Contact map guided ab initio structure prediction
|
|
|
- Angelica Williams
- 5 years ago
- Views:
Transcription

1 Contact map guided ab initio structure prediction S M Golam Mortuza Postdoctoral Research Fellow I-TASSER Workshop 2017 North Carolina A&T State University, Greensboro, NC
2 Outline Ab initio structure prediction: QUARK Contact map prediction: NeBcon Contact guided ab initio structure prediction: C-QUARK Ab initio GPCR structure prediction: GPCR-AIM 3/20/2017 2
3 Ab initio structure prediction In the absence of homologous templates,i- TASSER based models are often less useful for biomedical studies due to less accuracy of the models Ab initio protein folding method assembles protein structures without using templates Ab initio structure modeling represents the most challenging problem in structure prediction 3/20/2017 3
4 QUARK: Ab initio structure prediction method Knowledge-based potentials: 3/20/2017 4
5 QUARK: Fragment generation and distance profile Xu et al., Proteins-Structure Function and Bioinformatics, 81(2), pp (2012) 3/20/2017 5
6 QUARK: Energy Function E tot = E prm + w 1 E prs + w 2 E ev + w 3 E hb + w 4 E sa + w 5 E dh + w 6 E dp + w 7 E rg + w 8 E dab + w 9 E hp + w 10 E bp 1. Backbone atomic pair-wise potential (E prm ) 7. Fragment-based distance profile (E dp ) 2. Side-chain center pair-wise potential (E prs ) 8. Radius of gyration (E rg ) 3. Excluded volume (E ev ) 4. Hydrogen bonding (E hb ) 9. Strand-helix-strand packing (E dab ) 5. Solvent accessibility (E sa ) 6. Backbone torsion potential (E dh ) 10. Helix packing (E hp ) 11. Strand packing (E bp ) 6

7 Problems with Metropolis Monte Carlo E Low Temperature p accept ~ e de/t 1. Possibility of getting trapped at local energy basin 2. Increasing T can overcome local energy barrier, but it cannot detect low-energy regions E X X High Temperature 3/20/2017 7
8 Replica Exchange Monte Carlo Initial Random Configuration Initial Random Configuration Initial Random Configuration Make Random Change Make Random Change Make Random Change Calculate de Calculate de Calculate de p accept = e de/t 1 p accept = e de/t 2 p accept = e de/t 3 Pswap i,j = e E i E j 1 t i 1 t j T 1 T 2 T 3 3/20/ T max = L T min = L
9 Benchmark Results: QUARK vs. Rosetta Data set: 51 small proteins ( AA) and 94 medium proteins ( AA) RMSD: 96/145 targets QUARK models are better than Rosetta (pvalue: 1.51X10-4 ) TM-score: 95/145 targets QUARK models are better than Rosetta (p-value: 2.87X10-7 ) Xu et al., Proteins-Structure Function and 3/20/2017 Bioinformatics, 80(7), pp (2012) 9
10 Benchmark Results: QUARK vs. Rosetta Data set 51 small proteins with ( residues) 94 medium proteins with ( residues) Methods First (best in top five) cluster center model RMSD TM-score Rosetta 10.1 (8.5) (0.393) QUARK 9.1 (7.7) (0.441) Rosetta 13.0 (11.5) (0.346) QUARK 12.5 (10.7) (0.374) Xu et al., Proteins-Structure Function and Bioinformatics, 80(7), pp (2012) 3/20/


11 Benchmark Results: QUARK vs. Rosetta Red: Native Blue: Rosetta Green: QUARK Xu et al., Proteins- Structure Function and Bioinformatics, 80(7), pp (2012) 3/20/
12 QUARK in CASP Experiments CASP9 CASP10 CASP11 Groups Z Groups Z Groups Z QUARK 31.6 QUARK 17.1 QUARK 33.5 Multicon-Refine 22.4 TASSER-VMT 13.9 RBO_Aleph 29.6 Chunk-TASSER 20.7 Pcons-net 13.7 Multicom-con 21.4 RaptorX 19.8 PMS 11.7 RaptorX-FM 17.6 Baker-Rosetta 19.0 RaptorX-Roll 11.3 myprotein-me 15.9 Jiang_Assembly 14.7 HHpred-thread 10.9 TASSER-VMT 15.8 Gws 13.9 Multicom-clust 10.6 Baker-Rosetta 15.7 BioSerf 13.6 RBO-MBS 9.1 Seok-server 15.6 SAM-T08-server 12.7 MUFold_CRF 8.8 FUSION 15.5 Seok-server 12.6 Baker-Rosetta 8.1 nns 15.4 Here, Z-score (Z) represents the significance of the structure predictions by 3/20/2017 each group compared to the average performance 12
 in CASP 11 QUARK fragments")


13 QUARK modeling of T0837-D1 (128 AA) in CASP 11 QUARK fragments RMSD ~ A Assessor s comment: T0837-D1_499_1 represents the FM model with 13 biggest improvement for PDB templates in CASP11 experiment
14 Why Zhang-Server performs better than QUARK in CASP experiments?? Models built by QUARK are compared with threading templates by LOMETS The templates are then re-ranked by their similarity to the QUARK models before they are subjected to the I-TASSER structure-assembly simulations. Zhang et al., Proteins, 84, pp (2015) 3/20/


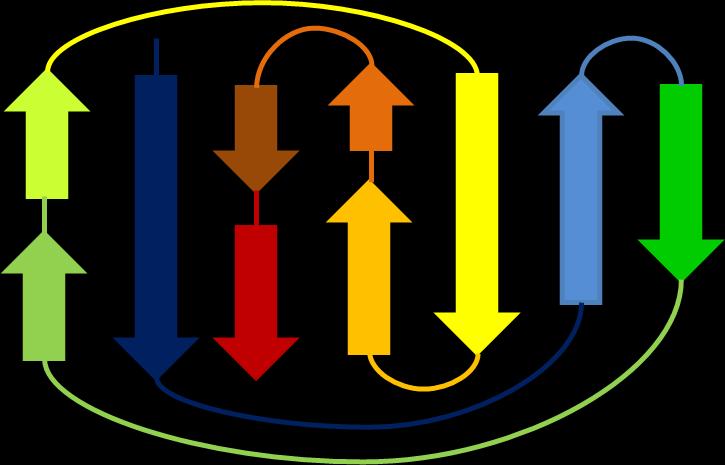




15 Limitations in current methods Fold small proteins (<150 residues) Can only fold beta-protein with simple topology R0014 CASP10 3/20/
16 Contact maps in ab initio protein structure prediction Sequence-based contact map prediction can be useful for 3D structure folding of larger size proteins that have complicated topologies Incorrectly predicted contacts can be harmful to 3D structure construction. Contact prediction should have an accuracy of at least 22% to generate a positive effect to the ab initio structure prediction 3/20/

17 Basic information on contact maps Residues are in contact if the distance between C α or C β atoms of the residues is < 8 Å Contact classification: Short range: Sequence separation 6-11 residues Medium range: Sequence separation residues Long range: Sequence separation >24 residues TTSQKHRDFVAEPGEKPVGSLAGIGEVLGKKLEERG 1 Short range Medium range Long range 3/20/
18 Programs for predicting contact maps Machine Learning: o BETACON o SVMcon o SVMSEQ Coevolution: o PSICOV o CCMpred o mfdca o Gremlin Meta: ostructch ometapsicov opconsc2, PconsC31 3/20/
19 NeBcon (Neural network and Bayesclassifier based contact prediction) 3/20/
20 Naïve Bayes Classifier (NBC) X ij = (X ij 1, X ij 2,, X ij m) is the confidence score for the ith and jth residues to be in contact as predicted by mth contact predictor. X ij m P C X ij = P 0 X ij = = P C P(X ij C) P(X ij ) Under the naïve assumption, the confidence scores from different contact predictors are independent from each other P C X ij = P C N m=1 P X ij m C P X ij N P C N m=1 P X ij m C P 0 m=1 P X m ij 0 + P 1 m=1 P X m ij 1 N N P 0 P X m ij 0 m=1 P 0 m=1 P X m ij 0 + P 1 m=1 P X m ij 1 N N 0 =in contact 1 =not in contact 3/20/
21 Accuracy Accuracy Contact prediction accuracy comparison Accuracy of the prediction: Acc = N corr /N T N corr = # of correctly predicted contacts in the contact map N T = # of predicted contacts in the contact map easy targets Top L/5 long range 48 hard targets Top L/5 long range 3/20/
22 Contact prediction accuracy comparison (all ranges) Methods Short (6-11) Medium (12-24) Long (>24) BETACON ( ) ( ) ( ) SVMSEQ ( ) ( ) ( ) SVMcon ( ) ( ) ( ) PSICOV ( ) ( ) ( ) CCMpred ( ) ( ) ( ) FreeContact ( ) ( ) ( ) STRUCTCH ( ) ( ) ( ) MetaPSICOV ( ) ( ) ( ) NeBcon /20/
23 Contact prediction accuracy comparison (long range) Average ACC of MetaPSICOV = Average ACC of NBC = P-value= 0.03 Average ACC of NeBcon= Average ACC of NBC = P-value= /20/2017 He et al., Bioinformatics (2017) 23

 All contacts are evenly distributed when L>100")
24 Diversity of contact maps 100 H = p i log 2 p i i p i is the fraction of the top-l contacts at ith cell, where L is the length of the protein H min = 0 All contacts are accumulated in one cell H max =6.64 (=log 2 100) All contacts are evenly distributed when L>100 3/20/2017
25 Diversity of contact maps Methods Long All BETACON (8.4*10-16 ) (6.9*10-25 ) SVMSEQ (4.9*10-7 ) (5.6*10-13 ) SVMcon (1.5*10-16 ) (1.2*10-24 ) PSICOV (6.2*10-2 ) (1.23*10-2 ) CCMpred (6.9*10-9 ) (1.1*10-6 ) FreeContact (4.5*10-10 ) (5.0*10-6 ) STRUCTCH (2.6*10-8 ) (7.7*10-17 ) MetaPSICOV (4.0*10-5 ) (9.7*10-6 ) NeBcon (6.5*10-5 ) (3.3*10-9 ) Native /20/
26 Example: diversity of contact maps He et al., Bioinformatics (2017) 3/20/



27 C-QUARK: Contact map guided ab initio structure prediction NeBcon Knowledge-based potentials: 3/20/
28 C-QUARK in CASP 12 Groups Z C-QUARK 65.1 Baker-Rosetta 60.3 GOAL 49.9 RaptorX 44.2 ToyPred_ 40.4 Multicom-Novl 19.4 Seok-server 9.2 IntFOLD4 9.1 FFAS-3D 8.4 FALCON_TOPO 6.3 Here, Z-score (Z) represents the significance of the structure predictions by each group compared to the average performance 3/20/

29 GPCR-AIM: Ab initio GPCR structure prediction 3/20/
30 References Xu, D., and Zhang, Y., "Ab initio protein structure assembly using continuous structure fragments and optimized knowledge-based force field," Proteins-Structure Function and Bioinformatics, 80(7), pp (2012) Xu, D., and Zhang, Y., Toward optimal fragment generation for ab initio protein structure assembly," Proteins-Structure Function and Bioinformatics, 81(2), pp (2012) Zhang et al., "Integration of QUARK and I-TASSER for Ab Initio Protein Structure Prediction in CASP11," Proteins, 84, pp (2015) He, B., Mortuza, S.M., Shen, H., Wang, Y., Zhang, Y. NeBcon: Protein contact map prediction using neural network training coupled with naïve Bayes classifiers. Bioinformatics (2017) (In press) He, B., Mortuza, S.M., Wang, Y., Zhang, Y. NeBcon used to improve structure prediction. (2017) (In preparation) Wu, H., Zhang, C., Zhang, Y., Assemble atomic structure of G proteincoupled receptors from primary sequences. (2017) (In preparation) 3/20/








31 Thank You!! umich.edu/quark/ umich.edu/nebcon/ umich.edu/c-quark/ umich.edu/gpcr-aim/
proteins Ab initio protein structure assembly using continuous structure fragments and optimized knowledge-based force field
 proteins STRUCTURE O FUNCTION O BIOINFORMATICS Ab initio protein structure assembly using continuous structure fragments and optimized knowledge-based force field Dong Xu1 and Yang Zhang1,2* 1 Department
proteins STRUCTURE O FUNCTION O BIOINFORMATICS Ab initio protein structure assembly using continuous structure fragments and optimized knowledge-based force field Dong Xu1 and Yang Zhang1,2* 1 Department
Prediction and refinement of NMR structures from sparse experimental data
 Prediction and refinement of NMR structures from sparse experimental data Jeff Skolnick Director Center for the Study of Systems Biology School of Biology Georgia Institute of Technology Overview of talk
Prediction and refinement of NMR structures from sparse experimental data Jeff Skolnick Director Center for the Study of Systems Biology School of Biology Georgia Institute of Technology Overview of talk
Protein Structure Prediction
 Protein Structure Prediction Michael Feig MMTSB/CTBP 2009 Summer Workshop From Sequence to Structure SEALGDTIVKNA Folding with All-Atom Models AAQAAAAQAAAAQAA All-atom MD in general not succesful for real
Protein Structure Prediction Michael Feig MMTSB/CTBP 2009 Summer Workshop From Sequence to Structure SEALGDTIVKNA Folding with All-Atom Models AAQAAAAQAAAAQAA All-atom MD in general not succesful for real
Protein Structure Prediction, Engineering & Design CHEM 430
 Protein Structure Prediction, Engineering & Design CHEM 430 Eero Saarinen The free energy surface of a protein Protein Structure Prediction & Design Full Protein Structure from Sequence - High Alignment
Protein Structure Prediction, Engineering & Design CHEM 430 Eero Saarinen The free energy surface of a protein Protein Structure Prediction & Design Full Protein Structure from Sequence - High Alignment
Template-Based Modeling of Protein Structure
 Template-Based Modeling of Protein Structure David Constant Biochemistry 218 December 11, 2011 Introduction. Much can be learned about the biology of a protein from its structure. Simply put, structure
Template-Based Modeling of Protein Structure David Constant Biochemistry 218 December 11, 2011 Introduction. Much can be learned about the biology of a protein from its structure. Simply put, structure
Improving De novo Protein Structure Prediction using Contact Maps Information
 CIBCB 2017 IEEE Conference on Computational Intelligence in Bioinformatics and Computational Biology Improving De novo Protein Structure Prediction using Contact Maps Information Karina Baptista dos Santos
CIBCB 2017 IEEE Conference on Computational Intelligence in Bioinformatics and Computational Biology Improving De novo Protein Structure Prediction using Contact Maps Information Karina Baptista dos Santos
proteins Prediction Methods and Reports
 proteins STRUCTURE O FUNCTION O BIOINFORMATICS Prediction Methods and Reports Automated protein structure modeling in CASP9 by I-TASSER pipeline combined with QUARK-based ab initio folding and FG-MD-based
proteins STRUCTURE O FUNCTION O BIOINFORMATICS Prediction Methods and Reports Automated protein structure modeling in CASP9 by I-TASSER pipeline combined with QUARK-based ab initio folding and FG-MD-based
CMPS 3110: Bioinformatics. Tertiary Structure Prediction
 CMPS 3110: Bioinformatics Tertiary Structure Prediction Tertiary Structure Prediction Why Should Tertiary Structure Prediction Be Possible? Molecules obey the laws of physics! Conformation space is finite
CMPS 3110: Bioinformatics Tertiary Structure Prediction Tertiary Structure Prediction Why Should Tertiary Structure Prediction Be Possible? Molecules obey the laws of physics! Conformation space is finite
CMPS 6630: Introduction to Computational Biology and Bioinformatics. Tertiary Structure Prediction
 CMPS 6630: Introduction to Computational Biology and Bioinformatics Tertiary Structure Prediction Tertiary Structure Prediction Why Should Tertiary Structure Prediction Be Possible? Molecules obey the
CMPS 6630: Introduction to Computational Biology and Bioinformatics Tertiary Structure Prediction Tertiary Structure Prediction Why Should Tertiary Structure Prediction Be Possible? Molecules obey the
Template Free Protein Structure Modeling Jianlin Cheng, PhD
 Template Free Protein Structure Modeling Jianlin Cheng, PhD Professor Department of EECS Informatics Institute University of Missouri, Columbia 2018 Protein Energy Landscape & Free Sampling http://pubs.acs.org/subscribe/archive/mdd/v03/i09/html/willis.html
Template Free Protein Structure Modeling Jianlin Cheng, PhD Professor Department of EECS Informatics Institute University of Missouri, Columbia 2018 Protein Energy Landscape & Free Sampling http://pubs.acs.org/subscribe/archive/mdd/v03/i09/html/willis.html
Protein Structure Prediction
 Protein Structure Prediction Michael Feig MMTSB/CTBP 2006 Summer Workshop From Sequence to Structure SEALGDTIVKNA Ab initio Structure Prediction Protocol Amino Acid Sequence Conformational Sampling to
Protein Structure Prediction Michael Feig MMTSB/CTBP 2006 Summer Workshop From Sequence to Structure SEALGDTIVKNA Ab initio Structure Prediction Protocol Amino Acid Sequence Conformational Sampling to
Syllabus of BIOINF 528 (2017 Fall, Bioinformatics Program)
") Syllabus of BIOINF 528 (2017 Fall, Bioinformatics Program) Course Name: Structural Bioinformatics Course Description: Instructor: This course introduces fundamental concepts and methods for structural
Syllabus of BIOINF 528 (2017 Fall, Bioinformatics Program) Course Name: Structural Bioinformatics Course Description: Instructor: This course introduces fundamental concepts and methods for structural
3DRobot: automated generation of diverse and well-packed protein structure decoys
 Bioinformatics, 32(3), 2016, 378 387 doi: 10.1093/bioinformatics/btv601 Advance Access Publication Date: 14 October 2015 Original Paper Structural bioinformatics 3DRobot: automated generation of diverse
Bioinformatics, 32(3), 2016, 378 387 doi: 10.1093/bioinformatics/btv601 Advance Access Publication Date: 14 October 2015 Original Paper Structural bioinformatics 3DRobot: automated generation of diverse
Programme Last week s quiz results + Summary Fold recognition Break Exercise: Modelling remote homologues
 Programme 8.00-8.20 Last week s quiz results + Summary 8.20-9.00 Fold recognition 9.00-9.15 Break 9.15-11.20 Exercise: Modelling remote homologues 11.20-11.40 Summary & discussion 11.40-12.00 Quiz 1 Feedback
Programme 8.00-8.20 Last week s quiz results + Summary 8.20-9.00 Fold recognition 9.00-9.15 Break 9.15-11.20 Exercise: Modelling remote homologues 11.20-11.40 Summary & discussion 11.40-12.00 Quiz 1 Feedback
Template Free Protein Structure Modeling Jianlin Cheng, PhD
 Template Free Protein Structure Modeling Jianlin Cheng, PhD Associate Professor Computer Science Department Informatics Institute University of Missouri, Columbia 2013 Protein Energy Landscape & Free Sampling
Template Free Protein Structure Modeling Jianlin Cheng, PhD Associate Professor Computer Science Department Informatics Institute University of Missouri, Columbia 2013 Protein Energy Landscape & Free Sampling
Protein Structure Prediction
 Page 1 Protein Structure Prediction Russ B. Altman BMI 214 CS 274 Protein Folding is different from structure prediction --Folding is concerned with the process of taking the 3D shape, usually based on
Page 1 Protein Structure Prediction Russ B. Altman BMI 214 CS 274 Protein Folding is different from structure prediction --Folding is concerned with the process of taking the 3D shape, usually based on
Design of a Novel Globular Protein Fold with Atomic-Level Accuracy
 Design of a Novel Globular Protein Fold with Atomic-Level Accuracy Brian Kuhlman, Gautam Dantas, Gregory C. Ireton, Gabriele Varani, Barry L. Stoddard, David Baker Presented by Kate Stafford 4 May 05 Protein
Design of a Novel Globular Protein Fold with Atomic-Level Accuracy Brian Kuhlman, Gautam Dantas, Gregory C. Ireton, Gabriele Varani, Barry L. Stoddard, David Baker Presented by Kate Stafford 4 May 05 Protein
Protein structure prediction. CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror
 Protein structure prediction CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror 1 Outline Why predict protein structure? Can we use (pure) physics-based methods? Knowledge-based methods Two major
Protein structure prediction CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror 1 Outline Why predict protein structure? Can we use (pure) physics-based methods? Knowledge-based methods Two major
Improving the Physical Realism and Structural Accuracy of Protein Models by a Two-Step Atomic-Level Energy Minimization
 Biophysical Journal Volume 101 November 2011 2525 2534 2525 Improving the Physical Realism and Structural Accuracy of Protein Models by a Two-Step Atomic-Level Energy Minimization Dong Xu and Yang Zhang
Biophysical Journal Volume 101 November 2011 2525 2534 2525 Improving the Physical Realism and Structural Accuracy of Protein Models by a Two-Step Atomic-Level Energy Minimization Dong Xu and Yang Zhang
114 Grundlagen der Bioinformatik, SS 09, D. Huson, July 6, 2009
 114 Grundlagen der Bioinformatik, SS 09, D. Huson, July 6, 2009 9 Protein tertiary structure Sources for this chapter, which are all recommended reading: D.W. Mount. Bioinformatics: Sequences and Genome
114 Grundlagen der Bioinformatik, SS 09, D. Huson, July 6, 2009 9 Protein tertiary structure Sources for this chapter, which are all recommended reading: D.W. Mount. Bioinformatics: Sequences and Genome
Supporting Online Material for
 www.sciencemag.org/cgi/content/full/309/5742/1868/dc1 Supporting Online Material for Toward High-Resolution de Novo Structure Prediction for Small Proteins Philip Bradley, Kira M. S. Misura, David Baker*
www.sciencemag.org/cgi/content/full/309/5742/1868/dc1 Supporting Online Material for Toward High-Resolution de Novo Structure Prediction for Small Proteins Philip Bradley, Kira M. S. Misura, David Baker*
Protein structure prediction. CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror
 Protein structure prediction CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror 1 Outline Why predict protein structure? Can we use (pure) physics-based methods? Knowledge-based methods Two major
Protein structure prediction CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror 1 Outline Why predict protein structure? Can we use (pure) physics-based methods? Knowledge-based methods Two major
Statistical Machine Learning Methods for Bioinformatics IV. Neural Network & Deep Learning Applications in Bioinformatics
 Statistical Machine Learning Methods for Bioinformatics IV. Neural Network & Deep Learning Applications in Bioinformatics Jianlin Cheng, PhD Department of Computer Science University of Missouri, Columbia
Statistical Machine Learning Methods for Bioinformatics IV. Neural Network & Deep Learning Applications in Bioinformatics Jianlin Cheng, PhD Department of Computer Science University of Missouri, Columbia
Protein Structure Determination from Pseudocontact Shifts Using ROSETTA
 Supporting Information Protein Structure Determination from Pseudocontact Shifts Using ROSETTA Christophe Schmitz, Robert Vernon, Gottfried Otting, David Baker and Thomas Huber Table S0. Biological Magnetic
Supporting Information Protein Structure Determination from Pseudocontact Shifts Using ROSETTA Christophe Schmitz, Robert Vernon, Gottfried Otting, David Baker and Thomas Huber Table S0. Biological Magnetic
Giri Narasimhan. CAP 5510: Introduction to Bioinformatics. ECS 254; Phone: x3748
 CAP 5510: Introduction to Bioinformatics Giri Narasimhan ECS 254; Phone: x3748 giri@cis.fiu.edu www.cis.fiu.edu/~giri/teach/bioinfs07.html 2/15/07 CAP5510 1 EM Algorithm Goal: Find θ, Z that maximize Pr
CAP 5510: Introduction to Bioinformatics Giri Narasimhan ECS 254; Phone: x3748 giri@cis.fiu.edu www.cis.fiu.edu/~giri/teach/bioinfs07.html 2/15/07 CAP5510 1 EM Algorithm Goal: Find θ, Z that maximize Pr
Human and Server CAPRI Protein Docking Prediction Using LZerD with Combined Scoring Functions. Daisuke Kihara
 Human and Server CAPRI Protein Docking Prediction Using LZerD with Combined Scoring Functions Daisuke Kihara Department of Biological Sciences Department of Computer Science Purdue University, Indiana,
Human and Server CAPRI Protein Docking Prediction Using LZerD with Combined Scoring Functions Daisuke Kihara Department of Biological Sciences Department of Computer Science Purdue University, Indiana,
Presentation Outline. Prediction of Protein Secondary Structure using Neural Networks at Better than 70% Accuracy
 Prediction of Protein Secondary Structure using Neural Networks at Better than 70% Accuracy Burkhard Rost and Chris Sander By Kalyan C. Gopavarapu 1 Presentation Outline Major Terminology Problem Method
Prediction of Protein Secondary Structure using Neural Networks at Better than 70% Accuracy Burkhard Rost and Chris Sander By Kalyan C. Gopavarapu 1 Presentation Outline Major Terminology Problem Method
Protein Dynamics. The space-filling structures of myoglobin and hemoglobin show that there are no pathways for O 2 to reach the heme iron.
 Protein Dynamics The space-filling structures of myoglobin and hemoglobin show that there are no pathways for O 2 to reach the heme iron. Below is myoglobin hydrated with 350 water molecules. Only a small
Protein Dynamics The space-filling structures of myoglobin and hemoglobin show that there are no pathways for O 2 to reach the heme iron. Below is myoglobin hydrated with 350 water molecules. Only a small
Predicting protein contact map using evolutionary and physical constraints by integer programming (extended version)
") Predicting protein contact map using evolutionary and physical constraints by integer programming (extended version) Zhiyong Wang 1 and Jinbo Xu 1,* 1 Toyota Technological Institute at Chicago 6045 S Kenwood,
Predicting protein contact map using evolutionary and physical constraints by integer programming (extended version) Zhiyong Wang 1 and Jinbo Xu 1,* 1 Toyota Technological Institute at Chicago 6045 S Kenwood,
Procheck output. Bond angles (Procheck) Structure verification and validation Bond lengths (Procheck) Introduction to Bioinformatics.
 Structure verification and validation Bond lengths (Procheck) Introduction to Bioinformatics.") Structure verification and validation Bond lengths (Procheck) Introduction to Bioinformatics Iosif Vaisman Email: ivaisman@gmu.edu ----------------------------------------------------------------- Bond
Structure verification and validation Bond lengths (Procheck) Introduction to Bioinformatics Iosif Vaisman Email: ivaisman@gmu.edu ----------------------------------------------------------------- Bond
Chemical Shift Restraints Tools and Methods. Andrea Cavalli
 Chemical Shift Restraints Tools and Methods Andrea Cavalli Overview Methods Overview Methods Details Overview Methods Details Results/Discussion Overview Methods Methods Cheshire base solid-state Methods
Chemical Shift Restraints Tools and Methods Andrea Cavalli Overview Methods Overview Methods Details Overview Methods Details Results/Discussion Overview Methods Methods Cheshire base solid-state Methods
Can protein model accuracy be. identified? NO! CBS, BioCentrum, Morten Nielsen, DTU
 Can protein model accuracy be identified? Morten Nielsen, CBS, BioCentrum, DTU NO! Identification of Protein-model accuracy Why is it important? What is accuracy RMSD, fraction correct, Protein model correctness/quality
Can protein model accuracy be identified? Morten Nielsen, CBS, BioCentrum, DTU NO! Identification of Protein-model accuracy Why is it important? What is accuracy RMSD, fraction correct, Protein model correctness/quality
Ab-initio protein structure prediction
 Ab-initio protein structure prediction Jaroslaw Pillardy Computational Biology Service Unit Cornell Theory Center, Cornell University Ithaca, NY USA Methods for predicting protein structure 1. Homology
Ab-initio protein structure prediction Jaroslaw Pillardy Computational Biology Service Unit Cornell Theory Center, Cornell University Ithaca, NY USA Methods for predicting protein structure 1. Homology
Presenter: She Zhang
 Presenter: She Zhang Introduction Dr. David Baker Introduction Why design proteins de novo? It is not clear how non-covalent interactions favor one specific native structure over many other non-native
Presenter: She Zhang Introduction Dr. David Baker Introduction Why design proteins de novo? It is not clear how non-covalent interactions favor one specific native structure over many other non-native
ALL LECTURES IN SB Introduction
 1. Introduction 2. Molecular Architecture I 3. Molecular Architecture II 4. Molecular Simulation I 5. Molecular Simulation II 6. Bioinformatics I 7. Bioinformatics II 8. Prediction I 9. Prediction II ALL
1. Introduction 2. Molecular Architecture I 3. Molecular Architecture II 4. Molecular Simulation I 5. Molecular Simulation II 6. Bioinformatics I 7. Bioinformatics II 8. Prediction I 9. Prediction II ALL
Protein Structure Determination
 Protein Structure Determination Given a protein sequence, determine its 3D structure 1 MIKLGIVMDP IANINIKKDS SFAMLLEAQR RGYELHYMEM GDLYLINGEA 51 RAHTRTLNVK QNYEEWFSFV GEQDLPLADL DVILMRKDPP FDTEFIYATY 101
Protein Structure Determination Given a protein sequence, determine its 3D structure 1 MIKLGIVMDP IANINIKKDS SFAMLLEAQR RGYELHYMEM GDLYLINGEA 51 RAHTRTLNVK QNYEEWFSFV GEQDLPLADL DVILMRKDPP FDTEFIYATY 101
TASSER: An Automated Method for the Prediction of Protein Tertiary Structures in CASP6
 PROTEINS: Structure, Function, and Bioinformatics Suppl 7:91 98 (2005) TASSER: An Automated Method for the Prediction of Protein Tertiary Structures in CASP6 Yang Zhang, Adrian K. Arakaki, and Jeffrey
PROTEINS: Structure, Function, and Bioinformatics Suppl 7:91 98 (2005) TASSER: An Automated Method for the Prediction of Protein Tertiary Structures in CASP6 Yang Zhang, Adrian K. Arakaki, and Jeffrey
Protein Modeling Methods. Knowledge. Protein Modeling Methods. Fold Recognition. Knowledge-based methods. Introduction to Bioinformatics
 Protein Modeling Methods Introduction to Bioinformatics Iosif Vaisman Ab initio methods Energy-based methods Knowledge-based methods Email: ivaisman@gmu.edu Protein Modeling Methods Ab initio methods:
Protein Modeling Methods Introduction to Bioinformatics Iosif Vaisman Ab initio methods Energy-based methods Knowledge-based methods Email: ivaisman@gmu.edu Protein Modeling Methods Ab initio methods:
Bioinformatics III Structural Bioinformatics and Genome Analysis Part Protein Secondary Structure Prediction. Sepp Hochreiter
 Bioinformatics III Structural Bioinformatics and Genome Analysis Part Protein Secondary Structure Prediction Institute of Bioinformatics Johannes Kepler University, Linz, Austria Chapter 4 Protein Secondary
Bioinformatics III Structural Bioinformatics and Genome Analysis Part Protein Secondary Structure Prediction Institute of Bioinformatics Johannes Kepler University, Linz, Austria Chapter 4 Protein Secondary
Protein Folding Prof. Eugene Shakhnovich
 Protein Folding Eugene Shakhnovich Department of Chemistry and Chemical Biology Harvard University 1 Proteins are folded on various scales As of now we know hundreds of thousands of sequences (Swissprot)
Protein Folding Eugene Shakhnovich Department of Chemistry and Chemical Biology Harvard University 1 Proteins are folded on various scales As of now we know hundreds of thousands of sequences (Swissprot)
Computer simulations of protein folding with a small number of distance restraints
 Vol. 49 No. 3/2002 683 692 QUARTERLY Computer simulations of protein folding with a small number of distance restraints Andrzej Sikorski 1, Andrzej Kolinski 1,2 and Jeffrey Skolnick 2 1 Department of Chemistry,
Vol. 49 No. 3/2002 683 692 QUARTERLY Computer simulations of protein folding with a small number of distance restraints Andrzej Sikorski 1, Andrzej Kolinski 1,2 and Jeffrey Skolnick 2 1 Department of Chemistry,
Recognizing Protein Substructure Similarity Using Segmental Threading
 Article Recognizing Protein Substructure Similarity Using Sitao Wu 2,3 and Yang Zhang 1,2, * 1 Center for Computational Medicine and Bioinformatics, University of Michigan, 100 Washtenaw Avenue, Ann Arbor,
Article Recognizing Protein Substructure Similarity Using Sitao Wu 2,3 and Yang Zhang 1,2, * 1 Center for Computational Medicine and Bioinformatics, University of Michigan, 100 Washtenaw Avenue, Ann Arbor,
CS612 - Algorithms in Bioinformatics
 Fall 2017 Protein Structure Detection Methods October 30, 2017 Comparative Modeling Comparative modeling is modeling of the unknown based on comparison to what is known In the context of modeling or computing
Fall 2017 Protein Structure Detection Methods October 30, 2017 Comparative Modeling Comparative modeling is modeling of the unknown based on comparison to what is known In the context of modeling or computing
Introduction to Comparative Protein Modeling. Chapter 4 Part I
 Introduction to Comparative Protein Modeling Chapter 4 Part I 1 Information on Proteins Each modeling study depends on the quality of the known experimental data. Basis of the model Search in the literature
Introduction to Comparative Protein Modeling Chapter 4 Part I 1 Information on Proteins Each modeling study depends on the quality of the known experimental data. Basis of the model Search in the literature
Generalized ensemble methods for de novo structure prediction. 1 To whom correspondence may be addressed.
 Generalized ensemble methods for de novo structure prediction Alena Shmygelska 1 and Michael Levitt 1 Department of Structural Biology, Stanford University, Stanford, CA 94305-5126 Contributed by Michael
Generalized ensemble methods for de novo structure prediction Alena Shmygelska 1 and Michael Levitt 1 Department of Structural Biology, Stanford University, Stanford, CA 94305-5126 Contributed by Michael
SUPPLEMENTARY MATERIALS
 SUPPLEMENTARY MATERIALS Enhanced Recognition of Transmembrane Protein Domains with Prediction-based Structural Profiles Baoqiang Cao, Aleksey Porollo, Rafal Adamczak, Mark Jarrell and Jaroslaw Meller Contact:
SUPPLEMENTARY MATERIALS Enhanced Recognition of Transmembrane Protein Domains with Prediction-based Structural Profiles Baoqiang Cao, Aleksey Porollo, Rafal Adamczak, Mark Jarrell and Jaroslaw Meller Contact:
CAP 5510 Lecture 3 Protein Structures
 CAP 5510 Lecture 3 Protein Structures Su-Shing Chen Bioinformatics CISE 8/19/2005 Su-Shing Chen, CISE 1 Protein Conformation 8/19/2005 Su-Shing Chen, CISE 2 Protein Conformational Structures Hydrophobicity
CAP 5510 Lecture 3 Protein Structures Su-Shing Chen Bioinformatics CISE 8/19/2005 Su-Shing Chen, CISE 1 Protein Conformation 8/19/2005 Su-Shing Chen, CISE 2 Protein Conformational Structures Hydrophobicity
Chapter 11: Genome-wide protein structure prediction
 Chapter 11: Genome-wide protein structure prediction Srayanta Mukherjee a,b, Andras Szilagyi b,c, Ambrish Roy a,b, Yang Zhang a,b * a Center for Computational Medicine and Bioinformatics, University of
Chapter 11: Genome-wide protein structure prediction Srayanta Mukherjee a,b, Andras Szilagyi b,c, Ambrish Roy a,b, Yang Zhang a,b * a Center for Computational Medicine and Bioinformatics, University of
Prediction of Protein Backbone Structure by Preference Classification with SVM
 Prediction of Protein Backbone Structure by Preference Classification with SVM Kai-Yu Chen #, Chang-Biau Yang #1 and Kuo-Si Huang & # National Sun Yat-sen University, Kaohsiung, Taiwan & National Kaohsiung
Prediction of Protein Backbone Structure by Preference Classification with SVM Kai-Yu Chen #, Chang-Biau Yang #1 and Kuo-Si Huang & # National Sun Yat-sen University, Kaohsiung, Taiwan & National Kaohsiung
FlexPepDock In a nutshell
 FlexPepDock In a nutshell All Tutorial files are located in http://bit.ly/mxtakv FlexPepdock refinement Step 1 Step 3 - Refinement Step 4 - Selection of models Measure of fit FlexPepdock Ab-initio Step
FlexPepDock In a nutshell All Tutorial files are located in http://bit.ly/mxtakv FlexPepdock refinement Step 1 Step 3 - Refinement Step 4 - Selection of models Measure of fit FlexPepdock Ab-initio Step
Neural Networks for Protein Structure Prediction Brown, JMB CS 466 Saurabh Sinha
 Neural Networks for Protein Structure Prediction Brown, JMB 1999 CS 466 Saurabh Sinha Outline Goal is to predict secondary structure of a protein from its sequence Artificial Neural Network used for this
Neural Networks for Protein Structure Prediction Brown, JMB 1999 CS 466 Saurabh Sinha Outline Goal is to predict secondary structure of a protein from its sequence Artificial Neural Network used for this
Atomic-Level Protein Structure Refinement Using Fragment-Guided Molecular Dynamics Conformation Sampling
 Article Atomic-Level Protein Structure Refinement Using Fragment-Guided Molecular Dynamics Conformation Sampling Jian Zhang, 1 Yu Liang, 2 and Yang Zhang 1,3, * 1 Center for Computational Medicine and
Article Atomic-Level Protein Structure Refinement Using Fragment-Guided Molecular Dynamics Conformation Sampling Jian Zhang, 1 Yu Liang, 2 and Yang Zhang 1,3, * 1 Center for Computational Medicine and
proteins 3Drefine: Consistent protein structure refinement by optimizing hydrogen bonding network and atomic-level energy minimization
 proteins STRUCTURE O FUNCTION O BIOINFORMATICS 3Drefine: Consistent protein structure refinement by optimizing hydrogen bonding network and atomic-level energy minimization Debswapna Bhattacharya1 and
proteins STRUCTURE O FUNCTION O BIOINFORMATICS 3Drefine: Consistent protein structure refinement by optimizing hydrogen bonding network and atomic-level energy minimization Debswapna Bhattacharya1 and
CAP 5510: Introduction to Bioinformatics CGS 5166: Bioinformatics Tools. Giri Narasimhan
 CAP 5510: Introduction to Bioinformatics CGS 5166: Bioinformatics Tools Giri Narasimhan ECS 254; Phone: x3748 giri@cis.fiu.edu www.cis.fiu.edu/~giri/teach/bioinff18.html Proteins and Protein Structure
CAP 5510: Introduction to Bioinformatics CGS 5166: Bioinformatics Tools Giri Narasimhan ECS 254; Phone: x3748 giri@cis.fiu.edu www.cis.fiu.edu/~giri/teach/bioinff18.html Proteins and Protein Structure
Mass Spectrometry Coupled Experiments and Protein Structure Modeling Methods
 Int. J. Mol. Sci. 2013, 14, 20635-20657; doi:10.3390/ijms141020635 Review OPEN ACCESS International Journal of Molecular Sciences ISSN 1422-0067 www.mdpi.com/journal/ijms Mass Spectrometry Coupled Experiments
Int. J. Mol. Sci. 2013, 14, 20635-20657; doi:10.3390/ijms141020635 Review OPEN ACCESS International Journal of Molecular Sciences ISSN 1422-0067 www.mdpi.com/journal/ijms Mass Spectrometry Coupled Experiments
Development and Large Scale Benchmark Testing of the PROSPECTOR_3 Threading Algorithm
 PROTEINS: Structure, Function, and Bioinformatics 56:502 518 (2004) Development and Large Scale Benchmark Testing of the PROSPECTOR_3 Threading Algorithm Jeffrey Skolnick,* Daisuke Kihara, and Yang Zhang
PROTEINS: Structure, Function, and Bioinformatics 56:502 518 (2004) Development and Large Scale Benchmark Testing of the PROSPECTOR_3 Threading Algorithm Jeffrey Skolnick,* Daisuke Kihara, and Yang Zhang
Alpha-helical Topology and Tertiary Structure Prediction of Globular Proteins Scott R. McAllister Christodoulos A. Floudas Princeton University
 Alpha-helical Topology and Tertiary Structure Prediction of Globular Proteins Scott R. McAllister Christodoulos A. Floudas Princeton University Department of Chemical Engineering Program of Applied and
Alpha-helical Topology and Tertiary Structure Prediction of Globular Proteins Scott R. McAllister Christodoulos A. Floudas Princeton University Department of Chemical Engineering Program of Applied and
A new prediction strategy for long local protein. structures using an original description
 Author manuscript, published in "Proteins Structure Function and Bioinformatics 2009;76(3):570-87" DOI : 10.1002/prot.22370 A new prediction strategy for long local protein structures using an original
Author manuscript, published in "Proteins Structure Function and Bioinformatics 2009;76(3):570-87" DOI : 10.1002/prot.22370 A new prediction strategy for long local protein structures using an original
Protein quality assessment
 Protein quality assessment Speaker: Renzhi Cao Advisor: Dr. Jianlin Cheng Major: Computer Science May 17 th, 2013 1 Outline Introduction Paper1 Paper2 Paper3 Discussion and research plan Acknowledgement
Protein quality assessment Speaker: Renzhi Cao Advisor: Dr. Jianlin Cheng Major: Computer Science May 17 th, 2013 1 Outline Introduction Paper1 Paper2 Paper3 Discussion and research plan Acknowledgement
Coordinate Refinement on All Atoms of the Protein Backbone with Support Vector Regression
 Coordinate Refinement on All Atoms of the Protein Backbone with Support Vector Regression Ding-Yao Huang, Chiou-Yi Hor and Chang-Biau Yang Department of Computer Science and Engineering, National Sun Yat-sen
Coordinate Refinement on All Atoms of the Protein Backbone with Support Vector Regression Ding-Yao Huang, Chiou-Yi Hor and Chang-Biau Yang Department of Computer Science and Engineering, National Sun Yat-sen
As of December 30, 2003, 23,000 solved protein structures
 The protein structure prediction problem could be solved using the current PDB library Yang Zhang and Jeffrey Skolnick* Center of Excellence in Bioinformatics, University at Buffalo, 901 Washington Street,
The protein structure prediction problem could be solved using the current PDB library Yang Zhang and Jeffrey Skolnick* Center of Excellence in Bioinformatics, University at Buffalo, 901 Washington Street,
Molecular Modeling. Prediction of Protein 3D Structure from Sequence. Vimalkumar Velayudhan. May 21, 2007
 Molecular Modeling Prediction of Protein 3D Structure from Sequence Vimalkumar Velayudhan Jain Institute of Vocational and Advanced Studies May 21, 2007 Vimalkumar Velayudhan Molecular Modeling 1/23 Outline
Molecular Modeling Prediction of Protein 3D Structure from Sequence Vimalkumar Velayudhan Jain Institute of Vocational and Advanced Studies May 21, 2007 Vimalkumar Velayudhan Molecular Modeling 1/23 Outline
BIOINFORMATICS TOOLS & ANALYSIS OF PROTEIN STRUCTURE AND FUNCTION FEI JI. (Under the Direction of Ying Xu) ABSTRACT
 ABSTRACT") BIOINFORMATICS TOOLS & ANALYSIS OF PROTEIN STRUCTURE AND FUNCTION by FEI JI (Under the Direction of Ying Xu) ABSTRACT This dissertation mainly focuses on protein structure and functional studies from the
BIOINFORMATICS TOOLS & ANALYSIS OF PROTEIN STRUCTURE AND FUNCTION by FEI JI (Under the Direction of Ying Xu) ABSTRACT This dissertation mainly focuses on protein structure and functional studies from the
PROTEIN STRUCTURE PREDICTION II
 PROTEIN STRUCTURE PREDICTION II Jeffrey Skolnick 1,2 Yang Zhang 1 Because the molecular function of a protein depends on its three dimensional structure, which is often unknown, protein structure prediction
PROTEIN STRUCTURE PREDICTION II Jeffrey Skolnick 1,2 Yang Zhang 1 Because the molecular function of a protein depends on its three dimensional structure, which is often unknown, protein structure prediction
Structural Bioinformatics
 arxiv:1712.00425v1 [q-bio.bm] 1 Dec 2017 Structural Bioinformatics Sanne Abeln K. Anton Feenstra Centre for Integrative Bioinformatics (IBIVU), and Department of Computer Science, Vrije Universiteit, De
arxiv:1712.00425v1 [q-bio.bm] 1 Dec 2017 Structural Bioinformatics Sanne Abeln K. Anton Feenstra Centre for Integrative Bioinformatics (IBIVU), and Department of Computer Science, Vrije Universiteit, De
TOUCHSTONE: A Unified Approach to Protein Structure Prediction
 PROTEINS: Structure, Function, and Genetics 53:469 479 (2003) TOUCHSTONE: A Unified Approach to Protein Structure Prediction Jeffrey Skolnick, 1 * Yang Zhang, 1 Adrian K. Arakaki, 1 Andrzej Kolinski, 1,2
PROTEINS: Structure, Function, and Genetics 53:469 479 (2003) TOUCHSTONE: A Unified Approach to Protein Structure Prediction Jeffrey Skolnick, 1 * Yang Zhang, 1 Adrian K. Arakaki, 1 Andrzej Kolinski, 1,2
Bioinformatics. Macromolecular structure
 Bioinformatics Macromolecular structure Contents Determination of protein structure Structure databases Secondary structure elements (SSE) Tertiary structure Structure analysis Structure alignment Domain
Bioinformatics Macromolecular structure Contents Determination of protein structure Structure databases Secondary structure elements (SSE) Tertiary structure Structure analysis Structure alignment Domain
CMPS 6630: Introduction to Computational Biology and Bioinformatics. Structure Comparison
 CMPS 6630: Introduction to Computational Biology and Bioinformatics Structure Comparison Protein Structure Comparison Motivation Understand sequence and structure variability Understand Domain architecture
CMPS 6630: Introduction to Computational Biology and Bioinformatics Structure Comparison Protein Structure Comparison Motivation Understand sequence and structure variability Understand Domain architecture
Basics of protein structure
 Today: 1. Projects a. Requirements: i. Critical review of one paper ii. At least one computational result b. Noon, Dec. 3 rd written report and oral presentation are due; submit via email to bphys101@fas.harvard.edu
Today: 1. Projects a. Requirements: i. Critical review of one paper ii. At least one computational result b. Noon, Dec. 3 rd written report and oral presentation are due; submit via email to bphys101@fas.harvard.edu
1-D Predictions. Prediction of local features: Secondary structure & surface exposure
 1-D Predictions Prediction of local features: Secondary structure & surface exposure 1 Learning Objectives After today s session you should be able to: Explain the meaning and usage of the following local
1-D Predictions Prediction of local features: Secondary structure & surface exposure 1 Learning Objectives After today s session you should be able to: Explain the meaning and usage of the following local
Protein Structure Analysis with Sequential Monte Carlo Method. Jinfeng Zhang Computational Biology Lab Department of Statistics Harvard University
 Protein Structure Analysis with Sequential Monte Carlo Method Jinfeng Zhang Computational Biology Lab Department of Statistics Harvard University Introduction Structure Function & Interaction Protein structure
Protein Structure Analysis with Sequential Monte Carlo Method Jinfeng Zhang Computational Biology Lab Department of Statistics Harvard University Introduction Structure Function & Interaction Protein structure
Molecular modeling. A fragment sequence of 24 residues encompassing the region of interest of WT-
 SUPPLEMENTARY DATA Molecular dynamics Molecular modeling. A fragment sequence of 24 residues encompassing the region of interest of WT- KISS1R, i.e. the last intracellular domain (Figure S1a), has been
SUPPLEMENTARY DATA Molecular dynamics Molecular modeling. A fragment sequence of 24 residues encompassing the region of interest of WT- KISS1R, i.e. the last intracellular domain (Figure S1a), has been
Multi-Scale Hierarchical Structure Prediction of Helical Transmembrane Proteins
 Multi-Scale Hierarchical Structure Prediction of Helical Transmembrane Proteins Zhong Chen Dept. of Biochemistry and Molecular Biology University of Georgia, Athens, GA 30602 Email: zc@csbl.bmb.uga.edu
Multi-Scale Hierarchical Structure Prediction of Helical Transmembrane Proteins Zhong Chen Dept. of Biochemistry and Molecular Biology University of Georgia, Athens, GA 30602 Email: zc@csbl.bmb.uga.edu
Assignment 2 Atomic-Level Molecular Modeling
 Assignment 2 Atomic-Level Molecular Modeling CS/BIOE/CME/BIOPHYS/BIOMEDIN 279 Due: November 3, 2016 at 3:00 PM The goal of this assignment is to understand the biological and computational aspects of macromolecular
Assignment 2 Atomic-Level Molecular Modeling CS/BIOE/CME/BIOPHYS/BIOMEDIN 279 Due: November 3, 2016 at 3:00 PM The goal of this assignment is to understand the biological and computational aspects of macromolecular
Protein Structures. 11/19/2002 Lecture 24 1
 Protein Structures 11/19/2002 Lecture 24 1 All 3 figures are cartoons of an amino acid residue. 11/19/2002 Lecture 24 2 Peptide bonds in chains of residues 11/19/2002 Lecture 24 3 Angles φ and ψ in the
Protein Structures 11/19/2002 Lecture 24 1 All 3 figures are cartoons of an amino acid residue. 11/19/2002 Lecture 24 2 Peptide bonds in chains of residues 11/19/2002 Lecture 24 3 Angles φ and ψ in the
Homology Modeling. Roberto Lins EPFL - summer semester 2005
 Homology Modeling Roberto Lins EPFL - summer semester 2005 Disclaimer: course material is mainly taken from: P.E. Bourne & H Weissig, Structural Bioinformatics; C.A. Orengo, D.T. Jones & J.M. Thornton,
Homology Modeling Roberto Lins EPFL - summer semester 2005 Disclaimer: course material is mainly taken from: P.E. Bourne & H Weissig, Structural Bioinformatics; C.A. Orengo, D.T. Jones & J.M. Thornton,
HMM applications. Applications of HMMs. Gene finding with HMMs. Using the gene finder
 HMM applications Applications of HMMs Gene finding Pairwise alignment (pair HMMs) Characterizing protein families (profile HMMs) Predicting membrane proteins, and membrane protein topology Gene finding
HMM applications Applications of HMMs Gene finding Pairwise alignment (pair HMMs) Characterizing protein families (profile HMMs) Predicting membrane proteins, and membrane protein topology Gene finding
TOUCHSTONE II: A New Approach to Ab Initio Protein Structure Prediction
 Biophysical Journal Volume 85 August 2003 1145 1164 1145 TOUCHSTONE II: A New Approach to Ab Initio Protein Structure Prediction Yang Zhang,* Andrzej Kolinski,* y and Jeffrey Skolnick* *Center of Excellence
Biophysical Journal Volume 85 August 2003 1145 1164 1145 TOUCHSTONE II: A New Approach to Ab Initio Protein Structure Prediction Yang Zhang,* Andrzej Kolinski,* y and Jeffrey Skolnick* *Center of Excellence
proteins Michal Brylinski and Jeffrey Skolnick* INTRODUCTION
 proteins STRUCTURE O FUNCTION O BIOINFORMATICS FINDSITE-metal: Integrating evolutionary information and machine learning for structure-based metal-binding site prediction at the proteome level Michal Brylinski
proteins STRUCTURE O FUNCTION O BIOINFORMATICS FINDSITE-metal: Integrating evolutionary information and machine learning for structure-based metal-binding site prediction at the proteome level Michal Brylinski
All-atom ab initio folding of a diverse set of proteins
 All-atom ab initio folding of a diverse set of proteins Jae Shick Yang 1, William W. Chen 2,1, Jeffrey Skolnick 3, and Eugene I. Shakhnovich 1, * 1 Department of Chemistry and Chemical Biology 2 Department
All-atom ab initio folding of a diverse set of proteins Jae Shick Yang 1, William W. Chen 2,1, Jeffrey Skolnick 3, and Eugene I. Shakhnovich 1, * 1 Department of Chemistry and Chemical Biology 2 Department
) P = 1 if exp # " s. + 0 otherwise
 P = 1 if exp # s. + 0 otherwise") Supplementary Material Monte Carlo algorithm procedures. The Monte Carlo conformational search algorithm has been successfully applied by programs dedicated to finding new folds (Jones 2001; Rohl, Strauss,
Supplementary Material Monte Carlo algorithm procedures. The Monte Carlo conformational search algorithm has been successfully applied by programs dedicated to finding new folds (Jones 2001; Rohl, Strauss,
Predicting Continuous Local Structure and the Effect of Its Substitution for Secondary Structure in Fragment-Free Protein Structure Prediction
 Predicting Continuous Local Structure and the Effect of Its Substitution for Secondary Structure in Fragment-Free Protein Structure Prediction Author Faraggi, Eshel, Yang, Yuedong, Zhang, Shesheng, Zhou,
Predicting Continuous Local Structure and the Effect of Its Substitution for Secondary Structure in Fragment-Free Protein Structure Prediction Author Faraggi, Eshel, Yang, Yuedong, Zhang, Shesheng, Zhou,
SCOP. all-β class. all-α class, 3 different folds. T4 endonuclease V. 4-helical cytokines. Globin-like
 SCOP all-β class 4-helical cytokines T4 endonuclease V all-α class, 3 different folds Globin-like TIM-barrel fold α/β class Profilin-like fold α+β class http://scop.mrc-lmb.cam.ac.uk/scop CATH Class, Architecture,
SCOP all-β class 4-helical cytokines T4 endonuclease V all-α class, 3 different folds Globin-like TIM-barrel fold α/β class Profilin-like fold α+β class http://scop.mrc-lmb.cam.ac.uk/scop CATH Class, Architecture,
Protein Structure Prediction II Lecturer: Serafim Batzoglou Scribe: Samy Hamdouche
 Protein Structure Prediction II Lecturer: Serafim Batzoglou Scribe: Samy Hamdouche The molecular structure of a protein can be broken down hierarchically. The primary structure of a protein is simply its
Protein Structure Prediction II Lecturer: Serafim Batzoglou Scribe: Samy Hamdouche The molecular structure of a protein can be broken down hierarchically. The primary structure of a protein is simply its
Protein Contact Prediction by Integrating Joint Evolutionary Coupling Analysis and Supervised Learning
 Protein Contact Prediction by Integrating Joint Evolutionary Coupling Analysis and Supervised Learning Abstract Jianzhu Ma Sheng Wang Zhiyong Wang Jinbo Xu Toyota Technological Institute at Chicago {majianzhu,
Protein Contact Prediction by Integrating Joint Evolutionary Coupling Analysis and Supervised Learning Abstract Jianzhu Ma Sheng Wang Zhiyong Wang Jinbo Xu Toyota Technological Institute at Chicago {majianzhu,
Bioinformatics: Secondary Structure Prediction
 Bioinformatics: Secondary Structure Prediction Prof. David Jones d.jones@cs.ucl.ac.uk LMLSTQNPALLKRNIIYWNNVALLWEAGSD The greatest unsolved problem in molecular biology:the Protein Folding Problem? Entries
Bioinformatics: Secondary Structure Prediction Prof. David Jones d.jones@cs.ucl.ac.uk LMLSTQNPALLKRNIIYWNNVALLWEAGSD The greatest unsolved problem in molecular biology:the Protein Folding Problem? Entries
IT og Sundhed 2010/11
 IT og Sundhed 2010/11 Sequence based predictors. Secondary structure and surface accessibility Bent Petersen 13 January 2011 1 NetSurfP Real Value Solvent Accessibility predictions with amino acid associated
IT og Sundhed 2010/11 Sequence based predictors. Secondary structure and surface accessibility Bent Petersen 13 January 2011 1 NetSurfP Real Value Solvent Accessibility predictions with amino acid associated
Template Based Protein Structure Modeling Jianlin Cheng, PhD
 Template Based Protein Structure Modeling Jianlin Cheng, PhD Professor Department of EECS Informatics Institute University of Missouri, Columbia 2018 Sequence, Structure and Function AGCWY Cell Protein
Template Based Protein Structure Modeling Jianlin Cheng, PhD Professor Department of EECS Informatics Institute University of Missouri, Columbia 2018 Sequence, Structure and Function AGCWY Cell Protein
proteins Estimating quality of template-based protein models by alignment stability Hao Chen 1 and Daisuke Kihara 1,2,3,4 * INTRODUCTION
 proteins STRUCTURE O FUNCTION O BIOINFORMATICS Estimating quality of template-based protein models by alignment stability Hao Chen 1 and Daisuke Kihara 1,2,3,4 * 1 Department of Biological Sciences, College
proteins STRUCTURE O FUNCTION O BIOINFORMATICS Estimating quality of template-based protein models by alignment stability Hao Chen 1 and Daisuke Kihara 1,2,3,4 * 1 Department of Biological Sciences, College
Evolutionary design of energy functions for protein structure prediction
 Evolutionary design of energy functions for protein structure prediction Natalio Krasnogor nxk@ cs. nott. ac. uk Paweł Widera, Jonathan Garibaldi 7th Annual HUMIES Awards 2010-07-09 Protein structure prediction
Evolutionary design of energy functions for protein structure prediction Natalio Krasnogor nxk@ cs. nott. ac. uk Paweł Widera, Jonathan Garibaldi 7th Annual HUMIES Awards 2010-07-09 Protein structure prediction
Homology modeling. Dinesh Gupta ICGEB, New Delhi 1/27/2010 5:59 PM
 Homology modeling Dinesh Gupta ICGEB, New Delhi Protein structure prediction Methods: Homology (comparative) modelling Threading Ab-initio Protein Homology modeling Homology modeling is an extrapolation
Homology modeling Dinesh Gupta ICGEB, New Delhi Protein structure prediction Methods: Homology (comparative) modelling Threading Ab-initio Protein Homology modeling Homology modeling is an extrapolation
Homology Modeling (Comparative Structure Modeling) GBCB 5874: Problem Solving in GBCB
 GBCB 5874: Problem Solving in GBCB") Homology Modeling (Comparative Structure Modeling) Aims of Structural Genomics High-throughput 3D structure determination and analysis To determine or predict the 3D structures of all the proteins encoded
Homology Modeling (Comparative Structure Modeling) Aims of Structural Genomics High-throughput 3D structure determination and analysis To determine or predict the 3D structures of all the proteins encoded
Predicting backbone C# angles and dihedrals from protein sequences by stacked sparse auto-encoder deep neural network
 Predicting backbone C# angles and dihedrals from protein sequences by stacked sparse auto-encoder deep neural network Author Lyons, James, Dehzangi, Iman, Heffernan, Rhys, Sharma, Alok, Paliwal, Kuldip,
Predicting backbone C# angles and dihedrals from protein sequences by stacked sparse auto-encoder deep neural network Author Lyons, James, Dehzangi, Iman, Heffernan, Rhys, Sharma, Alok, Paliwal, Kuldip,
Conformational Sampling in Template-Free Protein Loop Structure Modeling: An Overview
 # of Loops, http://dx.doi.org/10.5936/csbj.201302003 CSBJ Conformational Sampling in Template-Free Protein Loop Structure Modeling: An Overview Yaohang Li a,* Abstract: Accurately modeling protein loops
# of Loops, http://dx.doi.org/10.5936/csbj.201302003 CSBJ Conformational Sampling in Template-Free Protein Loop Structure Modeling: An Overview Yaohang Li a,* Abstract: Accurately modeling protein loops
Reconstruction of Protein Backbone with the α-carbon Coordinates *
 JOURNAL OF INFORMATION SCIENCE AND ENGINEERING 26, 1107-1119 (2010) Reconstruction of Protein Backbone with the α-carbon Coordinates * JEN-HUI WANG, CHANG-BIAU YANG + AND CHIOU-TING TSENG Department of
JOURNAL OF INFORMATION SCIENCE AND ENGINEERING 26, 1107-1119 (2010) Reconstruction of Protein Backbone with the α-carbon Coordinates * JEN-HUI WANG, CHANG-BIAU YANG + AND CHIOU-TING TSENG Department of
Can a continuum solvent model reproduce the free energy landscape of a β-hairpin folding in water?
 Can a continuum solvent model reproduce the free energy landscape of a β-hairpin folding in water? Ruhong Zhou 1 and Bruce J. Berne 2 1 IBM Thomas J. Watson Research Center; and 2 Department of Chemistry,
Can a continuum solvent model reproduce the free energy landscape of a β-hairpin folding in water? Ruhong Zhou 1 and Bruce J. Berne 2 1 IBM Thomas J. Watson Research Center; and 2 Department of Chemistry,
3D Structure. Prediction & Assessment Pt. 2. David Wishart 3-41 Athabasca Hall
 3D Structure Prediction & Assessment Pt. 2 David Wishart 3-41 Athabasca Hall david.wishart@ualberta.ca Objectives Become familiar with methods and algorithms for secondary Structure Prediction Become familiar
3D Structure Prediction & Assessment Pt. 2 David Wishart 3-41 Athabasca Hall david.wishart@ualberta.ca Objectives Become familiar with methods and algorithms for secondary Structure Prediction Become familiar
Protein structure analysis. Risto Laakso 10th January 2005
 Protein structure analysis Risto Laakso risto.laakso@hut.fi 10th January 2005 1 1 Summary Various methods of protein structure analysis were examined. Two proteins, 1HLB (Sea cucumber hemoglobin) and 1HLM
Protein structure analysis Risto Laakso risto.laakso@hut.fi 10th January 2005 1 1 Summary Various methods of protein structure analysis were examined. Two proteins, 1HLB (Sea cucumber hemoglobin) and 1HLM
proteins Comparison of structure-based and threading-based approaches to protein functional annotation Michal Brylinski, and Jeffrey Skolnick*
 proteins STRUCTURE O FUNCTION O BIOINFORMATICS Comparison of structure-based and threading-based approaches to protein functional annotation Michal Brylinski, and Jeffrey Skolnick* Center for the Study
proteins STRUCTURE O FUNCTION O BIOINFORMATICS Comparison of structure-based and threading-based approaches to protein functional annotation Michal Brylinski, and Jeffrey Skolnick* Center for the Study
Outline. The ensemble folding kinetics of protein G from an all-atom Monte Carlo simulation. Unfolded Folded. What is protein folding?
 The ensemble folding kinetics of protein G from an all-atom Monte Carlo simulation By Jun Shimada and Eugine Shaknovich Bill Hawse Dr. Bahar Elisa Sandvik and Mehrdad Safavian Outline Background on protein
The ensemble folding kinetics of protein G from an all-atom Monte Carlo simulation By Jun Shimada and Eugine Shaknovich Bill Hawse Dr. Bahar Elisa Sandvik and Mehrdad Safavian Outline Background on protein