Study of Ozone in Tribhuvan University, Kathmandu, Nepal. Prof. S. Gurung Central Department of Physics, Tribhuvan University, Kathmandu, Nepal
|
|
|
- Ernest James Snow
- 6 years ago
- Views:
Transcription
1 Study of Ozone in Tribhuvan University, Kathmandu, Nepal Prof. S. Gurung Central Department of Physics, Tribhuvan University, Kathmandu, Nepal 1

2 Country of the Mt Everest 2


3 View of the Mt Everest 3
4 4
5 5

6 Central Department of Physics, Kathmandu 6




7 7





8 8

9 9
10 Dr. Ken Lamb Calibrating Brewer 10
11 11


12 Dr. Arne Dahlback at CDP, Kathmandu 12
13 13





14 14


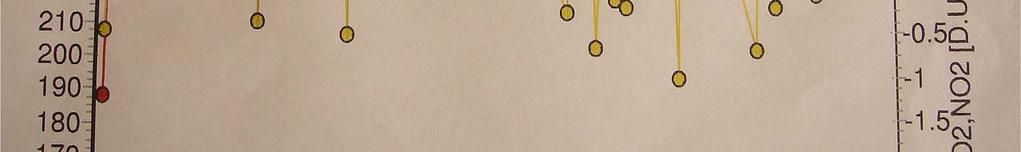

15 15
16 Data/ Years Production Consumption OMI Average O3 in DU Sunspot
17 17
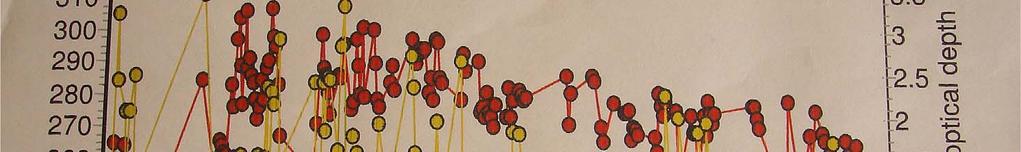
18 Comparison Between Brewer and OMI data 2002 Months Brewer DU OMI DU January February March April May June July August September October November December
19 Comparison between Brewer and OMI data Ozone in DU Brewer 150 OMI Months 19
20 20
21 First-Principles study of Ozone Group Memebers Prof. D.R. Mishra (Group Leader) Prof. M.M. Aryal Prof. S. Gurung Dr. N.P. Adhikari Mr. N. Subedi 21
22 First-Principles study of Ozone ab initio does not use empirical information (except for fundamental constants), may not be exact! In spite of necessary approximations, its successes and failures are more or less predictable 22
23 ab initio : an overview (contd ) Approximations (solving Schroedinger Equation (SE)): Time independence : Stationary states Neglect of relativistic effects Born-Oppenheimer approximation Orbital approximation: Electrons are confined to certain regions of space 23
24 ab initio : an overview (contd ) Hartree-Fock SCF Method: SE for an electron i in the field of other electrons and nuclei k is [Blinder(1965)]: OR, 2 ћ ћ Z i e i) i 2 m e 2 e i j 2 2 k () () i ( k 2mk k k i rik ZkZl ( i) e ( i) E ( i) r r ij H E k l kl Retaining 1 st, 3 rd and 4 th terms one gets HF equation. 24
25 ab initio : an overview (contd ) Hartree-Fock SCF Method: Independent particle approximation * Z N 2 j 2 j j j ri R j 1 s ri rj 2 ћ ( ) ( ) () () () i i e i e drj i 2 m ( j) ( j) N * 2 e drj i E i 1 s ri rj j () () j Coulomb Exchange 25
26 ab initio : an overview (contd ) HF SCF Method: Advantages: Variational, computationally efficient Limitations: Neglect of correlation energy Correlations are important even though it is ~1% of the total energy of a molecule (Cramer (2004)) Correlations are taken into account by CI, MP, DFT etc. 26
27 ab initio : an overview (contd ) Perturbation method (MP): The difference between the Fock operator and exact Hamiltonian can be considered as a perturbation Lowest level of perturbation is 2 nd order Speed of the same order of magnitude as HF Limitation: Not variational, the correlation energy could be overcorrected 27
28 ab initio : an overview (contd ) Configuration Interaction (CI): Uses wave function which is a linear combination of the HF determinant and determinants from excitations of electrons Variational and full CI is exact Computationally expensive and works only for small systems 28
29 ab initio : an overview (contd ) Density functional theory (DFT): The dynamical correlation effects due to electrons moving out of each other s way as a result of the coulomb repulsion between them are accounted for Energy is computed with density of electrons 29
30 ab initio : an overview (contd ) DFT: Many-body system Hamiltonian can be constructed only from the density of electrons (ρ) and their positions and atomic number of the nuclei Exchange-Correlation Functional 2 ћ Z ( ) 2 j r j H e dr i j V 2 m j ri R j ri rj xc [ ( r)] In principle, it s exact but in practice one must rely on approximations of exchange correlation functional 30
31 ab initio : an overview (contd ) LDA Local density approximation LSDA Local spin density approximation GGA Genaralized gradient approximation Hybrid MPW1PW91, B3LYP (better than others? depends upon system) Present work MPW1PW91 31
32 ab initio : an overview (contd ) Basis set : Compromise between accuracy and computational cost Gaussian 98 set of programs Basis set convergence, 6-311G** (* refers to the inclusion of polarization functions) Convergence : Energy a.u., Maximum displacement a.u. Maximum force a.u. 32
33 Results and discussion Oxygen atom : Triplet state is more stable than the singlet state Energy difference = 3.46 ev (HF) =2.63 ev (QCISD) = 3.00 ev (DFT) Ground state energy (in a.u.); (HF), (HF+MP2), (QCISD), (DFT), Basis set 6-311G** Basis set 6-311G** (Experimental) [Thijsen(2001)] Results of present work agree within 1% to the experimental value Correlation energy = ev in the QCISD approximation 33
 = 1.62 ev (QCISD) = 1.")
34 Results and discussion Oxygen molecule : Triplet state is more stable than the singlet state Energy difference = 2.31 ev (HF) = 1.62 ev (QCISD) = 1.78 ev (DFT) Basis set 6-311G** 34
35 Results and discussion Oxygen molecule Basis set 6-311G** Parameters Levels of Calculation Estimated values Experimental values a Bond length (Ǻ) HF (4%) 1.21 HF+MP (1%) QCISD (2%) DFT (1%) Binding Energy (ev) HF 1.35 (74%) 5.21 HF+MP (2%) QCISD 3.81 (27%) DFT 5.17 (<1%) a Experimental data are from Levine(2003) Mainali(2004) 35
36 Results and discussion Ozone molecule: Singlet state is more stable than the triplet state Energy difference =2.01 ev (HF+MP2) =1.11 ev (QCISD) =0.92 ev (DFT) = 0.36 ev (HF) Basis set 6-311G** 36
37 Results and discussion Ozone molecule: Ground state Isomeric excited state Bond length =1.26 Ǻ Bond angle = Total energy = a.u. Bond length =1.39 Ǻ Bond angle = 60 0 Total energy = a.u. At QCISD/6-311G** level of approximation 37
38 Results and discussion Ozone molecule: Ground state Isomeric excited state Binding Energy = kcal/mol (HF+MP2) [~1%] = kcal/mol (QCISD) = kcal/mol (DFT) No binding in the HF approximation Binding Energy = kcal/mol (HF+MP2) = kcal/mol (QCISD) = kcal/mol (DFT) No binding in the HF approximation 6-311G** basis set Experimental value142.2 kcal/mol [Foresman & Frisch (1996)] 38
39 Results and discussion Binding is due to correlation effects, Similar results observed in solid halogens, H 2 O 2, and B 2 H [Aryal et al. (2004), Lamsal(2004), Khanal(2005) ] 39
40 Results and discussion Dissociation energy: ΔE1=E(O)+E(O 2 )-E(O 3 ) HF+MP2/6-31G** O 3 -> O 2 +O ΔE1= KJ/mol (~1%) [105 KJ/mol, Baird (1995)] ΔE2= 3E(O 2 )-2E(O 3 ) 2O 3 -> 3O 2 +O [HF+MP2/6-31G**] ΔE2 = kcal/mol 40
41 Results and discussion Ozone cluster : dimer of ozone (equilibrium configuration) Distance between central atoms =3.85 Ǻ Binding Energy =2E(O3) - E(O3-O3) B.E. (DFT) = ev (4%), [ ev, Murai et. al, (2003)] B.E. (HF) = ev 41

 - E(O3-O3-O3) B.E. (DFT) = 0.115 ev (~10%) B.E. (HF) = 0.")
42 Results and discussion Ozone cluster : trimer of ozone (equilibrium configuration) Central atoms form an equilateral triangle having sides ~3.80 Ǻ Central atoms are in a straight line Distance between central consecutive atoms ~ 3.5 Ǻ Binding Energy =3E(O3) - E(O3-O3-O3) B.E. (DFT) = ev (~10%) B.E. (HF) = ev (<3%) [0.104 ev, Murai et al (2003)] B.E. (DFT) = ev 42
 - E(O3-O3-O3-O3) B.E. (DFT) = 0.151 ev B.E. (HF) = 0.103 ev B.E. (DFT) = 0.073 ev B.")
43 Results and discussion Ozone cluster : quadramer of ozone (equilibrium configuration) Central atoms form almost a parallelogram, with sides ~3.85 Ǻ and ~4.2 Ǻ Central atoms are in a straight line with distance between two consecutive atoms ~ 3.25 Ǻ Binding Energy =4E(O3) - E(O3-O3-O3-O3) B.E. (DFT) = ev B.E. (HF) = ev B.E. (DFT) = ev B.E. (HF) = ev 43
44 Conclusions The present work shows that ozone cluster with four molecules of ozone is stable with binding energy of ev and the equilibrium geometry as shown below. Previous studies (Murai et al (2003)) were unable to obtain the equilibrium configuration of ozone clusters with n=4 or more. We are studying the stability of ozone clusters with higher number (n 5) of ozone molecules and interaction of ozone with halogens. 44
45 References Aryal MM, Mishra DR, Byahut SP, Paudyal DD, Scheicher RH, Jeong J, Gaire C and Das TP, First principles investigation of binding and nuclear quadrupole interactions of Halogens molecules in solid halogens, Paper presented at the March meeting of APS, Montreal, Canada, 2004 Blinder SM, Am. J. Phys., 33,431(1965) Cramer CJ, Essentials of Computational Chemistry, John wiley & sons, Ltd., New York, 2002 Khanal K, M.Sc. Dissertation(2005), Tribhuvan University, Kathmandu, Nepal Lamsal C, M.Sc. Dissertation(2004), Tribhuvan University, Kathmandu, Nepal Levine IN, Quantum chemistry, Pearson education, Singapore, 2003 Mainali L, M.Sc. Dissertation (2004), Tribhuvan University, Kathmandu, Nepal Murai et. al, Ozone Science & Engineering, 25, 211(2003) Thijsen JM, Computational Physics, Cambridge University, Press, Cambridge,
46 Acknowledgment We acknowledge Prof. T.P. Das (State University of New York, Albany, NY, USA) for the support to carry out this research 46
First Principles Calculations to Study the Equilibrium Configuration of Ozone Molecule
 First Principles Calculations to Study the Equilibrium Configuration of Ozone Molecule Laxman Mainali 1,2*, Devendra Raj Mishra 2, Mukunda Mani Aryal 2 1 Department of Biophysics, Medical College of Wisconsin,
First Principles Calculations to Study the Equilibrium Configuration of Ozone Molecule Laxman Mainali 1,2*, Devendra Raj Mishra 2, Mukunda Mani Aryal 2 1 Department of Biophysics, Medical College of Wisconsin,
Session 1. Introduction to Computational Chemistry. Computational (chemistry education) and/or (Computational chemistry) education
 and/or (Computational chemistry) education") Session 1 Introduction to Computational Chemistry 1 Introduction to Computational Chemistry Computational (chemistry education) and/or (Computational chemistry) education First one: Use computational tools
Session 1 Introduction to Computational Chemistry 1 Introduction to Computational Chemistry Computational (chemistry education) and/or (Computational chemistry) education First one: Use computational tools
Computational Methods. Chem 561
 Computational Methods Chem 561 Lecture Outline 1. Ab initio methods a) HF SCF b) Post-HF methods 2. Density Functional Theory 3. Semiempirical methods 4. Molecular Mechanics Computational Chemistry " Computational
Computational Methods Chem 561 Lecture Outline 1. Ab initio methods a) HF SCF b) Post-HF methods 2. Density Functional Theory 3. Semiempirical methods 4. Molecular Mechanics Computational Chemistry " Computational
OVERVIEW OF QUANTUM CHEMISTRY METHODS
 OVERVIEW OF QUANTUM CHEMISTRY METHODS Outline I Generalities Correlation, basis sets Spin II Wavefunction methods Hartree-Fock Configuration interaction Coupled cluster Perturbative methods III Density
OVERVIEW OF QUANTUM CHEMISTRY METHODS Outline I Generalities Correlation, basis sets Spin II Wavefunction methods Hartree-Fock Configuration interaction Coupled cluster Perturbative methods III Density
Performance of Hartree-Fock and Correlated Methods
 Chemistry 460 Fall 2017 Dr. Jean M. Standard December 4, 2017 Performance of Hartree-Fock and Correlated Methods Hartree-Fock Methods Hartree-Fock methods generally yield optimized geomtries and molecular
Chemistry 460 Fall 2017 Dr. Jean M. Standard December 4, 2017 Performance of Hartree-Fock and Correlated Methods Hartree-Fock Methods Hartree-Fock methods generally yield optimized geomtries and molecular
Joint ICTP-IAEA Workshop on Fusion Plasma Modelling using Atomic and Molecular Data January 2012
 2327-3 Joint ICTP-IAEA Workshop on Fusion Plasma Modelling using Atomic and Molecular Data 23-27 January 2012 Qunatum Methods for Plasma-Facing Materials Alain ALLOUCHE Univ.de Provence, Lab.de la Phys.
2327-3 Joint ICTP-IAEA Workshop on Fusion Plasma Modelling using Atomic and Molecular Data 23-27 January 2012 Qunatum Methods for Plasma-Facing Materials Alain ALLOUCHE Univ.de Provence, Lab.de la Phys.
Computational Chemistry I
 Computational Chemistry I Text book Cramer: Essentials of Quantum Chemistry, Wiley (2 ed.) Chapter 3. Post Hartree-Fock methods (Cramer: chapter 7) There are many ways to improve the HF method. Most of
Computational Chemistry I Text book Cramer: Essentials of Quantum Chemistry, Wiley (2 ed.) Chapter 3. Post Hartree-Fock methods (Cramer: chapter 7) There are many ways to improve the HF method. Most of
Electronic structure theory: Fundamentals to frontiers. 2. Density functional theory
 Electronic structure theory: Fundamentals to frontiers. 2. Density functional theory MARTIN HEAD-GORDON, Department of Chemistry, University of California, and Chemical Sciences Division, Lawrence Berkeley
Electronic structure theory: Fundamentals to frontiers. 2. Density functional theory MARTIN HEAD-GORDON, Department of Chemistry, University of California, and Chemical Sciences Division, Lawrence Berkeley
DFT calculations of NMR indirect spin spin coupling constants
 DFT calculations of NMR indirect spin spin coupling constants Dalton program system Program capabilities Density functional theory Kohn Sham theory LDA, GGA and hybrid theories Indirect NMR spin spin coupling
DFT calculations of NMR indirect spin spin coupling constants Dalton program system Program capabilities Density functional theory Kohn Sham theory LDA, GGA and hybrid theories Indirect NMR spin spin coupling
Computational Modeling Software and their applications
 Computational Modeling Software and their applications June 21, 2011 Damilola Daramola Center for Electrochemical Engineering Research ABC s of electrochemistry Introduction Computational Modeling the
Computational Modeling Software and their applications June 21, 2011 Damilola Daramola Center for Electrochemical Engineering Research ABC s of electrochemistry Introduction Computational Modeling the
Quantum Mechanical Simulations
 Quantum Mechanical Simulations Prof. Yan Wang Woodruff School of Mechanical Engineering Georgia Institute of Technology Atlanta, GA 30332, U.S.A. yan.wang@me.gatech.edu Topics Quantum Monte Carlo Hartree-Fock
Quantum Mechanical Simulations Prof. Yan Wang Woodruff School of Mechanical Engineering Georgia Institute of Technology Atlanta, GA 30332, U.S.A. yan.wang@me.gatech.edu Topics Quantum Monte Carlo Hartree-Fock
Density Functional Theory - II part
 Density Functional Theory - II part antonino.polimeno@unipd.it Overview From theory to practice Implementation Functionals Local functionals Gradient Others From theory to practice From now on, if not
Density Functional Theory - II part antonino.polimeno@unipd.it Overview From theory to practice Implementation Functionals Local functionals Gradient Others From theory to practice From now on, if not
QMC dissociation energy of the water dimer: Time step errors and backflow calculations
 QMC dissociation energy of the water dimer: Time step errors and backflow calculations Idoia G. de Gurtubay and Richard J. Needs TCM group. Cavendish Laboratory University of Cambridge Idoia G. de Gurtubay.
QMC dissociation energy of the water dimer: Time step errors and backflow calculations Idoia G. de Gurtubay and Richard J. Needs TCM group. Cavendish Laboratory University of Cambridge Idoia G. de Gurtubay.
MO Calculation for a Diatomic Molecule. /4 0 ) i=1 j>i (1/r ij )
 i=1 j>i (1/r ij )") MO Calculation for a Diatomic Molecule Introduction The properties of any molecular system can in principle be found by looking at the solutions to the corresponding time independent Schrodinger equation
MO Calculation for a Diatomic Molecule Introduction The properties of any molecular system can in principle be found by looking at the solutions to the corresponding time independent Schrodinger equation
Introduction to Density Functional Theory
 1 Introduction to Density Functional Theory 21 February 2011; V172 P.Ravindran, FME-course on Ab initio Modelling of solar cell Materials 21 February 2011 Introduction to DFT 2 3 4 Ab initio Computational
1 Introduction to Density Functional Theory 21 February 2011; V172 P.Ravindran, FME-course on Ab initio Modelling of solar cell Materials 21 February 2011 Introduction to DFT 2 3 4 Ab initio Computational
An Introduction to Quantum Chemistry and Potential Energy Surfaces. Benjamin G. Levine
 An Introduction to Quantum Chemistry and Potential Energy Surfaces Benjamin G. Levine This Week s Lecture Potential energy surfaces What are they? What are they good for? How do we use them to solve chemical
An Introduction to Quantum Chemistry and Potential Energy Surfaces Benjamin G. Levine This Week s Lecture Potential energy surfaces What are they? What are they good for? How do we use them to solve chemical
Electron Correlation
 Electron Correlation Levels of QM Theory HΨ=EΨ Born-Oppenheimer approximation Nuclear equation: H n Ψ n =E n Ψ n Electronic equation: H e Ψ e =E e Ψ e Single determinant SCF Semi-empirical methods Correlation
Electron Correlation Levels of QM Theory HΨ=EΨ Born-Oppenheimer approximation Nuclear equation: H n Ψ n =E n Ψ n Electronic equation: H e Ψ e =E e Ψ e Single determinant SCF Semi-empirical methods Correlation
No. 2 lectronic state and potential energy function for UH where ρ = r r e, r being the interatomic distance and r e its equilibrium value. How
 Vol 12 No 2, February 2003 cfl 2003 Chin. Phys. Soc. 1009-1963/2003/12(02)/0154-05 Chinese Physics and IOP Publishing Ltd lectronic state and potential energy function for UH 2+* Wang Hong-Yan( Ψ) a)y,
Vol 12 No 2, February 2003 cfl 2003 Chin. Phys. Soc. 1009-1963/2003/12(02)/0154-05 Chinese Physics and IOP Publishing Ltd lectronic state and potential energy function for UH 2+* Wang Hong-Yan( Ψ) a)y,
Computational Chemistry. An Introduction to Molecular Dynamic Simulations
 Computational Chemistry An Introduction to Molecular Dynamic Simulations Computational chemistry simulates chemical structures and reactions numerically, based in full or in part on the fundamental laws
Computational Chemistry An Introduction to Molecular Dynamic Simulations Computational chemistry simulates chemical structures and reactions numerically, based in full or in part on the fundamental laws
Time-Dependent Density-Functional Theory
 Summer School on First Principles Calculations for Condensed Matter and Nanoscience August 21 September 3, 2005 Santa Barbara, California Time-Dependent Density-Functional Theory X. Gonze, Université Catholique
Summer School on First Principles Calculations for Condensed Matter and Nanoscience August 21 September 3, 2005 Santa Barbara, California Time-Dependent Density-Functional Theory X. Gonze, Université Catholique
Exchange Correlation Functional Investigation of RT-TDDFT on a Sodium Chloride. Dimer. Philip Straughn
 Exchange Correlation Functional Investigation of RT-TDDFT on a Sodium Chloride Dimer Philip Straughn Abstract Charge transfer between Na and Cl ions is an important problem in physical chemistry. However,
Exchange Correlation Functional Investigation of RT-TDDFT on a Sodium Chloride Dimer Philip Straughn Abstract Charge transfer between Na and Cl ions is an important problem in physical chemistry. However,
DENSITY FUNCTIONAL THEORY FOR NON-THEORISTS JOHN P. PERDEW DEPARTMENTS OF PHYSICS AND CHEMISTRY TEMPLE UNIVERSITY
 DENSITY FUNCTIONAL THEORY FOR NON-THEORISTS JOHN P. PERDEW DEPARTMENTS OF PHYSICS AND CHEMISTRY TEMPLE UNIVERSITY A TUTORIAL FOR PHYSICAL SCIENTISTS WHO MAY OR MAY NOT HATE EQUATIONS AND PROOFS REFERENCES
DENSITY FUNCTIONAL THEORY FOR NON-THEORISTS JOHN P. PERDEW DEPARTMENTS OF PHYSICS AND CHEMISTRY TEMPLE UNIVERSITY A TUTORIAL FOR PHYSICAL SCIENTISTS WHO MAY OR MAY NOT HATE EQUATIONS AND PROOFS REFERENCES
Density Functional Theory
 Chemistry 380.37 Fall 2015 Dr. Jean M. Standard October 28, 2015 Density Functional Theory What is a Functional? A functional is a general mathematical quantity that represents a rule to convert a function
Chemistry 380.37 Fall 2015 Dr. Jean M. Standard October 28, 2015 Density Functional Theory What is a Functional? A functional is a general mathematical quantity that represents a rule to convert a function
Solid State Theory: Band Structure Methods
 Solid State Theory: Band Structure Methods Lilia Boeri Wed., 11:15-12:45 HS P3 (PH02112) http://itp.tugraz.at/lv/boeri/ele/ Plan of the Lecture: DFT1+2: Hohenberg-Kohn Theorem and Kohn and Sham equations.
Solid State Theory: Band Structure Methods Lilia Boeri Wed., 11:15-12:45 HS P3 (PH02112) http://itp.tugraz.at/lv/boeri/ele/ Plan of the Lecture: DFT1+2: Hohenberg-Kohn Theorem and Kohn and Sham equations.
Density Functional Theory. Martin Lüders Daresbury Laboratory
 Density Functional Theory Martin Lüders Daresbury Laboratory Ab initio Calculations Hamiltonian: (without external fields, non-relativistic) impossible to solve exactly!! Electrons Nuclei Electron-Nuclei
Density Functional Theory Martin Lüders Daresbury Laboratory Ab initio Calculations Hamiltonian: (without external fields, non-relativistic) impossible to solve exactly!! Electrons Nuclei Electron-Nuclei
Oslo node. Highly accurate calculations benchmarking and extrapolations
 Oslo node Highly accurate calculations benchmarking and extrapolations Torgeir Ruden, with A. Halkier, P. Jørgensen, J. Olsen, W. Klopper, J. Gauss, P. Taylor Explicitly correlated methods Pål Dahle, collaboration
Oslo node Highly accurate calculations benchmarking and extrapolations Torgeir Ruden, with A. Halkier, P. Jørgensen, J. Olsen, W. Klopper, J. Gauss, P. Taylor Explicitly correlated methods Pål Dahle, collaboration
Density Functional Theory
 Density Functional Theory March 26, 2009 ? DENSITY FUNCTIONAL THEORY is a method to successfully describe the behavior of atomic and molecular systems and is used for instance for: structural prediction
Density Functional Theory March 26, 2009 ? DENSITY FUNCTIONAL THEORY is a method to successfully describe the behavior of atomic and molecular systems and is used for instance for: structural prediction
Introduction to Computational Chemistry
 Introduction to Computational Chemistry Vesa Hänninen Laboratory of Physical Chemistry Chemicum 4th floor vesa.hanninen@helsinki.fi September 10, 2013 Lecture 3. Electron correlation methods September
Introduction to Computational Chemistry Vesa Hänninen Laboratory of Physical Chemistry Chemicum 4th floor vesa.hanninen@helsinki.fi September 10, 2013 Lecture 3. Electron correlation methods September
Multi-reference Density Functional Theory. COLUMBUS Workshop Argonne National Laboratory 15 August 2005
 Multi-reference Density Functional Theory COLUMBUS Workshop Argonne National Laboratory 15 August 2005 Capt Eric V. Beck Air Force Institute of Technology Department of Engineering Physics 2950 Hobson
Multi-reference Density Functional Theory COLUMBUS Workshop Argonne National Laboratory 15 August 2005 Capt Eric V. Beck Air Force Institute of Technology Department of Engineering Physics 2950 Hobson
Introduction to Computational Quantum Chemistry: Theory
 Introduction to Computational Quantum Chemistry: Theory Dr Andrew Gilbert Rm 118, Craig Building, RSC 3108 Course Lectures 2007 Introduction Hartree Fock Theory Configuration Interaction Lectures 1 Introduction
Introduction to Computational Quantum Chemistry: Theory Dr Andrew Gilbert Rm 118, Craig Building, RSC 3108 Course Lectures 2007 Introduction Hartree Fock Theory Configuration Interaction Lectures 1 Introduction
QUANTUM CHEMISTRY FOR TRANSITION METALS
 QUANTUM CHEMISTRY FOR TRANSITION METALS Outline I Introduction II Correlation Static correlation effects MC methods DFT III Relativity Generalities From 4 to 1 components Effective core potential Outline
QUANTUM CHEMISTRY FOR TRANSITION METALS Outline I Introduction II Correlation Static correlation effects MC methods DFT III Relativity Generalities From 4 to 1 components Effective core potential Outline
GEM4 Summer School OpenCourseWare
 GEM4 Summer School OpenCourseWare http://gem4.educommons.net/ http://www.gem4.org/ Lecture: Molecular Mechanics by Ju Li. Given August 9, 2006 during the GEM4 session at MIT in Cambridge, MA. Please use
GEM4 Summer School OpenCourseWare http://gem4.educommons.net/ http://www.gem4.org/ Lecture: Molecular Mechanics by Ju Li. Given August 9, 2006 during the GEM4 session at MIT in Cambridge, MA. Please use
Principles of Quantum Mechanics
 Principles of Quantum Mechanics - indistinguishability of particles: bosons & fermions bosons: total wavefunction is symmetric upon interchange of particle coordinates (space,spin) fermions: total wavefuncftion
Principles of Quantum Mechanics - indistinguishability of particles: bosons & fermions bosons: total wavefunction is symmetric upon interchange of particle coordinates (space,spin) fermions: total wavefuncftion
Introduction to density-functional theory. Emmanuel Fromager
 Institut de Chimie, Strasbourg, France Page 1 Emmanuel Fromager Institut de Chimie de Strasbourg - Laboratoire de Chimie Quantique - Université de Strasbourg /CNRS M2 lecture, Strasbourg, France. Institut
Institut de Chimie, Strasbourg, France Page 1 Emmanuel Fromager Institut de Chimie de Strasbourg - Laboratoire de Chimie Quantique - Université de Strasbourg /CNRS M2 lecture, Strasbourg, France. Institut
CHEM6085: Density Functional Theory Lecture 10
 CHEM6085: Density Functional Theory Lecture 10 1) Spin-polarised calculations 2) Geometry optimisation C.-K. Skylaris 1 Unpaired electrons So far we have developed Kohn-Sham DFT for the case of paired
CHEM6085: Density Functional Theory Lecture 10 1) Spin-polarised calculations 2) Geometry optimisation C.-K. Skylaris 1 Unpaired electrons So far we have developed Kohn-Sham DFT for the case of paired
one ν im: transition state saddle point
 Hypothetical Potential Energy Surface Ethane conformations Hartree-Fock theory, basis set stationary points all ν s >0: minimum eclipsed one ν im: transition state saddle point multiple ν im: hilltop 1
Hypothetical Potential Energy Surface Ethane conformations Hartree-Fock theory, basis set stationary points all ν s >0: minimum eclipsed one ν im: transition state saddle point multiple ν im: hilltop 1
Ab initio asymptotic-expansion coefficients for pair energies in Møller-Plesset perturbation theory for atoms
 Ab initio asymptotic-expansion coefficients for pair energies in Møller-Plesset perturbation theory for atoms K. JANKOWSKI a, R. SŁUPSKI a, and J. R. FLORES b a Nicholas Copernicus University 87-100 Toruń,
Ab initio asymptotic-expansion coefficients for pair energies in Møller-Plesset perturbation theory for atoms K. JANKOWSKI a, R. SŁUPSKI a, and J. R. FLORES b a Nicholas Copernicus University 87-100 Toruń,
Chemistry 334 Part 2: Computational Quantum Chemistry
 Chemistry 334 Part 2: Computational Quantum Chemistry 1. Definition Louis Scudiero, Ben Shepler and Kirk Peterson Washington State University January 2006 Computational chemistry is an area of theoretical
Chemistry 334 Part 2: Computational Quantum Chemistry 1. Definition Louis Scudiero, Ben Shepler and Kirk Peterson Washington State University January 2006 Computational chemistry is an area of theoretical
The Basics of Theoretical and Computational Chemistry
 Bernd M. Rode, Thomas S. Hofer, and Michael D. Kugler The Basics of Theoretical and Computational Chemistry BICENTENNIA BICENTBNN I AL. WILEY-VCH Verlag GmbH & Co. KGaA V Contents Preface IX 1 Introduction
Bernd M. Rode, Thomas S. Hofer, and Michael D. Kugler The Basics of Theoretical and Computational Chemistry BICENTENNIA BICENTBNN I AL. WILEY-VCH Verlag GmbH & Co. KGaA V Contents Preface IX 1 Introduction
Example questions for Molecular modelling (Level 4) Dr. Adrian Mulholland
 Dr. Adrian Mulholland") Example questions for Molecular modelling (Level 4) Dr. Adrian Mulholland 1) Question. Two methods which are widely used for the optimization of molecular geometies are the Steepest descents and Newton-Raphson
Example questions for Molecular modelling (Level 4) Dr. Adrian Mulholland 1) Question. Two methods which are widely used for the optimization of molecular geometies are the Steepest descents and Newton-Raphson
Density Functional Theory for Electrons in Materials
 Density Functional Theory for Electrons in Materials Richard M. Martin Department of Physics and Materials Research Laboratory University of Illinois at Urbana-Champaign 1 Density Functional Theory for
Density Functional Theory for Electrons in Materials Richard M. Martin Department of Physics and Materials Research Laboratory University of Illinois at Urbana-Champaign 1 Density Functional Theory for
Teoría del Funcional de la Densidad (Density Functional Theory)
") Teoría del Funcional de la Densidad (Density Functional Theory) Motivation: limitations of the standard approach based on the wave function. The electronic density n(r) as the key variable: Functionals
Teoría del Funcional de la Densidad (Density Functional Theory) Motivation: limitations of the standard approach based on the wave function. The electronic density n(r) as the key variable: Functionals
Electronic structure calculations: fundamentals George C. Schatz Northwestern University
 Electronic structure calculations: fundamentals George C. Schatz Northwestern University Electronic Structure (often called Quantum Chemistry) calculations use quantum mechanics to determine the wavefunctions
Electronic structure calculations: fundamentals George C. Schatz Northwestern University Electronic Structure (often called Quantum Chemistry) calculations use quantum mechanics to determine the wavefunctions
Self-Consistent Implementation of Self-Interaction Corrected DFT and of the Exact Exchange Functionals in Plane-Wave DFT
 Self-Consistent Implementation of Self-Interaction Corrected DFT and of the Exact Exchange Functionals in Plane-Wave DFT Kiril Tsemekhman (a), Eric Bylaska (b), Hannes Jonsson (a,c) (a) Department of Chemistry,
Self-Consistent Implementation of Self-Interaction Corrected DFT and of the Exact Exchange Functionals in Plane-Wave DFT Kiril Tsemekhman (a), Eric Bylaska (b), Hannes Jonsson (a,c) (a) Department of Chemistry,
Electron Correlation - Methods beyond Hartree-Fock
 Electron Correlation - Methods beyond Hartree-Fock how to approach chemical accuracy Alexander A. Auer Max-Planck-Institute for Chemical Energy Conversion, Mülheim September 4, 2014 MMER Summerschool 2014
Electron Correlation - Methods beyond Hartree-Fock how to approach chemical accuracy Alexander A. Auer Max-Planck-Institute for Chemical Energy Conversion, Mülheim September 4, 2014 MMER Summerschool 2014
Introduction to Density Functional Theory
 Introduction to Density Functional Theory S. Sharma Institut für Physik Karl-Franzens-Universität Graz, Austria 19th October 2005 Synopsis Motivation 1 Motivation : where can one use DFT 2 : 1 Elementary
Introduction to Density Functional Theory S. Sharma Institut für Physik Karl-Franzens-Universität Graz, Austria 19th October 2005 Synopsis Motivation 1 Motivation : where can one use DFT 2 : 1 Elementary
Introduction to Computational Chemistry: Theory
 Introduction to Computational Chemistry: Theory Dr Andrew Gilbert Rm 118, Craig Building, RSC andrew.gilbert@anu.edu.au 3023 Course Lectures Introduction Hartree Fock Theory Basis Sets Lecture 1 1 Introduction
Introduction to Computational Chemistry: Theory Dr Andrew Gilbert Rm 118, Craig Building, RSC andrew.gilbert@anu.edu.au 3023 Course Lectures Introduction Hartree Fock Theory Basis Sets Lecture 1 1 Introduction
Molecular Mechanics: The Ab Initio Foundation
 Molecular Mechanics: The Ab Initio Foundation Ju Li GEM4 Summer School 2006 Cell and Molecular Mechanics in BioMedicine August 7 18, 2006, MIT, Cambridge, MA, USA 2 Outline Why are electrons quantum? Born-Oppenheimer
Molecular Mechanics: The Ab Initio Foundation Ju Li GEM4 Summer School 2006 Cell and Molecular Mechanics in BioMedicine August 7 18, 2006, MIT, Cambridge, MA, USA 2 Outline Why are electrons quantum? Born-Oppenheimer
Ab initio calculations for potential energy surfaces. D. Talbi GRAAL- Montpellier
 Ab initio calculations for potential energy surfaces D. Talbi GRAAL- Montpellier A theoretical study of a reaction is a two step process I-Electronic calculations : techniques of quantum chemistry potential
Ab initio calculations for potential energy surfaces D. Talbi GRAAL- Montpellier A theoretical study of a reaction is a two step process I-Electronic calculations : techniques of quantum chemistry potential
Introduction to Computational Chemistry
 Introduction to Computational Chemistry Vesa Hänninen Laboratory of Physical Chemistry room B430, Chemicum 4th floor vesa.hanninen@helsinki.fi September 3, 2013 Introduction and theoretical backround September
Introduction to Computational Chemistry Vesa Hänninen Laboratory of Physical Chemistry room B430, Chemicum 4th floor vesa.hanninen@helsinki.fi September 3, 2013 Introduction and theoretical backround September
Calculations of band structures
 Chemistry and Physics at Albany Planning for the Future Calculations of band structures using wave-function based correlation methods Elke Pahl Centre of Theoretical Chemistry and Physics Institute of
Chemistry and Physics at Albany Planning for the Future Calculations of band structures using wave-function based correlation methods Elke Pahl Centre of Theoretical Chemistry and Physics Institute of
AN INTRODUCTION TO QUANTUM CHEMISTRY. Mark S. Gordon Iowa State University
 AN INTRODUCTION TO QUANTUM CHEMISTRY Mark S. Gordon Iowa State University 1 OUTLINE Theoretical Background in Quantum Chemistry Overview of GAMESS Program Applications 2 QUANTUM CHEMISTRY In principle,
AN INTRODUCTION TO QUANTUM CHEMISTRY Mark S. Gordon Iowa State University 1 OUTLINE Theoretical Background in Quantum Chemistry Overview of GAMESS Program Applications 2 QUANTUM CHEMISTRY In principle,
CLIMBING THE LADDER OF DENSITY FUNCTIONAL APPROXIMATIONS JOHN P. PERDEW DEPARTMENT OF PHYSICS TEMPLE UNIVERSITY PHILADELPHIA, PA 19122
 CLIMBING THE LADDER OF DENSITY FUNCTIONAL APPROXIMATIONS JOHN P. PERDEW DEPARTMENT OF PHYSICS TEMPLE UNIVERSITY PHILADELPHIA, PA 191 THANKS TO MANY COLLABORATORS, INCLUDING SY VOSKO DAVID LANGRETH ALEX
CLIMBING THE LADDER OF DENSITY FUNCTIONAL APPROXIMATIONS JOHN P. PERDEW DEPARTMENT OF PHYSICS TEMPLE UNIVERSITY PHILADELPHIA, PA 191 THANKS TO MANY COLLABORATORS, INCLUDING SY VOSKO DAVID LANGRETH ALEX
Chemistry 3502/4502. Final Exam Part I. May 14, 2005
 Advocacy chit Chemistry 350/450 Final Exam Part I May 4, 005. For which of the below systems is = where H is the Hamiltonian operator and T is the kinetic-energy operator? (a) The free particle
Advocacy chit Chemistry 350/450 Final Exam Part I May 4, 005. For which of the below systems is = where H is the Hamiltonian operator and T is the kinetic-energy operator? (a) The free particle
Band calculations: Theory and Applications
 Band calculations: Theory and Applications Lecture 2: Different approximations for the exchange-correlation correlation functional in DFT Local density approximation () Generalized gradient approximation
Band calculations: Theory and Applications Lecture 2: Different approximations for the exchange-correlation correlation functional in DFT Local density approximation () Generalized gradient approximation
One-Electron Hamiltonians
 One-Electron Hamiltonians Hartree-Fock and Density Func7onal Theory Christopher J. Cramer @ChemProfCramer 2017 MSSC, July 10, 2017 REVIEW A One-Slide Summary of Quantum Mechanics Fundamental Postulate:
One-Electron Hamiltonians Hartree-Fock and Density Func7onal Theory Christopher J. Cramer @ChemProfCramer 2017 MSSC, July 10, 2017 REVIEW A One-Slide Summary of Quantum Mechanics Fundamental Postulate:
Simulation Methods II
 Simulation Methods II Maria Fyta Institute for Computational Physics Universität Stuttgart Summer Term 2018 SM II - contents First principles methods Hartree-Fock and beyond Density-funtional-theory Ab
Simulation Methods II Maria Fyta Institute for Computational Physics Universität Stuttgart Summer Term 2018 SM II - contents First principles methods Hartree-Fock and beyond Density-funtional-theory Ab
Molecular Magnetism. Magnetic Resonance Parameters. Trygve Helgaker
 Molecular Magnetism Magnetic Resonance Parameters Trygve Helgaker Centre for Theoretical and Computational Chemistry (CTCC), Department of Chemistry, University of Oslo, Norway Laboratoire de Chimie Théorique,
Molecular Magnetism Magnetic Resonance Parameters Trygve Helgaker Centre for Theoretical and Computational Chemistry (CTCC), Department of Chemistry, University of Oslo, Norway Laboratoire de Chimie Théorique,
Yingwei Wang Computational Quantum Chemistry 1 Hartree energy 2. 2 Many-body system 2. 3 Born-Oppenheimer approximation 2
 Purdue University CHM 67300 Computational Quantum Chemistry REVIEW Yingwei Wang October 10, 2013 Review: Prof Slipchenko s class, Fall 2013 Contents 1 Hartree energy 2 2 Many-body system 2 3 Born-Oppenheimer
Purdue University CHM 67300 Computational Quantum Chemistry REVIEW Yingwei Wang October 10, 2013 Review: Prof Slipchenko s class, Fall 2013 Contents 1 Hartree energy 2 2 Many-body system 2 3 Born-Oppenheimer
Module 6 1. Density functional theory
 Module 6 1. Density functional theory Updated May 12, 2016 B A DDFT C K A bird s-eye view of density-functional theory Authors: Klaus Capelle G http://arxiv.org/abs/cond-mat/0211443 R https://trac.cc.jyu.fi/projects/toolbox/wiki/dft
Module 6 1. Density functional theory Updated May 12, 2016 B A DDFT C K A bird s-eye view of density-functional theory Authors: Klaus Capelle G http://arxiv.org/abs/cond-mat/0211443 R https://trac.cc.jyu.fi/projects/toolbox/wiki/dft
Spring College on Computational Nanoscience May Variational Principles, the Hellmann-Feynman Theorem, Density Functional Theor
 2145-25 Spring College on Computational Nanoscience 17-28 May 2010 Variational Principles, the Hellmann-Feynman Theorem, Density Functional Theor Stefano BARONI SISSA & CNR-IOM DEMOCRITOS Simulation Center
2145-25 Spring College on Computational Nanoscience 17-28 May 2010 Variational Principles, the Hellmann-Feynman Theorem, Density Functional Theor Stefano BARONI SISSA & CNR-IOM DEMOCRITOS Simulation Center
Electric properties of molecules
 Electric properties of molecules For a molecule in a uniform electric fielde the Hamiltonian has the form: Ĥ(E) = Ĥ + E ˆµ x where we assume that the field is directed along the x axis and ˆµ x is the
Electric properties of molecules For a molecule in a uniform electric fielde the Hamiltonian has the form: Ĥ(E) = Ĥ + E ˆµ x where we assume that the field is directed along the x axis and ˆµ x is the
4 Post-Hartree Fock Methods: MPn and Configuration Interaction
 4 Post-Hartree Fock Methods: MPn and Configuration Interaction In the limit of a complete basis, the Hartree-Fock (HF) energy in the complete basis set limit (ECBS HF ) yields an upper boundary to the
4 Post-Hartree Fock Methods: MPn and Configuration Interaction In the limit of a complete basis, the Hartree-Fock (HF) energy in the complete basis set limit (ECBS HF ) yields an upper boundary to the
Lecture 5: More about one- Final words about the Hartree-Fock theory. First step above it by the Møller-Plesset perturbation theory.
 Lecture 5: More about one- determinant wave functions Final words about the Hartree-Fock theory. First step above it by the Møller-Plesset perturbation theory. Items from Lecture 4 Could the Koopmans theorem
Lecture 5: More about one- determinant wave functions Final words about the Hartree-Fock theory. First step above it by the Møller-Plesset perturbation theory. Items from Lecture 4 Could the Koopmans theorem
Same idea for polyatomics, keep track of identical atom e.g. NH 3 consider only valence electrons F(2s,2p) H(1s)
 H(1s)") XIII 63 Polyatomic bonding -09 -mod, Notes (13) Engel 16-17 Balance: nuclear repulsion, positive e-n attraction, neg. united atom AO ε i applies to all bonding, just more nuclei repulsion biggest at low
XIII 63 Polyatomic bonding -09 -mod, Notes (13) Engel 16-17 Balance: nuclear repulsion, positive e-n attraction, neg. united atom AO ε i applies to all bonding, just more nuclei repulsion biggest at low
NMR and IR spectra & vibrational analysis
 Lab 5: NMR and IR spectra & vibrational analysis A brief theoretical background 1 Some of the available chemical quantum methods for calculating NMR chemical shifts are based on the Hartree-Fock self-consistent
Lab 5: NMR and IR spectra & vibrational analysis A brief theoretical background 1 Some of the available chemical quantum methods for calculating NMR chemical shifts are based on the Hartree-Fock self-consistent
Introduction to Computational Chemistry Computational (chemistry education) and/or. (Computational chemistry) education
 and/or. (Computational chemistry) education") Introduction to Computational Chemistry Computational (chemistry education) and/or (Computational chemistry) education First one: Use computational tools to help increase student understanding of material
Introduction to Computational Chemistry Computational (chemistry education) and/or (Computational chemistry) education First one: Use computational tools to help increase student understanding of material
Handbook of Computational Quantum Chemistry. DAVID B. COOK The Department of Chemistry, University of Sheffield
 Handbook of Computational Quantum Chemistry DAVID B. COOK The Department of Chemistry, University of Sheffield Oxford New York Tokyo OXFORD UNIVERSITY PRESS 1998 CONTENTS 1 Mechanics and molecules 1 1.1
Handbook of Computational Quantum Chemistry DAVID B. COOK The Department of Chemistry, University of Sheffield Oxford New York Tokyo OXFORD UNIVERSITY PRESS 1998 CONTENTS 1 Mechanics and molecules 1 1.1
Supporting Information
 Supporting Information Roles of Zeolite Confinement and Cu O Cu Angle on the Direct Conversion of Methane to Methanol by [Cu 2 (µ-o)] 2+ -exchanged AEI, CHA, AFX, and MFI Zeolites M. Haris Mahyuddin,,
Supporting Information Roles of Zeolite Confinement and Cu O Cu Angle on the Direct Conversion of Methane to Methanol by [Cu 2 (µ-o)] 2+ -exchanged AEI, CHA, AFX, and MFI Zeolites M. Haris Mahyuddin,,
Section 3 Electronic Configurations, Term Symbols, and States
 Section 3 Electronic Configurations, Term Symbols, and States Introductory Remarks- The Orbital, Configuration, and State Pictures of Electronic Structure One of the goals of quantum chemistry is to allow
Section 3 Electronic Configurations, Term Symbols, and States Introductory Remarks- The Orbital, Configuration, and State Pictures of Electronic Structure One of the goals of quantum chemistry is to allow
QMC dissociation energies of three-electron hemibonded radical cation dimers... and water clusters
 QMC dissociation energies of three-electron hemibonded radical cation dimers... and water clusters Idoia G. de Gurtubay TCM group. Cavendish Laboratory University of Cambridge Idoia G. de Gurtubay. Quantum
QMC dissociation energies of three-electron hemibonded radical cation dimers... and water clusters Idoia G. de Gurtubay TCM group. Cavendish Laboratory University of Cambridge Idoia G. de Gurtubay. Quantum
Fixed-Node quantum Monte Carlo for Chemistry
 Fixed-Node quantum Monte Carlo for Chemistry Michel Caffarel Lab. Physique et Chimie Quantiques, CNRS-IRSAMC, Université de Toulouse e-mail : caffarel@irsamc.ups-tlse.fr. p.1/29 The N-body problem of Chemistry
Fixed-Node quantum Monte Carlo for Chemistry Michel Caffarel Lab. Physique et Chimie Quantiques, CNRS-IRSAMC, Université de Toulouse e-mail : caffarel@irsamc.ups-tlse.fr. p.1/29 The N-body problem of Chemistry
Chem 442 Review for Exam 2. Exact separation of the Hamiltonian of a hydrogenic atom into center-of-mass (3D) and relative (3D) components.
 and relative (3D) components.") Chem 44 Review for Exam Hydrogenic atoms: The Coulomb energy between two point charges Ze and e: V r Ze r Exact separation of the Hamiltonian of a hydrogenic atom into center-of-mass (3D) and relative
Chem 44 Review for Exam Hydrogenic atoms: The Coulomb energy between two point charges Ze and e: V r Ze r Exact separation of the Hamiltonian of a hydrogenic atom into center-of-mass (3D) and relative
Dept of Mechanical Engineering MIT Nanoengineering group
 1 Dept of Mechanical Engineering MIT Nanoengineering group » To calculate all the properties of a molecule or crystalline system knowing its atomic information: Atomic species Their coordinates The Symmetry
1 Dept of Mechanical Engineering MIT Nanoengineering group » To calculate all the properties of a molecule or crystalline system knowing its atomic information: Atomic species Their coordinates The Symmetry
The successful wavefunction can be written as a determinant: # 1 (2) # 2 (2) Electrons. This can be generalized to our 2N-electron wavefunction:
 # 2 (2) Electrons. This can be generalized to our 2N-electron wavefunction:") T2. CNDO to AM1: The Semiempirical Molecular Orbital Models The discussion in sections T2.1 T2.3 applies also to ab initio molecular orbital calculations. T2.1 Slater Determinants Consider the general
T2. CNDO to AM1: The Semiempirical Molecular Orbital Models The discussion in sections T2.1 T2.3 applies also to ab initio molecular orbital calculations. T2.1 Slater Determinants Consider the general
The Schrödinger equation for many-electron systems
 The Schrödinger equation for many-electron systems Ĥ!( x,, x ) = E!( x,, x ) 1 N 1 1 Z 1 Ĥ = " $ # " $ + $ 2 r 2 A j j A, j RAj i, j < i a linear differential equation in 4N variables (atomic units) (3
The Schrödinger equation for many-electron systems Ĥ!( x,, x ) = E!( x,, x ) 1 N 1 1 Z 1 Ĥ = " $ # " $ + $ 2 r 2 A j j A, j RAj i, j < i a linear differential equation in 4N variables (atomic units) (3
1 Density functional theory (DFT)
") 1 Density functional theory (DFT) 1.1 Introduction Density functional theory is an alternative to ab initio methods for solving the nonrelativistic, time-independent Schrödinger equation H Φ = E Φ. The
1 Density functional theory (DFT) 1.1 Introduction Density functional theory is an alternative to ab initio methods for solving the nonrelativistic, time-independent Schrödinger equation H Φ = E Φ. The
Exchange-Correlation Functional
 Exchange-Correlation Functional Aiichiro Nakano Collaboratory for Advanced Computing & Simulations Depts. of Computer Science, Physics & Astronomy, Chemical Engineering & Materials Science, and Biological
Exchange-Correlation Functional Aiichiro Nakano Collaboratory for Advanced Computing & Simulations Depts. of Computer Science, Physics & Astronomy, Chemical Engineering & Materials Science, and Biological
Lecture 9. Hartree Fock Method and Koopman s Theorem
 Lecture 9 Hartree Fock Method and Koopman s Theorem Ψ(N) is approximated as a single slater determinant Φ of N orthogonal One electron spin-orbitals. One electron orbital φ i = φ i (r) χ i (σ) χ i (σ)
Lecture 9 Hartree Fock Method and Koopman s Theorem Ψ(N) is approximated as a single slater determinant Φ of N orthogonal One electron spin-orbitals. One electron orbital φ i = φ i (r) χ i (σ) χ i (σ)
Jack Simons, Henry Eyring Scientist and Professor Chemistry Department University of Utah
 1. Born-Oppenheimer approx.- energy surfaces 2. Mean-field (Hartree-Fock) theory- orbitals 3. Pros and cons of HF- RHF, UHF 4. Beyond HF- why? 5. First, one usually does HF-how? 6. Basis sets and notations
1. Born-Oppenheimer approx.- energy surfaces 2. Mean-field (Hartree-Fock) theory- orbitals 3. Pros and cons of HF- RHF, UHF 4. Beyond HF- why? 5. First, one usually does HF-how? 6. Basis sets and notations
Feet on the potential energy surface, head in the π clouds
 Graduate Theses and Dissertations Iowa State University Capstones, Theses and Dissertations 2011 Feet on the potential energy surface, head in the π clouds Quentin Anthony Smith Iowa State University Follow
Graduate Theses and Dissertations Iowa State University Capstones, Theses and Dissertations 2011 Feet on the potential energy surface, head in the π clouds Quentin Anthony Smith Iowa State University Follow
LUMO + 1 LUMO. Tómas Arnar Guðmundsson Report 2 Reikniefnafræði G
 Q1: Display all the MOs for N2 in your report and classify each one of them as bonding, antibonding or non-bonding, and say whether the symmetry of the orbital is σ or π. Sketch a molecular orbital diagram
Q1: Display all the MOs for N2 in your report and classify each one of them as bonding, antibonding or non-bonding, and say whether the symmetry of the orbital is σ or π. Sketch a molecular orbital diagram
Handbook of Computational Quantum Chemistry
 Handbook of Computational Quantum Chemistry David B. Cook Dept. of Chemistry University of Sheffield DOVER PUBLICATIONS, INC. Mineola, New York F Contents 1 Mechanics and molecules 1 1.1 1.2 1.3 1.4 1.5
Handbook of Computational Quantum Chemistry David B. Cook Dept. of Chemistry University of Sheffield DOVER PUBLICATIONS, INC. Mineola, New York F Contents 1 Mechanics and molecules 1 1.1 1.2 1.3 1.4 1.5
Electronic structure theory: Fundamentals to frontiers. 1. Hartree-Fock theory
 Electronic structure theory: Fundamentals to frontiers. 1. Hartree-Fock theory MARTIN HEAD-GORDON, Department of Chemistry, University of California, and Chemical Sciences Division, Lawrence Berkeley National
Electronic structure theory: Fundamentals to frontiers. 1. Hartree-Fock theory MARTIN HEAD-GORDON, Department of Chemistry, University of California, and Chemical Sciences Division, Lawrence Berkeley National
Structure of Cement Phases from ab initio Modeling Crystalline C-S-HC
 Structure of Cement Phases from ab initio Modeling Crystalline C-S-HC Sergey V. Churakov sergey.churakov@psi.ch Paul Scherrer Institute Switzerland Cement Phase Composition C-S-H H Solid Solution Model
Structure of Cement Phases from ab initio Modeling Crystalline C-S-HC Sergey V. Churakov sergey.churakov@psi.ch Paul Scherrer Institute Switzerland Cement Phase Composition C-S-H H Solid Solution Model
Semi-Empirical MO Methods
 Semi-Empirical MO Methods the high cost of ab initio MO calculations is largely due to the many integrals that need to be calculated (esp. two electron integrals) semi-empirical MO methods start with the
Semi-Empirical MO Methods the high cost of ab initio MO calculations is largely due to the many integrals that need to be calculated (esp. two electron integrals) semi-empirical MO methods start with the
Joint ICTP-IAEA Workshop on Nuclear Structure Decay Data: Theory and Evaluation August Introduction to Nuclear Physics - 1
 2358-19 Joint ICTP-IAEA Workshop on Nuclear Structure Decay Data: Theory and Evaluation 6-17 August 2012 Introduction to Nuclear Physics - 1 P. Van Isacker GANIL, Grand Accelerateur National d'ions Lourds
2358-19 Joint ICTP-IAEA Workshop on Nuclear Structure Decay Data: Theory and Evaluation 6-17 August 2012 Introduction to Nuclear Physics - 1 P. Van Isacker GANIL, Grand Accelerateur National d'ions Lourds
Orbital dependent correlation potentials in ab initio density functional theory
 Orbital dependent correlation potentials in ab initio density functional theory noniterative - one step - calculations Ireneusz Grabowski Institute of Physics Nicolaus Copernicus University Toruń, Poland
Orbital dependent correlation potentials in ab initio density functional theory noniterative - one step - calculations Ireneusz Grabowski Institute of Physics Nicolaus Copernicus University Toruń, Poland
Session 7 Overview: Part A I. Prediction of Vibrational Frequencies (IR) Part B III. Prediction of Electronic Transitions (UV-Vis) IV.
 Part B III. Prediction of Electronic Transitions (UV-Vis) IV.") Session 7 Overview: Part A I. Prediction of Vibrational Frequencies (IR) II. Thermochemistry Part B III. Prediction of Electronic Transitions (UV-Vis) IV. NMR Predictions 1 I. Prediction of Vibrational
Session 7 Overview: Part A I. Prediction of Vibrational Frequencies (IR) II. Thermochemistry Part B III. Prediction of Electronic Transitions (UV-Vis) IV. NMR Predictions 1 I. Prediction of Vibrational
Relativistic and correlated calculations on the ground, excited, and ionized states of iodine
 Relativistic and correlated calculations on the ground, excited, and ionized states of iodine W. A. de Jong, L. Visscher, a) and W. C. Nieuwpoort Laboratory for Chemical Physics and Materials Science Centre,
Relativistic and correlated calculations on the ground, excited, and ionized states of iodine W. A. de Jong, L. Visscher, a) and W. C. Nieuwpoort Laboratory for Chemical Physics and Materials Science Centre,
Atom-molecule molecule collisions in spin-polarized polarized alkalis: potential energy surfaces and quantum dynamics
 Atom-molecule molecule collisions in spin-polarized polarized alkalis: potential energy surfaces and quantum dynamics Pavel Soldán, Marko T. Cvitaš and Jeremy M. Hutson University of Durham with Jean-Michel
Atom-molecule molecule collisions in spin-polarized polarized alkalis: potential energy surfaces and quantum dynamics Pavel Soldán, Marko T. Cvitaš and Jeremy M. Hutson University of Durham with Jean-Michel
Fine Structure Calculations of Atomic Data for Ar XVI
 Journal of Modern Physics, 2015, 6, 1609-1630 Published Online September 2015 in SciRes. http://www.scirp.org/journal/jmp http://dx.doi.org/10.4236/jmp.2015.611163 Fine Structure Calculations of Atomic
Journal of Modern Physics, 2015, 6, 1609-1630 Published Online September 2015 in SciRes. http://www.scirp.org/journal/jmp http://dx.doi.org/10.4236/jmp.2015.611163 Fine Structure Calculations of Atomic
Exam 4 Review. Exam Review: A exam review sheet for exam 4 will be posted on the course webpage. Additionally, a practice exam will also be posted.
 Chem 4502 Quantum Mechanics & Spectroscopy (Jason Goodpaster) Exam 4 Review Exam Review: A exam review sheet for exam 4 will be posted on the course webpage. Additionally, a practice exam will also be
Chem 4502 Quantum Mechanics & Spectroscopy (Jason Goodpaster) Exam 4 Review Exam Review: A exam review sheet for exam 4 will be posted on the course webpage. Additionally, a practice exam will also be
σ u * 1s g - gerade u - ungerade * - antibonding σ g 1s
 One of these two states is a repulsive (dissociative) state. Other excited states can be constructed using linear combinations of other orbitals. Some will be binding and others will be repulsive. Thus
One of these two states is a repulsive (dissociative) state. Other excited states can be constructed using linear combinations of other orbitals. Some will be binding and others will be repulsive. Thus
Additional background material on the Nobel Prize in Chemistry 1998
 Additional background material on the Nobel Prize in Chemistry 1998 The Royal Swedish Academy of Sciences has decided to award the 1998 Nobel Prize in Chemistry with one half to Professor WALTER KOHN,
Additional background material on the Nobel Prize in Chemistry 1998 The Royal Swedish Academy of Sciences has decided to award the 1998 Nobel Prize in Chemistry with one half to Professor WALTER KOHN,
Advanced Electronic Structure Theory Density functional theory. Dr Fred Manby
 Advanced Electronic Structure Theory Density functional theory Dr Fred Manby fred.manby@bris.ac.uk http://www.chm.bris.ac.uk/pt/manby/ 6 Strengths of DFT DFT is one of many theories used by (computational)
Advanced Electronic Structure Theory Density functional theory Dr Fred Manby fred.manby@bris.ac.uk http://www.chm.bris.ac.uk/pt/manby/ 6 Strengths of DFT DFT is one of many theories used by (computational)
Introduction to Electronic Structure Theory
 CSC/PRACE Spring School in Computational Chemistry 2017 Introduction to Electronic Structure Theory Mikael Johansson http://www.iki.fi/~mpjohans Objective: To get familiarised with the, subjectively chosen,
CSC/PRACE Spring School in Computational Chemistry 2017 Introduction to Electronic Structure Theory Mikael Johansson http://www.iki.fi/~mpjohans Objective: To get familiarised with the, subjectively chosen,
CHEM3023: Spins, Atoms and Molecules
 CHEM3023: Spins, Atoms and Molecules Lecture 5 The Hartree-Fock method C.-K. Skylaris Learning outcomes Be able to use the variational principle in quantum calculations Be able to construct Fock operators
CHEM3023: Spins, Atoms and Molecules Lecture 5 The Hartree-Fock method C.-K. Skylaris Learning outcomes Be able to use the variational principle in quantum calculations Be able to construct Fock operators
Quantum Theory of Many-Particle Systems, Phys. 540
 Quantum Theory of Many-Particle Systems, Phys. 540 Questions about organization Second quantization Questions about last class? Comments? Similar strategy N-particles Consider Two-body operators in Fock
Quantum Theory of Many-Particle Systems, Phys. 540 Questions about organization Second quantization Questions about last class? Comments? Similar strategy N-particles Consider Two-body operators in Fock
Algorithms and Computational Aspects of DFT Calculations
 Algorithms and Computational Aspects of DFT Calculations Part I Juan Meza and Chao Yang High Performance Computing Research Lawrence Berkeley National Laboratory IMA Tutorial Mathematical and Computational
Algorithms and Computational Aspects of DFT Calculations Part I Juan Meza and Chao Yang High Performance Computing Research Lawrence Berkeley National Laboratory IMA Tutorial Mathematical and Computational