxation analysis in a prototype RNA hairpin
 Structural fidelity and NMR relaxation analysis in a prototype RNA hairpin GEORGE M. GIAMBAŞU, DARRIN M. YORK, and DAVID A. CASE BioMaPS Institute for Quantitative Biology and Department of Chemistry and
Structural fidelity and NMR relaxation analysis in a prototype RNA hairpin GEORGE M. GIAMBAŞU, DARRIN M. YORK, and DAVID A. CASE BioMaPS Institute for Quantitative Biology and Department of Chemistry and
Supplementary Figures:
 Supplementary Figures: Supplementary Figure 1: The two strings converge to two qualitatively different pathways. A) Models of active (red) and inactive (blue) states used as end points for the string calculations
Supplementary Figures: Supplementary Figure 1: The two strings converge to two qualitatively different pathways. A) Models of active (red) and inactive (blue) states used as end points for the string calculations
DISCRETE TUTORIAL. Agustí Emperador. Institute for Research in Biomedicine, Barcelona APPLICATION OF DISCRETE TO FLEXIBLE PROTEIN-PROTEIN DOCKING:
 DISCRETE TUTORIAL Agustí Emperador Institute for Research in Biomedicine, Barcelona APPLICATION OF DISCRETE TO FLEXIBLE PROTEIN-PROTEIN DOCKING: STRUCTURAL REFINEMENT OF DOCKING CONFORMATIONS Emperador
DISCRETE TUTORIAL Agustí Emperador Institute for Research in Biomedicine, Barcelona APPLICATION OF DISCRETE TO FLEXIBLE PROTEIN-PROTEIN DOCKING: STRUCTURAL REFINEMENT OF DOCKING CONFORMATIONS Emperador
Magnetic Resonance Lectures for Chem 341 James Aramini, PhD. CABM 014A
 Magnetic Resonance Lectures for Chem 341 James Aramini, PhD. CABM 014A jma@cabm.rutgers.edu " J.A. 12/11/13 Dec. 4 Dec. 9 Dec. 11" " Outline" " 1. Introduction / Spectroscopy Overview 2. NMR Spectroscopy
Magnetic Resonance Lectures for Chem 341 James Aramini, PhD. CABM 014A jma@cabm.rutgers.edu " J.A. 12/11/13 Dec. 4 Dec. 9 Dec. 11" " Outline" " 1. Introduction / Spectroscopy Overview 2. NMR Spectroscopy
Comparing crystal structure of M.HhaI with and without DNA1, 2 (PDBID:1hmy and PDBID:2hmy),
,") Supporting Information 1. Constructing the starting structure Comparing crystal structure of M.HhaI with and without DNA1, 2 (PDBID:1hmy and PDBID:2hmy), we find that: the RMSD of overall structure and
Supporting Information 1. Constructing the starting structure Comparing crystal structure of M.HhaI with and without DNA1, 2 (PDBID:1hmy and PDBID:2hmy), we find that: the RMSD of overall structure and
CHEM 498Q / CHEM 630Q: Molecular Modeling of Proteins TUTORIAL #3a: Empirical force fields
 Department of Chemistry and Biochemistry, Concordia University page 1 of 6 CHEM 498Q / CHEM 630Q: Molecular Modeling of Proteins TUTORIAL #3a: Empirical force fields INTRODUCTION The goal of this tutorial
Department of Chemistry and Biochemistry, Concordia University page 1 of 6 CHEM 498Q / CHEM 630Q: Molecular Modeling of Proteins TUTORIAL #3a: Empirical force fields INTRODUCTION The goal of this tutorial
A conserved P-loop anchor limits the structural dynamics that mediate. nucleotide dissociation in EF-Tu.
 Supplemental Material for A conserved P-loop anchor limits the structural dynamics that mediate nucleotide dissociation in EF-Tu. Evan Mercier 1,2, Dylan Girodat 1, and Hans-Joachim Wieden 1 * 1 Alberta
Supplemental Material for A conserved P-loop anchor limits the structural dynamics that mediate nucleotide dissociation in EF-Tu. Evan Mercier 1,2, Dylan Girodat 1, and Hans-Joachim Wieden 1 * 1 Alberta
MINT version Users Manual. Anna Górska Maciej Jasiński Joanna Trylska
 MINT version 1.0.1 Users Manual Anna Górska Maciej Jasiński Joanna Trylska 1 MINT is a free software; you can redistribute it and/or modify it under the terms of the GNU General Public License version
MINT version 1.0.1 Users Manual Anna Górska Maciej Jasiński Joanna Trylska 1 MINT is a free software; you can redistribute it and/or modify it under the terms of the GNU General Public License version
Folding of a Small RNA Hairpin Based on Simulation with Replica Exchange Molecular Dynamics
 J. Phys. Chem. B 2010, 114, 5835 5839 5835 Folding of a Small RNA Hairpin Based on Simulation with Replica Exchange Molecular Dynamics Guanghong Zuo,, Wenfei Li,*, Jian Zhang, Jin Wang, and Wei Wang*,
J. Phys. Chem. B 2010, 114, 5835 5839 5835 Folding of a Small RNA Hairpin Based on Simulation with Replica Exchange Molecular Dynamics Guanghong Zuo,, Wenfei Li,*, Jian Zhang, Jin Wang, and Wei Wang*,
Biochemistry,530:,, Introduc5on,to,Structural,Biology, Autumn,Quarter,2015,
 Biochemistry,530:,, Introduc5on,to,Structural,Biology, Autumn,Quarter,2015, Course,Informa5on, BIOC%530% GraduateAlevel,discussion,of,the,structure,,func5on,,and,chemistry,of,proteins,and, nucleic,acids,,control,of,enzyma5c,reac5ons.,please,see,the,course,syllabus,and,
Biochemistry,530:,, Introduc5on,to,Structural,Biology, Autumn,Quarter,2015, Course,Informa5on, BIOC%530% GraduateAlevel,discussion,of,the,structure,,func5on,,and,chemistry,of,proteins,and, nucleic,acids,,control,of,enzyma5c,reac5ons.,please,see,the,course,syllabus,and,
Protein Structure. W. M. Grogan, Ph.D. OBJECTIVES
 Protein Structure W. M. Grogan, Ph.D. OBJECTIVES 1. Describe the structure and characteristic properties of typical proteins. 2. List and describe the four levels of structure found in proteins. 3. Relate
Protein Structure W. M. Grogan, Ph.D. OBJECTIVES 1. Describe the structure and characteristic properties of typical proteins. 2. List and describe the four levels of structure found in proteins. 3. Relate
Molecular dynamics simulation of Aquaporin-1. 4 nm
 Molecular dynamics simulation of Aquaporin-1 4 nm Molecular Dynamics Simulations Schrödinger equation i~@ t (r, R) =H (r, R) Born-Oppenheimer approximation H e e(r; R) =E e (R) e(r; R) Nucleic motion described
Molecular dynamics simulation of Aquaporin-1 4 nm Molecular Dynamics Simulations Schrödinger equation i~@ t (r, R) =H (r, R) Born-Oppenheimer approximation H e e(r; R) =E e (R) e(r; R) Nucleic motion described
How Tertiary Interactions Between the L2 and L3 Loops Affect the Dynamics of the Distant Ligand Binding Site in the Guanine Sensing Riboswitch
 How Tertiary Interactions Between the L2 and L3 Loops Affect the Dynamics of the Distant Ligand Binding Site in the Guanine Sensing Riboswitch Christian A. Hanke and Holger Gohlke Institute for Pharmaceutical
How Tertiary Interactions Between the L2 and L3 Loops Affect the Dynamics of the Distant Ligand Binding Site in the Guanine Sensing Riboswitch Christian A. Hanke and Holger Gohlke Institute for Pharmaceutical
Potential Energy (hyper)surface
surface") The Molecular Dynamics Method Thermal motion of a lipid bilayer Water permeation through channels Selective sugar transport Potential Energy (hyper)surface What is Force? Energy U(x) F = " d dx U(x) Conformation
The Molecular Dynamics Method Thermal motion of a lipid bilayer Water permeation through channels Selective sugar transport Potential Energy (hyper)surface What is Force? Energy U(x) F = " d dx U(x) Conformation
HOMOLOGY MODELING. The sequence alignment and template structure are then used to produce a structural model of the target.
 HOMOLOGY MODELING Homology modeling, also known as comparative modeling of protein refers to constructing an atomic-resolution model of the "target" protein from its amino acid sequence and an experimental
HOMOLOGY MODELING Homology modeling, also known as comparative modeling of protein refers to constructing an atomic-resolution model of the "target" protein from its amino acid sequence and an experimental
Computational Modeling of Protein Kinase A and Comparison with Nuclear Magnetic Resonance Data
 Computational Modeling of Protein Kinase A and Comparison with Nuclear Magnetic Resonance Data ABSTRACT Keyword Lei Shi 1 Advisor: Gianluigi Veglia 1,2 Department of Chemistry 1, & Biochemistry, Molecular
Computational Modeling of Protein Kinase A and Comparison with Nuclear Magnetic Resonance Data ABSTRACT Keyword Lei Shi 1 Advisor: Gianluigi Veglia 1,2 Department of Chemistry 1, & Biochemistry, Molecular
User Guide for LeDock
 User Guide for LeDock Hongtao Zhao, PhD Email: htzhao@lephar.com Website: www.lephar.com Copyright 2017 Hongtao Zhao. All rights reserved. Introduction LeDock is flexible small-molecule docking software,
User Guide for LeDock Hongtao Zhao, PhD Email: htzhao@lephar.com Website: www.lephar.com Copyright 2017 Hongtao Zhao. All rights reserved. Introduction LeDock is flexible small-molecule docking software,
NMR-Structure determination with the program CNS
 NMR-Structure determination with the program CNS Blockkurs 2013 Exercise 11.10.2013, room Mango? 1 NMR-Structure determination - Overview Amino acid sequence Topology file nef_seq.mtf loop cns_mtf_atom.id
NMR-Structure determination with the program CNS Blockkurs 2013 Exercise 11.10.2013, room Mango? 1 NMR-Structure determination - Overview Amino acid sequence Topology file nef_seq.mtf loop cns_mtf_atom.id
MARTINI simulation details
 S1 Appendix MARTINI simulation details MARTINI simulation initialization and equilibration In this section, we describe the initialization of simulations from Main Text section Residue-based coarsegrained
S1 Appendix MARTINI simulation details MARTINI simulation initialization and equilibration In this section, we describe the initialization of simulations from Main Text section Residue-based coarsegrained
Supporting Information. Copyright Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2009
 Supporting Information Copyright Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, 2009 Helical Hairpin Structure of a potent Antimicrobial Peptide MSI-594 in Lipopolysaccharide Micelles by NMR Anirban
Supporting Information Copyright Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, 2009 Helical Hairpin Structure of a potent Antimicrobial Peptide MSI-594 in Lipopolysaccharide Micelles by NMR Anirban
Structural Basis of Multivalent Binding to Wheat Germ Agglutinin
 Structural Basis of Multivalent Binding to Wheat Germ Agglutinin David Schwefel, Caroline Maierhofer, Johannes G. Beck, Sonja Seeberger, Kay Diederichs, Heiko M. Möller,*, Wolfram Welte,*, and Valentin
Structural Basis of Multivalent Binding to Wheat Germ Agglutinin David Schwefel, Caroline Maierhofer, Johannes G. Beck, Sonja Seeberger, Kay Diederichs, Heiko M. Möller,*, Wolfram Welte,*, and Valentin
Explicit Ion, Implicit Water Solvation for Molecular Dynamics of Nucleic Acids and Highly Charged Molecules
 Explicit Ion, Implicit Water Solvation for Molecular Dynamics of Nucleic Acids and Highly Charged Molecules NINAD V. PRABHU, 1 MANORANJAN PANDA, 2 QINGYI YANG, 3 KIM A. SHARP 3 1 Schering-Plough, Kenilworth,
Explicit Ion, Implicit Water Solvation for Molecular Dynamics of Nucleic Acids and Highly Charged Molecules NINAD V. PRABHU, 1 MANORANJAN PANDA, 2 QINGYI YANG, 3 KIM A. SHARP 3 1 Schering-Plough, Kenilworth,
SUPPLEMENTARY INFORMATION
 5 N 4 8 20 22 24 2 28 4 8 20 22 24 2 28 a b 0 9 8 7 H c (kda) 95 0 57 4 28 2 5.5 Precipitate before NMR expt. Supernatant before NMR expt. Precipitate after hrs NMR expt. Supernatant after hrs NMR expt.
5 N 4 8 20 22 24 2 28 4 8 20 22 24 2 28 a b 0 9 8 7 H c (kda) 95 0 57 4 28 2 5.5 Precipitate before NMR expt. Supernatant before NMR expt. Precipitate after hrs NMR expt. Supernatant after hrs NMR expt.
Outline. The ensemble folding kinetics of protein G from an all-atom Monte Carlo simulation. Unfolded Folded. What is protein folding?
 The ensemble folding kinetics of protein G from an all-atom Monte Carlo simulation By Jun Shimada and Eugine Shaknovich Bill Hawse Dr. Bahar Elisa Sandvik and Mehrdad Safavian Outline Background on protein
The ensemble folding kinetics of protein G from an all-atom Monte Carlo simulation By Jun Shimada and Eugine Shaknovich Bill Hawse Dr. Bahar Elisa Sandvik and Mehrdad Safavian Outline Background on protein
MOLECULAR DYNAMICS OF RNA STRUCTURAL MOTIF. TEMPERATURE ENHANCED SAMPLING OF THE CONFORMATIONAL SPACE
 COMPUTATIONAL METHODS IN SCIENCE AND TECHNOLOGY 8(2), 37-45 (2002) MOLECULAR DYNAMICS OF RNA STRUCTURAL MOTIF. TEMPERATURE ENHANCED SAMPLING OF THE CONFORMATIONAL SPACE TADEUSZ KULIŃSKI, KATARZYNA KULIŃSKA
COMPUTATIONAL METHODS IN SCIENCE AND TECHNOLOGY 8(2), 37-45 (2002) MOLECULAR DYNAMICS OF RNA STRUCTURAL MOTIF. TEMPERATURE ENHANCED SAMPLING OF THE CONFORMATIONAL SPACE TADEUSZ KULIŃSKI, KATARZYNA KULIŃSKA
3rd Advanced in silico Drug Design KFC/ADD Molecular mechanics intro Karel Berka, Ph.D. Martin Lepšík, Ph.D. Pavel Polishchuk, Ph.D.
 3rd Advanced in silico Drug Design KFC/ADD Molecular mechanics intro Karel Berka, Ph.D. Martin Lepšík, Ph.D. Pavel Polishchuk, Ph.D. Thierry Langer, Ph.D. Jana Vrbková, Ph.D. UP Olomouc, 23.1.-26.1. 2018
3rd Advanced in silico Drug Design KFC/ADD Molecular mechanics intro Karel Berka, Ph.D. Martin Lepšík, Ph.D. Pavel Polishchuk, Ph.D. Thierry Langer, Ph.D. Jana Vrbková, Ph.D. UP Olomouc, 23.1.-26.1. 2018
Supplementary information for cloud computing approaches for prediction of ligand binding poses and pathways
 Supplementary information for cloud computing approaches for prediction of ligand binding poses and pathways Morgan Lawrenz 1, Diwakar Shukla 1,2 & Vijay S. Pande 1,2 1 Department of Chemistry, Stanford
Supplementary information for cloud computing approaches for prediction of ligand binding poses and pathways Morgan Lawrenz 1, Diwakar Shukla 1,2 & Vijay S. Pande 1,2 1 Department of Chemistry, Stanford
How is molecular dynamics being used in life sciences? Davide Branduardi
 How is molecular dynamics being used in life sciences? Davide Branduardi davide.branduardi@schrodinger.com Exploring molecular processes with MD Drug discovery and design Protein-protein interactions Protein-DNA
How is molecular dynamics being used in life sciences? Davide Branduardi davide.branduardi@schrodinger.com Exploring molecular processes with MD Drug discovery and design Protein-protein interactions Protein-DNA
Carbazole Derivatives Binding to c-kit G-quadruplex DNA
 Supplementary Materials Carbazole Derivatives Binding to c-kit G-quadruplex DNA Agata Głuszyńska 1, *, Bernard Juskowiak 1, Martyna Kuta-Siejkowska 2, Marcin Hoffmann 2 and Shozeb Haider 3 1 Laboratory
Supplementary Materials Carbazole Derivatives Binding to c-kit G-quadruplex DNA Agata Głuszyńska 1, *, Bernard Juskowiak 1, Martyna Kuta-Siejkowska 2, Marcin Hoffmann 2 and Shozeb Haider 3 1 Laboratory
Supplementary Figure S1. Urea-mediated buffering mechanism of H. pylori. Gastric urea is funneled to a cytoplasmic urease that is presumably attached
 Supplementary Figure S1. Urea-mediated buffering mechanism of H. pylori. Gastric urea is funneled to a cytoplasmic urease that is presumably attached to HpUreI. Urea hydrolysis products 2NH 3 and 1CO 2
Supplementary Figure S1. Urea-mediated buffering mechanism of H. pylori. Gastric urea is funneled to a cytoplasmic urease that is presumably attached to HpUreI. Urea hydrolysis products 2NH 3 and 1CO 2
Hyeyoung Shin a, Tod A. Pascal ab, William A. Goddard III abc*, and Hyungjun Kim a* Korea
 The Scaled Effective Solvent Method for Predicting the Equilibrium Ensemble of Structures with Analysis of Thermodynamic Properties of Amorphous Polyethylene Glycol-Water Mixtures Hyeyoung Shin a, Tod
The Scaled Effective Solvent Method for Predicting the Equilibrium Ensemble of Structures with Analysis of Thermodynamic Properties of Amorphous Polyethylene Glycol-Water Mixtures Hyeyoung Shin a, Tod
Supporting Information
 Supporting Information Smith et al. 10.1073/pnas.1519609113 SI Methods Sample Preparation. Perdeuterated, 15 N-labeled WT and mutant ubiquitin was expressed in Escherichia coli adapted to 100% D 2 O Toronto
Supporting Information Smith et al. 10.1073/pnas.1519609113 SI Methods Sample Preparation. Perdeuterated, 15 N-labeled WT and mutant ubiquitin was expressed in Escherichia coli adapted to 100% D 2 O Toronto
Tailoring the Properties of Quadruplex Nucleobases for Biological and Nanomaterial Applications
 Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 2014 Supporting Information for: Tailoring the Properties of Quadruplex Nucleobases
Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 2014 Supporting Information for: Tailoring the Properties of Quadruplex Nucleobases
Multi-Scale Hierarchical Structure Prediction of Helical Transmembrane Proteins
 Multi-Scale Hierarchical Structure Prediction of Helical Transmembrane Proteins Zhong Chen Dept. of Biochemistry and Molecular Biology University of Georgia, Athens, GA 30602 Email: zc@csbl.bmb.uga.edu
Multi-Scale Hierarchical Structure Prediction of Helical Transmembrane Proteins Zhong Chen Dept. of Biochemistry and Molecular Biology University of Georgia, Athens, GA 30602 Email: zc@csbl.bmb.uga.edu
Bioengineering 215. An Introduction to Molecular Dynamics for Biomolecules
 Bioengineering 215 An Introduction to Molecular Dynamics for Biomolecules David Parker May 18, 2007 ntroduction A principal tool to study biological molecules is molecular dynamics simulations (MD). MD
Bioengineering 215 An Introduction to Molecular Dynamics for Biomolecules David Parker May 18, 2007 ntroduction A principal tool to study biological molecules is molecular dynamics simulations (MD). MD
Structure Investigation of Fam20C, a Golgi Casein Kinase
 Structure Investigation of Fam20C, a Golgi Casein Kinase Sharon Grubner National Taiwan University, Dr. Jung-Hsin Lin University of California San Diego, Dr. Rommie Amaro Abstract This research project
Structure Investigation of Fam20C, a Golgi Casein Kinase Sharon Grubner National Taiwan University, Dr. Jung-Hsin Lin University of California San Diego, Dr. Rommie Amaro Abstract This research project
With the advance of fast computers, explicit solvent
 pubs.acs.org/jpcl Terms of Use Molecular Dynamics Simulations of Nucleic Acids. From Tetranucleotides to the Ribosome Jir í S poner,*,, Pavel Banaś, Petr Jurecǩa, Marie Zgarbova, Petra Kuḧrova, Marek Havrila,,
pubs.acs.org/jpcl Terms of Use Molecular Dynamics Simulations of Nucleic Acids. From Tetranucleotides to the Ribosome Jir í S poner,*,, Pavel Banaś, Petr Jurecǩa, Marie Zgarbova, Petra Kuḧrova, Marek Havrila,,
Assignment 2: Conformation Searching (50 points)
") Chemistry 380.37 Fall 2015 Dr. Jean M. Standard September 16, 2015 Assignment 2: Conformation Searching (50 points) In this assignment, you will use the Spartan software package to investigate some conformation
Chemistry 380.37 Fall 2015 Dr. Jean M. Standard September 16, 2015 Assignment 2: Conformation Searching (50 points) In this assignment, you will use the Spartan software package to investigate some conformation
Chapter 6 Cyclic urea - a new central unit in bent-core compounds
 82 Chapter 6 Cyclic urea - a new central unit in bent-core compounds A new class of five-ring bent-core molecules with a cyclic urea group as a central unit was synthesized [94]. A significant difference
82 Chapter 6 Cyclic urea - a new central unit in bent-core compounds A new class of five-ring bent-core molecules with a cyclic urea group as a central unit was synthesized [94]. A significant difference
Supporting Information
 Supporting Information Micelle-Triggered b-hairpin to a-helix Transition in a 14-Residue Peptide from a Choline-Binding Repeat of the Pneumococcal Autolysin LytA HØctor Zamora-Carreras, [a] Beatriz Maestro,
Supporting Information Micelle-Triggered b-hairpin to a-helix Transition in a 14-Residue Peptide from a Choline-Binding Repeat of the Pneumococcal Autolysin LytA HØctor Zamora-Carreras, [a] Beatriz Maestro,
Dimer Dissociation of a Photoreceptor Protein from QM/MM and MD Simulations
 Dimer Dissociation of a Photoreceptor Protein from QM/MM and MD Simulations IMA University of Minnesota Minneapolis, MN, July 20, 2015 Haisheng Ren Advisor: Prof. Jiali Gao Department of Chemistry, University
Dimer Dissociation of a Photoreceptor Protein from QM/MM and MD Simulations IMA University of Minnesota Minneapolis, MN, July 20, 2015 Haisheng Ren Advisor: Prof. Jiali Gao Department of Chemistry, University
The Effect of Motional Averaging on the Calculation of NMR-Derived Structural Properties
 PROTEINS: Structure, Function, and Genetics 6:542 555 (1999) The Effect of Motional Averaging on the Calculation of NMR-Derived Structural Properties Xavier Daura, 1 Iris Antes, 1,2 Wilfred F. van Gunsteren,
PROTEINS: Structure, Function, and Genetics 6:542 555 (1999) The Effect of Motional Averaging on the Calculation of NMR-Derived Structural Properties Xavier Daura, 1 Iris Antes, 1,2 Wilfred F. van Gunsteren,
Computational Studies of Molecular Mechanisms Mediating Protein Adsorption on Material Surfaces
 Clemson University TigerPrints All Dissertations Dissertations 8-2016 Computational Studies of Molecular Mechanisms Mediating Protein Adsorption on Material Surfaces Tigran M. Abramyan Clemson University
Clemson University TigerPrints All Dissertations Dissertations 8-2016 Computational Studies of Molecular Mechanisms Mediating Protein Adsorption on Material Surfaces Tigran M. Abramyan Clemson University
Supporting Information
 Supporting Information The Predicted Ensemble of Low-Energy Conformations of Human Somatostatin Receptor Subtype 5 and the Binding of Antagonists Sijia S. Dong, [a] Ravinder Abrol, [a, b] and William A.
Supporting Information The Predicted Ensemble of Low-Energy Conformations of Human Somatostatin Receptor Subtype 5 and the Binding of Antagonists Sijia S. Dong, [a] Ravinder Abrol, [a, b] and William A.
THE TANGO ALGORITHM: SECONDARY STRUCTURE PROPENSITIES, STATISTICAL MECHANICS APPROXIMATION
 THE TANGO ALGORITHM: SECONDARY STRUCTURE PROPENSITIES, STATISTICAL MECHANICS APPROXIMATION AND CALIBRATION Calculation of turn and beta intrinsic propensities. A statistical analysis of a protein structure
THE TANGO ALGORITHM: SECONDARY STRUCTURE PROPENSITIES, STATISTICAL MECHANICS APPROXIMATION AND CALIBRATION Calculation of turn and beta intrinsic propensities. A statistical analysis of a protein structure
Medical Research, Medicinal Chemistry, University of Leuven, Leuven, Belgium.
 Supporting Information Towards peptide vaccines against Zika virus: Immunoinformatics combined with molecular dynamics simulations to predict antigenic epitopes of Zika viral proteins Muhammad Usman Mirza
Supporting Information Towards peptide vaccines against Zika virus: Immunoinformatics combined with molecular dynamics simulations to predict antigenic epitopes of Zika viral proteins Muhammad Usman Mirza
Supplementary Information Intrinsic Localized Modes in Proteins
 Supplementary Information Intrinsic Localized Modes in Proteins Adrien Nicolaï 1,, Patrice Delarue and Patrick Senet, 1 Department of Physics, Applied Physics and Astronomy, Rensselaer Polytechnic Institute,
Supplementary Information Intrinsic Localized Modes in Proteins Adrien Nicolaï 1,, Patrice Delarue and Patrick Senet, 1 Department of Physics, Applied Physics and Astronomy, Rensselaer Polytechnic Institute,
Practical Manual. General outline to use the structural information obtained from molecular alignment
 Practical Manual General outline to use the structural information obtained from molecular alignment 1. In order to use the information one needs to know the direction and the size of the tensor (susceptibility,
Practical Manual General outline to use the structural information obtained from molecular alignment 1. In order to use the information one needs to know the direction and the size of the tensor (susceptibility,
T H E J O U R N A L O F G E N E R A L P H Y S I O L O G Y. jgp
 S u p p l e m e n ta l m at e r i a l jgp Lee et al., http://www.jgp.org/cgi/content/full/jgp.201411219/dc1 T H E J O U R N A L O F G E N E R A L P H Y S I O L O G Y S u p p l e m e n ta l D I S C U S
S u p p l e m e n ta l m at e r i a l jgp Lee et al., http://www.jgp.org/cgi/content/full/jgp.201411219/dc1 T H E J O U R N A L O F G E N E R A L P H Y S I O L O G Y S u p p l e m e n ta l D I S C U S