Computational Chemistry I
|
|
|
- Sylvia Greer
- 5 years ago
- Views:
Transcription
1 Computational Chemistry I Text book Cramer: Essentials of Quantum Chemistry, Wiley (2 ed.) Chapter 3. Post Hartree-Fock methods (Cramer: chapter 7) There are many ways to improve the HF method. Most of them are very technical and in this course, the main ideas behind them are given. We can first define the correlation energy. The best (lowest) non-relativistic total energy within the Born- Oppenheimer approximation of the system is E tot, the correlation energy is the difference E corr = E tot - E HF < 0 As such, this is not very useful definition but if we have different well defined methods and basis sets to compute the correlation energy, the one which have lowest energy is the best. The variational principle works also here. The conceptually simplest post HF method is the Configuration Interaction (CI) method. In this method the wave functions is built from several determinants Ψ(,.. ) = Ψ +, Ψ +, Ψ +... The new determinants are built form the HF orbitals but the electrons are also placed on exited states. The notation Ψ means that one electron is excited from state i to state a, similarly Ψ means that two electrons are excited from states i and j to states a and b. This wave functions is denoted as CISD (CI with singlet and double excitations). The Ψ is the Slater determinant (or the HF wave functions). All the excited determinants are orthogonal to the HF wf (and to each other). Exercise: Show this.
2 In the CI calculations the HF orbitals are kept constant and the a coefficients are optimized. Again, the variational method can be used = min( Ψ Ψ + Ψ Ψ ), Or the matrix formalism can be used = Where mean any level of excited determinant. The matrix is very big but mostly empty (sparse) and efficient handling of it requires methods of sparse matrixes. The CISD method is a reasonable solution for the correlation energy for small molecules but for larger molecules, it become rather inefficient. Also for larger molecules CISD is not very accurate and higher terms would be useful but methods like CISDT become computationally very expensive. There will be a huge amount of excitations to be computed. In general the pure CI type methods become too expensive when the molecules size is increasing. The best CI method is called Full CI or FCI. In principle, it is an exact method but it is doable only for small molecules. For Full CI the number of determinant can be computed as = Where n is the number of orbital and k the number of electrons. This grows very rapidly, when n=2k (and k is big) = 2 16 if k=10 N det = 3.4*10 10!! This is impossible to compute. Scaling N 4 N 5 Method HF MP2
.")
3 N 6 N 7 N 8 N 10 MP3, CISD, CCSD MP4, CCSD(T) (MP5), CISDT, CCSDT (MP7), CISDTQ, CCSDTQ The CI has also a size consistency problem. This can be illustrated with an example of He dimer. The He atom have only two electrons and thus CISD is an exact method for it (CISD=FCI). When the He 2 is studied at CISD level the theory is not exact since the triple and quadrupole excitation are missing, (CISDTQ=FCI). In general finite-ci accuracy reduce with the size of the molecule and any calculation which deals with molecules association is biased with the size consistency. This is a rather large problem for computational binding energies and an unfortunately feature for many Post-HF methods. Active Space SCF Another approach to correlation is Active Space SCF. The first assumption is that not all possible excitations are needed. One can define limited amount of electrons and states in where all excitation are considered. This is called Complete Active Space (CAS). Then for larger amount electrons and states only N excitations are allowed. This is Restricted Active Space (RAS).
4 Then also the orbitals can be optimized for each excitation. This is called as Multi Reference method (MRSCF) (Note in CI the orbitals are not optimized). This causes more work but can significantly improve the total electronic energy. In general, the CAS methods are not easy to use. The good CAS depend strongly from the system. There also numerical problems related to the orbital optimizations. The CASSCF methods are not recommended for non-experts. On can also combine the CI and the MR approach. The MRCI methods, like MRCISD, are not much used. They are expensive and difficult to use. empty states RAS active states CAS filled states RAS Coupled Cluster method. The Coupled Cluster Methods is an advanced method to solve the CI wave functions. The CC wave function is computed as Ψ = Ψ where T=1+T 1 +T 2 +T 3 +, T n is an operator that makes n times excited states. For example =,,,
5 will make a determinant that have two exited electrons. The exponent of an operator is mathematically very complicated but the main point is that the CC wave functions are more complete than the CI wf s. The CC wave functions have contributions like C Ψ = Ψ = 1, =, = +, = + + = For example at CCSD level we have only operators T 1 and T 2 we will have higher order terms in the C 3 and C 4 etc. level. The solution of the amplitudes is the trick. It can be done effectively. Also the CC is size consistent which is also a bonus compared to CI. A good but far from easy presentation of CC can be found from Helgaker, Jorgensen and Olsen: Molecular Electronic-Structure Theory (Willey). For practical point of view the CC is the best black box quantum chemistry methods there exists. CCSD is a good method and CCSDT and CCSDTQ are excellent methods but the later are expensive. Using perturbation method the triplet excitations can be treated more effectively and the CCSD(T) method is also a very reliable methods. In practice it is often even better than CCSDT due to slight overestimation of the correlation. Today the CCSD(T) is considered the golden standard of quantum chemistry. This is means that it is almost as accurate (for medium size molecules) as FCI or any super accurate method but much cheaper. Compared to MRSCR methods the CC methods are easy to use and almost always reliable. Naturally the CCSD(T) is not cheap so it cannot be used for large molecules. Perturbation methods Another approach to correlation it the perturbation methods. The idea is very general and it can be used to several other quantum chemical problems. We assume that we can solve Hamiltonian H (0)
6 and the full Hamiltonian have a small perturbation V. The perturbation will be scale with parameter l. = ( ) + We can next write the energy and wave functions as a power series of l. = ( ) + ( ) + ( ) + = ( ) + ( ) + ( ) + Now we can write hierarchical equation for each power of l ( ) ( ) = ( ) ( ) ( ) ( ) + ( ) = ( ) ( ) + ( ) ( ) ( ) ( ) + ( ) = ( ) ( ) + ( ) ( ) + ( ) ( ) ( ) Note that notation means the ground state wave function at perturbation level n. The different level wave functions are orthogonal ( ) ( ) =. So we can solve ( ), Ψ ( ), ( ) etc. ( ) = ( ) ( ) ( ) = ( ) ( ) ( ) = ( ) ( ) ( ) = ( ) ( ) ( ) ( ) ( ) ( ) ( ) ( ( ) means non-ground state wave function at iteration level 0). The perturbation theory can be applied to correlation energy. This is called Moller-Plesset theory (MPn). Then the H (0) is the HF theory and the perturbation is = 1 ( 1 2 )
7 This is clever since it corrects the HF error. The level (2) correction is ( ) [( ) ( )] = +, The i,j denote occupied state and a,b unoccupied states. This the first real correction and it is called the MP2 theory. (The E (0) + E (1) is in fact the HF energy). The perturbation theory can be computed to arbitrary level. The different levels are denotes as MPn. Of these, the most important is the MP2. It is rather fast and causes a significant correction to HF. The MP3 is a disappointment since it does not corrects much the MP2. The MP4 is somewhat better but it is already very expensive. The higher terms are not used and in some model systems, it has been shown that the MPn series do not converge. Below are convergence of the HF molecule with respect of the MPn series. The energies are related to the FCI energy. (Picture From Helgaker et al, Molecular Electronic-Structure Theory). The molecules eq distance is R e (so 2.5 R e is stretched molecule). As one can see the results are strange.
8 Note the CC perturbation behaves better. RI-approximation Computationally the MP2 with small approximations can be implemented very effectively. The RI-MP2 method is much faster than and essentially as accurate as the true MP2. All practical MP2 calculations for larger systems are done with RI-MP2 (or its variants). In big RI-MP2 calculations the time consuming part is the HF. The basic idea behind RI-MP2 is to fit the expensive double wave functions (ia to new basis functions P) (auxiliary basis). Now the expensive (ij ab) term can be approximated ( ) ( ) ( ) this is more complex but computationally faster. The details are complex but the main point is that RI methods need an additional basis. The Orca input is simple! RHF RI-MP2 TZVP TZV/C Opt
9 See also Orca Manual (3.0 page 41 to 42) You see that nothing is lost in the optimized geometry through the RI approximation thanks to the efficient and accurate RI-auxiliary basis sets of the Karlsruhe group (in general the deviations in the geometries between standard MP2 and RI-MP2 are very small). Thus, RI-MP2 really is a substantial improvement in efficiency over standard MP2. The perturbation method is very general and it can used in several quantum chemical problems. One application is the CI and CC methods where the higher order excitations can be solved with perturbation theory. The CCSD(T) method is the golden standard of quantum chemistry and it has the same scaling behavior as MP4. Last, the basis set needed for well converged CI and MPn methods is larger than in the HF calculations. This is bad news since these post-hf methods have poorer scaling behavior than HF so the computations become quickly very time consuming. Summary: the MP2 method is a very important method for correlation. It can be used for large molecules and it is a significant improvement to HF. The RI-method speedup the MP2 significantly. Multilevel methods The systematic single method approach to solve the correlation problem is usually very time consuming (both human and computer time) so several multilevel extrapolation methods have been proposed. The multilevel methods are very complex looking but the main idea is to extrapolate both the correlation and basis. Below is the G2 and G3 multilevel methods. Note that Orca do not have the multilevel methods. It utilizes the F12-methods.

10 F12-method The multilevel methods are messy and they contain empirical parameters so a more systematic way is to extrapolate the correlation energy from smaller basis sets. The best extrapolation method for correlation energy at the moment is the F12-method. It clearly outperforms the old extrapolation methods. But nothing comes for free. The additional effort for the F12-MP2 calculation is rather high, a DZ F12 calculation is roughly as expensive as normal QZ calculation. This can be improve with RI-KJ methods. # # Correlation energies of the water molecule: extrapolation versus F12 # # cc-pvdz MP2:
11 # T : # Q : # T/Q : (normal extrapolation) # Q/5 : (normal extrapolation) # F12-DZ : # RI-F12-DZ : (cc-pvdz/c note: basis for RI) # (cc-pvtz/c) # F12-TZ : # RI-F12-TZ : (cc-pvqz/c) # F12-QZ : Note that F12-method is also available for CCSD(T). There the F12 is much more efficient than the traditional extrapolation. Density Functional Theory The traditional Quantum Chemical methods will approach systematically the exact wave functions and thus the solution of the correlation energy but they will lead to very heavy computational procedures. The Density Functional Theory will utilize another approach. In its hart is a proof that the ground state of an electronic system depend only on the electron density r(r). Y ( 2 [ ( )] r1, r,.., rn ) a Y r r This wave function is much much simpler than the true wf. The problem is that we do not know the equations to solve the wf. The basic ideas are form Kohn, Hohenberg and Sham (1963, 1964, Kohn Nobel prize 1998). Even we do not know the correct equation, it is useful to try to find a good approximation for it. Kohn and Sham showed that a single Slater determinant type wf with simple model for the Exchange and Correlation (XC) energy will give good results. The formulation is quite similar to the HF: = = +
12 = ( ) ħ ( ) = (, ( ),..) ; = ( ) = 1 2 ( ) ( ) ( ) =, ( ), The difficult part is the XC energy. It is not known but several approximation are done. Formally the E xc can be written as [ ] = [ ] + [ ] Where <T> is the exact kinetic energy, T s is the DFT K.E. <W> is the exact electron-electron interaction energy and J is the Coulomb energy of the electron density. The is often expressed as an integral [ ] = ( ) [ ] The simplest Local Density Approximation (LDA) depend only on density, E XC,LDA [r(r)]. The exchange part is easy (a = 1) [ ] = ( ) But the correlation is more complex. This functional can be obtained from exact calculations of homogenous electron gas. The LDA work reasonably well and it is a bit better than HF (with few exceptions) but electrons are NOT homogenously spread in molecules. The next level is the Generalized Gradient Approximation, E XC,GGA [ r(r), ρ( )]. The GGA models are much better but there is no simple (or even non-simple) procedure to build the GGA functionals. For this reason there are a huge amount of different GGA models. There is a very extensive library of the XC functionals. The Libxc which is implemented to many codes (but not Orca, see the Orca manual)
13 The most common are BLYP and PBE. Even there are far too many GGA models, the DFT-GGA method have one very strong advantage. It scales as N 3. It is much more accurate than HF but it is much faster. With DFT we cannot get systematic improvement of the accuracy but it can be applied to big systems are the results are almost always good (same level as MP2). One can also mix the HF and DFT theories. These are called hybrid methods and they are also very accurate. The most common of them is the B3LYP model also PBE0 model is good. = + (1 ) + For example PBE0, a=1/4, b=1. GGA=PBE; = + (1 ) + ( )+ +(1 ) B3LYP is more complex: a=0.2, b=0.72, c=0.81. The next GGA level is meta-gga where the kinetic energy density is included to the GGA, ( ) = 1/2 ( ) E XC,m-GGA [ r(r), ρ( ), ( )]. Good meta-gga s are TPSS (implemented to Orca), HCTC, M06L. Last one can also combine MP2 and GGA. These are called double hybrids. In Orca B2PLYP is implemented. As DFT cannot be improved systematically the results matters. The DFT wf s are variational but the models are not, so we cannot judge the quality of results with the correlation energy. The results can be compared to experiments or to more accurate quantum chemical methods. Below there are few tables.
14
15
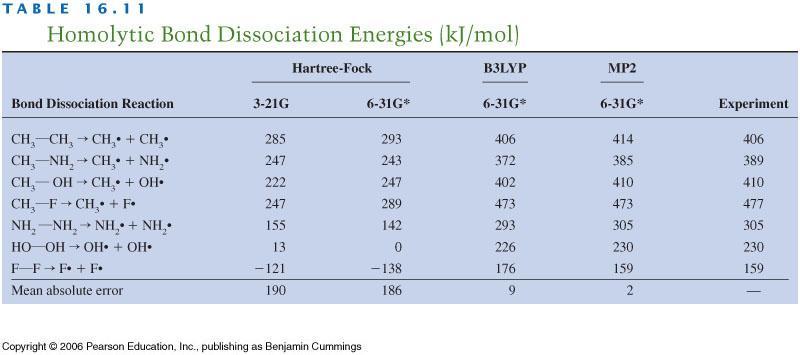
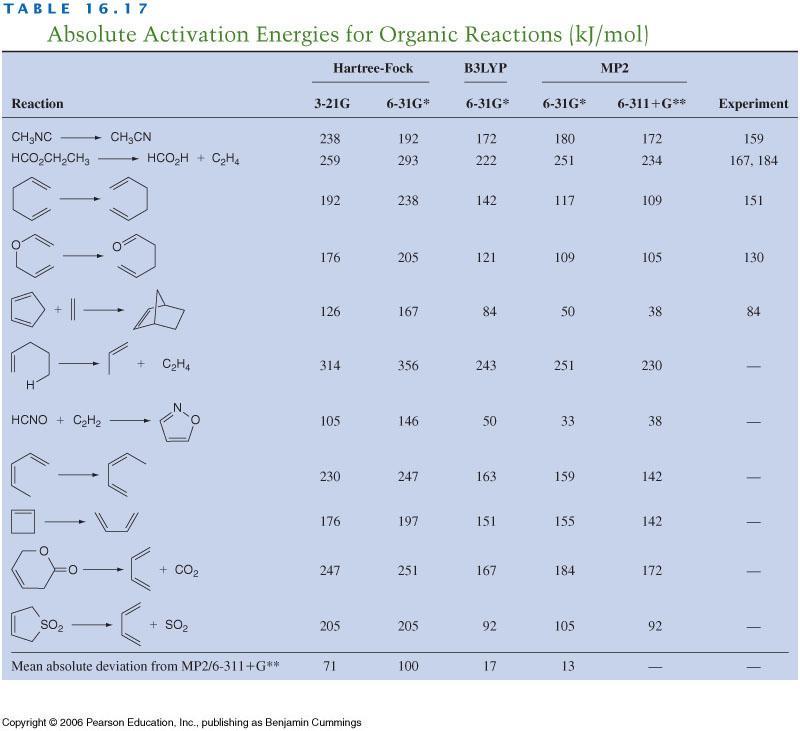
16 They show that OLYP and B3LYP method is usually as good as MP2. Unfortunately the results depend a (quite) bit from the property we are looking but in most cases the DFT methods are as good as or better than MP2. There are much more tables in the Cramers book. Almost all methods the bond distances are quite good. For energies the HF or LDA are not reliable whereas most GGA s and correlated quantum chemistry methods are good. The reaction barriers are challenging form computational methods and in them the GGA methods work rather well. Very crudely one can say that Double hybrid < Meta-GGA ~ Hybrid < pure GGA << LDA Naturally the GGA methods have some problems. The most obvious one is the difficulty to improve the XC-functionals. Another is the van der Waals interactions (or dispersion) which is important in weakly bonded systems. HF and by far most of the GGAs do not have them and they have to be added using additional (empirical) terms. In most cases the Grimme s D3 model works well. DFT have also problems to describe the hydrogen correctly. This is due to the Self Interaction Error (SIE). In DFT the density is the basic quantity and hydrogen sees its own density. This is not correct (think the Schrödinger equation of H atom). A good GGA will almost cancel this but not exactly. There are several Self Interaction Corrections (SIC) but they are difficult to use. Note that HF is exact for hydrogen so the hybrid functionals have smaller SIE and this is one reason of their success. Like HF the DFT models do not give good HOMO-LUMO gap or absolute energy levels for electronic states. Summary: It does not matter whether we like or dislike DFT, its accuracy compared to computational cost is superior to any of the traditional quantum chemistry methods. For large systems we need to use DFT until we get very efficient MP2-type methods. Computational development There is a lot of research focusing on development of the computational methods. In traditional quantum chemistry one area
17 is on development fast computational algorithms for the known methods. The RI-type methods have improve the MP2 calculations a lot. The very new DLPNO-CCSD(T)development of Orca team is very interesting, it makes CCSD(T)level calculations possible for large molecules. (See more from Orca manual.) In DFT the development is rather slow. Now there is quite a bit of interest of beyond-dft methods. Like RPA, MP2 etc. On the other hand both these methods have enormous amount of applications covering almost all fields of chemistry. DFT can be used to model single molecules, clusters of atoms, surfaces, solid systems and liquids. Systems up to valence electrons can be computed. An emerging research field is quantum mechanical materials screening where properties of materials (or molecules) will be evaluated computationally and this information is used to help the synthetic work.
18 Practical quantum chemistry calculations The first choice it the DFT with modern GGA or hybrid method. The basis should be TZ level. If possible test calculations (very often single point calculations are enough) with MP2 at least TZ basis should be done. The state-of-the-art basis set extrapolation methods like F12-RI-MP2 are efficient to estimate the infinite basis set limit. CCSD calculations are usually too expensive, but the DLPNO-CCSD(T)method looks very promising.
Electron Correlation
 Electron Correlation Levels of QM Theory HΨ=EΨ Born-Oppenheimer approximation Nuclear equation: H n Ψ n =E n Ψ n Electronic equation: H e Ψ e =E e Ψ e Single determinant SCF Semi-empirical methods Correlation
Electron Correlation Levels of QM Theory HΨ=EΨ Born-Oppenheimer approximation Nuclear equation: H n Ψ n =E n Ψ n Electronic equation: H e Ψ e =E e Ψ e Single determinant SCF Semi-empirical methods Correlation
Electron Correlation - Methods beyond Hartree-Fock
 Electron Correlation - Methods beyond Hartree-Fock how to approach chemical accuracy Alexander A. Auer Max-Planck-Institute for Chemical Energy Conversion, Mülheim September 4, 2014 MMER Summerschool 2014
Electron Correlation - Methods beyond Hartree-Fock how to approach chemical accuracy Alexander A. Auer Max-Planck-Institute for Chemical Energy Conversion, Mülheim September 4, 2014 MMER Summerschool 2014
Computational Methods. Chem 561
 Computational Methods Chem 561 Lecture Outline 1. Ab initio methods a) HF SCF b) Post-HF methods 2. Density Functional Theory 3. Semiempirical methods 4. Molecular Mechanics Computational Chemistry " Computational
Computational Methods Chem 561 Lecture Outline 1. Ab initio methods a) HF SCF b) Post-HF methods 2. Density Functional Theory 3. Semiempirical methods 4. Molecular Mechanics Computational Chemistry " Computational
Introduction to Computational Chemistry
 Introduction to Computational Chemistry Vesa Hänninen Laboratory of Physical Chemistry Chemicum 4th floor vesa.hanninen@helsinki.fi September 10, 2013 Lecture 3. Electron correlation methods September
Introduction to Computational Chemistry Vesa Hänninen Laboratory of Physical Chemistry Chemicum 4th floor vesa.hanninen@helsinki.fi September 10, 2013 Lecture 3. Electron correlation methods September
OVERVIEW OF QUANTUM CHEMISTRY METHODS
 OVERVIEW OF QUANTUM CHEMISTRY METHODS Outline I Generalities Correlation, basis sets Spin II Wavefunction methods Hartree-Fock Configuration interaction Coupled cluster Perturbative methods III Density
OVERVIEW OF QUANTUM CHEMISTRY METHODS Outline I Generalities Correlation, basis sets Spin II Wavefunction methods Hartree-Fock Configuration interaction Coupled cluster Perturbative methods III Density
Introduction to Electronic Structure Theory
 CSC/PRACE Spring School in Computational Chemistry 2017 Introduction to Electronic Structure Theory Mikael Johansson http://www.iki.fi/~mpjohans Objective: To get familiarised with the, subjectively chosen,
CSC/PRACE Spring School in Computational Chemistry 2017 Introduction to Electronic Structure Theory Mikael Johansson http://www.iki.fi/~mpjohans Objective: To get familiarised with the, subjectively chosen,
QUANTUM CHEMISTRY FOR TRANSITION METALS
 QUANTUM CHEMISTRY FOR TRANSITION METALS Outline I Introduction II Correlation Static correlation effects MC methods DFT III Relativity Generalities From 4 to 1 components Effective core potential Outline
QUANTUM CHEMISTRY FOR TRANSITION METALS Outline I Introduction II Correlation Static correlation effects MC methods DFT III Relativity Generalities From 4 to 1 components Effective core potential Outline
Module 6 1. Density functional theory
 Module 6 1. Density functional theory Updated May 12, 2016 B A DDFT C K A bird s-eye view of density-functional theory Authors: Klaus Capelle G http://arxiv.org/abs/cond-mat/0211443 R https://trac.cc.jyu.fi/projects/toolbox/wiki/dft
Module 6 1. Density functional theory Updated May 12, 2016 B A DDFT C K A bird s-eye view of density-functional theory Authors: Klaus Capelle G http://arxiv.org/abs/cond-mat/0211443 R https://trac.cc.jyu.fi/projects/toolbox/wiki/dft
Session 1. Introduction to Computational Chemistry. Computational (chemistry education) and/or (Computational chemistry) education
 and/or (Computational chemistry) education") Session 1 Introduction to Computational Chemistry 1 Introduction to Computational Chemistry Computational (chemistry education) and/or (Computational chemistry) education First one: Use computational tools
Session 1 Introduction to Computational Chemistry 1 Introduction to Computational Chemistry Computational (chemistry education) and/or (Computational chemistry) education First one: Use computational tools
Jack Simons, Henry Eyring Scientist and Professor Chemistry Department University of Utah
 1. Born-Oppenheimer approx.- energy surfaces 2. Mean-field (Hartree-Fock) theory- orbitals 3. Pros and cons of HF- RHF, UHF 4. Beyond HF- why? 5. First, one usually does HF-how? 6. Basis sets and notations
1. Born-Oppenheimer approx.- energy surfaces 2. Mean-field (Hartree-Fock) theory- orbitals 3. Pros and cons of HF- RHF, UHF 4. Beyond HF- why? 5. First, one usually does HF-how? 6. Basis sets and notations
Methods for Treating Electron Correlation CHEM 430
 Methods for Treating Electron Correlation CHEM 430 Electron Correlation Energy in the Hartree-Fock approximation, each electron sees the average density of all of the other electrons two electrons cannot
Methods for Treating Electron Correlation CHEM 430 Electron Correlation Energy in the Hartree-Fock approximation, each electron sees the average density of all of the other electrons two electrons cannot
High-level Quantum Chemistry Methods and Benchmark Datasets for Molecules
 High-level Quantum Chemistry Methods and Benchmark Datasets for Molecules Markus Schneider Fritz Haber Institute of the MPS, Berlin, Germany École Polytechnique Fédérale de Lausanne, Switzerland دانشگاه
High-level Quantum Chemistry Methods and Benchmark Datasets for Molecules Markus Schneider Fritz Haber Institute of the MPS, Berlin, Germany École Polytechnique Fédérale de Lausanne, Switzerland دانشگاه
Chemistry 334 Part 2: Computational Quantum Chemistry
 Chemistry 334 Part 2: Computational Quantum Chemistry 1. Definition Louis Scudiero, Ben Shepler and Kirk Peterson Washington State University January 2006 Computational chemistry is an area of theoretical
Chemistry 334 Part 2: Computational Quantum Chemistry 1. Definition Louis Scudiero, Ben Shepler and Kirk Peterson Washington State University January 2006 Computational chemistry is an area of theoretical
Exchange Correlation Functional Investigation of RT-TDDFT on a Sodium Chloride. Dimer. Philip Straughn
 Exchange Correlation Functional Investigation of RT-TDDFT on a Sodium Chloride Dimer Philip Straughn Abstract Charge transfer between Na and Cl ions is an important problem in physical chemistry. However,
Exchange Correlation Functional Investigation of RT-TDDFT on a Sodium Chloride Dimer Philip Straughn Abstract Charge transfer between Na and Cl ions is an important problem in physical chemistry. However,
Multi-reference Density Functional Theory. COLUMBUS Workshop Argonne National Laboratory 15 August 2005
 Multi-reference Density Functional Theory COLUMBUS Workshop Argonne National Laboratory 15 August 2005 Capt Eric V. Beck Air Force Institute of Technology Department of Engineering Physics 2950 Hobson
Multi-reference Density Functional Theory COLUMBUS Workshop Argonne National Laboratory 15 August 2005 Capt Eric V. Beck Air Force Institute of Technology Department of Engineering Physics 2950 Hobson
AN INTRODUCTION TO QUANTUM CHEMISTRY. Mark S. Gordon Iowa State University
 AN INTRODUCTION TO QUANTUM CHEMISTRY Mark S. Gordon Iowa State University 1 OUTLINE Theoretical Background in Quantum Chemistry Overview of GAMESS Program Applications 2 QUANTUM CHEMISTRY In principle,
AN INTRODUCTION TO QUANTUM CHEMISTRY Mark S. Gordon Iowa State University 1 OUTLINE Theoretical Background in Quantum Chemistry Overview of GAMESS Program Applications 2 QUANTUM CHEMISTRY In principle,
Density Functional Theory. Martin Lüders Daresbury Laboratory
 Density Functional Theory Martin Lüders Daresbury Laboratory Ab initio Calculations Hamiltonian: (without external fields, non-relativistic) impossible to solve exactly!! Electrons Nuclei Electron-Nuclei
Density Functional Theory Martin Lüders Daresbury Laboratory Ab initio Calculations Hamiltonian: (without external fields, non-relativistic) impossible to solve exactly!! Electrons Nuclei Electron-Nuclei
Density Functional Theory
 Density Functional Theory March 26, 2009 ? DENSITY FUNCTIONAL THEORY is a method to successfully describe the behavior of atomic and molecular systems and is used for instance for: structural prediction
Density Functional Theory March 26, 2009 ? DENSITY FUNCTIONAL THEORY is a method to successfully describe the behavior of atomic and molecular systems and is used for instance for: structural prediction
Electron Correlation Methods
 Electron Correlation Methods HF method: electron-electron interaction is replaced by an average interaction E HF c = E 0 E HF E 0 exact ground state energy E HF HF energy for a given basis set HF E c
Electron Correlation Methods HF method: electron-electron interaction is replaced by an average interaction E HF c = E 0 E HF E 0 exact ground state energy E HF HF energy for a given basis set HF E c
Ab initio calculations for potential energy surfaces. D. Talbi GRAAL- Montpellier
 Ab initio calculations for potential energy surfaces D. Talbi GRAAL- Montpellier A theoretical study of a reaction is a two step process I-Electronic calculations : techniques of quantum chemistry potential
Ab initio calculations for potential energy surfaces D. Talbi GRAAL- Montpellier A theoretical study of a reaction is a two step process I-Electronic calculations : techniques of quantum chemistry potential
DFT calculations of NMR indirect spin spin coupling constants
 DFT calculations of NMR indirect spin spin coupling constants Dalton program system Program capabilities Density functional theory Kohn Sham theory LDA, GGA and hybrid theories Indirect NMR spin spin coupling
DFT calculations of NMR indirect spin spin coupling constants Dalton program system Program capabilities Density functional theory Kohn Sham theory LDA, GGA and hybrid theories Indirect NMR spin spin coupling
Introduction to Density Functional Theory
 Introduction to Density Functional Theory S. Sharma Institut für Physik Karl-Franzens-Universität Graz, Austria 19th October 2005 Synopsis Motivation 1 Motivation : where can one use DFT 2 : 1 Elementary
Introduction to Density Functional Theory S. Sharma Institut für Physik Karl-Franzens-Universität Graz, Austria 19th October 2005 Synopsis Motivation 1 Motivation : where can one use DFT 2 : 1 Elementary
Exercise 1: Structure and dipole moment of a small molecule
 Introduction to computational chemistry Exercise 1: Structure and dipole moment of a small molecule Vesa Hänninen 1 Introduction In this exercise the equilibrium structure and the dipole moment of a small
Introduction to computational chemistry Exercise 1: Structure and dipole moment of a small molecule Vesa Hänninen 1 Introduction In this exercise the equilibrium structure and the dipole moment of a small
CLIMBING THE LADDER OF DENSITY FUNCTIONAL APPROXIMATIONS JOHN P. PERDEW DEPARTMENT OF PHYSICS TEMPLE UNIVERSITY PHILADELPHIA, PA 19122
 CLIMBING THE LADDER OF DENSITY FUNCTIONAL APPROXIMATIONS JOHN P. PERDEW DEPARTMENT OF PHYSICS TEMPLE UNIVERSITY PHILADELPHIA, PA 191 THANKS TO MANY COLLABORATORS, INCLUDING SY VOSKO DAVID LANGRETH ALEX
CLIMBING THE LADDER OF DENSITY FUNCTIONAL APPROXIMATIONS JOHN P. PERDEW DEPARTMENT OF PHYSICS TEMPLE UNIVERSITY PHILADELPHIA, PA 191 THANKS TO MANY COLLABORATORS, INCLUDING SY VOSKO DAVID LANGRETH ALEX
 3/23/2010 More basics of DFT Kieron Burke and friends UC Irvine Physics and Chemistry References for ground-state DFT ABC of DFT, by KB and Rudy Magyar, http://dft.uci.edu A Primer in Density Functional
3/23/2010 More basics of DFT Kieron Burke and friends UC Irvine Physics and Chemistry References for ground-state DFT ABC of DFT, by KB and Rudy Magyar, http://dft.uci.edu A Primer in Density Functional
This is a very succinct primer intended as supplementary material for an undergraduate course in physical chemistry.
 1 Computational Chemistry (Quantum Chemistry) Primer This is a very succinct primer intended as supplementary material for an undergraduate course in physical chemistry. TABLE OF CONTENTS Methods...1 Basis
1 Computational Chemistry (Quantum Chemistry) Primer This is a very succinct primer intended as supplementary material for an undergraduate course in physical chemistry. TABLE OF CONTENTS Methods...1 Basis
Teoría del Funcional de la Densidad (Density Functional Theory)
") Teoría del Funcional de la Densidad (Density Functional Theory) Motivation: limitations of the standard approach based on the wave function. The electronic density n(r) as the key variable: Functionals
Teoría del Funcional de la Densidad (Density Functional Theory) Motivation: limitations of the standard approach based on the wave function. The electronic density n(r) as the key variable: Functionals
MO Calculation for a Diatomic Molecule. /4 0 ) i=1 j>i (1/r ij )
 i=1 j>i (1/r ij )") MO Calculation for a Diatomic Molecule Introduction The properties of any molecular system can in principle be found by looking at the solutions to the corresponding time independent Schrodinger equation
MO Calculation for a Diatomic Molecule Introduction The properties of any molecular system can in principle be found by looking at the solutions to the corresponding time independent Schrodinger equation
Oslo node. Highly accurate calculations benchmarking and extrapolations
 Oslo node Highly accurate calculations benchmarking and extrapolations Torgeir Ruden, with A. Halkier, P. Jørgensen, J. Olsen, W. Klopper, J. Gauss, P. Taylor Explicitly correlated methods Pål Dahle, collaboration
Oslo node Highly accurate calculations benchmarking and extrapolations Torgeir Ruden, with A. Halkier, P. Jørgensen, J. Olsen, W. Klopper, J. Gauss, P. Taylor Explicitly correlated methods Pål Dahle, collaboration
Density Functional Theory
 Chemistry 380.37 Fall 2015 Dr. Jean M. Standard October 28, 2015 Density Functional Theory What is a Functional? A functional is a general mathematical quantity that represents a rule to convert a function
Chemistry 380.37 Fall 2015 Dr. Jean M. Standard October 28, 2015 Density Functional Theory What is a Functional? A functional is a general mathematical quantity that represents a rule to convert a function
( R)Ψ el ( r;r) = E el ( R)Ψ el ( r;r)
Ψ el ( r;r) = E el ( R)Ψ el ( r;r)") Born Oppenheimer Approximation: Ĥ el ( R)Ψ el ( r;r) = E el ( R)Ψ el ( r;r) For a molecule with N electrons and M nuclei: Ĥ el What is E el (R)? s* potential surface Reaction Barrier Unstable intermediate
Born Oppenheimer Approximation: Ĥ el ( R)Ψ el ( r;r) = E el ( R)Ψ el ( r;r) For a molecule with N electrons and M nuclei: Ĥ el What is E el (R)? s* potential surface Reaction Barrier Unstable intermediate
Supporting Information: Predicting the Ionic Product of Water
 Supporting Information: Predicting the Ionic Product of Water Eva Perlt 1,+, Michael von Domaros 1,+, Barbara Kirchner 1, Ralf Ludwig 2, and Frank Weinhold 3,* 1 Mulliken Center for Theoretical Chemistry,
Supporting Information: Predicting the Ionic Product of Water Eva Perlt 1,+, Michael von Domaros 1,+, Barbara Kirchner 1, Ralf Ludwig 2, and Frank Weinhold 3,* 1 Mulliken Center for Theoretical Chemistry,
Electronic structure theory: Fundamentals to frontiers. 2. Density functional theory
 Electronic structure theory: Fundamentals to frontiers. 2. Density functional theory MARTIN HEAD-GORDON, Department of Chemistry, University of California, and Chemical Sciences Division, Lawrence Berkeley
Electronic structure theory: Fundamentals to frontiers. 2. Density functional theory MARTIN HEAD-GORDON, Department of Chemistry, University of California, and Chemical Sciences Division, Lawrence Berkeley
Highly accurate quantum-chemical calculations
 1 Highly accurate quantum-chemical calculations T. Helgaker Centre for Theoretical and Computational Chemistry, Department of Chemistry, University of Oslo, Norway A. C. Hennum and T. Ruden, University
1 Highly accurate quantum-chemical calculations T. Helgaker Centre for Theoretical and Computational Chemistry, Department of Chemistry, University of Oslo, Norway A. C. Hennum and T. Ruden, University
Introduction to Density Functional Theory
 1 Introduction to Density Functional Theory 21 February 2011; V172 P.Ravindran, FME-course on Ab initio Modelling of solar cell Materials 21 February 2011 Introduction to DFT 2 3 4 Ab initio Computational
1 Introduction to Density Functional Theory 21 February 2011; V172 P.Ravindran, FME-course on Ab initio Modelling of solar cell Materials 21 February 2011 Introduction to DFT 2 3 4 Ab initio Computational
Walter Kohn was awarded with the Nobel Prize in Chemistry in 1998 for his development of the density functional theory.
 Walter Kohn was awarded with the Nobel Prize in Chemistry in 1998 for his development of the density functional theory. Walter Kohn receiving his Nobel Prize from His Majesty the King at the Stockholm
Walter Kohn was awarded with the Nobel Prize in Chemistry in 1998 for his development of the density functional theory. Walter Kohn receiving his Nobel Prize from His Majesty the King at the Stockholm
Density Func,onal Theory (Chapter 6, Jensen)
") Chem 580: DFT Density Func,onal Theory (Chapter 6, Jensen) Hohenberg- Kohn Theorem (Phys. Rev., 136,B864 (1964)): For molecules with a non degenerate ground state, the ground state molecular energy and
Chem 580: DFT Density Func,onal Theory (Chapter 6, Jensen) Hohenberg- Kohn Theorem (Phys. Rev., 136,B864 (1964)): For molecules with a non degenerate ground state, the ground state molecular energy and
Beyond the Hartree-Fock Approximation: Configuration Interaction
 Beyond the Hartree-Fock Approximation: Configuration Interaction The Hartree-Fock (HF) method uses a single determinant (single electronic configuration) description of the electronic wavefunction. For
Beyond the Hartree-Fock Approximation: Configuration Interaction The Hartree-Fock (HF) method uses a single determinant (single electronic configuration) description of the electronic wavefunction. For
GEM4 Summer School OpenCourseWare
 GEM4 Summer School OpenCourseWare http://gem4.educommons.net/ http://www.gem4.org/ Lecture: Molecular Mechanics by Ju Li. Given August 9, 2006 during the GEM4 session at MIT in Cambridge, MA. Please use
GEM4 Summer School OpenCourseWare http://gem4.educommons.net/ http://www.gem4.org/ Lecture: Molecular Mechanics by Ju Li. Given August 9, 2006 during the GEM4 session at MIT in Cambridge, MA. Please use
Introduction to density-functional theory. Emmanuel Fromager
 Institut de Chimie, Strasbourg, France Page 1 Emmanuel Fromager Institut de Chimie de Strasbourg - Laboratoire de Chimie Quantique - Université de Strasbourg /CNRS M2 lecture, Strasbourg, France. Institut
Institut de Chimie, Strasbourg, France Page 1 Emmanuel Fromager Institut de Chimie de Strasbourg - Laboratoire de Chimie Quantique - Université de Strasbourg /CNRS M2 lecture, Strasbourg, France. Institut
Chemistry 3502/4502. Final Exam Part I. May 14, 2005
 Advocacy chit Chemistry 350/450 Final Exam Part I May 4, 005. For which of the below systems is = where H is the Hamiltonian operator and T is the kinetic-energy operator? (a) The free particle
Advocacy chit Chemistry 350/450 Final Exam Part I May 4, 005. For which of the below systems is = where H is the Hamiltonian operator and T is the kinetic-energy operator? (a) The free particle
Molecular Mechanics: The Ab Initio Foundation
 Molecular Mechanics: The Ab Initio Foundation Ju Li GEM4 Summer School 2006 Cell and Molecular Mechanics in BioMedicine August 7 18, 2006, MIT, Cambridge, MA, USA 2 Outline Why are electrons quantum? Born-Oppenheimer
Molecular Mechanics: The Ab Initio Foundation Ju Li GEM4 Summer School 2006 Cell and Molecular Mechanics in BioMedicine August 7 18, 2006, MIT, Cambridge, MA, USA 2 Outline Why are electrons quantum? Born-Oppenheimer
Introduction to computational chemistry Exercise I: Structure and electronic energy of a small molecule. Vesa Hänninen
 Introduction to computational chemistry Exercise I: Structure and electronic energy of a small molecule Vesa Hänninen 1 Introduction In this exercise the equilibrium structure and the electronic energy
Introduction to computational chemistry Exercise I: Structure and electronic energy of a small molecule Vesa Hänninen 1 Introduction In this exercise the equilibrium structure and the electronic energy
DENSITY FUNCTIONAL THEORY FOR NON-THEORISTS JOHN P. PERDEW DEPARTMENTS OF PHYSICS AND CHEMISTRY TEMPLE UNIVERSITY
 DENSITY FUNCTIONAL THEORY FOR NON-THEORISTS JOHN P. PERDEW DEPARTMENTS OF PHYSICS AND CHEMISTRY TEMPLE UNIVERSITY A TUTORIAL FOR PHYSICAL SCIENTISTS WHO MAY OR MAY NOT HATE EQUATIONS AND PROOFS REFERENCES
DENSITY FUNCTIONAL THEORY FOR NON-THEORISTS JOHN P. PERDEW DEPARTMENTS OF PHYSICS AND CHEMISTRY TEMPLE UNIVERSITY A TUTORIAL FOR PHYSICAL SCIENTISTS WHO MAY OR MAY NOT HATE EQUATIONS AND PROOFS REFERENCES
Density Functional Theory - II part
 Density Functional Theory - II part antonino.polimeno@unipd.it Overview From theory to practice Implementation Functionals Local functionals Gradient Others From theory to practice From now on, if not
Density Functional Theory - II part antonino.polimeno@unipd.it Overview From theory to practice Implementation Functionals Local functionals Gradient Others From theory to practice From now on, if not
MODERN ELECTRONIC STRUCTURE THEORY: Electron Correlation
 5.61 Physical Chemistry Lecture #30 1 MODERN ELECTRONIC STRUCTURE THEORY: Electron Correlation In the previous lecture, we covered all the ingredients necessary to choose a good atomic orbital basis set.
5.61 Physical Chemistry Lecture #30 1 MODERN ELECTRONIC STRUCTURE THEORY: Electron Correlation In the previous lecture, we covered all the ingredients necessary to choose a good atomic orbital basis set.
Lecture 5: More about one- Final words about the Hartree-Fock theory. First step above it by the Møller-Plesset perturbation theory.
 Lecture 5: More about one- determinant wave functions Final words about the Hartree-Fock theory. First step above it by the Møller-Plesset perturbation theory. Items from Lecture 4 Could the Koopmans theorem
Lecture 5: More about one- determinant wave functions Final words about the Hartree-Fock theory. First step above it by the Møller-Plesset perturbation theory. Items from Lecture 4 Could the Koopmans theorem
Practical Advice for Quantum Chemistry Computations. C. David Sherrill School of Chemistry and Biochemistry Georgia Institute of Technology
 Practical Advice for Quantum Chemistry Computations C. David Sherrill School of Chemistry and Biochemistry Georgia Institute of Technology Choice of Basis Set STO-3G is too small 6-31G* or 6-31G** 6 probably
Practical Advice for Quantum Chemistry Computations C. David Sherrill School of Chemistry and Biochemistry Georgia Institute of Technology Choice of Basis Set STO-3G is too small 6-31G* or 6-31G** 6 probably
Study of Ozone in Tribhuvan University, Kathmandu, Nepal. Prof. S. Gurung Central Department of Physics, Tribhuvan University, Kathmandu, Nepal
 Study of Ozone in Tribhuvan University, Kathmandu, Nepal Prof. S. Gurung Central Department of Physics, Tribhuvan University, Kathmandu, Nepal 1 Country of the Mt Everest 2 View of the Mt Everest 3 4 5
Study of Ozone in Tribhuvan University, Kathmandu, Nepal Prof. S. Gurung Central Department of Physics, Tribhuvan University, Kathmandu, Nepal 1 Country of the Mt Everest 2 View of the Mt Everest 3 4 5
Electric properties of molecules
 Electric properties of molecules For a molecule in a uniform electric fielde the Hamiltonian has the form: Ĥ(E) = Ĥ + E ˆµ x where we assume that the field is directed along the x axis and ˆµ x is the
Electric properties of molecules For a molecule in a uniform electric fielde the Hamiltonian has the form: Ĥ(E) = Ĥ + E ˆµ x where we assume that the field is directed along the x axis and ˆµ x is the
Performance of Hartree-Fock and Correlated Methods
 Chemistry 460 Fall 2017 Dr. Jean M. Standard December 4, 2017 Performance of Hartree-Fock and Correlated Methods Hartree-Fock Methods Hartree-Fock methods generally yield optimized geomtries and molecular
Chemistry 460 Fall 2017 Dr. Jean M. Standard December 4, 2017 Performance of Hartree-Fock and Correlated Methods Hartree-Fock Methods Hartree-Fock methods generally yield optimized geomtries and molecular
Introduction to Computational Chemistry: Theory
 Introduction to Computational Chemistry: Theory Dr Andrew Gilbert Rm 118, Craig Building, RSC andrew.gilbert@anu.edu.au 3023 Course Lectures Introduction Hartree Fock Theory Basis Sets Lecture 1 1 Introduction
Introduction to Computational Chemistry: Theory Dr Andrew Gilbert Rm 118, Craig Building, RSC andrew.gilbert@anu.edu.au 3023 Course Lectures Introduction Hartree Fock Theory Basis Sets Lecture 1 1 Introduction
Calculations of band structures
 Chemistry and Physics at Albany Planning for the Future Calculations of band structures using wave-function based correlation methods Elke Pahl Centre of Theoretical Chemistry and Physics Institute of
Chemistry and Physics at Albany Planning for the Future Calculations of band structures using wave-function based correlation methods Elke Pahl Centre of Theoretical Chemistry and Physics Institute of
Introduction to Computational Quantum Chemistry: Theory
 Introduction to Computational Quantum Chemistry: Theory Dr Andrew Gilbert Rm 118, Craig Building, RSC 3108 Course Lectures 2007 Introduction Hartree Fock Theory Configuration Interaction Lectures 1 Introduction
Introduction to Computational Quantum Chemistry: Theory Dr Andrew Gilbert Rm 118, Craig Building, RSC 3108 Course Lectures 2007 Introduction Hartree Fock Theory Configuration Interaction Lectures 1 Introduction
4 Post-Hartree Fock Methods: MPn and Configuration Interaction
 4 Post-Hartree Fock Methods: MPn and Configuration Interaction In the limit of a complete basis, the Hartree-Fock (HF) energy in the complete basis set limit (ECBS HF ) yields an upper boundary to the
4 Post-Hartree Fock Methods: MPn and Configuration Interaction In the limit of a complete basis, the Hartree-Fock (HF) energy in the complete basis set limit (ECBS HF ) yields an upper boundary to the
Chemistry 3502/4502. Final Exam Part I. May 14, 2005
 Chemistry 3502/4502 Final Exam Part I May 14, 2005 1. For which of the below systems is = where H is the Hamiltonian operator and T is the kinetic-energy operator? (a) The free particle (e) The
Chemistry 3502/4502 Final Exam Part I May 14, 2005 1. For which of the below systems is = where H is the Hamiltonian operator and T is the kinetic-energy operator? (a) The free particle (e) The
Orbital dependent correlation potentials in ab initio density functional theory
 Orbital dependent correlation potentials in ab initio density functional theory noniterative - one step - calculations Ireneusz Grabowski Institute of Physics Nicolaus Copernicus University Toruń, Poland
Orbital dependent correlation potentials in ab initio density functional theory noniterative - one step - calculations Ireneusz Grabowski Institute of Physics Nicolaus Copernicus University Toruń, Poland
Self-Consistent Implementation of Self-Interaction Corrected DFT and of the Exact Exchange Functionals in Plane-Wave DFT
 Self-Consistent Implementation of Self-Interaction Corrected DFT and of the Exact Exchange Functionals in Plane-Wave DFT Kiril Tsemekhman (a), Eric Bylaska (b), Hannes Jonsson (a,c) (a) Department of Chemistry,
Self-Consistent Implementation of Self-Interaction Corrected DFT and of the Exact Exchange Functionals in Plane-Wave DFT Kiril Tsemekhman (a), Eric Bylaska (b), Hannes Jonsson (a,c) (a) Department of Chemistry,
Electronic structure theory: Fundamentals to frontiers. 1. Hartree-Fock theory
 Electronic structure theory: Fundamentals to frontiers. 1. Hartree-Fock theory MARTIN HEAD-GORDON, Department of Chemistry, University of California, and Chemical Sciences Division, Lawrence Berkeley National
Electronic structure theory: Fundamentals to frontiers. 1. Hartree-Fock theory MARTIN HEAD-GORDON, Department of Chemistry, University of California, and Chemical Sciences Division, Lawrence Berkeley National
Introduction to Density Functional Theory with Applications to Graphene Branislav K. Nikolić
 Introduction to Density Functional Theory with Applications to Graphene Branislav K. Nikolić Department of Physics and Astronomy, University of Delaware, Newark, DE 19716, U.S.A. http://wiki.physics.udel.edu/phys824
Introduction to Density Functional Theory with Applications to Graphene Branislav K. Nikolić Department of Physics and Astronomy, University of Delaware, Newark, DE 19716, U.S.A. http://wiki.physics.udel.edu/phys824
An Introduction to Quantum Chemistry and Potential Energy Surfaces. Benjamin G. Levine
 An Introduction to Quantum Chemistry and Potential Energy Surfaces Benjamin G. Levine This Week s Lecture Potential energy surfaces What are they? What are they good for? How do we use them to solve chemical
An Introduction to Quantum Chemistry and Potential Energy Surfaces Benjamin G. Levine This Week s Lecture Potential energy surfaces What are they? What are they good for? How do we use them to solve chemical
Electronic band structure, sx-lda, Hybrid DFT, LDA+U and all that. Keith Refson STFC Rutherford Appleton Laboratory
 Electronic band structure, sx-lda, Hybrid DFT, LDA+U and all that Keith Refson STFC Rutherford Appleton Laboratory LDA/GGA DFT is good but... Naive LDA/GGA calculation severely underestimates band-gaps.
Electronic band structure, sx-lda, Hybrid DFT, LDA+U and all that Keith Refson STFC Rutherford Appleton Laboratory LDA/GGA DFT is good but... Naive LDA/GGA calculation severely underestimates band-gaps.
Molecular Magnetism. Magnetic Resonance Parameters. Trygve Helgaker
 Molecular Magnetism Magnetic Resonance Parameters Trygve Helgaker Centre for Theoretical and Computational Chemistry (CTCC), Department of Chemistry, University of Oslo, Norway Laboratoire de Chimie Théorique,
Molecular Magnetism Magnetic Resonance Parameters Trygve Helgaker Centre for Theoretical and Computational Chemistry (CTCC), Department of Chemistry, University of Oslo, Norway Laboratoire de Chimie Théorique,
Exchange-Correlation Functional
 Exchange-Correlation Functional Aiichiro Nakano Collaboratory for Advanced Computing & Simulations Depts. of Computer Science, Physics & Astronomy, Chemical Engineering & Materials Science, and Biological
Exchange-Correlation Functional Aiichiro Nakano Collaboratory for Advanced Computing & Simulations Depts. of Computer Science, Physics & Astronomy, Chemical Engineering & Materials Science, and Biological
The Accurate Calculation of Molecular Energies and Properties: A Tour of High-Accuracy Quantum-Chemical Methods
 1 The Accurate Calculation of Molecular Energies and Properties: A Tour of High-Accuracy Quantum-Chemical Methods T. Helgaker Centre for Theoretical and Computational Chemistry Department of Chemistry,
1 The Accurate Calculation of Molecular Energies and Properties: A Tour of High-Accuracy Quantum-Chemical Methods T. Helgaker Centre for Theoretical and Computational Chemistry Department of Chemistry,
Chemistry 4560/5560 Molecular Modeling Fall 2014
 Final Exam Name:. User s guide: 1. Read questions carefully and make sure you understand them before answering (if not, ask). 2. Answer only the question that is asked, not a different question. 3. Unless
Final Exam Name:. User s guide: 1. Read questions carefully and make sure you understand them before answering (if not, ask). 2. Answer only the question that is asked, not a different question. 3. Unless
Density-functional theory in quantum chemistry. Trygve Helgaker. From Quarks to the Nuclear Many-Body Problem
 1 Density-functional theory in quantum chemistry Trygve Helgaker Centre for Theoretical and Computational Chemistry, University of Oslo, Norway From Quarks to the Nuclear Many-Body Problem A conference
1 Density-functional theory in quantum chemistry Trygve Helgaker Centre for Theoretical and Computational Chemistry, University of Oslo, Norway From Quarks to the Nuclear Many-Body Problem A conference
Advanced Quantum Chemistry III: Part 3. Haruyuki Nakano. Kyushu University
 Advanced Quantum Chemistry III: Part 3 Haruyuki Nakano Kyushu University 2013 Winter Term 1. Hartree-Fock theory Density Functional Theory 2. Hohenberg-Kohn theorem 3. Kohn-Sham method 4. Exchange-correlation
Advanced Quantum Chemistry III: Part 3 Haruyuki Nakano Kyushu University 2013 Winter Term 1. Hartree-Fock theory Density Functional Theory 2. Hohenberg-Kohn theorem 3. Kohn-Sham method 4. Exchange-correlation
Quantum Mechanical Simulations
 Quantum Mechanical Simulations Prof. Yan Wang Woodruff School of Mechanical Engineering Georgia Institute of Technology Atlanta, GA 30332, U.S.A. yan.wang@me.gatech.edu Topics Quantum Monte Carlo Hartree-Fock
Quantum Mechanical Simulations Prof. Yan Wang Woodruff School of Mechanical Engineering Georgia Institute of Technology Atlanta, GA 30332, U.S.A. yan.wang@me.gatech.edu Topics Quantum Monte Carlo Hartree-Fock
Advanced Electronic Structure Theory Density functional theory. Dr Fred Manby
 Advanced Electronic Structure Theory Density functional theory Dr Fred Manby fred.manby@bris.ac.uk http://www.chm.bris.ac.uk/pt/manby/ 6 Strengths of DFT DFT is one of many theories used by (computational)
Advanced Electronic Structure Theory Density functional theory Dr Fred Manby fred.manby@bris.ac.uk http://www.chm.bris.ac.uk/pt/manby/ 6 Strengths of DFT DFT is one of many theories used by (computational)
DFT: Exchange-Correlation
 DFT: Local functionals, exact exchange and other post-dft methods Stewart Clark University of Outline Introduction What is exchange and correlation? Quick tour of XC functionals (Semi-)local: LDA, PBE,
DFT: Local functionals, exact exchange and other post-dft methods Stewart Clark University of Outline Introduction What is exchange and correlation? Quick tour of XC functionals (Semi-)local: LDA, PBE,
Post Hartree-Fock: MP2 and RPA in CP2K
 Post Hartree-Fock: MP2 and RPA in CP2K A tutorial Jan Wilhelm jan.wilhelm@chem.uzh.ch 4 September 2015 Further reading MP2 and RPA by Mauro Del Ben, Jürg Hutter, Joost VandeVondele Del Ben, M; Hutter,
Post Hartree-Fock: MP2 and RPA in CP2K A tutorial Jan Wilhelm jan.wilhelm@chem.uzh.ch 4 September 2015 Further reading MP2 and RPA by Mauro Del Ben, Jürg Hutter, Joost VandeVondele Del Ben, M; Hutter,
DFT: Exchange-Correlation
 DFT: Exchange-Correlation Local functionals, exact exchange and other post-dft methods Paul Tulip Centre for Materials Physics Department of Physics University of Durham Outline Introduction What is exchange
DFT: Exchange-Correlation Local functionals, exact exchange and other post-dft methods Paul Tulip Centre for Materials Physics Department of Physics University of Durham Outline Introduction What is exchange
Reactivity and Organocatalysis. (Adalgisa Sinicropi and Massimo Olivucci)
") Reactivity and Organocatalysis (Adalgisa Sinicropi and Massimo Olivucci) The Aldol Reaction - O R 1 O R 1 + - O O OH * * H R 2 R 1 R 2 The (1957) Zimmerman-Traxler Transition State Counterion (e.g. Li
Reactivity and Organocatalysis (Adalgisa Sinicropi and Massimo Olivucci) The Aldol Reaction - O R 1 O R 1 + - O O OH * * H R 2 R 1 R 2 The (1957) Zimmerman-Traxler Transition State Counterion (e.g. Li
T. Helgaker, Department of Chemistry, University of Oslo, Norway. T. Ruden, University of Oslo, Norway. W. Klopper, University of Karlsruhe, Germany
 1 The a priori calculation of molecular properties to chemical accuarcy T. Helgaker, Department of Chemistry, University of Oslo, Norway T. Ruden, University of Oslo, Norway W. Klopper, University of Karlsruhe,
1 The a priori calculation of molecular properties to chemical accuarcy T. Helgaker, Department of Chemistry, University of Oslo, Norway T. Ruden, University of Oslo, Norway W. Klopper, University of Karlsruhe,
Joint ICTP-IAEA Workshop on Fusion Plasma Modelling using Atomic and Molecular Data January 2012
 2327-3 Joint ICTP-IAEA Workshop on Fusion Plasma Modelling using Atomic and Molecular Data 23-27 January 2012 Qunatum Methods for Plasma-Facing Materials Alain ALLOUCHE Univ.de Provence, Lab.de la Phys.
2327-3 Joint ICTP-IAEA Workshop on Fusion Plasma Modelling using Atomic and Molecular Data 23-27 January 2012 Qunatum Methods for Plasma-Facing Materials Alain ALLOUCHE Univ.de Provence, Lab.de la Phys.
Molecular Magnetic Properties
 Molecular Magnetic Properties Trygve Helgaker Hylleraas Centre, Department of Chemistry, University of Oslo, Norway and Centre for Advanced Study at the Norwegian Academy of Science and Letters, Oslo,
Molecular Magnetic Properties Trygve Helgaker Hylleraas Centre, Department of Chemistry, University of Oslo, Norway and Centre for Advanced Study at the Norwegian Academy of Science and Letters, Oslo,
Hartree, Hartree-Fock and post-hf methods
 Hartree, Hartree-Fock and post-hf methods MSE697 fall 2015 Nicolas Onofrio School of Materials Engineering DLR 428 Purdue University nonofrio@purdue.edu 1 The curse of dimensionality Let s consider a multi
Hartree, Hartree-Fock and post-hf methods MSE697 fall 2015 Nicolas Onofrio School of Materials Engineering DLR 428 Purdue University nonofrio@purdue.edu 1 The curse of dimensionality Let s consider a multi
Solid State Theory: Band Structure Methods
 Solid State Theory: Band Structure Methods Lilia Boeri Wed., 11:15-12:45 HS P3 (PH02112) http://itp.tugraz.at/lv/boeri/ele/ Plan of the Lecture: DFT1+2: Hohenberg-Kohn Theorem and Kohn and Sham equations.
Solid State Theory: Band Structure Methods Lilia Boeri Wed., 11:15-12:45 HS P3 (PH02112) http://itp.tugraz.at/lv/boeri/ele/ Plan of the Lecture: DFT1+2: Hohenberg-Kohn Theorem and Kohn and Sham equations.
1 Density functional theory (DFT)
") 1 Density functional theory (DFT) 1.1 Introduction Density functional theory is an alternative to ab initio methods for solving the nonrelativistic, time-independent Schrödinger equation H Φ = E Φ. The
1 Density functional theory (DFT) 1.1 Introduction Density functional theory is an alternative to ab initio methods for solving the nonrelativistic, time-independent Schrödinger equation H Φ = E Φ. The
Computational Chemistry. An Introduction to Molecular Dynamic Simulations
 Computational Chemistry An Introduction to Molecular Dynamic Simulations Computational chemistry simulates chemical structures and reactions numerically, based in full or in part on the fundamental laws
Computational Chemistry An Introduction to Molecular Dynamic Simulations Computational chemistry simulates chemical structures and reactions numerically, based in full or in part on the fundamental laws
Intro to ab initio methods
 Lecture 2 Part A Intro to ab initio methods Recommended reading: Leach, Chapters 2 & 3 for QM methods For more QM methods: Essentials of Computational Chemistry by C.J. Cramer, Wiley (2002) 1 ab initio
Lecture 2 Part A Intro to ab initio methods Recommended reading: Leach, Chapters 2 & 3 for QM methods For more QM methods: Essentials of Computational Chemistry by C.J. Cramer, Wiley (2002) 1 ab initio
Electronic Structure Calculations and Density Functional Theory
 Electronic Structure Calculations and Density Functional Theory Rodolphe Vuilleumier Pôle de chimie théorique Département de chimie de l ENS CNRS Ecole normale supérieure UPMC Formation ModPhyChem Lyon,
Electronic Structure Calculations and Density Functional Theory Rodolphe Vuilleumier Pôle de chimie théorique Département de chimie de l ENS CNRS Ecole normale supérieure UPMC Formation ModPhyChem Lyon,
one ν im: transition state saddle point
 Hypothetical Potential Energy Surface Ethane conformations Hartree-Fock theory, basis set stationary points all ν s >0: minimum eclipsed one ν im: transition state saddle point multiple ν im: hilltop 1
Hypothetical Potential Energy Surface Ethane conformations Hartree-Fock theory, basis set stationary points all ν s >0: minimum eclipsed one ν im: transition state saddle point multiple ν im: hilltop 1
2~:J~ -ryej- r- 2 Jr. A - f3. sr(djk nv~tor rn~ +~ rvjs (::-CJ) ::;-1-.'--~ -. rhd. ('-.Ji.L.~ )- r'-d)c, -r/~ JJr - 2~d ~2-Jr fn'6.
 ::;-1-.'--~ -. rhd. ('-.Ji.L.~ )- r'-d)c, -r/~ JJr - 2~d ~2-Jr fn'6.") .~, ~ I, sr(djk nv~tor rn~ +~ rvjs (::-CJ) ::;-1-.'--~ -. rhd. ('-.Ji.L.~ )- r'-d)c, -r/~ JJr - 2~d ~2-Jr fn'6.)1e'" 21t-ol Je C'...-------- lj-vi, J? Jr Jr \Ji 2~:J~ -ryej- r- 2 Jr A - f3 c _,~,= ~,.,w._..._.
.~, ~ I, sr(djk nv~tor rn~ +~ rvjs (::-CJ) ::;-1-.'--~ -. rhd. ('-.Ji.L.~ )- r'-d)c, -r/~ JJr - 2~d ~2-Jr fn'6.)1e'" 21t-ol Je C'...-------- lj-vi, J? Jr Jr \Ji 2~:J~ -ryej- r- 2 Jr A - f3 c _,~,= ~,.,w._..._.
Many electrons: Density functional theory Part II. Bedřich Velický VI.
 Many electrons: Density functional theory Part II. Bedřich Velický velicky@karlov.mff.cuni.cz VI. NEVF 514 Surface Physics Winter Term 013-014 Troja 1 st November 013 This class is the second devoted to
Many electrons: Density functional theory Part II. Bedřich Velický velicky@karlov.mff.cuni.cz VI. NEVF 514 Surface Physics Winter Term 013-014 Troja 1 st November 013 This class is the second devoted to
One-Electron Hamiltonians
 One-Electron Hamiltonians Hartree-Fock and Density Func7onal Theory Christopher J. Cramer @ChemProfCramer 2017 MSSC, July 10, 2017 REVIEW A One-Slide Summary of Quantum Mechanics Fundamental Postulate:
One-Electron Hamiltonians Hartree-Fock and Density Func7onal Theory Christopher J. Cramer @ChemProfCramer 2017 MSSC, July 10, 2017 REVIEW A One-Slide Summary of Quantum Mechanics Fundamental Postulate:
Density Functional Theory (DFT)
") Density Functional Theory (DFT) An Introduction by A.I. Al-Sharif Irbid, Aug, 2 nd, 2009 Density Functional Theory Revolutionized our approach to the electronic structure of atoms, molecules and solid
Density Functional Theory (DFT) An Introduction by A.I. Al-Sharif Irbid, Aug, 2 nd, 2009 Density Functional Theory Revolutionized our approach to the electronic structure of atoms, molecules and solid
Intermediate DFT. Kieron Burke and Lucas Wagner. Departments of Physics and of Chemistry, University of California, Irvine, CA 92697, USA
 Intermediate DFT Kieron Burke and Lucas Wagner Departments of Physics and of Chemistry, University of California, Irvine, CA 92697, USA October 10-19th, 2012 Kieron (UC Irvine) Intermediate DFT Lausanne12
Intermediate DFT Kieron Burke and Lucas Wagner Departments of Physics and of Chemistry, University of California, Irvine, CA 92697, USA October 10-19th, 2012 Kieron (UC Irvine) Intermediate DFT Lausanne12
The calculation of the universal density functional by Lieb maximization
 The calculation of the universal density functional by Lieb maximization Trygve Helgaker, Andy Teale, and Sonia Coriani Centre for Theoretical and Computational Chemistry (CTCC), Department of Chemistry,
The calculation of the universal density functional by Lieb maximization Trygve Helgaker, Andy Teale, and Sonia Coriani Centre for Theoretical and Computational Chemistry (CTCC), Department of Chemistry,
Pseudopotentials for hybrid density functionals and SCAN
 Pseudopotentials for hybrid density functionals and SCAN Jing Yang, Liang Z. Tan, Julian Gebhardt, and Andrew M. Rappe Department of Chemistry University of Pennsylvania Why do we need pseudopotentials?
Pseudopotentials for hybrid density functionals and SCAN Jing Yang, Liang Z. Tan, Julian Gebhardt, and Andrew M. Rappe Department of Chemistry University of Pennsylvania Why do we need pseudopotentials?
XYZ of ground-state DFT
 XYZ of ground-state DFT Kieron Burke and Lucas Wagner Departments of Physics and of Chemistry, University of California, Irvine, CA 92697, USA January 5-9th, 2014 Kieron (UC Irvine) XYZ of ground-state
XYZ of ground-state DFT Kieron Burke and Lucas Wagner Departments of Physics and of Chemistry, University of California, Irvine, CA 92697, USA January 5-9th, 2014 Kieron (UC Irvine) XYZ of ground-state
Institut Néel Institut Laue Langevin. Introduction to electronic structure calculations
 Institut Néel Institut Laue Langevin Introduction to electronic structure calculations 1 Institut Néel - 25 rue des Martyrs - Grenoble - France 2 Institut Laue Langevin - 71 avenue des Martyrs - Grenoble
Institut Néel Institut Laue Langevin Introduction to electronic structure calculations 1 Institut Néel - 25 rue des Martyrs - Grenoble - France 2 Institut Laue Langevin - 71 avenue des Martyrs - Grenoble
Jack Simons, Henry Eyring Scientist and Professor Chemistry Department University of Utah
 1. Born-Oppenheimer approx.- energy surfaces 2. Mean-field (Hartree-Fock) theory- orbitals 3. Pros and cons of HF- RHF, UHF 4. Beyond HF- why? 5. First, one usually does HF-how? 6. Basis sets and notations
1. Born-Oppenheimer approx.- energy surfaces 2. Mean-field (Hartree-Fock) theory- orbitals 3. Pros and cons of HF- RHF, UHF 4. Beyond HF- why? 5. First, one usually does HF-how? 6. Basis sets and notations
Quantum Chemical Simulations and Descriptors. Dr. Antonio Chana, Dr. Mosè Casalegno
 Quantum Chemical Simulations and Descriptors Dr. Antonio Chana, Dr. Mosè Casalegno Classical Mechanics: basics It models real-world objects as point particles, objects with negligible size. The motion
Quantum Chemical Simulations and Descriptors Dr. Antonio Chana, Dr. Mosè Casalegno Classical Mechanics: basics It models real-world objects as point particles, objects with negligible size. The motion
Example questions for Molecular modelling (Level 4) Dr. Adrian Mulholland
 Dr. Adrian Mulholland") Example questions for Molecular modelling (Level 4) Dr. Adrian Mulholland 1) Question. Two methods which are widely used for the optimization of molecular geometies are the Steepest descents and Newton-Raphson
Example questions for Molecular modelling (Level 4) Dr. Adrian Mulholland 1) Question. Two methods which are widely used for the optimization of molecular geometies are the Steepest descents and Newton-Raphson
Jack Simons, Henry Eyring Scientist and Professor Chemistry Department University of Utah
 1. Born-Oppenheimer approx.- energy surfaces 2. Mean-field (Hartree-Fock) theory- orbitals 3. Pros and cons of HF- RHF, UHF 4. Beyond HF- why? 5. First, one usually does HF-how? 6. Basis sets and notations
1. Born-Oppenheimer approx.- energy surfaces 2. Mean-field (Hartree-Fock) theory- orbitals 3. Pros and cons of HF- RHF, UHF 4. Beyond HF- why? 5. First, one usually does HF-how? 6. Basis sets and notations
The Schrödinger equation for many-electron systems
 The Schrödinger equation for many-electron systems Ĥ!( x,, x ) = E!( x,, x ) 1 N 1 1 Z 1 Ĥ = " $ # " $ + $ 2 r 2 A j j A, j RAj i, j < i a linear differential equation in 4N variables (atomic units) (3
The Schrödinger equation for many-electron systems Ĥ!( x,, x ) = E!( x,, x ) 1 N 1 1 Z 1 Ĥ = " $ # " $ + $ 2 r 2 A j j A, j RAj i, j < i a linear differential equation in 4N variables (atomic units) (3
Quantum Monte Carlo methods
 Quantum Monte Carlo methods Lubos Mitas North Carolina State University Urbana, August 2006 Lubos_Mitas@ncsu.edu H= 1 2 i i 2 i, I Z I r ii i j 1 r ij E ion ion H r 1, r 2,... =E r 1, r 2,... - ground
Quantum Monte Carlo methods Lubos Mitas North Carolina State University Urbana, August 2006 Lubos_Mitas@ncsu.edu H= 1 2 i i 2 i, I Z I r ii i j 1 r ij E ion ion H r 1, r 2,... =E r 1, r 2,... - ground