CHAPTER 4 Structure, Optical and Reactivity Analysis of Platinum Cluster 4.1 Introduction
|
|
|
- Todd George
- 6 years ago
- Views:
Transcription
1 CHAPTER 4 Structure, Optical and Reactivity Analysis of Platinum Cluster 4.1 Introduction Platinum clusters and other transition metal clusters like, Rh, and Pd are used as catalysts for reducing toxic pollutants, such as CO, NO, and hydrocarbons in automotive exhaust systems [1]. Compared with other metal clusters, Pt clusters have small hydrogenation energy, which make them suitable catalysts in many reactions such as hydrogenation, dehydrogenation, and cracking of various hydrocarbons [2]. Pt nanoclustrs are the preferred oxidation and reduction catalyst for low-temperature proton exchange membrane (PEM) fuel cells [3]. It also acts as an excellent catalyst for a number of electrochemical reactions related to fuel cells [4, 5] and for oxygen reduction reaction [6]. With the decrease of size, the catalytic activities of Pt tend to increase because of the increased surface area of smaller particle and the structural sensitivity of some reactions [7]. Reactivity of cluster is highly sensitive to its size and shape [8, 9]. The study of the mechanical, chemical and electromagnetic properties of the platinum clusters and their relationships to size and shape will enhance our understanding of these small clusters. This knowledge will aso be helpful in designing more efficient materials using clusters, thereby, increasing their applications in the chemical and material industry. The first step towards this goal is to determine their structure, which is very difficult to access directly using present experimental techniques. Structure prediction of platinum clusters becomes quite challenging because of competition between spin exchange stabilization and electron correlation effects [10]. These two effects tend to alter the electronic states and the structural properties as a function of theoretical method. Moreover, the open d shell of these clusters generates several electronic states with varied spin multiplicities and geometries, which make the investigation of structural and the spectroscopic properties of these clusters difficult [11]. The cluster size [12] and sometimes their symmetries [13] could be inferred from experiments, but the microscopic descriptions of their structure are far from definitive. As agreement between theoretical and experimental spectra confirms that the theoretically predicted structures are true; accurate prediction of vibrational frequency, IR intensity and Raman activity will facilitate in identifying the true cluster geometries. Despite many reports 69
2 on Pt clusters, the study of the optical response of small Pt clusters has not been performed to our knowledge. The investigation of optical response may provide much useful information about the dynamic properties of Pt clusters. Here, we have determined the ground state structures of Pt clusters and calculated their properties based on DFT methods. The absorption spectra are calculated using time dependent density functionl theory (TDDFT) on the ground state geometries of Pt clusters up to 10 atoms. Our aim is to understand the structure, stability, reactivity of small platinum clusters (Pt n, n=2 to 10) and shed light on the unique nature and properties of these transition metal clusters. 4.2 Computational details Initial geometries for platinum clusters Pt n (n=2-10) are generated using a constant temperature ab-initio molecular dynamics smulation using SIESTA 3.1 [14] near the melting temperature of bulk platinum. Ab-initio molecular dynamics are carried out in the NVT ensemble and temperature is controlled using Nose thermostate [15]. During the simulation process, the structure of the cluster goes through various changes. Based on low energy criteria, we chose some of these structures with reasonably different geometry. We have also considered those ground state geometries reported in the literature for Pt cluster. The calculations are performed within the framework of density functional theory employing GAUSSIAN 09 suite of program [16]. In this study, we have used B3LYP [17, 18] and B3PW91 [17, 19] functionals. The relativistic Los Alamos National Laboratory effective core potentials as well as DZ atomic basis set (LanL2DZ) [20], which uses relativistic effective core potentials (ECP) to reduce the number electrons are used in the calculation. In order to see the effects of basis set on the calculated properties, we also used LanL2MB [20-22] basis set. We have first performed optimization of the neutral clusters. It was followed by single point calculations of the positively and negatively charged clusters having the same optimized geometry as the neutral clusters to determine the vertical ionization potentials and electron affinities. The TDDFT calculations are performed on the ground state geometries of DFT calculations. The photoabsorption cross section has been calculated using the relation [23] (4.1) 70

3 where m is the mass of an electron, e is its charge, c is the velocity of light, is the vacuum permittivity, is the radiation energy with angular frequency. The transition energy and oscillator strengths corresponding to transitions from initial state i to final state l are computed for each point in the ensemble. We have convoluted the intensity with a normalized line shape function peaked at the transition energy. The calculation of photoabsorption cross section has been carried out using the methods implemented in NEWTON-X program [24, 25]. 4.3 Results and discussion To test the reliability of our method, we first calculated the IP for single platinum atom. The experimental value for the single Pt atom is reported as ev [26] and our calculated IP with B3LYP functional and LanL2DZ basis set is 8.75 ev. Other calculation levels give IP values as 8.51 ev (B3LYP/LanL2MB), 8.70 ev (B3PW91/LanL2DZ) and 8.44 ev (B3PW91/LanL2MB). We can see, IP value given by B3LYP/LanL2DZ is closest to the experimental value. The discussed properties in the following sections are calculated using B3LYP/LanL2DZ method, otherwise specifically mentioned. The most stable structures and some low-lying isomers of platinum clusters up to 10 atoms are shown in Figure Cluster geometries Most stable geometry Higher energy isomers E=0.00 ev 71








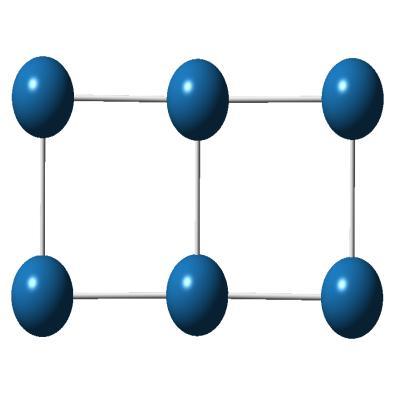
4 Most stable geometry Higher energy isomers E=0.00 ev E=0.10 ev E=2.10 ev E=0.00 ev E=0.31 ev E=0.36 ev E=0.00 ev E=0.06 ev E=0.11 ev E=0.00 ev E=0.29 ev E=0.39 ev 72







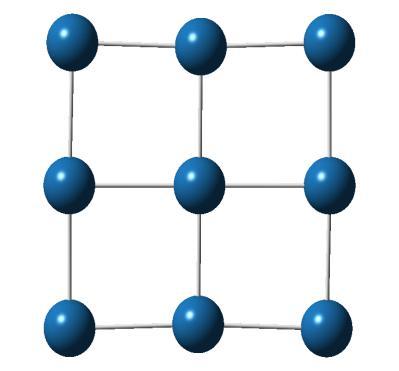
5 Most stable geometry Higher energy isomers E=0.00 ev E=0.13 ev E=0.92 ev E=0.00 ev E=0.64 ev E=1.23 ev E=0.00 ev E=0.90 ev E=2.04 ev 73


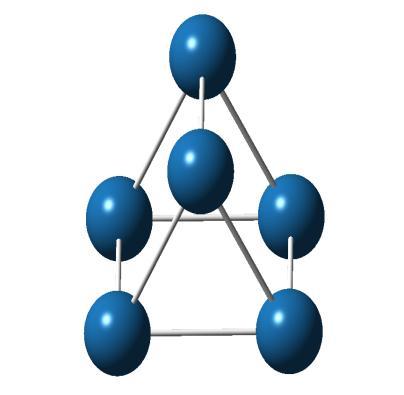
![Experimentally, Grushov and Ervin [28] have reported the bond length of Pt 2 as 2.33 Å and E b = 1.57 ev/atom. Pt 3 has the lowest energy structure of an equilateral triangle with bond length 2.](/docs-images/71/65948187/images/6-2.jpg "52 Å, E b =1.85 ev/atom and triplet spin multiplicity. An isosceles triangle is 0.10 ev and linear shape structure is 2.10 ev higher in energy than the equilateral triangle. Yang et al.")
6 Most stable geometry Higher energy isomers E=0.00 ev E=0.63 ev E=0.74 ev Figure 4.1 Low-lying isomers of Pt n (n=2 to 10) clusters. Structures on the leftmost column correspond to the most stable geometries. E is the energy difference of the given geometry from the most stable structure. The reported energy differences are calculated at B3LYP/LanL2DZ method. We have found that Pt 2 has a triplet ground state with a bond length of 2.37 Å same as obtained by Fortunelli [27] using DFT method. It is found in the D αh symmetry and has a binding energy per atom, E b = 1.40 ev/atom. Experimentally, Grushov and Ervin [28] have reported the bond length of Pt 2 as 2.33 Å and E b = 1.57 ev/atom. Pt 3 has the lowest energy structure of an equilateral triangle with bond length 2.52 Å, E b =1.85 ev/atom and triplet spin multiplicity. An isosceles triangle is 0.10 ev and linear shape structure is 2.10 ev higher in energy than the equilateral triangle. Yang et al. [29] have found an equilateral triangle with bond length 2.58 Å as the ground state of Pt 3 having E b =2.40 ev/atom. Kumar and Kawazoe [30] have also found an equilateral triangle with bond length 2.49 Å as the ground state of Pt 3. For Pt 4, the structure having minimum energy is a bent rhombus with bond length 2.51 Å and singlet spin multiplicity with E b =2.11 ev/atom. Kumar and Kawazoe [30] have also reported a bent rhombus with bond length 2.51 Å as the lowest energy structure for Pt 4. A tetrahedron is 0.31 ev higher in energy and a square lies 0.36 ev higher than bent rhombus. The lowest energy structure of Pt 5 is a planar side-capped square which has triplet spin and binding energy E b =2.14 ev/atom. Kumar and Kawazoe [30] have also reported side-capped square as the most favoured geometry for Pt 5. A square pyramid lies 0.06 ev higher in energy and a trigonal 74
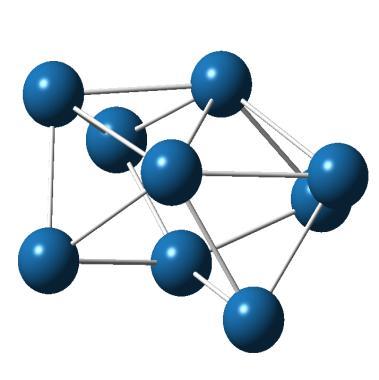
7 bipyramid is 0.11 ev higher in energy. We have found the most stable structure of Pt 6 as planar triangular-shaped structure, same as other DFT calculations [29, 30]. It has pentet spin multiplicity and E b =2.23 ev/atom. A double square, prism shaped and octahedron are 0.29, 0.39 and 0.45 ev higher in energy than the most stable structure. The lowest energy structure of Pt 7 is of the shape of two fused square pyramids and has quintet state with E b =2.41 ev/atom. Tian et al. [31] have also obtained similar ground state structure for Pt 7. A prism capped on triangular face lies only 0.13 ev higher in energy, while a planar side capped double square which was observed as the most stable structure by Kumar and Kawazoe [30] lies 0.92 ev higher in energy than the most stable structure. The lowest energy structure of Pt 8 is a hexagonal bipyramid and has septet spin with E b =2.47 ev/atom. Kumar and Kawazoe [30] have also reported hexagonal bipyramid as the lowest energy structure for Pt 8. A cubic structure, a bicapped octahedron and a double square bicapped on the long edge are 0.64, 0.80 and 1.23 ev higher in energy. The most stable structure of Pt 9 is tricapped octahedron, having nonet spin and E b =2.59 ev/atom. An isomer formed of fused square pyramids lies 0.90 ev higher in energy. A planar isomer with four squares which Kumar and Kawazoe [30] reported as the minimum energy structure for Pt 9 lies 2.04 ev higher. The lowest energy structure of Pt 10 is a tetracapped octahedron, which is in agreement with the geometries of Kumar and Kawazoe [30] having nonet spin multiplicity and E b =2.71 ev/atom. An isomer which can be considered as a distorted tetrahedron formed from fused square pyramid and prism lies 0.63 ev higher in energy and a bicapped cube isomer lies 0.74 ev higher. Planar isomers are obviously not favoured for Pt n clusters with n > 6. The binding energy of the cluster is increasing with cluster size Stability and reactivity properties Atom addition energy change (ΔE 1 ) and stability function (ΔE 2 ) are plotted as a function of cluster size in Figure 4.2a. It is seen that Pt 7 have high ΔE 2 value and large negative value of ΔE 1 indicating it is more stable than other clusters. The electron affinity (EA) and electrophilicity (ω) values for Pt n clusters as a function of cluster size are shown in Figure 4.2b. The experimental value of EA reported for Pt atom is 2.12 ev [32], which is in reasonable agreement with our B3LYP/LanL2DZ calculated value of 2.21 ev. The EA for Pt 2 is calculated to be 1.94 ev which is very close to the experimental result of 1.898±0.008 ev, reported by Ho 75
8 et al. [33]. EA for the Pt 3 is 2.09 ev, which is consistent with the experimental observation 1.89 ev by Ervin et al. [34] and 1.8± 0.10eV by Pontius et al. [35]. Our calculated EA value 2.22 ev, for Pt 4 is in agreement with the experimentally reported value 2.5 ± 0.10eV [35]. EA for Pt 5 is calculated as 2.54 ev which agrees well with the experimental value of 2.63 ± 0.1eV [35]. EA values for Pt 6, Pt 7, Pt 8, Pt 9 and Pt 10 are calculated as 3.53, 2.94, 2.93, 1.95 and 1.91 ev respectively. The ionization potential (IP) values, tabulated in Table 4.1, for Pt n (n=2-10) are in the range between 9.31 and 7.36 ev. Overall, the IP values are seen to decrease with increasing cluster size. A study based on image-charge model also reported that the electron removal energy decreases with increasing cluster size [36]. It may be mentioned here that experimentally determined IP for Pt atom is ev [28] and work function for bulk Pt metal is 6.10 ± 0.6 ev [37]. (a) (b) Figure 4.2 (a) Variation of atom addition energy change (ΔE 1 ) and stability function (ΔE 2 ) with cluster size. Red color represents atom addition energy change (ΔE 1 ) and black color represents stability function (ΔE 2 ) (b) Variation of electron affinity (EA) and electrophilicity (ω) with cluster size. Red and black color represent electron affinity (EA) and electrophilicity (ω) respectively. Irrespective of color, solid triangle ( ) represents B3LYP/LanL2DZ, solid rectangle ( ) represents B3PW91/LanL2DZ, hollow triangle ( ) represent B3LYP/LanL2MB and hollow rectangle ( ) represent B3PW91/LanL2MB. 76
9 The electrophilicity is a measure of the stabilization in energy when the cluster acquires an additional electronic charge from the surroundings. It is evident from Figure 4.2b that ω is closely related to EA and shows similar variation in changing cluster size. The ω value of Pt 7 is lower than its neighboring clusters, which supports its relative higher stability as seen from (ΔE 1 ) and (ΔE 2 ). The observed electrophilicity value of Pt 7 clusters is in agreement with the minimum electrophilicity principle (MEP) [38-40] which states that electrophilicity will be a minimum (maximum) when both chemical potential and hardness are maxima (minima). Table 4.1 Ionization potential (IP), HOMO-LUMO gap (E g ) and binding energy per atom (E b ) of Pt n (n=2 to 10) clusters calculated using B3LYP/LanL2DZ method. Cluster IP (ev) E g (ev) E b (ev/atom) Pt Pt Pt Pt Pt Pt Pt Pt Pt (a) (b) Figure 4.3 (a) Static polarizability per atom as a function of Pt n cluster size (b) Correlation between cube root of polarizabity and inverse of ionization potential for platinum clusters. 77
10 Polarizability can provide information about the electronic properties and structural geometry of the clusters as they are very sensitive to the delocalization of valence electrons and structural geometry. The theoretical prediction of polarizabilities and comparison with the experimental values, enable one to identify which cluster is being observed in the experiment. The polarizabity for single Pt atom is 44 a. u. (atomic units) [41], which is very close to our calculated result of a.u. The calculated static polarizabilities per atom of the platinum clusters are presented in Figure 4.3a. The polarizabilty per atom drops when cluster size changes from n=6 to n=7, indicating more stability of Pt 7. This observation is consistent with previous results of stability function and electrophilicity and also in accordance with the minimum polarizability principle [42]. Mohajeri et al. [43] have also showed that ZnS clusters which have lower polarizability per atom with respect to the neighbouring clusters are relatively more stable than the neighboring clusters. To check the correlation between the static polarizability and ionization potential, we have plotted cube root of the polarizability as a function of the inverse of ionization potential in Figure 4.3b. It is evident from Figure 4.3b, that the cube root of the polarizability and inverse of the ionization potential correlates well, with correlation coefficient of (B3LYP/LanL2DZ), (B3LYP/LanL2MB), (B3PW91/LanL2DZ) and (B3PW91/LanL2MB), thereby indicating the reliability of the correlation. It is an interesting finding, paving a way to calculate polarizabilty of larger clusters from their ionization potential Raman spectra 78
11 Figure 4.4 Raman spectra of platinum clusters. The vibrational frequency of Pt 2 in our calculation has been found as ω c = cm -1 and it corresponds to the stretching mode between the two platinum atoms. Jansson and Scullman [44] measured vibrational spectrum in Ar matrix, and observed single vibronic transition at ω c = cm -1. Using ab initio calculation, Yang et al. [29] have computed the vibrational frequency of Pt 2 as ω c = cm -1. We found the vibrational modes of the ground state of Pt 3 at and cm -1. The first mode appearing at cm -1 is due to the stretching mode of two Pt-Pt bonds, while the third bond remains fixed (asymmetric 79
12 stretching). Another vibrational mode at cm -1 corresponds to the symmetric stretching mode of the three bonds in the ground state of Pt 3 cluster. Photoelectron spectroscopic study of Pt 3 by Ervin et al. [34] confirmed peaks at frequencies 105±30 cm -1 and 225±30 cm -1 while ab initio study by Yang et al. [29] obtained the vibrational modes at , and cm -1. For Pt 4, the peaks of vibrational frequency are observed at 27.66, 46.21, , , cm -1. The lowest vibrational frequencies for Pt 5, Pt 6, Pt 7, Pt 8, Pt 9 and Pt 10 are observed at 18.76, 18.78, 11.43, 17.63, and cm -1 respectively, and the highest vibrational frequencies are found at , , , , and cm -1 respectively. Since Raman spectroscopy is very sensitive to the structure, we have used it in this study and is presented in Figure 4.4. There is only one peak for Pt 2, which is observed at cm -1. This Raman peak corresponds to the stretching mode between the two platinum atoms of Pt 2. The Raman spectra for the Pt 3 has two distinct peaks at cm -1 and cm -1. The first peak appearing at cm -1 is due to the stretching mode of two Pt-Pt bonds, while the third bond remains fixed (asymmetric stretching). The highest peak at cm -1 corresponds to the symmetric stretching mode of the three bonds in the ground state of Pt 3 cluster. The Pt 4 has four dominant peaks emerging in between and cm -1. The highest peak observed at cm -1 is assigned to the symmetric stretching mode of the Pt atoms. The other distinct peak appearing at cm -1 is assigned to the asymmetric stretching mode. For Pt 5, the observed Raman spectra shows a number of peaks in between cm -1 and cm -1, with the highest peak at cm -1. In the case of Pt 6, the maximum peak at cm 1 is assigned to the symmetric stretching of all the bonds. The Raman spectra for Pt 7, Pt 8, Pt 9, and Pt 10 show maximum peaks at , , , and cm -1 respectively Optical absorption spectra Optical spectroscopy is one the most powerful instrument to investigate the electronic properties. Theoretical investigations of optical properties are usually done by employing TDDFT methods, in which all or at least, the valence electrons are treated accurately. We have calculated the spectra for cluster geometries having lowest energy. Each cluster is observed to show their characteristic optical absorption bands as the geometry of the clusters are drastically different from one another. In Figure 4.5, the oscillator strength (f) and molar absorption 80
13 coefficient (Ɛ) of the ground state platinum clusters are presented. The absorption spectrum of Pt 2 shows maximum peak at λ= nm which lies in the visible region and for Pt 3, the maximum peak is observed at λ= nm of the visible region followed by another significant peak at nm. The absorption spectra for Pt 4 shows two significant peaks: one at nm and another at nm. The former one having the highest peak with oscillator strength and the latter one with diminished peak having oscillator strength For larger clusters with n > 4, the peaks are observed in the infra red region. The overall evolutionary trend of the most prominent peak occur at higher wavelength (or low energy) side once we move towards larger cluster size and also the onset of absorption shifted towards the low energy side with increasing the size of the cluster. Pt 2 Pt 3 Pt 4 Pt 5 Pt 6 Pt 7 81
14 Pt 8 Pt 9 Pt 10 Figure 4.5 Oscillator strength (atomic unit) and molar absorption coefficient (mol -1 dm 3 cm -1 ) of stable platinum clusters. Table 4.2 Calculated energy (E in ev) and oscillator strengths (f in ev) for excitations from the ground state. Pt n Cluster Pt 2 Pt 3 Single Photon excitation State 1 with E =0.156 ev (and f = ev) State 2 with E =0.156 ev (and f = ev) State 4 with E =0.191 ev (and f = ev) State 2 with E =0.457 ev (and f = ev) State 3 with E =0.456 ev (and f = ev) 82 Multiple photon excitation State 3 with E = ev. State 5 with E = ev. State 1 with E = ev. State 4 with E = ev. State 5 with E = ev. Pt 4 State 4 with E= ev (and f= ev). State 1 with E = ev. State 2 with E = ev. State 3 with E = ev. State 5 with E = ev. Pt 5 State 4 with E= ev (and f= ev). State 1 with E = ev. State 2 with E = ev. State 3 with E = ev. State 5 with E = ev. Pt 6 State 4 with E =0.547 ev (and f = ev) State 1 with E = 0.263eV. State 2 with E = 0.444eV. State 3 with E = ev. State 5 with E = ev. Pt 7 State 1 with E =0.341 ev (and f = ev) State 2 with E = ev.
15 State 4 with E =0.648 ev (and f = ev) State 3 with E = ev. State 5 with E =0.679 ev (and f = ev) Pt 8 State 4 with E =0.465 ev (and f = ev) State 1 with E = ev. State 2 with E = ev. State 3 with E = ev. State 5 with E = ev. Pt 9 State 7 with E =0.719 ev (and f = ev) State 1 with E = 0.524eV. State 2 with E = ev. State 3 with E = ev. State 4 with E = ev. State 5 with E = ev. Pt 10 State 7 with E =0.878 ev (and f = ev) State 1 with E = ev. State 2 with E = ev. State 3 with E = ev. State 4 with E = ev. State 5 with E = ev. The photoabsorption spectra of clusters are mainly determined by the collective plasmon excitation of delocalized electrons and the resonant transition of electrons bounded to the atom. In this study, we report transition up to first five excited states which include both single photon and multi-photon transitions. Table 4.2 represents single and multi-photon excitation energies. In Pt 2, the single photon transitions are observed for the excited states, 1 and 2, with same transition energy ev and oscillator strength ev. Single photon transition is also observed for excited state 4 with energy ev and oscillator strength ev. Multi photon transitions are observed for excited states 3 and 5 with transition energies and ev respectively. Pt 3 shows two single-photon excitations with energy ev (oscillator strength ev) at excited states 2 and 3. Excited states 1 (energy ev), 4 (energy ev) and 5 (energy ev) are multi-photon excitations. In case of Pt 4, Pt 5 and Pt 6, only one single-photon transition has been observed at excited state 4 with oscillator strength , , ev and energy , 0.448, ev respectively. Others are multi-photon transitions. The transitions for excited states 1, 2, 3 and 5 of Pt 5 are with energies 0.312, 0.353, and ev respectively, and that of Pt 6 are 0.263, 0.444, and ev respectively. Single-photon excitations are observed for excited states 1, 4 and 5 with energies 0.341, and respectively and multi-photon transitions are observed for excited states 2 and 83
16 3 with energies and ev respectively for Pt 7 cluster. Pt 8 has one single-photon transition at excited state 4 with energy ev. The multi-photon transitions for Pt 8 have been observed at energies 0.311, 0.418, and ev for excited states 1, 2, 3 and 5 respectively. In the case of Pt 9, all the excitations up to excited state 5 are multi-photon excitations with energies 0.524, 0.543, 0.568, and ev respectively. First singlephoton excitation is seen at excited state 7 with energy ev. Pt 10 has single-photon transitions at excited states 7 with energy ev, all the excitations up to excited state 5 are multi-photon excitations with energies 0.467, 0.489, 0.517, and ev respectively. The sum of the oscillator strength, which characterizes the valence electron delocalization rate, for the clusters are obtained as (Pt 2 ), (Pt 3 ), (Pt 4 ), (Pt 5 ), (Pt 6 ), (Pt 7 ), (Pt 8 ), (Pt 9 ), (Pt 10 ) ev. It may be noted here that the valence electron delocalization rate for the smaller clusters (Pt 2, Pt 3 and Pt 4 ) are higher than those of larger clusters. 84
17 Figure 4.6 Photoabsorption cross section calculated for the most stable platinum clusters. The photoabsorption cross sections are presented in Figure 4.6. The peaks of the photoabsorption curves are observed in the region 0.15 to 0.80 ev and may be due to the resonant excitation of the plasma oscillations of the cluster electrons [45]. From Figure 4.6, we can see that the photoabsorption spectra of Pt 2 show strong resonances near 0.15 ev, Pt 3 near 0.47 ev and Pt 4 near 0.82 ev. Apart from the main photoabsorption peak, Pt 3 shows additional secondary peaks in the energy range 1.05 to 1.40 ev, and Pt 4 shows an additional secondary peak near 0.67 ev. For Pt 5 and Pt 8, the absorption spectra show strong resonances in the vicinity of 0.47 ev. The absorption spectra of Pt 6, Pt 7 and Pt 9 show strong resonances around 0.70 ev. In the case of Pt 10, the photoabsorption curve strong peak at 0.66 ev, another very prominent peak is also observed at 0.44 ev. 85

18 4.3.5 Density of states Figure 4.7 Total and projected density of states for the most stable platinum clusters. 86
19 The reactivity of transition metal clusters depends on the arrangement of electrons because of their incomplete d shell electronic structure and there exists a size induced metal insulator transition in metal clusters. When the size of the metal cluster reduces, the energy level spacing become prominent resulting a large HOMO-LUMO gap. The density of states gives the detailed electronic structure of the clusters and therefore, can shed light on the reactivity of the clusters. Only the energy states close to the HOMO and LUMO have an appreciable influence on the chemical reactivity of the cluster. The discreteness of the DOS for Pt n cluster is observed in Figure 4.7. The DOS of Pt 2 cluster has high intense peaks, distributed around to ev. For Pt 2, the HOMO energy is at ev and LUMO energy is at ev. The main contribution of HOMO is coming from s and d orbitals; d orbital s contribution is highest and s orbital is also reasonably contributed. In case of LUMO, opposite trend has been found; here s orbital s contribution is higher than d orbital s contribution. In the case of Pt 3, the high intensity peaks in the energy range to 5.70 ev are mainly contributed by d orbital with a small contribution from s and p. The HOMO energy is at ev and LUMO energy is at ev. In the HOMO region, the contribution from d orbital is highest, with very small contributions from s and p orbitals. But in the LUMO region, the dominant orbital which contributes most is the p orbital and contribution from s and d orbitals are almost negligible. For Pt 4, the HOMO and LUMO energies are at ev and ev respectively. A similar trend (as in Pt 3 ) is seen for the HOMO region of Pt 4 with d orbital s contribution being highest in this region whereas, s and p orbital s contribution comparatively very less. In the LUMO region, contribution comes from all the s, p and d orbitals and the contribution is in the order d > s > p COOP analysis In order to understand the nature of bonding in the cluster, the crystal orbital overlap population (COOP) [46] analysis has been carried out. COOP can be used to quantify the bonding between two orbitals where positive and negative value signifies bonding and antibonding interactions. The COOP for platinum clusters are shown in Figure 4.8. For Pt 2, there is strong bonding interaction with large overlap between p and d orbitals in the energy range to ev. Interaction of s with p orbital and p with d orbital results to antibonding states in the energy range from to ev including the HOMO (-6.90 ev) 87
.")
 level, very weak bonding interaction is seen between p and d orbitals. On the other hand, if we move towards higher energy side from LUMO (in the energy region -3.80 to -3.")
20 level. As we move a little more above the HOMO, the energy states from to ev have bonding interaction due to s and p orbital overlapping. Antibonding interactions are originated from the overlap of s with d orbital and p with d orbital in this energy range (i.e to ev). Bonding interactions are observed between s with d orbital and p with d orbital at the lower energy side of LUMO (in the energy range to ev). At the LUMO (-3.89 ev) level, very weak bonding interaction is seen between p and d orbitals. On the other hand, if we move towards higher energy side from LUMO (in the energy region to ev), antibonding interaction is observed between s and p orbitals. 88

21 Figure 4.8 Crystal orbital overlap population (COOP) of the most stable platinum cluster. In the COOP diagram for Pt 3 cluster, it is seen that weak bonding interactions, because of overlapping of s with p and p with d, are formed in the energy region to ev i.e. on the lower energy side of HOMO. In the same region, antibonding states are also seen due to s and d orbital interaction. At the HOMO (-5.88 ev) region, weak bonding interactions are formed due to overlapping of s with p orbital, s with d orbital and p with d orbital. In contrary, no bonding or antibonding interaction is observed at the LUMO (-4.03 ev) position. But if we look above LUMO (at about to ev), strong bonding interaction of s with p orbital and moderate antibonding interaction of s with d orbital are observed. The COOP of Pt 4 shows that bonding interactions of s with p, s with d and p with d orbitals are observed from to ev, which includes the HOMO (-5.88 ev) level. At the LUMO position (-3.92 ev), we found strong bonding interactions due to overlap of s and p orbitals. Antibonding interactions are also observed at LUMO position whose primary contribution is coming from the overlap of s with d orbital and a small contribution from the overlap of p with d orbital. Above the LUMO region, strong bonding interaction is seen due to overlapping of s orbital with p orbital in the energy range to ev. Antibonding states, though feeble compared to its bonding counterpart, are formed in the same region due to overlapping of s with d orbital and p with d orbital in the energy range to ev. For clusters n>4, the most significant contribution of antibonding state is originated due to the p and d orbital overlapping followed by s and d orbital overlapping. However, in case of bonding 89
22 state, s and p orbital overlapping plays the fundamental role. It should be noted here that neither bonding nor antibonding states are formed in the LUMO position of Pt 6, Pt 7 and Pt Conclusions In summary, we have studied the structure, stability, reactivity and optical properties of small platinum clusters, Pt n (n=2 to 10). Stability is interpreted in terms of reactivity parameters. With cluster size, the binding energy per atom of the cluster increases. Our calculated electron affinity values and vibrational frequencies are in agreement with the available experimental results. Electron affinity and electrophilicity show similar variation with cluster size. The highest peak of the Raman spectra is shifted towards the low frequency region with increasing size of the cluster. By paying attention to the molar absorption coefficient, we see that the overall evolutionary trend of the most prominent peak occur at high wavelength (or low energy) side once we move towards larger cluster size. The total DOS upto HOMO position is primarily contributed by d orbital with a small contribution from s orbital, however in the above LUMO region the main contribution is coming from s orbital along with p orbital which was missing in the former case. Our COOP analysis suggest that below LUMO level the bonding states are formed due the overlapping of s orbital with p orbital and sometimes augmented by the overlapping of p with d, however, above the LUMO level it is due the overlapping of s orbital with p orbital. Except for small clusters, major contribution for the antibonding states is coming from the overlapping of p and d orbitals, and minor contribution is from either the overlapping of s and d orbitals (below LUMO level) or from the overlapping of both s with p and s with d ( above LUMO level) orbitals. The next chapter discusses gold-palladium bimetallic clusters. The properties of bimetallic clusters are compared with those of pure clusters. 90
23 4.5 References [1] Bell, A.T. (2003). Science. 299: [2] Bond, G.C. (1991). Chemistry of the platinum group metals: Recent developments. Elsevier, Amsterdam. [3] Vielstich, W.; Lamm, A.; Gasteiger, H. (2003). Handbook of fuel cells: Fundamentals, Technology, Applications. Sussex, Wiley West. [4] Yuan, Q.; Zhou, Z.; Zhuang, J.; Wang, X. (2010). Chem. Commun. 46: [5] Tian, N.; Zhou, Z.Y.; Sun, S.G.; Ding, Y.; Wang, Z.L. (2007). Science. 316: [6] Zhang, J.; Sasaki, K.; Sutter, E.; Adzic, R.R. (2007). Science. 315: [7] Xu, Y.; Shelton, W.A.; Schneider, W.F. (2006). J. Phys. Chem. A. 110: [8] Ghanty, T.K.; Ghosh, S.K. (1993). J. Phys. Chem. 97: [9] Bedamani, N.; Sarkar, U. (2014). Molecular Simulation. 40: [10] Dai, D.; Balasubramanian, K. (1992). J. Phys. Chem. 96: [11] Morse, M.D. (1986). Chem. Rev. 86: [12] Duncan, T.M.; Zilm, K.W.; Hamilton, D.A.; Root, T.W. (1989). J. Phys. Chem. 93: [13] Martin, T.P.; Bergmann, T.; Golich, G.; Lange, T. (1991). Chem. Phys. Lett. 176: [14] Soler, J.M.; Artacho, E.; Gale, J.D.; García, A.; Junquera, J.; Ordejón, P.; Sánchez-Portal, D. (2002). J. Phys.: Condens. Matter. 14: [15] Evans, D.J.; Holian, B.L. (1985). J. Chem. Phys. 83: [16] Frisch, M.J. et al. (2010). Gaussian 09, Revision C.01, Gaussian, Inc., Wallingford CT. [17] Becke, A.D. (1993). J. Chem. Phys. 98: [18] Lee, C.; Yang, W.; Parr, R.G. (1988). Phys. Rev. B. 37: [19] Perdew, J.J.; Yang, W. (1992). Phys. Rev. B. 45: [20] Hay, P.J.; Wadt, W.R. (1985). J. Chem. Phys. 82: [21] Wadt, W.R.; Hay, P.J. (1985). J. Chem. Phys. 82: [22] Hay, P.J.; Wadt, W.R. (1985). J. Chem. Phys. 82: [23] Barbatti, M.; Aquino, A.J.A.; Liscka, H. (2010). Phys. Che. Chem. Phys. 12: [24] Barbatti, M.; Granucci, G.; Persico, M.; Ruckenbauer, M.; Vazdar, M.; Eckert-Maksić, M.; Lischka, H. (2007). J. Photochem. Photobio. A. 190:
24 [25] Barbatti, M.; Granucci, G.; Ruckenbauer, M.; Plasser, F.; Pittner, J.; Persico, M.; Lischka, H. (2013). NEWTON-X: a package for Newtonian dynamics close to the crossing seam, version 1.4, [26] Taylor, S.; Lemire, G.W.; Hamrick, Y.M.; Fu, Z.; Morse, M.D. (1988). J. Chem. Phys. 89: [27] Fortunelli, A. (1999). J. Mol. Struct.: Theochem. 493: [28] Grushow, A.; Ervin, K.M. (1997). J. Chem. Phys. 106: [29] Yang, S.H.; Drabold, D.A.; Adams, J.B.; Ordejon, P.; Glassford, K. (1997). J. Phys.: Condens. Matter. 9:L39-L45. [30] Kumar, V.; Kawazoe, Y. (2008). Phys. Rev. B. 77: [31] Tian, W.Q.; Ge, M.; Sahu, B.R.; Wang, D.; Yamada, T.; Mashiko, S. (2004). J. Phys. Chem. A. 108: [32] Gibson, N.D.; Davies, B.J.; Larson, D.J. (1993). J. Chem. Phys. 98: [33] Ho, J.; Polak, M.L.; Ervin, K.M.; Lineberger, W.C. (1993). J. Chem. Phys. 99: [34] Ervin, K.M..; Ho, J.; Lineberger, W.C. (1988). J. Chem. Phys. 89: [35] Pontius, N.; Bechthold, P.S.; Neeb, M.; Eberhardt, W. (2000). J. Electron. Spectrosc. Relat. Phenom. 106: [36] Wong, K.; Vongehr, S.; Kresin, V.V. (2003). Phys. Rev. B. 67: [37] Derry, G.N.; Zhong, Z.J. (1989). Phys. Rev. B. 39: [38] Chamorro, E.; Chattaraj, P.K.; Fuentealba, P. (2003). J. Phys. Chem. A. 107: [39] Parthasarathi, R.; Elango, M.; Subramanian, V. (2005). Theor. Chem. Acc. 113: [40] Pan, S.; Sola, M.; Chattaraj, P.K. (2013). J. Phys. Chem. A. 117: [41] Miller, T.M. (2002). CRC Handbook of Chemistry and Physics. CRC Press, New York. [42] Chattaraj, P.K.; Sengupta, S. (1996). J. Phys. Chem. 100: [43] Mohajeri, A.; Alipour, M. (2011). Int. J. Quant. Chem. 111: [44] Jansson, K.; Scullman, R. (1976). J. Mol. Spectrosc. 61: [45] Solov yov, I.A.; Solov yov, A.V.; Greiner, W. (2004). J. Phys. B: At. Mol. Opt. Phys. 37: L137 L145. [46] Hughbanks, T.; Hoffmann, R. (1983). J. Am. Chem. Soc. 105:
Quantum-Mechanical Study of Small Au 2 Pd n (n = 1 4) Clusters
 Clusters") Commun. Theor. Phys. (Beijing, China) 46 (2006) pp. 155 160 c International Academic Publishers Vol. 46, No. 1, July 15, 2006 Quantum-Mechanical Study of Small Au 2 Pd n (n = 1 4) Clusters GUO Jian-Jun,
Commun. Theor. Phys. (Beijing, China) 46 (2006) pp. 155 160 c International Academic Publishers Vol. 46, No. 1, July 15, 2006 Quantum-Mechanical Study of Small Au 2 Pd n (n = 1 4) Clusters GUO Jian-Jun,
Electronic structures of one-dimension carbon nano wires and rings
 IOP Publishing Journal of Physics: Conference Series 61 (2007) 252 256 doi:10.1088/1742-6596/61/1/051 International Conference on Nanoscience and Technology (ICN&T 2006) Electronic structures of one-dimension
IOP Publishing Journal of Physics: Conference Series 61 (2007) 252 256 doi:10.1088/1742-6596/61/1/051 International Conference on Nanoscience and Technology (ICN&T 2006) Electronic structures of one-dimension
Structural and Electronic Properties of Small Silicon Nanoclusters
 Structural and Electronic Properties of Small Silicon Nanoclusters Prabodh Sahai Saxena a* and Amerendra Singh Sanger b a Physics Department, Lakshmi Narain College of Technology Excellence, Bhopal, India.
Structural and Electronic Properties of Small Silicon Nanoclusters Prabodh Sahai Saxena a* and Amerendra Singh Sanger b a Physics Department, Lakshmi Narain College of Technology Excellence, Bhopal, India.
Periodic Trends in Properties of Homonuclear
 Chapter 8 Periodic Trends in Properties of Homonuclear Diatomic Molecules Up to now, we have discussed various physical properties of nanostructures, namely, two-dimensional - graphene-like structures:
Chapter 8 Periodic Trends in Properties of Homonuclear Diatomic Molecules Up to now, we have discussed various physical properties of nanostructures, namely, two-dimensional - graphene-like structures:
Density Functional Theory (DFT) modelling of C60 and
 modelling of C60 and") ISPUB.COM The Internet Journal of Nanotechnology Volume 3 Number 1 Density Functional Theory (DFT) modelling of C60 and N@C60 N Kuganathan Citation N Kuganathan. Density Functional Theory (DFT) modelling
ISPUB.COM The Internet Journal of Nanotechnology Volume 3 Number 1 Density Functional Theory (DFT) modelling of C60 and N@C60 N Kuganathan Citation N Kuganathan. Density Functional Theory (DFT) modelling
Theoretical study of sequential oxidation of clusters of gallium oxide: Ga 3 O n (n: 4 8)
") Chemical Physics Letters 431 (2006) 358 363 www.elsevier.com/locate/cplett Theoretical study of sequential oxidation of clusters of gallium oxide: Ga 3 O n (n: 4 8) S. Gowtham a, Aurora Costales b, Ravindra
Chemical Physics Letters 431 (2006) 358 363 www.elsevier.com/locate/cplett Theoretical study of sequential oxidation of clusters of gallium oxide: Ga 3 O n (n: 4 8) S. Gowtham a, Aurora Costales b, Ravindra
Quantum chemical origin of high ionization potential and low electron affinity of Tungsten Hexafluoride
 Journal of Computational Methods in Molecular Design, 2015, 5 (4):142-146 Scholars Research Library (http://scholarsresearchlibrary.com/archive.html) ISSN : 2231-3176 CODEN (USA): JCMMDA Quantum chemical
Journal of Computational Methods in Molecular Design, 2015, 5 (4):142-146 Scholars Research Library (http://scholarsresearchlibrary.com/archive.html) ISSN : 2231-3176 CODEN (USA): JCMMDA Quantum chemical
RDCH 702 Lecture 4: Orbitals and energetics
 RDCH 702 Lecture 4: Orbitals and energetics Molecular symmetry Bonding and structure Molecular orbital theory Crystal field theory Ligand field theory Provide fundamental understanding of chemistry dictating
RDCH 702 Lecture 4: Orbitals and energetics Molecular symmetry Bonding and structure Molecular orbital theory Crystal field theory Ligand field theory Provide fundamental understanding of chemistry dictating
Structural, vibrational and electronic properties of small group IV oxide clusters in lower and higher spin state A DFT study
 J. At. Mol. Sci. doi: 10.4208/jams.052511.070411a Vol. x, No. x, pp. 1-10 xxx 2011 ½ ¾ Structural, vibrational and electronic properties of small group IV oxide clusters in lower and higher spin state
J. At. Mol. Sci. doi: 10.4208/jams.052511.070411a Vol. x, No. x, pp. 1-10 xxx 2011 ½ ¾ Structural, vibrational and electronic properties of small group IV oxide clusters in lower and higher spin state
Supporting Information
 Electronic Supplementary Material (ESI) for Dalton Transactions. This journal is The Royal Society of Chemistry 2016 Supporting Information Enhanced Catalytic Activity and Magnetization of Encapsulated
Electronic Supplementary Material (ESI) for Dalton Transactions. This journal is The Royal Society of Chemistry 2016 Supporting Information Enhanced Catalytic Activity and Magnetization of Encapsulated
Excited States Calculations for Protonated PAHs
 52 Chapter 3 Excited States Calculations for Protonated PAHs 3.1 Introduction Protonated PAHs are closed shell ions. Their electronic structure should therefore be similar to that of neutral PAHs, but
52 Chapter 3 Excited States Calculations for Protonated PAHs 3.1 Introduction Protonated PAHs are closed shell ions. Their electronic structure should therefore be similar to that of neutral PAHs, but
Fragmentation Behavior and Ionization Potentials of Lead Clusters Pb n (n 30)
") CHEM. RES. CHINESE UNIVERSITIES 2010, 26(6), 996 1001 Fragmentation Behavior and Ionization Potentials of Lead Clusters Pb n (n 30) LI Xiao-ping 1, ZHANG Wei 1, LÜ Wen-cai 1,2*, WANG Cai-zhuang 3 and HO
CHEM. RES. CHINESE UNIVERSITIES 2010, 26(6), 996 1001 Fragmentation Behavior and Ionization Potentials of Lead Clusters Pb n (n 30) LI Xiao-ping 1, ZHANG Wei 1, LÜ Wen-cai 1,2*, WANG Cai-zhuang 3 and HO
A density functional study on equilibrium geometries, stabilities and electronic properties of Au 5 Li binary clusters
 Appl Nanosci (2012) 2:359 364 DOI 10.1007/s13204-012-0092-x ORIGINAL ARTICLE A density functional study on equilibrium geometries, stabilities and electronic properties of Au 5 Li binary clusters Ajanta
Appl Nanosci (2012) 2:359 364 DOI 10.1007/s13204-012-0092-x ORIGINAL ARTICLE A density functional study on equilibrium geometries, stabilities and electronic properties of Au 5 Li binary clusters Ajanta
Calculate a rate given a species concentration change.
 Kinetics Define a rate for a given process. Change in concentration of a reagent with time. A rate is always positive, and is usually referred to with only magnitude (i.e. no sign) Reaction rates can be
Kinetics Define a rate for a given process. Change in concentration of a reagent with time. A rate is always positive, and is usually referred to with only magnitude (i.e. no sign) Reaction rates can be
First-principle studies of the geometries and electronic properties of Cu m Si n (2 m + n 7) clusters
 clusters") Vol 16 No 11, November 2007 c 2007 Chin. Phys. Soc. 1009-1963/2007/16(11)/3359-11 Chinese Physics and IOP Publishing Ltd First-principle studies of the geometries and electronic properties of Cu m Si n
Vol 16 No 11, November 2007 c 2007 Chin. Phys. Soc. 1009-1963/2007/16(11)/3359-11 Chinese Physics and IOP Publishing Ltd First-principle studies of the geometries and electronic properties of Cu m Si n
Journal of Chemical and Pharmaceutical Research
 Available on line www.jocpr.com Journal of Chemical and Pharmaceutical Research ISSN No: 0975-7384 CODEN(USA): JCPRC5 J. Chem. Pharm. Res., 2011, 3(4): 589-595 Altering the electronic properties of adamantane
Available on line www.jocpr.com Journal of Chemical and Pharmaceutical Research ISSN No: 0975-7384 CODEN(USA): JCPRC5 J. Chem. Pharm. Res., 2011, 3(4): 589-595 Altering the electronic properties of adamantane
Journal of Computational Methods in Molecular Design, 2013, 3 (1):1-8. Scholars Research Library (
:1-8. Scholars Research Library (") Journal of Computational Methods in Molecular Design, 2013, 3 (1):1-8 Scholars Research Library (http://scholarsresearchlibrary.com/archive.html) ISSN : 2231-3176 CODEN (USA): JCMMDA Theoretical study
Journal of Computational Methods in Molecular Design, 2013, 3 (1):1-8 Scholars Research Library (http://scholarsresearchlibrary.com/archive.html) ISSN : 2231-3176 CODEN (USA): JCMMDA Theoretical study
Electronic Supplementary Information
 Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 2014 Electronic Supplementary Information Rational modifications on champion porphyrin
Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 2014 Electronic Supplementary Information Rational modifications on champion porphyrin
Density Functional Theory Calculation of the Electronic Structure for C 20 Cage Fullerene حسابات نظرية دالة الكثافة للتركيب االلكتروني للفوليرين C 20
 Density Functional Theory Calculation of the Electronic Structure for C 20 Cage Fullerene حسابات نظرية دالة الكثافة للتركيب االلكتروني للفوليرين C 20 محمد اف ارح هادي هاشم نضال هادي طالب ليث عبيس اب ارهيم
Density Functional Theory Calculation of the Electronic Structure for C 20 Cage Fullerene حسابات نظرية دالة الكثافة للتركيب االلكتروني للفوليرين C 20 محمد اف ارح هادي هاشم نضال هادي طالب ليث عبيس اب ارهيم
Reduction of Nitrogen Oxides (NO x ) by Superalkalis
 by Superalkalis") Reduction of Nitrogen Oxides (NO x ) by Superalkalis Ambrish Kumar Srivastava P. G. Department of Physics, Veer Kunwar Singh University, Ara-802301, Bihar, India E-mail: ambrishphysics@gmail.com 1 Abstract
Reduction of Nitrogen Oxides (NO x ) by Superalkalis Ambrish Kumar Srivastava P. G. Department of Physics, Veer Kunwar Singh University, Ara-802301, Bihar, India E-mail: ambrishphysics@gmail.com 1 Abstract
Photoelectron spectroscopy of mixed metal cluster anions: NiCu, NiAg, NiAg 2, and Ni 2 Ag
 Photoelectron spectroscopy of mixed metal cluster anions: NiCu, NiAg, NiAg 2, and Ni 2 Ag St. J. Dixon-Warren, a) R. F. Gunion, and W. C. Lineberger JILA and Department of Chemistry and Biochemistry, University
Photoelectron spectroscopy of mixed metal cluster anions: NiCu, NiAg, NiAg 2, and Ni 2 Ag St. J. Dixon-Warren, a) R. F. Gunion, and W. C. Lineberger JILA and Department of Chemistry and Biochemistry, University
Analysis of the ultrafast dynamics of the silver trimer upon photodetachment
 J. Phys. B: At. Mol. Opt. Phys. 29 (1996) L545 L549. Printed in the UK LETTER TO THE EDITOR Analysis of the ultrafast dynamics of the silver trimer upon photodetachment H O Jeschke, M E Garcia and K H
J. Phys. B: At. Mol. Opt. Phys. 29 (1996) L545 L549. Printed in the UK LETTER TO THE EDITOR Analysis of the ultrafast dynamics of the silver trimer upon photodetachment H O Jeschke, M E Garcia and K H
Structural, Electronic, and Magnetic Properties of Bimetallic Ni m Nb n (m + n 8) Clusters: First Principle Study
 Clusters: First Principle Study") J Supercond Nov Magn (2017) 30:251 260 DOI 10.1007/s10948-016-3710-0 ORIGINAL PAPER Structural, Electronic, and Magnetic Properties of Bimetallic Ni m Nb n (m + n 8) Clusters: First Principle Study Mihai
J Supercond Nov Magn (2017) 30:251 260 DOI 10.1007/s10948-016-3710-0 ORIGINAL PAPER Structural, Electronic, and Magnetic Properties of Bimetallic Ni m Nb n (m + n 8) Clusters: First Principle Study Mihai
Structure determination of small vanadium clusters by density-functional theory in comparison with experimental far-infrared spectra
 THE JOURNAL OF CHEMICAL PHYSICS 122, 124302 2005 Structure determination of small vanadium clusters by density-functional theory in comparison with experimental far-infrared spectra C. Ratsch a Fritz-Haber-Institut
THE JOURNAL OF CHEMICAL PHYSICS 122, 124302 2005 Structure determination of small vanadium clusters by density-functional theory in comparison with experimental far-infrared spectra C. Ratsch a Fritz-Haber-Institut
Quantum chemical studies on the structures of some heterocyclic azo disperse dyes
 Quantum chemical studies on the structures of some heterocyclic azo disperse dyes Nesrin Tokay, a* Zeynel Seferoğlu, b Cemil Öğretir, c and Nermin Ertan b a Hacettepe University, Faculty of Science, Chemistry
Quantum chemical studies on the structures of some heterocyclic azo disperse dyes Nesrin Tokay, a* Zeynel Seferoğlu, b Cemil Öğretir, c and Nermin Ertan b a Hacettepe University, Faculty of Science, Chemistry
Chapter 10: Modern Atomic Theory and the Periodic Table. How does atomic structure relate to the periodic table? 10.1 Electromagnetic Radiation
 Chapter 10: Modern Atomic Theory and the Periodic Table How does atomic structure relate to the periodic table? 10.1 Electromagnetic Radiation Electromagnetic (EM) radiation is a form of energy that exhibits
Chapter 10: Modern Atomic Theory and the Periodic Table How does atomic structure relate to the periodic table? 10.1 Electromagnetic Radiation Electromagnetic (EM) radiation is a form of energy that exhibits
Supplementary Information
 Supplementary Information Enhancing the Double Exchange Interaction in Mixed Valence {V III -V II } Pair: A Theoretical Perspective Soumen Ghosh, Saurabh Kumar Singh and Gopalan Rajaraman* a Computational
Supplementary Information Enhancing the Double Exchange Interaction in Mixed Valence {V III -V II } Pair: A Theoretical Perspective Soumen Ghosh, Saurabh Kumar Singh and Gopalan Rajaraman* a Computational
Structural and Electronic Properties of Neutral and Ionic Ga n O n Clusters with n ) 4-7
 4-7") 3814 J. Phys. Chem. A 2006, 110, 3814-3819 Structural and Electronic Properties of Neutral and Ionic Ga n O n Clusters with n ) 4-7 Mrinalini Deshpande, D. G. Kanhere, and Ravindra Pandey* Michigan Technological
3814 J. Phys. Chem. A 2006, 110, 3814-3819 Structural and Electronic Properties of Neutral and Ionic Ga n O n Clusters with n ) 4-7 Mrinalini Deshpande, D. G. Kanhere, and Ravindra Pandey* Michigan Technological
Properties of Individual Nanoparticles
 TIGP Introduction technology (I) October 15, 2007 Properties of Individual Nanoparticles Clusters 1. Very small -- difficult to image individual nanoparticles. 2. New physical and/or chemical properties
TIGP Introduction technology (I) October 15, 2007 Properties of Individual Nanoparticles Clusters 1. Very small -- difficult to image individual nanoparticles. 2. New physical and/or chemical properties
Doped Quantum Sized Gold Nanoclusters
 Doped Quantum Sized Gold Nanoclusters Sumali Bansal 1*, Priyanka 2, Rajiv Bhandari 3, Keya Dharamvir 4 1 DAV College, Sector 10, Chandigarh, India 2 Guru Gobind Singh College for Women, Sector 26, Chandigarh,
Doped Quantum Sized Gold Nanoclusters Sumali Bansal 1*, Priyanka 2, Rajiv Bhandari 3, Keya Dharamvir 4 1 DAV College, Sector 10, Chandigarh, India 2 Guru Gobind Singh College for Women, Sector 26, Chandigarh,
Design of nanomechanical sensors based on carbon nanoribbons and nanotubes in a distributed computing system
 Design of nanomechanical sensors based on carbon nanoribbons and nanotubes in a distributed computing system T. L. Mitran a, C. M. Visan, G. A. Nemnes, I. T. Vasile, M. A. Dulea Computational Physics and
Design of nanomechanical sensors based on carbon nanoribbons and nanotubes in a distributed computing system T. L. Mitran a, C. M. Visan, G. A. Nemnes, I. T. Vasile, M. A. Dulea Computational Physics and
Orbitals and energetics
 Orbitals and energetics Bonding and structure Molecular orbital theory Crystal field theory Ligand field theory Provide fundamental understanding of chemistry dictating radionuclide complexes Structure
Orbitals and energetics Bonding and structure Molecular orbital theory Crystal field theory Ligand field theory Provide fundamental understanding of chemistry dictating radionuclide complexes Structure
G. Gantefdr and W. Eberhardt Institut fiir Festkiirperforschung, Forschungszentrum Jiilich, 5170 Jiilich, Germany
 Shell structure and s-p hybridization in small aluminum clusters G. Gantefdr and W. Eberhardt Institut fiir Festkiirperforschung, Forschungszentrum Jiilich, 5170 Jiilich, Germany Photoelectron spectra
Shell structure and s-p hybridization in small aluminum clusters G. Gantefdr and W. Eberhardt Institut fiir Festkiirperforschung, Forschungszentrum Jiilich, 5170 Jiilich, Germany Photoelectron spectra
Supplementary Note 1 Cleanliness during FM-TERS measurement Initial cleanliness of the whole system (before TERS measurement) can be assured using
 can be assured using") Supplementary Note 1 Cleanliness during FM-TERS measurement Initial cleanliness of the whole system (before TERS measurement) can be assured using the protocol in the Methods sections concerning tip and
Supplementary Note 1 Cleanliness during FM-TERS measurement Initial cleanliness of the whole system (before TERS measurement) can be assured using the protocol in the Methods sections concerning tip and
Supporting information
 Supporting information Toward a Janus Cluster: Regiospecific Decarboxylation of Ag 44 (4- MBA) 30 @Ag Nanoparticles Indranath Chakraborty, Anirban Som, Tuhina Adit Maark, Biswajit Mondal, Depanjan Sarkar
Supporting information Toward a Janus Cluster: Regiospecific Decarboxylation of Ag 44 (4- MBA) 30 @Ag Nanoparticles Indranath Chakraborty, Anirban Som, Tuhina Adit Maark, Biswajit Mondal, Depanjan Sarkar
Molecular Orbital Theory. Molecular Orbital Theory: Electrons are located in the molecule, not held in discrete regions between two bonded atoms
 Molecular Orbital Theory Valence Bond Theory: Electrons are located in discrete pairs between specific atoms Molecular Orbital Theory: Electrons are located in the molecule, not held in discrete regions
Molecular Orbital Theory Valence Bond Theory: Electrons are located in discrete pairs between specific atoms Molecular Orbital Theory: Electrons are located in the molecule, not held in discrete regions
Structural and electronic properties of Al 7 I n (n = 1,2,3)
") Chemical Physics Letters 420 (2006) 125 129 www.elsevier.com/locate/cplett Structural and electronic properties of Al 7 I n (n = 1,2,3) Shi-Hao Wei, Li Huang, M. Ji, X.G. Gong * Surface Physics Laboratory,
Chemical Physics Letters 420 (2006) 125 129 www.elsevier.com/locate/cplett Structural and electronic properties of Al 7 I n (n = 1,2,3) Shi-Hao Wei, Li Huang, M. Ji, X.G. Gong * Surface Physics Laboratory,
AB INITIO MODELING OF THE STRUCTURAL DEFECTS IN AMIDES
 Int. J. Chem. Sci.: 9(4), 2011, 1564-1568 ISSN 0972-768X www.sadgurupublications.com AB INITIO MODELING OF THE STRUCTURAL DEFECTS IN AMIDES M. FATHIMA BEGUM, HEMA TRESA VARGHESE a, Y. SHEENA MARY a, C.
Int. J. Chem. Sci.: 9(4), 2011, 1564-1568 ISSN 0972-768X www.sadgurupublications.com AB INITIO MODELING OF THE STRUCTURAL DEFECTS IN AMIDES M. FATHIMA BEGUM, HEMA TRESA VARGHESE a, Y. SHEENA MARY a, C.
Competition between Alkalide Characteristics and Nonlinear Optical Properties in OLi 3 M Li 3 O (M = Li, Na, and K) Complexes
 Complexes") Competition between Alkalide Characteristics and Nonlinear Optical Properties in OLi 3 M Li 3 O (M = Li, Na, and K) Complexes Ambrish Kumar Srivastava and Neeraj Misra * Department of Physics, University
Competition between Alkalide Characteristics and Nonlinear Optical Properties in OLi 3 M Li 3 O (M = Li, Na, and K) Complexes Ambrish Kumar Srivastava and Neeraj Misra * Department of Physics, University
The Boron Buckyball has an Unexpected T h Symmetry
 The Boron Buckyball has an Unexpected T h Symmetry G. Gopakumar, Minh Tho Nguyen, and Arnout Ceulemans* Department of Chemistry and Institute for Nanoscale Physics and Chemistry, University of Leuven,
The Boron Buckyball has an Unexpected T h Symmetry G. Gopakumar, Minh Tho Nguyen, and Arnout Ceulemans* Department of Chemistry and Institute for Nanoscale Physics and Chemistry, University of Leuven,
Electronegativity is a very useful concept for the explanation or understanding of chemical reactivity throughout the periodic table.
 1.6. Review of Electronegativity (χ) CONCEPT: Electronegativity is a very useful concept for the explanation or understanding of chemical reactivity throughout the periodic table. There are many definitions
1.6. Review of Electronegativity (χ) CONCEPT: Electronegativity is a very useful concept for the explanation or understanding of chemical reactivity throughout the periodic table. There are many definitions
Vacuum-Ultraviolet-Excited and CH 2 Cl 2 /H 2 O-Amplified Ionization- Coupled Mass Spectrometry for Oxygenated Organics Analysis
 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 Supporting Information for Vacuum-Ultraviolet-Excited and CH 2 Cl 2 /H 2 O-Amplified Ionization- Coupled Mass Spectrometry
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 Supporting Information for Vacuum-Ultraviolet-Excited and CH 2 Cl 2 /H 2 O-Amplified Ionization- Coupled Mass Spectrometry
Electronic shells or molecular orbitals: Photoelectron spectra of Ag n clusters
 Electronic shells or molecular orbitals: Photoelectron spectra of Ag n clusters H. Handschuh, Chia-Yen Cha, P. S. Bechthold, G. Ganteför, and W. Eberhardt Institut für Festkörperforschung, Forschungszentrum
Electronic shells or molecular orbitals: Photoelectron spectra of Ag n clusters H. Handschuh, Chia-Yen Cha, P. S. Bechthold, G. Ganteför, and W. Eberhardt Institut für Festkörperforschung, Forschungszentrum
Adsorption of Atomic H and O on the (111) Surface of Pt 3 Ni Alloys
 Surface of Pt 3 Ni Alloys") J. Phys. Chem. B 2004, 108, 8311-8323 8311 Adsorption of Atomic H and O on the (111) Surface of Pt 3 Ni Alloys Timo Jacob and William A. Goddard, III* Materials and Process Simulation Center, Beckman Institute
J. Phys. Chem. B 2004, 108, 8311-8323 8311 Adsorption of Atomic H and O on the (111) Surface of Pt 3 Ni Alloys Timo Jacob and William A. Goddard, III* Materials and Process Simulation Center, Beckman Institute
Structural Properties, Natural Bond Orbital, Theory Functional Calculations (DFT), and Energies for the α Halorganic Compounds
, and Energies for the α Halorganic Compounds") Current World Environment Vol. 7(2), 221-226 (2012) Structural Properties, Natural Bond Orbital, Theory Functional Calculations (DFT), and Energies for the α Halorganic Compounds NAJLA SEIDY and SHAHRIAR
Current World Environment Vol. 7(2), 221-226 (2012) Structural Properties, Natural Bond Orbital, Theory Functional Calculations (DFT), and Energies for the α Halorganic Compounds NAJLA SEIDY and SHAHRIAR
MOLECULES. ENERGY LEVELS electronic vibrational rotational
 MOLECULES BONDS Ionic: closed shell (+) or open shell (-) Covalent: both open shells neutral ( share e) Other (skip): van der Waals (He-He) Hydrogen bonds (in DNA, proteins, etc) ENERGY LEVELS electronic
MOLECULES BONDS Ionic: closed shell (+) or open shell (-) Covalent: both open shells neutral ( share e) Other (skip): van der Waals (He-He) Hydrogen bonds (in DNA, proteins, etc) ENERGY LEVELS electronic
Lecture 16, February 25, 2015 Metallic bonding
 Lecture 16, February 25, 2015 Metallic bonding Elements of Quantum Chemistry with Applications to Chemical Bonding and Properties of Molecules and Solids Course number: Ch125a; Room 115 BI Hours: 11-11:50am
Lecture 16, February 25, 2015 Metallic bonding Elements of Quantum Chemistry with Applications to Chemical Bonding and Properties of Molecules and Solids Course number: Ch125a; Room 115 BI Hours: 11-11:50am
Vibrationally resolved photoelectron spectroscopy of silicon cluster anions
 Vibrationally resolved photoelectron spectroscopy of silicon cluster anions Si n n3 7 Cangshan Xu, a) Travis R. Taylor, Gordon R. Burton, b) and Daniel M. Neumark Department of Chemistry, University of
Vibrationally resolved photoelectron spectroscopy of silicon cluster anions Si n n3 7 Cangshan Xu, a) Travis R. Taylor, Gordon R. Burton, b) and Daniel M. Neumark Department of Chemistry, University of
Cluster-π electronic interaction in a superatomic Au 13 cluster bearing σ-bonded acetylide ligands
 Electronic Supplementary Material (ESI) for Chemical Communications. This journal is The Royal Society of Chemistry 2015 SUPPORTING INFORMATION Cluster-π electronic interaction in a superatomic Au 13 cluster
Electronic Supplementary Material (ESI) for Chemical Communications. This journal is The Royal Society of Chemistry 2015 SUPPORTING INFORMATION Cluster-π electronic interaction in a superatomic Au 13 cluster
Part III: Theoretical Surface Science Adsorption at Surfaces
 Technische Universität München Part III: Theoretical Surface Science Adsorption at Surfaces Karsten Reuter Lecture course: Solid State Theory Adsorption at surfaces (T,p) Phase II Phase I Corrosion Growth
Technische Universität München Part III: Theoretical Surface Science Adsorption at Surfaces Karsten Reuter Lecture course: Solid State Theory Adsorption at surfaces (T,p) Phase II Phase I Corrosion Growth
Australian Journal of Basic and Applied Sciences
 AENSI Journals Australian Journal of Basic and Applied Sciences ISSN:1991-8178 Journal home page: www.ajbasweb.com Theoretical Study for the Effect of Hydroxyl Radical on the Electronic Properties of Cyclobutadiene
AENSI Journals Australian Journal of Basic and Applied Sciences ISSN:1991-8178 Journal home page: www.ajbasweb.com Theoretical Study for the Effect of Hydroxyl Radical on the Electronic Properties of Cyclobutadiene
CHEM 344 Molecular Modeling
 CHEM 344 Molecular Modeling The Use of Computational Chemistry to Support Experimental Organic Chemistry Part 1: Molecular Orbital Theory, Hybridization, & Formal Charge * all calculation data obtained
CHEM 344 Molecular Modeling The Use of Computational Chemistry to Support Experimental Organic Chemistry Part 1: Molecular Orbital Theory, Hybridization, & Formal Charge * all calculation data obtained
Structural and electronic properties of small titanium clusters: A density functional theory and anion photoelectron spectroscopy study
 JOURNAL OF CHEMICAL PHYSICS VOLUME 118, NUMBER 5 1 FEBRUARY 2003 Structural and electronic properties of small titanium clusters: A density functional theory and anion photoelectron spectroscopy study
JOURNAL OF CHEMICAL PHYSICS VOLUME 118, NUMBER 5 1 FEBRUARY 2003 Structural and electronic properties of small titanium clusters: A density functional theory and anion photoelectron spectroscopy study
Spectroscopy at nanometer scale
 Spectroscopy at nanometer scale 1. Physics of the spectroscopies 2. Spectroscopies for the bulk materials 3. Experimental setups for the spectroscopies 4. Physics and Chemistry of nanomaterials Various
Spectroscopy at nanometer scale 1. Physics of the spectroscopies 2. Spectroscopies for the bulk materials 3. Experimental setups for the spectroscopies 4. Physics and Chemistry of nanomaterials Various
Density functional study of structural trends for late-transition-metal 13-atom clusters
 Iowa State University From the SelectedWorks of Duane D. Johnson June 6, 2007 Density functional study of structural trends for late-transition-metal 13-atom clusters Lin-Lin Wang, University of Illinois
Iowa State University From the SelectedWorks of Duane D. Johnson June 6, 2007 Density functional study of structural trends for late-transition-metal 13-atom clusters Lin-Lin Wang, University of Illinois
Electronic Supporting Information for
 Electronic Supplementary Material (ESI) for Materials Horizons. This journal is The Royal Society of Chemistry 2015 Electronic Supporting Information for Probing the Energy Levels in Hole-doped Molecular
Electronic Supplementary Material (ESI) for Materials Horizons. This journal is The Royal Society of Chemistry 2015 Electronic Supporting Information for Probing the Energy Levels in Hole-doped Molecular
Ultraviolet and Visible Spectroscopy. interaction of materials with light at different electronic levels and the extent, to which such
 Surname 1 Ultraviolet and Visible Spectroscopy Introduction This experiment was carried out to demonstrate the effect of atomic structure on the interaction of materials with light at different electronic
Surname 1 Ultraviolet and Visible Spectroscopy Introduction This experiment was carried out to demonstrate the effect of atomic structure on the interaction of materials with light at different electronic
A critical approach toward resonance-assistance in the intramolecular hydrogen bond
 Electronic Supplementary Material (ESI) for Photochemical & Photobiological Sciences. This journal is The Royal Society of Chemistry and Owner Societies 2015 Supplementary Information for A critical approach
Electronic Supplementary Material (ESI) for Photochemical & Photobiological Sciences. This journal is The Royal Society of Chemistry and Owner Societies 2015 Supplementary Information for A critical approach
Geometry and energetics of Si 60 isomers
 Science and Technology of Advanced Materials 4 (2003) 361 365 www.elsevier.com/locate/stam Geometry and energetics of Si 60 isomers Q. Sun a,b, *, Q. Wang b, P. Jena b, J.Z. Yu a, Y. Kawazoe a a Kawazoe
Science and Technology of Advanced Materials 4 (2003) 361 365 www.elsevier.com/locate/stam Geometry and energetics of Si 60 isomers Q. Sun a,b, *, Q. Wang b, P. Jena b, J.Z. Yu a, Y. Kawazoe a a Kawazoe
DFT and TDDFT calculation of lead chalcogenide clusters up to (PbX) 32
 32") DFT and TDDFT calculation of lead chalcogenide clusters up to (PbX) 32 V. S. Gurin Research Institute for Physical Chemical Problems, Belarusian State University, Leningradskaya 14, 220006, Minsk, Belarus
DFT and TDDFT calculation of lead chalcogenide clusters up to (PbX) 32 V. S. Gurin Research Institute for Physical Chemical Problems, Belarusian State University, Leningradskaya 14, 220006, Minsk, Belarus
PBS: FROM SOLIDS TO CLUSTERS
 PBS: FROM SOLIDS TO CLUSTERS E. HOFFMANN AND P. ENTEL Theoretische Tieftemperaturphysik Gerhard-Mercator-Universität Duisburg, Lotharstraße 1 47048 Duisburg, Germany Semiconducting nanocrystallites like
PBS: FROM SOLIDS TO CLUSTERS E. HOFFMANN AND P. ENTEL Theoretische Tieftemperaturphysik Gerhard-Mercator-Universität Duisburg, Lotharstraße 1 47048 Duisburg, Germany Semiconducting nanocrystallites like
Geometry and electronic structure of V-n(Bz)(m) complexes
(m) complexes") Virginia Commonwealth University VCU Scholars Compass Physics Publications Dept. of Physics 2004 Geometry and electronic structure of V-n(Bz)(m) complexes Anil K. Kandalam Virginia Commonwealth University
Virginia Commonwealth University VCU Scholars Compass Physics Publications Dept. of Physics 2004 Geometry and electronic structure of V-n(Bz)(m) complexes Anil K. Kandalam Virginia Commonwealth University
PAPER No. : 8 (PHYSICAL SPECTROSCOPY) MODULE No. : 5 (TRANSITION PROBABILITIES AND TRANSITION DIPOLE MOMENT. OVERVIEW OF SELECTION RULES)
 MODULE No. : 5 (TRANSITION PROBABILITIES AND TRANSITION DIPOLE MOMENT. OVERVIEW OF SELECTION RULES)") Subject Chemistry Paper No and Title Module No and Title Module Tag 8 and Physical Spectroscopy 5 and Transition probabilities and transition dipole moment, Overview of selection rules CHE_P8_M5 TABLE
Subject Chemistry Paper No and Title Module No and Title Module Tag 8 and Physical Spectroscopy 5 and Transition probabilities and transition dipole moment, Overview of selection rules CHE_P8_M5 TABLE
DFT Study of Small Gold Clusters Au n (n=2-13): The Structural, Electronic, Thermodynamic and Spectral Properties
: The Structural, Electronic, Thermodynamic and Spectral Properties") DFT Study of Small Gold Clusters Au n (n=2-13): The Structural, Electronic, Thermodynamic and Spectral Properties Noha Jabr Department of chemistry, Faculty of Science, University of Al-Baath Homs, Syria
DFT Study of Small Gold Clusters Au n (n=2-13): The Structural, Electronic, Thermodynamic and Spectral Properties Noha Jabr Department of chemistry, Faculty of Science, University of Al-Baath Homs, Syria
Interaction of O 2 with Gold Clusters: Molecular and Dissociative Adsorption
 4066 J. Phys. Chem. A 2003, 107, 4066-4071 Interaction of O 2 with Gold Clusters: Molecular and Dissociative Adsorption Bokwon Yoon, Hannu Ha1kkinen,* and Uzi Landman School of Physics, Georgia Institute
4066 J. Phys. Chem. A 2003, 107, 4066-4071 Interaction of O 2 with Gold Clusters: Molecular and Dissociative Adsorption Bokwon Yoon, Hannu Ha1kkinen,* and Uzi Landman School of Physics, Georgia Institute
Structural, electronic and magnetic properties of vacancies in single-walled carbon nanotubes
 Structural, electronic and magnetic properties of vacancies in single-walled carbon nanotubes W. Orellana and P. Fuentealba Departamento de Física, Facultad de Ciencias, Universidad de Chile, Casilla 653,
Structural, electronic and magnetic properties of vacancies in single-walled carbon nanotubes W. Orellana and P. Fuentealba Departamento de Física, Facultad de Ciencias, Universidad de Chile, Casilla 653,
Supplementary Materials
 Supplementary Materials Sample characterization The presence of Si-QDs is established by Transmission Electron Microscopy (TEM), by which the average QD diameter of d QD 2.2 ± 0.5 nm has been determined
Supplementary Materials Sample characterization The presence of Si-QDs is established by Transmission Electron Microscopy (TEM), by which the average QD diameter of d QD 2.2 ± 0.5 nm has been determined
Geometry and electronic structure of Vn(Bz)m complexes
m complexes") West Chester University Digital Commons @ West Chester University Physics College of Arts & Sciences 6-8-2004 Geometry and electronic structure of Vn(Bz)m complexes Anil K. Kandalam West Chester University
West Chester University Digital Commons @ West Chester University Physics College of Arts & Sciences 6-8-2004 Geometry and electronic structure of Vn(Bz)m complexes Anil K. Kandalam West Chester University
Supporting Information
 Supporting Information Uniformly Sized (112) Facet Co 2 P on Graphene for Highly Effective Photocatalytic Hydrogen Evolution Bin Tian, a, b Zhen Li, a, b Wenlong Zhen c and Gongxuan Lu *a a State Key Laboratory
Supporting Information Uniformly Sized (112) Facet Co 2 P on Graphene for Highly Effective Photocatalytic Hydrogen Evolution Bin Tian, a, b Zhen Li, a, b Wenlong Zhen c and Gongxuan Lu *a a State Key Laboratory
Quantum Mechanical Study on the Adsorption of Drug Gentamicin onto γ-fe 2
 ORIENTAL JOURNAL OF CHEMISTRY An International Open Free Access, Peer Reviewed Research Journal www.orientjchem.org ISSN: 0970-00 X CODEN: OJCHEG 015, Vol. 31, No. (3): Pg. 1509-1513 Quantum Mechanical
ORIENTAL JOURNAL OF CHEMISTRY An International Open Free Access, Peer Reviewed Research Journal www.orientjchem.org ISSN: 0970-00 X CODEN: OJCHEG 015, Vol. 31, No. (3): Pg. 1509-1513 Quantum Mechanical
Uptake of OH radical to aqueous aerosol: a computational study
 Uptake of OH radical to aqueous aerosol: a computational study Grigory Andreev Karpov Institute of Physical Chemistry 10 Vorontsovo pole, Moscow, 105064, Russia Institute of Physical Chemistry and Electrochemistry
Uptake of OH radical to aqueous aerosol: a computational study Grigory Andreev Karpov Institute of Physical Chemistry 10 Vorontsovo pole, Moscow, 105064, Russia Institute of Physical Chemistry and Electrochemistry
Physical Chemistry Lab II CHEM 4644 Spring 2011 Final Exam 5 questions at 3 points each equals 15 total points possible.
 Physical Chemistry Lab II Name: KEY CHEM 4644 Spring 2011 Final Exam 5 questions at 3 points each equals 15 total points possible. Constants: c = 3.00 10 8 m/s h = 6.63 10-34 J s 1 Hartree = 4.36 10-18
Physical Chemistry Lab II Name: KEY CHEM 4644 Spring 2011 Final Exam 5 questions at 3 points each equals 15 total points possible. Constants: c = 3.00 10 8 m/s h = 6.63 10-34 J s 1 Hartree = 4.36 10-18
Ionic Bonding. Example: Atomic Radius: Na (r = 0.192nm) Cl (r = 0.099nm) Ionic Radius : Na (r = 0.095nm) Cl (r = 0.181nm)
 Cl (r = 0.099nm) Ionic Radius : Na (r = 0.095nm) Cl (r = 0.181nm)") Ionic Bonding Ion: an atom or molecule that gains or loses electrons (acquires an electrical charge). Atoms form cations (+charge), when they lose electrons, or anions (- charge), when they gain electrons.
Ionic Bonding Ion: an atom or molecule that gains or loses electrons (acquires an electrical charge). Atoms form cations (+charge), when they lose electrons, or anions (- charge), when they gain electrons.
Photon Interaction. Spectroscopy
 Photon Interaction Incident photon interacts with electrons Core and Valence Cross Sections Photon is Adsorbed Elastic Scattered Inelastic Scattered Electron is Emitted Excitated Dexcitated Stöhr, NEXAPS
Photon Interaction Incident photon interacts with electrons Core and Valence Cross Sections Photon is Adsorbed Elastic Scattered Inelastic Scattered Electron is Emitted Excitated Dexcitated Stöhr, NEXAPS
Theoretical Chemistry - Level II - Practical Class Molecular Orbitals in Diatomics
 Theoretical Chemistry - Level II - Practical Class Molecular Orbitals in Diatomics Problem 1 Draw molecular orbital diagrams for O 2 and O 2 +. E / ev dioxygen molecule, O 2 dioxygenyl cation, O 2 + 25
Theoretical Chemistry - Level II - Practical Class Molecular Orbitals in Diatomics Problem 1 Draw molecular orbital diagrams for O 2 and O 2 +. E / ev dioxygen molecule, O 2 dioxygenyl cation, O 2 + 25
Photoabsorption Spectra of Si n and Si n O (n 5)
") Commun. Theor. Phys. (Beijing, China) 51 (2009) pp. 751 755 c Chinese Physical Society and IOP Publishing Ltd Vol. 51, No. 4, April 15, 2009 Photoabsorption Spectra of Si n and Si n O (n 5) AN Fang-Fang,
Commun. Theor. Phys. (Beijing, China) 51 (2009) pp. 751 755 c Chinese Physical Society and IOP Publishing Ltd Vol. 51, No. 4, April 15, 2009 Photoabsorption Spectra of Si n and Si n O (n 5) AN Fang-Fang,
Supporting Information for. Predicting the Stability of Fullerene Allotropes Throughout the Periodic Table MA 02139
 Supporting Information for Predicting the Stability of Fullerene Allotropes Throughout the Periodic Table Qing Zhao 1, 2, Stanley S. H. Ng 1, and Heather J. Kulik 1, * 1 Department of Chemical Engineering,
Supporting Information for Predicting the Stability of Fullerene Allotropes Throughout the Periodic Table Qing Zhao 1, 2, Stanley S. H. Ng 1, and Heather J. Kulik 1, * 1 Department of Chemical Engineering,
DFT-TDDFT Computational Study of Three Different Chlorophyllous as Dye sensitized solar cells (DSSCs)
") DFT-TDDFT Computational Study of Three Different Chlorophyllous as Dye sensitized solar cells (DSSCs) Rahmatollah Rahimi 1, *, Mahboube Rabbani 1, Rahim Rahimi 2, Morteza Moghimi Waskasi 1 Rahimi_rah@iust.ac.ir
DFT-TDDFT Computational Study of Three Different Chlorophyllous as Dye sensitized solar cells (DSSCs) Rahmatollah Rahimi 1, *, Mahboube Rabbani 1, Rahim Rahimi 2, Morteza Moghimi Waskasi 1 Rahimi_rah@iust.ac.ir
Supplementary Note 1: Dark field measurements and Scattering properties of NPoM geometries
 Supplementary Note 1: Dark field measurements and Scattering properties of NPoM geometries Supplementary Figure 1: Dark field scattering properties of individual nanoparticle on mirror geometries separated
Supplementary Note 1: Dark field measurements and Scattering properties of NPoM geometries Supplementary Figure 1: Dark field scattering properties of individual nanoparticle on mirror geometries separated
Supporting Information. Revealing the Size Effect of Platinum Cocatalyst for Photocatalytic
 Supporting Information Revealing the Size Effect of Platinum Cocatalyst for Photocatalytic Hydrogen Evolution on TiO2 Support: A DFT Study Dong Wang,, Zhi-Pan Liu,*, Wei-Min Yang*, State Key Laboratory
Supporting Information Revealing the Size Effect of Platinum Cocatalyst for Photocatalytic Hydrogen Evolution on TiO2 Support: A DFT Study Dong Wang,, Zhi-Pan Liu,*, Wei-Min Yang*, State Key Laboratory
Chapter 9. Molecular Geometry and Bonding Theories
 Chapter 9. Molecular Geometry and Bonding Theories PART I Molecular Shapes Lewis structures give atomic connectivity: they tell us which atoms are physically connected to which atoms. The shape of a molecule
Chapter 9. Molecular Geometry and Bonding Theories PART I Molecular Shapes Lewis structures give atomic connectivity: they tell us which atoms are physically connected to which atoms. The shape of a molecule
Improved Electronic Structure and Optical Properties of sp-hybridized Semiconductors Using LDA+U SIC
 286 Brazilian Journal of Physics, vol. 36, no. 2A, June, 2006 Improved Electronic Structure and Optical Properties of sp-hybridized Semiconductors Using LDA+U SIC Clas Persson and Susanne Mirbt Department
286 Brazilian Journal of Physics, vol. 36, no. 2A, June, 2006 Improved Electronic Structure and Optical Properties of sp-hybridized Semiconductors Using LDA+U SIC Clas Persson and Susanne Mirbt Department
Calculation of Ionization Energy and Electron Affinity of Molecule of Hydro- and Fluorinefullerenes C 60 H(F) n (0 n 60)
 n (0 n 60)") 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 FULLERENES, NANOTUBES, AND CARBON NANOSTRUCTURES Vol. 12, No. 1, pp. 513 519, 2004 Calculation
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 FULLERENES, NANOTUBES, AND CARBON NANOSTRUCTURES Vol. 12, No. 1, pp. 513 519, 2004 Calculation
NPTEL/IITM. Molecular Spectroscopy Lectures 1 & 2. Prof.K. Mangala Sunder Page 1 of 15. Topics. Part I : Introductory concepts Topics
 Molecular Spectroscopy Lectures 1 & 2 Part I : Introductory concepts Topics Why spectroscopy? Introduction to electromagnetic radiation Interaction of radiation with matter What are spectra? Beer-Lambert
Molecular Spectroscopy Lectures 1 & 2 Part I : Introductory concepts Topics Why spectroscopy? Introduction to electromagnetic radiation Interaction of radiation with matter What are spectra? Beer-Lambert
1 Supporting information
 Electronic Supplementary Material (ESI) for Nanoscale. This journal is The Royal Society of Chemistry 2018 1 Supporting information 1.1 Separation of the chemical potentials of electrons and protons in
Electronic Supplementary Material (ESI) for Nanoscale. This journal is The Royal Society of Chemistry 2018 1 Supporting information 1.1 Separation of the chemical potentials of electrons and protons in
The Electronic Structure of Dye- Sensitized TiO 2 Clusters from Many- Body Perturbation Theory
 The Electronic Structure of Dye- Sensitized TiO 2 Clusters from Many- Body Perturbation Theory Noa Marom Center for Computational Materials Institute for Computational Engineering and Sciences The University
The Electronic Structure of Dye- Sensitized TiO 2 Clusters from Many- Body Perturbation Theory Noa Marom Center for Computational Materials Institute for Computational Engineering and Sciences The University
Density Functional Theory Investigation of The Physical Properties of Dicyano Pyridazine Molecules
 Density Functional Theory Investigation of The Physical Properties of Dicyano Pyridazine Molecules Fouad N. Ajeel 1, Alaa M. Khudhair 2, Anees A. Mohammed 3 1,2,3 Department of Physics, College of Sciences,
Density Functional Theory Investigation of The Physical Properties of Dicyano Pyridazine Molecules Fouad N. Ajeel 1, Alaa M. Khudhair 2, Anees A. Mohammed 3 1,2,3 Department of Physics, College of Sciences,
A study of nickel monoxide (NiO), nickel dioxide (ONiO), and Ni(O 2 ) complex by anion photoelectron spectroscopy
, nickel dioxide (ONiO), and Ni(O 2 ) complex by anion photoelectron spectroscopy") A study of nickel monoxide (NiO), nickel dioxide (ONiO), and Ni(O 2 ) complex by anion photoelectron spectroscopy Hongbin Wu and Lai-Sheng Wang Department of Physics, Washington State University, Richland,
A study of nickel monoxide (NiO), nickel dioxide (ONiO), and Ni(O 2 ) complex by anion photoelectron spectroscopy Hongbin Wu and Lai-Sheng Wang Department of Physics, Washington State University, Richland,
Bonding and Elastic Properties in Ti 2 AC (A = Ga or Tl)
") Printed in the Republic of Korea http://dx.doi.org/10.5012/jkcs.2013.57.1.35 Bonding and Elastic Properties in Ti 2 AC (A = Ga or Tl) Dae-Bok Kang* Department of Chemistry, Kyungsung University, Busan
Printed in the Republic of Korea http://dx.doi.org/10.5012/jkcs.2013.57.1.35 Bonding and Elastic Properties in Ti 2 AC (A = Ga or Tl) Dae-Bok Kang* Department of Chemistry, Kyungsung University, Busan
Ch. 9- Molecular Geometry and Bonding Theories
 Ch. 9- Molecular Geometry and Bonding Theories 9.0 Introduction A. Lewis structures do not show one of the most important aspects of molecules- their overall shapes B. The shape and size of molecules-
Ch. 9- Molecular Geometry and Bonding Theories 9.0 Introduction A. Lewis structures do not show one of the most important aspects of molecules- their overall shapes B. The shape and size of molecules-
DENSITY FUNCTIONAL THEORY STUDIES ON IR SPECTRA OF THE TRIPHENYLENE DERIVATIVES. A SCALED QUANTUM MECHANICAL FORCE FIELD APPROACH
 Vol. 98 (2000) ACTA PHYSICA POLONICA A No. 5 Proceedings of the International Conference "Condensed Matter Physics", Jaszowiec 2000 DENSITY FUNCTIONAL THEORY STUDIES ON IR SPECTRA OF THE TRIPHENYLENE DERIVATIVES.
Vol. 98 (2000) ACTA PHYSICA POLONICA A No. 5 Proceedings of the International Conference "Condensed Matter Physics", Jaszowiec 2000 DENSITY FUNCTIONAL THEORY STUDIES ON IR SPECTRA OF THE TRIPHENYLENE DERIVATIVES.
Supplemental Material: Experimental and Theoretical Investigations of the Electronic Band Structure of Metal-Organic Framework of HKUST-1 Type
 Supplemental Material: Experimental and Theoretical Investigations of the Electronic Band Structure of Metal-Organic Framework of HKUST-1 Type Zhigang Gu, a Lars Heinke, a,* Christof Wöll a, Tobias Neumann,
Supplemental Material: Experimental and Theoretical Investigations of the Electronic Band Structure of Metal-Organic Framework of HKUST-1 Type Zhigang Gu, a Lars Heinke, a,* Christof Wöll a, Tobias Neumann,
NUCLEAR MAGNETIC RESONANCE AND INTRODUCTION TO MASS SPECTROMETRY
 NUCLEAR MAGNETIC RESONANCE AND INTRODUCTION TO MASS SPECTROMETRY A STUDENT SHOULD BE ABLE TO: 1. Identify and explain the processes involved in proton ( 1 H) and carbon-13 ( 13 C) nuclear magnetic resonance
NUCLEAR MAGNETIC RESONANCE AND INTRODUCTION TO MASS SPECTROMETRY A STUDENT SHOULD BE ABLE TO: 1. Identify and explain the processes involved in proton ( 1 H) and carbon-13 ( 13 C) nuclear magnetic resonance
 CHAPTER 6 CHEMICAL BONDING SHORT QUESTION WITH ANSWERS Q.1 Dipole moments of chlorobenzene is 1.70 D and of chlorobenzene is 2.5 D while that of paradichlorbenzene is zero; why? Benzene has zero dipole
CHAPTER 6 CHEMICAL BONDING SHORT QUESTION WITH ANSWERS Q.1 Dipole moments of chlorobenzene is 1.70 D and of chlorobenzene is 2.5 D while that of paradichlorbenzene is zero; why? Benzene has zero dipole
Electronic structure of calcium clusters
 PHYSICAL REVIEW A, VOLUME 63, 023202 Electronic structure of calcium clusters Jeffrey W. Mirick, Chang-Hong Chien, and Estela Blaisten-Barojas* School of Computational Sciences, George Mason University,
PHYSICAL REVIEW A, VOLUME 63, 023202 Electronic structure of calcium clusters Jeffrey W. Mirick, Chang-Hong Chien, and Estela Blaisten-Barojas* School of Computational Sciences, George Mason University,
Particle Behavior of Light 1. Calculate the energy of a photon, mole of photons 2. Find binding energy of an electron (know KE) 3. What is a quanta?
 3. What is a quanta?") Properties of Electromagnetic Radiation 1. What is spectroscopy, a continuous spectrum, a line spectrum, differences and similarities 2. Relationship of wavelength to frequency, relationship of E to λ
Properties of Electromagnetic Radiation 1. What is spectroscopy, a continuous spectrum, a line spectrum, differences and similarities 2. Relationship of wavelength to frequency, relationship of E to λ
Nanotechnology and Solar Energy. Solar Electricity Photovoltaics. Fuel from the Sun Photosynthesis Biofuels Split Water Fuel Cells
 Nanotechnology and Solar Energy Solar Electricity Photovoltaics Fuel from the Sun Photosynthesis Biofuels Split Water Fuel Cells Solar cell A photon from the Sun generates an electron-hole pair in a semiconductor.
Nanotechnology and Solar Energy Solar Electricity Photovoltaics Fuel from the Sun Photosynthesis Biofuels Split Water Fuel Cells Solar cell A photon from the Sun generates an electron-hole pair in a semiconductor.
with the larger dimerization energy also exhibits the larger structural changes.
 A7. Looking at the image and table provided below, it is apparent that the monomer and dimer are structurally almost identical. Although angular and dihedral data were not included, these data are also
A7. Looking at the image and table provided below, it is apparent that the monomer and dimer are structurally almost identical. Although angular and dihedral data were not included, these data are also
Spectroscopy at nanometer scale
 Spectroscopy at nanometer scale 1. Physics of the spectroscopies 2. Spectroscopies for the bulk materials 3. Experimental setups for the spectroscopies 4. Physics and Chemistry of nanomaterials Various
Spectroscopy at nanometer scale 1. Physics of the spectroscopies 2. Spectroscopies for the bulk materials 3. Experimental setups for the spectroscopies 4. Physics and Chemistry of nanomaterials Various
Structures and photoelectron spectroscopy of Cun(BO2)(m) - (n, m=1, 2) clusters: Observation of hyperhalogen behavior
(m) - (n, m=1, 2) clusters: Observation of hyperhalogen behavior") Virginia Commonwealth University VCU Scholars Compass Physics Publications Dept. of Physics 2011 Structures and photoelectron spectroscopy of Cun(BO2)(m) - (n, m=1, 2) clusters: Observation of hyperhalogen
Virginia Commonwealth University VCU Scholars Compass Physics Publications Dept. of Physics 2011 Structures and photoelectron spectroscopy of Cun(BO2)(m) - (n, m=1, 2) clusters: Observation of hyperhalogen