Orbital Density Dependent Functionals
|
|
|
- Ross Sherman
- 5 years ago
- Views:
Transcription

1 Orbital Density Dependent Functionals S. Kluepfel1, P. Kluepfel1, Hildur Guðmundsdóttir1 and Hannes Jónsson1,2 1. Univ. of Iceland; 2. Aalto University Outline: Problems with GGA approximation (PBE, RPBE,...) Orbital density dependent (ODD) functionals: in particular Perdew-Zunger self-interaction correction; CPU time can scale as for DFT/GGA; Codes: Quantice (Gaussian orbitals), GPAW Application to atoms, molecules and solids
 Exact solution obtained in")
 Meta GGA GGA LDA Improved accuracy needed, keeping scaling of effort low (~ N3 for GGA which")
2 Electronic Structure Calculations Wave function based Methods DFT and beyond... GW MPn perturbation Multi-Configuration HF Hartree-Fock (HF) Exact solution obtained in principle, but effort scales ~ N7. Good for small systems (N<30) Hybrid?? (PBE0,HSE...) Meta GGA GGA LDA Improved accuracy needed, keeping scaling of effort low (~ N3 for GGA which can be applied to large systems even N~1000)
3 Hartree-Fock approximation Each electron is subject to the average interaction with the other electrons a mean field approximation orbital orbital energy
.")
4 Hartree-Fock calculations of atoms Compare energy per electron with high level estimate of total energy,eref, by Chakravorty & Davidson, JPC 100, 6167 (1996). HF gives poor estimate of the total energy But, HOMO energy agrees quite well with experimental ionization energy (Koopman). Orbital energies correspond remarkably well will photo electron spectra ( orbitals are real???).
 LDA worse than HF, but GGA (the PBE functional) which includes gradients")
 is only marginally better.")
5 KS-DFT (cont.) LDA worse than HF, but GGA (the PBE functional) which includes gradients is more accurate than HF. Hybrid functional (PBE0) is only marginally better. But, KS-DFT orbital energies are not good estimates of ionization energy PBE0 hybrid functional again only marginally better than PBE




 delocalized")
6 DFT/GGA and LDA calculations have several shortcomings fractional charges in dissociated molecules transition energy / reaction paths: orbital energy / band structure: underestimated ionization potentials and band-gaps Energy (ev) electron distribution: activation energy underestimated orbital shape / chemical bonds molecular geometry / crystal structure Localized electronic defects destabilized (e.g., self-trapped holes) orbitals are not well-defined (post-process into natural / Wannier ) delocalized molecular orbitals vs. localized and chemically intuitive
7 + Na0.4 +Cl0.4-
8

9 Examples of problems with GGA: Too delocalized spin density Properly localized spin density Lægsgaard & Stokbro, PRL (2001)

10 Kohn-Sham density functional theory (KS-DFT)
11 EH EH
12 for a single electron
13 Back to H2+
14





15 But, how good is PZ-SIC? Calculate the Energy of atoms when real expansion coefficients are used in the orbitals: SIC-PBE improves total energy for Z < 7 SIC-PBE but, for Z 7 the total energy estimate becomes worse!!! consistent with results reported by Vydrov and Scuseria, JCP 121 (2004).
 Rather, use complex expansion coefficients SIC-PBE improves total energies for all studied atoms S IC-PBE error reduced to about 0.")
16 Test SIC in atoms, (cont.) Rather, use complex expansion coefficients SIC-PBE improves total energies for all studied atoms S IC-PBE error reduced to about 0.15 ev / electron Minimization of ODD functionals requires complex orbitals (see S. Klüpfel, P. Klüpfel and HJ, Phys.Rev.A (RC) 84, (2011))

 gives some, but small")


17 Ionization potentials of atoms Obtained from the highest occupied orbital (HOMO) PBE has ~40% errors PBE0 (hybrid) gives some, but small improvement, error remains ~30% SIC-PBE has errors of ~5%, can give good estimates of orbital energies, also for deeper ionization (HOMO-1, HOMO-2, )

18 Bond energy of diatomics
19 Kluepfel, Kluepfel and Jónsson, JCP 137, (2012)
20
21 Kluepfel, Kluepfel and Jónsson, JCP 137, (2012)
22 Kluepfel, Kluepfel and Jónsson, JCP 137, (2012)
23 Molecular geometry can become wrong when SIC is applied with real orbitals, resolved when complex orbitals are used Kluepfel, Kluepfel and Jónsson, JCP 137, (2012)
. Compare with EPR measurements, hyperfine constants: Al: Si: 0.32 0.3 80 85 0.18 0.50 0.4 0.")
24 Apply to defects in oxides: Electron hole in quartz with Al The DFT-SIC/2 correctly gives localized spin density and only one of the Al-O bonds is lengthened (by 0.3 A). Compare with EPR measurements, hyperfine constants: Al: Si: PBE-SIC/2 Exp PBE-SIC/2 Exp

25 Application to excited states of large molecules With SIC, the right 1/r dependence of the long range potential is built in, get the right Rydberg series of unoccupied states. Use solid-state approach to excited states of molecules, DFT-SIC energy functional, OEP for virtual. 3pxy orbital of trimethylamine Collaboration with Peter Weber at Brown University. Test: NH3 Orb. Exp. Calc. 3s 3pxy pz



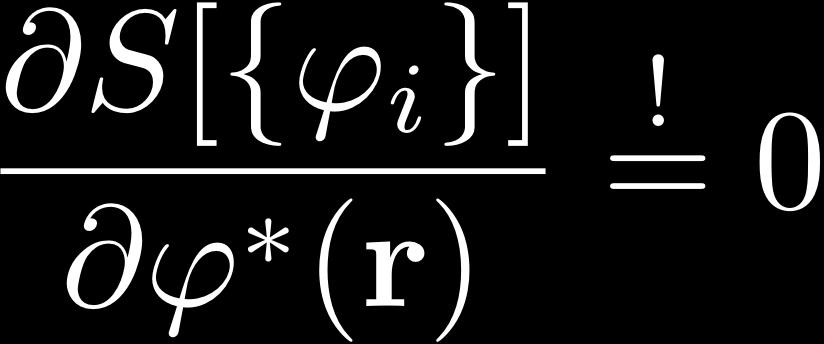
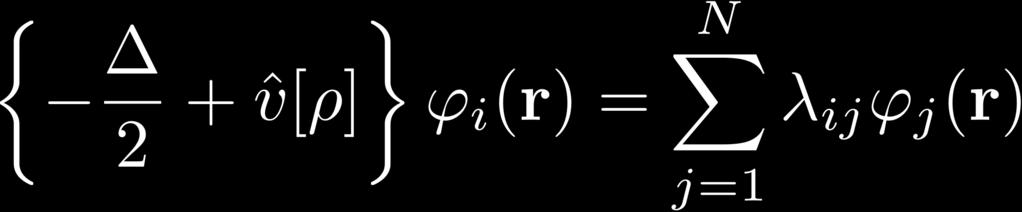


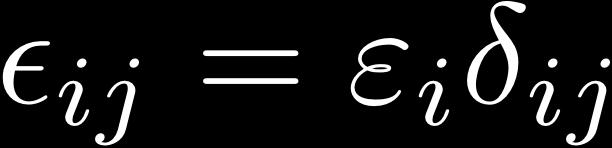
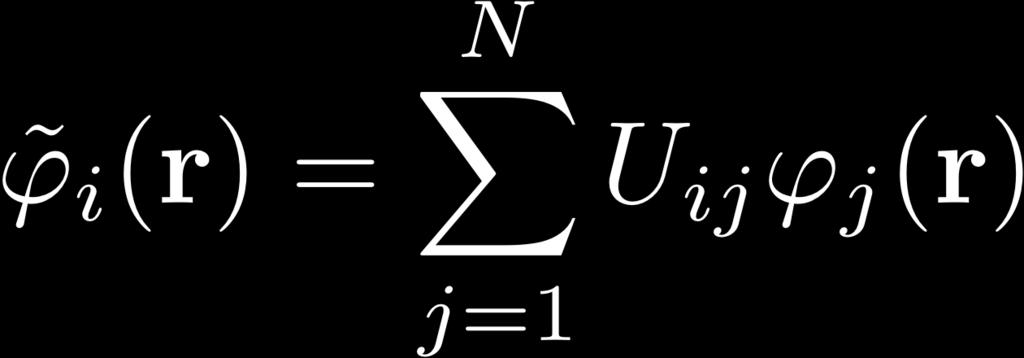
26 Minimization of ODD Energy Functionals minimization under constrained orthonormalization of the orbitals: condition for minimal energy KS-DFT: Unitary invariance: Constraint matrix always Hermitian: Choose canonical orbitals, diagonalize λ: Effective Schrödinger equation, eigenvalue problem: & HF:


 The lack of unitary invariance represents a problem in the minimization.")
27 Minimization of PZ-SIC energy functional Energy/potential is not unitary invariant Localized orbitals give larger SIC than delocalized orbitals The equations for the orbitals are coupled cannot choose orbitals to diagonalize Hamiltonian depends on orbital index This is in contrast to Kohn-Sham and HF where the set of equations can be reduced to several one-electron eigen value problems. Umrigar ) The lack of unitary invariance represents a problem in the minimization. But, a set of unique orbitals is obtained, possibly more meaningful than MOs.
28






29 Minimization of ODD functionals Evaluate densities/potentials Optimize unitary transformation Evaluate Lagrange Multipliers Orthonormalization Evaluate Residual Vector Correction of the orbitals (Line Search, RMM-DIIS, ) Preconditioning of the search direction (Conjugate Gradients, Davidson Methods, )
30 Orbital Density Dependent (ODD) Functionals Pros: scaling of CPU time same as GGA localized electronic states not penalized improved total/single-particle energy can give meaningful orbitals directly Cons: inefficient minimization (not eigen value problem) several local minima Progress: Improved minimization procedure: P. Klüpfel, S. Klüpfel, K. Tsemekhman and HJ, Lecture Notes In Computer Science (2012)). Stage set for the development of an optimal ODD functional!
31 Summary - PBE-SIC (or, better, PW91-SIC) is an alternative to hybrid functionals such as PBE0 giving better estimate of total energy of atoms and ionization potentials. - Complex wave functions have to be used in self-consistent minimization of ODD functionals. - Orbital-Density-Dependent functionals can give meaningful, well-defined, localized orbitals and this extended functional form opens the possibility for higher accuracy (defect states, band gaps, bond energy ) at computational cost that scales with N as DFT/GGA. - Further development of ODD: Construct new exchange correlation functional consistent with self-interaction free Hartree energy (need new PAW projectors). Need to ensure size consistency, especially for solid state applications. Improve the minimization algorithm (get stuck in local minima, large systems don't converge...)
Self-Consistent Implementation of Self-Interaction Corrected DFT and of the Exact Exchange Functionals in Plane-Wave DFT
 Self-Consistent Implementation of Self-Interaction Corrected DFT and of the Exact Exchange Functionals in Plane-Wave DFT Kiril Tsemekhman (a), Eric Bylaska (b), Hannes Jonsson (a,c) (a) Department of Chemistry,
Self-Consistent Implementation of Self-Interaction Corrected DFT and of the Exact Exchange Functionals in Plane-Wave DFT Kiril Tsemekhman (a), Eric Bylaska (b), Hannes Jonsson (a,c) (a) Department of Chemistry,
OVERVIEW OF QUANTUM CHEMISTRY METHODS
 OVERVIEW OF QUANTUM CHEMISTRY METHODS Outline I Generalities Correlation, basis sets Spin II Wavefunction methods Hartree-Fock Configuration interaction Coupled cluster Perturbative methods III Density
OVERVIEW OF QUANTUM CHEMISTRY METHODS Outline I Generalities Correlation, basis sets Spin II Wavefunction methods Hartree-Fock Configuration interaction Coupled cluster Perturbative methods III Density
Answers Quantum Chemistry NWI-MOL406 G. C. Groenenboom and G. A. de Wijs, HG00.307, 8:30-11:30, 21 jan 2014
 Answers Quantum Chemistry NWI-MOL406 G. C. Groenenboom and G. A. de Wijs, HG00.307, 8:30-11:30, 21 jan 2014 Question 1: Basis sets Consider the split valence SV3-21G one electron basis set for formaldehyde
Answers Quantum Chemistry NWI-MOL406 G. C. Groenenboom and G. A. de Wijs, HG00.307, 8:30-11:30, 21 jan 2014 Question 1: Basis sets Consider the split valence SV3-21G one electron basis set for formaldehyde
Electronic band structure, sx-lda, Hybrid DFT, LDA+U and all that. Keith Refson STFC Rutherford Appleton Laboratory
 Electronic band structure, sx-lda, Hybrid DFT, LDA+U and all that Keith Refson STFC Rutherford Appleton Laboratory LDA/GGA DFT is good but... Naive LDA/GGA calculation severely underestimates band-gaps.
Electronic band structure, sx-lda, Hybrid DFT, LDA+U and all that Keith Refson STFC Rutherford Appleton Laboratory LDA/GGA DFT is good but... Naive LDA/GGA calculation severely underestimates band-gaps.
A FRESH LOOK AT THE BAND-GAP PROBLEM IN DENSITY FUNCTIONAL THEORY
 A FRESH LOOK AT THE BAND-GAP PROBLEM IN DENSITY FUNCTIONAL THEORY JOHN P. PERDEW PHYSICS & CHEMISTRY, TEMPLE UNIVERSITY PHILADELPHIA, PENNSYLVANIA, USA SUPPORTED BY THE U.S. DEPARTMENT OF ENERGY, EFRC
A FRESH LOOK AT THE BAND-GAP PROBLEM IN DENSITY FUNCTIONAL THEORY JOHN P. PERDEW PHYSICS & CHEMISTRY, TEMPLE UNIVERSITY PHILADELPHIA, PENNSYLVANIA, USA SUPPORTED BY THE U.S. DEPARTMENT OF ENERGY, EFRC
Electronic structure theory: Fundamentals to frontiers. 2. Density functional theory
 Electronic structure theory: Fundamentals to frontiers. 2. Density functional theory MARTIN HEAD-GORDON, Department of Chemistry, University of California, and Chemical Sciences Division, Lawrence Berkeley
Electronic structure theory: Fundamentals to frontiers. 2. Density functional theory MARTIN HEAD-GORDON, Department of Chemistry, University of California, and Chemical Sciences Division, Lawrence Berkeley
XYZ of ground-state DFT
 XYZ of ground-state DFT Kieron Burke and Lucas Wagner Departments of Physics and of Chemistry, University of California, Irvine, CA 92697, USA January 5-9th, 2014 Kieron (UC Irvine) XYZ of ground-state
XYZ of ground-state DFT Kieron Burke and Lucas Wagner Departments of Physics and of Chemistry, University of California, Irvine, CA 92697, USA January 5-9th, 2014 Kieron (UC Irvine) XYZ of ground-state
Teoría del Funcional de la Densidad (Density Functional Theory)
") Teoría del Funcional de la Densidad (Density Functional Theory) Motivation: limitations of the standard approach based on the wave function. The electronic density n(r) as the key variable: Functionals
Teoría del Funcional de la Densidad (Density Functional Theory) Motivation: limitations of the standard approach based on the wave function. The electronic density n(r) as the key variable: Functionals
DENSITY FUNCTIONAL THEORY FOR NON-THEORISTS JOHN P. PERDEW DEPARTMENTS OF PHYSICS AND CHEMISTRY TEMPLE UNIVERSITY
 DENSITY FUNCTIONAL THEORY FOR NON-THEORISTS JOHN P. PERDEW DEPARTMENTS OF PHYSICS AND CHEMISTRY TEMPLE UNIVERSITY A TUTORIAL FOR PHYSICAL SCIENTISTS WHO MAY OR MAY NOT HATE EQUATIONS AND PROOFS REFERENCES
DENSITY FUNCTIONAL THEORY FOR NON-THEORISTS JOHN P. PERDEW DEPARTMENTS OF PHYSICS AND CHEMISTRY TEMPLE UNIVERSITY A TUTORIAL FOR PHYSICAL SCIENTISTS WHO MAY OR MAY NOT HATE EQUATIONS AND PROOFS REFERENCES
Advanced Quantum Chemistry III: Part 3. Haruyuki Nakano. Kyushu University
 Advanced Quantum Chemistry III: Part 3 Haruyuki Nakano Kyushu University 2013 Winter Term 1. Hartree-Fock theory Density Functional Theory 2. Hohenberg-Kohn theorem 3. Kohn-Sham method 4. Exchange-correlation
Advanced Quantum Chemistry III: Part 3 Haruyuki Nakano Kyushu University 2013 Winter Term 1. Hartree-Fock theory Density Functional Theory 2. Hohenberg-Kohn theorem 3. Kohn-Sham method 4. Exchange-correlation
Pseudopotentials for hybrid density functionals and SCAN
 Pseudopotentials for hybrid density functionals and SCAN Jing Yang, Liang Z. Tan, Julian Gebhardt, and Andrew M. Rappe Department of Chemistry University of Pennsylvania Why do we need pseudopotentials?
Pseudopotentials for hybrid density functionals and SCAN Jing Yang, Liang Z. Tan, Julian Gebhardt, and Andrew M. Rappe Department of Chemistry University of Pennsylvania Why do we need pseudopotentials?
Exchange Correlation Functional Investigation of RT-TDDFT on a Sodium Chloride. Dimer. Philip Straughn
 Exchange Correlation Functional Investigation of RT-TDDFT on a Sodium Chloride Dimer Philip Straughn Abstract Charge transfer between Na and Cl ions is an important problem in physical chemistry. However,
Exchange Correlation Functional Investigation of RT-TDDFT on a Sodium Chloride Dimer Philip Straughn Abstract Charge transfer between Na and Cl ions is an important problem in physical chemistry. However,
Intermediate DFT. Kieron Burke and Lucas Wagner. Departments of Physics and of Chemistry, University of California, Irvine, CA 92697, USA
 Intermediate DFT Kieron Burke and Lucas Wagner Departments of Physics and of Chemistry, University of California, Irvine, CA 92697, USA October 10-19th, 2012 Kieron (UC Irvine) Intermediate DFT Lausanne12
Intermediate DFT Kieron Burke and Lucas Wagner Departments of Physics and of Chemistry, University of California, Irvine, CA 92697, USA October 10-19th, 2012 Kieron (UC Irvine) Intermediate DFT Lausanne12
Many electrons: Density functional theory Part II. Bedřich Velický VI.
 Many electrons: Density functional theory Part II. Bedřich Velický velicky@karlov.mff.cuni.cz VI. NEVF 514 Surface Physics Winter Term 013-014 Troja 1 st November 013 This class is the second devoted to
Many electrons: Density functional theory Part II. Bedřich Velický velicky@karlov.mff.cuni.cz VI. NEVF 514 Surface Physics Winter Term 013-014 Troja 1 st November 013 This class is the second devoted to
Density Functional Theory for Electrons in Materials
 Density Functional Theory for Electrons in Materials Richard M. Martin Department of Physics and Materials Research Laboratory University of Illinois at Urbana-Champaign 1 Density Functional Theory for
Density Functional Theory for Electrons in Materials Richard M. Martin Department of Physics and Materials Research Laboratory University of Illinois at Urbana-Champaign 1 Density Functional Theory for
 3/23/2010 More basics of DFT Kieron Burke and friends UC Irvine Physics and Chemistry References for ground-state DFT ABC of DFT, by KB and Rudy Magyar, http://dft.uci.edu A Primer in Density Functional
3/23/2010 More basics of DFT Kieron Burke and friends UC Irvine Physics and Chemistry References for ground-state DFT ABC of DFT, by KB and Rudy Magyar, http://dft.uci.edu A Primer in Density Functional
Calculations of band structures
 Chemistry and Physics at Albany Planning for the Future Calculations of band structures using wave-function based correlation methods Elke Pahl Centre of Theoretical Chemistry and Physics Institute of
Chemistry and Physics at Albany Planning for the Future Calculations of band structures using wave-function based correlation methods Elke Pahl Centre of Theoretical Chemistry and Physics Institute of
CLIMBING THE LADDER OF DENSITY FUNCTIONAL APPROXIMATIONS JOHN P. PERDEW DEPARTMENT OF PHYSICS TEMPLE UNIVERSITY PHILADELPHIA, PA 19122
 CLIMBING THE LADDER OF DENSITY FUNCTIONAL APPROXIMATIONS JOHN P. PERDEW DEPARTMENT OF PHYSICS TEMPLE UNIVERSITY PHILADELPHIA, PA 191 THANKS TO MANY COLLABORATORS, INCLUDING SY VOSKO DAVID LANGRETH ALEX
CLIMBING THE LADDER OF DENSITY FUNCTIONAL APPROXIMATIONS JOHN P. PERDEW DEPARTMENT OF PHYSICS TEMPLE UNIVERSITY PHILADELPHIA, PA 191 THANKS TO MANY COLLABORATORS, INCLUDING SY VOSKO DAVID LANGRETH ALEX
DFT: Exchange-Correlation
 DFT: Exchange-Correlation Local functionals, exact exchange and other post-dft methods Paul Tulip Centre for Materials Physics Department of Physics University of Durham Outline Introduction What is exchange
DFT: Exchange-Correlation Local functionals, exact exchange and other post-dft methods Paul Tulip Centre for Materials Physics Department of Physics University of Durham Outline Introduction What is exchange
DFT: Exchange-Correlation
 DFT: Local functionals, exact exchange and other post-dft methods Stewart Clark University of Outline Introduction What is exchange and correlation? Quick tour of XC functionals (Semi-)local: LDA, PBE,
DFT: Local functionals, exact exchange and other post-dft methods Stewart Clark University of Outline Introduction What is exchange and correlation? Quick tour of XC functionals (Semi-)local: LDA, PBE,
The Self Interaction Correction revisited
 The Self Interaction Correction revisited Explicit dynamics of clusters and molecules under irradiation Spectroscopic accuracy at low energy SIC problem : one electron interacts with its own mean-field!
The Self Interaction Correction revisited Explicit dynamics of clusters and molecules under irradiation Spectroscopic accuracy at low energy SIC problem : one electron interacts with its own mean-field!
The effect of the Perdew-Zunger self-interaction correction to density functionals on the energetics of small molecules
 THE JOURNAL OF CHEMICAL PHYSICS 137,124102(2012) The effect of the Perdew-Zunger self-interaction correction to density functionals on the energetics of small molecules Simon Klüpfel, 1,a) Peter Klüpfel,
THE JOURNAL OF CHEMICAL PHYSICS 137,124102(2012) The effect of the Perdew-Zunger self-interaction correction to density functionals on the energetics of small molecules Simon Klüpfel, 1,a) Peter Klüpfel,
The effect of the Perdew-Zunger self-interaction correction to density functionals on the energetics of small molecules
 The effect of the Perdew-Zunger self-interaction correction to density functionals on the energetics of small molecules Simon Klüpfel, 1, a) Peter Klüpfel, 2 and Hannes Jónsson 2 1) Science Institute of
The effect of the Perdew-Zunger self-interaction correction to density functionals on the energetics of small molecules Simon Klüpfel, 1, a) Peter Klüpfel, 2 and Hannes Jónsson 2 1) Science Institute of
Density Functional Theory
 Chemistry 380.37 Fall 2015 Dr. Jean M. Standard October 28, 2015 Density Functional Theory What is a Functional? A functional is a general mathematical quantity that represents a rule to convert a function
Chemistry 380.37 Fall 2015 Dr. Jean M. Standard October 28, 2015 Density Functional Theory What is a Functional? A functional is a general mathematical quantity that represents a rule to convert a function
Exchange-Correlation Functional
 Exchange-Correlation Functional Aiichiro Nakano Collaboratory for Advanced Computing & Simulations Depts. of Computer Science, Physics & Astronomy, Chemical Engineering & Materials Science, and Biological
Exchange-Correlation Functional Aiichiro Nakano Collaboratory for Advanced Computing & Simulations Depts. of Computer Science, Physics & Astronomy, Chemical Engineering & Materials Science, and Biological
Independent electrons in an effective potential
 ABC of DFT Adiabatic approximation Independent electrons in an effective potential Hartree Fock Density Functional Theory MBPT - GW Density Functional Theory in a nutshell Every observable quantity of
ABC of DFT Adiabatic approximation Independent electrons in an effective potential Hartree Fock Density Functional Theory MBPT - GW Density Functional Theory in a nutshell Every observable quantity of
Generalized generalized gradient approximation: An improved density-functional theory for accurate orbital eigenvalues
 PHYSICAL REVIEW B VOLUME 55, NUMBER 24 15 JUNE 1997-II Generalized generalized gradient approximation: An improved density-functional theory for accurate orbital eigenvalues Xinlei Hua, Xiaojie Chen, and
PHYSICAL REVIEW B VOLUME 55, NUMBER 24 15 JUNE 1997-II Generalized generalized gradient approximation: An improved density-functional theory for accurate orbital eigenvalues Xinlei Hua, Xiaojie Chen, and
Spring College on Computational Nanoscience
 2145-29 Spring College on Computational Nanoscience 17-28 May 2010 At the Fifth Rung of Jacob's Ladder: A Discussion of Exact Exchange plus Local- and Nonlocal-density Approximations to the Correlation
2145-29 Spring College on Computational Nanoscience 17-28 May 2010 At the Fifth Rung of Jacob's Ladder: A Discussion of Exact Exchange plus Local- and Nonlocal-density Approximations to the Correlation
Introduction to DFT and its Application to Defects in Semiconductors
 Introduction to DFT and its Application to Defects in Semiconductors Noa Marom Physics and Engineering Physics Tulane University New Orleans The Future: Computer-Aided Materials Design Can access the space
Introduction to DFT and its Application to Defects in Semiconductors Noa Marom Physics and Engineering Physics Tulane University New Orleans The Future: Computer-Aided Materials Design Can access the space
Introduction to Density Functional Theory with Applications to Graphene Branislav K. Nikolić
 Introduction to Density Functional Theory with Applications to Graphene Branislav K. Nikolić Department of Physics and Astronomy, University of Delaware, Newark, DE 19716, U.S.A. http://wiki.physics.udel.edu/phys824
Introduction to Density Functional Theory with Applications to Graphene Branislav K. Nikolić Department of Physics and Astronomy, University of Delaware, Newark, DE 19716, U.S.A. http://wiki.physics.udel.edu/phys824
Comparison of DFT Methods for Molecular Orbital Eigenvalue Calculations
 1554 J. Phys. Chem. A 2007, 111, 1554-1561 Comparison of DFT Methods for Molecular Orbital Eigenvalue Calculations Gang Zhang and Charles B. Musgrave* Department of Chemical Engineering, Stanford UniVersity,
1554 J. Phys. Chem. A 2007, 111, 1554-1561 Comparison of DFT Methods for Molecular Orbital Eigenvalue Calculations Gang Zhang and Charles B. Musgrave* Department of Chemical Engineering, Stanford UniVersity,
Computational Methods. Chem 561
 Computational Methods Chem 561 Lecture Outline 1. Ab initio methods a) HF SCF b) Post-HF methods 2. Density Functional Theory 3. Semiempirical methods 4. Molecular Mechanics Computational Chemistry " Computational
Computational Methods Chem 561 Lecture Outline 1. Ab initio methods a) HF SCF b) Post-HF methods 2. Density Functional Theory 3. Semiempirical methods 4. Molecular Mechanics Computational Chemistry " Computational
Magnetism in transition metal oxides by post-dft methods
 Magnetism in transition metal oxides by post-dft methods Cesare Franchini Faculty of Physics & Center for Computational Materials Science University of Vienna, Austria Workshop on Magnetism in Complex
Magnetism in transition metal oxides by post-dft methods Cesare Franchini Faculty of Physics & Center for Computational Materials Science University of Vienna, Austria Workshop on Magnetism in Complex
arxiv: v1 [cond-mat.other] 4 Apr 2008
![arxiv: v1 [cond-mat.other] 4 Apr 2008](/thumbs/92/108476259.jpg "arxiv: v1 [cond-mat.other] 4 Apr 2008") Self-interaction correction in a simple model P. M. Dinh a, J. Messud a, P.-G. Reinhard b, and E. Suraud a arxiv:0804.0684v1 [cond-mat.other] 4 Apr 2008 Abstract a Laboratoire de Physique Théorique, Université
Self-interaction correction in a simple model P. M. Dinh a, J. Messud a, P.-G. Reinhard b, and E. Suraud a arxiv:0804.0684v1 [cond-mat.other] 4 Apr 2008 Abstract a Laboratoire de Physique Théorique, Université
Role of van der Waals Interactions in Physics, Chemistry, and Biology
 Role of van der Waals Interactions in Physics, Chemistry, and Biology How can we describe vdw forces in materials accurately? Failure of DFT Approximations for (Long-Range) Van der Waals Interactions 1
Role of van der Waals Interactions in Physics, Chemistry, and Biology How can we describe vdw forces in materials accurately? Failure of DFT Approximations for (Long-Range) Van der Waals Interactions 1
arxiv: v2 [cond-mat.other] 27 Jun 2008
![arxiv: v2 [cond-mat.other] 27 Jun 2008](/thumbs/92/110117177.jpg "arxiv: v2 [cond-mat.other] 27 Jun 2008") Self-interaction correction in a simple model arxiv:0804.0684v [cond-mat.other] 7 Jun 008 P. M. Dinh a, J. Messud a, P.-G. Reinhard b, and E. Suraud a Abstract a Laboratoire de Physique Théorique, Université
Self-interaction correction in a simple model arxiv:0804.0684v [cond-mat.other] 7 Jun 008 P. M. Dinh a, J. Messud a, P.-G. Reinhard b, and E. Suraud a Abstract a Laboratoire de Physique Théorique, Université
QUANTUM CHEMISTRY FOR TRANSITION METALS
 QUANTUM CHEMISTRY FOR TRANSITION METALS Outline I Introduction II Correlation Static correlation effects MC methods DFT III Relativity Generalities From 4 to 1 components Effective core potential Outline
QUANTUM CHEMISTRY FOR TRANSITION METALS Outline I Introduction II Correlation Static correlation effects MC methods DFT III Relativity Generalities From 4 to 1 components Effective core potential Outline
ABC of ground-state DFT
 ABC of ground-state DFT Kieron Burke and Lucas Wagner Departments of Physics and of Chemistry, University of California, Irvine, CA 92697, USA January 5-9th, 2014 Kieron (UC Irvine) ABC of ground-state
ABC of ground-state DFT Kieron Burke and Lucas Wagner Departments of Physics and of Chemistry, University of California, Irvine, CA 92697, USA January 5-9th, 2014 Kieron (UC Irvine) ABC of ground-state
Dept of Mechanical Engineering MIT Nanoengineering group
 1 Dept of Mechanical Engineering MIT Nanoengineering group » To calculate all the properties of a molecule or crystalline system knowing its atomic information: Atomic species Their coordinates The Symmetry
1 Dept of Mechanical Engineering MIT Nanoengineering group » To calculate all the properties of a molecule or crystalline system knowing its atomic information: Atomic species Their coordinates The Symmetry
Bayesian Error Estimation in Density Functional Theory
 Bayesian Error Estimation in Density Functional Theory Karsten W. Jacobsen Jens Jørgen Mortensen Kristen Kaasbjerg Søren L. Frederiksen Jens K. Nørskov CAMP, Dept. of Physics, DTU James P. Sethna LASSP,
Bayesian Error Estimation in Density Functional Theory Karsten W. Jacobsen Jens Jørgen Mortensen Kristen Kaasbjerg Søren L. Frederiksen Jens K. Nørskov CAMP, Dept. of Physics, DTU James P. Sethna LASSP,
CHEM6085: Density Functional Theory Lecture 10
 CHEM6085: Density Functional Theory Lecture 10 1) Spin-polarised calculations 2) Geometry optimisation C.-K. Skylaris 1 Unpaired electrons So far we have developed Kohn-Sham DFT for the case of paired
CHEM6085: Density Functional Theory Lecture 10 1) Spin-polarised calculations 2) Geometry optimisation C.-K. Skylaris 1 Unpaired electrons So far we have developed Kohn-Sham DFT for the case of paired
Multi-Scale Modeling from First Principles
 m mm Multi-Scale Modeling from First Principles μm nm m mm μm nm space space Predictive modeling and simulations must address all time and Continuum Equations, densityfunctional space scales Rate Equations
m mm Multi-Scale Modeling from First Principles μm nm m mm μm nm space space Predictive modeling and simulations must address all time and Continuum Equations, densityfunctional space scales Rate Equations
Density Functional Theory. Martin Lüders Daresbury Laboratory
 Density Functional Theory Martin Lüders Daresbury Laboratory Ab initio Calculations Hamiltonian: (without external fields, non-relativistic) impossible to solve exactly!! Electrons Nuclei Electron-Nuclei
Density Functional Theory Martin Lüders Daresbury Laboratory Ab initio Calculations Hamiltonian: (without external fields, non-relativistic) impossible to solve exactly!! Electrons Nuclei Electron-Nuclei
Basics of DFT. Kieron Burke and Lucas Wagner. Departments of Physics and of Chemistry, University of California, Irvine, CA 92697, USA
 Basics of DFT Kieron Burke and Lucas Wagner Departments of Physics and of Chemistry, University of California, Irvine, CA 92697, USA October 10-19th, 2012 Kieron (UC Irvine) Basics of DFT Lausanne12 1
Basics of DFT Kieron Burke and Lucas Wagner Departments of Physics and of Chemistry, University of California, Irvine, CA 92697, USA October 10-19th, 2012 Kieron (UC Irvine) Basics of DFT Lausanne12 1
Band calculations: Theory and Applications
 Band calculations: Theory and Applications Lecture 2: Different approximations for the exchange-correlation correlation functional in DFT Local density approximation () Generalized gradient approximation
Band calculations: Theory and Applications Lecture 2: Different approximations for the exchange-correlation correlation functional in DFT Local density approximation () Generalized gradient approximation
Electronic structure theory: Fundamentals to frontiers. 1. Hartree-Fock theory
 Electronic structure theory: Fundamentals to frontiers. 1. Hartree-Fock theory MARTIN HEAD-GORDON, Department of Chemistry, University of California, and Chemical Sciences Division, Lawrence Berkeley National
Electronic structure theory: Fundamentals to frontiers. 1. Hartree-Fock theory MARTIN HEAD-GORDON, Department of Chemistry, University of California, and Chemical Sciences Division, Lawrence Berkeley National
The electronic structure of materials 2 - DFT
 Quantum mechanics 2 - Lecture 9 December 19, 2012 1 Density functional theory (DFT) 2 Literature Contents 1 Density functional theory (DFT) 2 Literature Historical background The beginnings: L. de Broglie
Quantum mechanics 2 - Lecture 9 December 19, 2012 1 Density functional theory (DFT) 2 Literature Contents 1 Density functional theory (DFT) 2 Literature Historical background The beginnings: L. de Broglie
Density functional theory in the solid state
 Density functional theory in the solid state Ari P Seitsonen IMPMC, CNRS & Universités 6 et 7 Paris, IPGP Department of Applied Physics, Helsinki University of Technology Physikalisch-Chemisches Institut
Density functional theory in the solid state Ari P Seitsonen IMPMC, CNRS & Universités 6 et 7 Paris, IPGP Department of Applied Physics, Helsinki University of Technology Physikalisch-Chemisches Institut
Introduction to Density Functional Theory
 1 Introduction to Density Functional Theory 21 February 2011; V172 P.Ravindran, FME-course on Ab initio Modelling of solar cell Materials 21 February 2011 Introduction to DFT 2 3 4 Ab initio Computational
1 Introduction to Density Functional Theory 21 February 2011; V172 P.Ravindran, FME-course on Ab initio Modelling of solar cell Materials 21 February 2011 Introduction to DFT 2 3 4 Ab initio Computational
Density Functional Theory - II part
 Density Functional Theory - II part antonino.polimeno@unipd.it Overview From theory to practice Implementation Functionals Local functionals Gradient Others From theory to practice From now on, if not
Density Functional Theory - II part antonino.polimeno@unipd.it Overview From theory to practice Implementation Functionals Local functionals Gradient Others From theory to practice From now on, if not
An Approximate DFT Method: The Density-Functional Tight-Binding (DFTB) Method
 Method") Fakultät für Mathematik und Naturwissenschaften - Lehrstuhl für Physikalische Chemie I / Theoretische Chemie An Approximate DFT Method: The Density-Functional Tight-Binding (DFTB) Method Jan-Ole Joswig
Fakultät für Mathematik und Naturwissenschaften - Lehrstuhl für Physikalische Chemie I / Theoretische Chemie An Approximate DFT Method: The Density-Functional Tight-Binding (DFTB) Method Jan-Ole Joswig
A self-interaction free non-local correlation functional derived from exact criteria
 A self-interaction free non-local correlation functional derived from exact criteria Tobias Schmidt and Stephan Kümmel Theoretical Physics IV University of Bayreuth Germany - International Workshop DFT
A self-interaction free non-local correlation functional derived from exact criteria Tobias Schmidt and Stephan Kümmel Theoretical Physics IV University of Bayreuth Germany - International Workshop DFT
Modified Becke-Johnson (mbj) exchange potential
 exchange potential") Modified Becke-Johnson (mbj) exchange potential Hideyuki Jippo Fujitsu Laboratories LTD. 2015.12.21-22 OpenMX developer s meeting @ Kobe Overview: mbj potential The semilocal exchange potential adding
Modified Becke-Johnson (mbj) exchange potential Hideyuki Jippo Fujitsu Laboratories LTD. 2015.12.21-22 OpenMX developer s meeting @ Kobe Overview: mbj potential The semilocal exchange potential adding
Quantum Monte Carlo Benchmarks Density Functionals: Si Defects
 Quantum Monte Carlo Benchmarks Density Functionals: Si Defects K P Driver, W D Parker, R G Hennig, J W Wilkins (OSU) C J Umrigar (Cornell), R Martin, E Batista, B Uberuaga (LANL), J Heyd, G Scuseria (Rice)
Quantum Monte Carlo Benchmarks Density Functionals: Si Defects K P Driver, W D Parker, R G Hennig, J W Wilkins (OSU) C J Umrigar (Cornell), R Martin, E Batista, B Uberuaga (LANL), J Heyd, G Scuseria (Rice)
Lecture 4: Hartree-Fock Theory
 Lecture 4: Hartree-Fock Theory One determinant to rule them all, One determinant to find them, One determinant to bring them all and in the darkness bind them Second quantization rehearsal The formalism
Lecture 4: Hartree-Fock Theory One determinant to rule them all, One determinant to find them, One determinant to bring them all and in the darkness bind them Second quantization rehearsal The formalism
Multi-reference Density Functional Theory. COLUMBUS Workshop Argonne National Laboratory 15 August 2005
 Multi-reference Density Functional Theory COLUMBUS Workshop Argonne National Laboratory 15 August 2005 Capt Eric V. Beck Air Force Institute of Technology Department of Engineering Physics 2950 Hobson
Multi-reference Density Functional Theory COLUMBUS Workshop Argonne National Laboratory 15 August 2005 Capt Eric V. Beck Air Force Institute of Technology Department of Engineering Physics 2950 Hobson
v(r i r j ) = h(r i )+ 1 N
 = h(r i )+ 1 N") Chapter 1 Hartree-Fock Theory 1.1 Formalism For N electrons in an external potential V ext (r), the many-electron Hamiltonian can be written as follows: N H = [ p i i=1 m +V ext(r i )]+ 1 N N v(r i r j
Chapter 1 Hartree-Fock Theory 1.1 Formalism For N electrons in an external potential V ext (r), the many-electron Hamiltonian can be written as follows: N H = [ p i i=1 m +V ext(r i )]+ 1 N N v(r i r j
The Basics of Theoretical and Computational Chemistry
 Bernd M. Rode, Thomas S. Hofer, and Michael D. Kugler The Basics of Theoretical and Computational Chemistry BICENTENNIA BICENTBNN I AL. WILEY-VCH Verlag GmbH & Co. KGaA V Contents Preface IX 1 Introduction
Bernd M. Rode, Thomas S. Hofer, and Michael D. Kugler The Basics of Theoretical and Computational Chemistry BICENTENNIA BICENTBNN I AL. WILEY-VCH Verlag GmbH & Co. KGaA V Contents Preface IX 1 Introduction
We also deduced that transformations between Slater determinants are always of the form
 .3 Hartree-Fock The Hartree-Fock method assumes that the true N-body ground state wave function can be approximated by a single Slater determinant and minimizes the energy among all possible choices of
.3 Hartree-Fock The Hartree-Fock method assumes that the true N-body ground state wave function can be approximated by a single Slater determinant and minimizes the energy among all possible choices of
Computational Chemistry I
 Computational Chemistry I Text book Cramer: Essentials of Quantum Chemistry, Wiley (2 ed.) Chapter 3. Post Hartree-Fock methods (Cramer: chapter 7) There are many ways to improve the HF method. Most of
Computational Chemistry I Text book Cramer: Essentials of Quantum Chemistry, Wiley (2 ed.) Chapter 3. Post Hartree-Fock methods (Cramer: chapter 7) There are many ways to improve the HF method. Most of
Random-phase approximation and beyond for materials: concepts, practice, and future perspectives. Xinguo Ren
 Random-phase approximation and beyond for materials: concepts, practice, and future perspectives Xinguo Ren University of Science and Technology of China, Hefei USTC-FHI workshop on frontiers of Advanced
Random-phase approximation and beyond for materials: concepts, practice, and future perspectives Xinguo Ren University of Science and Technology of China, Hefei USTC-FHI workshop on frontiers of Advanced
Joint ICTP-IAEA Workshop on Fusion Plasma Modelling using Atomic and Molecular Data January 2012
 2327-3 Joint ICTP-IAEA Workshop on Fusion Plasma Modelling using Atomic and Molecular Data 23-27 January 2012 Qunatum Methods for Plasma-Facing Materials Alain ALLOUCHE Univ.de Provence, Lab.de la Phys.
2327-3 Joint ICTP-IAEA Workshop on Fusion Plasma Modelling using Atomic and Molecular Data 23-27 January 2012 Qunatum Methods for Plasma-Facing Materials Alain ALLOUCHE Univ.de Provence, Lab.de la Phys.
Performance ofhybrid density functional methods,screened exchange and EXX-OEP methodsin the PAW approach p.1/26
 Performance of hybrid density functional methods, screened exchange and EXX-OEP methods in the PAW approach Georg Kresse, J Paier, R Hirschl, M Marsmann Institut für Materialphysik and Centre for Computational
Performance of hybrid density functional methods, screened exchange and EXX-OEP methods in the PAW approach Georg Kresse, J Paier, R Hirschl, M Marsmann Institut für Materialphysik and Centre for Computational
VASP: running on HPC resources. University of Vienna, Faculty of Physics and Center for Computational Materials Science, Vienna, Austria
 VASP: running on HPC resources University of Vienna, Faculty of Physics and Center for Computational Materials Science, Vienna, Austria The Many-Body Schrödinger equation 0 @ 1 2 X i i + X i Ĥ (r 1,...,r
VASP: running on HPC resources University of Vienna, Faculty of Physics and Center for Computational Materials Science, Vienna, Austria The Many-Body Schrödinger equation 0 @ 1 2 X i i + X i Ĥ (r 1,...,r
Handbook of Computational Quantum Chemistry. DAVID B. COOK The Department of Chemistry, University of Sheffield
 Handbook of Computational Quantum Chemistry DAVID B. COOK The Department of Chemistry, University of Sheffield Oxford New York Tokyo OXFORD UNIVERSITY PRESS 1998 CONTENTS 1 Mechanics and molecules 1 1.1
Handbook of Computational Quantum Chemistry DAVID B. COOK The Department of Chemistry, University of Sheffield Oxford New York Tokyo OXFORD UNIVERSITY PRESS 1998 CONTENTS 1 Mechanics and molecules 1 1.1
The Electronic Structure of Dye- Sensitized TiO 2 Clusters from Many- Body Perturbation Theory
 The Electronic Structure of Dye- Sensitized TiO 2 Clusters from Many- Body Perturbation Theory Noa Marom Center for Computational Materials Institute for Computational Engineering and Sciences The University
The Electronic Structure of Dye- Sensitized TiO 2 Clusters from Many- Body Perturbation Theory Noa Marom Center for Computational Materials Institute for Computational Engineering and Sciences The University
Advanced Solid State Theory SS Roser Valentí and Harald Jeschke Institut für Theoretische Physik, Goethe-Universität Frankfurt
 Advanced Solid State Theory SS 2010 Roser Valentí and Harald Jeschke Institut für Theoretische Physik, Goethe-Universität Frankfurt i 0. Literatur R. M. Martin, Electronic Structure: Basic Theory and
Advanced Solid State Theory SS 2010 Roser Valentí and Harald Jeschke Institut für Theoretische Physik, Goethe-Universität Frankfurt i 0. Literatur R. M. Martin, Electronic Structure: Basic Theory and
Practical Guide to Density Functional Theory (DFT)
") Practical Guide to Density Functional Theory (DFT) Brad Malone, Sadas Shankar Quick recap of where we left off last time BD Malone, S Shankar Therefore there is a direct one-to-one correspondence between
Practical Guide to Density Functional Theory (DFT) Brad Malone, Sadas Shankar Quick recap of where we left off last time BD Malone, S Shankar Therefore there is a direct one-to-one correspondence between
1 Density functional theory (DFT)
") 1 Density functional theory (DFT) 1.1 Introduction Density functional theory is an alternative to ab initio methods for solving the nonrelativistic, time-independent Schrödinger equation H Φ = E Φ. The
1 Density functional theory (DFT) 1.1 Introduction Density functional theory is an alternative to ab initio methods for solving the nonrelativistic, time-independent Schrödinger equation H Φ = E Φ. The
Time-dependent density functional theory (TDDFT)
") Advanced Workshop on High-Performance & High-Throughput Materials Simulations using Quantum ESPRESSO ICTP, Trieste, Italy, January 16 to 27, 2017 Time-dependent density functional theory (TDDFT) Ralph
Advanced Workshop on High-Performance & High-Throughput Materials Simulations using Quantum ESPRESSO ICTP, Trieste, Italy, January 16 to 27, 2017 Time-dependent density functional theory (TDDFT) Ralph
Introduction to DFT and Density Functionals. by Michel Côté Université de Montréal Département de physique
 Introduction to DFT and Density Functionals by Michel Côté Université de Montréal Département de physique Eamples Carbazole molecule Inside of diamant Réf: Jean-François Brière http://www.phys.umontreal.ca/~michel_
Introduction to DFT and Density Functionals by Michel Côté Université de Montréal Département de physique Eamples Carbazole molecule Inside of diamant Réf: Jean-François Brière http://www.phys.umontreal.ca/~michel_
Ensemble-generalization approach:
 Ensemble-generalization approach: addressing some long-standing problems in DFT Eli Kraisler Max Planck Institute of Microstructure Physics, Halle (Saale), Germany DFT2016 summer school, Los Angeles, August
Ensemble-generalization approach: addressing some long-standing problems in DFT Eli Kraisler Max Planck Institute of Microstructure Physics, Halle (Saale), Germany DFT2016 summer school, Los Angeles, August
Discontinuity of the chemical potential in reduced-density-matrix-functional theory for open-shell systems
 PHYSICAL REVIEW A 79, 022504 2009 Discontinuity of the chemical potential in reduced-density-matrix-functional theory for open-shell systems N. Helbig, 1,2,3 N. N. Lathiotakis, 1,3,4 and E. K. U. Gross
PHYSICAL REVIEW A 79, 022504 2009 Discontinuity of the chemical potential in reduced-density-matrix-functional theory for open-shell systems N. Helbig, 1,2,3 N. N. Lathiotakis, 1,3,4 and E. K. U. Gross
Institut Néel Institut Laue Langevin. Introduction to electronic structure calculations
 Institut Néel Institut Laue Langevin Introduction to electronic structure calculations 1 Institut Néel - 25 rue des Martyrs - Grenoble - France 2 Institut Laue Langevin - 71 avenue des Martyrs - Grenoble
Institut Néel Institut Laue Langevin Introduction to electronic structure calculations 1 Institut Néel - 25 rue des Martyrs - Grenoble - France 2 Institut Laue Langevin - 71 avenue des Martyrs - Grenoble
ABC of ground-state DFT
 ABC of ground-state DFT Kieron Burke and Lucas Wagner Departments of Physics and of Chemistry, University of California, Irvine, CA 92697, USA July 31, 2014 Kieron (UC Irvine) ABC of ground-state DFT HoW
ABC of ground-state DFT Kieron Burke and Lucas Wagner Departments of Physics and of Chemistry, University of California, Irvine, CA 92697, USA July 31, 2014 Kieron (UC Irvine) ABC of ground-state DFT HoW
Module 6 1. Density functional theory
 Module 6 1. Density functional theory Updated May 12, 2016 B A DDFT C K A bird s-eye view of density-functional theory Authors: Klaus Capelle G http://arxiv.org/abs/cond-mat/0211443 R https://trac.cc.jyu.fi/projects/toolbox/wiki/dft
Module 6 1. Density functional theory Updated May 12, 2016 B A DDFT C K A bird s-eye view of density-functional theory Authors: Klaus Capelle G http://arxiv.org/abs/cond-mat/0211443 R https://trac.cc.jyu.fi/projects/toolbox/wiki/dft
Molecular Magnetism. Magnetic Resonance Parameters. Trygve Helgaker
 Molecular Magnetism Magnetic Resonance Parameters Trygve Helgaker Centre for Theoretical and Computational Chemistry (CTCC), Department of Chemistry, University of Oslo, Norway Laboratoire de Chimie Théorique,
Molecular Magnetism Magnetic Resonance Parameters Trygve Helgaker Centre for Theoretical and Computational Chemistry (CTCC), Department of Chemistry, University of Oslo, Norway Laboratoire de Chimie Théorique,
Introduction to Computational Chemistry
 Introduction to Computational Chemistry Vesa Hänninen Laboratory of Physical Chemistry Chemicum 4th floor vesa.hanninen@helsinki.fi September 10, 2013 Lecture 3. Electron correlation methods September
Introduction to Computational Chemistry Vesa Hänninen Laboratory of Physical Chemistry Chemicum 4th floor vesa.hanninen@helsinki.fi September 10, 2013 Lecture 3. Electron correlation methods September
Handbook of Computational Quantum Chemistry
 Handbook of Computational Quantum Chemistry David B. Cook Dept. of Chemistry University of Sheffield DOVER PUBLICATIONS, INC. Mineola, New York F Contents 1 Mechanics and molecules 1 1.1 1.2 1.3 1.4 1.5
Handbook of Computational Quantum Chemistry David B. Cook Dept. of Chemistry University of Sheffield DOVER PUBLICATIONS, INC. Mineola, New York F Contents 1 Mechanics and molecules 1 1.1 1.2 1.3 1.4 1.5
College of Chemistry, Peking University, Beijing, China. Fritz-Haber-Institut der MPG, Berlin, Germany
 KITP Program Excitations in Condensed Matter Localized and Itinerant States in a Unified Picture beyond Density Functional Theory Hong Jiang 1, Patrick Rinke 2 and Matthias Scheffler 2 1 College of Chemistry,
KITP Program Excitations in Condensed Matter Localized and Itinerant States in a Unified Picture beyond Density Functional Theory Hong Jiang 1, Patrick Rinke 2 and Matthias Scheffler 2 1 College of Chemistry,
Lecture 5. Hartree-Fock Theory. WS2010/11: Introduction to Nuclear and Particle Physics
 Lecture 5 Hartree-Fock Theory WS2010/11: Introduction to Nuclear and Particle Physics Particle-number representation: General formalism The simplest starting point for a many-body state is a system of
Lecture 5 Hartree-Fock Theory WS2010/11: Introduction to Nuclear and Particle Physics Particle-number representation: General formalism The simplest starting point for a many-body state is a system of
The LDA-1/2 method in exciting
 http://exciting-code.org The LDA-1/2 method in exciting Ronaldo Rodrigues Pelá Humboldt Universität zu Berlin Instituto Tecnológico de Aeronáutica Outline DFT-1/2 Exchange-correlation functionals Exact
http://exciting-code.org The LDA-1/2 method in exciting Ronaldo Rodrigues Pelá Humboldt Universität zu Berlin Instituto Tecnológico de Aeronáutica Outline DFT-1/2 Exchange-correlation functionals Exact
One-Electron Properties of Solids
 One-Electron Properties of Solids Alessandro Erba Università di Torino alessandro.erba@unito.it most slides are courtesy of R. Orlando and B. Civalleri Energy vs Wave-function Energy vs Wave-function Density
One-Electron Properties of Solids Alessandro Erba Università di Torino alessandro.erba@unito.it most slides are courtesy of R. Orlando and B. Civalleri Energy vs Wave-function Energy vs Wave-function Density
ABC of DFT: Hands-on session 1 Introduction into calculations on molecules
 ABC of DFT: Hands-on session 1 Introduction into calculations on molecules Tutor: Alexej Bagrets Wann? 09.11.2012, 11:30-13:00 Wo? KIT Campus Nord, Flachbau Physik, Geb. 30.22, Computerpool, Raum FE-6
ABC of DFT: Hands-on session 1 Introduction into calculations on molecules Tutor: Alexej Bagrets Wann? 09.11.2012, 11:30-13:00 Wo? KIT Campus Nord, Flachbau Physik, Geb. 30.22, Computerpool, Raum FE-6
Density Func,onal Theory (Chapter 6, Jensen)
") Chem 580: DFT Density Func,onal Theory (Chapter 6, Jensen) Hohenberg- Kohn Theorem (Phys. Rev., 136,B864 (1964)): For molecules with a non degenerate ground state, the ground state molecular energy and
Chem 580: DFT Density Func,onal Theory (Chapter 6, Jensen) Hohenberg- Kohn Theorem (Phys. Rev., 136,B864 (1964)): For molecules with a non degenerate ground state, the ground state molecular energy and
CHEM3023: Spins, Atoms and Molecules
 CHEM3023: Spins, Atoms and Molecules Lecture 5 The Hartree-Fock method C.-K. Skylaris Learning outcomes Be able to use the variational principle in quantum calculations Be able to construct Fock operators
CHEM3023: Spins, Atoms and Molecules Lecture 5 The Hartree-Fock method C.-K. Skylaris Learning outcomes Be able to use the variational principle in quantum calculations Be able to construct Fock operators
Supplementary Figure 1 Two-dimensional map of the spin-orbit coupling correction to the scalar-relativistic DFT/LDA band gap. The calculations were
 Supplementary Figure 1 Two-dimensional map of the spin-orbit coupling correction to the scalar-relativistic DFT/LDA band gap. The calculations were performed for the Platonic model of PbI 3 -based perovskites
Supplementary Figure 1 Two-dimensional map of the spin-orbit coupling correction to the scalar-relativistic DFT/LDA band gap. The calculations were performed for the Platonic model of PbI 3 -based perovskites
Time-dependent density functional theory (TDDFT)
") 04/05/16 Hands-on workshop and Humboldt-Kolleg: Density-Functional Theory and Beyond - Basic Principles and Modern Insights Isfahan University of Technology, Isfahan, Iran, May 2 to 13, 2016 Time-dependent
04/05/16 Hands-on workshop and Humboldt-Kolleg: Density-Functional Theory and Beyond - Basic Principles and Modern Insights Isfahan University of Technology, Isfahan, Iran, May 2 to 13, 2016 Time-dependent
Improved Electronic Structure and Optical Properties of sp-hybridized Semiconductors Using LDA+U SIC
 286 Brazilian Journal of Physics, vol. 36, no. 2A, June, 2006 Improved Electronic Structure and Optical Properties of sp-hybridized Semiconductors Using LDA+U SIC Clas Persson and Susanne Mirbt Department
286 Brazilian Journal of Physics, vol. 36, no. 2A, June, 2006 Improved Electronic Structure and Optical Properties of sp-hybridized Semiconductors Using LDA+U SIC Clas Persson and Susanne Mirbt Department
Introduction and Overview of the Reduced Density Matrix Functional Theory
 Introduction and Overview of the Reduced Density Matrix Functional Theory N. N. Lathiotakis Theoretical and Physical Chemistry Institute, National Hellenic Research Foundation, Athens April 13, 2016 Outline
Introduction and Overview of the Reduced Density Matrix Functional Theory N. N. Lathiotakis Theoretical and Physical Chemistry Institute, National Hellenic Research Foundation, Athens April 13, 2016 Outline
Explaining the apparent arbitrariness of the LDA-1/2 self-energy. correction method applied to purely covalent systems
 Explaining the apparent arbitrariness of the LDA-1/2 self-energy correction method applied to purely covalent systems Kan-Hao Xue, 1,2 Leonardo R. C. Fonseca, 3 and Xiang-Shui Miao 1,2 1 School of Optical
Explaining the apparent arbitrariness of the LDA-1/2 self-energy correction method applied to purely covalent systems Kan-Hao Xue, 1,2 Leonardo R. C. Fonseca, 3 and Xiang-Shui Miao 1,2 1 School of Optical
Orbital dependent correlation potentials in ab initio density functional theory
 Orbital dependent correlation potentials in ab initio density functional theory noniterative - one step - calculations Ireneusz Grabowski Institute of Physics Nicolaus Copernicus University Toruń, Poland
Orbital dependent correlation potentials in ab initio density functional theory noniterative - one step - calculations Ireneusz Grabowski Institute of Physics Nicolaus Copernicus University Toruń, Poland
article Finding electron affinities with approximate density functionals
 Molecular Physics Vol. 00, No. 00, Month 2009, 1 21 article Finding electron affinities with approximate density functionals Donghyung Lee a and Kieron Burke a a Department of Chemistry, University of
Molecular Physics Vol. 00, No. 00, Month 2009, 1 21 article Finding electron affinities with approximate density functionals Donghyung Lee a and Kieron Burke a a Department of Chemistry, University of
Supplemental Material: Experimental and Theoretical Investigations of the Electronic Band Structure of Metal-Organic Framework of HKUST-1 Type
 Supplemental Material: Experimental and Theoretical Investigations of the Electronic Band Structure of Metal-Organic Framework of HKUST-1 Type Zhigang Gu, a Lars Heinke, a,* Christof Wöll a, Tobias Neumann,
Supplemental Material: Experimental and Theoretical Investigations of the Electronic Band Structure of Metal-Organic Framework of HKUST-1 Type Zhigang Gu, a Lars Heinke, a,* Christof Wöll a, Tobias Neumann,
Time-Dependent Density-Functional Theory
 Summer School on First Principles Calculations for Condensed Matter and Nanoscience August 21 September 3, 2005 Santa Barbara, California Time-Dependent Density-Functional Theory X. Gonze, Université Catholique
Summer School on First Principles Calculations for Condensed Matter and Nanoscience August 21 September 3, 2005 Santa Barbara, California Time-Dependent Density-Functional Theory X. Gonze, Université Catholique
Basics of density-functional theory and fast guide to actual calculations Matthias Scheffler
 Basics of density-functional theory and fast guide to actual calculations Matthias Scheffler http://www.fhi-berlin.mpg.de/th/th.html I. From the many-particle problem to the Kohn-Sham functional II. From
Basics of density-functional theory and fast guide to actual calculations Matthias Scheffler http://www.fhi-berlin.mpg.de/th/th.html I. From the many-particle problem to the Kohn-Sham functional II. From
arxiv: v1 [cond-mat.mtrl-sci] 26 Jan 2014
![arxiv: v1 [cond-mat.mtrl-sci] 26 Jan 2014](/thumbs/73/68153161.jpg "arxiv: v1 [cond-mat.mtrl-sci] 26 Jan 2014") Self Interaction Correction with Unitary Invariance Mark R Pederson 1, Adrienn Ruzsinszky 2 and John P. Perdew 2 1 Department of Chemistry, Johns Hopkins University, Baltimore MD, 21218 and Office of Basic
Self Interaction Correction with Unitary Invariance Mark R Pederson 1, Adrienn Ruzsinszky 2 and John P. Perdew 2 1 Department of Chemistry, Johns Hopkins University, Baltimore MD, 21218 and Office of Basic
Lecture 5: More about one- Final words about the Hartree-Fock theory. First step above it by the Møller-Plesset perturbation theory.
 Lecture 5: More about one- determinant wave functions Final words about the Hartree-Fock theory. First step above it by the Møller-Plesset perturbation theory. Items from Lecture 4 Could the Koopmans theorem
Lecture 5: More about one- determinant wave functions Final words about the Hartree-Fock theory. First step above it by the Møller-Plesset perturbation theory. Items from Lecture 4 Could the Koopmans theorem
The calculation of the universal density functional by Lieb maximization
 The calculation of the universal density functional by Lieb maximization Trygve Helgaker, Andy Teale, and Sonia Coriani Centre for Theoretical and Computational Chemistry (CTCC), Department of Chemistry,
The calculation of the universal density functional by Lieb maximization Trygve Helgaker, Andy Teale, and Sonia Coriani Centre for Theoretical and Computational Chemistry (CTCC), Department of Chemistry,