MODULE 2: QUANTUM MECHANICS. Principles and Theory
|
|
|
- Charity Norris
- 5 years ago
- Views:
Transcription
1 MODULE 2: QUANTUM MECHANICS Principles and Theory
2 You are here 2
3 Short Review of Quantum Mechanics Why do we need quantum mechanics? Bonding and structure Electronic, magnetic and optical properties of materials Chemistry and reactions Standard model of matter Matter consists of atoms Atoms consist of Massive, point-like nuclei (protons + neutrons) That are surrounded by tightly bound core electrons And held together in molecules, liquid and solids by the bonds formed by valence electrons 3
 + V (r, t) (r, t) =")
4 Short Review of Quantum Mechanics Wave-particle duality Planck s uncertainty relationship p = h Schrödinger equation Time dependent 2 2m 2 (r, t) + V (r, t) (r, t) = i (r, t) t 4
 = exp (i k r t) E k = 2 k 2 2m =")
5 Solutions of the Schrödinger Equation (1) Free particle V(r) = 0 Solutions are plane waves k(r, t) = exp (i k r t) E k = 2 k 2 2m = 5
6 Solutions of the Schrödinger Equation (2) Infinite square well Plane waves that vanish at the boundary V (x) = n (x) = E n = 0 if 0 < x < a otherwise 2 a sin n x a 2 2 n 2 2ma 2 Energy E n /E n = 4 n = 3 n = 2 n = 1!(x)! 1! 4! 3! a x 6
 Metal surface")

7 Solutions of the Schrödinger Equation (3) Metal surface Potential step Plane wave inside metal, exponential decay outside 7
8 Solutions for the Coulomb Potential Spherical Symmetry 2 = 2 x y z 2 = 1 r 2 r r 2 r + 1 r 2 sin sin r 2 sin 2 2 Separation of variables nlm(r) = R nl (r) Y lm (, ) Equation for radial wave functions 2 d 2 2m dr d l(l + 1) + r dr 2mr 2 + V (r) R nl (r) = E R nl (r) 8

9 The Periodic System of Elements 9
10 Basis Set Expansion (Matrix Formulation) Expand wave function in set of n orthogonal functions = k k n=1 c n n Plugging this solution into the Schrödinger equation yields and multiplying with function ϕm yields m H = E m n=1 c n m H n = E c m k H mn c n = E c m n=1 H 11 H 1k c 1... = E c 1... H k1 H kk c k c k 10
11 Variational Principle E[ ] = H E 0 E[ ] = E 0 If is the groundstate Example: The hydrogen atom = c exp( r) E[ ] = H 11
12 The Hydrogen Atom Ansatz: = c exp( r) Calculate: Use: = E[ ] = H r 2...d 3 r 0 1 r r n exp( ar) = n! a n+1 2 = 2 x y z 2 = 1 r 2 r r 2 r + 1 r 2 sin sin r 2 sin
13 The Hydrogen Atom Result: = c2 3, = c2 2, 1 r = c2 2 Write out the energy as a function of α and minimize with respect to α: E( )= c 2 c2 2 2 c2 3 = d d E( )= 1=0 13
14 Atomic Units Quantity Name Symbol SI value Energy Hartree energy Ha (75) J Length Bohr radius a (18) m Mass Electron rest mass me (16) kg Electric charge Elementary charge e (14) C Electrostatic force constant Coulombs constant 1/4πε C -2 Nm
15 The Many-Electron Problem 1 2 n i=1 2 i n i=1 Z r i + n 1 i=1 n j=i+1 1 r i r j (r 1,..., r n ) = E (r 1,..., r n ) Example: Fe atom Fe has 26 electrons wave function has 3 26 = 78 variables Store wave function on a grid Use a coarse grid of only 10 points along each direction To store wave function would require storage of numbers Single precision 1 number = 4 Bytes Compare that to all the data stored worldwide 1 zettabyte = Bytes 15
16 The Hartree Method Independent electron method Assume that electrons move independently of each other Each electron moves in an effective potential that consists of - Attraction of nuclei - Average repulsive interaction of other electrons Many-body wave function as product of single-particle orbitals (r 1, r 2,..., r n ) = 1 (r 1 ) 2 (r 2 )... n (r n ) 16

17 The Hartree Method 1 2 V (RI i + 2 I ri ) + j=i j (rj ) d rj i (ri ) = i (ri ) ri rj Hartree potential Douglas Hartree and Arthur Porter s Meccano differential analyzer built in 1934 at a cost of 20. It achieved an accuracy of about 2%. 17


18 Illustration of Electron Correlations Uncorrelated Cars are smeared out Correlated Cars avoid each other Uncorrelated Electrons described by their independent density, electrons can get arbitrary close Correlated Electrons avoid each other to due to the Coulomb interaction between them 18
19 The Hartree-Fock Method What is missing in the Hartree approximation Wave function is not antisymmetric Does not include electron correlation Antisymmetry for Fermions Exchanging two identical (indistinguishable) fermions changes the sign of the wave function (r 1, r 2,..., r j,..., r k,..., r n ) = (r 1, r 2,..., r k,..., r j,..., r n ) Pauli Exclusion Principle Two electrons cannot be in the same quantum state Consequence of the antisymmetry 19
20 Slater Determinants Slater determinant Antisymmetric product of single particle orbitals (r 1, r 2,..., r n ) = 1 n! 1 (r 1 ) 2 (r 1 ) n (r 1 ) 1 (r 2 ) 2 (r 2 ) n (r 2 ) (r n ) 2 (r n ) n (r n ) Swapping rows in a determinant changes the sign 20
21 The Hartree-Fock Method Hartree-Fock equation for orbitals φλ Use of variational principle leads to set of equations for φλ i + I V (R I r i ) (r i ) + µ µ 1 µ (r j ) r i r j µ(r j )d 3 r j (r i ) 1 µ (r j ) r i r j (r j)d 3 r j µ (r i ) = (r i ) 21
22 The Exchange Term 1 µ µ (r j ) r i r j (r j)d 3 r j µ (r i ) Describes effect of exchange of electrons Cannot be written in the form x V x (r i ) (r i ) Instead it is of the form x V x (r i, r j ) (r j )d 3 r j This is called a non-local potential 22
23 Successes and Limitations of Hartree-Fock Successes Good for atomic properties Self-interaction free Good starting point for correlated-electron methods Limitations Schrödinger equation: Hartree-Fock equations: Any effect beyond HF is called correlation Size of correlation energy Example: N2 molecule: However: binding energy N2 N + N is De(Hartree-Fock) = 5.1 ev, De(exp) = 9.9 ev H Ψ = E Ψ Εexact F φi = εi φi ΕHF Ecorr = Eexact - EHF Ecorr < 1% of Eexact Ecorr = 14.9 ev < 1% of Eexact Thus, there are large contribution from the correlation energy to relative energies, i.e. chemical reaction energies. 23
24 Beyond Hartree-Fock Hartree Fock configuration HF Slater determinant is built from lowest energy 1-e orbitals 0 =... K HF 1 2 Slater determinant is also called a configuration since it refers to certain filled orbitals Configuration interaction method Add additional configurations to the wave functions that mix in excited states Excite electron from orbital i to orbital K+1 1 HF = K+1... K CI = c c c
25 Density Functional Theory Theory for the ground state energy of a system as a function of the electron density instead of the wave function Walter Kohn received the Nobel prize in 1998 for his development of density functional theory Motivation (r 1, r 2,...,r n ) (r) Function of 3N variables Function of 3 variables Electron density Locally homogeneous electron gas 25 Position
26 The Hohenberg-Kohn Theorems The external potential and the number of electrons define the problem Schrödinger s equation in principle uniquely determines the wave functions All system properties follow from the wave functions HK Thm I: The energy and everything else (incl. the density) is a determined by Vext and Nel Schrödinger equation Hohenberg-Kohn Theorem V ext (r)!! 0 (r) # $ Integration of! 0 2 over N-1 electron coordinates! i (r 1,... r N ) "! 0 (r 1,... r N ) 26
27 The Universal Functional Since the ground state density determines all properties, the ground state energy and its components are a functional of the density Vext is known E 0 tot = E kin [ 0 ] + V ext [ 0 ] + V el el [ 0 ] V ext [ 0 ] = V ext (r) (r)d 3 r For the kinetic energy and the e-e interaction F [ 0 ] = E kin [ 0 ] + V el el [ 0 ] Form of this functional is the same for any molecule or solid HK Thm II: A universal energy functional can be defined in terms of the density, F[ρ], and the exact ground state is the functional minimum (But, the functional form is unknown!) 27
28 2 nd Hohenberg-Kohn Theorem The groundstate energy can be obtained variationally. The density that minimizes the total energy is the exact groundstate density. E[ (r)] = F [ (r)] + V ext (r) (r)d 3 r E 0 28

![T S [ (r)] = 1 2 Universal functional now takes the form N i=1 N i=1 i (r) 2 Electron-electron interaction is separated](/docs-images/84/89136963/images/29-2.jpg "into two terms, the Hartree term (Coulomb interaction) and the unknown(!")
29 The Kohn-Sham Equations Mapping to a non-interacting system (r) = Why? The kinetic energy of the non-interacting system is well defined. T S [ (r)] = 1 2 Universal functional now takes the form N i=1 N i=1 i (r) 2 Electron-electron interaction is separated into two terms, the Hartree term (Coulomb interaction) and the unknown(!) exchange-correlation energy term i (r) 2 i (r) F [ (r)] = T S [ (r)] + E H [ (r)] + E xc [ (r)] = 1 (r) (r ) E d 3 r d 3 H [ (r)] r 2 r r E xc [ (r)] = 29
30 Euler-Lagrange Equations Minimize energy with respect to variations of the density F [ (r)] + Vext (r) (r)d 3 r µ (r)d 3 r N =0 F [ (r) (r) + V ext = µ Resulting equations have a similar form as the Schrödinger equation and are known as the Kohn-Sham equations V H (r) + V xc (r) + V ext (r) H KS i (r) = i i (r) 30
! i=1,n (r)!! i (r) E[{ i (r)}] = N i=1 1 2 i (r) 2 i (r)d 3 r + E H [ (r)] + E xc [ (r)] + V ext (r) (r)d 3 r But exchange-correlation functional is still not known!")
31 Summary Hohenberg-Kohn theorem V ext (r)!! 0 (r) # $ Kohn-Sham Hohenberg-Kohn theorem applied to non-interacting electrons! 0 (r) " $ V ext (r) #! i (r 1,... r N ) "! 0 (r 1,... r N )! i=1,n (r)!! i (r) E[{ i (r)}] = N i=1 1 2 i (r) 2 i (r)d 3 r + E H [ (r)] + E xc [ (r)] + V ext (r) (r)d 3 r But exchange-correlation functional is still not known! 31
32 What about Exc? The Local Density Approximation D. M. Ceperley and B. J. Alder, Ground State of the Electron Gas by a Stochastic Method Phys. Rev. Lett. 45, 566 (1980). 32
33 The Phases of Silicon Under pressure Si displays 11 crystal phases LDA correctly predicts the energetic order of all these phases Compression 11 GPa 13 GPa 16 GPa 36 GPa 42 GPa 79 GPa Si(I) diamond Z=4 Si(II)! tin Z=6 Si(XI) Imma Si(V) hexagonal Si(VI) orthorhombic Z=6 Z=8 Z=10 Si(VII) hcp Si(X) fcc Z=12 Z=12 Decompression Slow pressure release Fast pressure release 9 GPa 2 GPa >480 K Si(VIII) and Si(IX) tetragonal Si(II)! tin Z=6 Si(XII) R8 Z=4 Si(III) BC 8 Z=4 Si(IV) hex. diamond Z=4 Phys. Rev. B 24, 7210 (1981), ibid. 49, (1994), ibid. 69, (2004)
34 Exchange-Correlation Functionals Local density approximation (LDA) Based on Ceperley & Alder s calculations for the uniform electron gas by quantum Monte Carlo (a stochastic method for quantum particles) Generalized gradient approximations (GGA: PW91, PBE) Gradients of the density are introduced Preserve analytic scaling features of the unknown exact functional Meta-GGA (TPSS) Include information about curvature of the density Hybrid density functionals (B3LYP, HSE) Based on GGA or meta-gga approximations Add some non-local Hartree-Fock exchange to the functional 34
35 Density Functional Theory in Practice 1. Remove tightly bound core electrons: the pseudopotential approach 2. Represent orbitals with a basis (plane waves or Gaussians) 3. Calculate total energy for trial orbitals - Kinetic and Hartree energy in reciprocal space - Exchange-correlation energy and external potential in real space - Method can take advantage of Fast Fourier Transformations - Sum over all states: BZ integrations 4. Minimize energy and iterate charge density to self-consistency 35
36 Pseudopotentials Electrons in the inner shells do not contribute to bonding Core electrons are effectively frozen Replace Coulomb potential between electrons and nuclei with effective potential, the pseudopotential 3p 3s -2.7 ev -7.8 ev Valence states 2p 1s -2.7 ev -7.8 ev Al Z = 13 2p 2s 1s ev -108 ev ev Core states Pseudo Al Z = V e 1 i = i i V pseudo e pseudo i = i pseudo i 36
37 Pseudopotentials The pseudopotential and the wave function Real potential and wave function are shown in blue Pseudopotential and pseudo wave function in red Outside the cutoff region (vertical black line) the two are identical 37
38 Bloch Theorem G s and k s Basis set choices For molecules: use atomic orbitals, or localized functions like Gaussians For solids, periodic functions such as sines and cosines (i.e., plane waves) Use Bloch Theorem for periodic solids: plane waves wavevector k ~ electron momentum (k point in 1 st Brillouin Zone) ( [H, T R ] = 0 (r) = u nk (r) exp(ik r) u nk ( reciprocal lattice vector G G R = 2 m Hamiltonian and translation operator commute u nk (r) = wavefunction of electron n at wavevector k in Brillouin Zone (Wigner-Seitz cell in reciprocal space) 38 G G max c nk (G) exp(i G r) function with periodicity of xtal lattice expanded in PW basis set u nk (r)+ R) = u nk (r)
 = c nk (G) exp(i G r) G G max Increase size of basis set to approach completeness (i.e., convergence) 39")
39 G s: Plane Wave Basis Set Superposition of plane waves to represent orbitals with xtal lattice periodicity u nk (r) = c nk (G) exp(i G r) G G max Increase size of basis set to approach completeness (i.e., convergence) 39
40 k s: 1 st Brillouin Zone (1BZ) Crystal structures defined by Bravais lattice {ai} and basis Reciprocal lattice b k =2 a l a m a k (a l a m ) Brillouin zone is the Wigner-Seitz cell of the reciprocal lattice 40
41 k s: 1 st Brillouin Zone (1BZ) The wavefunction (and energy) of each electron depends on both its quantum number n and its position k (i.e., its momentum) within the 1BZ We must solve a different Kohn-Sham equation for each n and k Bloch Thm has allowed us to replace analysis of ~infinite number of electrons in the xtal with only those in BZ but at an infinite number of k points! Payoff is that wavefunctions change smoothly in k so in practice consider finite number of k-points sampling over a k-space grid, refine grid until convergence Real-space quantities are computed by a discrete sum over n and integration over k within the Brillouin Zone (approximated over a grid at finite k-points) e.g. density, n(r) 41
 = u nk (r) exp(ik r) W L! \" # X Z W K path in BZ (i.e., k) Nearly free electron s-band dominates at low and high energies Hybridization of nearly-free s and atomic-like d orbitals at intermediate energies 42")
42 k s: Band Structure Plot εn,k Copper: Band structure calculated with Wien2k band (i.e., n) E F Energy (ev) s-d hybridization u nk (r) = u nk (r) exp(ik r) W L! " # X Z W K path in BZ (i.e., k) Nearly free electron s-band dominates at low and high energies Hybridization of nearly-free s and atomic-like d orbitals at intermediate energies 42
43 Successes and Failures of DFT Structural and elastic properties Lattice parameters are typically within a few percent of experimental values and often accurate to better than 1% The bulk modulus and other elastic constants are usually within 10% Vibrational Properties Forces = 1 st derivative of energy with respect to atomic displacement Forces are accurate to better than 10% (similar to elastic constants) Vibrational frequencies are the 2 nd derivatives Their accuracy is about 1/2 the accuracy of the forces or about 5% 43 Frequency [THz] ! [00"] Ni fcc X [0"1] W X K [0""] EAM (NRL) EAM (Voter&Chen) DFT (PW91) Neutron diffr. (296K) ["""]! L
44 Successes and Failures of DFT Defect energies In many cases such as for metals, vacancy and interstitial energies are highly accurate (within 0.1 ev) In some cases such as interstitial defects in silicon, DFT is too low by about 1 ev predicting a ev formation energy instead of the 4.5 ev of experiments and QMC [Phys. Rev. B 74, (R) (2006)] Excited states and gaps Local density approximation fails for excited states Bandgaps in LDA and GGA are usually underestimated by 20 50% In some cases such as Ge, LDA predicts a metallic instead of semiconducting state Hybrid functionals (e.g. B3LYP and HSE) improve the accuracy to about 10% 44
45 The bandgap problem of DFT Example: Bandstructure of InAs LDA, no gap: ev PBE, no gap: ev B3LYP 20%, gap: 0.54 ev 5 E-E Fermi [ev] Wien2k Crystal03 L Γ X W K Γ L Γ X W K Γ L Γ X W K Γ Experimental bandgap: 0.41 ev Band gap problem: LDA and GGA yield a metallic ground state! Practical solution: Hybrid functionals B3LYP & HSE (0.39 ev) Better solution: GW approximation or QMC methods 45
46 Beyond DFT The GW approximation Si Ge diamond 3C SiC LiCl!-C 3 N 4 BN BP BAs AlN AlP AlAs AlSb GaN GaP GaAs GaSb InP InAs InSb Al 0.5 Ga 0.5 As In 0.53 Ga 0.47 As GaAs 0.5 N 0.5 GaAs 0.75 N 0.25 ZnO ZnS ZnSe ZnTe CdS CdSe CdTe MgO MnO NiO CaCuO 2 Li 2 O ZrO 2 SnO 2 Energy gap (ev) LDA GWA Expt., indirect gap Expt., direct gap
47 Summary of Density Functional Theory LDA Lattice constants: 1-3% too small Cohesive Energies: 5-20% too strongly bound Bulk Modulus: 5-20% (largest errors for late TM) Bandgaps: too small GGA Improves cohesive energies Often but not always better for lattice parameters Important for magnetic systems Hybrid functionals Improved band gaps, often very accurate Always check the accuracy of the computational method by benchmarking against experimental data or more accurate theory. 47
André Schleife Department of Materials Science and Engineering
 André Schleife Department of Materials Science and Engineering Yesterday you (should have) learned this: http://upload.wikimedia.org/wikipedia/commons/e/ea/ Simple_Harmonic_Motion_Orbit.gif 1. deterministic
André Schleife Department of Materials Science and Engineering Yesterday you (should have) learned this: http://upload.wikimedia.org/wikipedia/commons/e/ea/ Simple_Harmonic_Motion_Orbit.gif 1. deterministic
The electronic structure of materials 2 - DFT
 Quantum mechanics 2 - Lecture 9 December 19, 2012 1 Density functional theory (DFT) 2 Literature Contents 1 Density functional theory (DFT) 2 Literature Historical background The beginnings: L. de Broglie
Quantum mechanics 2 - Lecture 9 December 19, 2012 1 Density functional theory (DFT) 2 Literature Contents 1 Density functional theory (DFT) 2 Literature Historical background The beginnings: L. de Broglie
Teoría del Funcional de la Densidad (Density Functional Theory)
") Teoría del Funcional de la Densidad (Density Functional Theory) Motivation: limitations of the standard approach based on the wave function. The electronic density n(r) as the key variable: Functionals
Teoría del Funcional de la Densidad (Density Functional Theory) Motivation: limitations of the standard approach based on the wave function. The electronic density n(r) as the key variable: Functionals
An Approximate DFT Method: The Density-Functional Tight-Binding (DFTB) Method
 Method") Fakultät für Mathematik und Naturwissenschaften - Lehrstuhl für Physikalische Chemie I / Theoretische Chemie An Approximate DFT Method: The Density-Functional Tight-Binding (DFTB) Method Jan-Ole Joswig
Fakultät für Mathematik und Naturwissenschaften - Lehrstuhl für Physikalische Chemie I / Theoretische Chemie An Approximate DFT Method: The Density-Functional Tight-Binding (DFTB) Method Jan-Ole Joswig
Density Functional Theory. Martin Lüders Daresbury Laboratory
 Density Functional Theory Martin Lüders Daresbury Laboratory Ab initio Calculations Hamiltonian: (without external fields, non-relativistic) impossible to solve exactly!! Electrons Nuclei Electron-Nuclei
Density Functional Theory Martin Lüders Daresbury Laboratory Ab initio Calculations Hamiltonian: (without external fields, non-relativistic) impossible to solve exactly!! Electrons Nuclei Electron-Nuclei
Dept of Mechanical Engineering MIT Nanoengineering group
 1 Dept of Mechanical Engineering MIT Nanoengineering group » To calculate all the properties of a molecule or crystalline system knowing its atomic information: Atomic species Their coordinates The Symmetry
1 Dept of Mechanical Engineering MIT Nanoengineering group » To calculate all the properties of a molecule or crystalline system knowing its atomic information: Atomic species Their coordinates The Symmetry
Density Functional Theory (DFT)
") Density Functional Theory (DFT) An Introduction by A.I. Al-Sharif Irbid, Aug, 2 nd, 2009 Density Functional Theory Revolutionized our approach to the electronic structure of atoms, molecules and solid
Density Functional Theory (DFT) An Introduction by A.I. Al-Sharif Irbid, Aug, 2 nd, 2009 Density Functional Theory Revolutionized our approach to the electronic structure of atoms, molecules and solid
MODULE 2: QUANTUM MECHANICS. Practice: Quantum ESPRESSO
 MODULE 2: QUANTUM MECHANICS Practice: Quantum ESPRESSO I. What is Quantum ESPRESSO? 2 DFT software PW-DFT, PP, US-PP, PAW http://www.quantum-espresso.org FREE PW-DFT, PP, PAW http://www.abinit.org FREE
MODULE 2: QUANTUM MECHANICS Practice: Quantum ESPRESSO I. What is Quantum ESPRESSO? 2 DFT software PW-DFT, PP, US-PP, PAW http://www.quantum-espresso.org FREE PW-DFT, PP, PAW http://www.abinit.org FREE
Electronic Structure Theory for Periodic Systems: The Concepts. Christian Ratsch
 Electronic Structure Theory for Periodic Systems: The Concepts Christian Ratsch Institute for Pure and Applied Mathematics and Department of Mathematics, UCLA Motivation There are 10 20 atoms in 1 mm 3
Electronic Structure Theory for Periodic Systems: The Concepts Christian Ratsch Institute for Pure and Applied Mathematics and Department of Mathematics, UCLA Motivation There are 10 20 atoms in 1 mm 3
From Quantum Mechanics to Materials Design
 The Basics of Density Functional Theory Volker Eyert Center for Electronic Correlations and Magnetism Institute of Physics, University of Augsburg December 03, 2010 Outline Formalism 1 Formalism Definitions
The Basics of Density Functional Theory Volker Eyert Center for Electronic Correlations and Magnetism Institute of Physics, University of Augsburg December 03, 2010 Outline Formalism 1 Formalism Definitions
Exchange-Correlation Functional
 Exchange-Correlation Functional Aiichiro Nakano Collaboratory for Advanced Computing & Simulations Depts. of Computer Science, Physics & Astronomy, Chemical Engineering & Materials Science, and Biological
Exchange-Correlation Functional Aiichiro Nakano Collaboratory for Advanced Computing & Simulations Depts. of Computer Science, Physics & Astronomy, Chemical Engineering & Materials Science, and Biological
Dept of Mechanical Engineering MIT Nanoengineering group
 1 Dept of Mechanical Engineering MIT Nanoengineering group » Recap of HK theorems and KS equations» The physical meaning of the XC energy» Solution of a one-particle Schroedinger equation» Pseudo Potentials»
1 Dept of Mechanical Engineering MIT Nanoengineering group » Recap of HK theorems and KS equations» The physical meaning of the XC energy» Solution of a one-particle Schroedinger equation» Pseudo Potentials»
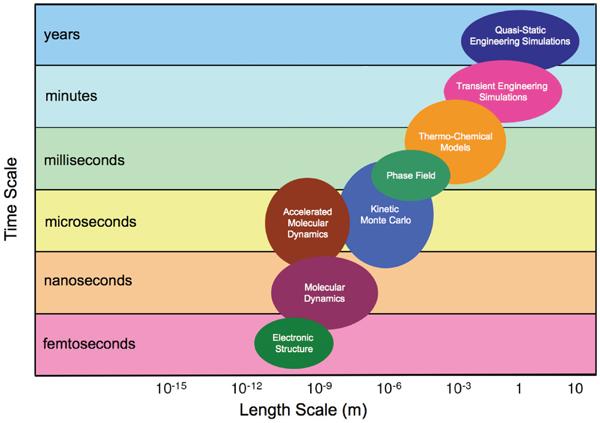
Multi-Scale Modeling from First Principles
 m mm Multi-Scale Modeling from First Principles μm nm m mm μm nm space space Predictive modeling and simulations must address all time and Continuum Equations, densityfunctional space scales Rate Equations
m mm Multi-Scale Modeling from First Principles μm nm m mm μm nm space space Predictive modeling and simulations must address all time and Continuum Equations, densityfunctional space scales Rate Equations
Introduction to Density Functional Theory
 1 Introduction to Density Functional Theory 21 February 2011; V172 P.Ravindran, FME-course on Ab initio Modelling of solar cell Materials 21 February 2011 Introduction to DFT 2 3 4 Ab initio Computational
1 Introduction to Density Functional Theory 21 February 2011; V172 P.Ravindran, FME-course on Ab initio Modelling of solar cell Materials 21 February 2011 Introduction to DFT 2 3 4 Ab initio Computational
Quantum Monte Carlo methods
 Quantum Monte Carlo methods Lubos Mitas North Carolina State University Urbana, August 2006 Lubos_Mitas@ncsu.edu H= 1 2 i i 2 i, I Z I r ii i j 1 r ij E ion ion H r 1, r 2,... =E r 1, r 2,... - ground
Quantum Monte Carlo methods Lubos Mitas North Carolina State University Urbana, August 2006 Lubos_Mitas@ncsu.edu H= 1 2 i i 2 i, I Z I r ii i j 1 r ij E ion ion H r 1, r 2,... =E r 1, r 2,... - ground
Introduction to Density Functional Theory with Applications to Graphene Branislav K. Nikolić
 Introduction to Density Functional Theory with Applications to Graphene Branislav K. Nikolić Department of Physics and Astronomy, University of Delaware, Newark, DE 19716, U.S.A. http://wiki.physics.udel.edu/phys824
Introduction to Density Functional Theory with Applications to Graphene Branislav K. Nikolić Department of Physics and Astronomy, University of Delaware, Newark, DE 19716, U.S.A. http://wiki.physics.udel.edu/phys824
Density Functional Theory for Electrons in Materials
 Density Functional Theory for Electrons in Materials Richard M. Martin Department of Physics and Materials Research Laboratory University of Illinois at Urbana-Champaign 1 Density Functional Theory for
Density Functional Theory for Electrons in Materials Richard M. Martin Department of Physics and Materials Research Laboratory University of Illinois at Urbana-Champaign 1 Density Functional Theory for
Practical Guide to Density Functional Theory (DFT)
") Practical Guide to Density Functional Theory (DFT) Brad Malone, Sadas Shankar Quick recap of where we left off last time BD Malone, S Shankar Therefore there is a direct one-to-one correspondence between
Practical Guide to Density Functional Theory (DFT) Brad Malone, Sadas Shankar Quick recap of where we left off last time BD Malone, S Shankar Therefore there is a direct one-to-one correspondence between
1 Density functional theory (DFT)
") 1 Density functional theory (DFT) 1.1 Introduction Density functional theory is an alternative to ab initio methods for solving the nonrelativistic, time-independent Schrödinger equation H Φ = E Φ. The
1 Density functional theory (DFT) 1.1 Introduction Density functional theory is an alternative to ab initio methods for solving the nonrelativistic, time-independent Schrödinger equation H Φ = E Φ. The
All electron optimized effective potential method for solids
 All electron optimized effective potential method for solids Institut für Theoretische Physik Freie Universität Berlin, Germany and Fritz Haber Institute of the Max Planck Society, Berlin, Germany. 22
All electron optimized effective potential method for solids Institut für Theoretische Physik Freie Universität Berlin, Germany and Fritz Haber Institute of the Max Planck Society, Berlin, Germany. 22
Quantum Monte Carlo Benchmarks Density Functionals: Si Defects
 Quantum Monte Carlo Benchmarks Density Functionals: Si Defects K P Driver, W D Parker, R G Hennig, J W Wilkins (OSU) C J Umrigar (Cornell), R Martin, E Batista, B Uberuaga (LANL), J Heyd, G Scuseria (Rice)
Quantum Monte Carlo Benchmarks Density Functionals: Si Defects K P Driver, W D Parker, R G Hennig, J W Wilkins (OSU) C J Umrigar (Cornell), R Martin, E Batista, B Uberuaga (LANL), J Heyd, G Scuseria (Rice)
Module 6 1. Density functional theory
 Module 6 1. Density functional theory Updated May 12, 2016 B A DDFT C K A bird s-eye view of density-functional theory Authors: Klaus Capelle G http://arxiv.org/abs/cond-mat/0211443 R https://trac.cc.jyu.fi/projects/toolbox/wiki/dft
Module 6 1. Density functional theory Updated May 12, 2016 B A DDFT C K A bird s-eye view of density-functional theory Authors: Klaus Capelle G http://arxiv.org/abs/cond-mat/0211443 R https://trac.cc.jyu.fi/projects/toolbox/wiki/dft
Pseudopotentials for hybrid density functionals and SCAN
 Pseudopotentials for hybrid density functionals and SCAN Jing Yang, Liang Z. Tan, Julian Gebhardt, and Andrew M. Rappe Department of Chemistry University of Pennsylvania Why do we need pseudopotentials?
Pseudopotentials for hybrid density functionals and SCAN Jing Yang, Liang Z. Tan, Julian Gebhardt, and Andrew M. Rappe Department of Chemistry University of Pennsylvania Why do we need pseudopotentials?
v(r i r j ) = h(r i )+ 1 N
 = h(r i )+ 1 N") Chapter 1 Hartree-Fock Theory 1.1 Formalism For N electrons in an external potential V ext (r), the many-electron Hamiltonian can be written as follows: N H = [ p i i=1 m +V ext(r i )]+ 1 N N v(r i r j
Chapter 1 Hartree-Fock Theory 1.1 Formalism For N electrons in an external potential V ext (r), the many-electron Hamiltonian can be written as follows: N H = [ p i i=1 m +V ext(r i )]+ 1 N N v(r i r j
The Schrödinger equation for many-electron systems
 The Schrödinger equation for many-electron systems Ĥ!( x,, x ) = E!( x,, x ) 1 N 1 1 Z 1 Ĥ = " $ # " $ + $ 2 r 2 A j j A, j RAj i, j < i a linear differential equation in 4N variables (atomic units) (3
The Schrödinger equation for many-electron systems Ĥ!( x,, x ) = E!( x,, x ) 1 N 1 1 Z 1 Ĥ = " $ # " $ + $ 2 r 2 A j j A, j RAj i, j < i a linear differential equation in 4N variables (atomic units) (3
Density Functional Theory: from theory to Applications
 Density Functional Theory: from theory to Applications Uni Mainz November 29, 2010 The self interaction error and its correction Perdew-Zunger SIC Average-density approximation Weighted density approximation
Density Functional Theory: from theory to Applications Uni Mainz November 29, 2010 The self interaction error and its correction Perdew-Zunger SIC Average-density approximation Weighted density approximation
Fundamentals and applications of Density Functional Theory Astrid Marthinsen PhD candidate, Department of Materials Science and Engineering
 Fundamentals and applications of Density Functional Theory Astrid Marthinsen PhD candidate, Department of Materials Science and Engineering Outline PART 1: Fundamentals of Density functional theory (DFT)
Fundamentals and applications of Density Functional Theory Astrid Marthinsen PhD candidate, Department of Materials Science and Engineering Outline PART 1: Fundamentals of Density functional theory (DFT)
DFT: Exchange-Correlation
 DFT: Local functionals, exact exchange and other post-dft methods Stewart Clark University of Outline Introduction What is exchange and correlation? Quick tour of XC functionals (Semi-)local: LDA, PBE,
DFT: Local functionals, exact exchange and other post-dft methods Stewart Clark University of Outline Introduction What is exchange and correlation? Quick tour of XC functionals (Semi-)local: LDA, PBE,
Three Most Important Topics (MIT) Today
 Today") Three Most Important Topics (MIT) Today Electrons in periodic potential Energy gap nearly free electron Bloch Theorem Energy gap tight binding Chapter 1 1 Electrons in Periodic Potential We now know the
Three Most Important Topics (MIT) Today Electrons in periodic potential Energy gap nearly free electron Bloch Theorem Energy gap tight binding Chapter 1 1 Electrons in Periodic Potential We now know the
Institut Néel Institut Laue Langevin. Introduction to electronic structure calculations
 Institut Néel Institut Laue Langevin Introduction to electronic structure calculations 1 Institut Néel - 25 rue des Martyrs - Grenoble - France 2 Institut Laue Langevin - 71 avenue des Martyrs - Grenoble
Institut Néel Institut Laue Langevin Introduction to electronic structure calculations 1 Institut Néel - 25 rue des Martyrs - Grenoble - France 2 Institut Laue Langevin - 71 avenue des Martyrs - Grenoble
Key concepts in Density Functional Theory
 From the many body problem to the Kohn-Sham scheme ILM (LPMCN) CNRS, Université Lyon 1 - France European Theoretical Spectroscopy Facility (ETSF) December 12, 2012 Lyon Outline 1 The many-body problem
From the many body problem to the Kohn-Sham scheme ILM (LPMCN) CNRS, Université Lyon 1 - France European Theoretical Spectroscopy Facility (ETSF) December 12, 2012 Lyon Outline 1 The many-body problem
Quantum Mechanical Simulations
 Quantum Mechanical Simulations Prof. Yan Wang Woodruff School of Mechanical Engineering Georgia Institute of Technology Atlanta, GA 30332, U.S.A. yan.wang@me.gatech.edu Topics Quantum Monte Carlo Hartree-Fock
Quantum Mechanical Simulations Prof. Yan Wang Woodruff School of Mechanical Engineering Georgia Institute of Technology Atlanta, GA 30332, U.S.A. yan.wang@me.gatech.edu Topics Quantum Monte Carlo Hartree-Fock
Principles of Quantum Mechanics
 Principles of Quantum Mechanics - indistinguishability of particles: bosons & fermions bosons: total wavefunction is symmetric upon interchange of particle coordinates (space,spin) fermions: total wavefuncftion
Principles of Quantum Mechanics - indistinguishability of particles: bosons & fermions bosons: total wavefunction is symmetric upon interchange of particle coordinates (space,spin) fermions: total wavefuncftion
Improved Electronic Structure and Optical Properties of sp-hybridized Semiconductors Using LDA+U SIC
 286 Brazilian Journal of Physics, vol. 36, no. 2A, June, 2006 Improved Electronic Structure and Optical Properties of sp-hybridized Semiconductors Using LDA+U SIC Clas Persson and Susanne Mirbt Department
286 Brazilian Journal of Physics, vol. 36, no. 2A, June, 2006 Improved Electronic Structure and Optical Properties of sp-hybridized Semiconductors Using LDA+U SIC Clas Persson and Susanne Mirbt Department
Key concepts in Density Functional Theory (I) Silvana Botti
 Silvana Botti") From the many body problem to the Kohn-Sham scheme European Theoretical Spectroscopy Facility (ETSF) CNRS - Laboratoire des Solides Irradiés Ecole Polytechnique, Palaiseau - France Temporary Address: Centre
From the many body problem to the Kohn-Sham scheme European Theoretical Spectroscopy Facility (ETSF) CNRS - Laboratoire des Solides Irradiés Ecole Polytechnique, Palaiseau - France Temporary Address: Centre
Electronic Structure of Crystalline Solids
 Electronic Structure of Crystalline Solids Computing the electronic structure of electrons in solid materials (insulators, conductors, semiconductors, superconductors) is in general a very difficult problem
Electronic Structure of Crystalline Solids Computing the electronic structure of electrons in solid materials (insulators, conductors, semiconductors, superconductors) is in general a very difficult problem
Introduction to Density Functional Theory
 Introduction to Density Functional Theory S. Sharma Institut für Physik Karl-Franzens-Universität Graz, Austria 19th October 2005 Synopsis Motivation 1 Motivation : where can one use DFT 2 : 1 Elementary
Introduction to Density Functional Theory S. Sharma Institut für Physik Karl-Franzens-Universität Graz, Austria 19th October 2005 Synopsis Motivation 1 Motivation : where can one use DFT 2 : 1 Elementary
Introduction to DFT and Density Functionals. by Michel Côté Université de Montréal Département de physique
 Introduction to DFT and Density Functionals by Michel Côté Université de Montréal Département de physique Eamples Carbazole molecule Inside of diamant Réf: Jean-François Brière http://www.phys.umontreal.ca/~michel_
Introduction to DFT and Density Functionals by Michel Côté Université de Montréal Département de physique Eamples Carbazole molecule Inside of diamant Réf: Jean-François Brière http://www.phys.umontreal.ca/~michel_
DENSITY FUNCTIONAL THEORY FOR NON-THEORISTS JOHN P. PERDEW DEPARTMENTS OF PHYSICS AND CHEMISTRY TEMPLE UNIVERSITY
 DENSITY FUNCTIONAL THEORY FOR NON-THEORISTS JOHN P. PERDEW DEPARTMENTS OF PHYSICS AND CHEMISTRY TEMPLE UNIVERSITY A TUTORIAL FOR PHYSICAL SCIENTISTS WHO MAY OR MAY NOT HATE EQUATIONS AND PROOFS REFERENCES
DENSITY FUNCTIONAL THEORY FOR NON-THEORISTS JOHN P. PERDEW DEPARTMENTS OF PHYSICS AND CHEMISTRY TEMPLE UNIVERSITY A TUTORIAL FOR PHYSICAL SCIENTISTS WHO MAY OR MAY NOT HATE EQUATIONS AND PROOFS REFERENCES
Pseudopotentials: design, testing, typical errors
 Pseudopotentials: design, testing, typical errors Kevin F. Garrity Part 1 National Institute of Standards and Technology (NIST) Uncertainty Quantification in Materials Modeling 2015 Parameter free calculations.
Pseudopotentials: design, testing, typical errors Kevin F. Garrity Part 1 National Institute of Standards and Technology (NIST) Uncertainty Quantification in Materials Modeling 2015 Parameter free calculations.
Electronic band structure, sx-lda, Hybrid DFT, LDA+U and all that. Keith Refson STFC Rutherford Appleton Laboratory
 Electronic band structure, sx-lda, Hybrid DFT, LDA+U and all that Keith Refson STFC Rutherford Appleton Laboratory LDA/GGA DFT is good but... Naive LDA/GGA calculation severely underestimates band-gaps.
Electronic band structure, sx-lda, Hybrid DFT, LDA+U and all that Keith Refson STFC Rutherford Appleton Laboratory LDA/GGA DFT is good but... Naive LDA/GGA calculation severely underestimates band-gaps.
1.1 Atoms. 1.1 Atoms
 1. Chemical bonding and crystal structure 19 21 Hydrogen atom Scanning electron microscopy Ni surface Cleaved surface ZnO, TiO 2, NiO, NaCl, Si, Ge, GaAs, InP Crystals are build by small repeating units
1. Chemical bonding and crystal structure 19 21 Hydrogen atom Scanning electron microscopy Ni surface Cleaved surface ZnO, TiO 2, NiO, NaCl, Si, Ge, GaAs, InP Crystals are build by small repeating units
Electronic Structure Calculations and Density Functional Theory
 Electronic Structure Calculations and Density Functional Theory Rodolphe Vuilleumier Pôle de chimie théorique Département de chimie de l ENS CNRS Ecole normale supérieure UPMC Formation ModPhyChem Lyon,
Electronic Structure Calculations and Density Functional Theory Rodolphe Vuilleumier Pôle de chimie théorique Département de chimie de l ENS CNRS Ecole normale supérieure UPMC Formation ModPhyChem Lyon,
CHEM6085: Density Functional Theory
 Lecture 5 CHEM6085: Density Functional Theory Orbital-free (or pure ) DFT C.-K. Skylaris 1 Consists of three terms The electronic Hamiltonian operator Electronic kinetic energy operator Electron-Electron
Lecture 5 CHEM6085: Density Functional Theory Orbital-free (or pure ) DFT C.-K. Skylaris 1 Consists of three terms The electronic Hamiltonian operator Electronic kinetic energy operator Electron-Electron
CHEM6085: Density Functional Theory
 Lecture 11 CHEM6085: Density Functional Theory DFT for periodic crystalline solids C.-K. Skylaris 1 Electron in a one-dimensional periodic box (in atomic units) Schrödinger equation Energy eigenvalues
Lecture 11 CHEM6085: Density Functional Theory DFT for periodic crystalline solids C.-K. Skylaris 1 Electron in a one-dimensional periodic box (in atomic units) Schrödinger equation Energy eigenvalues
2. Surface geometric and electronic structure: a primer
 2. Surface geometric and electronic structure: a primer 2.1 Surface crystallography 2.1.1. Crystal structures - A crystal structure is made up of two basic elements: lattice + basis Basis: Lattice: simplest
2. Surface geometric and electronic structure: a primer 2.1 Surface crystallography 2.1.1. Crystal structures - A crystal structure is made up of two basic elements: lattice + basis Basis: Lattice: simplest
Introduction to density-functional theory. Emmanuel Fromager
 Institut de Chimie, Strasbourg, France Page 1 Emmanuel Fromager Institut de Chimie de Strasbourg - Laboratoire de Chimie Quantique - Université de Strasbourg /CNRS M2 lecture, Strasbourg, France. Institut
Institut de Chimie, Strasbourg, France Page 1 Emmanuel Fromager Institut de Chimie de Strasbourg - Laboratoire de Chimie Quantique - Université de Strasbourg /CNRS M2 lecture, Strasbourg, France. Institut
Joint ICTP-IAEA Workshop on Fusion Plasma Modelling using Atomic and Molecular Data January 2012
 2327-3 Joint ICTP-IAEA Workshop on Fusion Plasma Modelling using Atomic and Molecular Data 23-27 January 2012 Qunatum Methods for Plasma-Facing Materials Alain ALLOUCHE Univ.de Provence, Lab.de la Phys.
2327-3 Joint ICTP-IAEA Workshop on Fusion Plasma Modelling using Atomic and Molecular Data 23-27 January 2012 Qunatum Methods for Plasma-Facing Materials Alain ALLOUCHE Univ.de Provence, Lab.de la Phys.
Bonding in solids The interaction of electrons in neighboring atoms of a solid serves the very important function of holding the crystal together.
 Bonding in solids The interaction of electrons in neighboring atoms of a solid serves the very important function of holding the crystal together. For example Nacl In the Nacl lattice, each Na atom is
Bonding in solids The interaction of electrons in neighboring atoms of a solid serves the very important function of holding the crystal together. For example Nacl In the Nacl lattice, each Na atom is
Density functional theory in the solid state
 Density functional theory in the solid state Ari P Seitsonen IMPMC, CNRS & Universités 6 et 7 Paris, IPGP Department of Applied Physics, Helsinki University of Technology Physikalisch-Chemisches Institut
Density functional theory in the solid state Ari P Seitsonen IMPMC, CNRS & Universités 6 et 7 Paris, IPGP Department of Applied Physics, Helsinki University of Technology Physikalisch-Chemisches Institut
Chapter 3. The (L)APW+lo Method. 3.1 Choosing A Basis Set
APW+lo Method. 3.1 Choosing A Basis Set") Chapter 3 The (L)APW+lo Method 3.1 Choosing A Basis Set The Kohn-Sham equations (Eq. (2.17)) provide a formulation of how to practically find a solution to the Hohenberg-Kohn functional (Eq. (2.15)). Nevertheless
Chapter 3 The (L)APW+lo Method 3.1 Choosing A Basis Set The Kohn-Sham equations (Eq. (2.17)) provide a formulation of how to practically find a solution to the Hohenberg-Kohn functional (Eq. (2.15)). Nevertheless
Integrated Computational Materials Engineering Education
 Integrated Computational Materials Engineering Education Lecture on Density Functional Theory An Introduction Mark Asta Dept. of Materials Science and Engineering, University of California, Berkeley &
Integrated Computational Materials Engineering Education Lecture on Density Functional Theory An Introduction Mark Asta Dept. of Materials Science and Engineering, University of California, Berkeley &
Computational Methods. Chem 561
 Computational Methods Chem 561 Lecture Outline 1. Ab initio methods a) HF SCF b) Post-HF methods 2. Density Functional Theory 3. Semiempirical methods 4. Molecular Mechanics Computational Chemistry " Computational
Computational Methods Chem 561 Lecture Outline 1. Ab initio methods a) HF SCF b) Post-HF methods 2. Density Functional Theory 3. Semiempirical methods 4. Molecular Mechanics Computational Chemistry " Computational
Supporting Information for. Predicting the Stability of Fullerene Allotropes Throughout the Periodic Table MA 02139
 Supporting Information for Predicting the Stability of Fullerene Allotropes Throughout the Periodic Table Qing Zhao 1, 2, Stanley S. H. Ng 1, and Heather J. Kulik 1, * 1 Department of Chemical Engineering,
Supporting Information for Predicting the Stability of Fullerene Allotropes Throughout the Periodic Table Qing Zhao 1, 2, Stanley S. H. Ng 1, and Heather J. Kulik 1, * 1 Department of Chemical Engineering,
Band calculations: Theory and Applications
 Band calculations: Theory and Applications Lecture 2: Different approximations for the exchange-correlation correlation functional in DFT Local density approximation () Generalized gradient approximation
Band calculations: Theory and Applications Lecture 2: Different approximations for the exchange-correlation correlation functional in DFT Local density approximation () Generalized gradient approximation
Many electrons: Density functional theory Part II. Bedřich Velický VI.
 Many electrons: Density functional theory Part II. Bedřich Velický velicky@karlov.mff.cuni.cz VI. NEVF 514 Surface Physics Winter Term 013-014 Troja 1 st November 013 This class is the second devoted to
Many electrons: Density functional theory Part II. Bedřich Velický velicky@karlov.mff.cuni.cz VI. NEVF 514 Surface Physics Winter Term 013-014 Troja 1 st November 013 This class is the second devoted to
Modified Becke-Johnson (mbj) exchange potential
 exchange potential") Modified Becke-Johnson (mbj) exchange potential Hideyuki Jippo Fujitsu Laboratories LTD. 2015.12.21-22 OpenMX developer s meeting @ Kobe Overview: mbj potential The semilocal exchange potential adding
Modified Becke-Johnson (mbj) exchange potential Hideyuki Jippo Fujitsu Laboratories LTD. 2015.12.21-22 OpenMX developer s meeting @ Kobe Overview: mbj potential The semilocal exchange potential adding
Quantum Condensed Matter Physics Lecture 4
 Quantum Condensed Matter Physics Lecture 4 David Ritchie QCMP Lent/Easter 2019 http://www.sp.phy.cam.ac.uk/drp2/home 4.1 Quantum Condensed Matter Physics 1. Classical and Semi-classical models for electrons
Quantum Condensed Matter Physics Lecture 4 David Ritchie QCMP Lent/Easter 2019 http://www.sp.phy.cam.ac.uk/drp2/home 4.1 Quantum Condensed Matter Physics 1. Classical and Semi-classical models for electrons
The electronic structure of materials 1
 Quantum mechanics 2 - Lecture 9 December 18, 2013 1 An overview 2 Literature Contents 1 An overview 2 Literature Electronic ground state Ground state cohesive energy equilibrium crystal structure phase
Quantum mechanics 2 - Lecture 9 December 18, 2013 1 An overview 2 Literature Contents 1 An overview 2 Literature Electronic ground state Ground state cohesive energy equilibrium crystal structure phase
Walter Kohn was awarded with the Nobel Prize in Chemistry in 1998 for his development of the density functional theory.
 Walter Kohn was awarded with the Nobel Prize in Chemistry in 1998 for his development of the density functional theory. Walter Kohn receiving his Nobel Prize from His Majesty the King at the Stockholm
Walter Kohn was awarded with the Nobel Prize in Chemistry in 1998 for his development of the density functional theory. Walter Kohn receiving his Nobel Prize from His Majesty the King at the Stockholm
Molecular Mechanics: The Ab Initio Foundation
 Molecular Mechanics: The Ab Initio Foundation Ju Li GEM4 Summer School 2006 Cell and Molecular Mechanics in BioMedicine August 7 18, 2006, MIT, Cambridge, MA, USA 2 Outline Why are electrons quantum? Born-Oppenheimer
Molecular Mechanics: The Ab Initio Foundation Ju Li GEM4 Summer School 2006 Cell and Molecular Mechanics in BioMedicine August 7 18, 2006, MIT, Cambridge, MA, USA 2 Outline Why are electrons quantum? Born-Oppenheimer
Band Structure Calculations; Electronic and Optical Properties
 ; Electronic and Optical Properties Stewart Clark University of Outline Introduction to band structures Calculating band structures using Castep Calculating optical properties Examples results Some applications
; Electronic and Optical Properties Stewart Clark University of Outline Introduction to band structures Calculating band structures using Castep Calculating optical properties Examples results Some applications
Modelowanie Nanostruktur
 Chair of Condensed Matter Physics Institute of Theoretical Physics Faculty of Physics, Universityof Warsaw Modelowanie Nanostruktur, 2012/2013 Jacek A. Majewski Semester Zimowy 2012/2013 Wykad Modelowanie
Chair of Condensed Matter Physics Institute of Theoretical Physics Faculty of Physics, Universityof Warsaw Modelowanie Nanostruktur, 2012/2013 Jacek A. Majewski Semester Zimowy 2012/2013 Wykad Modelowanie
Electrochemistry project, Chemistry Department, November Ab-initio Molecular Dynamics Simulation
 Electrochemistry project, Chemistry Department, November 2006 Ab-initio Molecular Dynamics Simulation Outline Introduction Ab-initio concepts Total energy concepts Adsorption energy calculation Project
Electrochemistry project, Chemistry Department, November 2006 Ab-initio Molecular Dynamics Simulation Outline Introduction Ab-initio concepts Total energy concepts Adsorption energy calculation Project
Advanced Quantum Chemistry III: Part 3. Haruyuki Nakano. Kyushu University
 Advanced Quantum Chemistry III: Part 3 Haruyuki Nakano Kyushu University 2013 Winter Term 1. Hartree-Fock theory Density Functional Theory 2. Hohenberg-Kohn theorem 3. Kohn-Sham method 4. Exchange-correlation
Advanced Quantum Chemistry III: Part 3 Haruyuki Nakano Kyushu University 2013 Winter Term 1. Hartree-Fock theory Density Functional Theory 2. Hohenberg-Kohn theorem 3. Kohn-Sham method 4. Exchange-correlation
DFT / SIESTA algorithms
 DFT / SIESTA algorithms Javier Junquera José M. Soler References http://siesta.icmab.es Documentation Tutorials Atomic units e = m e = =1 atomic mass unit = m e atomic length unit = 1 Bohr = 0.5292 Ang
DFT / SIESTA algorithms Javier Junquera José M. Soler References http://siesta.icmab.es Documentation Tutorials Atomic units e = m e = =1 atomic mass unit = m e atomic length unit = 1 Bohr = 0.5292 Ang
OVERVIEW OF QUANTUM CHEMISTRY METHODS
 OVERVIEW OF QUANTUM CHEMISTRY METHODS Outline I Generalities Correlation, basis sets Spin II Wavefunction methods Hartree-Fock Configuration interaction Coupled cluster Perturbative methods III Density
OVERVIEW OF QUANTUM CHEMISTRY METHODS Outline I Generalities Correlation, basis sets Spin II Wavefunction methods Hartree-Fock Configuration interaction Coupled cluster Perturbative methods III Density
First Principles Investigation into the Atom in Jellium Model System. Andrew Ian Duff
 First Principles Investigation into the Atom in Jellium Model System Andrew Ian Duff H. H. Wills Physics Laboratory University of Bristol A thesis submitted to the University of Bristol in accordance with
First Principles Investigation into the Atom in Jellium Model System Andrew Ian Duff H. H. Wills Physics Laboratory University of Bristol A thesis submitted to the University of Bristol in accordance with
GEM4 Summer School OpenCourseWare
 GEM4 Summer School OpenCourseWare http://gem4.educommons.net/ http://www.gem4.org/ Lecture: Molecular Mechanics by Ju Li. Given August 9, 2006 during the GEM4 session at MIT in Cambridge, MA. Please use
GEM4 Summer School OpenCourseWare http://gem4.educommons.net/ http://www.gem4.org/ Lecture: Molecular Mechanics by Ju Li. Given August 9, 2006 during the GEM4 session at MIT in Cambridge, MA. Please use
High pressure core structures of Si nanoparticles for solar energy conversion
 High pressure core structures of Si nanoparticles for solar energy conversion S. Wippermann, M. Vörös, D. Rocca, A. Gali, G. Zimanyi, G. Galli [Phys. Rev. Lett. 11, 4684 (213)] NSF/Solar DMR-135468 NISE-project
High pressure core structures of Si nanoparticles for solar energy conversion S. Wippermann, M. Vörös, D. Rocca, A. Gali, G. Zimanyi, G. Galli [Phys. Rev. Lett. 11, 4684 (213)] NSF/Solar DMR-135468 NISE-project
Preface Introduction to the electron liquid
 Table of Preface page xvii 1 Introduction to the electron liquid 1 1.1 A tale of many electrons 1 1.2 Where the electrons roam: physical realizations of the electron liquid 5 1.2.1 Three dimensions 5 1.2.2
Table of Preface page xvii 1 Introduction to the electron liquid 1 1.1 A tale of many electrons 1 1.2 Where the electrons roam: physical realizations of the electron liquid 5 1.2.1 Three dimensions 5 1.2.2
Performance ofhybrid density functional methods,screened exchange and EXX-OEP methodsin the PAW approach p.1/26
 Performance of hybrid density functional methods, screened exchange and EXX-OEP methods in the PAW approach Georg Kresse, J Paier, R Hirschl, M Marsmann Institut für Materialphysik and Centre for Computational
Performance of hybrid density functional methods, screened exchange and EXX-OEP methods in the PAW approach Georg Kresse, J Paier, R Hirschl, M Marsmann Institut für Materialphysik and Centre for Computational
Practical calculations with the GW approximation and Bethe-Salpeter equation in BerkeleyGW
 Practical calculations with the GW approximation and Bethe-Salpeter equation in BerkeleyGW David A. Strubbe Department of Physics, University of California, Merced Benasque, Spain 23 August 2018 Band gaps:
Practical calculations with the GW approximation and Bethe-Salpeter equation in BerkeleyGW David A. Strubbe Department of Physics, University of California, Merced Benasque, Spain 23 August 2018 Band gaps:
Basic cell design. Si cell
 Basic cell design Si cell 1 Concepts needed to describe photovoltaic device 1. energy bands in semiconductors: from bonds to bands 2. free carriers: holes and electrons, doping 3. electron and hole current:
Basic cell design Si cell 1 Concepts needed to describe photovoltaic device 1. energy bands in semiconductors: from bonds to bands 2. free carriers: holes and electrons, doping 3. electron and hole current:
Exchange Correlation Functional Investigation of RT-TDDFT on a Sodium Chloride. Dimer. Philip Straughn
 Exchange Correlation Functional Investigation of RT-TDDFT on a Sodium Chloride Dimer Philip Straughn Abstract Charge transfer between Na and Cl ions is an important problem in physical chemistry. However,
Exchange Correlation Functional Investigation of RT-TDDFT on a Sodium Chloride Dimer Philip Straughn Abstract Charge transfer between Na and Cl ions is an important problem in physical chemistry. However,
Hartree, Hartree-Fock and post-hf methods
 Hartree, Hartree-Fock and post-hf methods MSE697 fall 2015 Nicolas Onofrio School of Materials Engineering DLR 428 Purdue University nonofrio@purdue.edu 1 The curse of dimensionality Let s consider a multi
Hartree, Hartree-Fock and post-hf methods MSE697 fall 2015 Nicolas Onofrio School of Materials Engineering DLR 428 Purdue University nonofrio@purdue.edu 1 The curse of dimensionality Let s consider a multi
Electronic Structure Methodology 1
 Electronic Structure Methodology 1 Chris J. Pickard Lecture Two Working with Density Functional Theory In the last lecture we learnt how to write the total energy as a functional of the density n(r): E
Electronic Structure Methodology 1 Chris J. Pickard Lecture Two Working with Density Functional Theory In the last lecture we learnt how to write the total energy as a functional of the density n(r): E
DFT EXERCISES. FELIPE CERVANTES SODI January 2006
 DFT EXERCISES FELIPE CERVANTES SODI January 2006 http://www.csanyi.net/wiki/space/dftexercises Dr. Gábor Csányi 1 Hydrogen atom Place a single H atom in the middle of a largish unit cell (start with a
DFT EXERCISES FELIPE CERVANTES SODI January 2006 http://www.csanyi.net/wiki/space/dftexercises Dr. Gábor Csányi 1 Hydrogen atom Place a single H atom in the middle of a largish unit cell (start with a
with cubic symmetry Supplementary Material
 Prediction uncertainty of density functional approximations for properties of crystals with cubic symmetry Supplementary Material Pascal Pernot,,, Bartolomeo Civalleri, Davide Presti, and Andreas Savin,
Prediction uncertainty of density functional approximations for properties of crystals with cubic symmetry Supplementary Material Pascal Pernot,,, Bartolomeo Civalleri, Davide Presti, and Andreas Savin,
Answers Quantum Chemistry NWI-MOL406 G. C. Groenenboom and G. A. de Wijs, HG00.307, 8:30-11:30, 21 jan 2014
 Answers Quantum Chemistry NWI-MOL406 G. C. Groenenboom and G. A. de Wijs, HG00.307, 8:30-11:30, 21 jan 2014 Question 1: Basis sets Consider the split valence SV3-21G one electron basis set for formaldehyde
Answers Quantum Chemistry NWI-MOL406 G. C. Groenenboom and G. A. de Wijs, HG00.307, 8:30-11:30, 21 jan 2014 Question 1: Basis sets Consider the split valence SV3-21G one electron basis set for formaldehyde
Kevin Driver 1 Shuai Zhang 1 Burkhard Militzer 1 R. E. Cohen 2.
 Quantum Monte Carlo Simulations of a Single Iron Impurity in MgO Kevin Driver 1 Shuai Zhang 1 Burkhard Militzer 1 R. E. Cohen 2 1 Department of Earth & Planetary Science University of California, Berkeley
Quantum Monte Carlo Simulations of a Single Iron Impurity in MgO Kevin Driver 1 Shuai Zhang 1 Burkhard Militzer 1 R. E. Cohen 2 1 Department of Earth & Planetary Science University of California, Berkeley
ECE440 Nanoelectronics. Lecture 07 Atomic Orbitals
 ECE44 Nanoelectronics Lecture 7 Atomic Orbitals Atoms and atomic orbitals It is instructive to compare the simple model of a spherically symmetrical potential for r R V ( r) for r R and the simplest hydrogen
ECE44 Nanoelectronics Lecture 7 Atomic Orbitals Atoms and atomic orbitals It is instructive to compare the simple model of a spherically symmetrical potential for r R V ( r) for r R and the simplest hydrogen
7/29/2014. Electronic Structure. Electrons in Momentum Space. Electron Density Matrices FKF FKF. Ulrich Wedig
 Electron Density Matrices Density matrices Γ, an alternative to the wavefunction Ψ, for the description of a quantum system Electronic Structure The N-particle density matrix Electrons in Momentum Space
Electron Density Matrices Density matrices Γ, an alternative to the wavefunction Ψ, for the description of a quantum system Electronic Structure The N-particle density matrix Electrons in Momentum Space
MEASURING THE PERFORMANCE OF RECENT GENERALIZED GRADIENT APPROXIMATIONS TO DENSITY FUNCTIONAL THEORY IN MOLECULES AND SOLIDS A THESIS
 MEASURING THE PERFORMANCE OF RECENT GENERALIZED GRADIENT APPROXIMATIONS TO DENSITY FUNCTIONAL THEORY IN MOLECULES AND SOLIDS A THESIS SUBMITTED TO THE GRADUATE SCHOOL IN PARTIAL FULFILLMENT OF THE REQUIREMENTS
MEASURING THE PERFORMANCE OF RECENT GENERALIZED GRADIENT APPROXIMATIONS TO DENSITY FUNCTIONAL THEORY IN MOLECULES AND SOLIDS A THESIS SUBMITTED TO THE GRADUATE SCHOOL IN PARTIAL FULFILLMENT OF THE REQUIREMENTS
Computational Physics. J. M. Thijssen
 Computational Physics J. M. Thijssen Delft University of Technology CAMBRIDGE UNIVERSITY PRESS Contents Preface xi 1 Introduction 1 1.1 Physics and computational physics 1 1.2 Classical mechanics and statistical
Computational Physics J. M. Thijssen Delft University of Technology CAMBRIDGE UNIVERSITY PRESS Contents Preface xi 1 Introduction 1 1.1 Physics and computational physics 1 1.2 Classical mechanics and statistical
3: Density Functional Theory
 The Nuts and Bolts of First-Principles Simulation 3: Density Functional Theory CASTEP Developers Group with support from the ESF ψ k Network Density functional theory Mike Gillan, University College London
The Nuts and Bolts of First-Principles Simulation 3: Density Functional Theory CASTEP Developers Group with support from the ESF ψ k Network Density functional theory Mike Gillan, University College London
Lecture 8: Introduction to Density Functional Theory
 Lecture 8: Introduction to Density Functional Theory Marie Curie Tutorial Series: Modeling Biomolecules December 6-11, 2004 Mark Tuckerman Dept. of Chemistry and Courant Institute of Mathematical Science
Lecture 8: Introduction to Density Functional Theory Marie Curie Tutorial Series: Modeling Biomolecules December 6-11, 2004 Mark Tuckerman Dept. of Chemistry and Courant Institute of Mathematical Science
Electron bands in crystals Pseudopotentials, Plane Waves, Local Orbitals
 Electron bands in crystals Pseudopotentials, Plane Waves, Local Orbitals Richard M. Martin UIUC Lecture at Summer School Hands-on introduction to Electronic Structure Materials Computation Center University
Electron bands in crystals Pseudopotentials, Plane Waves, Local Orbitals Richard M. Martin UIUC Lecture at Summer School Hands-on introduction to Electronic Structure Materials Computation Center University
The GW Approximation. Manish Jain 1. July 8, Department of Physics Indian Institute of Science Bangalore 1/36
 1/36 The GW Approximation Manish Jain 1 Department of Physics Indian Institute of Science Bangalore July 8, 2014 Ground-state properties 2/36 Properties that are intrinsic to a system with all its electrons
1/36 The GW Approximation Manish Jain 1 Department of Physics Indian Institute of Science Bangalore July 8, 2014 Ground-state properties 2/36 Properties that are intrinsic to a system with all its electrons
Basics of density-functional theory and fast guide to actual calculations Matthias Scheffler
 Basics of density-functional theory and fast guide to actual calculations Matthias Scheffler http://www.fhi-berlin.mpg.de/th/th.html I. From the many-particle problem to the Kohn-Sham functional II. From
Basics of density-functional theory and fast guide to actual calculations Matthias Scheffler http://www.fhi-berlin.mpg.de/th/th.html I. From the many-particle problem to the Kohn-Sham functional II. From
Quantum Modeling of Solids: Basic Properties
 1.021, 3.021, 10.333, 22.00 : Introduction to Modeling and Simulation : Spring 2011 Part II Quantum Mechanical Methods : Lecture 5 Quantum Modeling of Solids: Basic Properties Jeffrey C. Grossman Department
1.021, 3.021, 10.333, 22.00 : Introduction to Modeling and Simulation : Spring 2011 Part II Quantum Mechanical Methods : Lecture 5 Quantum Modeling of Solids: Basic Properties Jeffrey C. Grossman Department
Independent electrons in an effective potential
 ABC of DFT Adiabatic approximation Independent electrons in an effective potential Hartree Fock Density Functional Theory MBPT - GW Density Functional Theory in a nutshell Every observable quantity of
ABC of DFT Adiabatic approximation Independent electrons in an effective potential Hartree Fock Density Functional Theory MBPT - GW Density Functional Theory in a nutshell Every observable quantity of
CLIMBING THE LADDER OF DENSITY FUNCTIONAL APPROXIMATIONS JOHN P. PERDEW DEPARTMENT OF PHYSICS TEMPLE UNIVERSITY PHILADELPHIA, PA 19122
 CLIMBING THE LADDER OF DENSITY FUNCTIONAL APPROXIMATIONS JOHN P. PERDEW DEPARTMENT OF PHYSICS TEMPLE UNIVERSITY PHILADELPHIA, PA 191 THANKS TO MANY COLLABORATORS, INCLUDING SY VOSKO DAVID LANGRETH ALEX
CLIMBING THE LADDER OF DENSITY FUNCTIONAL APPROXIMATIONS JOHN P. PERDEW DEPARTMENT OF PHYSICS TEMPLE UNIVERSITY PHILADELPHIA, PA 191 THANKS TO MANY COLLABORATORS, INCLUDING SY VOSKO DAVID LANGRETH ALEX
Kohn Sham density functional theory [1 3] is. Role of the Exchange Correlation Energy: Nature s Glue STEFAN KURTH, JOHN P. PERDEW.
![Kohn Sham density functional theory [1 3] is. Role of the Exchange Correlation Energy: Nature s Glue STEFAN KURTH, JOHN P. PERDEW.](/thumbs/80/82235561.jpg "Kohn Sham density functional theory [1 3] is. Role of the Exchange Correlation Energy: Nature s Glue STEFAN KURTH, JOHN P. PERDEW.") Role of the Exchange Correlation Energy: Nature s Glue STEFAN KURTH, JOHN P. PERDEW Department of Physics and Quantum Theory Group, Tulane University, New Orleans, Louisiana 70118 Received 11 March 1999;
Role of the Exchange Correlation Energy: Nature s Glue STEFAN KURTH, JOHN P. PERDEW Department of Physics and Quantum Theory Group, Tulane University, New Orleans, Louisiana 70118 Received 11 March 1999;
DFT: Exchange-Correlation
 DFT: Exchange-Correlation Local functionals, exact exchange and other post-dft methods Paul Tulip Centre for Materials Physics Department of Physics University of Durham Outline Introduction What is exchange
DFT: Exchange-Correlation Local functionals, exact exchange and other post-dft methods Paul Tulip Centre for Materials Physics Department of Physics University of Durham Outline Introduction What is exchange
Chapter 1 Overview of Semiconductor Materials and Physics
 Chapter 1 Overview of Semiconductor Materials and Physics Professor Paul K. Chu Conductivity / Resistivity of Insulators, Semiconductors, and Conductors Semiconductor Elements Period II III IV V VI 2 B
Chapter 1 Overview of Semiconductor Materials and Physics Professor Paul K. Chu Conductivity / Resistivity of Insulators, Semiconductors, and Conductors Semiconductor Elements Period II III IV V VI 2 B
Many electrons: Density functional theory
 Many electrons: Density functional theory Bedřich Velický V. NEVF 54 Surface Physics Winter Term 0-0 Troja, 8 th November 0 This class is devoted to the many-electron aspects of the solid state (computational
Many electrons: Density functional theory Bedřich Velický V. NEVF 54 Surface Physics Winter Term 0-0 Troja, 8 th November 0 This class is devoted to the many-electron aspects of the solid state (computational
Physics 541: Condensed Matter Physics
 Physics 541: Condensed Matter Physics In-class Midterm Exam Wednesday, October 26, 2011 / 14:00 15:20 / CCIS 4-285 Student s Name: Instructions There are 23 questions. You should attempt all of them. Mark
Physics 541: Condensed Matter Physics In-class Midterm Exam Wednesday, October 26, 2011 / 14:00 15:20 / CCIS 4-285 Student s Name: Instructions There are 23 questions. You should attempt all of them. Mark
Solid State Physics. Lecture 10 Band Theory. Professor Stephen Sweeney
 Solid State Physics Lecture 10 Band Theory Professor Stephen Sweeney Advanced Technology Institute and Department of Physics University of Surrey, Guildford, GU2 7XH, UK s.sweeney@surrey.ac.uk Recap from
Solid State Physics Lecture 10 Band Theory Professor Stephen Sweeney Advanced Technology Institute and Department of Physics University of Surrey, Guildford, GU2 7XH, UK s.sweeney@surrey.ac.uk Recap from
Is the homogeneous electron gas homogeneous?
 Is the homogeneous electron gas homogeneous? Electron gas (jellium): simplest way to view a metal homogeneous and normal Hartree-Fock: simplest method for many-electron systems a single Slater determinant
Is the homogeneous electron gas homogeneous? Electron gas (jellium): simplest way to view a metal homogeneous and normal Hartree-Fock: simplest method for many-electron systems a single Slater determinant