Homology modeling. Dinesh Gupta ICGEB, New Delhi 1/27/2010 5:59 PM
|
|
|
- Bernadette Stone
- 5 years ago
- Views:
Transcription
1 Homology modeling Dinesh Gupta ICGEB, New Delhi
2 Protein structure prediction Methods: Homology (comparative) modelling Threading Ab-initio
3 Protein Homology modeling Homology modeling is an extrapolation of protein structure for a target sequence using the known 3D structure of similar sequence as a template. Basis: proteins with similar sequences are likely to assume same folding Certain proteins with as low as 25% similarity have been observed to assume same 3D structure
4 The accuracy of modeling is proportional to the similarity in primary sequences
5 Steps Given: A query sequence Q A database of known protein structures Find protein P such that P has high sequence similarity to Q Return P s structure as an approximation to Q s structure Energy minimization
6 Sofware for homology molecular modelling Freeware: available for all OS Downloadable Modeller (Sali, 1998) DeepView (SwissPDB viewer) WHATIF (Krieger et al. 2003) Web based: SWISS MODEL server ( MODEL.html) CPH model server ( SDSC1 server (

7
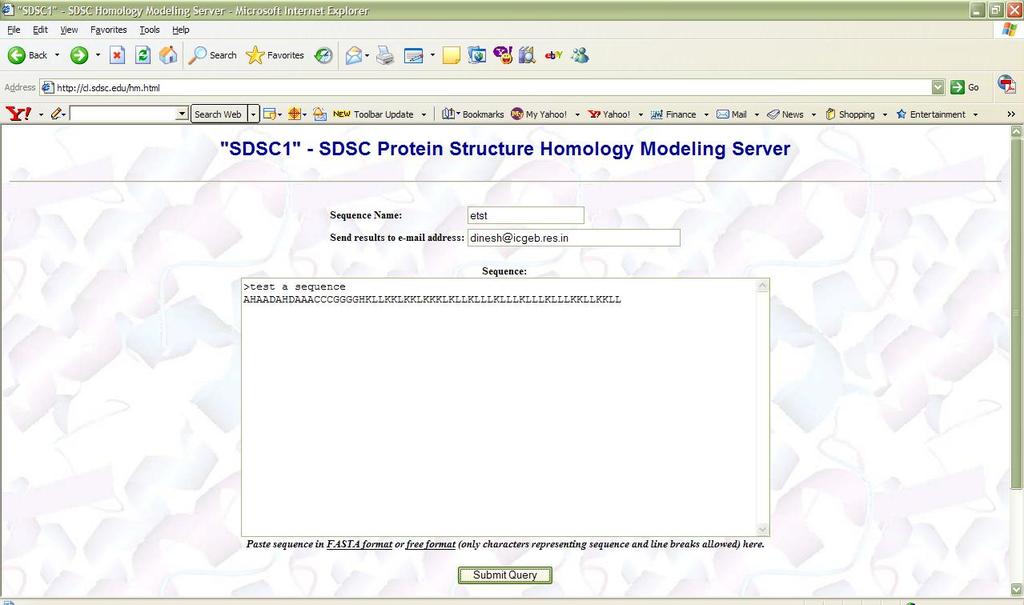
8

9
10 Protein structure prediction Methods: Homology (comparative) modelling Threading Ab-initio
11 Threading Structure prediction that picks up where homology modelling leaves off. Recognize folds in proteins having no similarity to known proteins structures Very approximate models Check by forcing a sequence of structure into known folds checking the packing of aa residues, including sides chains, in each fold.
12 2 kinds of threading Three dimensional threading Distance Based Method (DBM) Two dimensional threading Prediction Based Methods (PBM)
13
14 Threading software EVA: SAMt99: o/hmm-apps/t99-model-librarysearch.html 3DPSSM: FUGUE: Metaservers:

15

16


17

18
19 Protein structure prediction Methods: Homology (comparative) modelling Threading Ab-initio
20 Ab initio structure prediction Still experimental ROSETTA (David Baker)
21 Energy minimization (Molecular Mechanics, MM) Energy minimization is an important part of both empirical and predicted structures MM could be used to calculate large scale conformational changes over long periods of time, but currently computationally infeasible.
22 How does MM work? Three aspects: Functions that describe the forces acting on the atoms Numerical integration methods, to calculate the motion of the atoms due to the forces acting on them Long time propagation of the equations of motion Computational demands are intense Accuracy (small errors propagate!) Stability Lots of techniques for approximation (e.g. rigid bodies) and handling artifacts (resonance).
23 The Force Fields How do atoms stretch, vibrate, rotate, etc.? Must represent the constraints on atomic motion (e.g. van der Waals, electrostatic, bonds, etc.) Must also represent solvation effects etc. Quantum solutions exist, but are too complex to calculate for such large systems Empirical (approximate) energy functions must be used. No single best function exists.
24 Real energetics Steric (conformational) energy. Additive combination of Bonded: stretching, bending, stretching and bending Non-bonded: Van der Waals, electrostatic and torsional Minimum energy conformation minimizes these energies Rosetta energy function is an empirical attempt to capture most of this energy function without having to calculate it fully.
25 Bond length Spring-like term for energy based on distance E str = ½k s,ij (r ij -r o ) 2 where k s,ij is the stretching force constant for the bond between i and j, r ij is the length, and r o is the equilibrium bond length
26 Bond bend Same basic idea for bending E bend = ½k b,ij ( ij o ) 2 where where k b,ij is the bending force constant, ij is the instantaneous bond angle, and o is the equilibrium bond angle
27 Stretch-bend When a bond is bent, the two associated bond lengths increase, with interaction term: E str-bend =½k sb,ijk (r ij -r o )( ik - o ) where k sb,ijk is the stretch-bend force constant for the bond between atoms i and j with the bend between atoms i, j, and k.
28 Van der Waals A non-bonded interaction capturing the preferred distance between atoms where A and B are constants depending on the atoms. For two hydrogen atoms, A=70.4kCÅ 6 and B=6286kCÅ 12
29 Electrostatics If bonds in the molecule are polar, some atoms will have partial electrostatic charges, which attract if opposite and repel otherwise. where Q i and Q j are the partial atomic charges for i and j separated by distance r ij, is the dielectric constant of the solute, and k is a units constant (k=2086 kcal/mol)
30 Torsional energy Torsion is the energy needed to rotate about bonds. Only relevant to single bonds, since others are too stiff to rotate at all E tor = ½k tor,1 (1 - cos ) + ½k tor,2 (1-2cos ) + ½k tor,3 (1-3cos ) where is the dihedral angle around the bond, and k tor,1, k tor,2 and k tor,3 are constants for one-, two- and three-fold barriers. energy of 3-fold torsional barrier in ethane
31 Energy minimization Given some energy function and initial conditions, we want to find the minimum energy conformation. Optimization problem, various methods: Steepest descent Conjugate gradient descent Newton-Raphson Various programs: Charmm, Amber are two most widely used (and packaged)
32 Time steps Need time steps of roughly 1/10 the period of the smallest time scale of interest, or about a femtosecond (10-15 s). A million computational steps per nanosecond of simulation...
33 Issues in Molecular Mechanics Solvation models: water & salt are very important to molecular behaviour. Must model as many water atoms as protein atoms. Initial conditions: velocity & position Equilibration: simulated heating and cooling Chaos: sensitivity to initial conditions, and statistical characterization of states Computational issues (e.g. parallelization)
34 Molecular Dynamics Molecules, especially proteins, are not static. Dynamics can be important to function Trajectories, not just minimum energy state. MM ignores kinetic energy, does only potential energy MD takes same force model, but calculates F=ma and calculates velocities of all atoms (as well as positions)
35 Docking Computation to assess binding affinity Looks for conformational and electrostatic "fit" between proteins and other molecules e.g. inhibitors Optimization again: what position and orientation of the two molecules minimizes energy? Large computations, since there are many possible positions to check, and the energy for each position may involve many atoms
36 Virtual Screening Docking small ligands to proteins is a way to find potential drugs. Industrially important A small region of interest (pharmacophore) can be identified, reducing computation Empirical scoring functions are not universal Various search methods: Rigid provides score for whole ligand (accurate) Flexible breaks ligands into pieces and docks them individually

37 Docking example Benzamidine binding to beta-trypsin 3ptb,
38 Macromolecular docking Docking of proteins to proteins or to DNA Important to understanding macromolecular recognition, genetic regulation, etc. Conceptually similar to small molecule docking, but practically much more difficult Score function can't realistically compute energies Use either shape complementarities alone or some kind of mean field approximation
39 Docking Resources AutoDock FlexX and commercially at Dock 3D-Dock which uses an unusual Fourier correlation method and is aimed at protein-protein 1/27/2010 interactions 5:59 PM
40 Lab Exercise-1 Install: MDL chime RasMol SwissPDBviewer Cn3D Explore few protein/dna structures
41 Lab exercise-2 Download sequence file for S. cerevisiae endoplasmic reticulum mannosidase Generate a homology model using SWISS-model server Download the template structure from Compare the model and template structures Repeat the exercise for other protein sequences of your choice

42

43
Molecular Mechanics, Dynamics & Docking
 Molecular Mechanics, Dynamics & Docking Lawrence Hunter, Ph.D. Director, Computational Bioscience Program University of Colorado School of Medicine Larry.Hunter@uchsc.edu http://compbio.uchsc.edu/hunter
Molecular Mechanics, Dynamics & Docking Lawrence Hunter, Ph.D. Director, Computational Bioscience Program University of Colorado School of Medicine Larry.Hunter@uchsc.edu http://compbio.uchsc.edu/hunter
Structural Bioinformatics (C3210) Molecular Mechanics
 Molecular Mechanics") Structural Bioinformatics (C3210) Molecular Mechanics How to Calculate Energies Calculation of molecular energies is of key importance in protein folding, molecular modelling etc. There are two main computational
Structural Bioinformatics (C3210) Molecular Mechanics How to Calculate Energies Calculation of molecular energies is of key importance in protein folding, molecular modelling etc. There are two main computational
Why study protein dynamics?
 Why study protein dynamics? Protein flexibility is crucial for function. One average structure is not enough. Proteins constantly sample configurational space. Transport - binding and moving molecules
Why study protein dynamics? Protein flexibility is crucial for function. One average structure is not enough. Proteins constantly sample configurational space. Transport - binding and moving molecules
Why Proteins Fold? (Parts of this presentation are based on work of Ashok Kolaskar) CS490B: Introduction to Bioinformatics Mar.
 CS490B: Introduction to Bioinformatics Mar.") Why Proteins Fold? (Parts of this presentation are based on work of Ashok Kolaskar) CS490B: Introduction to Bioinformatics Mar. 25, 2002 Molecular Dynamics: Introduction At physiological conditions, the
Why Proteins Fold? (Parts of this presentation are based on work of Ashok Kolaskar) CS490B: Introduction to Bioinformatics Mar. 25, 2002 Molecular Dynamics: Introduction At physiological conditions, the
Example questions for Molecular modelling (Level 4) Dr. Adrian Mulholland
 Dr. Adrian Mulholland") Example questions for Molecular modelling (Level 4) Dr. Adrian Mulholland 1) Question. Two methods which are widely used for the optimization of molecular geometies are the Steepest descents and Newton-Raphson
Example questions for Molecular modelling (Level 4) Dr. Adrian Mulholland 1) Question. Two methods which are widely used for the optimization of molecular geometies are the Steepest descents and Newton-Raphson
Potential Energy (hyper)surface
surface") The Molecular Dynamics Method Thermal motion of a lipid bilayer Water permeation through channels Selective sugar transport Potential Energy (hyper)surface What is Force? Energy U(x) F = " d dx U(x) Conformation
The Molecular Dynamics Method Thermal motion of a lipid bilayer Water permeation through channels Selective sugar transport Potential Energy (hyper)surface What is Force? Energy U(x) F = " d dx U(x) Conformation
Bioengineering 215. An Introduction to Molecular Dynamics for Biomolecules
 Bioengineering 215 An Introduction to Molecular Dynamics for Biomolecules David Parker May 18, 2007 ntroduction A principal tool to study biological molecules is molecular dynamics simulations (MD). MD
Bioengineering 215 An Introduction to Molecular Dynamics for Biomolecules David Parker May 18, 2007 ntroduction A principal tool to study biological molecules is molecular dynamics simulations (MD). MD
An introduction to Molecular Dynamics. EMBO, June 2016
 An introduction to Molecular Dynamics EMBO, June 2016 What is MD? everything that living things do can be understood in terms of the jiggling and wiggling of atoms. The Feynman Lectures in Physics vol.
An introduction to Molecular Dynamics EMBO, June 2016 What is MD? everything that living things do can be understood in terms of the jiggling and wiggling of atoms. The Feynman Lectures in Physics vol.
Molecular dynamics simulation. CS/CME/BioE/Biophys/BMI 279 Oct. 5 and 10, 2017 Ron Dror
 Molecular dynamics simulation CS/CME/BioE/Biophys/BMI 279 Oct. 5 and 10, 2017 Ron Dror 1 Outline Molecular dynamics (MD): The basic idea Equations of motion Key properties of MD simulations Sample applications
Molecular dynamics simulation CS/CME/BioE/Biophys/BMI 279 Oct. 5 and 10, 2017 Ron Dror 1 Outline Molecular dynamics (MD): The basic idea Equations of motion Key properties of MD simulations Sample applications
Kd = koff/kon = [R][L]/[RL]
![Kd = koff/kon = [R][L]/[RL]](/thumbs/96/127564193.jpg "Kd = koff/kon = [R][L]/[RL]") Taller de docking y cribado virtual: Uso de herramientas computacionales en el diseño de fármacos Docking program GLIDE El programa de docking GLIDE Sonsoles Martín-Santamaría Shrödinger is a scientific
Taller de docking y cribado virtual: Uso de herramientas computacionales en el diseño de fármacos Docking program GLIDE El programa de docking GLIDE Sonsoles Martín-Santamaría Shrödinger is a scientific
ENERGY MINIMIZATION AND CONFORMATION SEARCH ANALYSIS OF TYPE-2 ANTI-DIABETES DRUGS
 Int. J. Chem. Sci.: 6(2), 2008, 982-992 EERGY MIIMIZATI AD CFRMATI SEARC AALYSIS F TYPE-2 ATI-DIABETES DRUGS R. PRASAA LAKSMI a, C. ARASIMA KUMAR a, B. VASATA LAKSMI, K. AGA SUDA, K. MAJA, V. JAYA LAKSMI
Int. J. Chem. Sci.: 6(2), 2008, 982-992 EERGY MIIMIZATI AD CFRMATI SEARC AALYSIS F TYPE-2 ATI-DIABETES DRUGS R. PRASAA LAKSMI a, C. ARASIMA KUMAR a, B. VASATA LAKSMI, K. AGA SUDA, K. MAJA, V. JAYA LAKSMI
Building 3D models of proteins
 Building 3D models of proteins Why make a structural model for your protein? The structure can provide clues to the function through structural similarity with other proteins With a structure it is easier
Building 3D models of proteins Why make a structural model for your protein? The structure can provide clues to the function through structural similarity with other proteins With a structure it is easier
Dihedral Angles. Homayoun Valafar. Department of Computer Science and Engineering, USC 02/03/10 CSCE 769
 Dihedral Angles Homayoun Valafar Department of Computer Science and Engineering, USC The precise definition of a dihedral or torsion angle can be found in spatial geometry Angle between to planes Dihedral
Dihedral Angles Homayoun Valafar Department of Computer Science and Engineering, USC The precise definition of a dihedral or torsion angle can be found in spatial geometry Angle between to planes Dihedral
3. An Introduction to Molecular Mechanics
 3. An Introduction to Molecular Mechanics Introduction When you use Chem3D to draw molecules, the program assigns bond lengths and bond angles based on experimental data. The program does not contain real
3. An Introduction to Molecular Mechanics Introduction When you use Chem3D to draw molecules, the program assigns bond lengths and bond angles based on experimental data. The program does not contain real
Programme Last week s quiz results + Summary Fold recognition Break Exercise: Modelling remote homologues
 Programme 8.00-8.20 Last week s quiz results + Summary 8.20-9.00 Fold recognition 9.00-9.15 Break 9.15-11.20 Exercise: Modelling remote homologues 11.20-11.40 Summary & discussion 11.40-12.00 Quiz 1 Feedback
Programme 8.00-8.20 Last week s quiz results + Summary 8.20-9.00 Fold recognition 9.00-9.15 Break 9.15-11.20 Exercise: Modelling remote homologues 11.20-11.40 Summary & discussion 11.40-12.00 Quiz 1 Feedback
Molecular Dynamics Simulation of HIV-1 Reverse. Transcriptase
 Molecular Dynamics Simulation of HIV-1 Reverse Transcriptase Abderrahmane Benghanem 1 Maria Kurnikova 2 1 Rensselaer Polytechnic Institute, Troy, NY 2 Carnegie Mellon University, Pittsburgh, PA June 16,
Molecular Dynamics Simulation of HIV-1 Reverse Transcriptase Abderrahmane Benghanem 1 Maria Kurnikova 2 1 Rensselaer Polytechnic Institute, Troy, NY 2 Carnegie Mellon University, Pittsburgh, PA June 16,
Molecular Mechanics. I. Quantum mechanical treatment of molecular systems
 Molecular Mechanics I. Quantum mechanical treatment of molecular systems The first principle approach for describing the properties of molecules, including proteins, involves quantum mechanics. For example,
Molecular Mechanics I. Quantum mechanical treatment of molecular systems The first principle approach for describing the properties of molecules, including proteins, involves quantum mechanics. For example,
ALL LECTURES IN SB Introduction
 1. Introduction 2. Molecular Architecture I 3. Molecular Architecture II 4. Molecular Simulation I 5. Molecular Simulation II 6. Bioinformatics I 7. Bioinformatics II 8. Prediction I 9. Prediction II ALL
1. Introduction 2. Molecular Architecture I 3. Molecular Architecture II 4. Molecular Simulation I 5. Molecular Simulation II 6. Bioinformatics I 7. Bioinformatics II 8. Prediction I 9. Prediction II ALL
Protein Dynamics. The space-filling structures of myoglobin and hemoglobin show that there are no pathways for O 2 to reach the heme iron.
 Protein Dynamics The space-filling structures of myoglobin and hemoglobin show that there are no pathways for O 2 to reach the heme iron. Below is myoglobin hydrated with 350 water molecules. Only a small
Protein Dynamics The space-filling structures of myoglobin and hemoglobin show that there are no pathways for O 2 to reach the heme iron. Below is myoglobin hydrated with 350 water molecules. Only a small
Examples of Protein Modeling. Protein Modeling. Primary Structure. Protein Structure Description. Protein Sequence Sources. Importing Sequences to MOE
 Examples of Protein Modeling Protein Modeling Visualization Examination of an experimental structure to gain insight about a research question Dynamics To examine the dynamics of protein structures To
Examples of Protein Modeling Protein Modeling Visualization Examination of an experimental structure to gain insight about a research question Dynamics To examine the dynamics of protein structures To
T6.2 Molecular Mechanics
 T6.2 Molecular Mechanics We have seen that Benson group additivities are capable of giving heats of formation of molecules with accuracies comparable to those of the best ab initio procedures. However,
T6.2 Molecular Mechanics We have seen that Benson group additivities are capable of giving heats of formation of molecules with accuracies comparable to those of the best ab initio procedures. However,
Softwares for Molecular Docking. Lokesh P. Tripathi NCBS 17 December 2007
 Softwares for Molecular Docking Lokesh P. Tripathi NCBS 17 December 2007 Molecular Docking Attempt to predict structures of an intermolecular complex between two or more molecules Receptor-ligand (or drug)
Softwares for Molecular Docking Lokesh P. Tripathi NCBS 17 December 2007 Molecular Docking Attempt to predict structures of an intermolecular complex between two or more molecules Receptor-ligand (or drug)
Protein structure prediction. CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror
 Protein structure prediction CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror 1 Outline Why predict protein structure? Can we use (pure) physics-based methods? Knowledge-based methods Two major
Protein structure prediction CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror 1 Outline Why predict protein structure? Can we use (pure) physics-based methods? Knowledge-based methods Two major
3. An Introduction to Molecular Mechanics
 3. An Introduction to Molecular Mechanics Introduction When you use Chem3D to draw molecules, the program assigns bond lengths and bond angles based on experimental data. The program does not contain real
3. An Introduction to Molecular Mechanics Introduction When you use Chem3D to draw molecules, the program assigns bond lengths and bond angles based on experimental data. The program does not contain real
Lecture 11: Potential Energy Functions
 Lecture 11: Potential Energy Functions Dr. Ronald M. Levy ronlevy@temple.edu Originally contributed by Lauren Wickstrom (2011) Microscopic/Macroscopic Connection The connection between microscopic interactions
Lecture 11: Potential Energy Functions Dr. Ronald M. Levy ronlevy@temple.edu Originally contributed by Lauren Wickstrom (2011) Microscopic/Macroscopic Connection The connection between microscopic interactions
Exercise 2: Solvating the Structure Before you continue, follow these steps: Setting up Periodic Boundary Conditions
 Exercise 2: Solvating the Structure HyperChem lets you place a molecular system in a periodic box of water molecules to simulate behavior in aqueous solution, as in a biological system. In this exercise,
Exercise 2: Solvating the Structure HyperChem lets you place a molecular system in a periodic box of water molecules to simulate behavior in aqueous solution, as in a biological system. In this exercise,
Molecular Modeling. Prediction of Protein 3D Structure from Sequence. Vimalkumar Velayudhan. May 21, 2007
 Molecular Modeling Prediction of Protein 3D Structure from Sequence Vimalkumar Velayudhan Jain Institute of Vocational and Advanced Studies May 21, 2007 Vimalkumar Velayudhan Molecular Modeling 1/23 Outline
Molecular Modeling Prediction of Protein 3D Structure from Sequence Vimalkumar Velayudhan Jain Institute of Vocational and Advanced Studies May 21, 2007 Vimalkumar Velayudhan Molecular Modeling 1/23 Outline
Molecular Simulation II. Classical Mechanical Treatment
 Molecular Simulation II Quantum Chemistry Classical Mechanics E = Ψ H Ψ ΨΨ U = E bond +E angle +E torsion +E non-bond Jeffry D. Madura Department of Chemistry & Biochemistry Center for Computational Sciences
Molecular Simulation II Quantum Chemistry Classical Mechanics E = Ψ H Ψ ΨΨ U = E bond +E angle +E torsion +E non-bond Jeffry D. Madura Department of Chemistry & Biochemistry Center for Computational Sciences
Atomistic Modeling of Small-Angle Scattering Data Using SASSIE-web
 Course Introduction Atomistic Modeling of Small-Angle Scattering Data Using SASSIE-web September 21-23, 2016 Advanced Photon Source Argonne National Laboratory, Argonne, IL ccpsas.org Scatters & Simulators
Course Introduction Atomistic Modeling of Small-Angle Scattering Data Using SASSIE-web September 21-23, 2016 Advanced Photon Source Argonne National Laboratory, Argonne, IL ccpsas.org Scatters & Simulators
The Molecular Dynamics Method
 The Molecular Dynamics Method Thermal motion of a lipid bilayer Water permeation through channels Selective sugar transport Potential Energy (hyper)surface What is Force? Energy U(x) F = d dx U(x) Conformation
The Molecular Dynamics Method Thermal motion of a lipid bilayer Water permeation through channels Selective sugar transport Potential Energy (hyper)surface What is Force? Energy U(x) F = d dx U(x) Conformation
Energy functions and their relationship to molecular conformation. CS/CME/BioE/Biophys/BMI 279 Oct. 3 and 5, 2017 Ron Dror
 Energy functions and their relationship to molecular conformation CS/CME/BioE/Biophys/BMI 279 Oct. 3 and 5, 2017 Ron Dror Yesterday s Nobel Prize: single-particle cryoelectron microscopy 2 Outline Energy
Energy functions and their relationship to molecular conformation CS/CME/BioE/Biophys/BMI 279 Oct. 3 and 5, 2017 Ron Dror Yesterday s Nobel Prize: single-particle cryoelectron microscopy 2 Outline Energy
Multiscale Materials Modeling
 Multiscale Materials Modeling Lecture 09 Quantum Mechanics/Molecular Mechanics (QM/MM) Techniques Fundamentals of Sustainable Technology These notes created by David Keffer, University of Tennessee, Knoxville,
Multiscale Materials Modeling Lecture 09 Quantum Mechanics/Molecular Mechanics (QM/MM) Techniques Fundamentals of Sustainable Technology These notes created by David Keffer, University of Tennessee, Knoxville,
Homology Modeling (Comparative Structure Modeling) GBCB 5874: Problem Solving in GBCB
 GBCB 5874: Problem Solving in GBCB") Homology Modeling (Comparative Structure Modeling) Aims of Structural Genomics High-throughput 3D structure determination and analysis To determine or predict the 3D structures of all the proteins encoded
Homology Modeling (Comparative Structure Modeling) Aims of Structural Genomics High-throughput 3D structure determination and analysis To determine or predict the 3D structures of all the proteins encoded
CMPS 6630: Introduction to Computational Biology and Bioinformatics. Tertiary Structure Prediction
 CMPS 6630: Introduction to Computational Biology and Bioinformatics Tertiary Structure Prediction Tertiary Structure Prediction Why Should Tertiary Structure Prediction Be Possible? Molecules obey the
CMPS 6630: Introduction to Computational Biology and Bioinformatics Tertiary Structure Prediction Tertiary Structure Prediction Why Should Tertiary Structure Prediction Be Possible? Molecules obey the
CMPS 3110: Bioinformatics. Tertiary Structure Prediction
 CMPS 3110: Bioinformatics Tertiary Structure Prediction Tertiary Structure Prediction Why Should Tertiary Structure Prediction Be Possible? Molecules obey the laws of physics! Conformation space is finite
CMPS 3110: Bioinformatics Tertiary Structure Prediction Tertiary Structure Prediction Why Should Tertiary Structure Prediction Be Possible? Molecules obey the laws of physics! Conformation space is finite
CE 530 Molecular Simulation
 1 CE 530 Molecular Simulation Lecture 14 Molecular Models David A. Kofke Department of Chemical Engineering SUNY Buffalo kofke@eng.buffalo.edu 2 Review Monte Carlo ensemble averaging, no dynamics easy
1 CE 530 Molecular Simulation Lecture 14 Molecular Models David A. Kofke Department of Chemical Engineering SUNY Buffalo kofke@eng.buffalo.edu 2 Review Monte Carlo ensemble averaging, no dynamics easy
Modeling for 3D structure prediction
 Modeling for 3D structure prediction What is a predicted structure? A structure that is constructed using as the sole source of information data obtained from computer based data-mining. However, mixing
Modeling for 3D structure prediction What is a predicted structure? A structure that is constructed using as the sole source of information data obtained from computer based data-mining. However, mixing
Can a continuum solvent model reproduce the free energy landscape of a β-hairpin folding in water?
 Can a continuum solvent model reproduce the free energy landscape of a β-hairpin folding in water? Ruhong Zhou 1 and Bruce J. Berne 2 1 IBM Thomas J. Watson Research Center; and 2 Department of Chemistry,
Can a continuum solvent model reproduce the free energy landscape of a β-hairpin folding in water? Ruhong Zhou 1 and Bruce J. Berne 2 1 IBM Thomas J. Watson Research Center; and 2 Department of Chemistry,
Protein Structure Prediction
 Page 1 Protein Structure Prediction Russ B. Altman BMI 214 CS 274 Protein Folding is different from structure prediction --Folding is concerned with the process of taking the 3D shape, usually based on
Page 1 Protein Structure Prediction Russ B. Altman BMI 214 CS 274 Protein Folding is different from structure prediction --Folding is concerned with the process of taking the 3D shape, usually based on
The protein folding problem consists of two parts:
 Energetics and kinetics of protein folding The protein folding problem consists of two parts: 1)Creating a stable, well-defined structure that is significantly more stable than all other possible structures.
Energetics and kinetics of protein folding The protein folding problem consists of two parts: 1)Creating a stable, well-defined structure that is significantly more stable than all other possible structures.
Fondamenti di Chimica Farmaceutica. Computer Chemistry in Drug Research: Introduction
 Fondamenti di Chimica Farmaceutica Computer Chemistry in Drug Research: Introduction Introduction Introduction Introduction Computer Chemistry in Drug Design Drug Discovery: Target identification Lead
Fondamenti di Chimica Farmaceutica Computer Chemistry in Drug Research: Introduction Introduction Introduction Introduction Computer Chemistry in Drug Design Drug Discovery: Target identification Lead
Session 1. Introduction to Computational Chemistry. Computational (chemistry education) and/or (Computational chemistry) education
 and/or (Computational chemistry) education") Session 1 Introduction to Computational Chemistry 1 Introduction to Computational Chemistry Computational (chemistry education) and/or (Computational chemistry) education First one: Use computational tools
Session 1 Introduction to Computational Chemistry 1 Introduction to Computational Chemistry Computational (chemistry education) and/or (Computational chemistry) education First one: Use computational tools
Docking. GBCB 5874: Problem Solving in GBCB
 Docking Benzamidine Docking to Trypsin Relationship to Drug Design Ligand-based design QSAR Pharmacophore modeling Can be done without 3-D structure of protein Receptor/Structure-based design Molecular
Docking Benzamidine Docking to Trypsin Relationship to Drug Design Ligand-based design QSAR Pharmacophore modeling Can be done without 3-D structure of protein Receptor/Structure-based design Molecular
Using AutoDock 4 with ADT: A Tutorial
 Using AutoDock 4 with ADT: A Tutorial Ruth Huey Sargis Dallakyan Alex Perryman David S. Goodsell (Garrett Morris) 9/2/08 Using AutoDock 4 with ADT 1 What is Docking? Predicting the best ways two molecules
Using AutoDock 4 with ADT: A Tutorial Ruth Huey Sargis Dallakyan Alex Perryman David S. Goodsell (Garrett Morris) 9/2/08 Using AutoDock 4 with ADT 1 What is Docking? Predicting the best ways two molecules
Bioinformatics. Macromolecular structure
 Bioinformatics Macromolecular structure Contents Determination of protein structure Structure databases Secondary structure elements (SSE) Tertiary structure Structure analysis Structure alignment Domain
Bioinformatics Macromolecular structure Contents Determination of protein structure Structure databases Secondary structure elements (SSE) Tertiary structure Structure analysis Structure alignment Domain
Lecture 2-3: Review of forces (ctd.) and elementary statistical mechanics. Contributions to protein stability
 and elementary statistical mechanics. Contributions to protein stability") Lecture 2-3: Review of forces (ctd.) and elementary statistical mechanics. Contributions to protein stability Part I. Review of forces Covalent bonds Non-covalent Interactions Van der Waals Interactions
Lecture 2-3: Review of forces (ctd.) and elementary statistical mechanics. Contributions to protein stability Part I. Review of forces Covalent bonds Non-covalent Interactions Van der Waals Interactions
Molecular Modelling for Medicinal Chemistry (F13MMM) Room A36
 Room A36") Molecular Modelling for Medicinal Chemistry (F13MMM) jonathan.hirst@nottingham.ac.uk Room A36 http://comp.chem.nottingham.ac.uk Assisted reading Molecular Modelling: Principles and Applications. Andrew
Molecular Modelling for Medicinal Chemistry (F13MMM) jonathan.hirst@nottingham.ac.uk Room A36 http://comp.chem.nottingham.ac.uk Assisted reading Molecular Modelling: Principles and Applications. Andrew
Protein Modeling. Generating, Evaluating and Refining Protein Homology Models
 Protein Modeling Generating, Evaluating and Refining Protein Homology Models Troy Wymore and Kristen Messinger Biomedical Initiatives Group Pittsburgh Supercomputing Center Homology Modeling of Proteins
Protein Modeling Generating, Evaluating and Refining Protein Homology Models Troy Wymore and Kristen Messinger Biomedical Initiatives Group Pittsburgh Supercomputing Center Homology Modeling of Proteins
What is Protein-Ligand Docking?
 MOLECULAR DOCKING Definition: What is Protein-Ligand Docking? Computationally predict the structures of protein-ligand complexes from their conformations and orientations. The orientation that maximizes
MOLECULAR DOCKING Definition: What is Protein-Ligand Docking? Computationally predict the structures of protein-ligand complexes from their conformations and orientations. The orientation that maximizes
Protein Structures. 11/19/2002 Lecture 24 1
 Protein Structures 11/19/2002 Lecture 24 1 All 3 figures are cartoons of an amino acid residue. 11/19/2002 Lecture 24 2 Peptide bonds in chains of residues 11/19/2002 Lecture 24 3 Angles φ and ψ in the
Protein Structures 11/19/2002 Lecture 24 1 All 3 figures are cartoons of an amino acid residue. 11/19/2002 Lecture 24 2 Peptide bonds in chains of residues 11/19/2002 Lecture 24 3 Angles φ and ψ in the
Molecular Mechanics. Yohann Moreau. November 26, 2015
 Molecular Mechanics Yohann Moreau yohann.moreau@ujf-grenoble.fr November 26, 2015 Yohann Moreau (UJF) Molecular Mechanics, Label RFCT 2015 November 26, 2015 1 / 29 Introduction A so-called Force-Field
Molecular Mechanics Yohann Moreau yohann.moreau@ujf-grenoble.fr November 26, 2015 Yohann Moreau (UJF) Molecular Mechanics, Label RFCT 2015 November 26, 2015 1 / 29 Introduction A so-called Force-Field
Molecular Dynamics, Monte Carlo and Docking. Lecture 21. Introduction to Bioinformatics MNW2
 Molecular Dynamics, Monte Carlo and Docking Lecture 21 Introduction to Bioinformatics MNW2 Allowed phi-psi angles Red areas are preferred, yellow areas are allowed, and white is avoided 2.3a Hamiltonian
Molecular Dynamics, Monte Carlo and Docking Lecture 21 Introduction to Bioinformatics MNW2 Allowed phi-psi angles Red areas are preferred, yellow areas are allowed, and white is avoided 2.3a Hamiltonian
DISCRETE TUTORIAL. Agustí Emperador. Institute for Research in Biomedicine, Barcelona APPLICATION OF DISCRETE TO FLEXIBLE PROTEIN-PROTEIN DOCKING:
 DISCRETE TUTORIAL Agustí Emperador Institute for Research in Biomedicine, Barcelona APPLICATION OF DISCRETE TO FLEXIBLE PROTEIN-PROTEIN DOCKING: STRUCTURAL REFINEMENT OF DOCKING CONFORMATIONS Emperador
DISCRETE TUTORIAL Agustí Emperador Institute for Research in Biomedicine, Barcelona APPLICATION OF DISCRETE TO FLEXIBLE PROTEIN-PROTEIN DOCKING: STRUCTURAL REFINEMENT OF DOCKING CONFORMATIONS Emperador
Chemical properties that affect binding of enzyme-inhibiting drugs to enzymes
 Chemical properties that affect binding of enzyme-inhibiting drugs to enzymes Introduction The production of new drugs requires time for development and testing, and can result in large prohibitive costs
Chemical properties that affect binding of enzyme-inhibiting drugs to enzymes Introduction The production of new drugs requires time for development and testing, and can result in large prohibitive costs
Course Introduction. SASSIE CCP-SAS Workshop. January 23-25, ISIS Neutron and Muon Source Rutherford Appleton Laboratory UK
 Course Introduction SASSIE CCP-SAS Workshop January 23-25, 2017 ISIS Neutron and Muon Source Rutherford Appleton Laboratory UK ccpsas.org Scatters & Simulators Develop a community of users and developers
Course Introduction SASSIE CCP-SAS Workshop January 23-25, 2017 ISIS Neutron and Muon Source Rutherford Appleton Laboratory UK ccpsas.org Scatters & Simulators Develop a community of users and developers
Structural Bioinformatics (C3210) Molecular Docking
 Molecular Docking") Structural Bioinformatics (C3210) Molecular Docking Molecular Recognition, Molecular Docking Molecular recognition is the ability of biomolecules to recognize other biomolecules and selectively interact
Structural Bioinformatics (C3210) Molecular Docking Molecular Recognition, Molecular Docking Molecular recognition is the ability of biomolecules to recognize other biomolecules and selectively interact
High Throughput In-Silico Screening Against Flexible Protein Receptors
 John von Neumann Institute for Computing High Throughput In-Silico Screening Against Flexible Protein Receptors H. Sánchez, B. Fischer, H. Merlitz, W. Wenzel published in From Computational Biophysics
John von Neumann Institute for Computing High Throughput In-Silico Screening Against Flexible Protein Receptors H. Sánchez, B. Fischer, H. Merlitz, W. Wenzel published in From Computational Biophysics
ONETEP PB/SA: Application to G-Quadruplex DNA Stability. Danny Cole
 ONETEP PB/SA: Application to G-Quadruplex DNA Stability Danny Cole Introduction Historical overview of structure and free energy calculation of complex molecules using molecular mechanics and continuum
ONETEP PB/SA: Application to G-Quadruplex DNA Stability Danny Cole Introduction Historical overview of structure and free energy calculation of complex molecules using molecular mechanics and continuum
Protein structure prediction. CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror
 Protein structure prediction CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror 1 Outline Why predict protein structure? Can we use (pure) physics-based methods? Knowledge-based methods Two major
Protein structure prediction CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror 1 Outline Why predict protein structure? Can we use (pure) physics-based methods? Knowledge-based methods Two major
Chemical properties that affect binding of enzyme-inhibiting drugs to enzymes
 Introduction Chemical properties that affect binding of enzyme-inhibiting drugs to enzymes The production of new drugs requires time for development and testing, and can result in large prohibitive costs
Introduction Chemical properties that affect binding of enzyme-inhibiting drugs to enzymes The production of new drugs requires time for development and testing, and can result in large prohibitive costs
CAP 5510 Lecture 3 Protein Structures
 CAP 5510 Lecture 3 Protein Structures Su-Shing Chen Bioinformatics CISE 8/19/2005 Su-Shing Chen, CISE 1 Protein Conformation 8/19/2005 Su-Shing Chen, CISE 2 Protein Conformational Structures Hydrophobicity
CAP 5510 Lecture 3 Protein Structures Su-Shing Chen Bioinformatics CISE 8/19/2005 Su-Shing Chen, CISE 1 Protein Conformation 8/19/2005 Su-Shing Chen, CISE 2 Protein Conformational Structures Hydrophobicity
THE TANGO ALGORITHM: SECONDARY STRUCTURE PROPENSITIES, STATISTICAL MECHANICS APPROXIMATION
 THE TANGO ALGORITHM: SECONDARY STRUCTURE PROPENSITIES, STATISTICAL MECHANICS APPROXIMATION AND CALIBRATION Calculation of turn and beta intrinsic propensities. A statistical analysis of a protein structure
THE TANGO ALGORITHM: SECONDARY STRUCTURE PROPENSITIES, STATISTICAL MECHANICS APPROXIMATION AND CALIBRATION Calculation of turn and beta intrinsic propensities. A statistical analysis of a protein structure
Solutions to Assignment #4 Getting Started with HyperChem
 Solutions to Assignment #4 Getting Started with HyperChem 1. This first exercise is meant to familiarize you with the different methods for visualizing molecules available in HyperChem. (a) Create a molecule
Solutions to Assignment #4 Getting Started with HyperChem 1. This first exercise is meant to familiarize you with the different methods for visualizing molecules available in HyperChem. (a) Create a molecule
Molecular Dynamics, Monte Carlo and Docking. Lecture 21. Introduction to Bioinformatics MNW2
 Molecular Dynamics, Monte Carlo and Docking Lecture 21 Introduction to Bioinformatics MNW2 If you throw up a stone, it is Physics. If you throw up a stone, it is Physics. If it lands on your head, it is
Molecular Dynamics, Monte Carlo and Docking Lecture 21 Introduction to Bioinformatics MNW2 If you throw up a stone, it is Physics. If you throw up a stone, it is Physics. If it lands on your head, it is
STRUCTURAL BIOINFORMATICS II. Spring 2018
 STRUCTURAL BIOINFORMATICS II Spring 2018 Syllabus Course Number - Classification: Chemistry 5412 Class Schedule: Monday 5:30-7:50 PM, SERC Room 456 (4 th floor) Instructors: Ronald Levy, SERC 718 (ronlevy@temple.edu)
STRUCTURAL BIOINFORMATICS II Spring 2018 Syllabus Course Number - Classification: Chemistry 5412 Class Schedule: Monday 5:30-7:50 PM, SERC Room 456 (4 th floor) Instructors: Ronald Levy, SERC 718 (ronlevy@temple.edu)
Protein structure (and biomolecular structure more generally) CS/CME/BioE/Biophys/BMI 279 Sept. 28 and Oct. 3, 2017 Ron Dror
 CS/CME/BioE/Biophys/BMI 279 Sept. 28 and Oct. 3, 2017 Ron Dror") Protein structure (and biomolecular structure more generally) CS/CME/BioE/Biophys/BMI 279 Sept. 28 and Oct. 3, 2017 Ron Dror Please interrupt if you have questions, and especially if you re confused! Assignment
Protein structure (and biomolecular structure more generally) CS/CME/BioE/Biophys/BMI 279 Sept. 28 and Oct. 3, 2017 Ron Dror Please interrupt if you have questions, and especially if you re confused! Assignment
Computational Methods. Chem 561
 Computational Methods Chem 561 Lecture Outline 1. Ab initio methods a) HF SCF b) Post-HF methods 2. Density Functional Theory 3. Semiempirical methods 4. Molecular Mechanics Computational Chemistry " Computational
Computational Methods Chem 561 Lecture Outline 1. Ab initio methods a) HF SCF b) Post-HF methods 2. Density Functional Theory 3. Semiempirical methods 4. Molecular Mechanics Computational Chemistry " Computational
User Guide for LeDock
 User Guide for LeDock Hongtao Zhao, PhD Email: htzhao@lephar.com Website: www.lephar.com Copyright 2017 Hongtao Zhao. All rights reserved. Introduction LeDock is flexible small-molecule docking software,
User Guide for LeDock Hongtao Zhao, PhD Email: htzhao@lephar.com Website: www.lephar.com Copyright 2017 Hongtao Zhao. All rights reserved. Introduction LeDock is flexible small-molecule docking software,
Gromacs Workshop Spring CSC
 Gromacs Workshop Spring 2007 @ CSC Erik Lindahl Center for Biomembrane Research Stockholm University, Sweden David van der Spoel Dept. Cell & Molecular Biology Uppsala University, Sweden Berk Hess Max-Planck-Institut
Gromacs Workshop Spring 2007 @ CSC Erik Lindahl Center for Biomembrane Research Stockholm University, Sweden David van der Spoel Dept. Cell & Molecular Biology Uppsala University, Sweden Berk Hess Max-Planck-Institut
Chem 1140; Molecular Modeling
 P. Wipf 1 Chem 1140 $E = -! (q a + q b )" ab S ab + ab!k
P. Wipf 1 Chem 1140 $E = -! (q a + q b )" ab S ab + ab!k
Protein Modeling Methods. Knowledge. Protein Modeling Methods. Fold Recognition. Knowledge-based methods. Introduction to Bioinformatics
 Protein Modeling Methods Introduction to Bioinformatics Iosif Vaisman Ab initio methods Energy-based methods Knowledge-based methods Email: ivaisman@gmu.edu Protein Modeling Methods Ab initio methods:
Protein Modeling Methods Introduction to Bioinformatics Iosif Vaisman Ab initio methods Energy-based methods Knowledge-based methods Email: ivaisman@gmu.edu Protein Modeling Methods Ab initio methods:
OpenDiscovery: Automated Docking of Ligands to Proteins and Molecular Simulation
 OpenDiscovery: Automated Docking of Ligands to Proteins and Molecular Simulation Gareth Price Computational MiniProject OpenDiscovery Aims + Achievements Produce a high-throughput protocol to screen a
OpenDiscovery: Automated Docking of Ligands to Proteins and Molecular Simulation Gareth Price Computational MiniProject OpenDiscovery Aims + Achievements Produce a high-throughput protocol to screen a
Lecture 11: Protein Folding & Stability
 Structure - Function Protein Folding: What we know Lecture 11: Protein Folding & Stability 1). Amino acid sequence dictates structure. 2). The native structure represents the lowest energy state for a
Structure - Function Protein Folding: What we know Lecture 11: Protein Folding & Stability 1). Amino acid sequence dictates structure. 2). The native structure represents the lowest energy state for a
Protein Folding & Stability. Lecture 11: Margaret A. Daugherty. Fall Protein Folding: What we know. Protein Folding
 Lecture 11: Protein Folding & Stability Margaret A. Daugherty Fall 2003 Structure - Function Protein Folding: What we know 1). Amino acid sequence dictates structure. 2). The native structure represents
Lecture 11: Protein Folding & Stability Margaret A. Daugherty Fall 2003 Structure - Function Protein Folding: What we know 1). Amino acid sequence dictates structure. 2). The native structure represents
Template Free Protein Structure Modeling Jianlin Cheng, PhD
 Template Free Protein Structure Modeling Jianlin Cheng, PhD Associate Professor Computer Science Department Informatics Institute University of Missouri, Columbia 2013 Protein Energy Landscape & Free Sampling
Template Free Protein Structure Modeling Jianlin Cheng, PhD Associate Professor Computer Science Department Informatics Institute University of Missouri, Columbia 2013 Protein Energy Landscape & Free Sampling
Francisco Melo, Damien Devos, Eric Depiereux and Ernest Feytmans
 From: ISMB-97 Proceedings. Copyright 1997, AAAI (www.aaai.org). All rights reserved. ANOLEA: A www Server to Assess Protein Structures Francisco Melo, Damien Devos, Eric Depiereux and Ernest Feytmans Facultés
From: ISMB-97 Proceedings. Copyright 1997, AAAI (www.aaai.org). All rights reserved. ANOLEA: A www Server to Assess Protein Structures Francisco Melo, Damien Devos, Eric Depiereux and Ernest Feytmans Facultés
Energy functions and their relationship to molecular conformation. CS/CME/BioE/Biophys/BMI 279 Oct. 3 and 5, 2017 Ron Dror
 Energy functions and their relationship to molecular conformation CS/CME/BioE/Biophys/BMI 279 Oct. 3 and 5, 2017 Ron Dror Outline Energy functions for proteins (or biomolecular systems more generally)
Energy functions and their relationship to molecular conformation CS/CME/BioE/Biophys/BMI 279 Oct. 3 and 5, 2017 Ron Dror Outline Energy functions for proteins (or biomolecular systems more generally)
Contents 1 Open-Source Tools, Techniques, and Data in Chemoinformatics
 Contents 1 Open-Source Tools, Techniques, and Data in Chemoinformatics... 1 1.1 Chemoinformatics... 2 1.1.1 Open-Source Tools... 2 1.1.2 Introduction to Programming Languages... 3 1.2 Chemical Structure
Contents 1 Open-Source Tools, Techniques, and Data in Chemoinformatics... 1 1.1 Chemoinformatics... 2 1.1.1 Open-Source Tools... 2 1.1.2 Introduction to Programming Languages... 3 1.2 Chemical Structure
Molecular Modelling. part of Bioinformatik von RNA- und Proteinstrukturen. Sonja Prohaska. Leipzig, SS Computational EvoDevo University Leipzig
 part of Bioinformatik von RNA- und Proteinstrukturen Computational EvoDevo University Leipzig Leipzig, SS 2011 Protein Structure levels or organization Primary structure: sequence of amino acids (from
part of Bioinformatik von RNA- und Proteinstrukturen Computational EvoDevo University Leipzig Leipzig, SS 2011 Protein Structure levels or organization Primary structure: sequence of amino acids (from
Molecular Simulation II
 Molecular Simulation II Quantum Chemistry Classical Mechanics E = Ψ H Ψ ΨΨ U = E bond +E angle +E torsion +E non-bond Jeffry D. Madura Department of Chemistry & Biochemistry Center for Computational Sciences
Molecular Simulation II Quantum Chemistry Classical Mechanics E = Ψ H Ψ ΨΨ U = E bond +E angle +E torsion +E non-bond Jeffry D. Madura Department of Chemistry & Biochemistry Center for Computational Sciences
BCB410 Protein-Ligand Docking Exercise Set Shirin Shahsavand December 11, 2011
 BCB410 Protein-Ligand Docking Exercise Set Shirin Shahsavand December 11, 2011 1. Describe the search algorithm(s) AutoDock uses for solving protein-ligand docking problems. AutoDock uses 3 different approaches
BCB410 Protein-Ligand Docking Exercise Set Shirin Shahsavand December 11, 2011 1. Describe the search algorithm(s) AutoDock uses for solving protein-ligand docking problems. AutoDock uses 3 different approaches
General Chemistry Lab Molecular Modeling
 PURPOSE The objectives of this experiment are PROCEDURE General Chemistry Lab Molecular Modeling To learn how to use molecular modeling software, a commonly used tool in chemical research and industry.
PURPOSE The objectives of this experiment are PROCEDURE General Chemistry Lab Molecular Modeling To learn how to use molecular modeling software, a commonly used tool in chemical research and industry.
Today s lecture. Molecular Mechanics and docking. Lecture 22. Introduction to Bioinformatics Docking - ZDOCK. Protein-protein docking
 C N F O N G A V B O N F O M A C S V U Molecular Mechanics and docking Lecture 22 ntroduction to Bioinformatics 2007 oday s lecture 1. Protein interaction and docking a) Zdock method 2. Molecular motion
C N F O N G A V B O N F O M A C S V U Molecular Mechanics and docking Lecture 22 ntroduction to Bioinformatics 2007 oday s lecture 1. Protein interaction and docking a) Zdock method 2. Molecular motion
3rd Advanced in silico Drug Design KFC/ADD Molecular mechanics intro Karel Berka, Ph.D. Martin Lepšík, Ph.D. Pavel Polishchuk, Ph.D.
 3rd Advanced in silico Drug Design KFC/ADD Molecular mechanics intro Karel Berka, Ph.D. Martin Lepšík, Ph.D. Pavel Polishchuk, Ph.D. Thierry Langer, Ph.D. Jana Vrbková, Ph.D. UP Olomouc, 23.1.-26.1. 2018
3rd Advanced in silico Drug Design KFC/ADD Molecular mechanics intro Karel Berka, Ph.D. Martin Lepšík, Ph.D. Pavel Polishchuk, Ph.D. Thierry Langer, Ph.D. Jana Vrbková, Ph.D. UP Olomouc, 23.1.-26.1. 2018
All-atom Molecular Mechanics. Trent E. Balius AMS 535 / CHE /27/2010
 All-atom Molecular Mechanics Trent E. Balius AMS 535 / CHE 535 09/27/2010 Outline Molecular models Molecular mechanics Force Fields Potential energy function functional form parameters and parameterization
All-atom Molecular Mechanics Trent E. Balius AMS 535 / CHE 535 09/27/2010 Outline Molecular models Molecular mechanics Force Fields Potential energy function functional form parameters and parameterization
Colby College Molecular Mechanics Tutorial. Thomas W. Shattuck Department of Chemistry Colby College Waterville, Maine 04901
 Colby College Molecular Mechanics Tutorial Thomas W. Shattuck Department of Chemistry Colby College Waterville, Maine 04901 Please, feel free to use this tutorial in any way you wish, provided that you
Colby College Molecular Mechanics Tutorial Thomas W. Shattuck Department of Chemistry Colby College Waterville, Maine 04901 Please, feel free to use this tutorial in any way you wish, provided that you
The Molecular Dynamics Method
 H-bond energy (kcal/mol) - 4.0 The Molecular Dynamics Method Fibronectin III_1, a mechanical protein that glues cells together in wound healing and in preventing tumor metastasis 0 ATPase, a molecular
H-bond energy (kcal/mol) - 4.0 The Molecular Dynamics Method Fibronectin III_1, a mechanical protein that glues cells together in wound healing and in preventing tumor metastasis 0 ATPase, a molecular
Lecture 2 and 3: Review of forces (ctd.) and elementary statistical mechanics. Contributions to protein stability
 and elementary statistical mechanics. Contributions to protein stability") Lecture 2 and 3: Review of forces (ctd.) and elementary statistical mechanics. Contributions to protein stability Part I. Review of forces Covalent bonds Non-covalent Interactions: Van der Waals Interactions
Lecture 2 and 3: Review of forces (ctd.) and elementary statistical mechanics. Contributions to protein stability Part I. Review of forces Covalent bonds Non-covalent Interactions: Van der Waals Interactions
FlexPepDock In a nutshell
 FlexPepDock In a nutshell All Tutorial files are located in http://bit.ly/mxtakv FlexPepdock refinement Step 1 Step 3 - Refinement Step 4 - Selection of models Measure of fit FlexPepdock Ab-initio Step
FlexPepDock In a nutshell All Tutorial files are located in http://bit.ly/mxtakv FlexPepdock refinement Step 1 Step 3 - Refinement Step 4 - Selection of models Measure of fit FlexPepdock Ab-initio Step
Molecular Simulation III
 Molecular Simulation III Quantum Chemistry Classical Mechanics E = Ψ H Ψ ΨΨ U = E bond +E angle +E torsion +E non-bond Molecular Dynamics Jeffry D. Madura Department of Chemistry & Biochemistry Center
Molecular Simulation III Quantum Chemistry Classical Mechanics E = Ψ H Ψ ΨΨ U = E bond +E angle +E torsion +E non-bond Molecular Dynamics Jeffry D. Madura Department of Chemistry & Biochemistry Center
Assignment 2 Atomic-Level Molecular Modeling
 Assignment 2 Atomic-Level Molecular Modeling CS/BIOE/CME/BIOPHYS/BIOMEDIN 279 Due: November 3, 2016 at 3:00 PM The goal of this assignment is to understand the biological and computational aspects of macromolecular
Assignment 2 Atomic-Level Molecular Modeling CS/BIOE/CME/BIOPHYS/BIOMEDIN 279 Due: November 3, 2016 at 3:00 PM The goal of this assignment is to understand the biological and computational aspects of macromolecular
5.1. Hardwares, Softwares and Web server used in Molecular modeling
 5. EXPERIMENTAL The tools, techniques and procedures/methods used for carrying out research work reported in this thesis have been described as follows: 5.1. Hardwares, Softwares and Web server used in
5. EXPERIMENTAL The tools, techniques and procedures/methods used for carrying out research work reported in this thesis have been described as follows: 5.1. Hardwares, Softwares and Web server used in
Medicinal Chemistry/ CHEM 458/658 Chapter 4- Computer-Aided Drug Design
 Medicinal Chemistry/ CHEM 458/658 Chapter 4- Computer-Aided Drug Design Bela Torok Department of Chemistry University of Massachusetts Boston Boston, MA 1 Computer Aided Drug Design - Introduction Development
Medicinal Chemistry/ CHEM 458/658 Chapter 4- Computer-Aided Drug Design Bela Torok Department of Chemistry University of Massachusetts Boston Boston, MA 1 Computer Aided Drug Design - Introduction Development
CS612 - Algorithms in Bioinformatics
 Fall 2017 Protein Structure Detection Methods October 30, 2017 Comparative Modeling Comparative modeling is modeling of the unknown based on comparison to what is known In the context of modeling or computing
Fall 2017 Protein Structure Detection Methods October 30, 2017 Comparative Modeling Comparative modeling is modeling of the unknown based on comparison to what is known In the context of modeling or computing
SCOP. all-β class. all-α class, 3 different folds. T4 endonuclease V. 4-helical cytokines. Globin-like
 SCOP all-β class 4-helical cytokines T4 endonuclease V all-α class, 3 different folds Globin-like TIM-barrel fold α/β class Profilin-like fold α+β class http://scop.mrc-lmb.cam.ac.uk/scop CATH Class, Architecture,
SCOP all-β class 4-helical cytokines T4 endonuclease V all-α class, 3 different folds Globin-like TIM-barrel fold α/β class Profilin-like fold α+β class http://scop.mrc-lmb.cam.ac.uk/scop CATH Class, Architecture,
Protein Folding & Stability. Lecture 11: Margaret A. Daugherty. Fall How do we go from an unfolded polypeptide chain to a
 Lecture 11: Protein Folding & Stability Margaret A. Daugherty Fall 2004 How do we go from an unfolded polypeptide chain to a compact folded protein? (Folding of thioredoxin, F. Richards) Structure - Function
Lecture 11: Protein Folding & Stability Margaret A. Daugherty Fall 2004 How do we go from an unfolded polypeptide chain to a compact folded protein? (Folding of thioredoxin, F. Richards) Structure - Function
Other Cells. Hormones. Viruses. Toxins. Cell. Bacteria
 Other Cells Hormones Viruses Toxins Cell Bacteria ΔH < 0 reaction is exothermic, tells us nothing about the spontaneity of the reaction Δ H > 0 reaction is endothermic, tells us nothing about the spontaneity
Other Cells Hormones Viruses Toxins Cell Bacteria ΔH < 0 reaction is exothermic, tells us nothing about the spontaneity of the reaction Δ H > 0 reaction is endothermic, tells us nothing about the spontaneity
4 th Advanced in silico Drug Design KFC/ADD Molecular Modelling Intro. Karel Berka, Ph.D.
 4 th Advanced in silico Drug Design KFC/ADD Molecular Modelling Intro Karel Berka, Ph.D. UP Olomouc, 21.1.-25.1. 2019 Motto A theory is something nobody believes, except the person who made it An experiment
4 th Advanced in silico Drug Design KFC/ADD Molecular Modelling Intro Karel Berka, Ph.D. UP Olomouc, 21.1.-25.1. 2019 Motto A theory is something nobody believes, except the person who made it An experiment
Type II Kinase Inhibitors Show an Unexpected Inhibition Mode against Parkinson s Disease-Linked LRRK2 Mutant G2019S.
 Type II Kinase Inhibitors Show an Unexpected Inhibition Mode against Parkinson s Disease-Linked LRRK2 Mutant G219S. Min Liu@&*, Samantha A. Bender%*, Gregory D Cuny@, Woody Sherman, Marcie Glicksman@ Soumya
Type II Kinase Inhibitors Show an Unexpected Inhibition Mode against Parkinson s Disease-Linked LRRK2 Mutant G219S. Min Liu@&*, Samantha A. Bender%*, Gregory D Cuny@, Woody Sherman, Marcie Glicksman@ Soumya
Improving Protein Function Prediction with Molecular Dynamics Simulations. Dariya Glazer Russ Altman
 Improving Protein Function Prediction with Molecular Dynamics Simulations Dariya Glazer Russ Altman Motivation Sometimes the 3D structure doesn t score well for a known function. The experimental structure
Improving Protein Function Prediction with Molecular Dynamics Simulations Dariya Glazer Russ Altman Motivation Sometimes the 3D structure doesn t score well for a known function. The experimental structure