Ab Initio modelling of structural and electronic. Matt Probert University of York
|
|
|
- Derek Scott Craig
- 6 years ago
- Views:
Transcription
1 Ab Initio modelling of structural and electronic properties of semiconductors Matt Probert University of York
2 Overview of Talk What is Ab Initio? What can we model? How does it all work? Semiconductor examples Summary
3 Ab Initio = From the beginning What is Ab Initio? i.e. first principles (quantum mechanics) no fitting to experimental data In quantum chemistry, usually taken to mean Hartree-Fock theory or beyond (eg MP2, CI, etc) In physics, usually taken to mean DFT or beyond (eg GW, QMC, etc)
4 Advantages of Ab Initio High accuracy and predictive ability not limited by the fitting data-set can cope with unusual environments, and bond breaking/making wide range of properties can be calculated BUT computationally expensive limited in size of system can study need powerful computers
5 Which Ab Initio? In this talk, will focus in particular on DFT Density Functional Theory widely used see many of the talks in rest of workshop good balance of accuracy and speed has some limitations one uncontrolled approximation at its heart GW uses many-body perturbation theory to improve excited state properties QMC avoids this altogether
6 Quantum Mechanics and Density Functional Theory
7 Quantum Mechanics General Time Dependent Schrödinger Equation: Atomic Time Independent Schrödinger Equation: t i V z y x m Atomic Time Independent Schrödinger Equation: where N=Nuclei at positions {R}, e=electrons at positions {r}, e is the eigenenergy and i J e N e N i J e N e N H r R r R,, ˆ,,,, e e e N N N e N e N V V V T T H ˆ ˆ ˆ ˆ ˆ ˆ,
8 Density Functional Theory (I) Uses Born-Oppenheimer Approximation decouples electron and nuclear problems assumes electrons respond instantly to the current nuclear coordinates Uses charge density n(r) instead of manybody wavefunction Y(r 1,r 2 r N ) Hohenberg-Kohn proved 2 key theorems: 1. total energy is a unique functional of n(r) 2. the density which minimises the energy is the ground state density and the minimum energy is the ground state energy
9 Density Functional Theory (II) Hohenberg-Kohn (1964) proved that there was a universal functional E[n] which could be minimised to obtain the exact groundstate density and energy but not what the form of E[n] was! Kohn-Sham (1965) derived an expression for E[n] with one key unknown the exchange-correlation functional E xc [n] and introduced the Local Density Approximation (LDA)
10 Do not know the unknown functional DFT Problems write in terms of things we do know, e.g. electronelectron interaction and hope the things we do not know are small and hence can be easily approximated hence the LDA etc. Do not know how to calculate the Kinetic Energy T of a density! only for a wavefunction so introduce fictitious single-particle orbitals that give the correct density and for which a Kinetic Energy can be calculated.
11 Kohn-Sham Equations Write density in terms of KS orbitals Hence KS equations: ) ( ) ( ) ( ' ) ' ( ) ( 1 2 r r r r r r V d n V N i i n 2 ) ( ) ( r r where and E xc is the Exchange-Correlation functional the only approximation! ) ( ) ( ) ( ' ' ) ' ( ) ( r r r r r r r r i i i ext V xc d n V ) ( ] [ ) ( r r n n E V xc xc N atoms ext Z V R r
12 Exchange-Correlation Functional DFT is formally exact but in practice we need to approximate E xc the LDA assumes that E xc [n(r)] at some point r is the same as if every point had the same density hence LDA is fitted to Homogenous Electron Gas data calculated with high level QM methods expected to be good for metals but actually works well for many systems including semiconductors tends to overbind energies, shorten bonds can also use density gradient information, i.e. Generalised Gradient Approximation (GGA) various GGAs available, all tend to underbind can also use exact exchange from HF theory to create hybrid functionals but not so rigorous see talks on Thursday
13 Many-body QM is hard Why Bother? It is impossible to solve the Schrödinger equation exactly for all but the most simple problems Numerical approaches expand unknown wavefunction in terms of known basis functions and unknown coefficients a simple spin system with 20 particles needs 2 20 coefficients (spin up & down) exponential scaling is bad! DFT is easy uses a 3D scalar field instead much better!
14 What Do You Get? Key output of any DFT calculation is the ground state energy and density of the electrons hence can get total energy of system at T=0 can extend to finite temperature With other ab initio methods can also get spectrum of excited states
15 What Can We Model?
16 Ground State Properties Many experimental measurements can be derived from ground state total energy and how it varies with some parameter
17 Forces and Stresses Classically, can derive forces, stresses, from derivative of energy Hellman-Feynman theorem shows how to R F U U do this with QM: and so for a position-independent basis set R R F H E R R R R H H H E
18 Implications If we have analytic forces then it is much more efficient to calculate optimal bonding arrangements Ditto stresses and cell parameters Can also extend this to molecular dynamics time and/or temperature variation And can extend general approach to do linear response (e.g. phonons, electric field response, magnetic field response, etc)
19 Introducing CASTEP CASTEP is an example of a general purpose DFT code uses plane-wave basis set (position independent, easy to improve accuracy) use pseudopotentials (replaces nuclei and inner electrons with pseudo-ion) can calculate wide range of properties will feature in some talks tomorrow freely available to UK academics
20 Total energies CASTEP Abilities forces and stresses, with LDA/GGA/sX/EXX/LDA+U/ etc. Electronic structure electronic charge, potential, band structure, DOS, atomic populations Geometry Optimisation atomic positions, cell parameters, external pressure/stress Molecular dynamics finite temperature, zero-point and non-equilibrium properties Transition state searches chemical reaction pathways, diffusion barriers Phonons perturbation theory, finite differences Electric field response polarisability, dielectric constants, Born charges, LO/TO splitting Magnetic Response NMR, Chemical shifts, electric field gradients, hyperfine constants, etc. ELNES, EELS, Raman, Wannier Functions, and more


21 2eV indirect bandgap CaS 2 TeP
22 How Does It All Work? So what is going on in CASTEP or other plane-wave DFT codes? To get accurate answers from such codes, we need to understand a little about what is going on. There are several key parameters that must be carefully converged before good quality answers will emerge!
23 Basis Sets, Periodic Systems, Bloch s Theorem, k-points and Supercells
24 Plane Waves The Kohn-Sham equations can be written as 2 V r r 1 2 We are interested in periodic systems eff hence expand the K-S orbitals in terms of a periodic basis set: i i i ( r) j cg, j g e ig.r
25 Basic Crystallography The atomic structure of a crystal can be described by the unit cell periodically repeated along the lattice vectors. The smallest cell is spanned by the primitive vectors: a 1, a 2 and a 3 that connect the nearest neighbors. Any point in the lattice is connected to another point by a lattice vector: R=n 1 a 1 + n 2 a 2 +n 3 a 3 The unit cell of face centered cubic (FCC) system, with the primitive cell also shown.
 u ( r) e nk k i kr k Bloch s Theorem where u nk (r) = u nk (r + R) is a periodic function that fits inside the unit cell and e ik.r is a complex phase-factor.")
26 The potential in the crystal is periodic V(r + R) =V(r) so any crystal Hamiltonian is periodic: 1 2 V ( r ) 2 k The wave functions Y nk in a crystal are then quasi-periodic and can be written as: nk ( r) u ( r) e nk k i kr k Bloch s Theorem where u nk (r) = u nk (r + R) is a periodic function that fits inside the unit cell and e ik.r is a complex phase-factor. FCC unit cell
27 Reciprocal Space The set of wave vectors {g} that give plane waves with the wavelength having the periodicity of a given lattice with lattice vectors {R} is defined as the reciprocal space: ig. R e 1 The reciprocal basis vectors are defined by: b l 2 a a ( a l m m a a A reciprocal lattice vector can be written as a linear combination of the basis vectors: g g b g b n n ) g3 b 3
28 The Brillouin Zone The region in reciprocal space that is closer to the reference lattice point than any other reciprocal lattice point is defined as the first Brillouin Zone: The first Brillouin zone in a 2D rectangular lattice. The first Brillouin zone for the 3D FCC lattice, Bouckaert et al., Phys. Rev 50, 58 (1938).
29 Plane Waves Revisited Hence the set of plane waves is finite: r i( gk). r k( ) ck, ge g with the smallest g given by the Brillouin zone, and the longest g determined by the planewave cutoff energy, E cut : g k 2 2 E cut which therefore defines a length-scale for smallest features captured in the calculation: E cut
30 Si8 Convergence with E cut Tota al Energy (ev) Cut-off Energy (ev) Variational principle monotonic decrease in E total with E cut
31 Band Structure The solutions to the free electron Schrödinger equation: 1 2 are plane waves: 2 k ( r) k ) e r i k. r k k k (k) (ev) with the eigenvalues ( k) k 2 2 k (Å -1 ) The energy of the electrons has a quadratic dispersion (k dependence).
32 Zone Schemes All properties of the crystal can be described in the first Brillouin zone. Fold the bands outside the first Brillouin zone back inside the first Brillouin zone. The extended zone scheme The folded zone scheme (k) (ev) First BZ Second BZ First BZ
33 BZ Integration Many observables are calculated as an integral over all k- points within the 1st Brillouin zone, for example: Etot E( k) d k 3 n( r) nk( r) d k V V BZ 1stBZ 1stBZ So must make sure have enough sampling points in the 1 st BZ (k-points) for convergence a non-variational parameter so E total may go up as well as down as the density of points is increased standard method is a regular Monkhorst-Pack grid must be dense for metals to capture discontinuity in occupation of bands at E=E f hence use of smearing can use crystal symmetry to reduce the number of points BZ
34 Band Structure of Si Silicon has diamond structure. Bouckaert et al., Phys. Rev 50, 58 (1938). High symmetry points in the Brillouin zone: G=center of the Brillouin zone L=mid point on the zone boundary plane in the {111}-directions K=mid point on the edge between two hexagons {110}-direction X= mid point on the zone boundary plane in the {100}-direction
35 Band Structure of Si Primitive cell of Si has 2 atoms and hence four valence electrons: four bands below the Fermi level. Indirect bandgap.
36 Supercell Approximation What if want to calculate properties of a crystal defect? or an isolated molecule? or a surface? Use a supercell e.g. put 1 defect into a 2x2x2 cell e.g. add vacuum around molecule e.g. add vacuum above surface


37 Nanotube Supercell

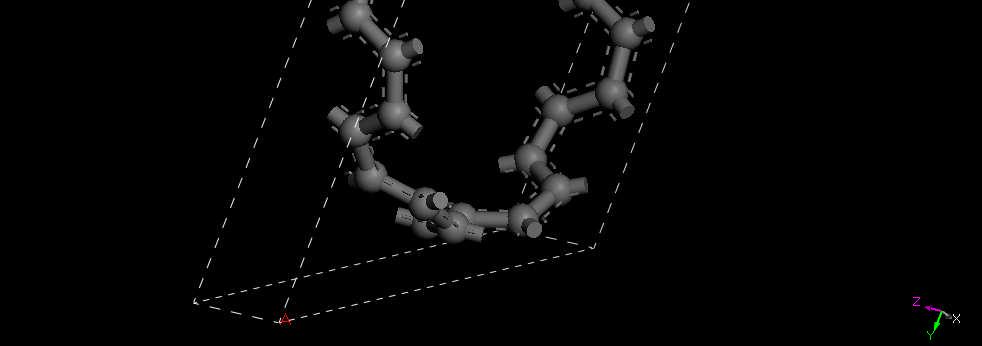
38 Nanotube Primitive Cell

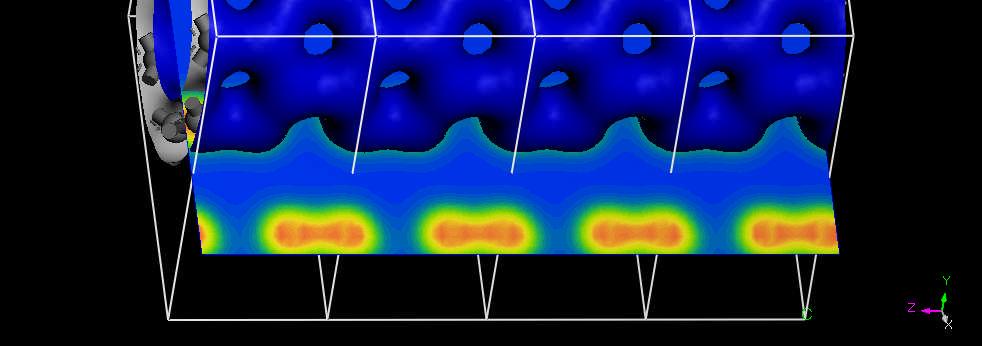
39 Nanotube Charge Density
40 Key Ideas Summary DFT replaces many-body wavefunction with density and Kohn-Sham equation LDA the simplest approximation to the unknown physics in the Kohn-Sham equation Bands single particle solutions to K-S equation Plane wave basis set bands represented in reciprocal space with cut-off energy K-points integration grid in 1st Brillouin zone Pseudopotentials a smooth representation of the nucleus and core electrons Supercells enable you to study non-periodic systems
41 Example 1 Si(100) Surface Reconstruction




42 Set up Supercell Si(100) supercell with vacuum gap with added hydrogen passivation 9 layers of Silicon and 9 Ǻ vacuum
=best lit.")
43 Convergence Tests Converge cut-off energy 370 ev Converge k-point sampling 9 k-points Converge number of bulk layers 9 layers Converge vacuum gap 9 Å only then see asymmetric dimerisation: (x)=best lit. value


44 Si(100) The Movie
45 Example 2 Si Vacancy
46 Si Vacancy Simplest example of a point defect But there have been many different values given in literature, using DFT, for neutral defect formation energy: N 1 EV EN 1 EN 1 N Why? A detailed study showed various factors responsible, including long-range relaxations need large supercells shallow PES around vacancy high sensitivity to noise in forces and k-point sampling
47 System Size Dependence Clear effect of supercell symmetry due to (spurious) longrange vacancy interaction between periodic images. Comparison is with all atoms in unrelaxed configuration. Convergence confirmed with 864 atom BCC supercell

48 Long-range Interaction Charge density difference iso-surface for unrelaxed 216 and 215 atom SC supercells Clear effect of supercell symmetry on electronic structure spurious!
49 Structural Optimisation Spontaneous symmetry breaking during relaxation Jahn-Teller distortion lowering energy by 1.2 ev Initial state has Td-point symmetry, final has D2d
50 Long-Ranged Relaxation Convergence of the ionic displacement of successive shells of atoms, centred on the vacancy site, as move across the supercell.
51 Example 3 Si Bandstructure

52 XC Functionals CASTEP can do standard LDA and GGA functionals (PW91, PBE, rpbe, WC) fast and reasonably accurate for structures Lattice parameters Bulk modulus But details depend on choice of XC functional


53 LDA Overbinding Charge density difference plot of Si8 r(lda)-r(pw91)
54 XC Functionals But still the traditional problems with band structures LDA/GGA typically underestimate band-gap whereas Hartree-Fock overestimates gap hence demand for hybrid functionals Fundamental science what is true nature of XC-hole? Can we go beyond local/semi-local approximations?
55 sx-lda Fully non-local XC-functional Uses Thomas-Fermi screened exchange: E XNL * * r r 1 ktf r r ik ik jq r jq r drdr e 2 ikjq r r with LDA correlation Expensive with plane-waves! Requires a double-sum over bands, double-sum over k-points and a triple-sum over plane-waves! Clever FFT trick NB2NK2 NP log NP

56 Improved Bandstructure Si band-structure Solid=sX-LDA Dash=LDA See later talks for more details
57 Example 4 H in Si
58 Hydrogen Structures Stable / Metastable sites BC two-fold coordinated T four-fold coordinated Possible saddlepoint sites AB antibonding site C half-way to T H hexagonal (6fold) site
59 Spin Density Plot for Site Planes of silicon atoms seen edge-on BC site Spin density iso-surface due to single extra electron
60 Experimental Input Traditional T=0 calculations suggest is most stable but with small binding energy H is light and hence might be quite mobile even at room temperatures must include thermal effects Key experimental probe is msr which uses Muonium (Mu) instead of H with mass Mu ~ 1/9 Mass H and ZPM ~ 1/sqrt(mass) must include quantum effects such as zero-point motion and tunnelling etc. Yse Path Integral Molecular Dynamics (PIMD)
61 Molecular Dynamics ab initio Molecular Dynamics Use classical mechanics to move the atoms Born-Oppenheimer approximation decouples nucleus and electrons and have electrons always relaxed onto the instantaneous B-O surface but using forces and stresses derived from the electronic wavefunction hence ab initio MD can use to study dynamical properties or to simulate a statistical ensemble (e.g. NVE, NVT, NPH or NPT) with various thermostats and/or barostats, etc. But the nucleus is always treated classically hence no quantum fluctuations, tunneling, zero point motion, etc.
62 Path Integral MD Use Feynman Path Integral formulation of Quantum Mechanics for the nucleus now includes ZPM etc important for light defects and/or low temperatures beads on springs view with imaginary time axis computationally expensive! Path integral view of a single quantum particle.


63 PIMD Movie of H in Silicon
64 More Examples Briefly
65 Defect Level of H0 in Al2O3 CASTEP can also calculate electronic density of states across whole Brillouin Zone Or projected DOS onto any given atom
66 Magnetic Systems Can also do magnetic systems including some that are not natural! Example shows AFM state of BCC Iron
67 Optical Properties Can also calculate optical properties, such as dielectric constant and refractive index but best if apply a scissors to get good band gap!
68 Phonon Spectra of NaCl And hence can use harmonic approximation to calculate zeropoint energy, Helmholtz free energy, heat capacity, etc.
69 Equation of State TiO2 Which phase is stable at which pressure? calculate E(V) curve Forsterite is most stable of all the phases considered then Wadsleyite and then Ringwoodite as increase pressure Can use common-tangent to find transition pressure.
70 More Atomic/Bond Population analysis Transition state searches Electric field response polarisability, dielectric constants, Born charges, LO/TO splitting Magnetic Response chemical reaction pathways, diffusion barriers NMR, Chemical shifts, electric field gradients, hyperfine constants, etc. Spectroscopy ELNES, EELS, Raman, and more
71 Summary
72 Disadvantages of DFT It only applies to the electronic groundstate Have to use approximations to the true density functional or an electronic system in thermal equilibrium not possible to predict error in the value of any particular property not possible to systematically improve accuracy of calculation Might have to use a more accurate (and much more expensive) method such as GW or QMC good for excited states as well as ground state but no forces so have to use DFT structures
73 Advantages of DFT Kohn was awarded the Nobel prize for DFT makes it hard to criticise! It offers very good scaling of computational cost with system size. It allows calculations to be performed on large and complex systems. Given the very large number of DFT calculations, the likely property accuracy for many properties/systems is known.
74 Summary of CASTEP CASTEP is a robust and reliable implementation of DFT for periodic systems uses plane wave basis and ultrasoft pseudopotentials can be used to calculate ground state electron density and energy and hence many derived properties See for more
75 Useful References Hohenberg & Kohn, Phys. Rev. B 136, 864 (1964) Kohn & Sham, Phys. Rev. A 140, 1133 (1965) MC Payne et al., Rev. Mod. Phys 64, 1045 (1992) RM Martin, Electronic Structure: basic theory and practical methods, Cambridge University Press (2004) SJ Clark, MD Segall, CJ Pickard, PJ Hasnip, MIJ Probert, K Refson and MC Payne, First principles methods using CASTEP, Zeitschrift für Kristallographie 220, 567 (2005)
Electronic Structure Theory for Periodic Systems: The Concepts. Christian Ratsch
 Electronic Structure Theory for Periodic Systems: The Concepts Christian Ratsch Institute for Pure and Applied Mathematics and Department of Mathematics, UCLA Motivation There are 10 20 atoms in 1 mm 3
Electronic Structure Theory for Periodic Systems: The Concepts Christian Ratsch Institute for Pure and Applied Mathematics and Department of Mathematics, UCLA Motivation There are 10 20 atoms in 1 mm 3
Teoría del Funcional de la Densidad (Density Functional Theory)
") Teoría del Funcional de la Densidad (Density Functional Theory) Motivation: limitations of the standard approach based on the wave function. The electronic density n(r) as the key variable: Functionals
Teoría del Funcional de la Densidad (Density Functional Theory) Motivation: limitations of the standard approach based on the wave function. The electronic density n(r) as the key variable: Functionals
Dept of Mechanical Engineering MIT Nanoengineering group
 1 Dept of Mechanical Engineering MIT Nanoengineering group » To calculate all the properties of a molecule or crystalline system knowing its atomic information: Atomic species Their coordinates The Symmetry
1 Dept of Mechanical Engineering MIT Nanoengineering group » To calculate all the properties of a molecule or crystalline system knowing its atomic information: Atomic species Their coordinates The Symmetry
Geometry Optimisation
 Geometry Optimisation Matt Probert Condensed Matter Dynamics Group Department of Physics, University of York, UK http://www.cmt.york.ac.uk/cmd http://www.castep.org Motivation Overview of Talk Background
Geometry Optimisation Matt Probert Condensed Matter Dynamics Group Department of Physics, University of York, UK http://www.cmt.york.ac.uk/cmd http://www.castep.org Motivation Overview of Talk Background
The Plane-wave Pseudopotential Method
 The Plane-wave Pseudopotential Method k(r) = X G c k,g e i(g+k) r Chris J Pickard Electrons in a Solid Nearly Free Electrons Nearly Free Electrons Nearly Free Electrons Electronic Structures Methods Empirical
The Plane-wave Pseudopotential Method k(r) = X G c k,g e i(g+k) r Chris J Pickard Electrons in a Solid Nearly Free Electrons Nearly Free Electrons Nearly Free Electrons Electronic Structures Methods Empirical
DFT: Exchange-Correlation
 DFT: Exchange-Correlation Local functionals, exact exchange and other post-dft methods Paul Tulip Centre for Materials Physics Department of Physics University of Durham Outline Introduction What is exchange
DFT: Exchange-Correlation Local functionals, exact exchange and other post-dft methods Paul Tulip Centre for Materials Physics Department of Physics University of Durham Outline Introduction What is exchange
Multi-Scale Modeling from First Principles
 m mm Multi-Scale Modeling from First Principles μm nm m mm μm nm space space Predictive modeling and simulations must address all time and Continuum Equations, densityfunctional space scales Rate Equations
m mm Multi-Scale Modeling from First Principles μm nm m mm μm nm space space Predictive modeling and simulations must address all time and Continuum Equations, densityfunctional space scales Rate Equations
Electron energy loss spectroscopy (EELS)
") Electron energy loss spectroscopy (EELS) Phil Hasnip Condensed Matter Dynamics Group Department of Physics, University of York, U.K. http://www-users.york.ac.uk/~pjh503 Many slides courtesy of Jonathan
Electron energy loss spectroscopy (EELS) Phil Hasnip Condensed Matter Dynamics Group Department of Physics, University of York, U.K. http://www-users.york.ac.uk/~pjh503 Many slides courtesy of Jonathan
The electronic structure of materials 2 - DFT
 Quantum mechanics 2 - Lecture 9 December 19, 2012 1 Density functional theory (DFT) 2 Literature Contents 1 Density functional theory (DFT) 2 Literature Historical background The beginnings: L. de Broglie
Quantum mechanics 2 - Lecture 9 December 19, 2012 1 Density functional theory (DFT) 2 Literature Contents 1 Density functional theory (DFT) 2 Literature Historical background The beginnings: L. de Broglie
6: Plane waves, unit cells, k- points and all that
 The Nuts and Bolts of First-Principles Simulation 6: Plane waves, unit cells, k- points and all that Durham, 6th- 13th December 2001 CASTEP Developers Group with support from the ESF ψ k Network Overview
The Nuts and Bolts of First-Principles Simulation 6: Plane waves, unit cells, k- points and all that Durham, 6th- 13th December 2001 CASTEP Developers Group with support from the ESF ψ k Network Overview
DFT: Exchange-Correlation
 DFT: Local functionals, exact exchange and other post-dft methods Stewart Clark University of Outline Introduction What is exchange and correlation? Quick tour of XC functionals (Semi-)local: LDA, PBE,
DFT: Local functionals, exact exchange and other post-dft methods Stewart Clark University of Outline Introduction What is exchange and correlation? Quick tour of XC functionals (Semi-)local: LDA, PBE,
Density Functional Theory. Martin Lüders Daresbury Laboratory
 Density Functional Theory Martin Lüders Daresbury Laboratory Ab initio Calculations Hamiltonian: (without external fields, non-relativistic) impossible to solve exactly!! Electrons Nuclei Electron-Nuclei
Density Functional Theory Martin Lüders Daresbury Laboratory Ab initio Calculations Hamiltonian: (without external fields, non-relativistic) impossible to solve exactly!! Electrons Nuclei Electron-Nuclei
Electronic Structure Methodology 1
 Electronic Structure Methodology 1 Chris J. Pickard Lecture Two Working with Density Functional Theory In the last lecture we learnt how to write the total energy as a functional of the density n(r): E
Electronic Structure Methodology 1 Chris J. Pickard Lecture Two Working with Density Functional Theory In the last lecture we learnt how to write the total energy as a functional of the density n(r): E
First-Principles Vibrational spectroscopy and lattice dynamics of materials in the solid state
 First-Principles Vibrational spectroscopy and lattice dynamics of materials in the solid state Keith Refson Computational Science and Engineering Department STFC Rutherford Appleton Laboratory First principles
First-Principles Vibrational spectroscopy and lattice dynamics of materials in the solid state Keith Refson Computational Science and Engineering Department STFC Rutherford Appleton Laboratory First principles
Quantum (Path Integral) Molecular Dynamics
 Molecular Dynamics") Quantum (Path Integral) Molecular Dynamics Matt Probert Condensed Matter Dynamics Group Department of Physics, University of York, U.K. http://www-users.york.ac.uk/~mijp1 Overview of lecture n Background
Quantum (Path Integral) Molecular Dynamics Matt Probert Condensed Matter Dynamics Group Department of Physics, University of York, U.K. http://www-users.york.ac.uk/~mijp1 Overview of lecture n Background
Density Functional Theory
 Density Functional Theory Iain Bethune EPCC ibethune@epcc.ed.ac.uk Overview Background Classical Atomistic Simulation Essential Quantum Mechanics DFT: Approximations and Theory DFT: Implementation using
Density Functional Theory Iain Bethune EPCC ibethune@epcc.ed.ac.uk Overview Background Classical Atomistic Simulation Essential Quantum Mechanics DFT: Approximations and Theory DFT: Implementation using
Band Structure Calculations; Electronic and Optical Properties
 ; Electronic and Optical Properties Stewart Clark University of Outline Introduction to band structures Calculating band structures using Castep Calculating optical properties Examples results Some applications
; Electronic and Optical Properties Stewart Clark University of Outline Introduction to band structures Calculating band structures using Castep Calculating optical properties Examples results Some applications
Fundamentals and applications of Density Functional Theory Astrid Marthinsen PhD candidate, Department of Materials Science and Engineering
 Fundamentals and applications of Density Functional Theory Astrid Marthinsen PhD candidate, Department of Materials Science and Engineering Outline PART 1: Fundamentals of Density functional theory (DFT)
Fundamentals and applications of Density Functional Theory Astrid Marthinsen PhD candidate, Department of Materials Science and Engineering Outline PART 1: Fundamentals of Density functional theory (DFT)
Electronic structure, plane waves and pseudopotentials
 Electronic structure, plane waves and pseudopotentials P.J. Hasnip Spectroscopy Workshop 2009 We want to be able to predict what electrons and nuclei will do from first principles, without needing to know
Electronic structure, plane waves and pseudopotentials P.J. Hasnip Spectroscopy Workshop 2009 We want to be able to predict what electrons and nuclei will do from first principles, without needing to know
Practical calculations using first-principles QM Convergence, convergence, convergence
 Practical calculations using first-principles QM Convergence, convergence, convergence Keith Refson STFC Rutherford Appleton Laboratory September 18, 2007 Results of First-Principles Simulations..........................................................
Practical calculations using first-principles QM Convergence, convergence, convergence Keith Refson STFC Rutherford Appleton Laboratory September 18, 2007 Results of First-Principles Simulations..........................................................
Electronic band structure, sx-lda, Hybrid DFT, LDA+U and all that. Keith Refson STFC Rutherford Appleton Laboratory
 Electronic band structure, sx-lda, Hybrid DFT, LDA+U and all that Keith Refson STFC Rutherford Appleton Laboratory LDA/GGA DFT is good but... Naive LDA/GGA calculation severely underestimates band-gaps.
Electronic band structure, sx-lda, Hybrid DFT, LDA+U and all that Keith Refson STFC Rutherford Appleton Laboratory LDA/GGA DFT is good but... Naive LDA/GGA calculation severely underestimates band-gaps.
André Schleife Department of Materials Science and Engineering
 André Schleife Department of Materials Science and Engineering Yesterday you (should have) learned this: http://upload.wikimedia.org/wikipedia/commons/e/ea/ Simple_Harmonic_Motion_Orbit.gif 1. deterministic
André Schleife Department of Materials Science and Engineering Yesterday you (should have) learned this: http://upload.wikimedia.org/wikipedia/commons/e/ea/ Simple_Harmonic_Motion_Orbit.gif 1. deterministic
Practical Guide to Density Functional Theory (DFT)
") Practical Guide to Density Functional Theory (DFT) Brad Malone, Sadas Shankar Quick recap of where we left off last time BD Malone, S Shankar Therefore there is a direct one-to-one correspondence between
Practical Guide to Density Functional Theory (DFT) Brad Malone, Sadas Shankar Quick recap of where we left off last time BD Malone, S Shankar Therefore there is a direct one-to-one correspondence between
Many electrons: Density functional theory Part II. Bedřich Velický VI.
 Many electrons: Density functional theory Part II. Bedřich Velický velicky@karlov.mff.cuni.cz VI. NEVF 514 Surface Physics Winter Term 013-014 Troja 1 st November 013 This class is the second devoted to
Many electrons: Density functional theory Part II. Bedřich Velický velicky@karlov.mff.cuni.cz VI. NEVF 514 Surface Physics Winter Term 013-014 Troja 1 st November 013 This class is the second devoted to
Introduction to Density Functional Theory with Applications to Graphene Branislav K. Nikolić
 Introduction to Density Functional Theory with Applications to Graphene Branislav K. Nikolić Department of Physics and Astronomy, University of Delaware, Newark, DE 19716, U.S.A. http://wiki.physics.udel.edu/phys824
Introduction to Density Functional Theory with Applications to Graphene Branislav K. Nikolić Department of Physics and Astronomy, University of Delaware, Newark, DE 19716, U.S.A. http://wiki.physics.udel.edu/phys824
Joint ICTP-IAEA Workshop on Fusion Plasma Modelling using Atomic and Molecular Data January 2012
 2327-3 Joint ICTP-IAEA Workshop on Fusion Plasma Modelling using Atomic and Molecular Data 23-27 January 2012 Qunatum Methods for Plasma-Facing Materials Alain ALLOUCHE Univ.de Provence, Lab.de la Phys.
2327-3 Joint ICTP-IAEA Workshop on Fusion Plasma Modelling using Atomic and Molecular Data 23-27 January 2012 Qunatum Methods for Plasma-Facing Materials Alain ALLOUCHE Univ.de Provence, Lab.de la Phys.
Density Functional Theory (DFT)
") Density Functional Theory (DFT) An Introduction by A.I. Al-Sharif Irbid, Aug, 2 nd, 2009 Density Functional Theory Revolutionized our approach to the electronic structure of atoms, molecules and solid
Density Functional Theory (DFT) An Introduction by A.I. Al-Sharif Irbid, Aug, 2 nd, 2009 Density Functional Theory Revolutionized our approach to the electronic structure of atoms, molecules and solid
Three Most Important Topics (MIT) Today
 Today") Three Most Important Topics (MIT) Today Electrons in periodic potential Energy gap nearly free electron Bloch Theorem Energy gap tight binding Chapter 1 1 Electrons in Periodic Potential We now know the
Three Most Important Topics (MIT) Today Electrons in periodic potential Energy gap nearly free electron Bloch Theorem Energy gap tight binding Chapter 1 1 Electrons in Periodic Potential We now know the
Introduction to Density Functional Theory
 1 Introduction to Density Functional Theory 21 February 2011; V172 P.Ravindran, FME-course on Ab initio Modelling of solar cell Materials 21 February 2011 Introduction to DFT 2 3 4 Ab initio Computational
1 Introduction to Density Functional Theory 21 February 2011; V172 P.Ravindran, FME-course on Ab initio Modelling of solar cell Materials 21 February 2011 Introduction to DFT 2 3 4 Ab initio Computational
Chapter 3. The (L)APW+lo Method. 3.1 Choosing A Basis Set
APW+lo Method. 3.1 Choosing A Basis Set") Chapter 3 The (L)APW+lo Method 3.1 Choosing A Basis Set The Kohn-Sham equations (Eq. (2.17)) provide a formulation of how to practically find a solution to the Hohenberg-Kohn functional (Eq. (2.15)). Nevertheless
Chapter 3 The (L)APW+lo Method 3.1 Choosing A Basis Set The Kohn-Sham equations (Eq. (2.17)) provide a formulation of how to practically find a solution to the Hohenberg-Kohn functional (Eq. (2.15)). Nevertheless
Universities of Leeds, Sheffield and York
 promoting access to White Rose research papers Universities of Leeds, Sheffield and York http://eprints.whiterose.ac.uk/ Paper published in Zeitschrift für Kristallographie. White Rose Research Online
promoting access to White Rose research papers Universities of Leeds, Sheffield and York http://eprints.whiterose.ac.uk/ Paper published in Zeitschrift für Kristallographie. White Rose Research Online
Self-Consistent Implementation of Self-Interaction Corrected DFT and of the Exact Exchange Functionals in Plane-Wave DFT
 Self-Consistent Implementation of Self-Interaction Corrected DFT and of the Exact Exchange Functionals in Plane-Wave DFT Kiril Tsemekhman (a), Eric Bylaska (b), Hannes Jonsson (a,c) (a) Department of Chemistry,
Self-Consistent Implementation of Self-Interaction Corrected DFT and of the Exact Exchange Functionals in Plane-Wave DFT Kiril Tsemekhman (a), Eric Bylaska (b), Hannes Jonsson (a,c) (a) Department of Chemistry,
Phonon calculations with SCAN
 Workshop on the SCAN density functional: Fundamentals, practices, and extensions Temple university, Philadelphia May 18th, 2017 Hands-on tutorial 3 Phonon calculations with SCAN Yubo Zhang and Jianwei
Workshop on the SCAN density functional: Fundamentals, practices, and extensions Temple university, Philadelphia May 18th, 2017 Hands-on tutorial 3 Phonon calculations with SCAN Yubo Zhang and Jianwei
Minimal Update of Solid State Physics
 Minimal Update of Solid State Physics It is expected that participants are acquainted with basics of solid state physics. Therefore here we will refresh only those aspects, which are absolutely necessary
Minimal Update of Solid State Physics It is expected that participants are acquainted with basics of solid state physics. Therefore here we will refresh only those aspects, which are absolutely necessary
Geometry Optimization
 Geometry Optimization Matt Probert Condensed Matter Dynamics Group Department of Physics, University of York, U.K. http://www-users.york.ac.uk/~mijp1 Overview of lecture n Background Theory n Hellman-Feynman
Geometry Optimization Matt Probert Condensed Matter Dynamics Group Department of Physics, University of York, U.K. http://www-users.york.ac.uk/~mijp1 Overview of lecture n Background Theory n Hellman-Feynman
Structural Calculations phase stability, surfaces, interfaces etc
 Structural Calculations phase stability, surfaces, interfaces etc Keith Refson STFC Rutherford Appleton Laboratory September 19, 2007 Phase Equilibrium 2 Energy-Volume curves..................................................................
Structural Calculations phase stability, surfaces, interfaces etc Keith Refson STFC Rutherford Appleton Laboratory September 19, 2007 Phase Equilibrium 2 Energy-Volume curves..................................................................
MODULE 2: QUANTUM MECHANICS. Principles and Theory
 MODULE 2: QUANTUM MECHANICS Principles and Theory You are here http://www.lbl.gov/cs/html/exascale4energy/nuclear.html 2 Short Review of Quantum Mechanics Why do we need quantum mechanics? Bonding and
MODULE 2: QUANTUM MECHANICS Principles and Theory You are here http://www.lbl.gov/cs/html/exascale4energy/nuclear.html 2 Short Review of Quantum Mechanics Why do we need quantum mechanics? Bonding and
Introduction to first-principles modelling and CASTEP
 to first-principles modelling and Phil Hasnip to + Atomistic Simulations If we know what the bonding in a material is beforehand, then we can often find good expressions for the forces between atoms, e.g.
to first-principles modelling and Phil Hasnip to + Atomistic Simulations If we know what the bonding in a material is beforehand, then we can often find good expressions for the forces between atoms, e.g.
Institut Néel Institut Laue Langevin. Introduction to electronic structure calculations
 Institut Néel Institut Laue Langevin Introduction to electronic structure calculations 1 Institut Néel - 25 rue des Martyrs - Grenoble - France 2 Institut Laue Langevin - 71 avenue des Martyrs - Grenoble
Institut Néel Institut Laue Langevin Introduction to electronic structure calculations 1 Institut Néel - 25 rue des Martyrs - Grenoble - France 2 Institut Laue Langevin - 71 avenue des Martyrs - Grenoble
Electrochemistry project, Chemistry Department, November Ab-initio Molecular Dynamics Simulation
 Electrochemistry project, Chemistry Department, November 2006 Ab-initio Molecular Dynamics Simulation Outline Introduction Ab-initio concepts Total energy concepts Adsorption energy calculation Project
Electrochemistry project, Chemistry Department, November 2006 Ab-initio Molecular Dynamics Simulation Outline Introduction Ab-initio concepts Total energy concepts Adsorption energy calculation Project
Density Functional Theory for Electrons in Materials
 Density Functional Theory for Electrons in Materials Richard M. Martin Department of Physics and Materials Research Laboratory University of Illinois at Urbana-Champaign 1 Density Functional Theory for
Density Functional Theory for Electrons in Materials Richard M. Martin Department of Physics and Materials Research Laboratory University of Illinois at Urbana-Champaign 1 Density Functional Theory for
Introduction to First-Principles Method
 Joint ICTP/CAS/IAEA School & Workshop on Plasma-Materials Interaction in Fusion Devices, July 18-22, 2016, Hefei Introduction to First-Principles Method by Guang-Hong LU ( 吕广宏 ) Beihang University Computer
Joint ICTP/CAS/IAEA School & Workshop on Plasma-Materials Interaction in Fusion Devices, July 18-22, 2016, Hefei Introduction to First-Principles Method by Guang-Hong LU ( 吕广宏 ) Beihang University Computer
PBS: FROM SOLIDS TO CLUSTERS
 PBS: FROM SOLIDS TO CLUSTERS E. HOFFMANN AND P. ENTEL Theoretische Tieftemperaturphysik Gerhard-Mercator-Universität Duisburg, Lotharstraße 1 47048 Duisburg, Germany Semiconducting nanocrystallites like
PBS: FROM SOLIDS TO CLUSTERS E. HOFFMANN AND P. ENTEL Theoretische Tieftemperaturphysik Gerhard-Mercator-Universität Duisburg, Lotharstraße 1 47048 Duisburg, Germany Semiconducting nanocrystallites like
CLIMBING THE LADDER OF DENSITY FUNCTIONAL APPROXIMATIONS JOHN P. PERDEW DEPARTMENT OF PHYSICS TEMPLE UNIVERSITY PHILADELPHIA, PA 19122
 CLIMBING THE LADDER OF DENSITY FUNCTIONAL APPROXIMATIONS JOHN P. PERDEW DEPARTMENT OF PHYSICS TEMPLE UNIVERSITY PHILADELPHIA, PA 191 THANKS TO MANY COLLABORATORS, INCLUDING SY VOSKO DAVID LANGRETH ALEX
CLIMBING THE LADDER OF DENSITY FUNCTIONAL APPROXIMATIONS JOHN P. PERDEW DEPARTMENT OF PHYSICS TEMPLE UNIVERSITY PHILADELPHIA, PA 191 THANKS TO MANY COLLABORATORS, INCLUDING SY VOSKO DAVID LANGRETH ALEX
dynamics computer code, whiccan simulate the motion of atoms at a surface and We coembininewton's equation for the nuclei,
 DUI FILE COpy T DTIC ANNUAL LETTER REPORT BY 0. F. ELEC33 20KE AD-A224 069 P.1. FOR N00014-85-K-0442 U Theory of electronic states and formations energies of.defect comp exes, interstitial defects, and
DUI FILE COpy T DTIC ANNUAL LETTER REPORT BY 0. F. ELEC33 20KE AD-A224 069 P.1. FOR N00014-85-K-0442 U Theory of electronic states and formations energies of.defect comp exes, interstitial defects, and
1. Hydrogen atom in a box
 1. Hydrogen atom in a box Recall H atom problem, V(r) = -1/r e r exact answer solved by expanding in Gaussian basis set, had to solve secular matrix involving matrix elements of basis functions place atom
1. Hydrogen atom in a box Recall H atom problem, V(r) = -1/r e r exact answer solved by expanding in Gaussian basis set, had to solve secular matrix involving matrix elements of basis functions place atom
Outline. Introduction: graphene. Adsorption on graphene: - Chemisorption - Physisorption. Summary
 Outline Introduction: graphene Adsorption on graphene: - Chemisorption - Physisorption Summary 1 Electronic band structure: Electronic properties K Γ M v F = 10 6 ms -1 = c/300 massless Dirac particles!
Outline Introduction: graphene Adsorption on graphene: - Chemisorption - Physisorption Summary 1 Electronic band structure: Electronic properties K Γ M v F = 10 6 ms -1 = c/300 massless Dirac particles!
DFT EXERCISES. FELIPE CERVANTES SODI January 2006
 DFT EXERCISES FELIPE CERVANTES SODI January 2006 http://www.csanyi.net/wiki/space/dftexercises Dr. Gábor Csányi 1 Hydrogen atom Place a single H atom in the middle of a largish unit cell (start with a
DFT EXERCISES FELIPE CERVANTES SODI January 2006 http://www.csanyi.net/wiki/space/dftexercises Dr. Gábor Csányi 1 Hydrogen atom Place a single H atom in the middle of a largish unit cell (start with a
1 Density functional theory (DFT)
") 1 Density functional theory (DFT) 1.1 Introduction Density functional theory is an alternative to ab initio methods for solving the nonrelativistic, time-independent Schrödinger equation H Φ = E Φ. The
1 Density functional theory (DFT) 1.1 Introduction Density functional theory is an alternative to ab initio methods for solving the nonrelativistic, time-independent Schrödinger equation H Φ = E Φ. The
The CASTEP Story. Mike Payne. Cavendish Laboratory University of Cambridge
 The CASTEP Story Mike Payne Cavendish Laboratory University of Cambridge mcp1@cam.ac.uk Outline of Talk 1. The foundations of quantum mechanics. 2. The unfulfilled promise. 3. The new dawn. i. Density
The CASTEP Story Mike Payne Cavendish Laboratory University of Cambridge mcp1@cam.ac.uk Outline of Talk 1. The foundations of quantum mechanics. 2. The unfulfilled promise. 3. The new dawn. i. Density
DFT / SIESTA algorithms
 DFT / SIESTA algorithms Javier Junquera José M. Soler References http://siesta.icmab.es Documentation Tutorials Atomic units e = m e = =1 atomic mass unit = m e atomic length unit = 1 Bohr = 0.5292 Ang
DFT / SIESTA algorithms Javier Junquera José M. Soler References http://siesta.icmab.es Documentation Tutorials Atomic units e = m e = =1 atomic mass unit = m e atomic length unit = 1 Bohr = 0.5292 Ang
Key concepts in Density Functional Theory (II) Silvana Botti
 Silvana Botti") Kohn-Sham scheme, band structure and optical spectra European Theoretical Spectroscopy Facility (ETSF) CNRS - Laboratoire des Solides Irradiés Ecole Polytechnique, Palaiseau - France Temporary Address:
Kohn-Sham scheme, band structure and optical spectra European Theoretical Spectroscopy Facility (ETSF) CNRS - Laboratoire des Solides Irradiés Ecole Polytechnique, Palaiseau - France Temporary Address:
Solid State Theory: Band Structure Methods
 Solid State Theory: Band Structure Methods Lilia Boeri Wed., 11:15-12:45 HS P3 (PH02112) http://itp.tugraz.at/lv/boeri/ele/ Plan of the Lecture: DFT1+2: Hohenberg-Kohn Theorem and Kohn and Sham equations.
Solid State Theory: Band Structure Methods Lilia Boeri Wed., 11:15-12:45 HS P3 (PH02112) http://itp.tugraz.at/lv/boeri/ele/ Plan of the Lecture: DFT1+2: Hohenberg-Kohn Theorem and Kohn and Sham equations.
and strong interlayer quantum confinement
 Supporting Information GeP3: A small indirect band gap 2D crystal with high carrier mobility and strong interlayer quantum confinement Yu Jing 1,3, Yandong Ma 1, Yafei Li 2, *, Thomas Heine 1,3 * 1 Wilhelm-Ostwald-Institute
Supporting Information GeP3: A small indirect band gap 2D crystal with high carrier mobility and strong interlayer quantum confinement Yu Jing 1,3, Yandong Ma 1, Yafei Li 2, *, Thomas Heine 1,3 * 1 Wilhelm-Ostwald-Institute
All electron optimized effective potential method for solids
 All electron optimized effective potential method for solids Institut für Theoretische Physik Freie Universität Berlin, Germany and Fritz Haber Institute of the Max Planck Society, Berlin, Germany. 22
All electron optimized effective potential method for solids Institut für Theoretische Physik Freie Universität Berlin, Germany and Fritz Haber Institute of the Max Planck Society, Berlin, Germany. 22
An Approximate DFT Method: The Density-Functional Tight-Binding (DFTB) Method
 Method") Fakultät für Mathematik und Naturwissenschaften - Lehrstuhl für Physikalische Chemie I / Theoretische Chemie An Approximate DFT Method: The Density-Functional Tight-Binding (DFTB) Method Jan-Ole Joswig
Fakultät für Mathematik und Naturwissenschaften - Lehrstuhl für Physikalische Chemie I / Theoretische Chemie An Approximate DFT Method: The Density-Functional Tight-Binding (DFTB) Method Jan-Ole Joswig
Key concepts in Density Functional Theory (II)
") Kohn-Sham scheme and band structures European Theoretical Spectroscopy Facility (ETSF) CNRS - Laboratoire des Solides Irradiés Ecole Polytechnique, Palaiseau - France Present Address: LPMCN Université
Kohn-Sham scheme and band structures European Theoretical Spectroscopy Facility (ETSF) CNRS - Laboratoire des Solides Irradiés Ecole Polytechnique, Palaiseau - France Present Address: LPMCN Université
Basics of DFT applications to solids and surfaces
 Basics of DFT applications to solids and surfaces Peter Kratzer Physics Department, University Duisburg-Essen, Duisburg, Germany E-mail: Peter.Kratzer@uni-duisburg-essen.de Periodicity in real space and
Basics of DFT applications to solids and surfaces Peter Kratzer Physics Department, University Duisburg-Essen, Duisburg, Germany E-mail: Peter.Kratzer@uni-duisburg-essen.de Periodicity in real space and
OVERVIEW OF QUANTUM CHEMISTRY METHODS
 OVERVIEW OF QUANTUM CHEMISTRY METHODS Outline I Generalities Correlation, basis sets Spin II Wavefunction methods Hartree-Fock Configuration interaction Coupled cluster Perturbative methods III Density
OVERVIEW OF QUANTUM CHEMISTRY METHODS Outline I Generalities Correlation, basis sets Spin II Wavefunction methods Hartree-Fock Configuration interaction Coupled cluster Perturbative methods III Density
Prerequisites for reliable modeling with first-principles methods. P. Kratzer Fritz-Haber-Institut der MPG D Berlin-Dahlem, Germany
 Prerequisites for reliable modeling with first-principles methods P. Kratzer Fritz-Haber-Institut der MPG D-14195 Berlin-Dahlem, Germany Prerequisites for modeling (I) Issues to consider when applying
Prerequisites for reliable modeling with first-principles methods P. Kratzer Fritz-Haber-Institut der MPG D-14195 Berlin-Dahlem, Germany Prerequisites for modeling (I) Issues to consider when applying
First-principles calculations of structural, electronic and optical properties of HfZn 2
 ~ 1 ~ First-principles calculations of structural, electronic and optical properties of HfZn 2 Md. Atikur Rahman *1, Md. Afjalur Rahman 2, Md. Zahidur Rahaman 3 1, 2, 3 Department of Physics, Pabna University
~ 1 ~ First-principles calculations of structural, electronic and optical properties of HfZn 2 Md. Atikur Rahman *1, Md. Afjalur Rahman 2, Md. Zahidur Rahaman 3 1, 2, 3 Department of Physics, Pabna University
Ab initio Electronic Structure
 Ab initio Electronic Structure M. Alouani IPCMS, UMR 7504, Université Louis Pasteur, Strasbourg France http://www-ipcms.u-strasbg.fr In coll. with: B. Arnaud, O. Bengone, Y. Dappe, and S. Lebègue 1965
Ab initio Electronic Structure M. Alouani IPCMS, UMR 7504, Université Louis Pasteur, Strasbourg France http://www-ipcms.u-strasbg.fr In coll. with: B. Arnaud, O. Bengone, Y. Dappe, and S. Lebègue 1965
MD simulation: output
 Properties MD simulation: output Trajectory of atoms positions: e. g. diffusion, mass transport velocities: e. g. v-v autocorrelation spectrum Energies temperature displacement fluctuations Mean square
Properties MD simulation: output Trajectory of atoms positions: e. g. diffusion, mass transport velocities: e. g. v-v autocorrelation spectrum Energies temperature displacement fluctuations Mean square
Defects in Semiconductors
 Defects in Semiconductors Mater. Res. Soc. Symp. Proc. Vol. 1370 2011 Materials Research Society DOI: 10.1557/opl.2011. 771 Electronic Structure of O-vacancy in High-k Dielectrics and Oxide Semiconductors
Defects in Semiconductors Mater. Res. Soc. Symp. Proc. Vol. 1370 2011 Materials Research Society DOI: 10.1557/opl.2011. 771 Electronic Structure of O-vacancy in High-k Dielectrics and Oxide Semiconductors
DENSITY FUNCTIONAL THEORY FOR NON-THEORISTS JOHN P. PERDEW DEPARTMENTS OF PHYSICS AND CHEMISTRY TEMPLE UNIVERSITY
 DENSITY FUNCTIONAL THEORY FOR NON-THEORISTS JOHN P. PERDEW DEPARTMENTS OF PHYSICS AND CHEMISTRY TEMPLE UNIVERSITY A TUTORIAL FOR PHYSICAL SCIENTISTS WHO MAY OR MAY NOT HATE EQUATIONS AND PROOFS REFERENCES
DENSITY FUNCTIONAL THEORY FOR NON-THEORISTS JOHN P. PERDEW DEPARTMENTS OF PHYSICS AND CHEMISTRY TEMPLE UNIVERSITY A TUTORIAL FOR PHYSICAL SCIENTISTS WHO MAY OR MAY NOT HATE EQUATIONS AND PROOFS REFERENCES
Density Functional Theory Calculations of Defect Energies Using Supercells.
 Density Functional Theory Calculations of Defect Energies Using Supercells. Christopher WM Castleton School of Science and Technology, Nottingham Trent University, NG 8NS, UK E-mail: Christopher.Castleton@ntu.ac.uk
Density Functional Theory Calculations of Defect Energies Using Supercells. Christopher WM Castleton School of Science and Technology, Nottingham Trent University, NG 8NS, UK E-mail: Christopher.Castleton@ntu.ac.uk
Condensed matter physics FKA091
 Condensed matter physics FKA091 Ermin Malic Department of Physics Chalmers University of Technology Henrik Johannesson Department of Physics University of Gothenburg Teaching assistants: Roland Jago &
Condensed matter physics FKA091 Ermin Malic Department of Physics Chalmers University of Technology Henrik Johannesson Department of Physics University of Gothenburg Teaching assistants: Roland Jago &
Optical Properties of Solid from DFT
 Optical Properties of Solid from DFT 1 Prof.P. Ravindran, Department of Physics, Central University of Tamil Nadu, India & Center for Materials Science and Nanotechnology, University of Oslo, Norway http://folk.uio.no/ravi/cmt15
Optical Properties of Solid from DFT 1 Prof.P. Ravindran, Department of Physics, Central University of Tamil Nadu, India & Center for Materials Science and Nanotechnology, University of Oslo, Norway http://folk.uio.no/ravi/cmt15
Theoretical studies of EELS in carbon structures
 Theoretical studies of EELS in carbon structures Nobuyuki Fujita Submitted for the Degree of Master of Philosophy University of York Department of Physics October 2012 Abstract Graphene nano-ribbons (GNRs)
Theoretical studies of EELS in carbon structures Nobuyuki Fujita Submitted for the Degree of Master of Philosophy University of York Department of Physics October 2012 Abstract Graphene nano-ribbons (GNRs)
MD Thermodynamics. Lecture 12 3/26/18. Harvard SEAS AP 275 Atomistic Modeling of Materials Boris Kozinsky
 MD Thermodynamics Lecture 1 3/6/18 1 Molecular dynamics The force depends on positions only (not velocities) Total energy is conserved (micro canonical evolution) Newton s equations of motion (second order
MD Thermodynamics Lecture 1 3/6/18 1 Molecular dynamics The force depends on positions only (not velocities) Total energy is conserved (micro canonical evolution) Newton s equations of motion (second order
Algorithms and Computational Aspects of DFT Calculations
 Algorithms and Computational Aspects of DFT Calculations Part I Juan Meza and Chao Yang High Performance Computing Research Lawrence Berkeley National Laboratory IMA Tutorial Mathematical and Computational
Algorithms and Computational Aspects of DFT Calculations Part I Juan Meza and Chao Yang High Performance Computing Research Lawrence Berkeley National Laboratory IMA Tutorial Mathematical and Computational
First-principles studies of the structural, electronic, and optical properties of a novel thorium compound Rb 2 Th 7 Se 15
 First-principles studies of the structural, electronic, and optical properties of a novel thorium compound Rb 2 Th 7 Se 15 M.G. Brik 1 Institute of Physics, University of Tartu, Riia 142, Tartu 5114, Estonia
First-principles studies of the structural, electronic, and optical properties of a novel thorium compound Rb 2 Th 7 Se 15 M.G. Brik 1 Institute of Physics, University of Tartu, Riia 142, Tartu 5114, Estonia
Ab initio study of Mn doped BN nanosheets Tudor Luca Mitran
 Ab initio study of Mn doped BN nanosheets Tudor Luca Mitran MDEO Research Center University of Bucharest, Faculty of Physics, Bucharest-Magurele, Romania Oldenburg 20.04.2012 Table of contents 1. Density
Ab initio study of Mn doped BN nanosheets Tudor Luca Mitran MDEO Research Center University of Bucharest, Faculty of Physics, Bucharest-Magurele, Romania Oldenburg 20.04.2012 Table of contents 1. Density
Quantum Modeling of Solids: Basic Properties
 1.021, 3.021, 10.333, 22.00 : Introduction to Modeling and Simulation : Spring 2012 Part II Quantum Mechanical Methods : Lecture 7 Quantum Modeling of Solids: Basic Properties Jeffrey C. Grossman Department
1.021, 3.021, 10.333, 22.00 : Introduction to Modeling and Simulation : Spring 2012 Part II Quantum Mechanical Methods : Lecture 7 Quantum Modeling of Solids: Basic Properties Jeffrey C. Grossman Department
Spins and spin-orbit coupling in semiconductors, metals, and nanostructures
 B. Halperin Spin lecture 1 Spins and spin-orbit coupling in semiconductors, metals, and nanostructures Behavior of non-equilibrium spin populations. Spin relaxation and spin transport. How does one produce
B. Halperin Spin lecture 1 Spins and spin-orbit coupling in semiconductors, metals, and nanostructures Behavior of non-equilibrium spin populations. Spin relaxation and spin transport. How does one produce
College of Science, Xi an University of Science and Technology, Xi an *Corresponding author
 2016 International Conference on Advanced Manufacture Technology and Industrial Application (AMTIA 2016) ISBN: 978-1-60595-387-8 The Study of Coordination Adsorption Effect that CO Adsorption on 4H-SiC
2016 International Conference on Advanced Manufacture Technology and Industrial Application (AMTIA 2016) ISBN: 978-1-60595-387-8 The Study of Coordination Adsorption Effect that CO Adsorption on 4H-SiC
Density Functional Theory: from theory to Applications
 Density Functional Theory: from theory to Applications Uni Mainz November 29, 2010 The self interaction error and its correction Perdew-Zunger SIC Average-density approximation Weighted density approximation
Density Functional Theory: from theory to Applications Uni Mainz November 29, 2010 The self interaction error and its correction Perdew-Zunger SIC Average-density approximation Weighted density approximation
Optical Properties of Semiconductors. Prof.P. Ravindran, Department of Physics, Central University of Tamil Nadu, India
 Optical Properties of Semiconductors 1 Prof.P. Ravindran, Department of Physics, Central University of Tamil Nadu, India http://folk.uio.no/ravi/semi2013 Light Matter Interaction Response to external electric
Optical Properties of Semiconductors 1 Prof.P. Ravindran, Department of Physics, Central University of Tamil Nadu, India http://folk.uio.no/ravi/semi2013 Light Matter Interaction Response to external electric
DFT calculations of NMR indirect spin spin coupling constants
 DFT calculations of NMR indirect spin spin coupling constants Dalton program system Program capabilities Density functional theory Kohn Sham theory LDA, GGA and hybrid theories Indirect NMR spin spin coupling
DFT calculations of NMR indirect spin spin coupling constants Dalton program system Program capabilities Density functional theory Kohn Sham theory LDA, GGA and hybrid theories Indirect NMR spin spin coupling
Quantum Modeling of Solids: Basic Properties
 1.021, 3.021, 10.333, 22.00 : Introduction to Modeling and Simulation : Spring 2011 Part II Quantum Mechanical Methods : Lecture 5 Quantum Modeling of Solids: Basic Properties Jeffrey C. Grossman Department
1.021, 3.021, 10.333, 22.00 : Introduction to Modeling and Simulation : Spring 2011 Part II Quantum Mechanical Methods : Lecture 5 Quantum Modeling of Solids: Basic Properties Jeffrey C. Grossman Department
Lecture contents. A few concepts from Quantum Mechanics. Tight-binding model Solid state physics review
 Lecture contents A few concepts from Quantum Mechanics Particle in a well Two wells: QM perturbation theory Many wells (atoms) BAND formation Tight-binding model Solid state physics review Approximations
Lecture contents A few concepts from Quantum Mechanics Particle in a well Two wells: QM perturbation theory Many wells (atoms) BAND formation Tight-binding model Solid state physics review Approximations
2. Surface geometric and electronic structure: a primer
 2. Surface geometric and electronic structure: a primer 2.1 Surface crystallography 2.1.1. Crystal structures - A crystal structure is made up of two basic elements: lattice + basis Basis: Lattice: simplest
2. Surface geometric and electronic structure: a primer 2.1 Surface crystallography 2.1.1. Crystal structures - A crystal structure is made up of two basic elements: lattice + basis Basis: Lattice: simplest
The Gutzwiller Density Functional Theory
 The Gutzwiller Density Functional Theory Jörg Bünemann, BTU Cottbus I) Introduction 1. Model for an H 2 -molecule 2. Transition metals and their compounds II) Gutzwiller variational theory 1. Gutzwiller
The Gutzwiller Density Functional Theory Jörg Bünemann, BTU Cottbus I) Introduction 1. Model for an H 2 -molecule 2. Transition metals and their compounds II) Gutzwiller variational theory 1. Gutzwiller
Dept of Mechanical Engineering MIT Nanoengineering group
 1 Dept of Mechanical Engineering MIT Nanoengineering group » Recap of HK theorems and KS equations» The physical meaning of the XC energy» Solution of a one-particle Schroedinger equation» Pseudo Potentials»
1 Dept of Mechanical Engineering MIT Nanoengineering group » Recap of HK theorems and KS equations» The physical meaning of the XC energy» Solution of a one-particle Schroedinger equation» Pseudo Potentials»
MODULE 2: QUANTUM MECHANICS. Practice: Quantum ESPRESSO
 MODULE 2: QUANTUM MECHANICS Practice: Quantum ESPRESSO I. What is Quantum ESPRESSO? 2 DFT software PW-DFT, PP, US-PP, PAW http://www.quantum-espresso.org FREE PW-DFT, PP, PAW http://www.abinit.org FREE
MODULE 2: QUANTUM MECHANICS Practice: Quantum ESPRESSO I. What is Quantum ESPRESSO? 2 DFT software PW-DFT, PP, US-PP, PAW http://www.quantum-espresso.org FREE PW-DFT, PP, PAW http://www.abinit.org FREE
Preface Introduction to the electron liquid
 Table of Preface page xvii 1 Introduction to the electron liquid 1 1.1 A tale of many electrons 1 1.2 Where the electrons roam: physical realizations of the electron liquid 5 1.2.1 Three dimensions 5 1.2.2
Table of Preface page xvii 1 Introduction to the electron liquid 1 1.1 A tale of many electrons 1 1.2 Where the electrons roam: physical realizations of the electron liquid 5 1.2.1 Three dimensions 5 1.2.2
The Plane-Wave Pseudopotential Method
 Hands-on Workshop on Density Functional Theory and Beyond: Computational Materials Science for Real Materials Trieste, August 6-15, 2013 The Plane-Wave Pseudopotential Method Ralph Gebauer ICTP, Trieste
Hands-on Workshop on Density Functional Theory and Beyond: Computational Materials Science for Real Materials Trieste, August 6-15, 2013 The Plane-Wave Pseudopotential Method Ralph Gebauer ICTP, Trieste
Computational Methods. Chem 561
 Computational Methods Chem 561 Lecture Outline 1. Ab initio methods a) HF SCF b) Post-HF methods 2. Density Functional Theory 3. Semiempirical methods 4. Molecular Mechanics Computational Chemistry " Computational
Computational Methods Chem 561 Lecture Outline 1. Ab initio methods a) HF SCF b) Post-HF methods 2. Density Functional Theory 3. Semiempirical methods 4. Molecular Mechanics Computational Chemistry " Computational
Pseudopotentials for hybrid density functionals and SCAN
 Pseudopotentials for hybrid density functionals and SCAN Jing Yang, Liang Z. Tan, Julian Gebhardt, and Andrew M. Rappe Department of Chemistry University of Pennsylvania Why do we need pseudopotentials?
Pseudopotentials for hybrid density functionals and SCAN Jing Yang, Liang Z. Tan, Julian Gebhardt, and Andrew M. Rappe Department of Chemistry University of Pennsylvania Why do we need pseudopotentials?
Quantum Condensed Matter Physics Lecture 4
 Quantum Condensed Matter Physics Lecture 4 David Ritchie QCMP Lent/Easter 2019 http://www.sp.phy.cam.ac.uk/drp2/home 4.1 Quantum Condensed Matter Physics 1. Classical and Semi-classical models for electrons
Quantum Condensed Matter Physics Lecture 4 David Ritchie QCMP Lent/Easter 2019 http://www.sp.phy.cam.ac.uk/drp2/home 4.1 Quantum Condensed Matter Physics 1. Classical and Semi-classical models for electrons
References. Documentation Manuals Tutorials Publications
 References http://siesta.icmab.es Documentation Manuals Tutorials Publications Atomic units e = m e = =1 atomic mass unit = m e atomic length unit = 1 Bohr = 0.5292 Ang atomic energy unit = 1 Hartree =
References http://siesta.icmab.es Documentation Manuals Tutorials Publications Atomic units e = m e = =1 atomic mass unit = m e atomic length unit = 1 Bohr = 0.5292 Ang atomic energy unit = 1 Hartree =
Structure and Dynamics : An Atomic View of Materials
 Structure and Dynamics : An Atomic View of Materials MARTIN T. DOVE Department ofearth Sciences University of Cambridge OXFORD UNIVERSITY PRESS Contents 1 Introduction 1 1.1 Observations 1 1.1.1 Microscopic
Structure and Dynamics : An Atomic View of Materials MARTIN T. DOVE Department ofearth Sciences University of Cambridge OXFORD UNIVERSITY PRESS Contents 1 Introduction 1 1.1 Observations 1 1.1.1 Microscopic
Introduction to DFT and Density Functionals. by Michel Côté Université de Montréal Département de physique
 Introduction to DFT and Density Functionals by Michel Côté Université de Montréal Département de physique Eamples Carbazole molecule Inside of diamant Réf: Jean-François Brière http://www.phys.umontreal.ca/~michel_
Introduction to DFT and Density Functionals by Michel Côté Université de Montréal Département de physique Eamples Carbazole molecule Inside of diamant Réf: Jean-François Brière http://www.phys.umontreal.ca/~michel_
The electronic structure of materials 1
 Quantum mechanics 2 - Lecture 9 December 18, 2013 1 An overview 2 Literature Contents 1 An overview 2 Literature Electronic ground state Ground state cohesive energy equilibrium crystal structure phase
Quantum mechanics 2 - Lecture 9 December 18, 2013 1 An overview 2 Literature Contents 1 An overview 2 Literature Electronic ground state Ground state cohesive energy equilibrium crystal structure phase
Exchange Correlation Functional Investigation of RT-TDDFT on a Sodium Chloride. Dimer. Philip Straughn
 Exchange Correlation Functional Investigation of RT-TDDFT on a Sodium Chloride Dimer Philip Straughn Abstract Charge transfer between Na and Cl ions is an important problem in physical chemistry. However,
Exchange Correlation Functional Investigation of RT-TDDFT on a Sodium Chloride Dimer Philip Straughn Abstract Charge transfer between Na and Cl ions is an important problem in physical chemistry. However,
Electronic structure theory: Fundamentals to frontiers. 2. Density functional theory
 Electronic structure theory: Fundamentals to frontiers. 2. Density functional theory MARTIN HEAD-GORDON, Department of Chemistry, University of California, and Chemical Sciences Division, Lawrence Berkeley
Electronic structure theory: Fundamentals to frontiers. 2. Density functional theory MARTIN HEAD-GORDON, Department of Chemistry, University of California, and Chemical Sciences Division, Lawrence Berkeley
Intro to ab initio methods
 Lecture 2 Part A Intro to ab initio methods Recommended reading: Leach, Chapters 2 & 3 for QM methods For more QM methods: Essentials of Computational Chemistry by C.J. Cramer, Wiley (2002) 1 ab initio
Lecture 2 Part A Intro to ab initio methods Recommended reading: Leach, Chapters 2 & 3 for QM methods For more QM methods: Essentials of Computational Chemistry by C.J. Cramer, Wiley (2002) 1 ab initio
Lecture 4: Band theory
 Lecture 4: Band theory Very short introduction to modern computational solid state chemistry Band theory of solids Molecules vs. solids Band structures Analysis of chemical bonding in Reciprocal space
Lecture 4: Band theory Very short introduction to modern computational solid state chemistry Band theory of solids Molecules vs. solids Band structures Analysis of chemical bonding in Reciprocal space
Introduction to Density Functional Theory
 Introduction to Density Functional Theory S. Sharma Institut für Physik Karl-Franzens-Universität Graz, Austria 19th October 2005 Synopsis Motivation 1 Motivation : where can one use DFT 2 : 1 Elementary
Introduction to Density Functional Theory S. Sharma Institut für Physik Karl-Franzens-Universität Graz, Austria 19th October 2005 Synopsis Motivation 1 Motivation : where can one use DFT 2 : 1 Elementary
Pseudopotentials: design, testing, typical errors
 Pseudopotentials: design, testing, typical errors Kevin F. Garrity Part 1 National Institute of Standards and Technology (NIST) Uncertainty Quantification in Materials Modeling 2015 Parameter free calculations.
Pseudopotentials: design, testing, typical errors Kevin F. Garrity Part 1 National Institute of Standards and Technology (NIST) Uncertainty Quantification in Materials Modeling 2015 Parameter free calculations.