Can NOFT bridge the gap between DFT and WFT?
|
|
|
- Charity Morris
- 5 years ago
- Views:
Transcription



1 Kathmandu Workshop on Theoretical Chemistry Euskal Herriko Unibertsitatea, Kimika Fakultatea, P.K. 1072, Donostia. IKERBASQUE, Basque Foundation for Science, Bilbao, Spain. May 3, 2012
2 The exact energy functional of RDMs Outline The electronic energy E for N-electron systems E = ik H ik Γ ki + ijkl < ij kl > D kl,ij Γ ki : 1-RDM D kl,ij : 2-RDM H ik : core-hamiltonian < ij kl >: Coulomb integrals E[N, Γ, D] is an explicitly known functional of the 1- and 2-RDMs! Variational Methods: DFT (reconstruction) ρ (r) = RDMs Γ, D (contraction) = Γ N Ψ CI, MCSCF, CCSD,...


3 Volume 13 Number 45 7 December 2011 Pages COVER ARTICLE Matxain et al. Homolytic molecular dissociation in natural orbital functional theory HOT ARTICLE De Kepper et al. View Online / Journal Homepage / Table of Contents for this issue Sustained self-organizing ph patterns in hydrogen peroxide driven aqueous redox systems Outline Introduction The exact energy functional of RDMs Outline 1 Introduction to the DMFT and NOFT 2 - computational eciency of the method 3 - examples of systems, where DFT yields pathological failures - potentiality of the NOF theory. Downloaded by UNIVERSIDAD DEL PAIS VASCO on 15 November 2011 Published on 09 September 2011 on doi: /c1cp21696a Physical Chemistry Chemical Physics 4 Closing Remarks ISSN
4 1-RDM Functional Introduction 1-RDM Functional Natural Orbital Functional Two-particle cumulant - and Π-matrices Last term in the Energy: U [N, D] = ijkl < ij kl > D kl,ij can be replaced by an unknown functional of the 1-RDM: V ee [N, Γ] = min U [N, D] D D(Γ) D (Γ): family of N-representable 2-RDMs which contract to the Γ E[N, Γ, D] E[N, Γ] = ik H ik Γ ki + V ee [N, Γ] T. L. Gilbert, Phys. Rev. B 12, 2111 (1975); M. Levy, Proc. Natl. Acad. Sci. U.S.A. 76, 6062 (1979)
5 Natural Orbital Functional 1-RDM Functional Natural Orbital Functional Two-particle cumulant - and Π-matrices The 1-RDM can be diagonalized by a unitary transformation of the spin-orbitals {φ i (x)}: Γ ki = n i δ ki, Γ ( x 1 x 1 ) = i n i φ i (x ) 1 φ i (x 1 ) φ i (x) is the natural spin-orbital with the corresponding occupation number n i E[N, Γ] E [N, {n i, φ i }] = i n i H ii + V ee [N, {n i, φ i }]
6 Cumulant expansion of the 2-RDM D σσ,σσ pq,rt D αβ,αβ pq,rt 1-RDM Functional Natural Orbital Functional Two-particle cumulant - and Π-matrices = n σ p nσ q (δ 2 pr δ qt δ pt δ qr ) + λ σσ,σσ pq,rt (σ = α, β) = n α p nβ q δ 2 pr δ qt + λ αβ,αβ pq,rt λ σσ,σσ pq,rt λ αβ,αβ pq,rt = σσ pq 2 (δ pr δ qt δ pt δ qr ) = αβ pq 2 δ pr δ qt + Πpr 2 δ pqδ rt : real symmetric matrix ( σ 1σ 2 pq = σ 2σ 1 P Sum Rules: σσ pq = n σ p `1 σ P np, q q Π : spin-independent Hermitian matrix (Π αα pr = Π αβ pr = Π βα pr = Π ββ pr qp ) αβ pq = Π pr, Π pr = Π rp) = Π pp Int. J. Quantum Chem. 106, 1093 (2006)
7 1-RDM Functional Natural Orbital Functional Two-particle cumulant - and Π-matrices Conserving rule for Ŝ 2 and diagonal elements Assume N α N β and high-spin multiplet state M S = S Ŝ 2 = S (S + 1) + N β n α p nβ p 2 λ αβ,αβ pq,qp p pq conservation of the total spin 2 pq λ αβ,αβ pq,qp = N β p n α p nβ p ( λ αβ,αβ pq,qp = 1 2 Π pp αβ pp ) δ pq J. Chem. Phys. 131, (2009). for our reconstruction n α p = n p + m p, n β p = n p spin conserving rule: αβ pp = n α p nβ p = n 2 p + n p m p, Π pp = n p
8 1-RDM Functional Natural Orbital Functional Two-particle cumulant - and Π-matrices The N-representability and o-diagonal elements N-representability RDMs must be derivable from an N-particle wave function Ψ N-representability of Γ: 0 n i 1 ( i n i = N) lack of sucient conditions for N-representability of D One may approximate the unknown [n] and Π [n], in terms of the occupation numbers, considering the analytic constraints imposed by necessary N-representability conditions of the 2-RDM. D 0, Q 0 σ 1σ 2 qp n σ 1 q nσ 2 p, σ 1σ 2 qp h σ 1 q hσ 2 p G 0 Π 2 qp n q h q n p h p + qp (n q h p + h q n p ) + 2 qp
9 Implemented Approximations 1-RDM Functional Natural Orbital Functional Two-particle cumulant - and Π-matrices PNOF1: Int. J. Quantum Chem. 106, 1093, PNOF2: J. Chem. Phys. 126, , PNOF3: J. Chem. Phys. 132, , PNOF4: J. Chem. Phys. 133, , PNOF5: J. Chem. Phys. 134, , 2011.
10 1-RDM Functional Natural Orbital Functional Two-particle cumulant - and Π-matrices PNOF5: - and Π-matrices for singlet states S = 0 n 2 p, q = p n p, q = p qp = 0, q p, Π qp = 0, q p n p n p, q = p n p n p, q = p n p + n p = 1 E PNOF 5 = N [ np (2H pp + J pp ) ] n p n p K p p p=1 + N p,q=1 n q n p (2J pq K pq ) ( p = N p + 1 ; P : q p, p) J. Chem. Phys. 134, , 2011
11 Minimization of the energy functional and Euler equations The Hermitian matrix F General overview of the program architecture Parallelization of the bottleneck Minimization of the functional E PNOF 5 under constraints 1 Löwdin's normalization: 2 p n p = N (n p + n p = 1) 2 N representability of the 1-RDM: 0 n p 1 = n p = cos 2 γ p, n p = sin 2 γ p : Conjugate Gradient Method 3 Orthonormality of natural orbitals: ϕ p ϕ q = δ pq = Method of Lagrangian multipliers Ω = E 2 ε qp [< ϕ p ϕ q > δ pq ] pq
12 Minimization of the energy functional and Euler equations The Hermitian matrix F General overview of the program architecture Parallelization of the bottleneck Euler equations for the natural orbitals {ϕ p (r)} n p ˆVp ϕ p = q ε qp ϕ q, ε qp = n p ϕ q ˆVp ϕ p ˆV p (1) = Ĥ (1) + Ĵ p (1) n p n p ˆK p (1) + N q=1 ] n q [2Ĵ q (1) ˆKq (1) [Λ, Γ] 0 solution cannot be reduced to diagonalization of Λ Λ = {ε qp }, Γ = {n p δ pq } Self-consistent iterative diagonalization procedure J. Comp. Chem. 30, 2078 (2009)
13 Minimization of the energy functional and Euler equations The Hermitian matrix F General overview of the program architecture Parallelization of the bottleneck The Hermeticity of Λ and the Aufbau Principle The Lagrangian is Hermitian at the extremum: ε pq = ε qp - Dene a new Hermitian matrix F: (o-diagonal elements) F pq = θ (q p) [ε pq ε qp] + θ (p q) [ε qp ε pq ] - {F pp } cannot be determined from the Hermiticity of Λ First order perturbative theory (Hillier 1970, Saunders 1973) F 0 pq 2 E = E p<q pqf 0 = E pq p<q Fpp 0 F qq 0 { F 0 > F 0 qq pp} E is bound to drop upon diagonalization of F 0 Aufbau Principle for diagonal elements J. Comp. Chem. 30, 2078 (2009)

14 Minimization of the energy functional and Euler equations The Hermitian matrix F General overview of the program architecture Parallelization of the bottleneck
15 Parallel Eciency (H 2 O, cc-pv6z, 290 GBF) Minimization of the energy functional and Euler equations The Hermitian matrix F General overview of the program architecture Parallelization of the bottleneck cores (n) nodes Time (h) Total # Iter. Eciency (%) Maximum speedup that can be achieved is 1/(1-P) 40 P is the proportion of the program made parallel ( 97.5%) E n = T 1 nt n IT n IT1 100%
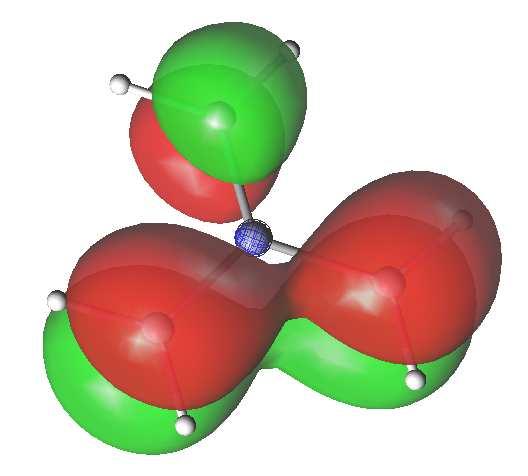





16 Planar trimethylenemethane TMM IMA OXA HOMO LUMO TMM: trimethylenemethane IMA: iminoallyl diradicaloid OXA: oxyallyl diradicaloid diradical character TMM > IMA > OXA
17 Relative Energies and Occupation Numbers Relative energy with respect to its cyclic isomer, in kcal/mol TMM IMA OXA CAS(12,12) PNOF CASPT2(12,12) Occupation numbers of the (pseudo)degenerate orbitals TMM IMA OXA PNOF4 1.07/ / /0.50 PNOF5 1.00/ / /0.54 CAS(12,12) 1.01/ / /0.55 ChemPhysChem 12, 1673, 2011




18 Ethylene Torsion Introduction Occupation γ 90 J. Chem. Phys. 134, , 2011
19 Ethylene Torsion. Energetics E (Hartrees) E (kcal/mol) Min(D2h) TS (D2d ) CASPT2(12,12) PNOF B3LYP PBE M06-2X cc-pvdz Basis Set. Optimized at the CASSCF(4,4)/cc-pVDZ level of theory. Broken symmetry energies for TS. S 2 = 1.01 J. Chem. Phys. 134, , 2011
20 cc-pvtz dissociation curves for diatomic molecules 0 Energies (kcal/mol) H2 LiH BH FH N2 CO BONDS covalent with dierent polarity H 2, FH, BH multiple bond CO, N 2 electrostatic LiH R (A) In all cases, dissociation limit implies an homolytic cleavage of the bond, high degree of near-degeneracy at the dissociation asymptote J. Chem. Phys. 134, , 2011
21 Dissociation for multiply bonded molecule: N 2 E(a.u.) N 2 ( 1 Σ) N( 4 S) + N( 4 S) PNOF5 CASSCF(10,8) CASSCF(14,14) Occupation PNOF5 - CASSCF(14,14) σ π π σ R (Å) R (Å) Phys. Chem. Chem. Phys. 13, 20129, 2011
22 cc-pvtz Ionization Potentials of N 2, in ev EKT: diagonalization of the matrix ν whose elements are ν qp = εqp nqn p Molecule MO KT PNOF5-EKT EXP N 2 σ g ( 1.63) (1.09) π u ( 0.00) (0.82) σ u ( 2.40) (1.67) J. Chem. Phys., 2012 (DOI: / )
23 14-electron isoelectronic series N 2 CN R e D e BO µ e q N R e D e BO µ e q N PNOF CAS(10,8) CAS(14,14) Exptl NO + CO R e D e BO µ e q N R e D e BO µ e q C PNOF / CAS(10,8) CAS(14,14) Exptl R e in Å, D e in kcal/mol and µ e in Debyes Phys. Chem. Chem. Phys. 13, 20129, 2011
24 Dissociation curves for O 2+ 2 E( a.u.) PNOF5 CAS (6,6) CAS (10,8) CAS (14,14) R (Å) R e (Å) BO R (Å) E ( kcal mol ) De ( kcal mol ) PNOF (6,6) (10,8) (14,14) MRCI R. H. Nobes, et. al. Chem. Phys. Lett. 182, 216 (1991) q O Phys. Chem. Chem. Phys. 13, 20129, 2011
 0.0608 (0.189) 0.1326 (0.603) 0.2010 (1.875) 0.")
 XY ( 1 Σ g ) X( 7 S3) + Y( 7 S3) CASSCF PNOF5 CASPT2")
 0.3552 (1.")
 Eective Bond Order: CASSCF/CASPT2:")
25 : Cr 2, CrMo, Mo 2 Cr 2 at R e = Å (0.125) (0.162) (0.189) (0.603) (1.875) (1.397) XY ( 1 Σ g ) X( 7 S3) + Y( 7 S3) CASSCF PNOF5 CASPT2 Exp. Cr a 1.56 CrMo b 2.09 Mo c (1.811) (1.838) a J. Chem. Theory Comput. 7, 1640 (2011) b Inorg. Chem. 50, 9219 (2011) c Chem. Phys. 343, 210 (2008) Eective Bond Order: CASSCF/CASPT2: ANO-RCC-QZ, PNOF5: 6-31G PNOF5=4.16, CASPT2=4.45
26 Canonical Orbitals Introduction E = N [n p H pp + ε pp ], ε pp = n p ϕ p ˆVp ϕ p p=1 the trace of square matrix is the sum of its diagonal elements, E = Tr (HΓ + Λ), Λ = {ε qp } the trace of a matrix is invariant under U (X = U X U) Tr (HΓ + Λ) = Tr ( H Γ + Λ ), U : Λ = U ΛU ε qp = ε p δ qp, Γ = U ΓU n qp n p δ qp {χ p (r)}: canonical orbitals
















27 Valence orbitals of methane (CH 4 ) Natural Orbital Representation Canonical Orbital Representation (0.0160) E (0.0160) (0.0160) (1.9840) (1.9840) (1.9840) ChemPhysChem 2012
28 Valence vertical ionization energies, in ev, for methane cc-pvdz cc-pvdz T 2 A1 T 2 A1 B3LYP BLYP BP M06-2X M06L M MPWPW O3LYP Experiment OLYP PBEPBE PBEHPBE PW91PW HF ε CanOrb pp EKT-PNOF OVGF Experiment Chem. Phys. Lett. 531, 272, 2012.


 Natural")









29 Valence orbitals of Benzene (C 6 H 6 ) Natural Orbital Representation Canonical Orbital Representation (0.0413) E (0.0413) (0.0427) (1.9573) (1.9587) (1.9587) ChemPhysChem 2012
30 Closing Remarks Introduction it is now feasible to perform expensive NOFT calculations. The parallelization of the bottlenecks of our code allows us to achieve an execution 37 times faster than the sequential one, in 48 processors, with an eciency of 86%. the functional N-representabilty plays a crucial role towards achieving chemical accuracy. The PNOF5 can describe in a balanced way chemical bonding situations that evolve gradually from non-degenerate to degenerate states. Integer number of electrons have been found on the dissociated atoms. two equivalent orbital representations are possible. PNOF5 could be a practical tool for the interpretation of the chemical bonding.
31 Acknowledgement Financial support comes from the Basque Government and the Spanish Oce for Scientic Research. The SGI/IZO-SGIker UPV/EHU is greatfully acknowledged for generous allocation of computational resources. Thank you for your attention!!!
NOF Theory of the molecular electronic structure
 M. Piris 1,2, J. M. Matxain 1, F. Ruipérez 1, X. Lopez 1, J. M. Ugalde 1, E. Matito 3 1 Kimika Fakultatea, UPV/EHU, and DIPC, P.K. 1072, 20080 Donostia 2 IKERBASQUE, Basque Foundation for Science, 48011
M. Piris 1,2, J. M. Matxain 1, F. Ruipérez 1, X. Lopez 1, J. M. Ugalde 1, E. Matito 3 1 Kimika Fakultatea, UPV/EHU, and DIPC, P.K. 1072, 20080 Donostia 2 IKERBASQUE, Basque Foundation for Science, 48011
QUANTUM CHEMISTRY FOR TRANSITION METALS
 QUANTUM CHEMISTRY FOR TRANSITION METALS Outline I Introduction II Correlation Static correlation effects MC methods DFT III Relativity Generalities From 4 to 1 components Effective core potential Outline
QUANTUM CHEMISTRY FOR TRANSITION METALS Outline I Introduction II Correlation Static correlation effects MC methods DFT III Relativity Generalities From 4 to 1 components Effective core potential Outline
Introduction to multiconfigurational quantum chemistry. Emmanuel Fromager
 Institut de Chimie, Strasbourg, France Page 1 Emmanuel Fromager Institut de Chimie de Strasbourg - Laboratoire de Chimie Quantique - Université de Strasbourg /CNRS M2 lecture, Strasbourg, France. Notations
Institut de Chimie, Strasbourg, France Page 1 Emmanuel Fromager Institut de Chimie de Strasbourg - Laboratoire de Chimie Quantique - Université de Strasbourg /CNRS M2 lecture, Strasbourg, France. Notations
Introduction and Overview of the Reduced Density Matrix Functional Theory
 Introduction and Overview of the Reduced Density Matrix Functional Theory N. N. Lathiotakis Theoretical and Physical Chemistry Institute, National Hellenic Research Foundation, Athens April 13, 2016 Outline
Introduction and Overview of the Reduced Density Matrix Functional Theory N. N. Lathiotakis Theoretical and Physical Chemistry Institute, National Hellenic Research Foundation, Athens April 13, 2016 Outline
Electron Correlation - Methods beyond Hartree-Fock
 Electron Correlation - Methods beyond Hartree-Fock how to approach chemical accuracy Alexander A. Auer Max-Planck-Institute for Chemical Energy Conversion, Mülheim September 4, 2014 MMER Summerschool 2014
Electron Correlation - Methods beyond Hartree-Fock how to approach chemical accuracy Alexander A. Auer Max-Planck-Institute for Chemical Energy Conversion, Mülheim September 4, 2014 MMER Summerschool 2014
Multiconfigurational Quantum Chemistry. Björn O. Roos as told by RL Department of Theoretical Chemistry Chemical Center Lund University Sweden
 Multiconfigurational Quantum Chemistry Björn O. Roos as told by RL Department of Theoretical Chemistry Chemical Center Lund University Sweden April 20, 2009 1 The Slater determinant Using the spin-orbitals,
Multiconfigurational Quantum Chemistry Björn O. Roos as told by RL Department of Theoretical Chemistry Chemical Center Lund University Sweden April 20, 2009 1 The Slater determinant Using the spin-orbitals,
AN INTRODUCTION TO QUANTUM CHEMISTRY. Mark S. Gordon Iowa State University
 AN INTRODUCTION TO QUANTUM CHEMISTRY Mark S. Gordon Iowa State University 1 OUTLINE Theoretical Background in Quantum Chemistry Overview of GAMESS Program Applications 2 QUANTUM CHEMISTRY In principle,
AN INTRODUCTION TO QUANTUM CHEMISTRY Mark S. Gordon Iowa State University 1 OUTLINE Theoretical Background in Quantum Chemistry Overview of GAMESS Program Applications 2 QUANTUM CHEMISTRY In principle,
Natural orbital functional for spin-polarized periodic systems
 Natural orbital functional for spin-polarized periodic systems Raul Quintero-Monsebaiz 1,, Ion Mitxelena,3, Mauricio Rodríguez-Mayorga, Alberto Vela 1, Mario Piris,3,4 1 Departamento de Química, Centro
Natural orbital functional for spin-polarized periodic systems Raul Quintero-Monsebaiz 1,, Ion Mitxelena,3, Mauricio Rodríguez-Mayorga, Alberto Vela 1, Mario Piris,3,4 1 Departamento de Química, Centro
Introduction to Computational Chemistry
 Introduction to Computational Chemistry Vesa Hänninen Laboratory of Physical Chemistry Chemicum 4th floor vesa.hanninen@helsinki.fi September 10, 2013 Lecture 3. Electron correlation methods September
Introduction to Computational Chemistry Vesa Hänninen Laboratory of Physical Chemistry Chemicum 4th floor vesa.hanninen@helsinki.fi September 10, 2013 Lecture 3. Electron correlation methods September
OVERVIEW OF QUANTUM CHEMISTRY METHODS
 OVERVIEW OF QUANTUM CHEMISTRY METHODS Outline I Generalities Correlation, basis sets Spin II Wavefunction methods Hartree-Fock Configuration interaction Coupled cluster Perturbative methods III Density
OVERVIEW OF QUANTUM CHEMISTRY METHODS Outline I Generalities Correlation, basis sets Spin II Wavefunction methods Hartree-Fock Configuration interaction Coupled cluster Perturbative methods III Density
Ab initio calculations for potential energy surfaces. D. Talbi GRAAL- Montpellier
 Ab initio calculations for potential energy surfaces D. Talbi GRAAL- Montpellier A theoretical study of a reaction is a two step process I-Electronic calculations : techniques of quantum chemistry potential
Ab initio calculations for potential energy surfaces D. Talbi GRAAL- Montpellier A theoretical study of a reaction is a two step process I-Electronic calculations : techniques of quantum chemistry potential
Coupled-Cluster Perturbative Triples for Bond Breaking
 Coupled-Cluster Perturbative Triples for Bond Breaking Andrew G. Taube and Rodney J. Bartlett Quantum Theory Project University of Florida INT CC Meeting Seattle July 8, 2008 Why does chemistry need triples?
Coupled-Cluster Perturbative Triples for Bond Breaking Andrew G. Taube and Rodney J. Bartlett Quantum Theory Project University of Florida INT CC Meeting Seattle July 8, 2008 Why does chemistry need triples?
The Overhauser Instability
 The Overhauser Instability Zoltán Radnai and Richard Needs TCM Group ESDG Talk 14th February 2007 Typeset by FoilTEX Introduction Hartree-Fock theory and Homogeneous Electron Gas Noncollinear spins and
The Overhauser Instability Zoltán Radnai and Richard Needs TCM Group ESDG Talk 14th February 2007 Typeset by FoilTEX Introduction Hartree-Fock theory and Homogeneous Electron Gas Noncollinear spins and
Answers Quantum Chemistry NWI-MOL406 G. C. Groenenboom and G. A. de Wijs, HG00.307, 8:30-11:30, 21 jan 2014
 Answers Quantum Chemistry NWI-MOL406 G. C. Groenenboom and G. A. de Wijs, HG00.307, 8:30-11:30, 21 jan 2014 Question 1: Basis sets Consider the split valence SV3-21G one electron basis set for formaldehyde
Answers Quantum Chemistry NWI-MOL406 G. C. Groenenboom and G. A. de Wijs, HG00.307, 8:30-11:30, 21 jan 2014 Question 1: Basis sets Consider the split valence SV3-21G one electron basis set for formaldehyde
Molecules in strong magnetic fields
 Molecules in strong magnetic fields Trygve Helgaker, Kai Lange, Alessandro Soncini, and Erik Tellgren Centre for Theoretical and Computational Chemistry Department of Chemistry, University of Oslo, Norway
Molecules in strong magnetic fields Trygve Helgaker, Kai Lange, Alessandro Soncini, and Erik Tellgren Centre for Theoretical and Computational Chemistry Department of Chemistry, University of Oslo, Norway
Valence electronic structure of isopropyl iodide investigated by electron momentum spectroscopy. --- Influence of intramolecular interactions
 Valence electronic structure of isopropyl iodide investigated by electron momentum spectroscopy --- Influence of intramolecular interactions Minfu Zhao, Xu Shan, Shanshan Niu, Yaguo Tang, Zhaohui Liu,
Valence electronic structure of isopropyl iodide investigated by electron momentum spectroscopy --- Influence of intramolecular interactions Minfu Zhao, Xu Shan, Shanshan Niu, Yaguo Tang, Zhaohui Liu,
Building a wavefunction within the Complete-Active. Cluster with Singles and Doubles formalism: straightforward description of quasidegeneracy
 Building a wavefunction within the Complete-Active Active-Space Coupled-Cluster Cluster with Singles and Doubles formalism: straightforward description of quasidegeneracy Dmitry I. Lyakh (Karazin Kharkiv
Building a wavefunction within the Complete-Active Active-Space Coupled-Cluster Cluster with Singles and Doubles formalism: straightforward description of quasidegeneracy Dmitry I. Lyakh (Karazin Kharkiv
Session 1. Introduction to Computational Chemistry. Computational (chemistry education) and/or (Computational chemistry) education
 and/or (Computational chemistry) education") Session 1 Introduction to Computational Chemistry 1 Introduction to Computational Chemistry Computational (chemistry education) and/or (Computational chemistry) education First one: Use computational tools
Session 1 Introduction to Computational Chemistry 1 Introduction to Computational Chemistry Computational (chemistry education) and/or (Computational chemistry) education First one: Use computational tools
Study of Ozone in Tribhuvan University, Kathmandu, Nepal. Prof. S. Gurung Central Department of Physics, Tribhuvan University, Kathmandu, Nepal
 Study of Ozone in Tribhuvan University, Kathmandu, Nepal Prof. S. Gurung Central Department of Physics, Tribhuvan University, Kathmandu, Nepal 1 Country of the Mt Everest 2 View of the Mt Everest 3 4 5
Study of Ozone in Tribhuvan University, Kathmandu, Nepal Prof. S. Gurung Central Department of Physics, Tribhuvan University, Kathmandu, Nepal 1 Country of the Mt Everest 2 View of the Mt Everest 3 4 5
3: Many electrons. Orbital symmetries. l =2 1. m l
 3: Many electrons Orbital symmetries Atomic orbitals are labelled according to the principal quantum number, n, and the orbital angular momentum quantum number, l. Electrons in a diatomic molecule experience
3: Many electrons Orbital symmetries Atomic orbitals are labelled according to the principal quantum number, n, and the orbital angular momentum quantum number, l. Electrons in a diatomic molecule experience
The calculation of the universal density functional by Lieb maximization
 The calculation of the universal density functional by Lieb maximization Trygve Helgaker, Andy Teale, and Sonia Coriani Centre for Theoretical and Computational Chemistry (CTCC), Department of Chemistry,
The calculation of the universal density functional by Lieb maximization Trygve Helgaker, Andy Teale, and Sonia Coriani Centre for Theoretical and Computational Chemistry (CTCC), Department of Chemistry,
Basis sets for electron correlation
 Basis sets for electron correlation Trygve Helgaker Centre for Theoretical and Computational Chemistry Department of Chemistry, University of Oslo, Norway The 12th Sostrup Summer School Quantum Chemistry
Basis sets for electron correlation Trygve Helgaker Centre for Theoretical and Computational Chemistry Department of Chemistry, University of Oslo, Norway The 12th Sostrup Summer School Quantum Chemistry
Density Functional Theory: from theory to Applications
 Density Functional Theory: from theory to Applications Uni Mainz November 29, 2010 The self interaction error and its correction Perdew-Zunger SIC Average-density approximation Weighted density approximation
Density Functional Theory: from theory to Applications Uni Mainz November 29, 2010 The self interaction error and its correction Perdew-Zunger SIC Average-density approximation Weighted density approximation
Conical Intersections. Spiridoula Matsika
 Conical Intersections Spiridoula Matsika The Born-Oppenheimer approximation Energy TS Nuclear coordinate R ν The study of chemical systems is based on the separation of nuclear and electronic motion The
Conical Intersections Spiridoula Matsika The Born-Oppenheimer approximation Energy TS Nuclear coordinate R ν The study of chemical systems is based on the separation of nuclear and electronic motion The
σ u * 1s g - gerade u - ungerade * - antibonding σ g 1s
 One of these two states is a repulsive (dissociative) state. Other excited states can be constructed using linear combinations of other orbitals. Some will be binding and others will be repulsive. Thus
One of these two states is a repulsive (dissociative) state. Other excited states can be constructed using linear combinations of other orbitals. Some will be binding and others will be repulsive. Thus
5.61 Physical Chemistry Exam III 11/29/12. MASSACHUSETTS INSTITUTE OF TECHNOLOGY Department of Chemistry Chemistry Physical Chemistry.
 MASSACHUSETTS INSTITUTE OF TECHNOLOGY Department of Chemistry Chemistry - 5.61 Physical Chemistry Exam III (1) PRINT your name on the cover page. (2) It is suggested that you READ THE ENTIRE EXAM before
MASSACHUSETTS INSTITUTE OF TECHNOLOGY Department of Chemistry Chemistry - 5.61 Physical Chemistry Exam III (1) PRINT your name on the cover page. (2) It is suggested that you READ THE ENTIRE EXAM before
Supporting Information
 Supporting Information Computational Evidence of Inversion of 1 L a and 1 L b -Derived Excited States in Naphthalene Excimer Formation from ab Initio Multireference Theory with Large Active Space: DMRG-CASPT2
Supporting Information Computational Evidence of Inversion of 1 L a and 1 L b -Derived Excited States in Naphthalene Excimer Formation from ab Initio Multireference Theory with Large Active Space: DMRG-CASPT2
List of Figures Page Figure No. Figure Caption No. Figure 1.1.
 List of Figures Figure No. Figure Caption Page No. Figure 1.1. Cation- interactions and their modulations. 4 Figure 1.2. Three conformations of benzene dimer, S is not a minimum on the potential energy
List of Figures Figure No. Figure Caption Page No. Figure 1.1. Cation- interactions and their modulations. 4 Figure 1.2. Three conformations of benzene dimer, S is not a minimum on the potential energy
Lecture 4: Hartree-Fock Theory
 Lecture 4: Hartree-Fock Theory One determinant to rule them all, One determinant to find them, One determinant to bring them all and in the darkness bind them Second quantization rehearsal The formalism
Lecture 4: Hartree-Fock Theory One determinant to rule them all, One determinant to find them, One determinant to bring them all and in the darkness bind them Second quantization rehearsal The formalism
Tutorial I: IQ MOL and Basic DFT and MP2 Calculations 1 / 30
 Tutorial I: IQ MOL and Basic DFT and MP2 Calculations Q-Chem User Workshop, Denver March 21, 2015 1 / 30 2 / 30 Introduction to IQMOL DFT and MP2 Calculations 3 / 30 IQMOL and Q-CHEM IQMOL is an open-source
Tutorial I: IQ MOL and Basic DFT and MP2 Calculations Q-Chem User Workshop, Denver March 21, 2015 1 / 30 2 / 30 Introduction to IQMOL DFT and MP2 Calculations 3 / 30 IQMOL and Q-CHEM IQMOL is an open-source
Advanced Electronic Structure Theory Density functional theory. Dr Fred Manby
 Advanced Electronic Structure Theory Density functional theory Dr Fred Manby fred.manby@bris.ac.uk http://www.chm.bris.ac.uk/pt/manby/ 6 Strengths of DFT DFT is one of many theories used by (computational)
Advanced Electronic Structure Theory Density functional theory Dr Fred Manby fred.manby@bris.ac.uk http://www.chm.bris.ac.uk/pt/manby/ 6 Strengths of DFT DFT is one of many theories used by (computational)
Quantum Chemistry Methods
 1 Quantum Chemistry Methods T. Helgaker, Department of Chemistry, University of Oslo, Norway The electronic Schrödinger equation Hartree Fock theory self-consistent field theory basis functions and basis
1 Quantum Chemistry Methods T. Helgaker, Department of Chemistry, University of Oslo, Norway The electronic Schrödinger equation Hartree Fock theory self-consistent field theory basis functions and basis
Multiconfigurational methods in MOLCAS
 Multiconfigurational methods in MOLCAS Valera Veryazov Valera.Veryazov@teokem.lu.se Division of Theoretical Chemistry Lund University Sweden CASSCF//CASPT2 p. 1/?? Overview What is correlation? CASPT2
Multiconfigurational methods in MOLCAS Valera Veryazov Valera.Veryazov@teokem.lu.se Division of Theoretical Chemistry Lund University Sweden CASSCF//CASPT2 p. 1/?? Overview What is correlation? CASPT2
4 Post-Hartree Fock Methods: MPn and Configuration Interaction
 4 Post-Hartree Fock Methods: MPn and Configuration Interaction In the limit of a complete basis, the Hartree-Fock (HF) energy in the complete basis set limit (ECBS HF ) yields an upper boundary to the
4 Post-Hartree Fock Methods: MPn and Configuration Interaction In the limit of a complete basis, the Hartree-Fock (HF) energy in the complete basis set limit (ECBS HF ) yields an upper boundary to the
Jack Simons, Henry Eyring Scientist and Professor Chemistry Department University of Utah
 1. Born-Oppenheimer approx.- energy surfaces 2. Mean-field (Hartree-Fock) theory- orbitals 3. Pros and cons of HF- RHF, UHF 4. Beyond HF- why? 5. First, one usually does HF-how? 6. Basis sets and notations
1. Born-Oppenheimer approx.- energy surfaces 2. Mean-field (Hartree-Fock) theory- orbitals 3. Pros and cons of HF- RHF, UHF 4. Beyond HF- why? 5. First, one usually does HF-how? 6. Basis sets and notations
Yingwei Wang Computational Quantum Chemistry 1 Hartree energy 2. 2 Many-body system 2. 3 Born-Oppenheimer approximation 2
 Purdue University CHM 67300 Computational Quantum Chemistry REVIEW Yingwei Wang October 10, 2013 Review: Prof Slipchenko s class, Fall 2013 Contents 1 Hartree energy 2 2 Many-body system 2 3 Born-Oppenheimer
Purdue University CHM 67300 Computational Quantum Chemistry REVIEW Yingwei Wang October 10, 2013 Review: Prof Slipchenko s class, Fall 2013 Contents 1 Hartree energy 2 2 Many-body system 2 3 Born-Oppenheimer
Pseudopotentials for hybrid density functionals and SCAN
 Pseudopotentials for hybrid density functionals and SCAN Jing Yang, Liang Z. Tan, Julian Gebhardt, and Andrew M. Rappe Department of Chemistry University of Pennsylvania Why do we need pseudopotentials?
Pseudopotentials for hybrid density functionals and SCAN Jing Yang, Liang Z. Tan, Julian Gebhardt, and Andrew M. Rappe Department of Chemistry University of Pennsylvania Why do we need pseudopotentials?
Electronic structure theory: Fundamentals to frontiers. 1. Hartree-Fock theory
 Electronic structure theory: Fundamentals to frontiers. 1. Hartree-Fock theory MARTIN HEAD-GORDON, Department of Chemistry, University of California, and Chemical Sciences Division, Lawrence Berkeley National
Electronic structure theory: Fundamentals to frontiers. 1. Hartree-Fock theory MARTIN HEAD-GORDON, Department of Chemistry, University of California, and Chemical Sciences Division, Lawrence Berkeley National
Lecture 9. Hartree Fock Method and Koopman s Theorem
 Lecture 9 Hartree Fock Method and Koopman s Theorem Ψ(N) is approximated as a single slater determinant Φ of N orthogonal One electron spin-orbitals. One electron orbital φ i = φ i (r) χ i (σ) χ i (σ)
Lecture 9 Hartree Fock Method and Koopman s Theorem Ψ(N) is approximated as a single slater determinant Φ of N orthogonal One electron spin-orbitals. One electron orbital φ i = φ i (r) χ i (σ) χ i (σ)
Optimization of quantum Monte Carlo wave functions by energy minimization
 Optimization of quantum Monte Carlo wave functions by energy minimization Julien Toulouse, Roland Assaraf, Cyrus J. Umrigar Laboratoire de Chimie Théorique, Université Pierre et Marie Curie and CNRS, Paris,
Optimization of quantum Monte Carlo wave functions by energy minimization Julien Toulouse, Roland Assaraf, Cyrus J. Umrigar Laboratoire de Chimie Théorique, Université Pierre et Marie Curie and CNRS, Paris,
Department of Physics, Chemistry and Biology
 Department of Physics, Chemistry and Biology Bachelor s Thesis Chemical bond analysis in the ten-electron series Thomas Fransson LITH-IFM-G-EX 09/2112 SE Department of Physics, Chemistry and Biology Linköpings
Department of Physics, Chemistry and Biology Bachelor s Thesis Chemical bond analysis in the ten-electron series Thomas Fransson LITH-IFM-G-EX 09/2112 SE Department of Physics, Chemistry and Biology Linköpings
TDDFT as a tool in biophysics
 TDDFT as a tool in biophysics The primary event in vision Robert Send Universität Karlsruhe 09.09.08 Robert Send TDDFT as a tool in biophysics 09.09.08 1 / 28 Outline 1 Human vision 2 The methods 3 The
TDDFT as a tool in biophysics The primary event in vision Robert Send Universität Karlsruhe 09.09.08 Robert Send TDDFT as a tool in biophysics 09.09.08 1 / 28 Outline 1 Human vision 2 The methods 3 The
Pseudo-Hermitian eigenvalue equations in linear-response electronic-structure theory
 1/11 Pseudo-Hermitian eigenvalue equations in linear-response electronic-structure theory Julien Toulouse Université Pierre & Marie Curie and CNRS, 4 place Jussieu, Paris, France Web page: www.lct.jussieu.fr/pagesperso/toulouse/
1/11 Pseudo-Hermitian eigenvalue equations in linear-response electronic-structure theory Julien Toulouse Université Pierre & Marie Curie and CNRS, 4 place Jussieu, Paris, France Web page: www.lct.jussieu.fr/pagesperso/toulouse/
ELECTRONIC STRUCTURE CALCULATIONS OF OPEN-SHELL MOLECULES. Lyudmila V. Slipchenko. A Dissertation Presented to the FACULTY OF THE GRADUATE SCHOOL
 ELECTRONIC STRUCTURE CALCULATIONS OF OPEN-SHELL MOLECULES by Lyudmila V. Slipchenko A Dissertation Presented to the FACULTY OF THE GRADUATE SCHOOL UNIVERSITY OF SOUTHERN CALIFORNIA In Partial Fulfillment
ELECTRONIC STRUCTURE CALCULATIONS OF OPEN-SHELL MOLECULES by Lyudmila V. Slipchenko A Dissertation Presented to the FACULTY OF THE GRADUATE SCHOOL UNIVERSITY OF SOUTHERN CALIFORNIA In Partial Fulfillment
Reactivity and Organocatalysis. (Adalgisa Sinicropi and Massimo Olivucci)
") Reactivity and Organocatalysis (Adalgisa Sinicropi and Massimo Olivucci) The Aldol Reaction - O R 1 O R 1 + - O O OH * * H R 2 R 1 R 2 The (1957) Zimmerman-Traxler Transition State Counterion (e.g. Li
Reactivity and Organocatalysis (Adalgisa Sinicropi and Massimo Olivucci) The Aldol Reaction - O R 1 O R 1 + - O O OH * * H R 2 R 1 R 2 The (1957) Zimmerman-Traxler Transition State Counterion (e.g. Li
Introduction to Electronic Structure Theory
 CSC/PRACE Spring School in Computational Chemistry 2017 Introduction to Electronic Structure Theory Mikael Johansson http://www.iki.fi/~mpjohans Objective: To get familiarised with the, subjectively chosen,
CSC/PRACE Spring School in Computational Chemistry 2017 Introduction to Electronic Structure Theory Mikael Johansson http://www.iki.fi/~mpjohans Objective: To get familiarised with the, subjectively chosen,
Importing ab-initio theory into DFT: Some applications of the Lieb variation principle
 Importing ab-initio theory into DFT: Some applications of the Lieb variation principle Trygve Helgaker, Andy Teale, and Sonia Coriani Centre for Theoretical and Computational Chemistry (CTCC), Department
Importing ab-initio theory into DFT: Some applications of the Lieb variation principle Trygve Helgaker, Andy Teale, and Sonia Coriani Centre for Theoretical and Computational Chemistry (CTCC), Department
with the larger dimerization energy also exhibits the larger structural changes.
 A7. Looking at the image and table provided below, it is apparent that the monomer and dimer are structurally almost identical. Although angular and dihedral data were not included, these data are also
A7. Looking at the image and table provided below, it is apparent that the monomer and dimer are structurally almost identical. Although angular and dihedral data were not included, these data are also
Molecular Magnetic Properties
 Molecular Magnetic Properties Trygve Helgaker Centre for Theoretical and Computational Chemistry (CTCC), Department of Chemistry, University of Oslo, Norway Raman Centre for Atomic, Molecular and Optical
Molecular Magnetic Properties Trygve Helgaker Centre for Theoretical and Computational Chemistry (CTCC), Department of Chemistry, University of Oslo, Norway Raman Centre for Atomic, Molecular and Optical
v(r i r j ) = h(r i )+ 1 N
 = h(r i )+ 1 N") Chapter 1 Hartree-Fock Theory 1.1 Formalism For N electrons in an external potential V ext (r), the many-electron Hamiltonian can be written as follows: N H = [ p i i=1 m +V ext(r i )]+ 1 N N v(r i r j
Chapter 1 Hartree-Fock Theory 1.1 Formalism For N electrons in an external potential V ext (r), the many-electron Hamiltonian can be written as follows: N H = [ p i i=1 m +V ext(r i )]+ 1 N N v(r i r j
H 2 in the minimal basis
 H 2 in the minimal basis Alston J. Misquitta Centre for Condensed Matter and Materials Physics Queen Mary, University of London January 27, 2016 Overview H 2 : The 1-electron basis. The two-electron basis
H 2 in the minimal basis Alston J. Misquitta Centre for Condensed Matter and Materials Physics Queen Mary, University of London January 27, 2016 Overview H 2 : The 1-electron basis. The two-electron basis
Hartree-Fock Theory. ˆf(r)χ i (x) = ɛ i χ i (x) (1) Z A K=1 I. DERIVATION OF THE HARTREE-FOCK EQUATIONS. A. The energy of a Slater determinant
χ i (x) = ɛ i χ i (x) (1) Z A K=1 I. DERIVATION OF THE HARTREE-FOCK EQUATIONS. A. The energy of a Slater determinant") Hartree-Fock Theory The HF approximation plays a crucial role in chemistry and constitutes the starting point for more elaborate treatments of electron correlation. Furthermore, many semi-empirical methods
Hartree-Fock Theory The HF approximation plays a crucial role in chemistry and constitutes the starting point for more elaborate treatments of electron correlation. Furthermore, many semi-empirical methods
SCF calculation on HeH +
 SCF calculation on HeH + Markus Meuwly Department of Chemistry, University of Basel, Basel, Switzerland Abstract This document describes the main steps involved in carrying out a SCF calculation on the
SCF calculation on HeH + Markus Meuwly Department of Chemistry, University of Basel, Basel, Switzerland Abstract This document describes the main steps involved in carrying out a SCF calculation on the
Noncollinear spins in QMC: spiral Spin Density Waves in the HEG
 Noncollinear spins in QMC: spiral Spin Density Waves in the HEG Zoltán Radnai and Richard J. Needs Workshop at The Towler Institute July 2006 Overview What are noncollinear spin systems and why are they
Noncollinear spins in QMC: spiral Spin Density Waves in the HEG Zoltán Radnai and Richard J. Needs Workshop at The Towler Institute July 2006 Overview What are noncollinear spin systems and why are they
Chapter 3: Relativistic Wave Equation
 Chapter 3: Relativistic Wave Equation Klein-Gordon Equation Dirac s Equation Free-electron Solutions of the Timeindependent Dirac Equation Hydrogen Solutions of the Timeindependent Dirac Equation (Angular
Chapter 3: Relativistic Wave Equation Klein-Gordon Equation Dirac s Equation Free-electron Solutions of the Timeindependent Dirac Equation Hydrogen Solutions of the Timeindependent Dirac Equation (Angular
Chem 442 Review for Exam 2. Exact separation of the Hamiltonian of a hydrogenic atom into center-of-mass (3D) and relative (3D) components.
 and relative (3D) components.") Chem 44 Review for Exam Hydrogenic atoms: The Coulomb energy between two point charges Ze and e: V r Ze r Exact separation of the Hamiltonian of a hydrogenic atom into center-of-mass (3D) and relative
Chem 44 Review for Exam Hydrogenic atoms: The Coulomb energy between two point charges Ze and e: V r Ze r Exact separation of the Hamiltonian of a hydrogenic atom into center-of-mass (3D) and relative
Size-extensive wave functions for QMC A linear-scaling GVB approach
 Size-extensive wave functions for QMC A linear-scaling GVB approach Claudia Filippi, University of Twente, The Netherlands Francesco Fracchia, University of Pisa, Italy Claudio Amovilli, University of
Size-extensive wave functions for QMC A linear-scaling GVB approach Claudia Filippi, University of Twente, The Netherlands Francesco Fracchia, University of Pisa, Italy Claudio Amovilli, University of
Beyond the Hartree-Fock Approximation: Configuration Interaction
 Beyond the Hartree-Fock Approximation: Configuration Interaction The Hartree-Fock (HF) method uses a single determinant (single electronic configuration) description of the electronic wavefunction. For
Beyond the Hartree-Fock Approximation: Configuration Interaction The Hartree-Fock (HF) method uses a single determinant (single electronic configuration) description of the electronic wavefunction. For
ABSTRACT. Takashi Tsuchimochi
 ABSTRACT Describing strong correlations with mean-field approximations by Takashi Tsuchimochi Strong electron correlations in electronic structure theory are purely quantum effects arising as a result
ABSTRACT Describing strong correlations with mean-field approximations by Takashi Tsuchimochi Strong electron correlations in electronic structure theory are purely quantum effects arising as a result
Jack Simons, Henry Eyring Scientist and Professor Chemistry Department University of Utah
 1. Born-Oppenheimer approx.- energy surfaces 2. Mean-field (Hartree-Fock) theory- orbitals 3. Pros and cons of HF- RHF, UHF 4. Beyond HF- why? 5. First, one usually does HF-how? 6. Basis sets and notations
1. Born-Oppenheimer approx.- energy surfaces 2. Mean-field (Hartree-Fock) theory- orbitals 3. Pros and cons of HF- RHF, UHF 4. Beyond HF- why? 5. First, one usually does HF-how? 6. Basis sets and notations
Practical Issues on the Use of the CASPT2/CASSCF Method in Modeling Photochemistry: the Selection and Protection of an Active Space
 Practical Issues on the Use of the CASPT2/CASSCF Method in Modeling Photochemistry: the Selection and Protection of an Active Space Roland Lindh Dept. of Chemistry Ångström The Theoretical Chemistry Programme
Practical Issues on the Use of the CASPT2/CASSCF Method in Modeling Photochemistry: the Selection and Protection of an Active Space Roland Lindh Dept. of Chemistry Ångström The Theoretical Chemistry Programme
LUMO + 1 LUMO. Tómas Arnar Guðmundsson Report 2 Reikniefnafræði G
 Q1: Display all the MOs for N2 in your report and classify each one of them as bonding, antibonding or non-bonding, and say whether the symmetry of the orbital is σ or π. Sketch a molecular orbital diagram
Q1: Display all the MOs for N2 in your report and classify each one of them as bonding, antibonding or non-bonding, and say whether the symmetry of the orbital is σ or π. Sketch a molecular orbital diagram
Quantum Physics III (8.06) Spring 2007 FINAL EXAMINATION Monday May 21, 9:00 am You have 3 hours.
 Spring 2007 FINAL EXAMINATION Monday May 21, 9:00 am You have 3 hours.") Quantum Physics III (8.06) Spring 2007 FINAL EXAMINATION Monday May 21, 9:00 am You have 3 hours. There are 10 problems, totalling 180 points. Do all problems. Answer all problems in the white books provided.
Quantum Physics III (8.06) Spring 2007 FINAL EXAMINATION Monday May 21, 9:00 am You have 3 hours. There are 10 problems, totalling 180 points. Do all problems. Answer all problems in the white books provided.
Jack Simons Henry Eyring Scientist and Professor Chemistry Department University of Utah
 1. Born-Oppenheimer approx.- energy surfaces 2. Mean-field (Hartree-Fock) theory- orbitals 3. Pros and cons of HF- RHF, UHF 4. Beyond HF- why? 5. First, one usually does HF-how? 6. Basis sets and notations
1. Born-Oppenheimer approx.- energy surfaces 2. Mean-field (Hartree-Fock) theory- orbitals 3. Pros and cons of HF- RHF, UHF 4. Beyond HF- why? 5. First, one usually does HF-how? 6. Basis sets and notations
Faddeev Random Phase Approximation (FRPA) Application to Molecules
 Application to Molecules") Faddeev Random Phase Approximation (FRPA) Application to Molecules Matthias Degroote Center for Molecular Modeling (CMM) Ghent University INT 2011 Spring Program Fermions from Cold Atoms to Neutron Stars:
Faddeev Random Phase Approximation (FRPA) Application to Molecules Matthias Degroote Center for Molecular Modeling (CMM) Ghent University INT 2011 Spring Program Fermions from Cold Atoms to Neutron Stars:
The wavefunction that describes a bonding pair of electrons:
 4.2. Molecular Properties from VB Theory a) Bonding and Bond distances The wavefunction that describes a bonding pair of electrons: Ψ b = a(h 1 ) + b(h 2 ) where h 1 and h 2 are HAOs on adjacent atoms
4.2. Molecular Properties from VB Theory a) Bonding and Bond distances The wavefunction that describes a bonding pair of electrons: Ψ b = a(h 1 ) + b(h 2 ) where h 1 and h 2 are HAOs on adjacent atoms
5.80 Small-Molecule Spectroscopy and Dynamics
 MIT OpenCourseWare http://ocw.mit.edu 5.80 Small-Molecule Spectroscopy and Dynamics Fall 2008 For information about citing these materials or our Terms of Use, visit: http://ocw.mit.edu/terms. 5.80 Lecture
MIT OpenCourseWare http://ocw.mit.edu 5.80 Small-Molecule Spectroscopy and Dynamics Fall 2008 For information about citing these materials or our Terms of Use, visit: http://ocw.mit.edu/terms. 5.80 Lecture
Accurate multireference configuration interaction calculations on the lowest 1 and 3 electronic states of C 2,CN, BN, and BO
 Accurate multireference configuration interaction calculations on the lowest 1 and 3 electronic states of C 2,CN, BN, and BO Kirk A. Peterson a) Department of Chemistry, Washington State University and
Accurate multireference configuration interaction calculations on the lowest 1 and 3 electronic states of C 2,CN, BN, and BO Kirk A. Peterson a) Department of Chemistry, Washington State University and
Semi-Empirical MO Methods
 Semi-Empirical MO Methods the high cost of ab initio MO calculations is largely due to the many integrals that need to be calculated (esp. two electron integrals) semi-empirical MO methods start with the
Semi-Empirical MO Methods the high cost of ab initio MO calculations is largely due to the many integrals that need to be calculated (esp. two electron integrals) semi-empirical MO methods start with the
Bonding in Molecules Prof John McGrady Michaelmas Term 2009
 Bonding in Molecules Prof John McGrady Michaelmas Term 2009 6 lectures building on material presented in Introduction to Molecular Orbitals (HT Year 1). Provides a basis for analysing the shapes, properties,
Bonding in Molecules Prof John McGrady Michaelmas Term 2009 6 lectures building on material presented in Introduction to Molecular Orbitals (HT Year 1). Provides a basis for analysing the shapes, properties,
Recent advances in quantum Monte Carlo for quantum chemistry: optimization of wave functions and calculation of observables
 Recent advances in quantum Monte Carlo for quantum chemistry: optimization of wave functions and calculation of observables Julien Toulouse 1, Cyrus J. Umrigar 2, Roland Assaraf 1 1 Laboratoire de Chimie
Recent advances in quantum Monte Carlo for quantum chemistry: optimization of wave functions and calculation of observables Julien Toulouse 1, Cyrus J. Umrigar 2, Roland Assaraf 1 1 Laboratoire de Chimie
Electronic structure theory: Fundamentals to frontiers. 2. Density functional theory
 Electronic structure theory: Fundamentals to frontiers. 2. Density functional theory MARTIN HEAD-GORDON, Department of Chemistry, University of California, and Chemical Sciences Division, Lawrence Berkeley
Electronic structure theory: Fundamentals to frontiers. 2. Density functional theory MARTIN HEAD-GORDON, Department of Chemistry, University of California, and Chemical Sciences Division, Lawrence Berkeley
Density matrix functional theory vis-á-vis density functional theory
 Density matrix functional theory vis-á-vis density functional theory 16.4.007 Ryan Requist Oleg Pankratov 1 Introduction Recently, there has been renewed interest in density matrix functional theory (DMFT)
Density matrix functional theory vis-á-vis density functional theory 16.4.007 Ryan Requist Oleg Pankratov 1 Introduction Recently, there has been renewed interest in density matrix functional theory (DMFT)
Electronic communication through molecular bridges Supporting Information
 Electronic communication through molecular bridges Supporting Information Carmen Herrmann and Jan Elmisz Institute of Inorganic and Applied Chemistry, University of Hamburg, Martin-Luther-King-Platz 6,
Electronic communication through molecular bridges Supporting Information Carmen Herrmann and Jan Elmisz Institute of Inorganic and Applied Chemistry, University of Hamburg, Martin-Luther-King-Platz 6,
MD simulation: output
 Properties MD simulation: output Trajectory of atoms positions: e. g. diffusion, mass transport velocities: e. g. v-v autocorrelation spectrum Energies temperature displacement fluctuations Mean square
Properties MD simulation: output Trajectory of atoms positions: e. g. diffusion, mass transport velocities: e. g. v-v autocorrelation spectrum Energies temperature displacement fluctuations Mean square
Computational Chemistry. An Introduction to Molecular Dynamic Simulations
 Computational Chemistry An Introduction to Molecular Dynamic Simulations Computational chemistry simulates chemical structures and reactions numerically, based in full or in part on the fundamental laws
Computational Chemistry An Introduction to Molecular Dynamic Simulations Computational chemistry simulates chemical structures and reactions numerically, based in full or in part on the fundamental laws
Orbital Density Dependent Functionals
 Orbital Density Dependent Functionals S. Kluepfel1, P. Kluepfel1, Hildur Guðmundsdóttir1 and Hannes Jónsson1,2 1. Univ. of Iceland; 2. Aalto University Outline: Problems with GGA approximation (PBE, RPBE,...)
Orbital Density Dependent Functionals S. Kluepfel1, P. Kluepfel1, Hildur Guðmundsdóttir1 and Hannes Jónsson1,2 1. Univ. of Iceland; 2. Aalto University Outline: Problems with GGA approximation (PBE, RPBE,...)
Second quantization. Emmanuel Fromager
 Institut de Chimie, Strasbourg, France Page 1 Emmanuel Fromager Institut de Chimie de Strasbourg - Laboratoire de Chimie Quantique - Université de Strasbourg /CNRS M2 lecture, Strasbourg, France. Institut
Institut de Chimie, Strasbourg, France Page 1 Emmanuel Fromager Institut de Chimie de Strasbourg - Laboratoire de Chimie Quantique - Université de Strasbourg /CNRS M2 lecture, Strasbourg, France. Institut
The symmetry properties & relative energies of atomic orbitals determine how they react to form molecular orbitals. These molecular orbitals are then
 1 The symmetry properties & relative energies of atomic orbitals determine how they react to form molecular orbitals. These molecular orbitals are then filled with the available electrons according to
1 The symmetry properties & relative energies of atomic orbitals determine how they react to form molecular orbitals. These molecular orbitals are then filled with the available electrons according to
Density-functional theory in quantum chemistry. Trygve Helgaker. From Quarks to the Nuclear Many-Body Problem
 1 Density-functional theory in quantum chemistry Trygve Helgaker Centre for Theoretical and Computational Chemistry, University of Oslo, Norway From Quarks to the Nuclear Many-Body Problem A conference
1 Density-functional theory in quantum chemistry Trygve Helgaker Centre for Theoretical and Computational Chemistry, University of Oslo, Norway From Quarks to the Nuclear Many-Body Problem A conference
Lecture 9 Electronic Spectroscopy
 Lecture 9 Electronic Spectroscopy Molecular Orbital Theory: A Review - LCAO approximaton & AO overlap - Variation Principle & Secular Determinant - Homonuclear Diatomic MOs - Energy Levels, Bond Order
Lecture 9 Electronic Spectroscopy Molecular Orbital Theory: A Review - LCAO approximaton & AO overlap - Variation Principle & Secular Determinant - Homonuclear Diatomic MOs - Energy Levels, Bond Order
Time-independent molecular properties
 Time-independent molecular properties Trygve Helgaker Hylleraas Centre, Department of Chemistry, University of Oslo, Norway and Centre for Advanced Study at the Norwegian Academy of Science and Letters,
Time-independent molecular properties Trygve Helgaker Hylleraas Centre, Department of Chemistry, University of Oslo, Norway and Centre for Advanced Study at the Norwegian Academy of Science and Letters,
Computational Chemistry I
 Computational Chemistry I Text book Cramer: Essentials of Quantum Chemistry, Wiley (2 ed.) Chapter 3. Post Hartree-Fock methods (Cramer: chapter 7) There are many ways to improve the HF method. Most of
Computational Chemistry I Text book Cramer: Essentials of Quantum Chemistry, Wiley (2 ed.) Chapter 3. Post Hartree-Fock methods (Cramer: chapter 7) There are many ways to improve the HF method. Most of
Negative anomalous dimensions in N=4 SYM
 13 Nov., 2015, YITP Workshop - Developments in String Theory and Quantum Field Theory Negative anomalous dimensions in N=4 SYM Yusuke Kimura (OIQP) 1503.0621 [hep-th] with Ryo Suzuki 1 1. Introduction
13 Nov., 2015, YITP Workshop - Developments in String Theory and Quantum Field Theory Negative anomalous dimensions in N=4 SYM Yusuke Kimura (OIQP) 1503.0621 [hep-th] with Ryo Suzuki 1 1. Introduction
"What are the electrons really doing in molecules?" *
 Basics of Electron Frank R. Wagner Max-Planck-Institut für Chemische Physik fester Stoffe, Dresden, Germany. "What are the electrons really doing in molecules?" * * Robert S. Mulliken, lecture title, in
Basics of Electron Frank R. Wagner Max-Planck-Institut für Chemische Physik fester Stoffe, Dresden, Germany. "What are the electrons really doing in molecules?" * * Robert S. Mulliken, lecture title, in
arxiv:quant-ph/ v1 29 Mar 2003
 Finite-Dimensional PT -Symmetric Hamiltonians arxiv:quant-ph/0303174v1 29 Mar 2003 Carl M. Bender, Peter N. Meisinger, and Qinghai Wang Department of Physics, Washington University, St. Louis, MO 63130,
Finite-Dimensional PT -Symmetric Hamiltonians arxiv:quant-ph/0303174v1 29 Mar 2003 Carl M. Bender, Peter N. Meisinger, and Qinghai Wang Department of Physics, Washington University, St. Louis, MO 63130,
Little Orthogonality Theorem (LOT)
") Little Orthogonality Theorem (LOT) Take diagonal elements of D matrices in RG * D R D R i j G ij mi N * D R D R N i j G G ij ij RG mi mi ( ) By definition, D j j j R TrD R ( R). Sum GOT over β: * * ( )
Little Orthogonality Theorem (LOT) Take diagonal elements of D matrices in RG * D R D R i j G ij mi N * D R D R N i j G G ij ij RG mi mi ( ) By definition, D j j j R TrD R ( R). Sum GOT over β: * * ( )
Supporting information
 Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 2014 Supporting information Resonant Raman Spectra of Molecules with Diradical Character:
Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 2014 Supporting information Resonant Raman Spectra of Molecules with Diradical Character:
5.4. Electronic structure of water
 5.4. Electronic structure of water Water belongs to C 2v point group, we have discussed the corresponding character table. Here it is again: C 2v E C 2 σ v (yz) σ v (xz) A 1 1 1 1 1 A 2 1 1-1 -1 B 1 1-1
5.4. Electronic structure of water Water belongs to C 2v point group, we have discussed the corresponding character table. Here it is again: C 2v E C 2 σ v (yz) σ v (xz) A 1 1 1 1 1 A 2 1 1-1 -1 B 1 1-1
(1/2) M α 2 α, ˆTe = i. 1 r i r j, ˆV NN = α>β
 M α 2 α, ˆTe = i. 1 r i r j, ˆV NN = α>β") Chemistry 26 Spectroscopy Week # The Born-Oppenheimer Approximation, H + 2. Born-Oppenheimer approximation As for atoms, all information about a molecule is contained in the wave function Ψ, which is the
Chemistry 26 Spectroscopy Week # The Born-Oppenheimer Approximation, H + 2. Born-Oppenheimer approximation As for atoms, all information about a molecule is contained in the wave function Ψ, which is the
I. CSFs Are Used to Express the Full N-Electron Wavefunction
 Chapter 11 One Must be Able to Evaluate the Matrix Elements Among Properly Symmetry Adapted N- Electron Configuration Functions for Any Operator, the Electronic Hamiltonian in Particular. The Slater-Condon
Chapter 11 One Must be Able to Evaluate the Matrix Elements Among Properly Symmetry Adapted N- Electron Configuration Functions for Any Operator, the Electronic Hamiltonian in Particular. The Slater-Condon
Introduction to Computational Chemistry: Theory
 Introduction to Computational Chemistry: Theory Dr Andrew Gilbert Rm 118, Craig Building, RSC andrew.gilbert@anu.edu.au 3023 Course Lectures Introduction Hartree Fock Theory Basis Sets Lecture 1 1 Introduction
Introduction to Computational Chemistry: Theory Dr Andrew Gilbert Rm 118, Craig Building, RSC andrew.gilbert@anu.edu.au 3023 Course Lectures Introduction Hartree Fock Theory Basis Sets Lecture 1 1 Introduction
The adiabatic connection
 The adiabatic connection Trygve Helgaker, Andy Teale, and Sonia Coriani Centre for Theoretical and Computational Chemistry (CTCC), Department of Chemistry, University of Oslo, Norway Dipartimento di Scienze
The adiabatic connection Trygve Helgaker, Andy Teale, and Sonia Coriani Centre for Theoretical and Computational Chemistry (CTCC), Department of Chemistry, University of Oslo, Norway Dipartimento di Scienze
Quantum Monte Carlo wave functions and their optimization for quantum chemistry
 Quantum Monte Carlo wave functions and their optimization for quantum chemistry Julien Toulouse Université Pierre & Marie Curie and CNRS, Paris, France CEA Saclay, SPhN Orme des Merisiers April 2015 Outline
Quantum Monte Carlo wave functions and their optimization for quantum chemistry Julien Toulouse Université Pierre & Marie Curie and CNRS, Paris, France CEA Saclay, SPhN Orme des Merisiers April 2015 Outline
Computational Methods. Chem 561
 Computational Methods Chem 561 Lecture Outline 1. Ab initio methods a) HF SCF b) Post-HF methods 2. Density Functional Theory 3. Semiempirical methods 4. Molecular Mechanics Computational Chemistry " Computational
Computational Methods Chem 561 Lecture Outline 1. Ab initio methods a) HF SCF b) Post-HF methods 2. Density Functional Theory 3. Semiempirical methods 4. Molecular Mechanics Computational Chemistry " Computational
Electron States of Diatomic Molecules
 IISER Pune March 2018 Hamiltonian for a Diatomic Molecule The hamiltonian for a diatomic molecule can be considered to be made up of three terms Ĥ = ˆT N + ˆT el + ˆV where ˆT N is the kinetic energy operator
IISER Pune March 2018 Hamiltonian for a Diatomic Molecule The hamiltonian for a diatomic molecule can be considered to be made up of three terms Ĥ = ˆT N + ˆT el + ˆV where ˆT N is the kinetic energy operator
Predictive Computing for Solids and Liquids
 Predictive Computing for Solids and Liquids So Hirata Department of Chemistry May 214 Blue Waters Symposium 1 Schrödinger equation for a water molecule 1-particle, 3-dimensional partial differential equation
Predictive Computing for Solids and Liquids So Hirata Department of Chemistry May 214 Blue Waters Symposium 1 Schrödinger equation for a water molecule 1-particle, 3-dimensional partial differential equation
Lec20 Fri 3mar17
 564-17 Lec20 Fri 3mar17 [PDF]GAUSSIAN 09W TUTORIAL www.molcalx.com.cn/wp-content/uploads/2015/01/gaussian09w_tutorial.pdf by A Tomberg - Cited by 8 - Related articles GAUSSIAN 09W TUTORIAL. AN INTRODUCTION
564-17 Lec20 Fri 3mar17 [PDF]GAUSSIAN 09W TUTORIAL www.molcalx.com.cn/wp-content/uploads/2015/01/gaussian09w_tutorial.pdf by A Tomberg - Cited by 8 - Related articles GAUSSIAN 09W TUTORIAL. AN INTRODUCTION
Practical Advice for Quantum Chemistry Computations. C. David Sherrill School of Chemistry and Biochemistry Georgia Institute of Technology
 Practical Advice for Quantum Chemistry Computations C. David Sherrill School of Chemistry and Biochemistry Georgia Institute of Technology Choice of Basis Set STO-3G is too small 6-31G* or 6-31G** 6 probably
Practical Advice for Quantum Chemistry Computations C. David Sherrill School of Chemistry and Biochemistry Georgia Institute of Technology Choice of Basis Set STO-3G is too small 6-31G* or 6-31G** 6 probably
4πε. me 1,2,3,... 1 n. H atom 4. in a.u. atomic units. energy: 1 a.u. = ev distance 1 a.u. = Å
 H atom 4 E a me =, n=,,3,... 8ε 0 0 π me e e 0 hn ε h = = 0.59Å E = me (4 πε ) 4 e 0 n n in a.u. atomic units E = r = Z n nao Z = e = me = 4πε = 0 energy: a.u. = 7. ev distance a.u. = 0.59 Å General results
H atom 4 E a me =, n=,,3,... 8ε 0 0 π me e e 0 hn ε h = = 0.59Å E = me (4 πε ) 4 e 0 n n in a.u. atomic units E = r = Z n nao Z = e = me = 4πε = 0 energy: a.u. = 7. ev distance a.u. = 0.59 Å General results