Evolution of Protein Structure in the Aminoacyl-tRNA Synthetases
|
|
|
- Jason Barber
- 5 years ago
- Views:
Transcription
1 Evolution of Protein Structure in the Aminoacyl-tRNA Synthetases class I class II P. O Donoghue and Z. Luthey-Schulten* Department of Chemistry, Beckman Institute, Center for Biophysics and Computational Biology University of Illinois at Urbana-Champaign
2 What can be learned from AARS? The aminoacyl-trna synthetases, perhaps better than any other molecules in the cell, eptiomize the current situation and help to under standard (the effects) of HGT Woese (PNAS, 2000; MMBR 2000)

3 Aminoacyl-tRNA synthetases Universal Tree of Life Woese PNAS 1990, 2002.

4 Structural Conservation in the Catalytic Domain of the AARSs


5 Why Study the Evolution of Protein Structure? 1. Important for Homology Modeling Better profiles improve database searches and give better alignments of distant homologs. Allows mixing of sequence and structure information systematically. 13% sequence id in the core (blue) 2. Learn how evolutionary dynamics changed protein shape. Mapping a protein of unknown structure onto a homologous protein of known structure is equivalent to defining the evolutionary pathway connecting the two proteins 3. Impact on protein structure prediction, folding, and function Evolutionary profiles increase the signal to noise ratio - Evolution is the foundation of bioinformatics.
6 Outline 1. Summarize evolutionary theory of the universal phylogenetic tree. Methods 2. Introduce a structure-based metric which accounts for gaps, and show that evolutionary information is encoded in protein structure. 3. Introduce multidimensional QR factorization for computing non-redundant representative multiple alignments in sequence or structure. Applications 4. Non-redundant multiple alignments which well represent the evolutionary history of a protein group provide better profiles for database searching. Eliminate bias inherited from structure or sequence databases. Important for bioinformatic analysis (substitution matrices, knowledge based potentials structure pred., genome annotation) and evolutionary analysis. 5. Depict the evolution of structure and function in Aspartyl-tRNA synthetase.
7 Universal Phylogenetic Tree three domains of life Archaea Eucarya Bacteria Leucyl-tRNA synthetase displays the full canonical phylogenetic distribution. for review see Woese PNAS 2000 Woese, Olsen, Ibba, Soll MMBR 2000

8 After W. Doolittle, modified by G. Olsen
9 Phylogenetic Distributions Full Canonical Basal Canonical Non-canonical E A A B B increasing inter-domain of life Horizontal Gene Transfer HGT erodes the historical trace, but does not completely erase it. G. Olsen
10 Protein Structure Similarity Measure Q H Structural Homology fraction of native contacts for aligned residues + presence and perturbation of gaps i i j j Gaps should count as a character but not dominate C. Woese O Donoghue & Luthey-Schulten MMBR.2003.
11 Protein structure encodes evolutionary information Sequence Phylogeny AspRS-AsnRS Group Structure Phylogeny Structure Phylogeny AspRS-AsnRS GroupClass II AARSs Da De N Db Woese, Olsen, Ibba, Soll MMBR 2000 O Donoghue & Luthey-Schulten MMBR.2003.


12 Horizontal Gene Transfer in Protein Structure Sequence Phylogeny AspRS-AsnRS Group Da archaeal helix archaeal helix extension De N Db bacterial insertions

13 Non-redundant Representative Sets Too much information 129 Structures Multidimensional QR factorization of alignment matrix, A. Economy of information 16 representatives QR computes a set of maximal linearly independent structures. P. O Donoghue and Z. Luthey-Schulten (2003) MMBR 67: , JMB (2004) in press.
14 Numerical Encoding of Proteins in a Multiple Alignment Encoding Structure Rotated Cartesian + Gap = 4-space Aligned position Gapped position Gap Scaling Sequence Space Orthogonal Encoding = 24-space 23 amino acids (20 + B, X, Z) + gap A = (1,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0) B = (0,1,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0) C = (0,0,1,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0) GAP = (0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,1) adjustable parameter Alignment Matrix A= m aligned positions n proteins d=1 d=2d=3 d=n encoded residue space
15 A Multiple Alignment is a Matrix with Linearly Dependent Columns redundancy is equivalent to linear dependence QR factorization Re-orders the columns of A, segregating the linearly independent columns from the dependent ones without scrambling the information in A. SVD not an option. Q T orthogonal matrix of product of Householder transformations. P permutation matrix encodes column pivoting which exchanges columns of A and puts the redundant or similar proteins to the right hand side. Multidimensional QR N simultaneous QR factorizations, one for each d-dimension. A minimal linearly dependent subset can be determined with respect to a threshold, e.g., similarity measure threshold. L. Heck, J. Olkin, and K. Nagshineh (1998) J. Vibration Acoustics 120:663. P. O Donoghue and Z. Luthey-Schulten (2003) MMBR. 67:
16 The QR establishes an order of linear dependence by applying Householder transformations and permutations Three 1-D (2 residue) proteins a b c. a is our measuring stick, reference frame. The transformation reveals that b is more linearly dependent on a, so the permutation swaps b with c. transformed original Given a, c adds more information to the system than b. Multiply aligned proteins exist in a higher dimensional space, so this magnitude is computed with a matrix p-norm: Householder, J. Assoc. Comput. Mach., adjustable parameter
17 What are the constraints on the parameters? Must maintain the evolutionary history of the protein group. This rule is used to determine the value of two adjustable parameters in our implementation of the QR.
18 Hierarchical Multidimensional QR -.

 γ (normalized) ordering")
19 Parameters Define the Measure of Linear Dependence AARS class I, Rossman fold AARS class II, Novel fold forbidden ordering p-norm allowed γ (normalized) γ (normalized) ordering norm gap scale
20 Class I AARSs evolutionary events 5 Subclasses Specificity 11 Amino acids Domain of life A, B, E
21 Profile of the ILMV Subclass How many sequences are needed to represent the Subclass ILMV? If each of ILMV was full canonical, then we would need 4x3=12 sequences. Since M and V are basal, we need at least 2x3 + 2x2 = 10 sequences. We have 6 structures.
22 Non-Redundant Profiles for Database Searching AARS Subclass ILMV HMMER Psi-Blast false positives Choosing the right 10 sequence makes all the difference. A. Sethi, P. O Donoghue, Z..Luthey-Schulten Starting with a non-redundant profile, accuracy diminishes with Psi-blast iterations which add in bias. Repair with QR filter.
23
24 The Economy of Information How many sequence are needed for profiles? A single profile for class I AARSs HisA and HisF Protein Family TIM Barrel fold PFAM profile of 113 sequences finds 3 additional sequence fragments compared to the non-redundant profile of 28 sequences. If the sequences well represent the evolutionary history of the protein family, a factor of 10 to 100 less information is required.
.")
25 Evolutionary Structure/Sequence Profiles Suggest Reaction Pathway R. Amaro and Z. Schulten, MD Simulations of Substrate Channeling, Chemical Physics Special Issue, 2004 (in press). FE Landscapes of Ammonia Channeling, PNAS 2003

26 Evolution of Structure and Function in AspRS


27 AARS domains have different Evolutionary Histories catalytic domains AARSs II catalytic domain accessory domain anticodon binding domain bacterial type aspartyl-trna synthetase E. coli, homodimer
28 Summary Evolutionary information is encoded in protein structure. Protein structure can be used to investigate early evolutionary events. Accounting for gaps is important for comparing homologous structures - structure metric Multidimensional QR factorization computes non-redundant sets from multiple sequence or structure alignments which well represent the evolutionary history of the group as expressed in phylogenetic tree Structure databases are limited, but multiple structural alignments provide accurate alignments, especially in the case of distant homologies Supplement the structures with an appropriate number and type of sequences (in accord with the phylogenetic topology) to produce minimal representative profiles. Search profiles for foldons!!




29 San Diego, 2004 Evolution of Protein Structure

30 VMD Multiple Sequence Display with Evolution Analysis Algorithms
31 Patrick O Donoghue Rommie Amaro Anurag Sethi John Eargle Corey Hardin Michael Baym Michael Janusyzk Felix Autenrieth Taras Pogorelov Brijeet Dhaliwal Acknowledgements Graphics Programmers VMD John Stone, Dan Wright, John Eargle Collaborators Evolutionary Studies Gary Olsen, Carl Woese (UIUC) Algorithms Mike Heath (UIUC) Rob Russell (EMBL) STAMP Protein Structure Prediction Peter Wolynes, Jose Onuchic, Ken Suslick Funding: NSF, NIH, NIH Resource for Macromolecular Modeling and Bioinformatics, NRAC NSF Supercomputer Centers
32 extra slides
33 Structure Phylogeny Class I AARSs Structure Phylogeny Class II AARSs
34 Structure Phylogeny Class I AARSs Structure Phylogeny Class II AARSs

35 Structural Overlap of the AARSs


36 Structural Conservation in trna T-loop acceptor stem D-loop anticodon loop
37 Representative set of OB folds involved in translation?
38
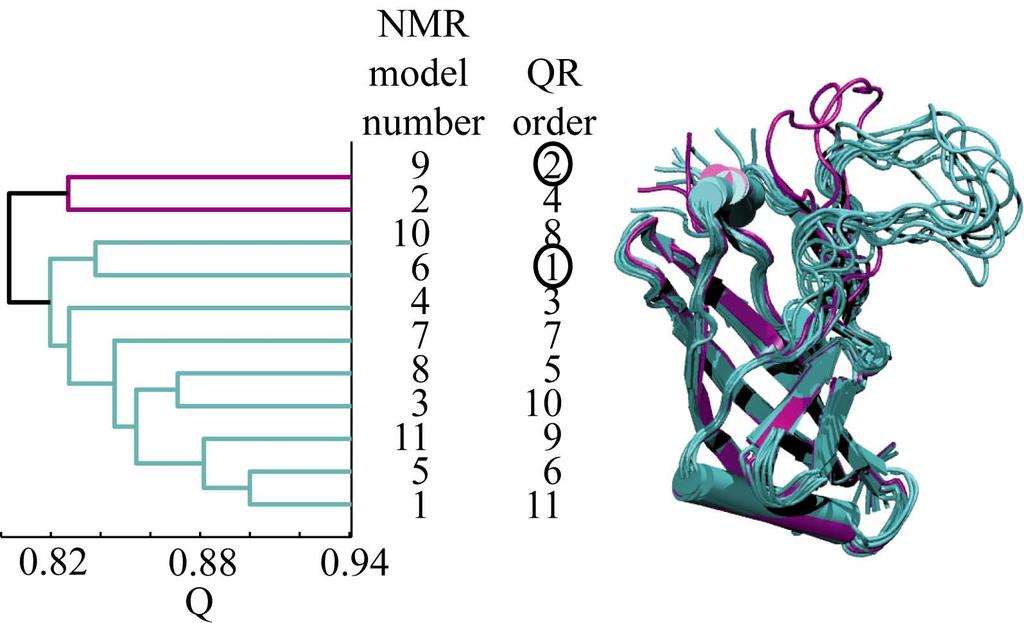
39
40 Only structure can reveal distant evolutionary relationships

41 Conservation of Sequence and Structure GlnRS AsnRS

42 Protein structure encodes evolutionary information Sequence Phylogeny Woese et al Structural Overlap Structure Phylogeny Class II AARSs Da De N Db T. thermophilus P. kodakaraensis

43 Sequence Phylogeny Woese et al Horizontal Gene Transfer and Protein Structure in ProRS Structural Overlap Structure Phylogeny Class II AARSs Pa Pb T. thermophilus M. thermoautotrophicus
44 Structural Homology Measure the effect of insertions Gaps influence the analysis But should not dominate it CW
45 Structural Homology Measure compare inserted residues to gap edges g a g a g a j j
46 Solve the least squares problem QR Factorization by triangularizing A with and orthogonal transformation. The system is now solved by back substitution, with a minimum residual of
47 Multi-Dimensional QR N-dimensional QR = N one-dimensional QRs. Permutation matrix is constant for each dimension, ordering norm is Frobenius-like matrix p-norm. Encoding Structure Aligned residues: Gap residues : Gap Scaling Encoding Sequence Orthogonal Encoding = 24-space 23 amino acids symbols (20 + B, X, Z + GAP) A=(1,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0) B=(0,1,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0) C=(0,0,1,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0) GAP=(0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,1) L. Heck, J. Olkin, and K. Nagshineh (1998) J. Vibration Acoustics 120:663. P. O Donoghue and Z. Luthey-Schulten (2003) Micro. Mol. Biol. Rev. 67:
48 QR Factorization with Column Pivoting 1. Calculate column norm of column i and all columns to the right. Ordering Norm 2. Swap column i with column to the right of maximum norm and record column permutation. 3. Construct and apply H k Golub, Numerische Mathematik, 1965 Original matrix, A, columns ordered by increasing linear dependence.
49 Protein Structure Prediction //backboneresidueresidueresidueresidueamcontacte Ab Initio protein folding N µ E AM = { } µ µ γ AM [P i, P j, P, P ] µ =1 i, j X exp (r ij µ r ) 2 i ' j ' 2σ ij 2 i ' j ' 1-D protein sequence SISSIRVKSKRIQLG 3-D protein structure Threading/Profile Alignment E = E + E match gap Target Sequence SISSRVKSKRIQLGLNQAELAQKV------GTTQ QFANEFKVRRIKLGYTQ----TNVGEALAAVHGS Known structure(s) Eastwood,Hardin,Luthey-Schulten,Wolynes (2001) IBM. J.RES.&DEV.45: Papoian, et.al. PNAS (2004)
 E profile = γ i i, i E contact = i, j 2 k = 1 ( A, SS SA ) γ ( ct) k i gap ( A, A ) U( r r ) i j 2.")
50 Target sequence Sequence-Structure Alignment Alignment between target(s) and scaffold(s) Scaffold structure A 1 A 2 A 3 A 4 A 5 1. Energy Based Threading* H + E = Econtact + E profile + EH bonds n ( p) E profile = γ i i, i E contact = i, j 2 k = 1 ( A, SS SA ) γ ( ct) k i gap ( A, A ) U( r r ) i j 2. Sequence Structure Profile Alignments k ij Clustal, Hidden Markov (HMMER, PSSM) with position dependent gap penalties *R. Goldstein, Z. Luthey-Schulten, P. Wolynes (1992, PNAS), K. Koretke et.al. (1996, Proteins)



51 CASP5 Fold Recognition/Threading Schulten-Wolynes Group The prediction is never better than the scaffold. Threading energy function/profiles requires improvement.

52 Why Study the Evolution of Protein Structure? In what specific ways has the evolutionary dynamic changed protein shape over time? Substitution Indel Domain Insertion What can studying the change in protein shape over time tell us about the evolutionary process? How did translation evolve? implications for protein structure prediction, protein design When, with respect to the root of the universal phylogenetic tree, was translation established in its modern form? What was the role of the AARSs in the evolution of the translation mechanism, development of the genetic code?
Introduction to Evolutionary Concepts
 Introduction to Evolutionary Concepts and VMD/MultiSeq - Part I Zaida (Zan) Luthey-Schulten Dept. Chemistry, Beckman Institute, Biophysics, Institute of Genomics Biology, & Physics NIH Workshop 2009 VMD/MultiSeq
Introduction to Evolutionary Concepts and VMD/MultiSeq - Part I Zaida (Zan) Luthey-Schulten Dept. Chemistry, Beckman Institute, Biophysics, Institute of Genomics Biology, & Physics NIH Workshop 2009 VMD/MultiSeq
Part III - Bioinformatics Study of Aminoacyl trna Synthetases. VMD Multiseq Tutorial Web tools. Perth, Australia 2004 Computational Biology Workshop
 Part III - Bioinformatics Study of Aminoacyl trna Synthetases VMD Multiseq Tutorial Web tools Perth, Australia 2004 Computational Biology Workshop Multiple Sequence Alignments The aminoacyl-trna synthetases,
Part III - Bioinformatics Study of Aminoacyl trna Synthetases VMD Multiseq Tutorial Web tools Perth, Australia 2004 Computational Biology Workshop Multiple Sequence Alignments The aminoacyl-trna synthetases,
Sequence and Structure Alignment Z. Luthey-Schulten, UIUC Pittsburgh, 2006 VMD 1.8.5
 Sequence and Structure Alignment Z. Luthey-Schulten, UIUC Pittsburgh, 2006 VMD 1.8.5 Why Look at More Than One Sequence? 1. Multiple Sequence Alignment shows patterns of conservation 2. What and how many
Sequence and Structure Alignment Z. Luthey-Schulten, UIUC Pittsburgh, 2006 VMD 1.8.5 Why Look at More Than One Sequence? 1. Multiple Sequence Alignment shows patterns of conservation 2. What and how many
Evolution of Protein Structure
 University of Illinois at Urbana-Champaign Luthey-Schulten Group Theoretical and Computational Biophysics Group Computational Biophysics Workshop Evolution of Protein Structure Aspartyl-tRNA Synthetase
University of Illinois at Urbana-Champaign Luthey-Schulten Group Theoretical and Computational Biophysics Group Computational Biophysics Workshop Evolution of Protein Structure Aspartyl-tRNA Synthetase
Evolution of Translation: Dynamics of Recognition in RNA:Protein Complexes
 Evolution of Translation: Dynamics of Recognition in RNA:Protein Complexes Zaida (Zan) Luthey-Schulten Dept. Chemistry, Beckman Institute, Biophysics, Institute of Genomics Biology, & Physics NIH Workshop
Evolution of Translation: Dynamics of Recognition in RNA:Protein Complexes Zaida (Zan) Luthey-Schulten Dept. Chemistry, Beckman Institute, Biophysics, Institute of Genomics Biology, & Physics NIH Workshop
Introduction to Evolutionary Concepts
 Introduction to Evolutionary Concepts and VMD/MultiSeq - Part I Zaida (Zan) Luthey-Schulten Dept. Chemistry, Beckman Institute, Biophysics, Institute of Genomics Biology, & Physics NIH Workshop 2010 VMD/MultiSeq
Introduction to Evolutionary Concepts and VMD/MultiSeq - Part I Zaida (Zan) Luthey-Schulten Dept. Chemistry, Beckman Institute, Biophysics, Institute of Genomics Biology, & Physics NIH Workshop 2010 VMD/MultiSeq
Characterizing molecular systems
 Introduction to evolutionary concepts and VMD/MultiSeq - Part I Characterizing molecular systems Zaida (Zan) Luthey-Schulten Dept. Chemistry, Physics, Beckman Institute, Institute of Genomics Biology,
Introduction to evolutionary concepts and VMD/MultiSeq - Part I Characterizing molecular systems Zaida (Zan) Luthey-Schulten Dept. Chemistry, Physics, Beckman Institute, Institute of Genomics Biology,
Supporting Information for Evolutionary profiles from the QR factorization of multiple sequence alignments
 Supporting Information for Evolutionary profiles from the QR factorization of multiple sequence alignments Anurag Sethi, Patrick O Donoghue and Zaida Luthey-Schulten Department of Chemistry University
Supporting Information for Evolutionary profiles from the QR factorization of multiple sequence alignments Anurag Sethi, Patrick O Donoghue and Zaida Luthey-Schulten Department of Chemistry University
On the Evolution of Structure in Aminoacyl-tRNA Synthetases
 MICROBIOLOGY AND MOLECULAR BIOLOGY REVIEWS, Dec. 2003, p. 550 573 Vol. 67, No. 4 1092-2172/03/$08.00 0 DOI: 10.1128/MMBR.67.4.550 573.2003 Copyright 2003, American Society for Microbiology. All Rights
MICROBIOLOGY AND MOLECULAR BIOLOGY REVIEWS, Dec. 2003, p. 550 573 Vol. 67, No. 4 1092-2172/03/$08.00 0 DOI: 10.1128/MMBR.67.4.550 573.2003 Copyright 2003, American Society for Microbiology. All Rights
ARTICLE IN PRESS. Evolutionary Profiles Derived from the QR Factorization of Multiple Structural Alignments Gives an Economy of Information
 doi:10.1016/j.jmb.2004.11.053 J. Mol. Biol. (2005) xx, 1 20 Evolutionary Profiles Derived from the QR Factorization of Multiple Structural Alignments Gives an Economy of Information Patrick O Donoghue
doi:10.1016/j.jmb.2004.11.053 J. Mol. Biol. (2005) xx, 1 20 Evolutionary Profiles Derived from the QR Factorization of Multiple Structural Alignments Gives an Economy of Information Patrick O Donoghue
Phylogenomics. Gene History, Genome History and Organismal Phylogeny (genealogy of cellular life)
") Phylogenomics Gene History, Genome History and Organismal Phylogeny (genealogy of cellular life) The time will come I believe, though I shall not live to see it,when we shall have very fairly true genealogical
Phylogenomics Gene History, Genome History and Organismal Phylogeny (genealogy of cellular life) The time will come I believe, though I shall not live to see it,when we shall have very fairly true genealogical
What can sequences tell us?
 Bioinformatics What can sequences tell us? AGACCTGAGATAACCGATAC By themselves? Not a heck of a lot...* *Indeed, one of the key results learned from the Human Genome Project is that disease is much more
Bioinformatics What can sequences tell us? AGACCTGAGATAACCGATAC By themselves? Not a heck of a lot...* *Indeed, one of the key results learned from the Human Genome Project is that disease is much more
Algorithms in Bioinformatics FOUR Pairwise Sequence Alignment. Pairwise Sequence Alignment. Convention: DNA Sequences 5. Sequence Alignment
 Algorithms in Bioinformatics FOUR Sami Khuri Department of Computer Science San José State University Pairwise Sequence Alignment Homology Similarity Global string alignment Local string alignment Dot
Algorithms in Bioinformatics FOUR Sami Khuri Department of Computer Science San José State University Pairwise Sequence Alignment Homology Similarity Global string alignment Local string alignment Dot
Introduction to Protein Structures - Molecular Graphics Tool
 Lecture 1a Introduction to Protein Structures - Molecular Graphics Tool amino acid tyrosine LH2 energetics traficking Ubiquitin enzymatic control BPTI Highlights of the VMD Molecular Graphics Program >
Lecture 1a Introduction to Protein Structures - Molecular Graphics Tool amino acid tyrosine LH2 energetics traficking Ubiquitin enzymatic control BPTI Highlights of the VMD Molecular Graphics Program >
Evolution of Biomolecular Structure
 University of Illinois at Urbana-Champaign Luthey-Schulten Group Theoretical and Computational Biophysics Group Evolution of Biomolecular Structure Class II trna-synthetases and trna MultiSeq Developers:
University of Illinois at Urbana-Champaign Luthey-Schulten Group Theoretical and Computational Biophysics Group Evolution of Biomolecular Structure Class II trna-synthetases and trna MultiSeq Developers:
Domain-based computational approaches to understand the molecular basis of diseases
 Domain-based computational approaches to understand the molecular basis of diseases Dr. Maricel G. Kann Assistant Professor Dept of Biological Sciences UMBC http://bioinf.umbc.edu Research at Kann s Lab.
Domain-based computational approaches to understand the molecular basis of diseases Dr. Maricel G. Kann Assistant Professor Dept of Biological Sciences UMBC http://bioinf.umbc.edu Research at Kann s Lab.
Evolution of Translation: Dynamics of Recognition in RNA:Protein Complexes Part II - Applications of MultiSeq
 Evolution of Translation: Dynamics of Recognition in RNA:Protein Complexes Part II - Applications of MultiSeq Zaida (Zan) Luthey-Schulten Dept. Chemistry, Physics, Beckman Institute, Biophysics, Institute
Evolution of Translation: Dynamics of Recognition in RNA:Protein Complexes Part II - Applications of MultiSeq Zaida (Zan) Luthey-Schulten Dept. Chemistry, Physics, Beckman Institute, Biophysics, Institute
NIH Center for Macromolecular Modeling and Bioinformatics Developer of VMD and NAMD. Beckman Institute
 NIH Center for Macromolecular Modeling and Bioinformatics Developer of VMD and NAMD 5 faculty members (2 physics, 1 chemistry, 1 biochemistry, 1 computer science); 8 developers; 1 system admin; 15 post
NIH Center for Macromolecular Modeling and Bioinformatics Developer of VMD and NAMD 5 faculty members (2 physics, 1 chemistry, 1 biochemistry, 1 computer science); 8 developers; 1 system admin; 15 post
Statistical Machine Learning Methods for Bioinformatics II. Hidden Markov Model for Biological Sequences
 Statistical Machine Learning Methods for Bioinformatics II. Hidden Markov Model for Biological Sequences Jianlin Cheng, PhD Department of Computer Science University of Missouri 2008 Free for Academic
Statistical Machine Learning Methods for Bioinformatics II. Hidden Markov Model for Biological Sequences Jianlin Cheng, PhD Department of Computer Science University of Missouri 2008 Free for Academic
Statistical Machine Learning Methods for Bioinformatics IV. Neural Network & Deep Learning Applications in Bioinformatics
 Statistical Machine Learning Methods for Bioinformatics IV. Neural Network & Deep Learning Applications in Bioinformatics Jianlin Cheng, PhD Department of Computer Science University of Missouri, Columbia
Statistical Machine Learning Methods for Bioinformatics IV. Neural Network & Deep Learning Applications in Bioinformatics Jianlin Cheng, PhD Department of Computer Science University of Missouri, Columbia
Bioinformatics (GLOBEX, Summer 2015) Pairwise sequence alignment
 Pairwise sequence alignment") Bioinformatics (GLOBEX, Summer 2015) Pairwise sequence alignment Substitution score matrices, PAM, BLOSUM Needleman-Wunsch algorithm (Global) Smith-Waterman algorithm (Local) BLAST (local, heuristic) E-value
Bioinformatics (GLOBEX, Summer 2015) Pairwise sequence alignment Substitution score matrices, PAM, BLOSUM Needleman-Wunsch algorithm (Global) Smith-Waterman algorithm (Local) BLAST (local, heuristic) E-value
SUPPLEMENTARY INFORMATION
 Supplementary information S3 (box) Methods Methods Genome weighting The currently available collection of archaeal and bacterial genomes has a highly biased distribution of isolates across taxa. For example,
Supplementary information S3 (box) Methods Methods Genome weighting The currently available collection of archaeal and bacterial genomes has a highly biased distribution of isolates across taxa. For example,
Basic Local Alignment Search Tool
 Basic Local Alignment Search Tool Alignments used to uncover homologies between sequences combined with phylogenetic studies o can determine orthologous and paralogous relationships Local Alignment uses
Basic Local Alignment Search Tool Alignments used to uncover homologies between sequences combined with phylogenetic studies o can determine orthologous and paralogous relationships Local Alignment uses
Sequence analysis and comparison
 The aim with sequence identification: Sequence analysis and comparison Marjolein Thunnissen Lund September 2012 Is there any known protein sequence that is homologous to mine? Are there any other species
The aim with sequence identification: Sequence analysis and comparison Marjolein Thunnissen Lund September 2012 Is there any known protein sequence that is homologous to mine? Are there any other species
Neural Networks for Protein Structure Prediction Brown, JMB CS 466 Saurabh Sinha
 Neural Networks for Protein Structure Prediction Brown, JMB 1999 CS 466 Saurabh Sinha Outline Goal is to predict secondary structure of a protein from its sequence Artificial Neural Network used for this
Neural Networks for Protein Structure Prediction Brown, JMB 1999 CS 466 Saurabh Sinha Outline Goal is to predict secondary structure of a protein from its sequence Artificial Neural Network used for this
Week 10: Homology Modelling (II) - HHpred
 - HHpred") Week 10: Homology Modelling (II) - HHpred Course: Tools for Structural Biology Fabian Glaser BKU - Technion 1 2 Identify and align related structures by sequence methods is not an easy task All comparative
Week 10: Homology Modelling (II) - HHpred Course: Tools for Structural Biology Fabian Glaser BKU - Technion 1 2 Identify and align related structures by sequence methods is not an easy task All comparative
Computational methods for predicting protein-protein interactions
 Computational methods for predicting protein-protein interactions Tomi Peltola T-61.6070 Special course in bioinformatics I 3.4.2008 Outline Biological background Protein-protein interactions Computational
Computational methods for predicting protein-protein interactions Tomi Peltola T-61.6070 Special course in bioinformatics I 3.4.2008 Outline Biological background Protein-protein interactions Computational
InDel 3-5. InDel 8-9. InDel 3-5. InDel 8-9. InDel InDel 8-9
 Lecture 5 Alignment I. Introduction. For sequence data, the process of generating an alignment establishes positional homologies; that is, alignment provides the identification of homologous phylogenetic
Lecture 5 Alignment I. Introduction. For sequence data, the process of generating an alignment establishes positional homologies; that is, alignment provides the identification of homologous phylogenetic
Pairwise & Multiple sequence alignments
 Pairwise & Multiple sequence alignments Urmila Kulkarni-Kale Bioinformatics Centre 411 007 urmila@bioinfo.ernet.in Basis for Sequence comparison Theory of evolution: gene sequences have evolved/derived
Pairwise & Multiple sequence alignments Urmila Kulkarni-Kale Bioinformatics Centre 411 007 urmila@bioinfo.ernet.in Basis for Sequence comparison Theory of evolution: gene sequences have evolved/derived
Protein Bioinformatics. Rickard Sandberg Dept. of Cell and Molecular Biology Karolinska Institutet sandberg.cmb.ki.
 Protein Bioinformatics Rickard Sandberg Dept. of Cell and Molecular Biology Karolinska Institutet rickard.sandberg@ki.se sandberg.cmb.ki.se Outline Protein features motifs patterns profiles signals 2 Protein
Protein Bioinformatics Rickard Sandberg Dept. of Cell and Molecular Biology Karolinska Institutet rickard.sandberg@ki.se sandberg.cmb.ki.se Outline Protein features motifs patterns profiles signals 2 Protein
MiGA: The Microbial Genome Atlas
 December 12 th 2017 MiGA: The Microbial Genome Atlas Jim Cole Center for Microbial Ecology Dept. of Plant, Soil & Microbial Sciences Michigan State University East Lansing, Michigan U.S.A. Where I m From
December 12 th 2017 MiGA: The Microbial Genome Atlas Jim Cole Center for Microbial Ecology Dept. of Plant, Soil & Microbial Sciences Michigan State University East Lansing, Michigan U.S.A. Where I m From
Sequence Alignment: A General Overview. COMP Fall 2010 Luay Nakhleh, Rice University
 Sequence Alignment: A General Overview COMP 571 - Fall 2010 Luay Nakhleh, Rice University Life through Evolution All living organisms are related to each other through evolution This means: any pair of
Sequence Alignment: A General Overview COMP 571 - Fall 2010 Luay Nakhleh, Rice University Life through Evolution All living organisms are related to each other through evolution This means: any pair of
Tools and Algorithms in Bioinformatics
 Tools and Algorithms in Bioinformatics GCBA815, Fall 2015 Week-4 BLAST Algorithm Continued Multiple Sequence Alignment Babu Guda, Ph.D. Department of Genetics, Cell Biology & Anatomy Bioinformatics and
Tools and Algorithms in Bioinformatics GCBA815, Fall 2015 Week-4 BLAST Algorithm Continued Multiple Sequence Alignment Babu Guda, Ph.D. Department of Genetics, Cell Biology & Anatomy Bioinformatics and
Chapter 19. Microbial Taxonomy
 Chapter 19 Microbial Taxonomy 12-17-2008 Taxonomy science of biological classification consists of three separate but interrelated parts classification arrangement of organisms into groups (taxa; s.,taxon)
Chapter 19 Microbial Taxonomy 12-17-2008 Taxonomy science of biological classification consists of three separate but interrelated parts classification arrangement of organisms into groups (taxa; s.,taxon)
09/06/25. Computergestützte Strukturbiologie (Strukturelle Bioinformatik) Non-uniform distribution of folds. Scheme of protein structure predicition
 Non-uniform distribution of folds. Scheme of protein structure predicition") Sequence identity Structural similarity Computergestützte Strukturbiologie (Strukturelle Bioinformatik) Fold recognition Sommersemester 2009 Peter Güntert Structural similarity X Sequence identity Non-uniform
Sequence identity Structural similarity Computergestützte Strukturbiologie (Strukturelle Bioinformatik) Fold recognition Sommersemester 2009 Peter Güntert Structural similarity X Sequence identity Non-uniform
CMPS 6630: Introduction to Computational Biology and Bioinformatics. Structure Comparison
 CMPS 6630: Introduction to Computational Biology and Bioinformatics Structure Comparison Protein Structure Comparison Motivation Understand sequence and structure variability Understand Domain architecture
CMPS 6630: Introduction to Computational Biology and Bioinformatics Structure Comparison Protein Structure Comparison Motivation Understand sequence and structure variability Understand Domain architecture
Large-Scale Genomic Surveys
 Bioinformatics Subtopics Fold Recognition Secondary Structure Prediction Docking & Drug Design Protein Geometry Protein Flexibility Homology Modeling Sequence Alignment Structure Classification Gene Prediction
Bioinformatics Subtopics Fold Recognition Secondary Structure Prediction Docking & Drug Design Protein Geometry Protein Flexibility Homology Modeling Sequence Alignment Structure Classification Gene Prediction
CONCEPT OF SEQUENCE COMPARISON. Natapol Pornputtapong 18 January 2018
 CONCEPT OF SEQUENCE COMPARISON Natapol Pornputtapong 18 January 2018 SEQUENCE ANALYSIS - A ROSETTA STONE OF LIFE Sequence analysis is the process of subjecting a DNA, RNA or peptide sequence to any of
CONCEPT OF SEQUENCE COMPARISON Natapol Pornputtapong 18 January 2018 SEQUENCE ANALYSIS - A ROSETTA STONE OF LIFE Sequence analysis is the process of subjecting a DNA, RNA or peptide sequence to any of
EECS730: Introduction to Bioinformatics
 EECS730: Introduction to Bioinformatics Lecture 07: profile Hidden Markov Model http://bibiserv.techfak.uni-bielefeld.de/sadr2/databasesearch/hmmer/profilehmm.gif Slides adapted from Dr. Shaojie Zhang
EECS730: Introduction to Bioinformatics Lecture 07: profile Hidden Markov Model http://bibiserv.techfak.uni-bielefeld.de/sadr2/databasesearch/hmmer/profilehmm.gif Slides adapted from Dr. Shaojie Zhang
Using Ensembles of Hidden Markov Models for Grand Challenges in Bioinformatics
 Using Ensembles of Hidden Markov Models for Grand Challenges in Bioinformatics Tandy Warnow Founder Professor of Engineering The University of Illinois at Urbana-Champaign http://tandy.cs.illinois.edu
Using Ensembles of Hidden Markov Models for Grand Challenges in Bioinformatics Tandy Warnow Founder Professor of Engineering The University of Illinois at Urbana-Champaign http://tandy.cs.illinois.edu
Computational Biology: Basics & Interesting Problems
 Computational Biology: Basics & Interesting Problems Summary Sources of information Biological concepts: structure & terminology Sequencing Gene finding Protein structure prediction Sources of information
Computational Biology: Basics & Interesting Problems Summary Sources of information Biological concepts: structure & terminology Sequencing Gene finding Protein structure prediction Sources of information
CMPS 6630: Introduction to Computational Biology and Bioinformatics. Tertiary Structure Prediction
 CMPS 6630: Introduction to Computational Biology and Bioinformatics Tertiary Structure Prediction Tertiary Structure Prediction Why Should Tertiary Structure Prediction Be Possible? Molecules obey the
CMPS 6630: Introduction to Computational Biology and Bioinformatics Tertiary Structure Prediction Tertiary Structure Prediction Why Should Tertiary Structure Prediction Be Possible? Molecules obey the
, Work in progress
 2012-2013, Work in progress Samuel Blanquart, CR2 INRIA, EPI Bonsai 10 juin 2013 2012 2013 : Researches, animation and teaching Teaching : Algorithm and Application for 20 master 1 students (24h). Teaching
2012-2013, Work in progress Samuel Blanquart, CR2 INRIA, EPI Bonsai 10 juin 2013 2012 2013 : Researches, animation and teaching Teaching : Algorithm and Application for 20 master 1 students (24h). Teaching
Homology Modeling. Roberto Lins EPFL - summer semester 2005
 Homology Modeling Roberto Lins EPFL - summer semester 2005 Disclaimer: course material is mainly taken from: P.E. Bourne & H Weissig, Structural Bioinformatics; C.A. Orengo, D.T. Jones & J.M. Thornton,
Homology Modeling Roberto Lins EPFL - summer semester 2005 Disclaimer: course material is mainly taken from: P.E. Bourne & H Weissig, Structural Bioinformatics; C.A. Orengo, D.T. Jones & J.M. Thornton,
Computational Biology From The Perspective Of A Physical Scientist
 Computational Biology From The Perspective Of A Physical Scientist Dr. Arthur Dong PP1@TUM 26 November 2013 Bioinformatics Education Curriculum Math, Physics, Computer Science (Statistics and Programming)
Computational Biology From The Perspective Of A Physical Scientist Dr. Arthur Dong PP1@TUM 26 November 2013 Bioinformatics Education Curriculum Math, Physics, Computer Science (Statistics and Programming)
Statistical Machine Learning Methods for Biomedical Informatics II. Hidden Markov Model for Biological Sequences
 Statistical Machine Learning Methods for Biomedical Informatics II. Hidden Markov Model for Biological Sequences Jianlin Cheng, PhD William and Nancy Thompson Missouri Distinguished Professor Department
Statistical Machine Learning Methods for Biomedical Informatics II. Hidden Markov Model for Biological Sequences Jianlin Cheng, PhD William and Nancy Thompson Missouri Distinguished Professor Department
Motivating the need for optimal sequence alignments...
 1 Motivating the need for optimal sequence alignments... 2 3 Note that this actually combines two objectives of optimal sequence alignments: (i) use the score of the alignment o infer homology; (ii) use
1 Motivating the need for optimal sequence alignments... 2 3 Note that this actually combines two objectives of optimal sequence alignments: (i) use the score of the alignment o infer homology; (ii) use
CMPS 3110: Bioinformatics. Tertiary Structure Prediction
 CMPS 3110: Bioinformatics Tertiary Structure Prediction Tertiary Structure Prediction Why Should Tertiary Structure Prediction Be Possible? Molecules obey the laws of physics! Conformation space is finite
CMPS 3110: Bioinformatics Tertiary Structure Prediction Tertiary Structure Prediction Why Should Tertiary Structure Prediction Be Possible? Molecules obey the laws of physics! Conformation space is finite
Quantifying sequence similarity
 Quantifying sequence similarity Bas E. Dutilh Systems Biology: Bioinformatic Data Analysis Utrecht University, February 16 th 2016 After this lecture, you can define homology, similarity, and identity
Quantifying sequence similarity Bas E. Dutilh Systems Biology: Bioinformatic Data Analysis Utrecht University, February 16 th 2016 After this lecture, you can define homology, similarity, and identity
CSCE555 Bioinformatics. Protein Function Annotation
 CSCE555 Bioinformatics Protein Function Annotation Why we need to do function annotation? Fig from: Network-based prediction of protein function. Molecular Systems Biology 3:88. 2007 What s function? The
CSCE555 Bioinformatics Protein Function Annotation Why we need to do function annotation? Fig from: Network-based prediction of protein function. Molecular Systems Biology 3:88. 2007 What s function? The
Algorithms in Bioinformatics
 Algorithms in Bioinformatics Sami Khuri Department of omputer Science San José State University San José, alifornia, USA khuri@cs.sjsu.edu www.cs.sjsu.edu/faculty/khuri Pairwise Sequence Alignment Homology
Algorithms in Bioinformatics Sami Khuri Department of omputer Science San José State University San José, alifornia, USA khuri@cs.sjsu.edu www.cs.sjsu.edu/faculty/khuri Pairwise Sequence Alignment Homology
3. SEQUENCE ANALYSIS BIOINFORMATICS COURSE MTAT
 3. SEQUENCE ANALYSIS BIOINFORMATICS COURSE MTAT.03.239 25.09.2012 SEQUENCE ANALYSIS IS IMPORTANT FOR... Prediction of function Gene finding the process of identifying the regions of genomic DNA that encode
3. SEQUENCE ANALYSIS BIOINFORMATICS COURSE MTAT.03.239 25.09.2012 SEQUENCE ANALYSIS IS IMPORTANT FOR... Prediction of function Gene finding the process of identifying the regions of genomic DNA that encode
NIH Center for Macromolecular Modeling and Bioinformatics Developer of VMD and NAMD. Beckman Institute
 NIH Center for Macromolecular Modeling and Bioinformatics Developer of VMD and NAMD 5 faculty members (2 physics, 1 chemistry, 1 biochemistry, 1 computer science); 8 developers; 1 system admin; 15 post
NIH Center for Macromolecular Modeling and Bioinformatics Developer of VMD and NAMD 5 faculty members (2 physics, 1 chemistry, 1 biochemistry, 1 computer science); 8 developers; 1 system admin; 15 post
Sequence Analysis, '18 -- lecture 9. Families and superfamilies. Sequence weights. Profiles. Logos. Building a representative model for a gene.
 Sequence Analysis, '18 -- lecture 9 Families and superfamilies. Sequence weights. Profiles. Logos. Building a representative model for a gene. How can I represent thousands of homolog sequences in a compact
Sequence Analysis, '18 -- lecture 9 Families and superfamilies. Sequence weights. Profiles. Logos. Building a representative model for a gene. How can I represent thousands of homolog sequences in a compact
SUPPLEMENTARY INFORMATION
 Supplementary information S1 (box). Supplementary Methods description. Prokaryotic Genome Database Archaeal and bacterial genome sequences were downloaded from the NCBI FTP site (ftp://ftp.ncbi.nlm.nih.gov/genomes/all/)
Supplementary information S1 (box). Supplementary Methods description. Prokaryotic Genome Database Archaeal and bacterial genome sequences were downloaded from the NCBI FTP site (ftp://ftp.ncbi.nlm.nih.gov/genomes/all/)
CISC 889 Bioinformatics (Spring 2004) Sequence pairwise alignment (I)
 Sequence pairwise alignment (I)") CISC 889 Bioinformatics (Spring 2004) Sequence pairwise alignment (I) Contents Alignment algorithms Needleman-Wunsch (global alignment) Smith-Waterman (local alignment) Heuristic algorithms FASTA BLAST
CISC 889 Bioinformatics (Spring 2004) Sequence pairwise alignment (I) Contents Alignment algorithms Needleman-Wunsch (global alignment) Smith-Waterman (local alignment) Heuristic algorithms FASTA BLAST
Syllabus of BIOINF 528 (2017 Fall, Bioinformatics Program)
") Syllabus of BIOINF 528 (2017 Fall, Bioinformatics Program) Course Name: Structural Bioinformatics Course Description: Instructor: This course introduces fundamental concepts and methods for structural
Syllabus of BIOINF 528 (2017 Fall, Bioinformatics Program) Course Name: Structural Bioinformatics Course Description: Instructor: This course introduces fundamental concepts and methods for structural
Bioinformatics. Proteins II. - Pattern, Profile, & Structure Database Searching. Robert Latek, Ph.D. Bioinformatics, Biocomputing
 Bioinformatics Proteins II. - Pattern, Profile, & Structure Database Searching Robert Latek, Ph.D. Bioinformatics, Biocomputing WIBR Bioinformatics Course, Whitehead Institute, 2002 1 Proteins I.-III.
Bioinformatics Proteins II. - Pattern, Profile, & Structure Database Searching Robert Latek, Ph.D. Bioinformatics, Biocomputing WIBR Bioinformatics Course, Whitehead Institute, 2002 1 Proteins I.-III.
Interpreting the Molecular Tree of Life: What Happened in Early Evolution? Norm Pace MCD Biology University of Colorado-Boulder
 Interpreting the Molecular Tree of Life: What Happened in Early Evolution? Norm Pace MCD Biology University of Colorado-Boulder nrpace@colorado.edu Outline What is the Tree of Life? -- Historical Conceptually
Interpreting the Molecular Tree of Life: What Happened in Early Evolution? Norm Pace MCD Biology University of Colorado-Boulder nrpace@colorado.edu Outline What is the Tree of Life? -- Historical Conceptually
Introduction to Protein Structures - Molecular Graphics Tool
 Lecture 1a Introduction to Protein Structures - Molecular Graphics Tool amino acid tyrosine LH2 energetics traficking Ubiquitin enzymatic control BPTI VMD - a Tool to Think Carl Woese Atomic, CG, Particle:
Lecture 1a Introduction to Protein Structures - Molecular Graphics Tool amino acid tyrosine LH2 energetics traficking Ubiquitin enzymatic control BPTI VMD - a Tool to Think Carl Woese Atomic, CG, Particle:
VMD: Visual Molecular Dynamics. Our Microscope is Made of...
 VMD: Visual Molecular Dynamics Computational Microscope / Tool to Think amino acid tyrosine enzymatic control BPTI VMD tutorial traficking Ubiquitin case study http://www.ks.uiuc.edu/training/casestudies/
VMD: Visual Molecular Dynamics Computational Microscope / Tool to Think amino acid tyrosine enzymatic control BPTI VMD tutorial traficking Ubiquitin case study http://www.ks.uiuc.edu/training/casestudies/
Multiple sequence alignment
 Multiple sequence alignment Multiple sequence alignment: today s goals to define what a multiple sequence alignment is and how it is generated; to describe profile HMMs to introduce databases of multiple
Multiple sequence alignment Multiple sequence alignment: today s goals to define what a multiple sequence alignment is and how it is generated; to describe profile HMMs to introduce databases of multiple
Exercise 5. Sequence Profiles & BLAST
 Exercise 5 Sequence Profiles & BLAST 1 Substitution Matrix (BLOSUM62) Likelihood to substitute one amino acid with another Figure taken from https://en.wikipedia.org/wiki/blosum 2 Substitution Matrix (BLOSUM62)
Exercise 5 Sequence Profiles & BLAST 1 Substitution Matrix (BLOSUM62) Likelihood to substitute one amino acid with another Figure taken from https://en.wikipedia.org/wiki/blosum 2 Substitution Matrix (BLOSUM62)
Computational Biology
 Computational Biology Lecture 6 31 October 2004 1 Overview Scoring matrices (Thanks to Shannon McWeeney) BLAST algorithm Start sequence alignment 2 1 What is a homologous sequence? A homologous sequence,
Computational Biology Lecture 6 31 October 2004 1 Overview Scoring matrices (Thanks to Shannon McWeeney) BLAST algorithm Start sequence alignment 2 1 What is a homologous sequence? A homologous sequence,
, Work in progress
 2011-2012, Work in progress Samuel Blanquart, CR2 INRIA, EPI Bonsai 12 juin 2012 Researches, animation and teaching Animation : Phylogeny-days, next week! Teaching : Algorithm and Application for 28 master
2011-2012, Work in progress Samuel Blanquart, CR2 INRIA, EPI Bonsai 12 juin 2012 Researches, animation and teaching Animation : Phylogeny-days, next week! Teaching : Algorithm and Application for 28 master
Page 1. References. Hidden Markov models and multiple sequence alignment. Markov chains. Probability review. Example. Markovian sequence
 Page Hidden Markov models and multiple sequence alignment Russ B Altman BMI 4 CS 74 Some slides borrowed from Scott C Schmidler (BMI graduate student) References Bioinformatics Classic: Krogh et al (994)
Page Hidden Markov models and multiple sequence alignment Russ B Altman BMI 4 CS 74 Some slides borrowed from Scott C Schmidler (BMI graduate student) References Bioinformatics Classic: Krogh et al (994)
Overview Multiple Sequence Alignment
 Overview Multiple Sequence Alignment Inge Jonassen Bioinformatics group Dept. of Informatics, UoB Inge.Jonassen@ii.uib.no Definition/examples Use of alignments The alignment problem scoring alignments
Overview Multiple Sequence Alignment Inge Jonassen Bioinformatics group Dept. of Informatics, UoB Inge.Jonassen@ii.uib.no Definition/examples Use of alignments The alignment problem scoring alignments
CISC 636 Computational Biology & Bioinformatics (Fall 2016)
") CISC 636 Computational Biology & Bioinformatics (Fall 2016) Predicting Protein-Protein Interactions CISC636, F16, Lec22, Liao 1 Background Proteins do not function as isolated entities. Protein-Protein
CISC 636 Computational Biology & Bioinformatics (Fall 2016) Predicting Protein-Protein Interactions CISC636, F16, Lec22, Liao 1 Background Proteins do not function as isolated entities. Protein-Protein
Alpha-helical Topology and Tertiary Structure Prediction of Globular Proteins Scott R. McAllister Christodoulos A. Floudas Princeton University
 Alpha-helical Topology and Tertiary Structure Prediction of Globular Proteins Scott R. McAllister Christodoulos A. Floudas Princeton University Department of Chemical Engineering Program of Applied and
Alpha-helical Topology and Tertiary Structure Prediction of Globular Proteins Scott R. McAllister Christodoulos A. Floudas Princeton University Department of Chemical Engineering Program of Applied and
THEORY. Based on sequence Length According to the length of sequence being compared it is of following two types
 Exp 11- THEORY Sequence Alignment is a process of aligning two sequences to achieve maximum levels of identity between them. This help to derive functional, structural and evolutionary relationships between
Exp 11- THEORY Sequence Alignment is a process of aligning two sequences to achieve maximum levels of identity between them. This help to derive functional, structural and evolutionary relationships between
Part II - Applications of MultiSeq Evolution of Translation: Dynamics of Recognition in RNA:Protein Complexes
 Part II - Applications of MultiSeq Evolution of Translation: Dynamics of Recognition in RNA:Protein Complexes Part III Towards in silico Cells: Simulating processes in entire cells Zaida (Zan) Luthey-Schulten!
Part II - Applications of MultiSeq Evolution of Translation: Dynamics of Recognition in RNA:Protein Complexes Part III Towards in silico Cells: Simulating processes in entire cells Zaida (Zan) Luthey-Schulten!
Homology Modeling (Comparative Structure Modeling) GBCB 5874: Problem Solving in GBCB
 GBCB 5874: Problem Solving in GBCB") Homology Modeling (Comparative Structure Modeling) Aims of Structural Genomics High-throughput 3D structure determination and analysis To determine or predict the 3D structures of all the proteins encoded
Homology Modeling (Comparative Structure Modeling) Aims of Structural Genomics High-throughput 3D structure determination and analysis To determine or predict the 3D structures of all the proteins encoded
08/21/2017 BLAST. Multiple Sequence Alignments: Clustal Omega
 BLAST Multiple Sequence Alignments: Clustal Omega What does basic BLAST do (e.g. what is input sequence and how does BLAST look for matches?) Susan Parrish McDaniel College Multiple Sequence Alignments
BLAST Multiple Sequence Alignments: Clustal Omega What does basic BLAST do (e.g. what is input sequence and how does BLAST look for matches?) Susan Parrish McDaniel College Multiple Sequence Alignments
Similarity searching summary (2)
") Similarity searching / sequence alignment summary Biol4230 Thurs, February 22, 2016 Bill Pearson wrp@virginia.edu 4-2818 Pinn 6-057 What have we covered? Homology excess similiarity but no excess similarity
Similarity searching / sequence alignment summary Biol4230 Thurs, February 22, 2016 Bill Pearson wrp@virginia.edu 4-2818 Pinn 6-057 What have we covered? Homology excess similiarity but no excess similarity
Lecture 14: Multiple Sequence Alignment (Gene Finding, Conserved Elements) Scribe: John Ekins
 Scribe: John Ekins") Lecture 14: Multiple Sequence Alignment (Gene Finding, Conserved Elements) 2 19 2015 Scribe: John Ekins Multiple Sequence Alignment Given N sequences x 1, x 2,, x N : Insert gaps in each of the sequences
Lecture 14: Multiple Sequence Alignment (Gene Finding, Conserved Elements) 2 19 2015 Scribe: John Ekins Multiple Sequence Alignment Given N sequences x 1, x 2,, x N : Insert gaps in each of the sequences
Sequence Alignment Techniques and Their Uses
 Sequence Alignment Techniques and Their Uses Sarah Fiorentino Since rapid sequencing technology and whole genomes sequencing, the amount of sequence information has grown exponentially. With all of this
Sequence Alignment Techniques and Their Uses Sarah Fiorentino Since rapid sequencing technology and whole genomes sequencing, the amount of sequence information has grown exponentially. With all of this
COMP 598 Advanced Computational Biology Methods & Research. Introduction. Jérôme Waldispühl School of Computer Science McGill University
 COMP 598 Advanced Computational Biology Methods & Research Introduction Jérôme Waldispühl School of Computer Science McGill University General informations (1) Office hours: by appointment Office: TR3018
COMP 598 Advanced Computational Biology Methods & Research Introduction Jérôme Waldispühl School of Computer Science McGill University General informations (1) Office hours: by appointment Office: TR3018
Taxonomy. Content. How to determine & classify a species. Phylogeny and evolution
 Taxonomy Content Why Taxonomy? How to determine & classify a species Domains versus Kingdoms Phylogeny and evolution Why Taxonomy? Classification Arrangement in groups or taxa (taxon = group) Nomenclature
Taxonomy Content Why Taxonomy? How to determine & classify a species Domains versus Kingdoms Phylogeny and evolution Why Taxonomy? Classification Arrangement in groups or taxa (taxon = group) Nomenclature
Bioinformatics Chapter 1. Introduction
 Bioinformatics Chapter 1. Introduction Outline! Biological Data in Digital Symbol Sequences! Genomes Diversity, Size, and Structure! Proteins and Proteomes! On the Information Content of Biological Sequences!
Bioinformatics Chapter 1. Introduction Outline! Biological Data in Digital Symbol Sequences! Genomes Diversity, Size, and Structure! Proteins and Proteomes! On the Information Content of Biological Sequences!
An Introduction to Bioinformatics Algorithms Hidden Markov Models
 Hidden Markov Models Outline 1. CG-Islands 2. The Fair Bet Casino 3. Hidden Markov Model 4. Decoding Algorithm 5. Forward-Backward Algorithm 6. Profile HMMs 7. HMM Parameter Estimation 8. Viterbi Training
Hidden Markov Models Outline 1. CG-Islands 2. The Fair Bet Casino 3. Hidden Markov Model 4. Decoding Algorithm 5. Forward-Backward Algorithm 6. Profile HMMs 7. HMM Parameter Estimation 8. Viterbi Training
An Introduction to Sequence Similarity ( Homology ) Searching
 Searching") An Introduction to Sequence Similarity ( Homology ) Searching Gary D. Stormo 1 UNIT 3.1 1 Washington University, School of Medicine, St. Louis, Missouri ABSTRACT Homologous sequences usually have the same,
An Introduction to Sequence Similarity ( Homology ) Searching Gary D. Stormo 1 UNIT 3.1 1 Washington University, School of Medicine, St. Louis, Missouri ABSTRACT Homologous sequences usually have the same,
Protein Structure Prediction II Lecturer: Serafim Batzoglou Scribe: Samy Hamdouche
 Protein Structure Prediction II Lecturer: Serafim Batzoglou Scribe: Samy Hamdouche The molecular structure of a protein can be broken down hierarchically. The primary structure of a protein is simply its
Protein Structure Prediction II Lecturer: Serafim Batzoglou Scribe: Samy Hamdouche The molecular structure of a protein can be broken down hierarchically. The primary structure of a protein is simply its
Chapter 5. Proteomics and the analysis of protein sequence Ⅱ
 Proteomics Chapter 5. Proteomics and the analysis of protein sequence Ⅱ 1 Pairwise similarity searching (1) Figure 5.5: manual alignment One of the amino acids in the top sequence has no equivalent and
Proteomics Chapter 5. Proteomics and the analysis of protein sequence Ⅱ 1 Pairwise similarity searching (1) Figure 5.5: manual alignment One of the amino acids in the top sequence has no equivalent and
BIOINFORMATICS: An Introduction
 BIOINFORMATICS: An Introduction What is Bioinformatics? The term was first coined in 1988 by Dr. Hwa Lim The original definition was : a collective term for data compilation, organisation, analysis and
BIOINFORMATICS: An Introduction What is Bioinformatics? The term was first coined in 1988 by Dr. Hwa Lim The original definition was : a collective term for data compilation, organisation, analysis and
Sara C. Madeira. Universidade da Beira Interior. (Thanks to Ana Teresa Freitas, IST for useful resources on this subject)
") Bioinformática Sequence Alignment Pairwise Sequence Alignment Universidade da Beira Interior (Thanks to Ana Teresa Freitas, IST for useful resources on this subject) 1 16/3/29 & 23/3/29 27/4/29 Outline
Bioinformática Sequence Alignment Pairwise Sequence Alignment Universidade da Beira Interior (Thanks to Ana Teresa Freitas, IST for useful resources on this subject) 1 16/3/29 & 23/3/29 27/4/29 Outline
ALL LECTURES IN SB Introduction
 1. Introduction 2. Molecular Architecture I 3. Molecular Architecture II 4. Molecular Simulation I 5. Molecular Simulation II 6. Bioinformatics I 7. Bioinformatics II 8. Prediction I 9. Prediction II ALL
1. Introduction 2. Molecular Architecture I 3. Molecular Architecture II 4. Molecular Simulation I 5. Molecular Simulation II 6. Bioinformatics I 7. Bioinformatics II 8. Prediction I 9. Prediction II ALL
Prediction of protein function from sequence analysis
 Prediction of protein function from sequence analysis Rita Casadio BIOCOMPUTING GROUP University of Bologna, Italy The omic era Genome Sequencing Projects: Archaea: 74 species In Progress:52 Bacteria:
Prediction of protein function from sequence analysis Rita Casadio BIOCOMPUTING GROUP University of Bologna, Italy The omic era Genome Sequencing Projects: Archaea: 74 species In Progress:52 Bacteria:
Computational Genomics and Molecular Biology, Fall
 Computational Genomics and Molecular Biology, Fall 2014 1 HMM Lecture Notes Dannie Durand and Rose Hoberman November 6th Introduction In the last few lectures, we have focused on three problems related
Computational Genomics and Molecular Biology, Fall 2014 1 HMM Lecture Notes Dannie Durand and Rose Hoberman November 6th Introduction In the last few lectures, we have focused on three problems related
Short Announcements. 1 st Quiz today: 15 minutes. Homework 3: Due next Wednesday.
 Short Announcements 1 st Quiz today: 15 minutes Homework 3: Due next Wednesday. Next Lecture, on Visualizing Molecular Dynamics (VMD) by Klaus Schulten Today s Lecture: Protein Folding, Misfolding, Aggregation
Short Announcements 1 st Quiz today: 15 minutes Homework 3: Due next Wednesday. Next Lecture, on Visualizing Molecular Dynamics (VMD) by Klaus Schulten Today s Lecture: Protein Folding, Misfolding, Aggregation
CAP 5510: Introduction to Bioinformatics CGS 5166: Bioinformatics Tools. Giri Narasimhan
 CAP 5510: Introduction to Bioinformatics CGS 5166: Bioinformatics Tools Giri Narasimhan ECS 254; Phone: x3748 giri@cis.fiu.edu www.cis.fiu.edu/~giri/teach/bioinfs15.html Describing & Modeling Patterns
CAP 5510: Introduction to Bioinformatics CGS 5166: Bioinformatics Tools Giri Narasimhan ECS 254; Phone: x3748 giri@cis.fiu.edu www.cis.fiu.edu/~giri/teach/bioinfs15.html Describing & Modeling Patterns
Supplementary Information
 Supplementary Information Supplementary Figure 1. Schematic pipeline for single-cell genome assembly, cleaning and annotation. a. The assembly process was optimized to account for multiple cells putatively
Supplementary Information Supplementary Figure 1. Schematic pipeline for single-cell genome assembly, cleaning and annotation. a. The assembly process was optimized to account for multiple cells putatively
Hidden Markov Models
 Hidden Markov Models Outline 1. CG-Islands 2. The Fair Bet Casino 3. Hidden Markov Model 4. Decoding Algorithm 5. Forward-Backward Algorithm 6. Profile HMMs 7. HMM Parameter Estimation 8. Viterbi Training
Hidden Markov Models Outline 1. CG-Islands 2. The Fair Bet Casino 3. Hidden Markov Model 4. Decoding Algorithm 5. Forward-Backward Algorithm 6. Profile HMMs 7. HMM Parameter Estimation 8. Viterbi Training
Title: Evolutionary dynamics of the protein structure-function relation and the origin of the genetic code
 Blue Waters Illinois Allocation Report 2016 Primary contact email and name: NAME: Dr. Gustavo Caetano-Anolles EMAIL: gca@illinois.edu Title: Evolutionary dynamics of the protein structure-function relation
Blue Waters Illinois Allocation Report 2016 Primary contact email and name: NAME: Dr. Gustavo Caetano-Anolles EMAIL: gca@illinois.edu Title: Evolutionary dynamics of the protein structure-function relation
Homology and Information Gathering and Domain Annotation for Proteins
 Homology and Information Gathering and Domain Annotation for Proteins Outline Homology Information Gathering for Proteins Domain Annotation for Proteins Examples and exercises The concept of homology The
Homology and Information Gathering and Domain Annotation for Proteins Outline Homology Information Gathering for Proteins Domain Annotation for Proteins Examples and exercises The concept of homology The
Syllabus BINF Computational Biology Core Course
 Course Description Syllabus BINF 701-702 Computational Biology Core Course BINF 701/702 is the Computational Biology core course developed at the KU Center for Computational Biology. The course is designed
Course Description Syllabus BINF 701-702 Computational Biology Core Course BINF 701/702 is the Computational Biology core course developed at the KU Center for Computational Biology. The course is designed
Sequence Bioinformatics. Multiple Sequence Alignment Waqas Nasir
 Sequence Bioinformatics Multiple Sequence Alignment Waqas Nasir 2010-11-12 Multiple Sequence Alignment One amino acid plays coy; a pair of homologous sequences whisper; many aligned sequences shout out
Sequence Bioinformatics Multiple Sequence Alignment Waqas Nasir 2010-11-12 Multiple Sequence Alignment One amino acid plays coy; a pair of homologous sequences whisper; many aligned sequences shout out
Genomics and bioinformatics summary. Finding genes -- computer searches
 Genomics and bioinformatics summary 1. Gene finding: computer searches, cdnas, ESTs, 2. Microarrays 3. Use BLAST to find homologous sequences 4. Multiple sequence alignments (MSAs) 5. Trees quantify sequence
Genomics and bioinformatics summary 1. Gene finding: computer searches, cdnas, ESTs, 2. Microarrays 3. Use BLAST to find homologous sequences 4. Multiple sequence alignments (MSAs) 5. Trees quantify sequence
Single alignment: Substitution Matrix. 16 march 2017
 Single alignment: Substitution Matrix 16 march 2017 BLOSUM Matrix BLOSUM Matrix [2] (Blocks Amino Acid Substitution Matrices ) It is based on the amino acids substitutions observed in ~2000 conserved block
Single alignment: Substitution Matrix 16 march 2017 BLOSUM Matrix BLOSUM Matrix [2] (Blocks Amino Acid Substitution Matrices ) It is based on the amino acids substitutions observed in ~2000 conserved block
CAP 5510 Lecture 3 Protein Structures
 CAP 5510 Lecture 3 Protein Structures Su-Shing Chen Bioinformatics CISE 8/19/2005 Su-Shing Chen, CISE 1 Protein Conformation 8/19/2005 Su-Shing Chen, CISE 2 Protein Conformational Structures Hydrophobicity
CAP 5510 Lecture 3 Protein Structures Su-Shing Chen Bioinformatics CISE 8/19/2005 Su-Shing Chen, CISE 1 Protein Conformation 8/19/2005 Su-Shing Chen, CISE 2 Protein Conformational Structures Hydrophobicity
HMMs and biological sequence analysis
 HMMs and biological sequence analysis Hidden Markov Model A Markov chain is a sequence of random variables X 1, X 2, X 3,... That has the property that the value of the current state depends only on the
HMMs and biological sequence analysis Hidden Markov Model A Markov chain is a sequence of random variables X 1, X 2, X 3,... That has the property that the value of the current state depends only on the