Fundamentals and applications of Density Functional Theory Astrid Marthinsen PhD candidate, Department of Materials Science and Engineering
|
|
|
- Ursula Patrick
- 6 years ago
- Views:
Transcription
1 Fundamentals and applications of Density Functional Theory Astrid Marthinsen PhD candidate, Department of Materials Science and Engineering

2 Outline PART 1: Fundamentals of Density functional theory (DFT) Solving the many body Schrödinger equation Application of DFT in crystalline solids PART 2: Doing DFT calculations An introduction to the VASP software 2




3 Computational material science Goal: Describe properties of matter from theoretical methods rooted in fundamental equations Vibrational properties Optics Structure Magnetic properties Electronic properties 3
4 4 PART 1 Fundamentals of DFT

5 Some definitions A wave function in quantum mechanics describes the quantum state of a set of particles in an isolated system. Operator: Does a mathematical operation on a variable e.g. a derivative x Quantum mechanical operator: Any observable in quantum mechanics is represented by an operator Kinentic energy: ħ2 2m 2 x 2 Ground state: The most stable state of a system lowest energy 5
 = Eψ( r i, R I")
 T: kinetic energy operator The Born Oppenheimer approximation:")
 Slow Fast Decouple into two")
6 Many particle problem in solids Find the ground state for a collection of atoms by solving the Schrödinger equation: Hψ r i,, {R I ) = Eψ( r i, R I ) H = T + V Coloumb V Coloumb = q iq j r i r j (pair of charged particles) T: kinetic energy operator The Born Oppenheimer approximation: m nuclei m e The dynamics of atomic nuclei and electrons can be separated. ψ {r i, }, {R I } ψ N R I ψ e ({r i }) Slow Fast Decouple into two wavefunctions 6
7 Many particle problem in solids: solve the Schrödinger equation for electrons Hψ(r 1, r 2, r 3 r N ) = Eψ(r 1, r 2, r 3 r N ) The electronic hamiltonian consists of three terms : N e H = ħ2 2 2m i e i N e N e + V ext r i + U(r i, r j ) i i=1 j>1 7


8 Many particle problem in solids CO 2 : 6+16 = 22 electrons Three spatial coordinates per electronic position Schrödinger equation becomes a 66 dimentional problem Pb nanocluster (82 electrons per atom) atoms -> 8200 electrons - Schrödinger equation becomes a dimentional proplem Solving the Schrödinger equation for materials is practically a bit of a hassle 8

9 Density functional theory from wavefunctions to electron density Define the electron density: n r = ψ r 1, r 2 r N ψ r 1, r 2 r N 3N dimensions reduces to 3 The j th electron is treated as a point charge in the field of all the other electrons. This simplifies the many-electron problem to many one electron problems: ψ r 1, r 2, r 3 r N = ψ r 1 ψ 2 r 2 ψ 3 r 3 ψ N (r N ) (Hartree product) Define the electron density in terms of the individual electron wave functions: n r = 2 ψ i r ψ i (r) i 9
] THEOREM 2: The electron density that minimizes the energy of the overall functional is the true ground state electron density.")
10 Density functional theory - from wave functions to electron density Hohenberg and Kohn at the heart of DFT THEOREM 1: The ground state energy E is a unique functional 1 of the electron density: E = E[n(r)] THEOREM 2: The electron density that minimizes the energy of the overall functional is the true ground state electron density. E[n(r)] > E 0 [n 0 (r)] 1 1 functional is a function of a function. I.e an integral: F f = f x dx 1 10
![The energy functional E ψ i = E known ψ i + E XC [{ψ i }] E known ψ i = ħ m e ψ i 2 ψ i d 3 r + V r n r d 3 r + e2 i 2 n r n r r r d 3 rd 3 r + E ion E XC [{ψ i }]: exchange-correlation](/docs-images/72/67510259/images/11-1.jpg "functional - Includes all quantum mechanical terms - Not known needs to be approximated Simplest XC-functionals: LDA : Local density approximation GGA: Generalized gradient approximation")
11 The energy functional E ψ i = E known ψ i + E XC [{ψ i }] E known ψ i = ħ m e ψ i 2 ψ i d 3 r + V r n r d 3 r + e2 i 2 n r n r r r d 3 rd 3 r + E ion E XC [{ψ i }]: exchange-correlation functional - Includes all quantum mechanical terms - Not known needs to be approximated Simplest XC-functionals: LDA : Local density approximation GGA: Generalized gradient approximation 11

 The")

12 The Kohn Sham scheme Solve a set of single-electron wave functions that only depend on three spatial variables, ψ(r) The exchange-correlation potential ħ 2m 2 + V r + V H r + V XC r ψ i r = ε i r ψ i Self-consistency scheme: 12

 the ionic")


13 Forces on atoms easily calculated when electron ground state is obtained F I = de dr I = ψ i H r I ψ i By moving along the ionic forces (steepest descent) the ionic ground can be calculated We can displace ions from the ionic groundstate, and determine the forces on all other ions Effective interatomic force constants and vibrational frequences 13
")
14 Crystalline solids and plane wave DFT A crystal: periodic arrangement atoms Free electrons: Plane waves: Electrons in a periodic potential are Bloch waves: ψ nk r = exp ik r u nk r (Perturbed free electrons) 14
15 Crystalline solids and plane wave DFT -Cutoff energy Reciprocal space: Real space F = Reciprocal space All periodic functions can be expanded by a Fourier series: ψ nk r = exp ik r u nk r = exp ik r c k exp(ig r) G = sum of plane waves Each plane wave in the sum have kinetic energy: E = ħ 2m k + G 2 Numerically we must define a cutoff energy for the expansion! The choice of cutoff energy must be tested with respect to energy convergence in calculations 15
 Any k-point that differ by a reciprocal lattice vector G are")

16 Crystalline solids and plane wave DFT - k-points Wavevectors k = 2π λ 1 m are reciprocal space The primitive unit cell in reciprocal space is called the 1st Brillouin Zone (BZ) Any k-point that differ by a reciprocal lattice vector G are equivalent : k`=k+g Integrals need only be evaluated in the 1st BZ Real space F = Reciprocal space Numerically we must chose an approprate number of k-points to sample the BZ The k-point sampling must be tested with respect to energy convergece 16
17 Pseudopotentials Chemical bonding and other characteristics of materials mainly characterized by valence (outer shell) electrons Core electrons thus less important Pseudopotentials replace the electron density from a chosen set of core electrons with a smoothed density Frozen core approximation Current DFT codes typically provide a library of potentials for different elements 17



18 Supercells and periodic boundary conditions Calculations are performed with periodic boundary conditions: But non-periodic entities can also be treated Interfaces Molecules Defects Vacancy 18
19 19 PART 2: Doing DFT calculations -practical aspects

20 What is VASP? One of the software packages that uses DFT to solve the quantum problem for materials Uses periodic boundary conditions Uses pseudopotential method with a plane waves basis set Can model systems with maximum no. of atoms in the range of Commercial software package Other DFT packages (public lisence) Quantum espresso ( Abinit( Siesta ( 20
21 VASP input files INCAR User spesified parameters that define the calculation Global break condition, Energy cutoff, Ionic and geometric relaxation degreses of freedom POSCAR Specifies the periodic simulation cell Information regarding the geometry of the system POTCAR Pseudopotential (PP) file Information on PP and XC functional KPOINTS defines k-point mesh for the Brilloun zone 21
22 Example: BaTiO 3 Lattice vectors: a = b= c = 4.01 Å Fractional ionic coordinates: Ba : (0 0 0) Ti : ( ) O1: ( ) O2: ( ) O3: ( ) Ba O Ti Visualization program (freeware): VESTA 22


23 The POSCAR file Scale factor for lattice vectors BaTiO Ba Ti Direct O Number of each element Cartesian lattice parameters Fractional ionic coordinates 23
24 The INCAR file ENCUT = 650 NSW = 100 EDIFFG = EDIFF = 1E-07 ISIF = 3 IBRION = 2 Many more tags can be set ENCUT tag: Sets the energy cutoff Energy convergence testing required NSW tag: Sets the maximum electronic steps To prevent a non-converging calculation to run forever EDIFFG tag: Sets the convergence criterion for ions Forces on ions or total energy EDIFF tag: Sets the global break conditions for electrons in the SC-loop Electronic relaxation will be stopped if the total energy change is less that this value ISIF tag: determines which degrees of freedom (ions, cell volume, cell shape) are allowed to change In accordance with the purpose of the calculation Number correspond to various degrees of freedom IBRION tag: Desides how ions are updated and moved Numbers 0,1,2 correspond to different algorithms 24
25 The KPOINTS file Automatic mesh 0 Gamma Number of k-points = 0 ->automatic generation scheme Generate Gamma-centered k-mesh Subdivisions N 1, N 2 and N 3 along recipr. l. vectors Subject for convergence testing Optional shift of the mesh 25
26 POTCAR file Library of pseudopotentials provided with VASP Type of pseudopotential The spesific XC potential Number of valence electrons 26
27 Standard output files CONTCAR Contains position of the system after the calculation has completed OSZICAR Contains data of electronic steps and ionic steps OUTCAR Complete output of run including input file data Total forces, charges on ions, symmetry CHGCAR Charge density of system after run 27

28 OSZICAR: Electronic minimization Total energy Energy difference between steps Electronic iterations before electron convergence reached Resulting energy from electronic steps 28
![Lattice parameter [Å] Total energy/atom [ev] total energy/atom (ev) Obtaining reliable total energies k-point density and cutoff energy must be sufficiently large to obtain converged energies](/docs-images/72/67510259/images/29-0.jpg "3,9855-8,375-8,38-8,385-8,39-8,395-8,4 0 5 10 15 k-points 3,985 3,9845 3,984 3,9835 3,983 3,9825 0 5 10 15 k-points -8,396-8,3965 400 500 600 700 800 900")
29 Lattice parameter [Å] Total energy/atom [ev] total energy/atom (ev) Obtaining reliable total energies k-point density and cutoff energy must be sufficiently large to obtain converged energies 3,9855-8,375-8,38-8,385-8,39-8,395-8, k-points 3,985 3,9845 3,984 3,9835 3,983 3, k-points -8,396-8, ,397-8,3975-8,398-8,3985-8,399-8,3995-8,4 Cutoff energy (ev) NB: Total energies are phyically quite meaningless in DFT, energy differences are relevant Energy resolution in DFT ~1meV/atom 29
30 DFT as a theoretical microscope DFT can obtain Structure beyond the current capabilities of experiments. Predict properties at a resolution and length scale currently inaccessible to experiments. 30


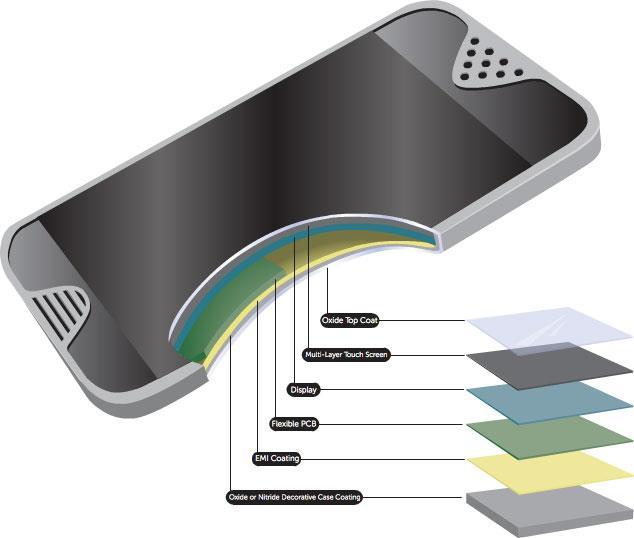

31 Modern electronics is all about thin film technology What s going on here?!? 31

32 With DFT We are in complete control of the degrees of freedom - We can isolate effects We know our model, so we know exactly what we «measure» - But does the model correpond to reality? 32


33 Looking through the DFT microscope Structural responses to strain and defects partial charge density: Orbital resolved density of states (DOS) 33


34 Summary Density functional theory is a powerful tool for predicting material properties Doing DFT calculations is not so scary VASP - commercial softwave for DFT calculations 34
35 References Book: David Sholl and Janice Steckel Denity functinal theory - A practical introduction Book: June Gunn Lee, Computational material science an introduction The vasp manual: 35
36 36 Thank you
1. Hydrogen atom in a box
 1. Hydrogen atom in a box Recall H atom problem, V(r) = -1/r e r exact answer solved by expanding in Gaussian basis set, had to solve secular matrix involving matrix elements of basis functions place atom
1. Hydrogen atom in a box Recall H atom problem, V(r) = -1/r e r exact answer solved by expanding in Gaussian basis set, had to solve secular matrix involving matrix elements of basis functions place atom
Due: since the calculation takes longer than before, we ll make it due on 02/05/2016, Friday
 Homework 3 Due: since the calculation takes longer than before, we ll make it due on 02/05/2016, Friday Email to: jqian@caltech.edu Introduction In this assignment, you will be using a commercial periodic
Homework 3 Due: since the calculation takes longer than before, we ll make it due on 02/05/2016, Friday Email to: jqian@caltech.edu Introduction In this assignment, you will be using a commercial periodic
Integrated Computational Materials Engineering Education
 Integrated Computational Materials Engineering Education Lecture on Density Functional Theory An Introduction Mark Asta Dept. of Materials Science and Engineering, University of California, Berkeley &
Integrated Computational Materials Engineering Education Lecture on Density Functional Theory An Introduction Mark Asta Dept. of Materials Science and Engineering, University of California, Berkeley &
Self Consistent Cycle
 Self Consistent Cycle Step 0 : defining your system namelist SYSTEM How to specify the System All periodic systems can be specified by a Bravais Lattice and and atomic basis How to specify the Bravais
Self Consistent Cycle Step 0 : defining your system namelist SYSTEM How to specify the System All periodic systems can be specified by a Bravais Lattice and and atomic basis How to specify the Bravais
Introduction to VASP (1)
") Introduction to VASP (1) Yun-Wen Chen http://cms.mpi.univie.ac.at/vasp/vasp/vasp.html https://www.vasp.at/ 1 What is VASP? The Vienna Ab initio Simulation Package (VASP) is a computer program for atomic
Introduction to VASP (1) Yun-Wen Chen http://cms.mpi.univie.ac.at/vasp/vasp/vasp.html https://www.vasp.at/ 1 What is VASP? The Vienna Ab initio Simulation Package (VASP) is a computer program for atomic
Practical Guide to Density Functional Theory (DFT)
") Practical Guide to Density Functional Theory (DFT) Brad Malone, Sadas Shankar Quick recap of where we left off last time BD Malone, S Shankar Therefore there is a direct one-to-one correspondence between
Practical Guide to Density Functional Theory (DFT) Brad Malone, Sadas Shankar Quick recap of where we left off last time BD Malone, S Shankar Therefore there is a direct one-to-one correspondence between
Density Functional Theory (DFT)
") Density Functional Theory (DFT) An Introduction by A.I. Al-Sharif Irbid, Aug, 2 nd, 2009 Density Functional Theory Revolutionized our approach to the electronic structure of atoms, molecules and solid
Density Functional Theory (DFT) An Introduction by A.I. Al-Sharif Irbid, Aug, 2 nd, 2009 Density Functional Theory Revolutionized our approach to the electronic structure of atoms, molecules and solid
Dept of Mechanical Engineering MIT Nanoengineering group
 1 Dept of Mechanical Engineering MIT Nanoengineering group » Recap of HK theorems and KS equations» The physical meaning of the XC energy» Solution of a one-particle Schroedinger equation» Pseudo Potentials»
1 Dept of Mechanical Engineering MIT Nanoengineering group » Recap of HK theorems and KS equations» The physical meaning of the XC energy» Solution of a one-particle Schroedinger equation» Pseudo Potentials»
Introduction to first-principles modelling and CASTEP
 to first-principles modelling and Phil Hasnip to + Atomistic Simulations If we know what the bonding in a material is beforehand, then we can often find good expressions for the forces between atoms, e.g.
to first-principles modelling and Phil Hasnip to + Atomistic Simulations If we know what the bonding in a material is beforehand, then we can often find good expressions for the forces between atoms, e.g.
The electronic structure of materials 2 - DFT
 Quantum mechanics 2 - Lecture 9 December 19, 2012 1 Density functional theory (DFT) 2 Literature Contents 1 Density functional theory (DFT) 2 Literature Historical background The beginnings: L. de Broglie
Quantum mechanics 2 - Lecture 9 December 19, 2012 1 Density functional theory (DFT) 2 Literature Contents 1 Density functional theory (DFT) 2 Literature Historical background The beginnings: L. de Broglie
Materials that you may find helpful when working through this exercise
 Detailed steps illustrating how to use VASP on the Suns in Fitz 177 For use in lab: 11/10/2009 (Original file by Dr. Rachel Getman, 11/18/2007. Editted for use by Dorrell McCalman 11/09/2009.) Note on
Detailed steps illustrating how to use VASP on the Suns in Fitz 177 For use in lab: 11/10/2009 (Original file by Dr. Rachel Getman, 11/18/2007. Editted for use by Dorrell McCalman 11/09/2009.) Note on
Hands on Session III
 Hands on Session III Andreas EICHLER Institut für Materialphysik and Center for Computational Materials Science Universität Wien, Sensengasse 8, A-1090 Wien, Austria b-initio ienna ackage imulation G.
Hands on Session III Andreas EICHLER Institut für Materialphysik and Center for Computational Materials Science Universität Wien, Sensengasse 8, A-1090 Wien, Austria b-initio ienna ackage imulation G.
Electronic structure, plane waves and pseudopotentials
 Electronic structure, plane waves and pseudopotentials P.J. Hasnip Spectroscopy Workshop 2009 We want to be able to predict what electrons and nuclei will do from first principles, without needing to know
Electronic structure, plane waves and pseudopotentials P.J. Hasnip Spectroscopy Workshop 2009 We want to be able to predict what electrons and nuclei will do from first principles, without needing to know
Intro to ab initio methods
 Lecture 2 Part A Intro to ab initio methods Recommended reading: Leach, Chapters 2 & 3 for QM methods For more QM methods: Essentials of Computational Chemistry by C.J. Cramer, Wiley (2002) 1 ab initio
Lecture 2 Part A Intro to ab initio methods Recommended reading: Leach, Chapters 2 & 3 for QM methods For more QM methods: Essentials of Computational Chemistry by C.J. Cramer, Wiley (2002) 1 ab initio
Introduction to Density Functional Theory
 Introduction to Density Functional Theory S. Sharma Institut für Physik Karl-Franzens-Universität Graz, Austria 19th October 2005 Synopsis Motivation 1 Motivation : where can one use DFT 2 : 1 Elementary
Introduction to Density Functional Theory S. Sharma Institut für Physik Karl-Franzens-Universität Graz, Austria 19th October 2005 Synopsis Motivation 1 Motivation : where can one use DFT 2 : 1 Elementary
Introduction to Density Functional Theory with Applications to Graphene Branislav K. Nikolić
 Introduction to Density Functional Theory with Applications to Graphene Branislav K. Nikolić Department of Physics and Astronomy, University of Delaware, Newark, DE 19716, U.S.A. http://wiki.physics.udel.edu/phys824
Introduction to Density Functional Theory with Applications to Graphene Branislav K. Nikolić Department of Physics and Astronomy, University of Delaware, Newark, DE 19716, U.S.A. http://wiki.physics.udel.edu/phys824
Electron levels in a periodic potential: general remarks. Daniele Toffoli January 11, / 46
 Electron levels in a periodic potential: general remarks Daniele Toffoli January 11, 2017 1 / 46 Outline 1 Mathematical tools 2 The periodic potential 3 Bloch s theorem and Born-von Karman boundary conditions
Electron levels in a periodic potential: general remarks Daniele Toffoli January 11, 2017 1 / 46 Outline 1 Mathematical tools 2 The periodic potential 3 Bloch s theorem and Born-von Karman boundary conditions
Electrochemistry project, Chemistry Department, November Ab-initio Molecular Dynamics Simulation
 Electrochemistry project, Chemistry Department, November 2006 Ab-initio Molecular Dynamics Simulation Outline Introduction Ab-initio concepts Total energy concepts Adsorption energy calculation Project
Electrochemistry project, Chemistry Department, November 2006 Ab-initio Molecular Dynamics Simulation Outline Introduction Ab-initio concepts Total energy concepts Adsorption energy calculation Project
Algorithms and Computational Aspects of DFT Calculations
 Algorithms and Computational Aspects of DFT Calculations Part I Juan Meza and Chao Yang High Performance Computing Research Lawrence Berkeley National Laboratory IMA Tutorial Mathematical and Computational
Algorithms and Computational Aspects of DFT Calculations Part I Juan Meza and Chao Yang High Performance Computing Research Lawrence Berkeley National Laboratory IMA Tutorial Mathematical and Computational
Computational Material Science Part II-1: introduction. Horng-Tay Jeng ( 鄭弘泰 ) Institute of Physics, Academia Sinica
 Institute of Physics, Academia Sinica") Computational Material Science Part II-1: introduction Horng-Tay Jeng ( 鄭弘泰 ) Institute of Physics, Academia Sinica Outline Introduction of Computational Material Science (CMS) Density Functional Theory
Computational Material Science Part II-1: introduction Horng-Tay Jeng ( 鄭弘泰 ) Institute of Physics, Academia Sinica Outline Introduction of Computational Material Science (CMS) Density Functional Theory
VASP Tutorial: Dielectric properties and the Random-Phase-Approximation (RPA)
") VASP Tutorial: Dielectric properties and the Random-Phase-Approximation (RPA) University of Vienna, Faculty of Physics and Center for Computational Materials Science, Vienna, Austria Setting up a VASP
VASP Tutorial: Dielectric properties and the Random-Phase-Approximation (RPA) University of Vienna, Faculty of Physics and Center for Computational Materials Science, Vienna, Austria Setting up a VASP
Introduction to First-Principles Method
 Joint ICTP/CAS/IAEA School & Workshop on Plasma-Materials Interaction in Fusion Devices, July 18-22, 2016, Hefei Introduction to First-Principles Method by Guang-Hong LU ( 吕广宏 ) Beihang University Computer
Joint ICTP/CAS/IAEA School & Workshop on Plasma-Materials Interaction in Fusion Devices, July 18-22, 2016, Hefei Introduction to First-Principles Method by Guang-Hong LU ( 吕广宏 ) Beihang University Computer
MODULE 2: QUANTUM MECHANICS. Practice: Quantum ESPRESSO
 MODULE 2: QUANTUM MECHANICS Practice: Quantum ESPRESSO I. What is Quantum ESPRESSO? 2 DFT software PW-DFT, PP, US-PP, PAW http://www.quantum-espresso.org FREE PW-DFT, PP, PAW http://www.abinit.org FREE
MODULE 2: QUANTUM MECHANICS Practice: Quantum ESPRESSO I. What is Quantum ESPRESSO? 2 DFT software PW-DFT, PP, US-PP, PAW http://www.quantum-espresso.org FREE PW-DFT, PP, PAW http://www.abinit.org FREE
Key concepts in Density Functional Theory (I) Silvana Botti
 Silvana Botti") From the many body problem to the Kohn-Sham scheme European Theoretical Spectroscopy Facility (ETSF) CNRS - Laboratoire des Solides Irradiés Ecole Polytechnique, Palaiseau - France Temporary Address: Centre
From the many body problem to the Kohn-Sham scheme European Theoretical Spectroscopy Facility (ETSF) CNRS - Laboratoire des Solides Irradiés Ecole Polytechnique, Palaiseau - France Temporary Address: Centre
Density Functional Theory
 Density Functional Theory Iain Bethune EPCC ibethune@epcc.ed.ac.uk Overview Background Classical Atomistic Simulation Essential Quantum Mechanics DFT: Approximations and Theory DFT: Implementation using
Density Functional Theory Iain Bethune EPCC ibethune@epcc.ed.ac.uk Overview Background Classical Atomistic Simulation Essential Quantum Mechanics DFT: Approximations and Theory DFT: Implementation using
Electronic Structure of MoS 2 Nanotubes
 Clemson University TigerPrints All Dissertations Dissertations 8-2007 Electronic Structure of MoS 2 Nanotubes Lingyun Xu Clemson University, neilxu@gmail.com Follow this and additional works at: http://tigerprints.clemson.edu/all_dissertations
Clemson University TigerPrints All Dissertations Dissertations 8-2007 Electronic Structure of MoS 2 Nanotubes Lingyun Xu Clemson University, neilxu@gmail.com Follow this and additional works at: http://tigerprints.clemson.edu/all_dissertations
Supplementary Materials for
 advances.sciencemag.org/cgi/content/full/3/7/e1700704/dc1 Supplementary Materials for Giant Rashba splitting in 2D organic-inorganic halide perovskites measured by transient spectroscopies Yaxin Zhai,
advances.sciencemag.org/cgi/content/full/3/7/e1700704/dc1 Supplementary Materials for Giant Rashba splitting in 2D organic-inorganic halide perovskites measured by transient spectroscopies Yaxin Zhai,
Electrons in Crystals. Chris J. Pickard
 Electrons in Crystals Chris J. Pickard Electrons in Crystals The electrons in a crystal experience a potential with the periodicity of the Bravais lattice: U(r + R) = U(r) The scale of the periodicity
Electrons in Crystals Chris J. Pickard Electrons in Crystals The electrons in a crystal experience a potential with the periodicity of the Bravais lattice: U(r + R) = U(r) The scale of the periodicity
Band Structure Calculations; Electronic and Optical Properties
 ; Electronic and Optical Properties Stewart Clark University of Outline Introduction to band structures Calculating band structures using Castep Calculating optical properties Examples results Some applications
; Electronic and Optical Properties Stewart Clark University of Outline Introduction to band structures Calculating band structures using Castep Calculating optical properties Examples results Some applications
Condensed matter physics FKA091
 Condensed matter physics FKA091 Ermin Malic Department of Physics Chalmers University of Technology Henrik Johannesson Department of Physics University of Gothenburg Teaching assistants: Roland Jago &
Condensed matter physics FKA091 Ermin Malic Department of Physics Chalmers University of Technology Henrik Johannesson Department of Physics University of Gothenburg Teaching assistants: Roland Jago &
Tutorial 1. Atoms, molecules, and bulk systems. Computational Materials Physics, Faculty of Physics, University Vienna, Vienna, Austria
 Tutorial 1 Atoms, molecules, and bulk systems Computational Materials Physics, Faculty of Physics, University Vienna, Vienna, Austria I. The INCAR file II. The POSCAR file III. The KPOINTS file IV. The
Tutorial 1 Atoms, molecules, and bulk systems Computational Materials Physics, Faculty of Physics, University Vienna, Vienna, Austria I. The INCAR file II. The POSCAR file III. The KPOINTS file IV. The
CHEM6085: Density Functional Theory
 Lecture 11 CHEM6085: Density Functional Theory DFT for periodic crystalline solids C.-K. Skylaris 1 Electron in a one-dimensional periodic box (in atomic units) Schrödinger equation Energy eigenvalues
Lecture 11 CHEM6085: Density Functional Theory DFT for periodic crystalline solids C.-K. Skylaris 1 Electron in a one-dimensional periodic box (in atomic units) Schrödinger equation Energy eigenvalues
Electronic Structure Theory for Periodic Systems: The Concepts. Christian Ratsch
 Electronic Structure Theory for Periodic Systems: The Concepts Christian Ratsch Institute for Pure and Applied Mathematics and Department of Mathematics, UCLA Motivation There are 10 20 atoms in 1 mm 3
Electronic Structure Theory for Periodic Systems: The Concepts Christian Ratsch Institute for Pure and Applied Mathematics and Department of Mathematics, UCLA Motivation There are 10 20 atoms in 1 mm 3
Electron bands in crystals Pseudopotentials, Plane Waves, Local Orbitals
 Electron bands in crystals Pseudopotentials, Plane Waves, Local Orbitals Richard M. Martin UIUC Lecture at Summer School Hands-on introduction to Electronic Structure Materials Computation Center University
Electron bands in crystals Pseudopotentials, Plane Waves, Local Orbitals Richard M. Martin UIUC Lecture at Summer School Hands-on introduction to Electronic Structure Materials Computation Center University
Density Functional Theory. Martin Lüders Daresbury Laboratory
 Density Functional Theory Martin Lüders Daresbury Laboratory Ab initio Calculations Hamiltonian: (without external fields, non-relativistic) impossible to solve exactly!! Electrons Nuclei Electron-Nuclei
Density Functional Theory Martin Lüders Daresbury Laboratory Ab initio Calculations Hamiltonian: (without external fields, non-relativistic) impossible to solve exactly!! Electrons Nuclei Electron-Nuclei
Phonon calculations with SCAN
 Workshop on the SCAN density functional: Fundamentals, practices, and extensions Temple university, Philadelphia May 18th, 2017 Hands-on tutorial 3 Phonon calculations with SCAN Yubo Zhang and Jianwei
Workshop on the SCAN density functional: Fundamentals, practices, and extensions Temple university, Philadelphia May 18th, 2017 Hands-on tutorial 3 Phonon calculations with SCAN Yubo Zhang and Jianwei
Multi-Scale Modeling from First Principles
 m mm Multi-Scale Modeling from First Principles μm nm m mm μm nm space space Predictive modeling and simulations must address all time and Continuum Equations, densityfunctional space scales Rate Equations
m mm Multi-Scale Modeling from First Principles μm nm m mm μm nm space space Predictive modeling and simulations must address all time and Continuum Equations, densityfunctional space scales Rate Equations
Teoría del Funcional de la Densidad (Density Functional Theory)
") Teoría del Funcional de la Densidad (Density Functional Theory) Motivation: limitations of the standard approach based on the wave function. The electronic density n(r) as the key variable: Functionals
Teoría del Funcional de la Densidad (Density Functional Theory) Motivation: limitations of the standard approach based on the wave function. The electronic density n(r) as the key variable: Functionals
Introduction to phonopy
 Introduction to phonopy Installation Download phonopy http://sourceforge.net/projects/phonopy/ Install Python libraries that phonopy relies Install phonopy % sudo apt-get install python-dev python-numpy
Introduction to phonopy Installation Download phonopy http://sourceforge.net/projects/phonopy/ Install Python libraries that phonopy relies Install phonopy % sudo apt-get install python-dev python-numpy
Key concepts in Density Functional Theory (II)
") Kohn-Sham scheme and band structures European Theoretical Spectroscopy Facility (ETSF) CNRS - Laboratoire des Solides Irradiés Ecole Polytechnique, Palaiseau - France Present Address: LPMCN Université
Kohn-Sham scheme and band structures European Theoretical Spectroscopy Facility (ETSF) CNRS - Laboratoire des Solides Irradiés Ecole Polytechnique, Palaiseau - France Present Address: LPMCN Université
Computational Modeling Software and their applications
 Computational Modeling Software and their applications June 21, 2011 Damilola Daramola Center for Electrochemical Engineering Research ABC s of electrochemistry Introduction Computational Modeling the
Computational Modeling Software and their applications June 21, 2011 Damilola Daramola Center for Electrochemical Engineering Research ABC s of electrochemistry Introduction Computational Modeling the
ALMA: All-scale predictive design of heat management material structures
 ALMA: All-scale predictive design of heat management material structures Version Date: 2015.11.13. Last updated 2015.12.02 Purpose of this document: Definition of a data organisation that is applicable
ALMA: All-scale predictive design of heat management material structures Version Date: 2015.11.13. Last updated 2015.12.02 Purpose of this document: Definition of a data organisation that is applicable
Introduction to density functional perturbation theory for lattice dynamics
 Introduction to density functional perturbation theory for lattice dynamics SISSA and DEMOCRITOS Trieste (Italy) Outline 1 Lattice dynamic of a solid: phonons Description of a solid Equations of motion
Introduction to density functional perturbation theory for lattice dynamics SISSA and DEMOCRITOS Trieste (Italy) Outline 1 Lattice dynamic of a solid: phonons Description of a solid Equations of motion
DFT / SIESTA algorithms
 DFT / SIESTA algorithms Javier Junquera José M. Soler References http://siesta.icmab.es Documentation Tutorials Atomic units e = m e = =1 atomic mass unit = m e atomic length unit = 1 Bohr = 0.5292 Ang
DFT / SIESTA algorithms Javier Junquera José M. Soler References http://siesta.icmab.es Documentation Tutorials Atomic units e = m e = =1 atomic mass unit = m e atomic length unit = 1 Bohr = 0.5292 Ang
Table of Contents. Table of Contents Spin-orbit splitting of semiconductor band structures
 Table of Contents Table of Contents Spin-orbit splitting of semiconductor band structures Relavistic effects in Kohn-Sham DFT Silicon band splitting with ATK-DFT LSDA initial guess for the ground state
Table of Contents Table of Contents Spin-orbit splitting of semiconductor band structures Relavistic effects in Kohn-Sham DFT Silicon band splitting with ATK-DFT LSDA initial guess for the ground state
Geometry Optimisation
 Geometry Optimisation Matt Probert Condensed Matter Dynamics Group Department of Physics, University of York, UK http://www.cmt.york.ac.uk/cmd http://www.castep.org Motivation Overview of Talk Background
Geometry Optimisation Matt Probert Condensed Matter Dynamics Group Department of Physics, University of York, UK http://www.cmt.york.ac.uk/cmd http://www.castep.org Motivation Overview of Talk Background
Half-Heusler Alloys as Promising Thermoelectric Materials. Alexander A. Page
 Half-Heusler Alloys as Promising Thermoelectric Materials by Alexander A. Page A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy (Physics) in the
Half-Heusler Alloys as Promising Thermoelectric Materials by Alexander A. Page A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy (Physics) in the
Determining elastic constants of graphene using density functional theory
 UNIVERSITY OF GRAZ Determining elastic constants of graphene using density functional theory by Max Niederreiter 01510759 A thesis submitted in partial fulfillment for the degree of Bachelor Of Science
UNIVERSITY OF GRAZ Determining elastic constants of graphene using density functional theory by Max Niederreiter 01510759 A thesis submitted in partial fulfillment for the degree of Bachelor Of Science
MODULE 2: QUANTUM MECHANICS. Principles and Theory
 MODULE 2: QUANTUM MECHANICS Principles and Theory You are here http://www.lbl.gov/cs/html/exascale4energy/nuclear.html 2 Short Review of Quantum Mechanics Why do we need quantum mechanics? Bonding and
MODULE 2: QUANTUM MECHANICS Principles and Theory You are here http://www.lbl.gov/cs/html/exascale4energy/nuclear.html 2 Short Review of Quantum Mechanics Why do we need quantum mechanics? Bonding and
Density Functional Theory
 Density Functional Theory March 26, 2009 ? DENSITY FUNCTIONAL THEORY is a method to successfully describe the behavior of atomic and molecular systems and is used for instance for: structural prediction
Density Functional Theory March 26, 2009 ? DENSITY FUNCTIONAL THEORY is a method to successfully describe the behavior of atomic and molecular systems and is used for instance for: structural prediction
3rd International Conference on Machinery, Materials and Information Technology Applications (ICMMITA 2015)
") 3rd International Conference on Machinery, Materials and Information Technology Applications (ICMMITA 2015) Study on Energy Band-gap Calculation of CuGaS 2 Liuyang YUa, Yong Xub, Kegao LIUc* School of
3rd International Conference on Machinery, Materials and Information Technology Applications (ICMMITA 2015) Study on Energy Band-gap Calculation of CuGaS 2 Liuyang YUa, Yong Xub, Kegao LIUc* School of
References. Documentation Manuals Tutorials Publications
 References http://siesta.icmab.es Documentation Manuals Tutorials Publications Atomic units e = m e = =1 atomic mass unit = m e atomic length unit = 1 Bohr = 0.5292 Ang atomic energy unit = 1 Hartree =
References http://siesta.icmab.es Documentation Manuals Tutorials Publications Atomic units e = m e = =1 atomic mass unit = m e atomic length unit = 1 Bohr = 0.5292 Ang atomic energy unit = 1 Hartree =
Introduction to Condensed Matter Physics
 Introduction to Condensed Matter Physics The Reciprocal Lattice M.P. Vaughan Overview Overview of the reciprocal lattice Periodic functions Reciprocal lattice vectors Bloch functions k-space Dispersion
Introduction to Condensed Matter Physics The Reciprocal Lattice M.P. Vaughan Overview Overview of the reciprocal lattice Periodic functions Reciprocal lattice vectors Bloch functions k-space Dispersion
Electronic Structure Methodology 1
 Electronic Structure Methodology 1 Chris J. Pickard Lecture Two Working with Density Functional Theory In the last lecture we learnt how to write the total energy as a functional of the density n(r): E
Electronic Structure Methodology 1 Chris J. Pickard Lecture Two Working with Density Functional Theory In the last lecture we learnt how to write the total energy as a functional of the density n(r): E
Key concepts in Density Functional Theory
 From the many body problem to the Kohn-Sham scheme ILM (LPMCN) CNRS, Université Lyon 1 - France European Theoretical Spectroscopy Facility (ETSF) December 12, 2012 Lyon Outline 1 The many-body problem
From the many body problem to the Kohn-Sham scheme ILM (LPMCN) CNRS, Université Lyon 1 - France European Theoretical Spectroscopy Facility (ETSF) December 12, 2012 Lyon Outline 1 The many-body problem
CLIMBING THE LADDER OF DENSITY FUNCTIONAL APPROXIMATIONS JOHN P. PERDEW DEPARTMENT OF PHYSICS TEMPLE UNIVERSITY PHILADELPHIA, PA 19122
 CLIMBING THE LADDER OF DENSITY FUNCTIONAL APPROXIMATIONS JOHN P. PERDEW DEPARTMENT OF PHYSICS TEMPLE UNIVERSITY PHILADELPHIA, PA 191 THANKS TO MANY COLLABORATORS, INCLUDING SY VOSKO DAVID LANGRETH ALEX
CLIMBING THE LADDER OF DENSITY FUNCTIONAL APPROXIMATIONS JOHN P. PERDEW DEPARTMENT OF PHYSICS TEMPLE UNIVERSITY PHILADELPHIA, PA 191 THANKS TO MANY COLLABORATORS, INCLUDING SY VOSKO DAVID LANGRETH ALEX
Structure of Cement Phases from ab initio Modeling Crystalline C-S-HC
 Structure of Cement Phases from ab initio Modeling Crystalline C-S-HC Sergey V. Churakov sergey.churakov@psi.ch Paul Scherrer Institute Switzerland Cement Phase Composition C-S-H H Solid Solution Model
Structure of Cement Phases from ab initio Modeling Crystalline C-S-HC Sergey V. Churakov sergey.churakov@psi.ch Paul Scherrer Institute Switzerland Cement Phase Composition C-S-H H Solid Solution Model
Electronic structure calculations with GPAW. Jussi Enkovaara CSC IT Center for Science, Finland
 Electronic structure calculations with GPAW Jussi Enkovaara CSC IT Center for Science, Finland Basics of density-functional theory Density-functional theory Many-body Schrödinger equation Can be solved
Electronic structure calculations with GPAW Jussi Enkovaara CSC IT Center for Science, Finland Basics of density-functional theory Density-functional theory Many-body Schrödinger equation Can be solved
Homework 5 Computational Chemistry (CBE 60547)
") Homework 5 Computational Chemistry (CBE 60547) Prof. William F. Schneider Due: 1 Not Ar again! As they say, there are many ways to skin a cat! You have computed the wavefunctions of Ar
Homework 5 Computational Chemistry (CBE 60547) Prof. William F. Schneider Due: 1 Not Ar again! As they say, there are many ways to skin a cat! You have computed the wavefunctions of Ar
Excercise : Ammonia Flipping
 Excercise : Ammonia Flipping Rennes, 1. September 2016 Faculty of Physics, AG-CMP, University of Vienna general remarks (1) this excercise consists of 4 steps which unfold if you untar the file ammonia
Excercise : Ammonia Flipping Rennes, 1. September 2016 Faculty of Physics, AG-CMP, University of Vienna general remarks (1) this excercise consists of 4 steps which unfold if you untar the file ammonia
Dept of Mechanical Engineering MIT Nanoengineering group
 1 Dept of Mechanical Engineering MIT Nanoengineering group » To calculate all the properties of a molecule or crystalline system knowing its atomic information: Atomic species Their coordinates The Symmetry
1 Dept of Mechanical Engineering MIT Nanoengineering group » To calculate all the properties of a molecule or crystalline system knowing its atomic information: Atomic species Their coordinates The Symmetry
Chapter 3. The (L)APW+lo Method. 3.1 Choosing A Basis Set
APW+lo Method. 3.1 Choosing A Basis Set") Chapter 3 The (L)APW+lo Method 3.1 Choosing A Basis Set The Kohn-Sham equations (Eq. (2.17)) provide a formulation of how to practically find a solution to the Hohenberg-Kohn functional (Eq. (2.15)). Nevertheless
Chapter 3 The (L)APW+lo Method 3.1 Choosing A Basis Set The Kohn-Sham equations (Eq. (2.17)) provide a formulation of how to practically find a solution to the Hohenberg-Kohn functional (Eq. (2.15)). Nevertheless
Is the homogeneous electron gas homogeneous?
 Is the homogeneous electron gas homogeneous? Electron gas (jellium): simplest way to view a metal homogeneous and normal Hartree-Fock: simplest method for many-electron systems a single Slater determinant
Is the homogeneous electron gas homogeneous? Electron gas (jellium): simplest way to view a metal homogeneous and normal Hartree-Fock: simplest method for many-electron systems a single Slater determinant
VASP Tutorial. Nodo Lee. Ph. D. Candidate
 VASP Tutorial July 17, 2014 @ SCENT HPC Summer School @ GIST Nodo Lee Ph. D. Candidate Molecular Modeling Laboratory (MML) School of Materials Science and Engineering (MSE) Gwangju Institute of Science
VASP Tutorial July 17, 2014 @ SCENT HPC Summer School @ GIST Nodo Lee Ph. D. Candidate Molecular Modeling Laboratory (MML) School of Materials Science and Engineering (MSE) Gwangju Institute of Science
The Plane-Wave Pseudopotential Method
 Hands-on Workshop on Density Functional Theory and Beyond: Computational Materials Science for Real Materials Trieste, August 6-15, 2013 The Plane-Wave Pseudopotential Method Ralph Gebauer ICTP, Trieste
Hands-on Workshop on Density Functional Theory and Beyond: Computational Materials Science for Real Materials Trieste, August 6-15, 2013 The Plane-Wave Pseudopotential Method Ralph Gebauer ICTP, Trieste
Pseudopotentials for hybrid density functionals and SCAN
 Pseudopotentials for hybrid density functionals and SCAN Jing Yang, Liang Z. Tan, Julian Gebhardt, and Andrew M. Rappe Department of Chemistry University of Pennsylvania Why do we need pseudopotentials?
Pseudopotentials for hybrid density functionals and SCAN Jing Yang, Liang Z. Tan, Julian Gebhardt, and Andrew M. Rappe Department of Chemistry University of Pennsylvania Why do we need pseudopotentials?
Electronic structure of solids: basic concepts and methods
 Electronic structure of solids: basic concepts and methods Ondřej Šipr II. NEVF 514 Surface Physics Winter Term 2016-2017 Troja, 21st October 2016 Outline A bit of formal mathematics for the beginning
Electronic structure of solids: basic concepts and methods Ondřej Šipr II. NEVF 514 Surface Physics Winter Term 2016-2017 Troja, 21st October 2016 Outline A bit of formal mathematics for the beginning
DFT and beyond: Hands-on Tutorial Workshop Tutorial 1: Basics of Electronic Structure Theory
 DFT and beyond: Hands-on Tutorial Workshop 2011 Tutorial 1: Basics of Electronic Structure Theory V. Atalla, O. T. Hofmann, S. V. Levchenko Theory Department, Fritz-Haber-Institut der MPG Berlin July 13,
DFT and beyond: Hands-on Tutorial Workshop 2011 Tutorial 1: Basics of Electronic Structure Theory V. Atalla, O. T. Hofmann, S. V. Levchenko Theory Department, Fritz-Haber-Institut der MPG Berlin July 13,
Session 1. Introduction to Computational Chemistry. Computational (chemistry education) and/or (Computational chemistry) education
 and/or (Computational chemistry) education") Session 1 Introduction to Computational Chemistry 1 Introduction to Computational Chemistry Computational (chemistry education) and/or (Computational chemistry) education First one: Use computational tools
Session 1 Introduction to Computational Chemistry 1 Introduction to Computational Chemistry Computational (chemistry education) and/or (Computational chemistry) education First one: Use computational tools
First-Principles Computational Investigations of. Titanium Carbide Nanocrystals. Qin Zhang. (Under the direction of Steven P.
 First-Principles Computational Investigations of Titanium Carbide Nanocrystals by Qin Zhang (Under the direction of Steven P. Lewis) Abstract Stable transition-metal carbide nanocrystals were observed
First-Principles Computational Investigations of Titanium Carbide Nanocrystals by Qin Zhang (Under the direction of Steven P. Lewis) Abstract Stable transition-metal carbide nanocrystals were observed
Ab Initio Study of the Mechanical, Electronic, Thermal and Optical Properties of Ge 2 Sb 2 Te 5
 Ab Initio Study of the Mechanical, Electronic, Thermal and Optical Properties of Ge 2 Sb 2 Te 5 Odhiambo H. 1, Amolo G. 2, Makau N. 2, Othieno H. 1, and Oduor A. 1 1 Department of Physics, Maseno University,
Ab Initio Study of the Mechanical, Electronic, Thermal and Optical Properties of Ge 2 Sb 2 Te 5 Odhiambo H. 1, Amolo G. 2, Makau N. 2, Othieno H. 1, and Oduor A. 1 1 Department of Physics, Maseno University,
GEM4 Summer School OpenCourseWare
 GEM4 Summer School OpenCourseWare http://gem4.educommons.net/ http://www.gem4.org/ Lecture: Molecular Mechanics by Ju Li. Given August 9, 2006 during the GEM4 session at MIT in Cambridge, MA. Please use
GEM4 Summer School OpenCourseWare http://gem4.educommons.net/ http://www.gem4.org/ Lecture: Molecular Mechanics by Ju Li. Given August 9, 2006 during the GEM4 session at MIT in Cambridge, MA. Please use
THE ELECTRONIC AND THERMODYNAMIC PROPERTIES USING DENSITY FUNCTIONAL THEORY. Davis George Daniel. A thesis. submitted in partial fulfillment
 THE ELECTRONIC AND THERMODYNAMIC PROPERTIES OF Ca DOPED LaFeO 3 A FIRST PRINCIPLES STUDY USING DENSITY FUNCTIONAL THEORY by Davis George Daniel A thesis submitted in partial fulfillment of the requirements
THE ELECTRONIC AND THERMODYNAMIC PROPERTIES OF Ca DOPED LaFeO 3 A FIRST PRINCIPLES STUDY USING DENSITY FUNCTIONAL THEORY by Davis George Daniel A thesis submitted in partial fulfillment of the requirements
Defects in TiO 2 Crystals
 , March 13-15, 2013, Hong Kong Defects in TiO 2 Crystals Richard Rivera, Arvids Stashans 1 Abstract-TiO 2 crystals, anatase and rutile, have been studied using Density Functional Theory (DFT) and the Generalized
, March 13-15, 2013, Hong Kong Defects in TiO 2 Crystals Richard Rivera, Arvids Stashans 1 Abstract-TiO 2 crystals, anatase and rutile, have been studied using Density Functional Theory (DFT) and the Generalized
Molecular Mechanics: The Ab Initio Foundation
 Molecular Mechanics: The Ab Initio Foundation Ju Li GEM4 Summer School 2006 Cell and Molecular Mechanics in BioMedicine August 7 18, 2006, MIT, Cambridge, MA, USA 2 Outline Why are electrons quantum? Born-Oppenheimer
Molecular Mechanics: The Ab Initio Foundation Ju Li GEM4 Summer School 2006 Cell and Molecular Mechanics in BioMedicine August 7 18, 2006, MIT, Cambridge, MA, USA 2 Outline Why are electrons quantum? Born-Oppenheimer
Ab-initio Electronic Structure Calculations β and γ KNO 3 Energetic Materials
 ISSN 0974-9373 Vol. 15 No.3 (2011) Journal of International Academy of Physical Sciences pp. 337-344 Ab-initio Electronic Structure Calculations of α, β and γ KNO 3 Energetic Materials Pradeep Jain and
ISSN 0974-9373 Vol. 15 No.3 (2011) Journal of International Academy of Physical Sciences pp. 337-344 Ab-initio Electronic Structure Calculations of α, β and γ KNO 3 Energetic Materials Pradeep Jain and
Ab Initio modelling of structural and electronic. Matt Probert University of York
 Ab Initio modelling of structural and electronic properties of semiconductors Matt Probert University of York http://www-users.york.ac.uk/~mijp1 Overview of Talk What is Ab Initio? What can we model? How
Ab Initio modelling of structural and electronic properties of semiconductors Matt Probert University of York http://www-users.york.ac.uk/~mijp1 Overview of Talk What is Ab Initio? What can we model? How
Tight-Binding Model of Electronic Structures
 Tight-Binding Model of Electronic Structures Consider a collection of N atoms. The electronic structure of this system refers to its electronic wave function and the description of how it is related to
Tight-Binding Model of Electronic Structures Consider a collection of N atoms. The electronic structure of this system refers to its electronic wave function and the description of how it is related to
Before we start: Important setup of your Computer
 Before we start: Important setup of your Computer change directory: cd /afs/ictp/public/shared/smr2475./setup-config.sh logout login again 1 st Tutorial: The Basics of DFT Lydia Nemec and Oliver T. Hofmann
Before we start: Important setup of your Computer change directory: cd /afs/ictp/public/shared/smr2475./setup-config.sh logout login again 1 st Tutorial: The Basics of DFT Lydia Nemec and Oliver T. Hofmann
CHAPTER 3 WIEN2k. Chapter 3 : WIEN2k 50
 CHAPTER 3 WIEN2k WIEN2k is one of the fastest and reliable simulation codes among computational methods. All the computational work presented on lanthanide intermetallic compounds has been performed by
CHAPTER 3 WIEN2k WIEN2k is one of the fastest and reliable simulation codes among computational methods. All the computational work presented on lanthanide intermetallic compounds has been performed by
How to run SIESTA. Introduction to input & output files
 How to run SIESTA Introduction to input & output files Linear-scaling DFT based on Numerical Atomic Orbitals (NAOs) Born-Oppenheimer DFT Pseudopotentials Numerical atomic orbitals relaxations, MD, phonons.
How to run SIESTA Introduction to input & output files Linear-scaling DFT based on Numerical Atomic Orbitals (NAOs) Born-Oppenheimer DFT Pseudopotentials Numerical atomic orbitals relaxations, MD, phonons.
Density-functional Theory Applied To Problems In Catalysis And Electrochemistry
 University of Central Florida Electronic Theses and Dissertations Masters Thesis (Open Access) Density-functional Theory Applied To Problems In Catalysis And Electrochemistry 2006 Santosh Kumar University
University of Central Florida Electronic Theses and Dissertations Masters Thesis (Open Access) Density-functional Theory Applied To Problems In Catalysis And Electrochemistry 2006 Santosh Kumar University
An Introduction to Quantum Chemistry and Potential Energy Surfaces. Benjamin G. Levine
 An Introduction to Quantum Chemistry and Potential Energy Surfaces Benjamin G. Levine This Week s Lecture Potential energy surfaces What are they? What are they good for? How do we use them to solve chemical
An Introduction to Quantum Chemistry and Potential Energy Surfaces Benjamin G. Levine This Week s Lecture Potential energy surfaces What are they? What are they good for? How do we use them to solve chemical
Recent advances in development of single-point orbital-free kinetic energy functionals
 PacifiChem 2010 p. 1/29 Recent advances in development of single-point orbital-free kinetic energy functionals Valentin V. Karasiev vkarasev@qtp.ufl.edu Quantum Theory Project, Departments of Physics and
PacifiChem 2010 p. 1/29 Recent advances in development of single-point orbital-free kinetic energy functionals Valentin V. Karasiev vkarasev@qtp.ufl.edu Quantum Theory Project, Departments of Physics and
Quantum Modeling of Solids: Basic Properties
 1.021, 3.021, 10.333, 22.00 : Introduction to Modeling and Simulation : Spring 2011 Part II Quantum Mechanical Methods : Lecture 5 Quantum Modeling of Solids: Basic Properties Jeffrey C. Grossman Department
1.021, 3.021, 10.333, 22.00 : Introduction to Modeling and Simulation : Spring 2011 Part II Quantum Mechanical Methods : Lecture 5 Quantum Modeling of Solids: Basic Properties Jeffrey C. Grossman Department
DFT EXERCISES. FELIPE CERVANTES SODI January 2006
 DFT EXERCISES FELIPE CERVANTES SODI January 2006 http://www.csanyi.net/wiki/space/dftexercises Dr. Gábor Csányi 1 Hydrogen atom Place a single H atom in the middle of a largish unit cell (start with a
DFT EXERCISES FELIPE CERVANTES SODI January 2006 http://www.csanyi.net/wiki/space/dftexercises Dr. Gábor Csányi 1 Hydrogen atom Place a single H atom in the middle of a largish unit cell (start with a
DFT: Exchange-Correlation
 DFT: Local functionals, exact exchange and other post-dft methods Stewart Clark University of Outline Introduction What is exchange and correlation? Quick tour of XC functionals (Semi-)local: LDA, PBE,
DFT: Local functionals, exact exchange and other post-dft methods Stewart Clark University of Outline Introduction What is exchange and correlation? Quick tour of XC functionals (Semi-)local: LDA, PBE,
André Schleife Department of Materials Science and Engineering
 André Schleife Department of Materials Science and Engineering Yesterday you (should have) learned this: http://upload.wikimedia.org/wikipedia/commons/e/ea/ Simple_Harmonic_Motion_Orbit.gif 1. deterministic
André Schleife Department of Materials Science and Engineering Yesterday you (should have) learned this: http://upload.wikimedia.org/wikipedia/commons/e/ea/ Simple_Harmonic_Motion_Orbit.gif 1. deterministic
Exchange Correlation Functional Investigation of RT-TDDFT on a Sodium Chloride. Dimer. Philip Straughn
 Exchange Correlation Functional Investigation of RT-TDDFT on a Sodium Chloride Dimer Philip Straughn Abstract Charge transfer between Na and Cl ions is an important problem in physical chemistry. However,
Exchange Correlation Functional Investigation of RT-TDDFT on a Sodium Chloride Dimer Philip Straughn Abstract Charge transfer between Na and Cl ions is an important problem in physical chemistry. However,
On the adaptive finite element analysis of the Kohn-Sham equations
 On the adaptive finite element analysis of the Kohn-Sham equations Denis Davydov, Toby Young, Paul Steinmann Denis Davydov, LTM, Erlangen, Germany August 2015 Denis Davydov, LTM, Erlangen, Germany College
On the adaptive finite element analysis of the Kohn-Sham equations Denis Davydov, Toby Young, Paul Steinmann Denis Davydov, LTM, Erlangen, Germany August 2015 Denis Davydov, LTM, Erlangen, Germany College
Problem Set 2: First-Principles Energy Methods
 Problem Set 2: First-Principles Energy Methods Problem 1 (10 points): Convergence of absolute energies with respect to cutoff energies. A Using the Quantum ESPRESSO PWscf package, calculate the energy
Problem Set 2: First-Principles Energy Methods Problem 1 (10 points): Convergence of absolute energies with respect to cutoff energies. A Using the Quantum ESPRESSO PWscf package, calculate the energy
Solid State Theory: Band Structure Methods
 Solid State Theory: Band Structure Methods Lilia Boeri Wed., 11:15-12:45 HS P3 (PH02112) http://itp.tugraz.at/lv/boeri/ele/ Plan of the Lecture: DFT1+2: Hohenberg-Kohn Theorem and Kohn and Sham equations.
Solid State Theory: Band Structure Methods Lilia Boeri Wed., 11:15-12:45 HS P3 (PH02112) http://itp.tugraz.at/lv/boeri/ele/ Plan of the Lecture: DFT1+2: Hohenberg-Kohn Theorem and Kohn and Sham equations.
Ab initio phonon calculations in mixed systems
 Ab initio phonon calculations in mixed systems Andrei Postnikov apostnik@uos.de Outline: Experiment vs. ab initio theory Ways of theory: linear response and frozen phonon approaches Applications: Be x
Ab initio phonon calculations in mixed systems Andrei Postnikov apostnik@uos.de Outline: Experiment vs. ab initio theory Ways of theory: linear response and frozen phonon approaches Applications: Be x
FYS Vår 2017 (Kondenserte fasers fysikk)
") FYS3410 - Vår 2017 (Kondenserte fasers fysikk) http://www.uio.no/studier/emner/matnat/fys/fys3410/v16/index.html Pensum: Introduction to Solid State Physics by Charles Kittel (Chapters 1-9, 11, 17, 18,
FYS3410 - Vår 2017 (Kondenserte fasers fysikk) http://www.uio.no/studier/emner/matnat/fys/fys3410/v16/index.html Pensum: Introduction to Solid State Physics by Charles Kittel (Chapters 1-9, 11, 17, 18,
6: Plane waves, unit cells, k- points and all that
 The Nuts and Bolts of First-Principles Simulation 6: Plane waves, unit cells, k- points and all that Durham, 6th- 13th December 2001 CASTEP Developers Group with support from the ESF ψ k Network Overview
The Nuts and Bolts of First-Principles Simulation 6: Plane waves, unit cells, k- points and all that Durham, 6th- 13th December 2001 CASTEP Developers Group with support from the ESF ψ k Network Overview
Electrons in periodic potential
 Chapter 9 Electrons in periodic potential The computation of electronic states in a solid is a nontrivial problem. A great simplification can be achieved if the solid is a crystal, i.e. if it can be described
Chapter 9 Electrons in periodic potential The computation of electronic states in a solid is a nontrivial problem. A great simplification can be achieved if the solid is a crystal, i.e. if it can be described
Ab Initio Structure Determination and Property Characterization of High-Pressure Ca-O Compounds and Li2(OH)Br Crystals
Br Crystals") UNLV Theses, Dissertations, Professional Papers, and Capstones 5-1-2016 Ab Initio Structure Determination and Property Characterization of High-Pressure Ca-O Compounds and Li2(OH)Br Crystals Christopher
UNLV Theses, Dissertations, Professional Papers, and Capstones 5-1-2016 Ab Initio Structure Determination and Property Characterization of High-Pressure Ca-O Compounds and Li2(OH)Br Crystals Christopher
Practical calculations using first-principles QM Convergence, convergence, convergence
 Practical calculations using first-principles QM Convergence, convergence, convergence Keith Refson STFC Rutherford Appleton Laboratory September 18, 2007 Results of First-Principles Simulations..........................................................
Practical calculations using first-principles QM Convergence, convergence, convergence Keith Refson STFC Rutherford Appleton Laboratory September 18, 2007 Results of First-Principles Simulations..........................................................
Comparison of various abinitio codes used in periodic calculations
 Comparison of various abinitio codes used in periodic calculations 1 Prof.P. Ravindran, Department of Physics, Central University of Tamil Nadu, India & Center for Materials Science and Nanotechnology,
Comparison of various abinitio codes used in periodic calculations 1 Prof.P. Ravindran, Department of Physics, Central University of Tamil Nadu, India & Center for Materials Science and Nanotechnology,
VALENCE Hilary Term 2018
 VALENCE Hilary Term 2018 8 Lectures Prof M. Brouard Valence is the theory of the chemical bond Outline plan 1. The Born-Oppenheimer approximation 2. Bonding in H + 2 the LCAO approximation 3. Many electron
VALENCE Hilary Term 2018 8 Lectures Prof M. Brouard Valence is the theory of the chemical bond Outline plan 1. The Born-Oppenheimer approximation 2. Bonding in H + 2 the LCAO approximation 3. Many electron