Spring College on Computational Nanoscience
|
|
|
- Charlotte York
- 5 years ago
- Views:
Transcription
1 Spring College on Computational Nanoscience May 2010 At the Fifth Rung of Jacob's Ladder: A Discussion of Exact Exchange plus Local- and Nonlocal-density Approximations to the Correlation Functional Matthias SCHEFFLER FHI der Max-Planck Gesellschaft Theory Department Berlin Germany



 have been very successful but there are problems for certain bonding situations (vdw,")
2 At the fifth rung of Jacob s ladder: A Discussion of Exact Exchange plus Local- and Nonlocal- Density Approximations to the Correlation Functional f spd Biophysics Surface Energies and Adsorption Df Defects in Mtl Metals, Semiconductors, and Insulators Lanthanide Oxides m mm m nm space densityfunctional theory and beyond ab initio Molecular ElectronicDynamics Structure Theory Predictive modeling and simulations must address all time and space scales ab initio kinetic Monte Carlo Continuum Equations, Rate Equations, and Finite Element Modeling Last Friday time f s p s n s s m s s hours years the base Approximate xc functionals (e.g. LDA and GGAs) have been very successful but there are problems for certain bonding situations (vdw, hydrogen bonding, certain covalent bonds) for highly correlated situations, and for excited states. 1

3 The zoo of approximate xc functional is getting hardly comprehensible. -- Patchwork or sound science? HL VWN BP86 PKZB PW91 PBE r-pbe RPBE revpbe B88 PBE0 How to clean up this mess? The challenges: Find practical ways to correct the xc approximation and/or to control the errors. Efficient all-electron code; DFT also with hybrid functionals and TDDFT, Hartree- Fock +MP2, EX+cRPA, GW selfenergies, etc. Perdew s Dream: Jacob s Ladder in DFT accuracy? computationa al cost 5 unoccupied i (r), e.g., ACFD-RPA 4 occupied i (r), hybrid functional (e.g., B3LYP, PBE0) 3 (r), meta-gga (e.g., TPSS) 2 n(r), GGA (e.g., PBE) 1 n(r), LDA (r) ( ) : KS kinetic energy density ACFD : adiabatic connection fluctuation dissipation theorem (Bohm, Pines (1953); Gell-Mann, Brueckner (1957); Langreth, Perdew (1977); Gunnarsson, Lunqvist (1975, 1976) RPA : random phase approximation 2
4 Exact Exchange plus Correlation in RPA The orbitals for evaluating E x are different in Hartree-Fock and Kohn-Sham DFT. --The numerical technique to evaluate E x is the same. Adding correlation: on top of Hartree-Fock exchange: Møller-Plesset perturbation theory (MP2) on top of DFT exact exchange : random phase approximation (RPA) 3
5 RPA Formulated within DFT Framework E xc RPA = E x + E c RPA 0 == dynamical-response function of the Kohn-Sham system The approach gives total energies but no information on how the Kohn-Sham energies,, i.e. spectroscopy, will change. This (corresponding) change is given by the GW self-energy. Pros and Cons of EX+cRPA The good aspects: Exchange is treated at the exact exchange level (so far with PBE or PBE0 orbitals) vdw interactions are included (automatically; seamlessly). Screening is taken into account. Thus EX+cRPA works for metals/small gap systems -- in contrast to MP2. Right decay behavior at metal surfaces (Rohlfing, Bredos, PRL 101 (2008). An A is still there! In contrast to LDA and GGA, EX+cRPA does not benefit from (fortuitous) error cancellation between the exchange and correlation terms. The A clearly shows up in atomization energies of small molecules: A non-selfconsistent (PBE-based) RPA does not always improve over hybrid functionals, e.g. B3LYP or PBE0. 4

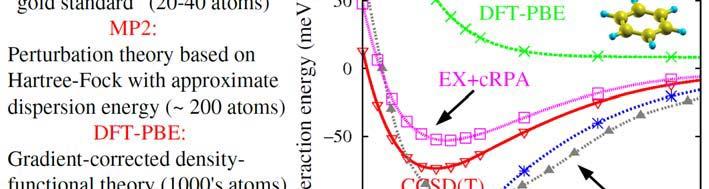
6 The RPA and vdw interactions in Comparison with Other Methods The RPA and vdw interactions in Comparison with Other Methods 5

7 The RPA and vdw interactions in Comparison with Other Methods EX + crpa is doing well, but the C 6 coefficient is slightly too small Another Example Where Present-Day xc Functionals Reveal (Severe) Limitations: CO adsorption at transition metal surfaces: LDA and GGA xc functionals dramatically fail to predict the correct adsorption site. For low coverage the theory gives the hollow site, but experimentally CO adsorbs on top. E.g.: For CO/Cu (111) the LDA error is 0.4 ev, and the GGA error is 02eV 0.2 ev. Feibelman, Hammer, Norskov, Wagner, Scheffler, Stumpf, Watwe, and Dumesic, The CO/Pt(111) puzzle. J. Phys. Chem. B 105, (2001). 6
 hollow top RPA was evaluated with LDA, PBE and PBE0 orbitals. The difference is only 0.02 ev. Xinguo Ren (1) Difference to Stroppa et al. PRB 76(2007) is less than 0.04 ev.")
 possibly corrected with hybrid XC s (B3LYP, PBE0, HSE03) definitely corrected by RPA (and also by MP2).")
8 EX+cRPA, MP2, and Others for the CO Adsorption Puzzle ) E fcc E top (ev LDA AM05 PBE0 PBE B3LYP & HSE exp. (1) (1) MP2 X. Ren, P. Rinke, M.S., PRB 80 (2009). RPA (2) hollow top RPA was evaluated with LDA, PBE and PBE0 orbitals. The difference is only 0.02 ev. Xinguo Ren (1) Difference to Stroppa et al. PRB 76(2007) is less than 0.04 ev. (2) Harl and Kresse, PRL 103 (2009) only 0.12 ev Patrick Rinke Wrong preference for the hollow site in LDA and GGA (PBE, AM05) possibly corrected with hybrid XC s (B3LYP, PBE0, HSE03) definitely corrected by RPA (and also by MP2). The CO Adsorption Puzzle gas phase 2 * lumo 5 homo O C O C adsorbed 2 * 5 2 * free CO clean Cu surface E F s-band d-band GW results: For details (RPA and GW) see X. Ren, P. Rinke, M.S., PRB 80 (2009). 7

.")
.")
9 Surface Energies from Y to Ag Theory: DFT-LDA M. Methfessel, D. Hennig, and M. S., Phys. Rev. B 46,, 4816 (1992) The trend in cohesive energies and surface energies is well understood (since long). Uncertainties in experimental estimates are significant (extrapolation from surface tension of liquids). For example, for Pd (111) the experimental value is 0.8 ± 0.15 ev/surface atom. 8
 E")




![(xc-better) E N cluster (LDA)] (1) Q.-M. Hu, K.](/docs-images/93/111716759/images/10-5.jpg "Reuter, and M. S.")

10 Surface Energies from Y to Ag We evaluate the total energy from a difference (1) E better-than-lda = E LDA + E xc. The total energy of a poly-atomic system can be written as Aloysius Soon E = N C N E N. C N is the number of atoms that are N-fold coordinated and E N is the energy contribution of each N-fold coordinated atom. We are not using this equation in full but only for the xc correction: E xc = N C N [ E N cluster (xc-better) E N cluster (LDA)] (1) Q.-M. Hu, K. Reuter, and M. S., PRL 98, (2007); and 99, (E) (2007). Some of the considered clusters: Pd 4 Pd 6 Pd 10 E = 4*E 3 E = 6*E 4 E = 6*E 4 + 3*E 5 + E 9 Pd 13 Pd 19 E = 12*E 5 + E 12 E = 6*E *E 7 + 1*E 12 9
 12 is PBE the difference of the PBEsol cohesive energy of AM05 xc-better btt and dthe BLYP LDA cohesive RPA: rpbe Harl&Kresse energy.")
11 Cohesive Energies and Surface Energies from Y to Ag differences with respect to LDA A. Soon et al., in preparation - xc(relative) 12 is PBE the difference of the PBEsol cohesive energy of AM05 xc-better btt and dthe BLYP LDA cohesive RPA: rpbe Harl&Kresse energy. (2009) RPA: ours B3LYP HSE03 experiment HSE06 RPA@PBE0 RPA@HSE06 For example: xc(relative) 12 - xc(relative) 9 is the difference of the fcc(111) surface energy of xc-better and the LDA. Cohesive Energies and Surface Energies from Y to Ag differences with respect to LDA A. Soon et al., in preparation PBE PBEsol AM05 BLYP rpbe B3LYP HSE03 experiment HSE06 RPA@PBE0 RPA@HSE06 Hybrids (HSE03, PBE0, B3LYP, M06 s) underestimate the cohesive energy severely. RPA is doing fine : For the cohesive energy it is worse than PBE; for the surface energies it is better than PBE (as good as LDA). 10
")

12 how about molecules? Hydrogen bonds and dispersion: S22 Jurecka, Sponer, Cerny, Hobza PCCP 8, 1985 (2006) MP2/ RPA (ev) Covalent bonds: small molecules CCSD(T) binding energies (ev) There is opportunity beyond the RPA F. Furche, Phys. Rev. B 64, (2001) The Role of vdw Forces in Organic Materials and Biophysics Alexandre Tkatchenko 11

13 EX + crpa is doing well, but the C 6 coefficient is slightly too small 12



14 13

15 14


16 15
17 16

18 17
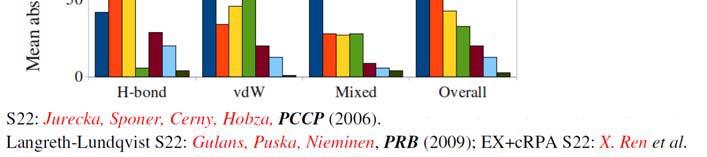

19 18




20 However, there are problems when applying the TS approach to bulk metals, as there are problems for all the other schemes, except EX+cRPA. Stability of the -helix structure of proteins (peptide chains): R O H N C C OH amino peptide carboxyl group bond group R 1 O O R 2 O O R 3 O R 4 O R n O H N C NC CN C NC CN C NC CN C... C N C C OH H H H H H H H H H H H H H H H -Faltblatt sheet Carboxylgruppe -Helix helix secondary structure 19
 Mariana Rossi Carvalho Volker Blum peptide: C, N, O Alanine: R = CH 3 Alexandre Tkatchenko")
21 The Critical Role of vdw Intereactions: Structure and Stability of Polypeptides Fully extended structure (FES) Mariana Rossi Carvalho Volker Blum R O peptide: H - N - C - C - OH Alanine: R = CH 3 H H Alexandre Tkatchenko Structure and Stability of Polypeptides Fully extended structure (FES) Mariana Rossi Carvalho Volker Blum peptide: C, N, O Alanine: R = CH 3 Alexandre Tkatchenko 20


22 21
 No of H-bomds 10 5 0 10 PBE+vdW 3 10")
 H-bond = 2.")


folding dynamics of polyalanine")

23 Role of vdw Interaction on (Un)folding; Comparing, 3 10, and Helices for Ac-Ala 15 LysH + s (*) No of H-bomds PBE+vdW 3 10 PBE-pure Time (pico seconds) PBE+vdW 1000K Mariana Rossi Carvalho Volker Blum (*) H-bond = 2.5 between CO and NH groups PBE-pure Alexandre Tkatchenko Summary Developed accurate and efficient first-principle method for the long-range van der Waal interaction. The method can be coupled with DFT (e.g., PBE+vdW) and MP2 (MP2+ vdw). Application for example to (un)folding dynamics of polyalanine helices: o vdw forces increase the stability of polyalanine helices by ~ 100%. o Qualitative i differences in unfolding dynamics between PBE-pure and PBE+vdW. o Remarkable stability of polyalanine helices in vacuo is attributed to synergy between H-bonds and vdw forces. 22
24 Summary EX+cRPA is promising. Though the improvement over lower-level xc functionals is (in principle) systematic, agreement with experiment is not always better. There is room (need) for improvements; we have to go to self-consistent RPA (in some cases) and to higher order in the expansion. An estimate of errors due to the xc approximation is becoming possible. 23
Role of van der Waals Interactions in Physics, Chemistry, and Biology
 Role of van der Waals Interactions in Physics, Chemistry, and Biology How can we describe vdw forces in materials accurately? Failure of DFT Approximations for (Long-Range) Van der Waals Interactions 1
Role of van der Waals Interactions in Physics, Chemistry, and Biology How can we describe vdw forces in materials accurately? Failure of DFT Approximations for (Long-Range) Van der Waals Interactions 1
5/27/2012. Role of van der Waals Interactions in Physics, Chemistry, and Biology
 Role of van der Waals Interactions in Physics, Chemistry, and Biology 1 Role of van der Waals Interactions in Physics, Chemistry, and Biology Karin Jacobs: Take van der Waals forces into account in theory,
Role of van der Waals Interactions in Physics, Chemistry, and Biology 1 Role of van der Waals Interactions in Physics, Chemistry, and Biology Karin Jacobs: Take van der Waals forces into account in theory,
Random-phase approximation and beyond for materials: concepts, practice, and future perspectives. Xinguo Ren
 Random-phase approximation and beyond for materials: concepts, practice, and future perspectives Xinguo Ren University of Science and Technology of China, Hefei USTC-FHI workshop on frontiers of Advanced
Random-phase approximation and beyond for materials: concepts, practice, and future perspectives Xinguo Ren University of Science and Technology of China, Hefei USTC-FHI workshop on frontiers of Advanced
Basic Concepts and First Principles Computations for Surface Science: Applications in Chemical Energy Conversion and Storage.
 International Summer School on Basic Concepts and First Principles Computations for Surface Science: Applications in Chemical Energy Conversion and Storage Norderney, Germany, July 21 26, 2013 Let s start
International Summer School on Basic Concepts and First Principles Computations for Surface Science: Applications in Chemical Energy Conversion and Storage Norderney, Germany, July 21 26, 2013 Let s start
Methods for van der Waals Interactions
 Methods for van der Waals Interactions Alexandre Tkatchenko Theory Department, Fritz Haber Institut der MPG Berlin, Germany tkatchen@fhi berlin.mpg.de Haber Institute FHI DFT and Beyond Workshop, Jul.
Methods for van der Waals Interactions Alexandre Tkatchenko Theory Department, Fritz Haber Institut der MPG Berlin, Germany tkatchen@fhi berlin.mpg.de Haber Institute FHI DFT and Beyond Workshop, Jul.
Bayesian Error Estimation in Density Functional Theory
 Bayesian Error Estimation in Density Functional Theory Karsten W. Jacobsen Jens Jørgen Mortensen Kristen Kaasbjerg Søren L. Frederiksen Jens K. Nørskov CAMP, Dept. of Physics, DTU James P. Sethna LASSP,
Bayesian Error Estimation in Density Functional Theory Karsten W. Jacobsen Jens Jørgen Mortensen Kristen Kaasbjerg Søren L. Frederiksen Jens K. Nørskov CAMP, Dept. of Physics, DTU James P. Sethna LASSP,
Role%of%van%der%Waals%Interac1ons%in%% Physics,%Chemistry,%and%Biology!
 Role%of%van%der%Waals%Interac1ons%in%% Physics,%Chemistry,%and%Biology! Role%of%van%der%Waals%Interac1ons%in%% Physics,%Chemistry,%and%Biology! Karin!Jacobs:! Take!van!der!Waals!forces!into! account!in!theory,!simula
Role%of%van%der%Waals%Interac1ons%in%% Physics,%Chemistry,%and%Biology! Role%of%van%der%Waals%Interac1ons%in%% Physics,%Chemistry,%and%Biology! Karin!Jacobs:! Take!van!der!Waals!forces!into! account!in!theory,!simula
DENSITY FUNCTIONAL THEORY FOR NON-THEORISTS JOHN P. PERDEW DEPARTMENTS OF PHYSICS AND CHEMISTRY TEMPLE UNIVERSITY
 DENSITY FUNCTIONAL THEORY FOR NON-THEORISTS JOHN P. PERDEW DEPARTMENTS OF PHYSICS AND CHEMISTRY TEMPLE UNIVERSITY A TUTORIAL FOR PHYSICAL SCIENTISTS WHO MAY OR MAY NOT HATE EQUATIONS AND PROOFS REFERENCES
DENSITY FUNCTIONAL THEORY FOR NON-THEORISTS JOHN P. PERDEW DEPARTMENTS OF PHYSICS AND CHEMISTRY TEMPLE UNIVERSITY A TUTORIAL FOR PHYSICAL SCIENTISTS WHO MAY OR MAY NOT HATE EQUATIONS AND PROOFS REFERENCES
Accurate van der Waals interactions from ground state electron density
 Accurate van der Waals interactions from ground state electron density Alexandre Tkatchenko Theory Department, Fritz Haber Institut der MPG Berlin, Germany tkatchen@fhi berlin.mpg.de Haber Institute EXCITCM09,
Accurate van der Waals interactions from ground state electron density Alexandre Tkatchenko Theory Department, Fritz Haber Institut der MPG Berlin, Germany tkatchen@fhi berlin.mpg.de Haber Institute EXCITCM09,
DFT: Exchange-Correlation
 DFT: Exchange-Correlation Local functionals, exact exchange and other post-dft methods Paul Tulip Centre for Materials Physics Department of Physics University of Durham Outline Introduction What is exchange
DFT: Exchange-Correlation Local functionals, exact exchange and other post-dft methods Paul Tulip Centre for Materials Physics Department of Physics University of Durham Outline Introduction What is exchange
DFT: Exchange-Correlation
 DFT: Local functionals, exact exchange and other post-dft methods Stewart Clark University of Outline Introduction What is exchange and correlation? Quick tour of XC functionals (Semi-)local: LDA, PBE,
DFT: Local functionals, exact exchange and other post-dft methods Stewart Clark University of Outline Introduction What is exchange and correlation? Quick tour of XC functionals (Semi-)local: LDA, PBE,
When (2 + 2) 4? Alexandre Tkatchenko Fritz Haber Institut der Max Planck Gesellschaft, Berlin, Germany
 4? Alexandre Tkatchenko Fritz Haber Institut der Max Planck Gesellschaft, Berlin, Germany") When (2 + 2) 4? Alexandre Tkatchenko Fritz Haber Institut der Max Planck Gesellschaft, Berlin, Germany QMC@TTI, Apuan Alps, Jul 29, 2013 When (2 + 2) 4 or Van der Waals Interactions in Complex (and Simple)
When (2 + 2) 4? Alexandre Tkatchenko Fritz Haber Institut der Max Planck Gesellschaft, Berlin, Germany QMC@TTI, Apuan Alps, Jul 29, 2013 When (2 + 2) 4 or Van der Waals Interactions in Complex (and Simple)
Towards a unified description of ground and excited state properties: RPA vs GW
 Towards a unified description of ground and excited state properties: RPA vs GW Patrick Rinke Fritz-Haber-Institut der Max-Planck-Gesellschaft, Berlin - Germany Towards Reality in Nanoscale Materials V
Towards a unified description of ground and excited state properties: RPA vs GW Patrick Rinke Fritz-Haber-Institut der Max-Planck-Gesellschaft, Berlin - Germany Towards Reality in Nanoscale Materials V
Electronic band structure, sx-lda, Hybrid DFT, LDA+U and all that. Keith Refson STFC Rutherford Appleton Laboratory
 Electronic band structure, sx-lda, Hybrid DFT, LDA+U and all that Keith Refson STFC Rutherford Appleton Laboratory LDA/GGA DFT is good but... Naive LDA/GGA calculation severely underestimates band-gaps.
Electronic band structure, sx-lda, Hybrid DFT, LDA+U and all that Keith Refson STFC Rutherford Appleton Laboratory LDA/GGA DFT is good but... Naive LDA/GGA calculation severely underestimates band-gaps.
The Electronic Structure of Dye- Sensitized TiO 2 Clusters from Many- Body Perturbation Theory
 The Electronic Structure of Dye- Sensitized TiO 2 Clusters from Many- Body Perturbation Theory Noa Marom Center for Computational Materials Institute for Computational Engineering and Sciences The University
The Electronic Structure of Dye- Sensitized TiO 2 Clusters from Many- Body Perturbation Theory Noa Marom Center for Computational Materials Institute for Computational Engineering and Sciences The University
Electronic-structure theory for data-driven materials discovery
 Monday, July 31: Tuesday, August 1: Wednesday, August 2: Thursday, August 3: Friday, August 4: Electronic-structure theory for data-driven materials discovery Implementing DFT Periodic systems Benchmark
Monday, July 31: Tuesday, August 1: Wednesday, August 2: Thursday, August 3: Friday, August 4: Electronic-structure theory for data-driven materials discovery Implementing DFT Periodic systems Benchmark
Chemisorption VIII. NEVF 514 Surface Physics. Winter Term Troja, 16th December 2016
 Chemisorption František Máca VIII. NEVF 514 Surface Physics Winter Term 2016-2017 Troja, 16th December 2016 Chemisorption The knowledge of chemisorption phenomena requires the determination of the geometrical
Chemisorption František Máca VIII. NEVF 514 Surface Physics Winter Term 2016-2017 Troja, 16th December 2016 Chemisorption The knowledge of chemisorption phenomena requires the determination of the geometrical
CLIMBING THE LADDER OF DENSITY FUNCTIONAL APPROXIMATIONS JOHN P. PERDEW DEPARTMENT OF PHYSICS TEMPLE UNIVERSITY PHILADELPHIA, PA 19122
 CLIMBING THE LADDER OF DENSITY FUNCTIONAL APPROXIMATIONS JOHN P. PERDEW DEPARTMENT OF PHYSICS TEMPLE UNIVERSITY PHILADELPHIA, PA 191 THANKS TO MANY COLLABORATORS, INCLUDING SY VOSKO DAVID LANGRETH ALEX
CLIMBING THE LADDER OF DENSITY FUNCTIONAL APPROXIMATIONS JOHN P. PERDEW DEPARTMENT OF PHYSICS TEMPLE UNIVERSITY PHILADELPHIA, PA 191 THANKS TO MANY COLLABORATORS, INCLUDING SY VOSKO DAVID LANGRETH ALEX
Orbital Density Dependent Functionals
 Orbital Density Dependent Functionals S. Kluepfel1, P. Kluepfel1, Hildur Guðmundsdóttir1 and Hannes Jónsson1,2 1. Univ. of Iceland; 2. Aalto University Outline: Problems with GGA approximation (PBE, RPBE,...)
Orbital Density Dependent Functionals S. Kluepfel1, P. Kluepfel1, Hildur Guðmundsdóttir1 and Hannes Jónsson1,2 1. Univ. of Iceland; 2. Aalto University Outline: Problems with GGA approximation (PBE, RPBE,...)
Multi-Scale Modeling from First Principles
 m mm Multi-Scale Modeling from First Principles μm nm m mm μm nm space space Predictive modeling and simulations must address all time and Continuum Equations, densityfunctional space scales Rate Equations
m mm Multi-Scale Modeling from First Principles μm nm m mm μm nm space space Predictive modeling and simulations must address all time and Continuum Equations, densityfunctional space scales Rate Equations
Electronic structure theory: Fundamentals to frontiers. 2. Density functional theory
 Electronic structure theory: Fundamentals to frontiers. 2. Density functional theory MARTIN HEAD-GORDON, Department of Chemistry, University of California, and Chemical Sciences Division, Lawrence Berkeley
Electronic structure theory: Fundamentals to frontiers. 2. Density functional theory MARTIN HEAD-GORDON, Department of Chemistry, University of California, and Chemical Sciences Division, Lawrence Berkeley
 3/23/2010 More basics of DFT Kieron Burke and friends UC Irvine Physics and Chemistry References for ground-state DFT ABC of DFT, by KB and Rudy Magyar, http://dft.uci.edu A Primer in Density Functional
3/23/2010 More basics of DFT Kieron Burke and friends UC Irvine Physics and Chemistry References for ground-state DFT ABC of DFT, by KB and Rudy Magyar, http://dft.uci.edu A Primer in Density Functional
Density functional theory in the solid state
 Density functional theory in the solid state Ari P Seitsonen IMPMC, CNRS & Universités 6 et 7 Paris, IPGP Department of Applied Physics, Helsinki University of Technology Physikalisch-Chemisches Institut
Density functional theory in the solid state Ari P Seitsonen IMPMC, CNRS & Universités 6 et 7 Paris, IPGP Department of Applied Physics, Helsinki University of Technology Physikalisch-Chemisches Institut
Advanced Quantum Chemistry III: Part 3. Haruyuki Nakano. Kyushu University
 Advanced Quantum Chemistry III: Part 3 Haruyuki Nakano Kyushu University 2013 Winter Term 1. Hartree-Fock theory Density Functional Theory 2. Hohenberg-Kohn theorem 3. Kohn-Sham method 4. Exchange-correlation
Advanced Quantum Chemistry III: Part 3 Haruyuki Nakano Kyushu University 2013 Winter Term 1. Hartree-Fock theory Density Functional Theory 2. Hohenberg-Kohn theorem 3. Kohn-Sham method 4. Exchange-correlation
Teoría del Funcional de la Densidad (Density Functional Theory)
") Teoría del Funcional de la Densidad (Density Functional Theory) Motivation: limitations of the standard approach based on the wave function. The electronic density n(r) as the key variable: Functionals
Teoría del Funcional de la Densidad (Density Functional Theory) Motivation: limitations of the standard approach based on the wave function. The electronic density n(r) as the key variable: Functionals
Dispersion Correcting Atom Centered Potentials (DCACP) and Many-Body Dispersion (MBD) contributions to interatomic vdw forces
 and Many-Body Dispersion (MBD) contributions to interatomic vdw forces") Dispersion Correcting Atom Centered Potentials (DCACP) and Many-Body Dispersion (MBD) contributions to interatomic vdw forces O. Anatole von Lilienfeld Current: Argonne Leadership Computing Facility From
Dispersion Correcting Atom Centered Potentials (DCACP) and Many-Body Dispersion (MBD) contributions to interatomic vdw forces O. Anatole von Lilienfeld Current: Argonne Leadership Computing Facility From
Modified Becke-Johnson (mbj) exchange potential
 exchange potential") Modified Becke-Johnson (mbj) exchange potential Hideyuki Jippo Fujitsu Laboratories LTD. 2015.12.21-22 OpenMX developer s meeting @ Kobe Overview: mbj potential The semilocal exchange potential adding
Modified Becke-Johnson (mbj) exchange potential Hideyuki Jippo Fujitsu Laboratories LTD. 2015.12.21-22 OpenMX developer s meeting @ Kobe Overview: mbj potential The semilocal exchange potential adding
Intermediate DFT. Kieron Burke and Lucas Wagner. Departments of Physics and of Chemistry, University of California, Irvine, CA 92697, USA
 Intermediate DFT Kieron Burke and Lucas Wagner Departments of Physics and of Chemistry, University of California, Irvine, CA 92697, USA October 10-19th, 2012 Kieron (UC Irvine) Intermediate DFT Lausanne12
Intermediate DFT Kieron Burke and Lucas Wagner Departments of Physics and of Chemistry, University of California, Irvine, CA 92697, USA October 10-19th, 2012 Kieron (UC Irvine) Intermediate DFT Lausanne12
XYZ of ground-state DFT
 XYZ of ground-state DFT Kieron Burke and Lucas Wagner Departments of Physics and of Chemistry, University of California, Irvine, CA 92697, USA January 5-9th, 2014 Kieron (UC Irvine) XYZ of ground-state
XYZ of ground-state DFT Kieron Burke and Lucas Wagner Departments of Physics and of Chemistry, University of California, Irvine, CA 92697, USA January 5-9th, 2014 Kieron (UC Irvine) XYZ of ground-state
Computational Methods. Chem 561
 Computational Methods Chem 561 Lecture Outline 1. Ab initio methods a) HF SCF b) Post-HF methods 2. Density Functional Theory 3. Semiempirical methods 4. Molecular Mechanics Computational Chemistry " Computational
Computational Methods Chem 561 Lecture Outline 1. Ab initio methods a) HF SCF b) Post-HF methods 2. Density Functional Theory 3. Semiempirical methods 4. Molecular Mechanics Computational Chemistry " Computational
Method development at SUNCAT in general
 Bayesian error estimation functionals and further method development at SUNCAT GPAW 2016 University of Jyväskylä June 8th, 2016 Johannes Voss vossj@stanford.edu Method development at SUNCAT in general
Bayesian error estimation functionals and further method development at SUNCAT GPAW 2016 University of Jyväskylä June 8th, 2016 Johannes Voss vossj@stanford.edu Method development at SUNCAT in general
Density Func,onal Theory (Chapter 6, Jensen)
") Chem 580: DFT Density Func,onal Theory (Chapter 6, Jensen) Hohenberg- Kohn Theorem (Phys. Rev., 136,B864 (1964)): For molecules with a non degenerate ground state, the ground state molecular energy and
Chem 580: DFT Density Func,onal Theory (Chapter 6, Jensen) Hohenberg- Kohn Theorem (Phys. Rev., 136,B864 (1964)): For molecules with a non degenerate ground state, the ground state molecular energy and
Conformational space and energetics of biomolecules: Physical concepts and performance of DFT based methods
 Conformational space and energetics of biomolecules: Physical concepts and performance of DFT based methods Alexandre Tkatchenko, Carsten Baldauf, Matti Ropo Practical Session III / Weekend Project Haber
Conformational space and energetics of biomolecules: Physical concepts and performance of DFT based methods Alexandre Tkatchenko, Carsten Baldauf, Matti Ropo Practical Session III / Weekend Project Haber
Pseudopotentials for hybrid density functionals and SCAN
 Pseudopotentials for hybrid density functionals and SCAN Jing Yang, Liang Z. Tan, Julian Gebhardt, and Andrew M. Rappe Department of Chemistry University of Pennsylvania Why do we need pseudopotentials?
Pseudopotentials for hybrid density functionals and SCAN Jing Yang, Liang Z. Tan, Julian Gebhardt, and Andrew M. Rappe Department of Chemistry University of Pennsylvania Why do we need pseudopotentials?
Performance of various density-functional approximations for cohesive properties of 64 bulk solids
 Performance of various density-functional approximations for cohesive properties of 64 bulk solids Guo-Xu Zhang 1,2, Anthony M. Reilly 1,3, Alexandre Tkatchenko 1,4, and Matthias Scheffler 1 1 Fritz-Haber-Institut
Performance of various density-functional approximations for cohesive properties of 64 bulk solids Guo-Xu Zhang 1,2, Anthony M. Reilly 1,3, Alexandre Tkatchenko 1,4, and Matthias Scheffler 1 1 Fritz-Haber-Institut
Comparison of exchange-correlation functionals: from LDA to GGA and beyond
 Comparison of ehange-correlation functionals: from LDA to GGA and beyond Martin Fuchs Fritz-Haber-Institut der MPG, Berlin, Germany Density-Functional Theory Calculations for Modeling Materials and Bio-Molecular
Comparison of ehange-correlation functionals: from LDA to GGA and beyond Martin Fuchs Fritz-Haber-Institut der MPG, Berlin, Germany Density-Functional Theory Calculations for Modeling Materials and Bio-Molecular
Computational Chemistry I
 Computational Chemistry I Text book Cramer: Essentials of Quantum Chemistry, Wiley (2 ed.) Chapter 3. Post Hartree-Fock methods (Cramer: chapter 7) There are many ways to improve the HF method. Most of
Computational Chemistry I Text book Cramer: Essentials of Quantum Chemistry, Wiley (2 ed.) Chapter 3. Post Hartree-Fock methods (Cramer: chapter 7) There are many ways to improve the HF method. Most of
Exchange-Correlation Functional
 Exchange-Correlation Functional Aiichiro Nakano Collaboratory for Advanced Computing & Simulations Depts. of Computer Science, Physics & Astronomy, Chemical Engineering & Materials Science, and Biological
Exchange-Correlation Functional Aiichiro Nakano Collaboratory for Advanced Computing & Simulations Depts. of Computer Science, Physics & Astronomy, Chemical Engineering & Materials Science, and Biological
Exchange Correlation Functional Investigation of RT-TDDFT on a Sodium Chloride. Dimer. Philip Straughn
 Exchange Correlation Functional Investigation of RT-TDDFT on a Sodium Chloride Dimer Philip Straughn Abstract Charge transfer between Na and Cl ions is an important problem in physical chemistry. However,
Exchange Correlation Functional Investigation of RT-TDDFT on a Sodium Chloride Dimer Philip Straughn Abstract Charge transfer between Na and Cl ions is an important problem in physical chemistry. However,
College of Chemistry, Peking University, Beijing, China. Fritz-Haber-Institut der MPG, Berlin, Germany
 KITP Program Excitations in Condensed Matter Localized and Itinerant States in a Unified Picture beyond Density Functional Theory Hong Jiang 1, Patrick Rinke 2 and Matthias Scheffler 2 1 College of Chemistry,
KITP Program Excitations in Condensed Matter Localized and Itinerant States in a Unified Picture beyond Density Functional Theory Hong Jiang 1, Patrick Rinke 2 and Matthias Scheffler 2 1 College of Chemistry,
Supporting information for Chemical and Electrochemical. Surfaces: Insights into the Mechanism and Selectivity from DFT.
 Electronic Supplementary Material (ESI) for RSC Advances. This journal is The Royal Society of Chemistry 2015 Supporting information for Chemical and Electrochemical Hydrogenation of CO 2 to hydrocarbons
Electronic Supplementary Material (ESI) for RSC Advances. This journal is The Royal Society of Chemistry 2015 Supporting information for Chemical and Electrochemical Hydrogenation of CO 2 to hydrocarbons
Electronic structure simulations of water solid interfaces
 Electronic structure simulations of water solid interfaces Angelos Michaelides London Centre for Nanotechnology & Department of Chemistry, University College London www.chem.ucl.ac.uk/ice Main co-workers:
Electronic structure simulations of water solid interfaces Angelos Michaelides London Centre for Nanotechnology & Department of Chemistry, University College London www.chem.ucl.ac.uk/ice Main co-workers:
A FRESH LOOK AT THE BAND-GAP PROBLEM IN DENSITY FUNCTIONAL THEORY
 A FRESH LOOK AT THE BAND-GAP PROBLEM IN DENSITY FUNCTIONAL THEORY JOHN P. PERDEW PHYSICS & CHEMISTRY, TEMPLE UNIVERSITY PHILADELPHIA, PENNSYLVANIA, USA SUPPORTED BY THE U.S. DEPARTMENT OF ENERGY, EFRC
A FRESH LOOK AT THE BAND-GAP PROBLEM IN DENSITY FUNCTIONAL THEORY JOHN P. PERDEW PHYSICS & CHEMISTRY, TEMPLE UNIVERSITY PHILADELPHIA, PENNSYLVANIA, USA SUPPORTED BY THE U.S. DEPARTMENT OF ENERGY, EFRC
Molecular Mechanics: The Ab Initio Foundation
 Molecular Mechanics: The Ab Initio Foundation Ju Li GEM4 Summer School 2006 Cell and Molecular Mechanics in BioMedicine August 7 18, 2006, MIT, Cambridge, MA, USA 2 Outline Why are electrons quantum? Born-Oppenheimer
Molecular Mechanics: The Ab Initio Foundation Ju Li GEM4 Summer School 2006 Cell and Molecular Mechanics in BioMedicine August 7 18, 2006, MIT, Cambridge, MA, USA 2 Outline Why are electrons quantum? Born-Oppenheimer
Independent electrons in an effective potential
 ABC of DFT Adiabatic approximation Independent electrons in an effective potential Hartree Fock Density Functional Theory MBPT - GW Density Functional Theory in a nutshell Every observable quantity of
ABC of DFT Adiabatic approximation Independent electrons in an effective potential Hartree Fock Density Functional Theory MBPT - GW Density Functional Theory in a nutshell Every observable quantity of
High-Capacity Hydrogen Storage in Metal-Free Organic Molecular Crystals. Abstract
 High-Capacity Hydrogen Storage in Metal-Free Organic Molecular Crystals Mina Yoon 1, 2 and Matthias Scheffler 1 1 Fritz-Haber-Institut der Max-Planck-Gesellschaft, Faradayweg 4-6, 14195 Berlin, Germany
High-Capacity Hydrogen Storage in Metal-Free Organic Molecular Crystals Mina Yoon 1, 2 and Matthias Scheffler 1 1 Fritz-Haber-Institut der Max-Planck-Gesellschaft, Faradayweg 4-6, 14195 Berlin, Germany
Big-Data Analytics for Materials Science: Concepts, Challenges, and Hype
 Big-Data Analytics for Materials Science: Concepts, Challenges, and Hype Matthias Scheffler (*) Fritz-Haber-Institut der Max-Planck-Gesellschaft, Berlin; http://th.fhi-berlin.mpg.de/ From the periodic
Big-Data Analytics for Materials Science: Concepts, Challenges, and Hype Matthias Scheffler (*) Fritz-Haber-Institut der Max-Planck-Gesellschaft, Berlin; http://th.fhi-berlin.mpg.de/ From the periodic
GEM4 Summer School OpenCourseWare
 GEM4 Summer School OpenCourseWare http://gem4.educommons.net/ http://www.gem4.org/ Lecture: Molecular Mechanics by Ju Li. Given August 9, 2006 during the GEM4 session at MIT in Cambridge, MA. Please use
GEM4 Summer School OpenCourseWare http://gem4.educommons.net/ http://www.gem4.org/ Lecture: Molecular Mechanics by Ju Li. Given August 9, 2006 during the GEM4 session at MIT in Cambridge, MA. Please use
Australian Journal of Basic and Applied Sciences
 AENSI Journals Australian Journal of Basic and Applied Sciences ISSN:1991-8178 Journal home page: www.ajbasweb.com Theoretical Study for the Effect of Hydroxyl Radical on the Electronic Properties of Cyclobutadiene
AENSI Journals Australian Journal of Basic and Applied Sciences ISSN:1991-8178 Journal home page: www.ajbasweb.com Theoretical Study for the Effect of Hydroxyl Radical on the Electronic Properties of Cyclobutadiene
Théorie de la fonctionnnelle de la densité avec séparation de portée pour les forces de van der Waals
 Théorie de la fonctionnnelle de la densité avec séparation de portée pour les forces de van der Waals Julien Toulouse 1 Iann Gerber 2, Georg Jansen 3, Andreas Savin 1, János Ángyán 4 1 Laboratoire de Chimie
Théorie de la fonctionnnelle de la densité avec séparation de portée pour les forces de van der Waals Julien Toulouse 1 Iann Gerber 2, Georg Jansen 3, Andreas Savin 1, János Ángyán 4 1 Laboratoire de Chimie
arxiv: v1 [cond-mat.mtrl-sci] 7 Nov 2011
![arxiv: v1 [cond-mat.mtrl-sci] 7 Nov 2011](/thumbs/91/106457150.jpg "arxiv: v1 [cond-mat.mtrl-sci] 7 Nov 2011") First Principles Study of Adsorption of O 2 on Al Surface with Hybrid Functionals Heng-Rui Liu, Hongjun Xiang and X. G. Gong 1 Key Laboratory for Computational Physical Sciences (MOE) and Surface Physics
First Principles Study of Adsorption of O 2 on Al Surface with Hybrid Functionals Heng-Rui Liu, Hongjun Xiang and X. G. Gong 1 Key Laboratory for Computational Physical Sciences (MOE) and Surface Physics
Progress & challenges with Luttinger-Ward approaches for going beyond DFT
 Progress & challenges with Luttinger-Ward approaches for going beyond DFT Sohrab Ismail-Beigi Yale University Dept. of Applied Physics and Physics & CRISP (NSF MRSEC) Ismail-Beigi, Phys. Rev. B (2010)
Progress & challenges with Luttinger-Ward approaches for going beyond DFT Sohrab Ismail-Beigi Yale University Dept. of Applied Physics and Physics & CRISP (NSF MRSEC) Ismail-Beigi, Phys. Rev. B (2010)
Outline. Introduction: graphene. Adsorption on graphene: - Chemisorption - Physisorption. Summary
 Outline Introduction: graphene Adsorption on graphene: - Chemisorption - Physisorption Summary 1 Electronic band structure: Electronic properties K Γ M v F = 10 6 ms -1 = c/300 massless Dirac particles!
Outline Introduction: graphene Adsorption on graphene: - Chemisorption - Physisorption Summary 1 Electronic band structure: Electronic properties K Γ M v F = 10 6 ms -1 = c/300 massless Dirac particles!
The LDA-1/2 method in exciting
 http://exciting-code.org The LDA-1/2 method in exciting Ronaldo Rodrigues Pelá Humboldt Universität zu Berlin Instituto Tecnológico de Aeronáutica Outline DFT-1/2 Exchange-correlation functionals Exact
http://exciting-code.org The LDA-1/2 method in exciting Ronaldo Rodrigues Pelá Humboldt Universität zu Berlin Instituto Tecnológico de Aeronáutica Outline DFT-1/2 Exchange-correlation functionals Exact
Adiabatic-connection fluctuation-dissipation density-functional theory based on range separation
 Adiabatic-connection fluctuation-dissipation density-functional theory based on range separation Julien Toulouse 1 I. Gerber 2, G. Jansen 3, A. Savin 1, W. Zhu 1, J. Ángyán 4 1 Laboratoire de Chimie Théorique,
Adiabatic-connection fluctuation-dissipation density-functional theory based on range separation Julien Toulouse 1 I. Gerber 2, G. Jansen 3, A. Savin 1, W. Zhu 1, J. Ángyán 4 1 Laboratoire de Chimie Théorique,
arxiv: v1 [cond-mat.mtrl-sci] 14 Jul 2018
![arxiv: v1 [cond-mat.mtrl-sci] 14 Jul 2018](/thumbs/82/85444144.jpg "arxiv: v1 [cond-mat.mtrl-sci] 14 Jul 2018") Re-thinking CO adsorption on transition-metal surfaces: Density-driven error? Abhirup Patra, 1 Jianwei Sun, 2 and John P. Perdew 1, 3 1 Department of Physics, Temple University, Philadelphia, PA 19122
Re-thinking CO adsorption on transition-metal surfaces: Density-driven error? Abhirup Patra, 1 Jianwei Sun, 2 and John P. Perdew 1, 3 1 Department of Physics, Temple University, Philadelphia, PA 19122
Band calculations: Theory and Applications
 Band calculations: Theory and Applications Lecture 2: Different approximations for the exchange-correlation correlation functional in DFT Local density approximation () Generalized gradient approximation
Band calculations: Theory and Applications Lecture 2: Different approximations for the exchange-correlation correlation functional in DFT Local density approximation () Generalized gradient approximation
André Schleife Department of Materials Science and Engineering
 André Schleife Department of Materials Science and Engineering Yesterday you (should have) learned this: http://upload.wikimedia.org/wikipedia/commons/e/ea/ Simple_Harmonic_Motion_Orbit.gif 1. deterministic
André Schleife Department of Materials Science and Engineering Yesterday you (should have) learned this: http://upload.wikimedia.org/wikipedia/commons/e/ea/ Simple_Harmonic_Motion_Orbit.gif 1. deterministic
DFT basée sur le théorème de fluctuation-dissipation avec séparation de portée pour les interactions de van der Waals
 DFT basée sur le théorème de fluctuation-dissipation avec séparation de portée pour les interactions de van der Waals Julien Toulouse 1 Iann Gerber 2, Georg Jansen 3, Andreas Savin 1, János Ángyán 4 1
DFT basée sur le théorème de fluctuation-dissipation avec séparation de portée pour les interactions de van der Waals Julien Toulouse 1 Iann Gerber 2, Georg Jansen 3, Andreas Savin 1, János Ángyán 4 1
ABC of ground-state DFT
 ABC of ground-state DFT Kieron Burke and Lucas Wagner Departments of Physics and of Chemistry, University of California, Irvine, CA 92697, USA July 31, 2014 Kieron (UC Irvine) ABC of ground-state DFT HoW
ABC of ground-state DFT Kieron Burke and Lucas Wagner Departments of Physics and of Chemistry, University of California, Irvine, CA 92697, USA July 31, 2014 Kieron (UC Irvine) ABC of ground-state DFT HoW
Intermolecular Forces in Density Functional Theory
 Intermolecular Forces in Density Functional Theory Problems of DFT Peter Pulay at WATOC2005: There are 3 problems with DFT 1. Accuracy does not converge 2. Spin states of open shell systems often incorrect
Intermolecular Forces in Density Functional Theory Problems of DFT Peter Pulay at WATOC2005: There are 3 problems with DFT 1. Accuracy does not converge 2. Spin states of open shell systems often incorrect
Combining quasiparticle energy calculations with exact-exchange density-functional theory
 Combining quasiparticle energy calculations with exact-exchange density-functional theory Patrick Rinke 1, Abdallah Qteish 1,2, Jörg Neugebauer 1,3,4, Christoph Freysoldt 1 and Matthias Scheffler 1 1 Fritz-Haber-Institut
Combining quasiparticle energy calculations with exact-exchange density-functional theory Patrick Rinke 1, Abdallah Qteish 1,2, Jörg Neugebauer 1,3,4, Christoph Freysoldt 1 and Matthias Scheffler 1 1 Fritz-Haber-Institut
Introduction to DFT and its Application to Defects in Semiconductors
 Introduction to DFT and its Application to Defects in Semiconductors Noa Marom Physics and Engineering Physics Tulane University New Orleans The Future: Computer-Aided Materials Design Can access the space
Introduction to DFT and its Application to Defects in Semiconductors Noa Marom Physics and Engineering Physics Tulane University New Orleans The Future: Computer-Aided Materials Design Can access the space
CO on Pt 111 puzzle: A possible solution
 JOURNAL OF CHEMICAL PHYSICS VOLUME 117, NUMBER 5 1 AUGUST 2002 CO on Pt 111 puzzle: A possible solution Ilya Grinberg, Yashar Yourdshahyan, and Andrew M. Rappe Department of Chemistry and Laboratory for
JOURNAL OF CHEMICAL PHYSICS VOLUME 117, NUMBER 5 1 AUGUST 2002 CO on Pt 111 puzzle: A possible solution Ilya Grinberg, Yashar Yourdshahyan, and Andrew M. Rappe Department of Chemistry and Laboratory for
Magnetism in transition metal oxides by post-dft methods
 Magnetism in transition metal oxides by post-dft methods Cesare Franchini Faculty of Physics & Center for Computational Materials Science University of Vienna, Austria Workshop on Magnetism in Complex
Magnetism in transition metal oxides by post-dft methods Cesare Franchini Faculty of Physics & Center for Computational Materials Science University of Vienna, Austria Workshop on Magnetism in Complex
Basics of DFT. Kieron Burke and Lucas Wagner. Departments of Physics and of Chemistry, University of California, Irvine, CA 92697, USA
 Basics of DFT Kieron Burke and Lucas Wagner Departments of Physics and of Chemistry, University of California, Irvine, CA 92697, USA October 10-19th, 2012 Kieron (UC Irvine) Basics of DFT Lausanne12 1
Basics of DFT Kieron Burke and Lucas Wagner Departments of Physics and of Chemistry, University of California, Irvine, CA 92697, USA October 10-19th, 2012 Kieron (UC Irvine) Basics of DFT Lausanne12 1
Quantum Monte Carlo Benchmarks Density Functionals: Si Defects
 Quantum Monte Carlo Benchmarks Density Functionals: Si Defects K P Driver, W D Parker, R G Hennig, J W Wilkins (OSU) C J Umrigar (Cornell), R Martin, E Batista, B Uberuaga (LANL), J Heyd, G Scuseria (Rice)
Quantum Monte Carlo Benchmarks Density Functionals: Si Defects K P Driver, W D Parker, R G Hennig, J W Wilkins (OSU) C J Umrigar (Cornell), R Martin, E Batista, B Uberuaga (LANL), J Heyd, G Scuseria (Rice)
ABC of ground-state DFT
 ABC of ground-state DFT Kieron Burke and Lucas Wagner Departments of Physics and of Chemistry, University of California, Irvine, CA 92697, USA January 5-9th, 2014 Kieron (UC Irvine) ABC of ground-state
ABC of ground-state DFT Kieron Burke and Lucas Wagner Departments of Physics and of Chemistry, University of California, Irvine, CA 92697, USA January 5-9th, 2014 Kieron (UC Irvine) ABC of ground-state
DFT calculations of NMR indirect spin spin coupling constants
 DFT calculations of NMR indirect spin spin coupling constants Dalton program system Program capabilities Density functional theory Kohn Sham theory LDA, GGA and hybrid theories Indirect NMR spin spin coupling
DFT calculations of NMR indirect spin spin coupling constants Dalton program system Program capabilities Density functional theory Kohn Sham theory LDA, GGA and hybrid theories Indirect NMR spin spin coupling
Spring College on Computational Nanoscience May Variational Principles, the Hellmann-Feynman Theorem, Density Functional Theor
 2145-25 Spring College on Computational Nanoscience 17-28 May 2010 Variational Principles, the Hellmann-Feynman Theorem, Density Functional Theor Stefano BARONI SISSA & CNR-IOM DEMOCRITOS Simulation Center
2145-25 Spring College on Computational Nanoscience 17-28 May 2010 Variational Principles, the Hellmann-Feynman Theorem, Density Functional Theor Stefano BARONI SISSA & CNR-IOM DEMOCRITOS Simulation Center
Electronic Structure and van der Waals Interactions in the Stability and Mobility of Point Defects in Semiconductors
 Electronic Structure and van der Waals Interactions in the Stability and Mobility of Point Defects in Semiconductors Wang Gao and Alexandre Tkatchenko Fritz-Haber-Institut der Max-Planck-Gesellschaft,
Electronic Structure and van der Waals Interactions in the Stability and Mobility of Point Defects in Semiconductors Wang Gao and Alexandre Tkatchenko Fritz-Haber-Institut der Max-Planck-Gesellschaft,
Supporting information for Polymer interactions with Reduced Graphene Oxide: Van der Waals binding energies of Benzene on defected Graphene
 Supporting information for Polymer interactions with Reduced Graphene Oxide: Van der Waals binding energies of Benzene on defected Graphene Mohamed Hassan, Michael Walter *,,, and Michael Moseler, Freiburg
Supporting information for Polymer interactions with Reduced Graphene Oxide: Van der Waals binding energies of Benzene on defected Graphene Mohamed Hassan, Michael Walter *,,, and Michael Moseler, Freiburg
An Introduction to the Theory of Crystalline Elemental Solids and their Surfaces
 An Introduction to the Theory of Crystalline Elemental Solids and their Surfaces Angelos Michaelides 1 and Matthias Scheffler 2 1 London Centre for Nanotechnology and Department of Chemistry, University
An Introduction to the Theory of Crystalline Elemental Solids and their Surfaces Angelos Michaelides 1 and Matthias Scheffler 2 1 London Centre for Nanotechnology and Department of Chemistry, University
Part III: Theoretical Surface Science Adsorption at Surfaces
 Technische Universität München Part III: Theoretical Surface Science Adsorption at Surfaces Karsten Reuter Lecture course: Solid State Theory Adsorption at surfaces (T,p) Phase II Phase I Corrosion Growth
Technische Universität München Part III: Theoretical Surface Science Adsorption at Surfaces Karsten Reuter Lecture course: Solid State Theory Adsorption at surfaces (T,p) Phase II Phase I Corrosion Growth
Periodic Trends in Properties of Homonuclear
 Chapter 8 Periodic Trends in Properties of Homonuclear Diatomic Molecules Up to now, we have discussed various physical properties of nanostructures, namely, two-dimensional - graphene-like structures:
Chapter 8 Periodic Trends in Properties of Homonuclear Diatomic Molecules Up to now, we have discussed various physical properties of nanostructures, namely, two-dimensional - graphene-like structures:
Basics of density-functional theory and fast guide to actual calculations Matthias Scheffler
 Basics of density-functional theory and fast guide to actual calculations Matthias Scheffler http://www.fhi-berlin.mpg.de/th/th.html I. From the many-particle problem to the Kohn-Sham functional II. From
Basics of density-functional theory and fast guide to actual calculations Matthias Scheffler http://www.fhi-berlin.mpg.de/th/th.html I. From the many-particle problem to the Kohn-Sham functional II. From
Introduction to Density Functional Theory
 1 Introduction to Density Functional Theory 21 February 2011; V172 P.Ravindran, FME-course on Ab initio Modelling of solar cell Materials 21 February 2011 Introduction to DFT 2 3 4 Ab initio Computational
1 Introduction to Density Functional Theory 21 February 2011; V172 P.Ravindran, FME-course on Ab initio Modelling of solar cell Materials 21 February 2011 Introduction to DFT 2 3 4 Ab initio Computational
Modeling Ultrafast Deactivation in Oligothiophenes via Nonadiabatic Dynamics
 Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 2015 Supplementary Data for Modeling Ultrafast Deactivation in Oligothiophenes via Nonadiabatic
Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 2015 Supplementary Data for Modeling Ultrafast Deactivation in Oligothiophenes via Nonadiabatic
2. Surface geometric and electronic structure: a primer
 2. Surface geometric and electronic structure: a primer 2.1 Surface crystallography 2.1.1. Crystal structures - A crystal structure is made up of two basic elements: lattice + basis Basis: Lattice: simplest
2. Surface geometric and electronic structure: a primer 2.1 Surface crystallography 2.1.1. Crystal structures - A crystal structure is made up of two basic elements: lattice + basis Basis: Lattice: simplest
The Gutzwiller Density Functional Theory
 The Gutzwiller Density Functional Theory Jörg Bünemann, BTU Cottbus I) Introduction 1. Model for an H 2 -molecule 2. Transition metals and their compounds II) Gutzwiller variational theory 1. Gutzwiller
The Gutzwiller Density Functional Theory Jörg Bünemann, BTU Cottbus I) Introduction 1. Model for an H 2 -molecule 2. Transition metals and their compounds II) Gutzwiller variational theory 1. Gutzwiller
OVERVIEW OF QUANTUM CHEMISTRY METHODS
 OVERVIEW OF QUANTUM CHEMISTRY METHODS Outline I Generalities Correlation, basis sets Spin II Wavefunction methods Hartree-Fock Configuration interaction Coupled cluster Perturbative methods III Density
OVERVIEW OF QUANTUM CHEMISTRY METHODS Outline I Generalities Correlation, basis sets Spin II Wavefunction methods Hartree-Fock Configuration interaction Coupled cluster Perturbative methods III Density
Ari P Seitsonen CNRS & Université Pierre et Marie Curie, Paris
 Self-organisation on noble metal surfaces Ari P Seitsonen CNRS & Université Pierre et Marie Curie, Paris Collaborations Alexandre Dmitriev, Nian Lin, Johannes Barth, Klaus Kern,... Thomas Greber, Jürg
Self-organisation on noble metal surfaces Ari P Seitsonen CNRS & Université Pierre et Marie Curie, Paris Collaborations Alexandre Dmitriev, Nian Lin, Johannes Barth, Klaus Kern,... Thomas Greber, Jürg
CO Adsorption Site Preference on Platinum: Charge Is the Essence
 Supporting Information CO Adsorption Site Preference on Platinum: Charge Is the Essence G.T. Kasun Kalhara Gunasooriya, and Mark Saeys *, Laboratory for Chemical Technology, Ghent University, Technologiepark
Supporting Information CO Adsorption Site Preference on Platinum: Charge Is the Essence G.T. Kasun Kalhara Gunasooriya, and Mark Saeys *, Laboratory for Chemical Technology, Ghent University, Technologiepark
The Nature of the Interlayer Interaction in Bulk. and Few-Layer Phosphorus
 Supporting Information for: The Nature of the Interlayer Interaction in Bulk and Few-Layer Phosphorus L. Shulenburger, A.D. Baczewski, Z. Zhu, J. Guan, and D. Tománek, Sandia National Laboratories, Albuquerque,
Supporting Information for: The Nature of the Interlayer Interaction in Bulk and Few-Layer Phosphorus L. Shulenburger, A.D. Baczewski, Z. Zhu, J. Guan, and D. Tománek, Sandia National Laboratories, Albuquerque,
Report on TS-vdW Method, and Code Development, and. Results
 Michael Pawley July, 21, 2010 Report on TS-vdW Method, and Code Development, and Results Introduction: Non-covalent Interactions have long been difficult to account for using Density Functional Theory(DFT),
Michael Pawley July, 21, 2010 Report on TS-vdW Method, and Code Development, and Results Introduction: Non-covalent Interactions have long been difficult to account for using Density Functional Theory(DFT),
Chris G. Van de Walle Materials Department, UCSB
 First-principles simulations of defects in oxides and nitrides Chris G. Van de Walle Materials Department, UCSB Acknowledgments: A. Janotti, J. Lyons, J. Varley, J. Weber (UCSB) P. Rinke (FHI), M. Scheffler
First-principles simulations of defects in oxides and nitrides Chris G. Van de Walle Materials Department, UCSB Acknowledgments: A. Janotti, J. Lyons, J. Varley, J. Weber (UCSB) P. Rinke (FHI), M. Scheffler
time (s) Present status of ab initio electronic structure calculations: length (m) Density functional theory
 Present status of ab initio electronic structure calculations: length (m) Density functional theory") Present status of ab initio electronic structure calculations: from the earth core to quantum dots to mad cow disease liquid 12 nm stress field at semiconductor nano structures Na Si Cl solid geophysics
Present status of ab initio electronic structure calculations: from the earth core to quantum dots to mad cow disease liquid 12 nm stress field at semiconductor nano structures Na Si Cl solid geophysics
Introduction to Density Functional Theory with Applications to Graphene Branislav K. Nikolić
 Introduction to Density Functional Theory with Applications to Graphene Branislav K. Nikolić Department of Physics and Astronomy, University of Delaware, Newark, DE 19716, U.S.A. http://wiki.physics.udel.edu/phys824
Introduction to Density Functional Theory with Applications to Graphene Branislav K. Nikolić Department of Physics and Astronomy, University of Delaware, Newark, DE 19716, U.S.A. http://wiki.physics.udel.edu/phys824
CCSD(T) benchmarks of non-equilibrium water clusters: the importance of monomer deformation
 benchmarks of non-equilibrium water clusters: the importance of monomer deformation") CCSD(T) benchmarks of non-equilibrium water clusters: the importance of monomer deformation Biswajit Santra 1, Angelos Michaelides 1,2, and Matthias Scheffler 1 1 Fritz-Haber-Institut der MPG, Berlin,
CCSD(T) benchmarks of non-equilibrium water clusters: the importance of monomer deformation Biswajit Santra 1, Angelos Michaelides 1,2, and Matthias Scheffler 1 1 Fritz-Haber-Institut der MPG, Berlin,
Supplemental Material: Experimental and Theoretical Investigations of the Electronic Band Structure of Metal-Organic Framework of HKUST-1 Type
 Supplemental Material: Experimental and Theoretical Investigations of the Electronic Band Structure of Metal-Organic Framework of HKUST-1 Type Zhigang Gu, a Lars Heinke, a,* Christof Wöll a, Tobias Neumann,
Supplemental Material: Experimental and Theoretical Investigations of the Electronic Band Structure of Metal-Organic Framework of HKUST-1 Type Zhigang Gu, a Lars Heinke, a,* Christof Wöll a, Tobias Neumann,
Variational Monte Carlo Optimization and Excited States
 Variational Monte Carlo Optimization and Excited States Eric Neuscamman August 9, 2018 motivation charge transfer core spectroscopy double excitations the menu aperitif: number counting Jastrows main course:
Variational Monte Carlo Optimization and Excited States Eric Neuscamman August 9, 2018 motivation charge transfer core spectroscopy double excitations the menu aperitif: number counting Jastrows main course:
Adsorption and dissociation of CO on Fe(110) from first principles
 from first principles") Surface Science 570 (2004) 167 177 www.elsevier.com/locate/susc Adsorption and dissociation of CO on Fe(110) from first principles D.E. Jiang, Emily A. Carter * Department of Chemistry and Biochemistry,
Surface Science 570 (2004) 167 177 www.elsevier.com/locate/susc Adsorption and dissociation of CO on Fe(110) from first principles D.E. Jiang, Emily A. Carter * Department of Chemistry and Biochemistry,
Prerequisites for reliable modeling with first-principles methods. P. Kratzer Fritz-Haber-Institut der MPG D Berlin-Dahlem, Germany
 Prerequisites for reliable modeling with first-principles methods P. Kratzer Fritz-Haber-Institut der MPG D-14195 Berlin-Dahlem, Germany Prerequisites for modeling (I) Issues to consider when applying
Prerequisites for reliable modeling with first-principles methods P. Kratzer Fritz-Haber-Institut der MPG D-14195 Berlin-Dahlem, Germany Prerequisites for modeling (I) Issues to consider when applying
Design of Efficient Catalysts with Double Transition Metal. Atoms on C 2 N Layer
 Supporting Information Design of Efficient Catalysts with Double Transition Metal Atoms on C 2 N Layer Xiyu Li, 1, Wenhui Zhong, 2, Peng Cui, 1 Jun Li, 1 Jun Jiang 1, * 1 Hefei National Laboratory for
Supporting Information Design of Efficient Catalysts with Double Transition Metal Atoms on C 2 N Layer Xiyu Li, 1, Wenhui Zhong, 2, Peng Cui, 1 Jun Li, 1 Jun Jiang 1, * 1 Hefei National Laboratory for
Many electrons: Density functional theory Part II. Bedřich Velický VI.
 Many electrons: Density functional theory Part II. Bedřich Velický velicky@karlov.mff.cuni.cz VI. NEVF 514 Surface Physics Winter Term 013-014 Troja 1 st November 013 This class is the second devoted to
Many electrons: Density functional theory Part II. Bedřich Velický velicky@karlov.mff.cuni.cz VI. NEVF 514 Surface Physics Winter Term 013-014 Troja 1 st November 013 This class is the second devoted to
QUANTUM CHEMISTRY FOR TRANSITION METALS
 QUANTUM CHEMISTRY FOR TRANSITION METALS Outline I Introduction II Correlation Static correlation effects MC methods DFT III Relativity Generalities From 4 to 1 components Effective core potential Outline
QUANTUM CHEMISTRY FOR TRANSITION METALS Outline I Introduction II Correlation Static correlation effects MC methods DFT III Relativity Generalities From 4 to 1 components Effective core potential Outline
Density functional theory (DFT) has revolutionized the role
 has revolutionized the role") Doubly hybrid density functional for accurate descriptions of nonbond interactions, thermochemistry, and thermochemical kinetics Ying Zhang a, Xin Xu a,1, and William A. Goddard III b,1 a Department of
Doubly hybrid density functional for accurate descriptions of nonbond interactions, thermochemistry, and thermochemical kinetics Ying Zhang a, Xin Xu a,1, and William A. Goddard III b,1 a Department of
Range-separated density-functional theory with long-range random phase approximation
 Range-separated density-functional theory with long-range random phase approximation Julien Toulouse 1 Wuming Zhu 1, Andreas Savin 1, János Ángyán2 1 Laboratoire de Chimie Théorique, UPMC Univ Paris 6
Range-separated density-functional theory with long-range random phase approximation Julien Toulouse 1 Wuming Zhu 1, Andreas Savin 1, János Ángyán2 1 Laboratoire de Chimie Théorique, UPMC Univ Paris 6
Van der Waals interactions in DFT
 Van der Waals interactions in DFT Maxime Dion*, Aaron Puzder*, T. Thonhauser,* Valentino R. Cooper*, Shen Li*, Eamonn Murray, Lingzhu Kong, Kyuho Lee, and David C. Langreth Department of Physics and Astronomy,
Van der Waals interactions in DFT Maxime Dion*, Aaron Puzder*, T. Thonhauser,* Valentino R. Cooper*, Shen Li*, Eamonn Murray, Lingzhu Kong, Kyuho Lee, and David C. Langreth Department of Physics and Astronomy,