Integrated Computational Materials Engineering Education
|
|
|
- Eugenia Small
- 5 years ago
- Views:
Transcription
1 Integrated Computational Materials Engineering Education Lecture on Density Functional Theory An Introduction Mark Asta Dept. of Materials Science and Engineering, University of California, Berkeley & Materials Sciences Division, Lawrence Berkeley National Laboratory Berkeley, CA 94720
2 Acknowledgements Portions of these lectures are based on material put together by Daryl Chrzan (UC Berkeley) and Jeff Grossman (MIT) as part of a TMS short course that the three of us taught at the 2010 annual meeting. The Division of Materials Research at the National Science Foundation is acknowledged for financial support in the development of the lecture and module

3 Use of DFT in Materials Research K. Thornton, S. Nola, R. E. Garcia, MA and G. B. Olson, Computational Materials Science and Engineering Education: A Survey of Trends and Needs, JOM (2009)
4 The Role of Electronic Structure Methods in ICME A wide variety of relevant properties can be calculated from knowledge of atomic numbers alone Elastic constants Finite-temperature thermodynamic and transport properties Energies of point, line and planar defects For many classes of systems accuracy is quite high Can be used to obtain missing properties in materials design when experimental data is lacking, hard to obtain, or controversial Can be used to discover new stable compounds with target properties The starting point for hierarchical multiscale modeling Enables development of interatomic potentials for larger-scale classical modeling
, 011002.")
5 Materials Data for Discovery & Design A. Jain, S.P. Ong, G. Hautier, W. Chen, W.D. Richards, S. Dacek, S. Cholia, D. Gunter, D. Skinner, G. Ceder, K.A. Persson, Applied Physics Letters Materials, 2013, 1(1),

6 Materials Data for Discovery & Design


7 Materials Data for Discovery & Design DFT Lecture, The 7th Summer School for Integrated Computational Materials Education
8 Materials Data for Discovery & Design


9 Materials Data for Discovery & Design
10 Formalism Hydrogen Atom Density Functional Theory Outline Exchange-Correlation Potentials Pseudopotentials and Related Approaches Some Commercial and Open Source Codes Practical Issues Implementation Periodic boundary conditions k-points Plane-wave basis sets Parameters controlling numerical precision Example Exercise
11 Introduction The Hydrogen Atom Proton with mass M 1, coordinate R 1 Electron with mass m 1, coordinate r 1 $ & % 2 2M m e2 r ' ) Ψ(R 1,r 2 ) = EΨ(R 1,r 2 ) ( r = r 1 r 2, R = M 1 R 1 + m 2 r 2 M 1 + m 2, m = M 1 m 2 M 1 + m 2, M = M 1 + m 2 $ & % 2 2M R 2 2 2m r 2 e2 r ' ) Ψ(R,r) = EΨ(R,r) ( Ψ(R,r) =ψ cm (R)ψ r (r) $ & % 2M ' $ 2 R ) ψ cm (R) = E cm ψ cm (R) & 2 ( 2m r % 2 2 e2 ' ) ψ r (r) = E r ψ r (r) r (
 = R nl (r)y l m (θ,φ) 2 2m # % $ 1 r 2 d # % dr $ r2 d & ( dr' l(l +1) r 2 e2 r & ( R nl (r) = E n R nl (r) ' θ φ E n = me4 1 2 2 n = 13.")
12 Hydrogen Atom Switch to Spherical Coordinates 2 2m % ' & 1 r 2 % ψ ( ' r & r2 * + r ) 1 % ψ ( ' sinθ * + r 2 sinθ θ & θ ) 1 2 ψ ( * e2 r 2 sin 2 θ ϕ 2 ) r ψ = Eψ ψ(r,θ,φ) = R nl (r)y l m (θ,φ) 2 2m # % $ 1 r 2 d # % dr $ r2 d & ( dr' l(l +1) r 2 e2 r & ( R nl (r) = E n R nl (r) ' θ φ E n = me n = 13.6 ev 2 n 2
 http://en.wikipedia.")
13 Hydrogen Atom Wavefunctions n = 1, 2, 3, l = 0 (s), 1(p), 2(d),, n-1 Probability densities through the xz-plane for the electron at different quantum numbers (l, across top; n, down side; m = 0)
14 The Many-Electron Problem Collection of N ions n electrons Analytical solution like that for hydrogen atom not available electrons ions
15 Born-Oppenheimer Approximation Mass of nuclei exceeds that of the electrons by a factor of 1000 or more we can neglect the kinetic energy of the nuclei treat the ion-ion interaction classically significantly simplifies the Hamiltonian for the electrons Consider Hamiltonian for n electrons in potential of N nuclei with atomic numbers Z i H = 2 2m n i=1 2 r i N n j=1 Z i e R i r j 2 i=1 n i=1 n j=1 i j e 2 r i r j external potential Vext ( r ) j
16 Density Functional Theory Hohenberg and Kohn (1964), Kohn and Sham (1965) For each external potential there is a unique groundstate electron density Energy can be obtained by minimizing of a density functional with respect to density of electrons n(r) E groundstate =min{e tot [n(r)]} [ ] = T [ n( r) ] + E [ int n( r) ] + drv ext r E tot n r ( ) ( ) n r ( ) + E ion ion Kinetic Energy Electron-Electron Interactions Electron-Ion Interactions
17 E[{φ i }] =! 2 Kohn-Sham Approach 2m n i=1 n(r) = e φ i (r) 2 n i= φ i * i2 φ i d 3 r + V ext (r)n(r)d 3 r n(r)n(r') d 3 rd 3 r'+ E xc [n(r)] r r' Many-Body Electron-Electron Interactions Lumped into E xc [n(r)] Exchange-Correlation Energy
18 Kohn-Sham Equations $! 2 & 2m 2 i +V nuclei (r)+ %& n(r') r r' ' d 3 r'+v xc (r)) () φ (r) = ε i iφ i (r) V xc (r) δe xc [n(r)] δn(r)
19 Local Density Approximation (e.g., J. P. Perdew and A. Zunger, Phys. Rev. B 23, 5048 (1981)) E xc [n(r)] = hom ε xc (n(r))n(r)d 3 r ε xc hom (n(r)) Exchange Correlation Energy of Homogeneous Electron Gas of Density n(r)
20 Generalized Gradient Approximation J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett. 77 (1996) E GGA xc [n(r)] = ε x hom (n(r))n(r)f xc (r s,ζ, s)d 3 r n = 3 / 4π r s 3 = k F 3 / 3π 2 ζ = (n n ) / n s = n /2k F n
21 A Note on Accuracy and Ongoing Research LDA often leads to overbinding Lattice constants can be 1-3 % too small, elastic constants % too stiff, cohesive energies 5-20 % too large BUT, errors are largely systematic Energy differences tend to be more accurate GGA corrects for overbinding Sometimes overcorrects Beyond DFT Approaches For highly correlated systems LDA & GGA perform worse and corrections required (DFT+U, Hybrid Hartree-Fock/DFT, Meta-GGA, DMFT, ) Non-bonded interactions, e.g., van der Waals interactions in graphite, require additional terms or functionals (e.g., vdw-df)
22 Pseudopotentials Potential due to ions is singular at ion core Eigenfunctions oscillate rapidly near singularity Eigenfunction in bonding region is smooth
23 Pseudopotentials For plane-wave basis sets, rapid oscillations require large number of basis functions expensive unnecessary these oscillations don't alter bonding properties Replace potential with nonsingular potential preserve bonding tails of eigenfunction preserve distribution of charge between core and tail regions reduces number of plane waves required for accurate expansion of wavefunction Transferable developed from properties of isolated atoms applied in other situations φ φ pseudo
24 Summary of Approaches Pseudopotentials Core electrons removed from problem and enter only in their effect of the pseudopotential felt by the valence electrons Kohn-Sham equations solved for valence electrons only Augment Plane Waves with atomic-like orbitals An efficient basis set that allows all electrons to be treated in the calculations Basis for all-electron codes Projector-Augmented-Wave method Combines features of both methods Generally accepted as the basis for the most accurate approach for calculations requiring consideration of valence electrons only
25 Some of the Widely Used Codes VASP ( Commercial, Plane-Wave Basis, Pseudopotentials and PAW PWSCF ( Free (and available to run on nanohub), Plane-Wave Basis, Pseudopotentials and PAW CASTEP ( Free in UK, licensed by Accelrys elsewhere, Plane-Wave Basis, Pseudopotentials ABINIT ( Free (and available to run on nanohub), plane-wave basis, pseudopotentials and PAW WIEN2K ( Commercial (modest license fee), all-electron augmented wave method
26 Formalism Hydrogen Atom Density Functional Theory Outline Exchange-Correlation Potentials Pseudopotentials and Related Approaches Some Commercial and Open Source Codes Practical Issues Implementation Periodic boundary conditions k-points Plane-wave basis sets Parameters controlling numerical precision Example Exercise
27 Total Energy in Density Functional Theory E[{φ i }] = 2 2m e N e i=1 φ * i 2 i φ i d 3 r + n(r)n(r') r r' Electron Density Electron Wavefunctions φ i (r) V ext (r)n(r)d 3 r n i=1 n(r) = e φ i (r) 2 d 3 rd 3 r'+e xc [n(r)] Potential Electrons Feel from Nuclei V ext (r) Exchange-Correlation Energy E xc [n(r)] Form depends on whether you use LDA or GGA
28 Kohn-Sham Equations Schrödinger Equation for Electron Wavefunctions $ & %& 2 2 i +V ext (r)+ 2m e n(r') r r' ' d 3 r'+v xc (r)) () φ (r) = ε i iφ i (r) Exchange-Correlation Potential V xc (r) δe xc [n(r)] δn(r) Electron Density n i=1 n(r) = e φ i (r) 2 Note: φ i depends on n(r) which depends on φ i è Solution of Kohn-Sham equations must be done iteratively
29 Self-Consistent Solution to DFT Equations Input Positions of Atoms for a Given Unit Cell and Lattice Constant different Energy for Given Lattice Constant guess charge density compute effective potential compute Kohn-Sham orbitals and density compare output and input charge densities same 1. Set up atom positions 2. Make initial guess of input charge density (often overlapping atomic charge densities) 3. Solve Kohn-Sham equations with this input charge density 4. Compute output charge density from resulting wavefunctions 5. If energy from input and output densities differ by amount greater than a chosen threshold, mix output and input density and go to step 2 6. Quit when energy from input and output densities agree to within prescribed tolerance (e.g., 10-5 ev) Note: In your exercise, positions of atoms are dictated by symmetry. If this is not the case another loop must be added to minimize energy with respect to atomic positions.
30 Implementation of DFT for a Single Crystal Crystal Structure Defined by Unit Cell Vectors and Positions of Basis Atoms Example: Diamond Cubic Structure of Si a a a Unit Cell Vectors a 1 = a (-1/2, 1/2, 0) a 2 = a (-1/2, 0, 1/2) a 3 = a (0, 1/2, 1/2) Basis Atom Positions ¼ ¼ ¼ All atoms in the crystal can be obtained by adding integer multiples of unit cell vectors to basis atom positions
31 Electron Density in Crystal Lattice a a a Unit-Cell Vectors a 1 = a (-1/2, 1/2, 0) a 2 = a (-1/2, 0, 1/2) a 3 = a (0, 1/2, 1/2) Electron density is periodic with periodicity given by R uvw n( r) = n( r + R uvw ) Translation Vectors: R uvw = ua 1 + va 2 + wa 3


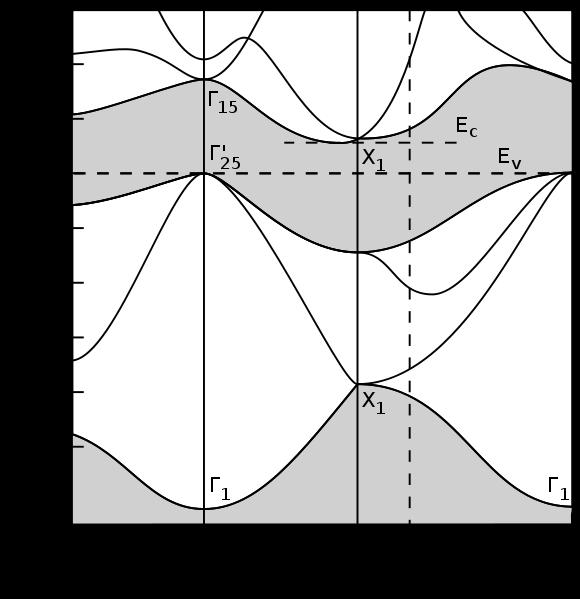
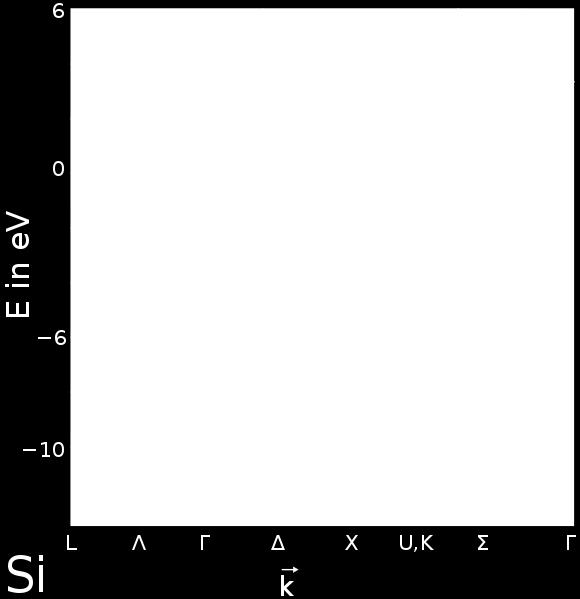
32 Brillouin Zone Electronic Bandstructure Example for Si Bandstructure Electronic wavefunctions in a crystal can be indexed by point in reciprocal space (k) and a band index (β)
33 Why? Wavefunctions in a Crystal Obey Bloch s Theorem For a given band β φ k β ( r) = exp ik r ( ) u β ( k r) ( ) Where u β is periodic in real space: u β k r k r ( ) = u β k ( r + R uvw ) Translation Vectors: R uvw = ua 1 + va 2 + wa 3
34 Representation of Electron Density φ k β ( r) = exp ik r ( ) u β ( k r) N e n(r) = e φ i (r) 2 i=1 ΩBZ ( ) n(r) = e φ k β r β 2 f (εk β ε F ) d 3 k Ω BZ In practice the integral over the Brillouin zone is replaced with a sum over a finite number of k-points (N kpt ) n(r) e Integral over k-points in first Brillouin zone f(ε-ε F ) is Fermi-Dirac distribution function with Fermi energy ε F β N kpt j=1 β w j φ k j ( r) 2 f (ε β k j ε F ) One parameter that needs to be checked for numerical convergence is number of k-points
35 Representation of Wavefunctions Fourier-Expansion as Series of Plane Waves For a given band: φ k β ( r) = exp ik r ( ) u β ( k r) Recall that u β is periodic in real space: u β k r k r u k β ( r) ( ) ( ) = u β k ( r + R uvw ) can be written as a Fourier Series: u β k ( r) = u β k ( G lmn )exp ig lmn r where the a i * G lmn = la * 1 + ma * * 2 + na 3 are primitive reciprocal lattice vectors lmn a * 1 a 1 = 2π a * 1 a 2 = 0 a * 1 a 3 = 0 a * 2 a 1 = 0 a * 2 a 2 = 2π a * 2 a 3 = 0 a * 3 a 1 = 0 a * 3 a 2 = 0 a * 3 a 3 = 2π ( )

36 Recall Properties of Fourier Series Black line = (exact) triangular wave Colored lines = Fourier series truncated at different orders General Form of Fourier Series: For Triangular Wave: Typically we expect the accuracy of a truncated Fourier series to improve as we increase the number of terms
37 Representation of Wavefunctions Plane-Wave Basis Set For a given band φ k β ( r) = exp ik r ( ) u β ( k r) Use Fourier Expansion φ k β ( r) = u β k ( G)exp"# i( G + k) r$ % G In practice the Fourier series is truncated to include all G for which: 2 2m G + k ( ) 2 < E cut Another parameter that needs to be checked for convergence is the plane-wave cutoff energy E cut

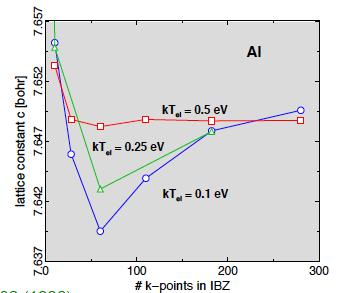
38 Examples of Convergence Checks Effect of E cut Effect of Number of k Points
39 Formalism Hydrogen Atom Density Functional Theory Outline Exchange-Correlation Potentials Pseudopotentials and Related Approaches Some Commercial and Open Source Codes Practical Issues Implementation Periodic boundary conditions k-points Plane-wave basis sets Parameters controlling numerical precision Example Exercise
40 Your Exercise: Part 1 Calculate equation of state of diamond cubic Si using Quantum Espresso on Nanohub ( You will compare accuracy of LDA and GGA You will check numerical convergence with respect to number of k-points and plane-wave cutoff You will make use of the following unit cell for diamond-cubic structure a Lattice Vectors a 1 = a (-1/2, 1/2, 0) a 2 = a (-1/2, 0, 1/2) a 3 = a (0, 1/2, 1/2) a a Basis Atom Positions ¼ ¼ ¼
 a a http://www.e6cvd.")
41 Equation of State A Probe of Interatomic Interactions Energy per atom Schematic Energy vs. Volume Relation Diamond Cubic Structure of Si a Volume per atom (=a 3 /8 for Si) a a pageid=361
42 P = E V Equation of State What Properties Can we Learn from It? Pressure versus Volume Relation Given E(V) one can compute P(V) by taking derivative Recall 1 st Law of Thermo: de = T ds - P dv and consider T = 0 K Equilibrium Volume (or Lattice Constant) Volume corresponding to zero pressure = Volume where slope of E(V) is zero Volume measured experimentally at P = 1 atm B = V P V = V 2 E V 2 Bulk Modulus B related to curvature of E(V) Function
43 Your Exercise: Part 2 Non-hydrostatic Stress and Strain Stress-Strain Relations in Linear Elasticity σ ij = k,l C ijkl ε kl Stress Strain C ijkl Single-Crystal Elastic Constants Stress-Strain Relations in Linear Elasticity Consider Single Strain ε 33 =ε σ 33 = C 11 ε σ 22 = C 12 ε Voigt Notation (for Cubic Crystal) C 3333 =C 2222 =C 1111 =C 11 C 2233 =C 1133 =C 1122 =C 2211 =C 3311 =C 3322 =C 12
The electronic structure of materials 2 - DFT
 Quantum mechanics 2 - Lecture 9 December 19, 2012 1 Density functional theory (DFT) 2 Literature Contents 1 Density functional theory (DFT) 2 Literature Historical background The beginnings: L. de Broglie
Quantum mechanics 2 - Lecture 9 December 19, 2012 1 Density functional theory (DFT) 2 Literature Contents 1 Density functional theory (DFT) 2 Literature Historical background The beginnings: L. de Broglie
Fundamentals and applications of Density Functional Theory Astrid Marthinsen PhD candidate, Department of Materials Science and Engineering
 Fundamentals and applications of Density Functional Theory Astrid Marthinsen PhD candidate, Department of Materials Science and Engineering Outline PART 1: Fundamentals of Density functional theory (DFT)
Fundamentals and applications of Density Functional Theory Astrid Marthinsen PhD candidate, Department of Materials Science and Engineering Outline PART 1: Fundamentals of Density functional theory (DFT)
Practical Guide to Density Functional Theory (DFT)
") Practical Guide to Density Functional Theory (DFT) Brad Malone, Sadas Shankar Quick recap of where we left off last time BD Malone, S Shankar Therefore there is a direct one-to-one correspondence between
Practical Guide to Density Functional Theory (DFT) Brad Malone, Sadas Shankar Quick recap of where we left off last time BD Malone, S Shankar Therefore there is a direct one-to-one correspondence between
Electronic Structure Methodology 1
 Electronic Structure Methodology 1 Chris J. Pickard Lecture Two Working with Density Functional Theory In the last lecture we learnt how to write the total energy as a functional of the density n(r): E
Electronic Structure Methodology 1 Chris J. Pickard Lecture Two Working with Density Functional Theory In the last lecture we learnt how to write the total energy as a functional of the density n(r): E
DFT: Exchange-Correlation
 DFT: Local functionals, exact exchange and other post-dft methods Stewart Clark University of Outline Introduction What is exchange and correlation? Quick tour of XC functionals (Semi-)local: LDA, PBE,
DFT: Local functionals, exact exchange and other post-dft methods Stewart Clark University of Outline Introduction What is exchange and correlation? Quick tour of XC functionals (Semi-)local: LDA, PBE,
MODULE 2: QUANTUM MECHANICS. Practice: Quantum ESPRESSO
 MODULE 2: QUANTUM MECHANICS Practice: Quantum ESPRESSO I. What is Quantum ESPRESSO? 2 DFT software PW-DFT, PP, US-PP, PAW http://www.quantum-espresso.org FREE PW-DFT, PP, PAW http://www.abinit.org FREE
MODULE 2: QUANTUM MECHANICS Practice: Quantum ESPRESSO I. What is Quantum ESPRESSO? 2 DFT software PW-DFT, PP, US-PP, PAW http://www.quantum-espresso.org FREE PW-DFT, PP, PAW http://www.abinit.org FREE
Pseudopotentials for hybrid density functionals and SCAN
 Pseudopotentials for hybrid density functionals and SCAN Jing Yang, Liang Z. Tan, Julian Gebhardt, and Andrew M. Rappe Department of Chemistry University of Pennsylvania Why do we need pseudopotentials?
Pseudopotentials for hybrid density functionals and SCAN Jing Yang, Liang Z. Tan, Julian Gebhardt, and Andrew M. Rappe Department of Chemistry University of Pennsylvania Why do we need pseudopotentials?
CLIMBING THE LADDER OF DENSITY FUNCTIONAL APPROXIMATIONS JOHN P. PERDEW DEPARTMENT OF PHYSICS TEMPLE UNIVERSITY PHILADELPHIA, PA 19122
 CLIMBING THE LADDER OF DENSITY FUNCTIONAL APPROXIMATIONS JOHN P. PERDEW DEPARTMENT OF PHYSICS TEMPLE UNIVERSITY PHILADELPHIA, PA 191 THANKS TO MANY COLLABORATORS, INCLUDING SY VOSKO DAVID LANGRETH ALEX
CLIMBING THE LADDER OF DENSITY FUNCTIONAL APPROXIMATIONS JOHN P. PERDEW DEPARTMENT OF PHYSICS TEMPLE UNIVERSITY PHILADELPHIA, PA 191 THANKS TO MANY COLLABORATORS, INCLUDING SY VOSKO DAVID LANGRETH ALEX
Density Functional Theory for Electrons in Materials
 Density Functional Theory for Electrons in Materials Richard M. Martin Department of Physics and Materials Research Laboratory University of Illinois at Urbana-Champaign 1 Density Functional Theory for
Density Functional Theory for Electrons in Materials Richard M. Martin Department of Physics and Materials Research Laboratory University of Illinois at Urbana-Champaign 1 Density Functional Theory for
Teoría del Funcional de la Densidad (Density Functional Theory)
") Teoría del Funcional de la Densidad (Density Functional Theory) Motivation: limitations of the standard approach based on the wave function. The electronic density n(r) as the key variable: Functionals
Teoría del Funcional de la Densidad (Density Functional Theory) Motivation: limitations of the standard approach based on the wave function. The electronic density n(r) as the key variable: Functionals
Electron bands in crystals Pseudopotentials, Plane Waves, Local Orbitals
 Electron bands in crystals Pseudopotentials, Plane Waves, Local Orbitals Richard M. Martin UIUC Lecture at Summer School Hands-on introduction to Electronic Structure Materials Computation Center University
Electron bands in crystals Pseudopotentials, Plane Waves, Local Orbitals Richard M. Martin UIUC Lecture at Summer School Hands-on introduction to Electronic Structure Materials Computation Center University
Computational Methods. Chem 561
 Computational Methods Chem 561 Lecture Outline 1. Ab initio methods a) HF SCF b) Post-HF methods 2. Density Functional Theory 3. Semiempirical methods 4. Molecular Mechanics Computational Chemistry " Computational
Computational Methods Chem 561 Lecture Outline 1. Ab initio methods a) HF SCF b) Post-HF methods 2. Density Functional Theory 3. Semiempirical methods 4. Molecular Mechanics Computational Chemistry " Computational
Density Functional Theory: from theory to Applications
 Density Functional Theory: from theory to Applications Uni Mainz November 29, 2010 The self interaction error and its correction Perdew-Zunger SIC Average-density approximation Weighted density approximation
Density Functional Theory: from theory to Applications Uni Mainz November 29, 2010 The self interaction error and its correction Perdew-Zunger SIC Average-density approximation Weighted density approximation
Key concepts in Density Functional Theory (II)
") Kohn-Sham scheme and band structures European Theoretical Spectroscopy Facility (ETSF) CNRS - Laboratoire des Solides Irradiés Ecole Polytechnique, Palaiseau - France Present Address: LPMCN Université
Kohn-Sham scheme and band structures European Theoretical Spectroscopy Facility (ETSF) CNRS - Laboratoire des Solides Irradiés Ecole Polytechnique, Palaiseau - France Present Address: LPMCN Université
Key concepts in Density Functional Theory
 From the many body problem to the Kohn-Sham scheme ILM (LPMCN) CNRS, Université Lyon 1 - France European Theoretical Spectroscopy Facility (ETSF) December 12, 2012 Lyon Outline 1 The many-body problem
From the many body problem to the Kohn-Sham scheme ILM (LPMCN) CNRS, Université Lyon 1 - France European Theoretical Spectroscopy Facility (ETSF) December 12, 2012 Lyon Outline 1 The many-body problem
nanohub.org learning module: Prelab lecture on bonding and band structure in Si
 nanohub.org learning module: Prelab lecture on bonding and band structure in Si Ravi Vedula, Janam Javerhi, Alejandro Strachan Center for Predictive Materials Modeling and Simulation, School of Materials
nanohub.org learning module: Prelab lecture on bonding and band structure in Si Ravi Vedula, Janam Javerhi, Alejandro Strachan Center for Predictive Materials Modeling and Simulation, School of Materials
Introduction to First-Principles Method
 Joint ICTP/CAS/IAEA School & Workshop on Plasma-Materials Interaction in Fusion Devices, July 18-22, 2016, Hefei Introduction to First-Principles Method by Guang-Hong LU ( 吕广宏 ) Beihang University Computer
Joint ICTP/CAS/IAEA School & Workshop on Plasma-Materials Interaction in Fusion Devices, July 18-22, 2016, Hefei Introduction to First-Principles Method by Guang-Hong LU ( 吕广宏 ) Beihang University Computer
1 Density functional theory (DFT)
") 1 Density functional theory (DFT) 1.1 Introduction Density functional theory is an alternative to ab initio methods for solving the nonrelativistic, time-independent Schrödinger equation H Φ = E Φ. The
1 Density functional theory (DFT) 1.1 Introduction Density functional theory is an alternative to ab initio methods for solving the nonrelativistic, time-independent Schrödinger equation H Φ = E Φ. The
First-principles modeling: The evolution of the field from Walter Kohn s seminal work to today s computer-aided materials design
 First-principles modeling: The evolution of the field from Walter Kohn s seminal work to today s computer-aided materials design Peter Kratzer 5/2/2018 Peter Kratzer Abeokuta School 5/2/2018 1 / 34 Outline
First-principles modeling: The evolution of the field from Walter Kohn s seminal work to today s computer-aided materials design Peter Kratzer 5/2/2018 Peter Kratzer Abeokuta School 5/2/2018 1 / 34 Outline
Key concepts in Density Functional Theory (II) Silvana Botti
 Silvana Botti") Kohn-Sham scheme, band structure and optical spectra European Theoretical Spectroscopy Facility (ETSF) CNRS - Laboratoire des Solides Irradiés Ecole Polytechnique, Palaiseau - France Temporary Address:
Kohn-Sham scheme, band structure and optical spectra European Theoretical Spectroscopy Facility (ETSF) CNRS - Laboratoire des Solides Irradiés Ecole Polytechnique, Palaiseau - France Temporary Address:
Density Functional Theory. Martin Lüders Daresbury Laboratory
 Density Functional Theory Martin Lüders Daresbury Laboratory Ab initio Calculations Hamiltonian: (without external fields, non-relativistic) impossible to solve exactly!! Electrons Nuclei Electron-Nuclei
Density Functional Theory Martin Lüders Daresbury Laboratory Ab initio Calculations Hamiltonian: (without external fields, non-relativistic) impossible to solve exactly!! Electrons Nuclei Electron-Nuclei
Pseudopotentials: design, testing, typical errors
 Pseudopotentials: design, testing, typical errors Kevin F. Garrity Part 1 National Institute of Standards and Technology (NIST) Uncertainty Quantification in Materials Modeling 2015 Parameter free calculations.
Pseudopotentials: design, testing, typical errors Kevin F. Garrity Part 1 National Institute of Standards and Technology (NIST) Uncertainty Quantification in Materials Modeling 2015 Parameter free calculations.
Key concepts in Density Functional Theory (I) Silvana Botti
 Silvana Botti") From the many body problem to the Kohn-Sham scheme European Theoretical Spectroscopy Facility (ETSF) CNRS - Laboratoire des Solides Irradiés Ecole Polytechnique, Palaiseau - France Temporary Address: Centre
From the many body problem to the Kohn-Sham scheme European Theoretical Spectroscopy Facility (ETSF) CNRS - Laboratoire des Solides Irradiés Ecole Polytechnique, Palaiseau - France Temporary Address: Centre
MODULE 2: QUANTUM MECHANICS. Principles and Theory
 MODULE 2: QUANTUM MECHANICS Principles and Theory You are here http://www.lbl.gov/cs/html/exascale4energy/nuclear.html 2 Short Review of Quantum Mechanics Why do we need quantum mechanics? Bonding and
MODULE 2: QUANTUM MECHANICS Principles and Theory You are here http://www.lbl.gov/cs/html/exascale4energy/nuclear.html 2 Short Review of Quantum Mechanics Why do we need quantum mechanics? Bonding and
Electrochemistry project, Chemistry Department, November Ab-initio Molecular Dynamics Simulation
 Electrochemistry project, Chemistry Department, November 2006 Ab-initio Molecular Dynamics Simulation Outline Introduction Ab-initio concepts Total energy concepts Adsorption energy calculation Project
Electrochemistry project, Chemistry Department, November 2006 Ab-initio Molecular Dynamics Simulation Outline Introduction Ab-initio concepts Total energy concepts Adsorption energy calculation Project
Dept of Mechanical Engineering MIT Nanoengineering group
 1 Dept of Mechanical Engineering MIT Nanoengineering group » Recap of HK theorems and KS equations» The physical meaning of the XC energy» Solution of a one-particle Schroedinger equation» Pseudo Potentials»
1 Dept of Mechanical Engineering MIT Nanoengineering group » Recap of HK theorems and KS equations» The physical meaning of the XC energy» Solution of a one-particle Schroedinger equation» Pseudo Potentials»
Session 1. Introduction to Computational Chemistry. Computational (chemistry education) and/or (Computational chemistry) education
 and/or (Computational chemistry) education") Session 1 Introduction to Computational Chemistry 1 Introduction to Computational Chemistry Computational (chemistry education) and/or (Computational chemistry) education First one: Use computational tools
Session 1 Introduction to Computational Chemistry 1 Introduction to Computational Chemistry Computational (chemistry education) and/or (Computational chemistry) education First one: Use computational tools
Chapter 3. The (L)APW+lo Method. 3.1 Choosing A Basis Set
APW+lo Method. 3.1 Choosing A Basis Set") Chapter 3 The (L)APW+lo Method 3.1 Choosing A Basis Set The Kohn-Sham equations (Eq. (2.17)) provide a formulation of how to practically find a solution to the Hohenberg-Kohn functional (Eq. (2.15)). Nevertheless
Chapter 3 The (L)APW+lo Method 3.1 Choosing A Basis Set The Kohn-Sham equations (Eq. (2.17)) provide a formulation of how to practically find a solution to the Hohenberg-Kohn functional (Eq. (2.15)). Nevertheless
DFT EXERCISES. FELIPE CERVANTES SODI January 2006
 DFT EXERCISES FELIPE CERVANTES SODI January 2006 http://www.csanyi.net/wiki/space/dftexercises Dr. Gábor Csányi 1 Hydrogen atom Place a single H atom in the middle of a largish unit cell (start with a
DFT EXERCISES FELIPE CERVANTES SODI January 2006 http://www.csanyi.net/wiki/space/dftexercises Dr. Gábor Csányi 1 Hydrogen atom Place a single H atom in the middle of a largish unit cell (start with a
Introduction to Density Functional Theory with Applications to Graphene Branislav K. Nikolić
 Introduction to Density Functional Theory with Applications to Graphene Branislav K. Nikolić Department of Physics and Astronomy, University of Delaware, Newark, DE 19716, U.S.A. http://wiki.physics.udel.edu/phys824
Introduction to Density Functional Theory with Applications to Graphene Branislav K. Nikolić Department of Physics and Astronomy, University of Delaware, Newark, DE 19716, U.S.A. http://wiki.physics.udel.edu/phys824
Supporting information. Origins of High Electrolyte-Electrode Interfacial Resistances in Lithium Cells. Containing Garnet Type LLZO Solid Electrolytes
 Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 2014 Supporting information Origins of High Electrolyte-Electrode Interfacial Resistances
Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 2014 Supporting information Origins of High Electrolyte-Electrode Interfacial Resistances
Introduction to density functional perturbation theory for lattice dynamics
 Introduction to density functional perturbation theory for lattice dynamics SISSA and DEMOCRITOS Trieste (Italy) Outline 1 Lattice dynamic of a solid: phonons Description of a solid Equations of motion
Introduction to density functional perturbation theory for lattice dynamics SISSA and DEMOCRITOS Trieste (Italy) Outline 1 Lattice dynamic of a solid: phonons Description of a solid Equations of motion
Answers Quantum Chemistry NWI-MOL406 G. C. Groenenboom and G. A. de Wijs, HG00.307, 8:30-11:30, 21 jan 2014
 Answers Quantum Chemistry NWI-MOL406 G. C. Groenenboom and G. A. de Wijs, HG00.307, 8:30-11:30, 21 jan 2014 Question 1: Basis sets Consider the split valence SV3-21G one electron basis set for formaldehyde
Answers Quantum Chemistry NWI-MOL406 G. C. Groenenboom and G. A. de Wijs, HG00.307, 8:30-11:30, 21 jan 2014 Question 1: Basis sets Consider the split valence SV3-21G one electron basis set for formaldehyde
Recent advances in development of single-point orbital-free kinetic energy functionals
 PacifiChem 2010 p. 1/29 Recent advances in development of single-point orbital-free kinetic energy functionals Valentin V. Karasiev vkarasev@qtp.ufl.edu Quantum Theory Project, Departments of Physics and
PacifiChem 2010 p. 1/29 Recent advances in development of single-point orbital-free kinetic energy functionals Valentin V. Karasiev vkarasev@qtp.ufl.edu Quantum Theory Project, Departments of Physics and
Electronic Structure Theory for Periodic Systems: The Concepts. Christian Ratsch
 Electronic Structure Theory for Periodic Systems: The Concepts Christian Ratsch Institute for Pure and Applied Mathematics and Department of Mathematics, UCLA Motivation There are 10 20 atoms in 1 mm 3
Electronic Structure Theory for Periodic Systems: The Concepts Christian Ratsch Institute for Pure and Applied Mathematics and Department of Mathematics, UCLA Motivation There are 10 20 atoms in 1 mm 3
Solid State Theory: Band Structure Methods
 Solid State Theory: Band Structure Methods Lilia Boeri Wed., 11:15-12:45 HS P3 (PH02112) http://itp.tugraz.at/lv/boeri/ele/ Plan of the Lecture: DFT1+2: Hohenberg-Kohn Theorem and Kohn and Sham equations.
Solid State Theory: Band Structure Methods Lilia Boeri Wed., 11:15-12:45 HS P3 (PH02112) http://itp.tugraz.at/lv/boeri/ele/ Plan of the Lecture: DFT1+2: Hohenberg-Kohn Theorem and Kohn and Sham equations.
Two implementations of the Projector Augmented Wave (PAW) formalism
 formalism") Introduction The tools available for detailed first-principles studies of materials have benefited enormously from the development of several international collaborations engaged in developing open source
Introduction The tools available for detailed first-principles studies of materials have benefited enormously from the development of several international collaborations engaged in developing open source
CHEM6085: Density Functional Theory
 Lecture 11 CHEM6085: Density Functional Theory DFT for periodic crystalline solids C.-K. Skylaris 1 Electron in a one-dimensional periodic box (in atomic units) Schrödinger equation Energy eigenvalues
Lecture 11 CHEM6085: Density Functional Theory DFT for periodic crystalline solids C.-K. Skylaris 1 Electron in a one-dimensional periodic box (in atomic units) Schrödinger equation Energy eigenvalues
Electronic structure, plane waves and pseudopotentials
 Electronic structure, plane waves and pseudopotentials P.J. Hasnip Spectroscopy Workshop 2009 We want to be able to predict what electrons and nuclei will do from first principles, without needing to know
Electronic structure, plane waves and pseudopotentials P.J. Hasnip Spectroscopy Workshop 2009 We want to be able to predict what electrons and nuclei will do from first principles, without needing to know
DFT / SIESTA algorithms
 DFT / SIESTA algorithms Javier Junquera José M. Soler References http://siesta.icmab.es Documentation Tutorials Atomic units e = m e = =1 atomic mass unit = m e atomic length unit = 1 Bohr = 0.5292 Ang
DFT / SIESTA algorithms Javier Junquera José M. Soler References http://siesta.icmab.es Documentation Tutorials Atomic units e = m e = =1 atomic mass unit = m e atomic length unit = 1 Bohr = 0.5292 Ang
Electrons in Crystals. Chris J. Pickard
 Electrons in Crystals Chris J. Pickard Electrons in Crystals The electrons in a crystal experience a potential with the periodicity of the Bravais lattice: U(r + R) = U(r) The scale of the periodicity
Electrons in Crystals Chris J. Pickard Electrons in Crystals The electrons in a crystal experience a potential with the periodicity of the Bravais lattice: U(r + R) = U(r) The scale of the periodicity
Algorithms and Computational Aspects of DFT Calculations
 Algorithms and Computational Aspects of DFT Calculations Part I Juan Meza and Chao Yang High Performance Computing Research Lawrence Berkeley National Laboratory IMA Tutorial Mathematical and Computational
Algorithms and Computational Aspects of DFT Calculations Part I Juan Meza and Chao Yang High Performance Computing Research Lawrence Berkeley National Laboratory IMA Tutorial Mathematical and Computational
Exchange-Correlation Functional
 Exchange-Correlation Functional Aiichiro Nakano Collaboratory for Advanced Computing & Simulations Depts. of Computer Science, Physics & Astronomy, Chemical Engineering & Materials Science, and Biological
Exchange-Correlation Functional Aiichiro Nakano Collaboratory for Advanced Computing & Simulations Depts. of Computer Science, Physics & Astronomy, Chemical Engineering & Materials Science, and Biological
DFT in practice : Part II. Ersen Mete
 pseudopotentials Department of Physics Balıkesir University, Balıkesir - Turkey August 13, 2009 - NanoDFT 09, İzmir Institute of Technology, İzmir Outline Pseudopotentials Basic Ideas Norm-conserving pseudopotentials
pseudopotentials Department of Physics Balıkesir University, Balıkesir - Turkey August 13, 2009 - NanoDFT 09, İzmir Institute of Technology, İzmir Outline Pseudopotentials Basic Ideas Norm-conserving pseudopotentials
Practical calculations using first-principles QM Convergence, convergence, convergence
 Practical calculations using first-principles QM Convergence, convergence, convergence Keith Refson STFC Rutherford Appleton Laboratory September 18, 2007 Results of First-Principles Simulations..........................................................
Practical calculations using first-principles QM Convergence, convergence, convergence Keith Refson STFC Rutherford Appleton Laboratory September 18, 2007 Results of First-Principles Simulations..........................................................
Electrons in a periodic potential
 Chapter 3 Electrons in a periodic potential 3.1 Bloch s theorem. We consider in this chapter electrons under the influence of a static, periodic potential V (x), i.e. such that it fulfills V (x) = V (x
Chapter 3 Electrons in a periodic potential 3.1 Bloch s theorem. We consider in this chapter electrons under the influence of a static, periodic potential V (x), i.e. such that it fulfills V (x) = V (x
Finite-Temperature Hartree-Fock Exchange and Exchange- Correlation Free Energy Functionals. Travis Sjostrom. IPAM 2012 Workshop IV
 1 of 45 Finite-Temperature Hartree-Fock Exchange and Exchange- Correlation Free Energy Functionals Travis Sjostrom Quantum Theory Project Depts. of Physics and Chemistry IPAM 2012 Workshop IV 2012 2 of
1 of 45 Finite-Temperature Hartree-Fock Exchange and Exchange- Correlation Free Energy Functionals Travis Sjostrom Quantum Theory Project Depts. of Physics and Chemistry IPAM 2012 Workshop IV 2012 2 of
The Plane-wave Pseudopotential Method
 The Plane-wave Pseudopotential Method k(r) = X G c k,g e i(g+k) r Chris J Pickard Electrons in a Solid Nearly Free Electrons Nearly Free Electrons Nearly Free Electrons Electronic Structures Methods Empirical
The Plane-wave Pseudopotential Method k(r) = X G c k,g e i(g+k) r Chris J Pickard Electrons in a Solid Nearly Free Electrons Nearly Free Electrons Nearly Free Electrons Electronic Structures Methods Empirical
Introduction to DFT and Density Functionals. by Michel Côté Université de Montréal Département de physique
 Introduction to DFT and Density Functionals by Michel Côté Université de Montréal Département de physique Eamples Carbazole molecule Inside of diamant Réf: Jean-François Brière http://www.phys.umontreal.ca/~michel_
Introduction to DFT and Density Functionals by Michel Côté Université de Montréal Département de physique Eamples Carbazole molecule Inside of diamant Réf: Jean-François Brière http://www.phys.umontreal.ca/~michel_
Basics of Density Functional Theory (DFT)
") Basics of Density Functional Theory (DFT) Ari Paavo SEITSONEN Ari.P.Seitsonen@iki.fi Département de Chimie École Normale Supérieure, Paris École de Sidi-BelAbbès de Nanomateriaux // Octobre 8-12, 2016
Basics of Density Functional Theory (DFT) Ari Paavo SEITSONEN Ari.P.Seitsonen@iki.fi Département de Chimie École Normale Supérieure, Paris École de Sidi-BelAbbès de Nanomateriaux // Octobre 8-12, 2016
All electron optimized effective potential method for solids
 All electron optimized effective potential method for solids Institut für Theoretische Physik Freie Universität Berlin, Germany and Fritz Haber Institute of the Max Planck Society, Berlin, Germany. 22
All electron optimized effective potential method for solids Institut für Theoretische Physik Freie Universität Berlin, Germany and Fritz Haber Institute of the Max Planck Society, Berlin, Germany. 22
Physics 211B : Problem Set #0
 Physics 211B : Problem Set #0 These problems provide a cross section of the sort of exercises I would have assigned had I taught 211A. Please take a look at all the problems, and turn in problems 1, 4,
Physics 211B : Problem Set #0 These problems provide a cross section of the sort of exercises I would have assigned had I taught 211A. Please take a look at all the problems, and turn in problems 1, 4,
An Approximate DFT Method: The Density-Functional Tight-Binding (DFTB) Method
 Method") Fakultät für Mathematik und Naturwissenschaften - Lehrstuhl für Physikalische Chemie I / Theoretische Chemie An Approximate DFT Method: The Density-Functional Tight-Binding (DFTB) Method Jan-Ole Joswig
Fakultät für Mathematik und Naturwissenschaften - Lehrstuhl für Physikalische Chemie I / Theoretische Chemie An Approximate DFT Method: The Density-Functional Tight-Binding (DFTB) Method Jan-Ole Joswig
Density Functional Theory (DFT)
") Density Functional Theory (DFT) An Introduction by A.I. Al-Sharif Irbid, Aug, 2 nd, 2009 Density Functional Theory Revolutionized our approach to the electronic structure of atoms, molecules and solid
Density Functional Theory (DFT) An Introduction by A.I. Al-Sharif Irbid, Aug, 2 nd, 2009 Density Functional Theory Revolutionized our approach to the electronic structure of atoms, molecules and solid
Density functional theory in the solid state
 Density functional theory in the solid state Ari P Seitsonen IMPMC, CNRS & Universités 6 et 7 Paris, IPGP Department of Applied Physics, Helsinki University of Technology Physikalisch-Chemisches Institut
Density functional theory in the solid state Ari P Seitsonen IMPMC, CNRS & Universités 6 et 7 Paris, IPGP Department of Applied Physics, Helsinki University of Technology Physikalisch-Chemisches Institut
Quantum Modeling of Solids: Basic Properties
 1.021, 3.021, 10.333, 22.00 : Introduction to Modeling and Simulation : Spring 2011 Part II Quantum Mechanical Methods : Lecture 5 Quantum Modeling of Solids: Basic Properties Jeffrey C. Grossman Department
1.021, 3.021, 10.333, 22.00 : Introduction to Modeling and Simulation : Spring 2011 Part II Quantum Mechanical Methods : Lecture 5 Quantum Modeling of Solids: Basic Properties Jeffrey C. Grossman Department
DENSITY FUNCTIONAL THEORY FOR NON-THEORISTS JOHN P. PERDEW DEPARTMENTS OF PHYSICS AND CHEMISTRY TEMPLE UNIVERSITY
 DENSITY FUNCTIONAL THEORY FOR NON-THEORISTS JOHN P. PERDEW DEPARTMENTS OF PHYSICS AND CHEMISTRY TEMPLE UNIVERSITY A TUTORIAL FOR PHYSICAL SCIENTISTS WHO MAY OR MAY NOT HATE EQUATIONS AND PROOFS REFERENCES
DENSITY FUNCTIONAL THEORY FOR NON-THEORISTS JOHN P. PERDEW DEPARTMENTS OF PHYSICS AND CHEMISTRY TEMPLE UNIVERSITY A TUTORIAL FOR PHYSICAL SCIENTISTS WHO MAY OR MAY NOT HATE EQUATIONS AND PROOFS REFERENCES
Density Functional Theory
 Density Functional Theory March 26, 2009 ? DENSITY FUNCTIONAL THEORY is a method to successfully describe the behavior of atomic and molecular systems and is used for instance for: structural prediction
Density Functional Theory March 26, 2009 ? DENSITY FUNCTIONAL THEORY is a method to successfully describe the behavior of atomic and molecular systems and is used for instance for: structural prediction
Introduction to first-principles modelling and CASTEP
 to first-principles modelling and Phil Hasnip to + Atomistic Simulations If we know what the bonding in a material is beforehand, then we can often find good expressions for the forces between atoms, e.g.
to first-principles modelling and Phil Hasnip to + Atomistic Simulations If we know what the bonding in a material is beforehand, then we can often find good expressions for the forces between atoms, e.g.
Self Consistent Cycle
 Self Consistent Cycle Step 0 : defining your system namelist SYSTEM How to specify the System All periodic systems can be specified by a Bravais Lattice and and atomic basis How to specify the Bravais
Self Consistent Cycle Step 0 : defining your system namelist SYSTEM How to specify the System All periodic systems can be specified by a Bravais Lattice and and atomic basis How to specify the Bravais
Electronic structure of solids: basic concepts and methods
 Electronic structure of solids: basic concepts and methods Ondřej Šipr II. NEVF 514 Surface Physics Winter Term 2016-2017 Troja, 21st October 2016 Outline A bit of formal mathematics for the beginning
Electronic structure of solids: basic concepts and methods Ondřej Šipr II. NEVF 514 Surface Physics Winter Term 2016-2017 Troja, 21st October 2016 Outline A bit of formal mathematics for the beginning
Density Functional Theory
 Density Functional Theory Iain Bethune EPCC ibethune@epcc.ed.ac.uk Overview Background Classical Atomistic Simulation Essential Quantum Mechanics DFT: Approximations and Theory DFT: Implementation using
Density Functional Theory Iain Bethune EPCC ibethune@epcc.ed.ac.uk Overview Background Classical Atomistic Simulation Essential Quantum Mechanics DFT: Approximations and Theory DFT: Implementation using
Electronic Structure Calculations, Density Functional Theory and its Modern Implementations
 Tutoriel Big RENOBLE Electronic Structure Calculations, Density Functional Theory and its Modern Implementations Thierry Deutsch L_Sim - CEA renoble 19 October 2011 Outline 1 of Atomistic calculations
Tutoriel Big RENOBLE Electronic Structure Calculations, Density Functional Theory and its Modern Implementations Thierry Deutsch L_Sim - CEA renoble 19 October 2011 Outline 1 of Atomistic calculations
Lecture 4: Basic elements of band theory
 Phys 769 Selected Topics in Condensed Matter Physics Summer 010 Lecture 4: Basic elements of band theory Lecturer: Anthony J. Leggett TA: Bill Coish 1 Introduction Most matter, in particular most insulating
Phys 769 Selected Topics in Condensed Matter Physics Summer 010 Lecture 4: Basic elements of band theory Lecturer: Anthony J. Leggett TA: Bill Coish 1 Introduction Most matter, in particular most insulating
Band calculations: Theory and Applications
 Band calculations: Theory and Applications Lecture 2: Different approximations for the exchange-correlation correlation functional in DFT Local density approximation () Generalized gradient approximation
Band calculations: Theory and Applications Lecture 2: Different approximations for the exchange-correlation correlation functional in DFT Local density approximation () Generalized gradient approximation
Module 6 1. Density functional theory
 Module 6 1. Density functional theory Updated May 12, 2016 B A DDFT C K A bird s-eye view of density-functional theory Authors: Klaus Capelle G http://arxiv.org/abs/cond-mat/0211443 R https://trac.cc.jyu.fi/projects/toolbox/wiki/dft
Module 6 1. Density functional theory Updated May 12, 2016 B A DDFT C K A bird s-eye view of density-functional theory Authors: Klaus Capelle G http://arxiv.org/abs/cond-mat/0211443 R https://trac.cc.jyu.fi/projects/toolbox/wiki/dft
Determining elastic constants of graphene using density functional theory
 UNIVERSITY OF GRAZ Determining elastic constants of graphene using density functional theory by Max Niederreiter 01510759 A thesis submitted in partial fulfillment for the degree of Bachelor Of Science
UNIVERSITY OF GRAZ Determining elastic constants of graphene using density functional theory by Max Niederreiter 01510759 A thesis submitted in partial fulfillment for the degree of Bachelor Of Science
Introduction to density-functional theory. Emmanuel Fromager
 Institut de Chimie, Strasbourg, France Page 1 Emmanuel Fromager Institut de Chimie de Strasbourg - Laboratoire de Chimie Quantique - Université de Strasbourg /CNRS M2 lecture, Strasbourg, France. Institut
Institut de Chimie, Strasbourg, France Page 1 Emmanuel Fromager Institut de Chimie de Strasbourg - Laboratoire de Chimie Quantique - Université de Strasbourg /CNRS M2 lecture, Strasbourg, France. Institut
Multi-Scale Modeling from First Principles
 m mm Multi-Scale Modeling from First Principles μm nm m mm μm nm space space Predictive modeling and simulations must address all time and Continuum Equations, densityfunctional space scales Rate Equations
m mm Multi-Scale Modeling from First Principles μm nm m mm μm nm space space Predictive modeling and simulations must address all time and Continuum Equations, densityfunctional space scales Rate Equations
CHAPTER 3 WIEN2k. Chapter 3 : WIEN2k 50
 CHAPTER 3 WIEN2k WIEN2k is one of the fastest and reliable simulation codes among computational methods. All the computational work presented on lanthanide intermetallic compounds has been performed by
CHAPTER 3 WIEN2k WIEN2k is one of the fastest and reliable simulation codes among computational methods. All the computational work presented on lanthanide intermetallic compounds has been performed by
Dept of Mechanical Engineering MIT Nanoengineering group
 1 Dept of Mechanical Engineering MIT Nanoengineering group » To calculate all the properties of a molecule or crystalline system knowing its atomic information: Atomic species Their coordinates The Symmetry
1 Dept of Mechanical Engineering MIT Nanoengineering group » To calculate all the properties of a molecule or crystalline system knowing its atomic information: Atomic species Their coordinates The Symmetry
Introduction to Computational Chemistry Computational (chemistry education) and/or. (Computational chemistry) education
 and/or. (Computational chemistry) education") Introduction to Computational Chemistry Computational (chemistry education) and/or (Computational chemistry) education First one: Use computational tools to help increase student understanding of material
Introduction to Computational Chemistry Computational (chemistry education) and/or (Computational chemistry) education First one: Use computational tools to help increase student understanding of material
DFT: Exchange-Correlation
 DFT: Exchange-Correlation Local functionals, exact exchange and other post-dft methods Paul Tulip Centre for Materials Physics Department of Physics University of Durham Outline Introduction What is exchange
DFT: Exchange-Correlation Local functionals, exact exchange and other post-dft methods Paul Tulip Centre for Materials Physics Department of Physics University of Durham Outline Introduction What is exchange
Introduction to Density Functional Theory
 1 Introduction to Density Functional Theory 21 February 2011; V172 P.Ravindran, FME-course on Ab initio Modelling of solar cell Materials 21 February 2011 Introduction to DFT 2 3 4 Ab initio Computational
1 Introduction to Density Functional Theory 21 February 2011; V172 P.Ravindran, FME-course on Ab initio Modelling of solar cell Materials 21 February 2011 Introduction to DFT 2 3 4 Ab initio Computational
CHEM6085: Density Functional Theory
 Lecture 5 CHEM6085: Density Functional Theory Orbital-free (or pure ) DFT C.-K. Skylaris 1 Consists of three terms The electronic Hamiltonian operator Electronic kinetic energy operator Electron-Electron
Lecture 5 CHEM6085: Density Functional Theory Orbital-free (or pure ) DFT C.-K. Skylaris 1 Consists of three terms The electronic Hamiltonian operator Electronic kinetic energy operator Electron-Electron
References. Documentation Manuals Tutorials Publications
 References http://siesta.icmab.es Documentation Manuals Tutorials Publications Atomic units e = m e = =1 atomic mass unit = m e atomic length unit = 1 Bohr = 0.5292 Ang atomic energy unit = 1 Hartree =
References http://siesta.icmab.es Documentation Manuals Tutorials Publications Atomic units e = m e = =1 atomic mass unit = m e atomic length unit = 1 Bohr = 0.5292 Ang atomic energy unit = 1 Hartree =
Homogeneous Electric and Magnetic Fields in Periodic Systems
 Electric and Magnetic Fields in Periodic Systems Josef W. idepartment of Chemistry and Institute for Research in Materials Dalhousie University Halifax, Nova Scotia June 2012 1/24 Acknowledgments NSERC,
Electric and Magnetic Fields in Periodic Systems Josef W. idepartment of Chemistry and Institute for Research in Materials Dalhousie University Halifax, Nova Scotia June 2012 1/24 Acknowledgments NSERC,
Electronic Structure of MoS 2 Nanotubes
 Clemson University TigerPrints All Dissertations Dissertations 8-2007 Electronic Structure of MoS 2 Nanotubes Lingyun Xu Clemson University, neilxu@gmail.com Follow this and additional works at: http://tigerprints.clemson.edu/all_dissertations
Clemson University TigerPrints All Dissertations Dissertations 8-2007 Electronic Structure of MoS 2 Nanotubes Lingyun Xu Clemson University, neilxu@gmail.com Follow this and additional works at: http://tigerprints.clemson.edu/all_dissertations
Set the initial conditions r i. Update neighborlist. Get new forces F i
 v Set the initial conditions r i ( t 0 ), v i ( t 0 ) Update neighborlist Quantum mechanical models Get new forces F i ( r i ) Solve the equations of motion numerically over time step Δt : r i ( t n )
v Set the initial conditions r i ( t 0 ), v i ( t 0 ) Update neighborlist Quantum mechanical models Get new forces F i ( r i ) Solve the equations of motion numerically over time step Δt : r i ( t n )
Exchange Correlation Functional Investigation of RT-TDDFT on a Sodium Chloride. Dimer. Philip Straughn
 Exchange Correlation Functional Investigation of RT-TDDFT on a Sodium Chloride Dimer Philip Straughn Abstract Charge transfer between Na and Cl ions is an important problem in physical chemistry. However,
Exchange Correlation Functional Investigation of RT-TDDFT on a Sodium Chloride Dimer Philip Straughn Abstract Charge transfer between Na and Cl ions is an important problem in physical chemistry. However,
FULL POTENTIAL LINEARIZED AUGMENTED PLANE WAVE (FP-LAPW) IN THE FRAMEWORK OF DENSITY FUNCTIONAL THEORY
 IN THE FRAMEWORK OF DENSITY FUNCTIONAL THEORY") FULL POTENTIAL LINEARIZED AUGMENTED PLANE WAVE (FP-LAPW) IN THE FRAMEWORK OF DENSITY FUNCTIONAL THEORY C.A. Madu and B.N Onwuagba Department of Physics, Federal University of Technology Owerri, Nigeria
FULL POTENTIAL LINEARIZED AUGMENTED PLANE WAVE (FP-LAPW) IN THE FRAMEWORK OF DENSITY FUNCTIONAL THEORY C.A. Madu and B.N Onwuagba Department of Physics, Federal University of Technology Owerri, Nigeria
Solving Many-Body Schrödinger Equation Using Density Functional Theory and Finite Elements
 Solving Many-Body Schrödinger Equation Using Density Functional Theory and Finite Elements Institute of Physics, Academy of Sciences of the Czech Republic June 21, 2008 Introduction Contens Density Functional
Solving Many-Body Schrödinger Equation Using Density Functional Theory and Finite Elements Institute of Physics, Academy of Sciences of the Czech Republic June 21, 2008 Introduction Contens Density Functional
Pseudopotential generation and test by the ld1.x atomic code: an introduction
 and test by the ld1.x atomic code: an introduction SISSA and DEMOCRITOS Trieste (Italy) Outline 1 2 3 Spherical symmetry - I The Kohn and Sham (KS) equation is (in atomic units): [ 1 ] 2 2 + V ext (r)
and test by the ld1.x atomic code: an introduction SISSA and DEMOCRITOS Trieste (Italy) Outline 1 2 3 Spherical symmetry - I The Kohn and Sham (KS) equation is (in atomic units): [ 1 ] 2 2 + V ext (r)
Structure of Cement Phases from ab initio Modeling Crystalline C-S-HC
 Structure of Cement Phases from ab initio Modeling Crystalline C-S-HC Sergey V. Churakov sergey.churakov@psi.ch Paul Scherrer Institute Switzerland Cement Phase Composition C-S-H H Solid Solution Model
Structure of Cement Phases from ab initio Modeling Crystalline C-S-HC Sergey V. Churakov sergey.churakov@psi.ch Paul Scherrer Institute Switzerland Cement Phase Composition C-S-H H Solid Solution Model
Improved Electronic Structure and Optical Properties of sp-hybridized Semiconductors Using LDA+U SIC
 286 Brazilian Journal of Physics, vol. 36, no. 2A, June, 2006 Improved Electronic Structure and Optical Properties of sp-hybridized Semiconductors Using LDA+U SIC Clas Persson and Susanne Mirbt Department
286 Brazilian Journal of Physics, vol. 36, no. 2A, June, 2006 Improved Electronic Structure and Optical Properties of sp-hybridized Semiconductors Using LDA+U SIC Clas Persson and Susanne Mirbt Department
GEM4 Summer School OpenCourseWare
 GEM4 Summer School OpenCourseWare http://gem4.educommons.net/ http://www.gem4.org/ Lecture: Molecular Mechanics by Ju Li. Given August 9, 2006 during the GEM4 session at MIT in Cambridge, MA. Please use
GEM4 Summer School OpenCourseWare http://gem4.educommons.net/ http://www.gem4.org/ Lecture: Molecular Mechanics by Ju Li. Given August 9, 2006 during the GEM4 session at MIT in Cambridge, MA. Please use
Electronic band structure, sx-lda, Hybrid DFT, LDA+U and all that. Keith Refson STFC Rutherford Appleton Laboratory
 Electronic band structure, sx-lda, Hybrid DFT, LDA+U and all that Keith Refson STFC Rutherford Appleton Laboratory LDA/GGA DFT is good but... Naive LDA/GGA calculation severely underestimates band-gaps.
Electronic band structure, sx-lda, Hybrid DFT, LDA+U and all that Keith Refson STFC Rutherford Appleton Laboratory LDA/GGA DFT is good but... Naive LDA/GGA calculation severely underestimates band-gaps.
Introduction to DFT and its Application to Defects in Semiconductors
 Introduction to DFT and its Application to Defects in Semiconductors Noa Marom Physics and Engineering Physics Tulane University New Orleans The Future: Computer-Aided Materials Design Can access the space
Introduction to DFT and its Application to Defects in Semiconductors Noa Marom Physics and Engineering Physics Tulane University New Orleans The Future: Computer-Aided Materials Design Can access the space
Electronic structure theory: Fundamentals to frontiers. 2. Density functional theory
 Electronic structure theory: Fundamentals to frontiers. 2. Density functional theory MARTIN HEAD-GORDON, Department of Chemistry, University of California, and Chemical Sciences Division, Lawrence Berkeley
Electronic structure theory: Fundamentals to frontiers. 2. Density functional theory MARTIN HEAD-GORDON, Department of Chemistry, University of California, and Chemical Sciences Division, Lawrence Berkeley
Magnetism in transition metal oxides by post-dft methods
 Magnetism in transition metal oxides by post-dft methods Cesare Franchini Faculty of Physics & Center for Computational Materials Science University of Vienna, Austria Workshop on Magnetism in Complex
Magnetism in transition metal oxides by post-dft methods Cesare Franchini Faculty of Physics & Center for Computational Materials Science University of Vienna, Austria Workshop on Magnetism in Complex
6.730 Physics for Solid State Applications
 6.730 Physics for Solid State Applications Lecture 29: Electron-phonon Scattering Outline Bloch Electron Scattering Deformation Potential Scattering LCAO Estimation of Deformation Potential Matrix Element
6.730 Physics for Solid State Applications Lecture 29: Electron-phonon Scattering Outline Bloch Electron Scattering Deformation Potential Scattering LCAO Estimation of Deformation Potential Matrix Element
Nonlocal exchange correlation in screened-exchange density functional methods
 Nonlocal exchange correlation in screened-exchange density functional methods Byounghak Lee and Lin-Wang Wang Computational Research Division, Lawrence Berkeley National Laboratory, Berkeley, California
Nonlocal exchange correlation in screened-exchange density functional methods Byounghak Lee and Lin-Wang Wang Computational Research Division, Lawrence Berkeley National Laboratory, Berkeley, California
Kronig-Penney model. 2m dx. Solutions can be found in region I and region II Match boundary conditions
 Kronig-Penney model I II d V( x) m dx E Solutions can be found in region I and region II Match boundary conditions Linear differential equations with periodic coefficients Have exponentially decaying solutions,
Kronig-Penney model I II d V( x) m dx E Solutions can be found in region I and region II Match boundary conditions Linear differential equations with periodic coefficients Have exponentially decaying solutions,
Lecture 8: Introduction to Density Functional Theory
 Lecture 8: Introduction to Density Functional Theory Marie Curie Tutorial Series: Modeling Biomolecules December 6-11, 2004 Mark Tuckerman Dept. of Chemistry and Courant Institute of Mathematical Science
Lecture 8: Introduction to Density Functional Theory Marie Curie Tutorial Series: Modeling Biomolecules December 6-11, 2004 Mark Tuckerman Dept. of Chemistry and Courant Institute of Mathematical Science
Three Most Important Topics (MIT) Today
 Today") Three Most Important Topics (MIT) Today Electrons in periodic potential Energy gap nearly free electron Bloch Theorem Energy gap tight binding Chapter 1 1 Electrons in Periodic Potential We now know the
Three Most Important Topics (MIT) Today Electrons in periodic potential Energy gap nearly free electron Bloch Theorem Energy gap tight binding Chapter 1 1 Electrons in Periodic Potential We now know the
6: Plane waves, unit cells, k- points and all that
 The Nuts and Bolts of First-Principles Simulation 6: Plane waves, unit cells, k- points and all that Durham, 6th- 13th December 2001 CASTEP Developers Group with support from the ESF ψ k Network Overview
The Nuts and Bolts of First-Principles Simulation 6: Plane waves, unit cells, k- points and all that Durham, 6th- 13th December 2001 CASTEP Developers Group with support from the ESF ψ k Network Overview
VASP: running on HPC resources. University of Vienna, Faculty of Physics and Center for Computational Materials Science, Vienna, Austria
 VASP: running on HPC resources University of Vienna, Faculty of Physics and Center for Computational Materials Science, Vienna, Austria The Many-Body Schrödinger equation 0 @ 1 2 X i i + X i Ĥ (r 1,...,r
VASP: running on HPC resources University of Vienna, Faculty of Physics and Center for Computational Materials Science, Vienna, Austria The Many-Body Schrödinger equation 0 @ 1 2 X i i + X i Ĥ (r 1,...,r
Lecture. Ref. Ihn Ch. 3, Yu&Cardona Ch. 2
 Lecture Review of quantum mechanics, statistical physics, and solid state Band structure of materials Semiconductor band structure Semiconductor nanostructures Ref. Ihn Ch. 3, Yu&Cardona Ch. 2 Reminder
Lecture Review of quantum mechanics, statistical physics, and solid state Band structure of materials Semiconductor band structure Semiconductor nanostructures Ref. Ihn Ch. 3, Yu&Cardona Ch. 2 Reminder
The Plane-Wave Pseudopotential Method
 Hands-on Workshop on Density Functional Theory and Beyond: Computational Materials Science for Real Materials Trieste, August 6-15, 2013 The Plane-Wave Pseudopotential Method Ralph Gebauer ICTP, Trieste
Hands-on Workshop on Density Functional Theory and Beyond: Computational Materials Science for Real Materials Trieste, August 6-15, 2013 The Plane-Wave Pseudopotential Method Ralph Gebauer ICTP, Trieste
Intro to ab initio methods
 Lecture 2 Part A Intro to ab initio methods Recommended reading: Leach, Chapters 2 & 3 for QM methods For more QM methods: Essentials of Computational Chemistry by C.J. Cramer, Wiley (2002) 1 ab initio
Lecture 2 Part A Intro to ab initio methods Recommended reading: Leach, Chapters 2 & 3 for QM methods For more QM methods: Essentials of Computational Chemistry by C.J. Cramer, Wiley (2002) 1 ab initio