Lecture 3, January 9, 2015 Bonding in H2+
|
|
|
- Anabel Reeves
- 5 years ago
- Views:
Transcription
1 Lecture 3, January 9, 2015 Bonding in H2+ Elements of Quantum Chemistry with Applications to Chemical Bonding and Properties of Molecules and Solids Course number: Ch125a; Room 147 Noyes Hours: 11-11:50am Monday, Wednesday, Friday William A. Goddard, III, 316 Beckman Institute, x3093 Charles and Mary Ferkel Professor of Chemistry, Materials Science, and Applied Physics, California Institute of Technology Special Instructor: Julius Su Teaching Assistants: Hai Xiao Mark Fornace 1
2 Now consider H 2 + molecule Bring a proton up to an H atom to form H 2 + Is the molecule bound? That is does it have a lower energy at finite R than at R = Several possibilities Electron is on the left proton, L L r L e r R R Electron is on the right proton, R Or we could combine them At R = these are have the same energy, but not for finite R In QM we always want the wavefunction with the lowest energy. Question: which combination is lowest? Ch125a-Goddard-L01 2
3 Combine Atomic Orbitals for H 2 + molecule Symmetric Two extreme combination possibilities Antisymmetric combination Which is best (lowest energy)? the D g = Sqrt[2(1+S)] and D u = Sqrt[2(1-S)] factors above are the constants needed to ensure that <Φ g Φ g > = Φ g Φ g dxdydz = 1 (normalized) <Φ u Φ u > = Φ u Φ u dxdydz = 1 (normalized) I will usually eschew writing such factors, leaving them to be understood Ch125a-Goddard-L01 3
4 Energies of of H 2 + Molecule g state is bound since starting the atoms at any distance between arrows, the molecule will stay bonded, with atoms vibrating forth and back Ungood state: u Good state: g LCAO = Linear Combination of Atomic Orbitals Ch125a-Goddard-L01 4
5 But WHY is the g state bound? Ch125a-Goddard-L01 5
6 Common rational : But WHY is the g state bound? Superimposing two orbitals and squaring to get the probability leads to moving charge into the bond region. This negative charge in the bond region attracts the two positive nuclei leading to the bond Sounds reasonable Ch125a-Goddard-L01 6
7 Common rational : But WHY is the g state bound? Superimposing two orbitals and squaring to get the probability leads to moving charge into the bond region. This negative charge in the bond region attracts the two positive nuclei leading to the bond Sounds reasonable, but increasing the density in bond region è decrease density near atoms, thus moves electrons from very attractive region near nuclei to less attractive region near bond midpoint, this INCREASES the PE Ch125a-Goddard-L01 7
8 Compare change in density with local PE function The local PE for the electron is PE(r) = -1/r a 1/r b lowered at the bond midpoint from the value of a single atom Since the best local PE is still near the nucleus The Φ g = χ L + χ R wavefunction moves charge to the bond region AT THE EXPENSE of the charge near the nuclei, causing an increase in the PE, and opposing bonding 8 Ch125a-Goddard-L01
9 The PE of H 2 + for g and u states The total PE of H 2 + for the Φ g = χ L + χ R and Φ u = χ L - χ R wavefunctions (relative to the values of V g = V u = -1 h 0 at R = ) Ch125a-Goddard-L01 9
10 If the bonding is not due to the PE, then it must be KE Ch125a-Goddard-L01 10
11 If the bonding is not due to the PE, then it must be KE The shape of the Φ g = χ L + χ R and Φ u = χ L - χ R wavefunctions compared to the pure atomic orbital (all normalized to a total probability of one). Ch125a-Goddard-L01 We see a dramatic decrease in the slope of the g orbital along the bond axis compared to the atomic orbital. This leads to a dramatic decrease in KE compared to the atomic orbital This decrease arises only in the bond region. It is this decrease in KE that is responsible for the bonding in H
12 The KE of g and u wavefunctions for H 2 + Use top part of 2-7 The change in the KE as a function of distance for the g and u wavefunctions of H 2 + (relative to the value at R= of KE g =KE u =+0.5 h 0 ) Ch125a-Goddard-L01 Comparison of the g and u wavefunctions of H + 2 (near the optimum bond distance for the g state), showing why g is so bonding and u is so antibonding 12
13 Why does KEg has an optimum? Ch125a-Goddard-L01 R too short leads to a big decrease in slope but over a very short region, è little bonding R is too large leads to a decrease in slope over a long region, but the change in slope is very small è little bonding Optimum bonding occurs when there is a large region where both atomic orbitals have large slopes in the opposite directions (contragradient). This leads to optimum bonding 13
14 KE dominates PE Ch125a-Goddard-L01 Ungood state: u Good state: g Changes in the total KE and PE for the g and u wavefunctions of H 2 + (relative to values at R= of KE :+0.5 h 0 PE: -1.0 h 0 E: -0.5 h 0 The g state is bound between R~1.5 a 0 and (starting the atoms at any distance in this range leads to atoms vibrating forth and back. Exciting to the u state leads to dissociation 14
15 KE dominates PE, leading to g as ground state Calculations show this, but how could we have predicted that g is better than u without calculations? Answer: the nodal theorem: The ground state of a QM systems has no nodes. Thus g state lower E than u state Ch125a-Goddard-L01 15
16 The nodal Theorem The ground state of a system has no nodes (more properly, the ground state never changes sign). This is often quite useful in reasoning about wavefunctions. For example the nodal theorem immediately implies that the g wavefunction for H 2 + is the ground state (not the u state) Ch125a-Goddard-L01 16
17 The nodal Theorem 1D Schrodinger equation, H Ψ k = E k Ψ k One dimensional: H =- ½ d 2 /dx 2 + V(x) Consider the best possible eigenstate of H with a node, Ψ 1 and construct a nonnegative function Ө 0 = Ψ 1 as in b For every value of x, V(x)[Ψ 1 ] 2 = V(x)[Ө 0 ] 2 so that a b Φ 1 Ө 0 V 0 = [Ө 0 ] * V(x)[Ө 0 ] = [Ψ 1 ] * V(x)[Ψ 1 ] 2 = V 1 c Φ 0 Also dө 0 /dx 2 = d Ψ 1 /dx 2 for every value of x except the single point at which the node occurs. Thus T 0 = ½ dө 0 /dx 2 = ½ dψ 1 /dx 2 = T 1. Hence E 0 = T 0 + V 0 = T 1 + V 1 = E 1. Ch125a-Goddard-L01 17
18 The nodal Theorem 1D We just showed that for the best possible eigenfunction of H with a node, H Ψ 1 = E 1 Ψ 1 a Φ 1 Ө 0 = Φ 1 has the same energy as Ψ 1 E 0 = T 0 + V 0 = T 1 + V 1 = E 1. However Ө 0 is just a special case of a nodeless wavefunction that happens to go to 0 at one point. In general not requiring it so go to zero will lower the energy. Thus we could smooth out Ө 0 in the region of the node as in c, decreasing the KE and lowering the energy. Thus the optimum nodeless wavefunction Ψ 0 leads to E 0 < E 1. Only for a potential so repulsive at some point, that all wavefunctions are 0, do we get E 0 = E 1 b Ө 0 c Φ 0 Ch125a-Goddard-L01 18
19 The nodal Theorem for excited states in 1D For one-dimensional finite systems, we can order all eigenstates by the number of nodes E 0 < E 1 < E 2... E n < E n+1 (where a sufficiently singular potential can lead to an = sign ) The argument is the same as for the ground state. Consider best wavefunction Ψ n with n nodes and flip the sign at one node to get a wavefunction Ө n-1 that changes sign only n-1 times. Show that E n-1 = E n But Ө n-1 is not the best with n-1 sign changes. Thus we can smooth out Ө n-1 in the region of the extra node to decrease the KE and lower the energy for the Ψ n-1,. Thus the optimum n-1 node wavefunction leads to E n-1 < E n. Ch125a-Goddard-L01 19
20 The nodal Theorem 3D In 2D a wavefunction that changes size once will have a line of points with Ψ 1 =0 (a nodal line) For 3D there will be a 2D nodal surface with Ψ 1 =0. In 3D the same argument as for 1D shows that the ground state is nodeless. We start with Ψ 1 the best possible eigenstate with a nodal surface and construct a nonnegative function Ө 0 = Ψ 1 For every value of x,y,z, V(x,y,z)[Ψ 1 ] 2 = V(x,y.z)[Ө 0 ] 2 so that V 0 = [Ө 0 ] * V(xyz)[Ө 0 ] = [Ψ 1 ] * V(xyz)[Ψ 1 ] 2 = V 1 Also Ө 0 2 = Ψ 1 2 everywhere except along a 2D plane (0 vol.) Thus T 0 = ½ Ө 0 2 = ½ Ψ 1 2 = T 1. Hence E 0 = T 0 + V 0 = T 1 + V 1 = E 1. As before E 1 is the best possible energy for an eigenstate with a nodal plane. However Ө 0 can be improved by smoothing Thus the optimum nodeless wavefunction Φ 0 leads to E 0 < E 1. Ch125a-Goddard-L01 20
21 The nodal Theorem for excited states in 3D For 2D and 3D, one cannot order all eigenstates by the number of nodes. Thus consider the 2D wavefunctions Φ 00 Φ Φ 01 Φ 20 Φ 11 Φ It is easy to show as in the earlier analysis that E 00 < E 10 < E 20 < E 21 E 00 < E 01 < E 11 < E 21 But the nodal argument does not indicate the relative energies of E 10 and E 20 versus E 01 Ch125a-Goddard-L01 21
22 Back to H 2 + g state u state Nodal theorem è The ground state must be the g wavefunction Ch125a-Goddard-L01 22
23 Ch120a-Goddard-L02 Symmetry Considerations The operation of inversion (denoted as ^ I ) through the origin of a coordinate system changes the coordinates as x è -x y è -y z è -z Taking the origin of the coordinate system as the bond midpoint, inversion changes the electronic coordinates as illustrated. After inversion the electron is remains a distance of r a from one of the nuclei and r b from the other, but their identities are transposed. Thus the potential energy, v(r) is unchanged by inversion, v(-x,-y,-z) v( ^ I r) = v(r) where r is considered as the 3D vector with components x,y,z Thus we say that v(r) is invariant under inversion 23
24 Considering symmetry Under inversion the kinetic energy terms in the Hamiltonian are also unchanged Hence the full Hamiltonian is invariant under inversion h(-x,-y,-z) h( ^ I r) = h(x,y,z) = h(r) Now consider that we had solved hφ=εφ for the exact wavefunction φ and apply the inversion to both sides ^ I hφ= ε ^ I φ Which we can rewrite as h(-r)φ(-r)=εφ(-r) But h(-r) = h(r) because of inversion symmetry Thus h(r)φ(-r)=εφ(-r) Ch120a-Goddard-L02 24
25 4 th postulate of QM Consider the exact eigenstate of a system HΦ = EΦ and multiply the Schrödinger equation by some CONSTANT phase factor (independent of position and time) exp(iα) = e iα e iα HΦ = H (e iα Φ) = E (e iα Φ) Thus Φ and (e iα Φ) lead to identical properties and we consider them to describe exactly the same state. 4th Postulate: wavefunctions differing only by a constant phase factor describe the same state Ch120a-Goddard-L02 25
26 Continue with symmetry discussion We just derived that because h(-r) = h(r) Then for any eigenfunction φ(r) of h h(r)φ(r) = εφ(r) It must be that φ(-r) also is an eigenfunction of the same h with the same energy, ε. h(r)φ(-r) = εφ(-r) There are two distinct possibilities here. (1) There is only ONE state with energy ε or (2) There is more than ONE state with energy ε. Ch120a-Goddard-L02 26
27 Case 1: Nondegenerate states Consider case 1 first in which only one state has energy ε. By the 4th postulate of QM, then at most, φ(-r) and φ(r) can differ by a phase factor, φ(-r) = e iα φ(r) Now consider what happens if the inversion is applied twice x è -x è x; y è -y è y z è -z è z Nothing changes! Thus applying inversion twice is equivalent to doing nothing. This do-nothing operator is called einheit (German for identity or unit) and denoted as e ^. We write ^ I ^ I = ( ^ I ) 2 = e ^ and say that the inversion operator is of order two Since ^ I φ(r) = φ(-r) = e iα φ(r) then applying a second inversion leads to ( ^ I ) 2 φ(r) = ^ I φ(-r) = ^ I e iα φ(r) = e i2α φ(r) But ( ^ I ) 2 φ(r) = e ^ φ(r) = φ(r); Thus e i2α φ(r) =φ(r) Hence e i2α = 1 or e iα = ±1 Ch120a-Goddard-L02 27
28 Case 1: Nondegenerate states: conclusion Since ^ I φ(r) = φ(-r) = e iα φ(r) and e iα = ±1 We conclude that either ^ I φ g (r) = + φ g (r) g for gerade or even or ^ I φ u (r) = - φ u (r) u for ungerade or odd Thus for a system with inversion, each nondegenerate eigenstate is of either g or u inversion symmetry. Indeed for H + φ 2 we found g (r) φ u (r) Ch120a-Goddard-L02 28
29 Case 2: Now consider the case where the state is degenerate In this case φ and ^ I φ can be independent states (not proportional to each other). Thus we can combine them to form two new wavefunctions and Since ^ I ( ) = ^ I φ + φ ^ and I ^ ( ) = I φ - φ Both are eigenfunctions of h with the same energy) we see that Since h(r)φ(r) = εφ(r) and h(r)φ(-r) = εφ(-r) We see that h(r)φ g (r) = εφ g (r) and h(r)φ u (r) = εφ u (r) Thus every degenerate eigenfunction of h can be rewritten so that each state is either g or u We say that all eigenstates of systems with inversion symmetry are either g or u Ch120a-Goddard-L02 29
 g for gerade or even φ u (r) = - φ u (r) u for ungerade or odd")
30 Apply symmetry principle to H 2 + Ch120a-Goddard-L02 φ g (r) = + φ g (r) g for gerade or even φ u (r) = - φ u (r) u for ungerade or odd 30
 The Hamiltonian for H2+ is (1) (2)")
31 More quantitative description of H2+ (from Ch chapter 5 (chapter 7) The Hamiltonian for H2+ is (1) (2) 31
32 Cylindrical Coordinates for H2+ where 32
33 Elliptic coordinates The solutions are where 33
34 Bates numerical solutions 34
35 Angular eigenfunctions for diatomic molecules 35
36 Inversion symmetry 36

37 The eigenstates of H2+ 37
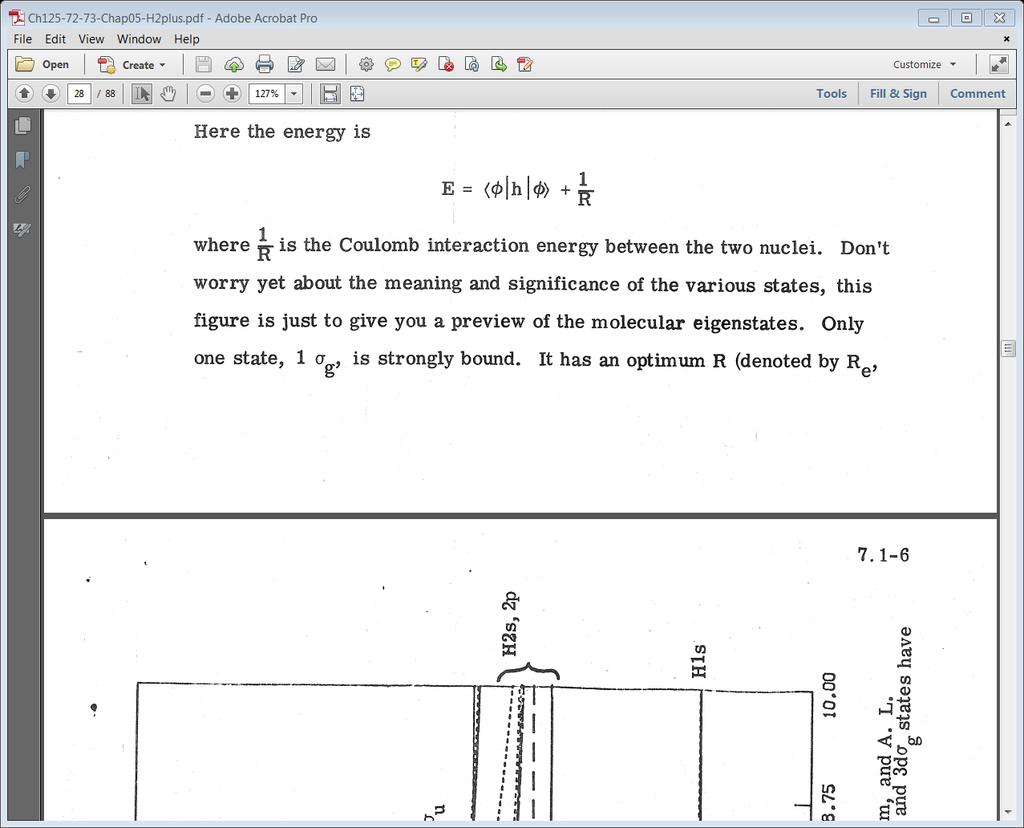
, 246, 215 (1953)] The 2pπ u and 3dσ g states have very small minima (~0.01 ev at large R (~7a0 and 9 a0)) H2s, 2p D e =0.")
38 Figure The total energy for various states of H2+ [DR Bates, K Ledsham, and AL Stewart; Phil Trans. Roy. Soc. (London), 246, 215 (1953)] The 2pπ u and 3dσ g states have very small minima (~0.01 ev at large R (~7a0 and 9 a0)) H2s, 2p D e =0.6 h 0 H1s R e =2.0 a 0 38
39 More quantitative description of the bonding in H2+ See (really chapter 5) Ch The Linear Combination of Atomic Orbitals (LCAO) description of bonding is where χ a and χ b are atomic orbitals on the left and right and S is their overlap The charge densities can be partitioned into a classical part (superposition of densities) and the exchange part (4) 39

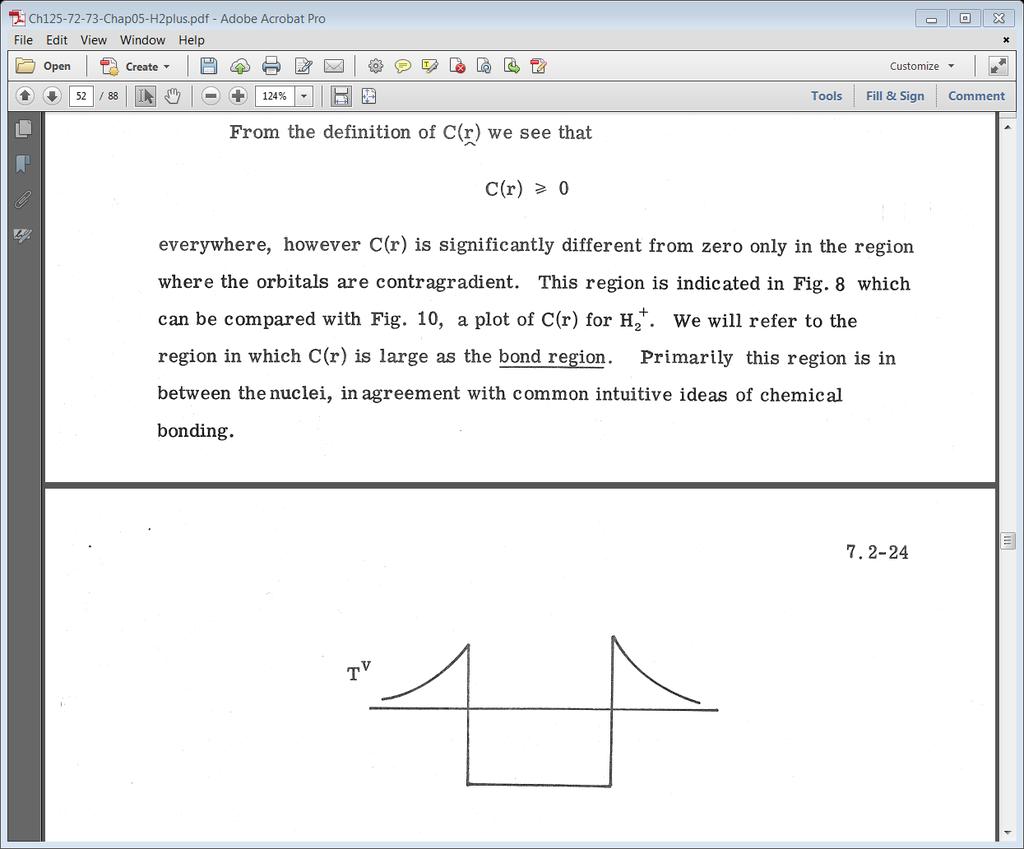
40 Classical and exchange density 40
41 Example numbers With normalization we get 41
42 Relation of bonding to classical and exchange density Table 1 shows that (12) which causes 42

43 Note ΔT is negative Note ΔV is positive 43
44 The scale optimized LCAO wavefunction The Virial Theorem paradox with ζ = 1 as for the H atom. Now we will optimize ζ For H2+ the potential energy (PE) operator is So that the scaled operator is if we also scale R. and hence the new PE is V ζ = ζ V 1 (note V is negative) and the new KE is T ζ = ζ 2 T 1 Thus the total scaled E = ζ 2 T 1 + ζ V 1 leading to an optimum value of 2ζ opt T 1 + V 1 = 0 or ζ opt= - V 1 / 2T 1 = Thus the optimum T opt = V 12 /4T 1 and Vopt = - V 12 /2T 1 Satisfying the Virial theorem, Eopt = -T opt = V opt /2, as expected. Starting with R = 2.5 a 0 (the Re for LCAO) this leads to a new R e = 2.02 a 0 which is nearly the exact value 44
 Re-LCAO Exact R e")
45 Table Energies for LCAO description of H2+ (atomic units) Re-LCAO Exact R e R exact = 2.0 D=Bond E D 1 = D opt = D exact =
46 Discussion of bonding of orbitals Optimizing the scale of the LCAO wavefunction leads to an excellent Re=2.02 a0 compared to the exact value of 2.0a0 The Bond energy, D, increases by 32% to D=0.086 compared to Dexact = 0.10 h0 Adding a 2p z basis function on each nucleus would lead to a further substantial improvement The fact that the Virial theorem must be satisfied at Re and at R= shows that the total bond energy D = -ΔT = ½ ΔV Confuses some to believe that the bonding is due to the PE In fact as shown clearly in Fig. 4, it is the large decrease in KE due to superposition of orbitals on two centers that is responsible for the bonding This big decrease in KE allows the contraction at shorter R of orbitals about each center until the Virial Theorem is satisfied at Re. This is illustrated in Fig
 T exact V LCAO E exact T LCAO E LCAO V exact For R>5a0, the LCAO and exact values of T, V, E are similar with the big decrease in T LCAO")
47 Energies for H2+ (Fig of Ch ) T exact V LCAO E exact T LCAO E LCAO V exact For R>5a0, the LCAO and exact values of T, V, E are similar with the big decrease in T LCAO dominating the bond For short R<4a0, the wavefunction contracts about each nucleus (ζ>1) Leading finally to the Virial Theorem values at R e 47
 to bonding in H2+ Evaluated at R=2a0 The decrease of 0.")
48 LCAO orbitals Figure Contributions (h0) to bonding in H2+ Evaluated at R=2a0 The decrease of 0.117h0 in T for the LCAO wavefunction at Re=2a0 dominates the final bond of 0.1 ho

49 Must have two hands to clap In trying to understand the origin of binding it is important to discuss simultaneously both the states that bind and the states that antibind (It takes 2 hands to clap) Our analysis using LCAO wavefunction does this since we examine both the g and the u states as in Fig. 7 Here no matter which atomic-like orbitals that we use, the g state is below the u state and the energy change from R= is dominated by the decrease in KE, showing that it is this decrease in KE that is responsible for the bond Fig. 7 (Fig from Ch =-73) 49
50 Classical and exchange contributions (14) where (15) For LCAO, T cl = 0.5, thus it is T x which gets more negative as R decreases that is responsible for bonding in H2+ 50
51 51
52 T x so that However the dot product quantity (19) 52

53 T x which Region over which negative, enhancing bonding is 53
54 contragradience (20) for all R. where The remainder of Tx is (21) (28) (29) LCAO (30) 54

55 55
56 Fig. 7,2-10 Fig. 7,2-9 Plots along the bond axis 56
57 Analytic analysis of bonding and antibonding in H2+ (5) 57

58 Analysis of Eg and Eu of H2+ where 58

59 We saw above Thus So that τ dominates the bonding and antibonding Put in analytic form for S and τ here Both S and τ go to zero exponentially with R so we can consider the bonding to be proportional to S 59
60 Energies (hartree) for the LCAO wavefunctions of H2+ 60

61 Energies (hartree) for the LCAO wavefunctions of H2+ 61
62 Higher excited states of H2+ 62

 (14)")
63 Splitting of degeneracies (7.3.2a Ch ) where (13b) (14) Hence 63
64 Since Breaking the 2s-2pz degeneracy and We have However (19) with energies and (20) 64
65 we obtain we obtain 65


66 2s,2p States of H perturbed by a proton at distance R. 66
67 where E a = E cl τ/(1-s) 67

68 68
69 Figure 3. Molecular orbitals from 2s,2p states 69

70 Total Energies of 2s,2p excited states of H2+ Fig from Ch

71 Electronic Energies of 2s,2p excited states of H2+ Fig a from Ch , same as Fig except omit 1/R nuclear term This shows the correlation from R~ to R=0 71

72 Enlargement of Fig. 5a 72
 Table 7.")
73 Energies for H2+ (Hartree) Table
74 The Molecular orbitals of 2s,2p states H c Fig. 6a Fig. 6a 74
75 Fig. 7 nodal patterns and names of 2s,2p derived states of H2+ United atom name at the right, separated atom name at the left and standard MO name 75
76 Energies near the united atom limit 7.3.2d 76
77 Correlation Diagrams


78 Correlation of MOs with Separated atom states 78

79 Fig. 9 Correlation diagram for H2+ 79
80 Heteronuclear Molecules 80
81 81

82 82
83 (6) 83
84 which leads to (7) 84
85 (11) (12) 85

86 Summary
 wag46 The Role of Kinetic Energy in Chemical Binding: I. The Nonclassical or Exchange Kinetic Energy C. W. Wilson, Jr., and W. A. Goddard III Theor. Chim. Acta.")
87 The Role of Kinetic Energy in Chemical Binding: II. Contragradience W. A. Goddard III and C. W. Wilson, Jr. Theor. Chim. Acta. 26, 211 (1972) wag46 The Role of Kinetic Energy in Chemical Binding: I. The Nonclassical or Exchange Kinetic Energy C. W. Wilson, Jr., and W. A. Goddard III Theor. Chim. Acta. 26, 195 (1972) wag45 has not been widely adopted 87
Lecture 2, January 9, 2012 Origin Binding H 2 +,nodal thm, H 2, QM post 2-4
 Lecture 2, January 9, 2012 Origin Binding H 2 +,nodal thm, H 2, QM post 2-4 Nature of the Chemical Bond with applications to catalysis, materials science, nanotechnology, surface science, bioinorganic
Lecture 2, January 9, 2012 Origin Binding H 2 +,nodal thm, H 2, QM post 2-4 Nature of the Chemical Bond with applications to catalysis, materials science, nanotechnology, surface science, bioinorganic
Lecture 1, January 4, 2012 Elements QM, stability H, H2+
 Lecture 1, January 4, 2012 Elements QM, stability H, H2+ Nature of the Chemical Bond with applications to catalysis, materials science, nanotechnology, surface science, bioinorganic chemistry, and energy
Lecture 1, January 4, 2012 Elements QM, stability H, H2+ Nature of the Chemical Bond with applications to catalysis, materials science, nanotechnology, surface science, bioinorganic chemistry, and energy
Lecture 1, January 3, 2011 Elements QM, stability H, H2+
 Lecture 1, January 3, 2011 Elements QM, stability H, H2+ Nature of the Chemical Bond with applications to catalysis, materials science, nanotechnology, surface science, bioinorganic chemistry, and energy
Lecture 1, January 3, 2011 Elements QM, stability H, H2+ Nature of the Chemical Bond with applications to catalysis, materials science, nanotechnology, surface science, bioinorganic chemistry, and energy
Lecture 4, January 12, 2015 Bonding in H2
 Lecture 4, January 12, 2015 Bonding in H2 Elements of Quantum Chemistry with Applications to Chemical Bonding and Properties of Molecules and Solids Course number: Ch125a; Room 147 Noyes Hours: 11-11:50am
Lecture 4, January 12, 2015 Bonding in H2 Elements of Quantum Chemistry with Applications to Chemical Bonding and Properties of Molecules and Solids Course number: Ch125a; Room 147 Noyes Hours: 11-11:50am
Lecture 16, February 25, 2015 Metallic bonding
 Lecture 16, February 25, 2015 Metallic bonding Elements of Quantum Chemistry with Applications to Chemical Bonding and Properties of Molecules and Solids Course number: Ch125a; Room 115 BI Hours: 11-11:50am
Lecture 16, February 25, 2015 Metallic bonding Elements of Quantum Chemistry with Applications to Chemical Bonding and Properties of Molecules and Solids Course number: Ch125a; Room 115 BI Hours: 11-11:50am
Lecture 14 February 7, 2014 Symmetry
 Lecture 14 February 7, 2014 Symmetry Nature of the Chemical Bond with applications to catalysis, materials science, nanotechnology, surface science, bioinorganic chemistry, and energy Course number: Ch120a
Lecture 14 February 7, 2014 Symmetry Nature of the Chemical Bond with applications to catalysis, materials science, nanotechnology, surface science, bioinorganic chemistry, and energy Course number: Ch120a
3: Many electrons. Orbital symmetries. l =2 1. m l
 3: Many electrons Orbital symmetries Atomic orbitals are labelled according to the principal quantum number, n, and the orbital angular momentum quantum number, l. Electrons in a diatomic molecule experience
3: Many electrons Orbital symmetries Atomic orbitals are labelled according to the principal quantum number, n, and the orbital angular momentum quantum number, l. Electrons in a diatomic molecule experience
Nature of the Chemical Bond with applications to catalysis, materials science, nanotechnology, surface science, bioinorganic chemistry, and energy
 Lecture 13, October 31, 2016 Transition metals, Pd and Pt Nature of the Chemical Bond with applications to catalysis, materials science, nanotechnology, surface science, bioinorganic chemistry, and energy
Lecture 13, October 31, 2016 Transition metals, Pd and Pt Nature of the Chemical Bond with applications to catalysis, materials science, nanotechnology, surface science, bioinorganic chemistry, and energy
Lecture 11 January 30, Transition metals, Pd and Pt
 Lecture 11 January 30, 2011 Nature of the Chemical Bond with applications to catalysis, materials science, nanotechnology, surface science, bioinorganic chemistry, and energy Course number: Ch120a Hours:
Lecture 11 January 30, 2011 Nature of the Chemical Bond with applications to catalysis, materials science, nanotechnology, surface science, bioinorganic chemistry, and energy Course number: Ch120a Hours:
General Physical Chemistry II
 General Physical Chemistry II Lecture 10 Aleksey Kocherzhenko October 7, 2014" Last time " promotion" Promotion and hybridization" [He] 2s 2 2p x 1 2p y 1 2p z0 " 2 unpaired electrons" [He] 2s 1 2p x 1
General Physical Chemistry II Lecture 10 Aleksey Kocherzhenko October 7, 2014" Last time " promotion" Promotion and hybridization" [He] 2s 2 2p x 1 2p y 1 2p z0 " 2 unpaired electrons" [He] 2s 1 2p x 1
MO theory is better for spectroscopy (Exited State Properties; Ionization)
") CHEM 2060 Lecture 25: MO Theory L25-1 Molecular Orbital Theory (MO theory) VB theory treats bonds as electron pairs. o There is a real emphasis on this point (over-emphasis actually). VB theory is very
CHEM 2060 Lecture 25: MO Theory L25-1 Molecular Orbital Theory (MO theory) VB theory treats bonds as electron pairs. o There is a real emphasis on this point (over-emphasis actually). VB theory is very
σ u * 1s g - gerade u - ungerade * - antibonding σ g 1s
 One of these two states is a repulsive (dissociative) state. Other excited states can be constructed using linear combinations of other orbitals. Some will be binding and others will be repulsive. Thus
One of these two states is a repulsive (dissociative) state. Other excited states can be constructed using linear combinations of other orbitals. Some will be binding and others will be repulsive. Thus
QUANTUM MECHANICS AND MOLECULAR STRUCTURE
 6 QUANTUM MECHANICS AND MOLECULAR STRUCTURE 6.1 Quantum Picture of the Chemical Bond 6.2 Exact Molecular Orbital for the Simplest Molecule: H + 2 6.3 Molecular Orbital Theory and the Linear Combination
6 QUANTUM MECHANICS AND MOLECULAR STRUCTURE 6.1 Quantum Picture of the Chemical Bond 6.2 Exact Molecular Orbital for the Simplest Molecule: H + 2 6.3 Molecular Orbital Theory and the Linear Combination
Molecular Structure Both atoms and molecules are quantum systems
 Molecular Structure Both atoms and molecules are quantum systems We need a method of describing molecules in a quantum mechanical way so that we can predict structure and properties The method we use is
Molecular Structure Both atoms and molecules are quantum systems We need a method of describing molecules in a quantum mechanical way so that we can predict structure and properties The method we use is
Lecture 16 February 20 Transition metals, Pd and Pt
 Lecture 16 February 20 Transition metals, Pd and Pt Nature of the Chemical Bond with applications to catalysis, materials science, nanotechnology, surface science, bioinorganic chemistry, and energy Course
Lecture 16 February 20 Transition metals, Pd and Pt Nature of the Chemical Bond with applications to catalysis, materials science, nanotechnology, surface science, bioinorganic chemistry, and energy Course
CHAPTER 11 MOLECULAR ORBITAL THEORY
 CHAPTER 11 MOLECULAR ORBITAL THEORY Molecular orbital theory is a conceptual extension of the orbital model, which was so successfully applied to atomic structure. As was once playfuly remarked, a molecue
CHAPTER 11 MOLECULAR ORBITAL THEORY Molecular orbital theory is a conceptual extension of the orbital model, which was so successfully applied to atomic structure. As was once playfuly remarked, a molecue
Nature of the Chemical Bond with applications to catalysis, materials science, nanotechnology, surface science, bioinorganic chemistry, and energy
 Lecture 12, October 21, 2016 Transition metals Heme-Fe Nature of the Chemical Bond with applications to catalysis, materials science, nanotechnology, surface science, bioinorganic chemistry, and energy
Lecture 12, October 21, 2016 Transition metals Heme-Fe Nature of the Chemical Bond with applications to catalysis, materials science, nanotechnology, surface science, bioinorganic chemistry, and energy
2 Electronic structure theory
 Electronic structure theory. Generalities.. Born-Oppenheimer approximation revisited In Sec..3 (lecture 3) the Born-Oppenheimer approximation was introduced (see also, for instance, [Tannor.]). We are
Electronic structure theory. Generalities.. Born-Oppenheimer approximation revisited In Sec..3 (lecture 3) the Born-Oppenheimer approximation was introduced (see also, for instance, [Tannor.]). We are
VALENCE Hilary Term 2018
 VALENCE Hilary Term 2018 8 Lectures Prof M. Brouard Valence is the theory of the chemical bond Outline plan 1. The Born-Oppenheimer approximation 2. Bonding in H + 2 the LCAO approximation 3. Many electron
VALENCE Hilary Term 2018 8 Lectures Prof M. Brouard Valence is the theory of the chemical bond Outline plan 1. The Born-Oppenheimer approximation 2. Bonding in H + 2 the LCAO approximation 3. Many electron
Lecture 9-10 January 25-27, 2012 Rules for Chem. React. - Woodward-Hoffmann
 Lecture 9-10 January 25-27, 2012 Rules for Chem. React. - Woodward-Hoffmann Nature of the Chemical Bond with applications to catalysis, materials science, nanotechnology, surface science, bioinorganic
Lecture 9-10 January 25-27, 2012 Rules for Chem. React. - Woodward-Hoffmann Nature of the Chemical Bond with applications to catalysis, materials science, nanotechnology, surface science, bioinorganic
Molecular-Orbital Theory
 Prof. Dr. I. Nasser atomic and molecular physics -551 (T-11) April 18, 01 Molecular-Orbital Theory You have to explain the following statements: 1- Helium is monatomic gas. - Oxygen molecule has a permanent
Prof. Dr. I. Nasser atomic and molecular physics -551 (T-11) April 18, 01 Molecular-Orbital Theory You have to explain the following statements: 1- Helium is monatomic gas. - Oxygen molecule has a permanent
CHAPTER 10 Tight-Binding Model
 CHAPTER 0 Tight-Binding Model Linear Combination of Atomic Orbitals (LCAO) Application to Bands from s-levels General Features of Tight-Binding Levels Wannier Functions 6 a : S S P 3S Core FE Semicore
CHAPTER 0 Tight-Binding Model Linear Combination of Atomic Orbitals (LCAO) Application to Bands from s-levels General Features of Tight-Binding Levels Wannier Functions 6 a : S S P 3S Core FE Semicore
Chemistry 2000 Lecture 1: Introduction to the molecular orbital theory
 Chemistry 2000 Lecture 1: Introduction to the molecular orbital theory Marc R. Roussel January 5, 2018 Marc R. Roussel Introduction to molecular orbitals January 5, 2018 1 / 24 Review: quantum mechanics
Chemistry 2000 Lecture 1: Introduction to the molecular orbital theory Marc R. Roussel January 5, 2018 Marc R. Roussel Introduction to molecular orbitals January 5, 2018 1 / 24 Review: quantum mechanics
Electron States of Diatomic Molecules
 IISER Pune March 2018 Hamiltonian for a Diatomic Molecule The hamiltonian for a diatomic molecule can be considered to be made up of three terms Ĥ = ˆT N + ˆT el + ˆV where ˆT N is the kinetic energy operator
IISER Pune March 2018 Hamiltonian for a Diatomic Molecule The hamiltonian for a diatomic molecule can be considered to be made up of three terms Ĥ = ˆT N + ˆT el + ˆV where ˆT N is the kinetic energy operator
Be H. Delocalized Bonding. Localized Bonding. σ 2. σ 1. Two (sp-1s) Be-H σ bonds. The two σ bonding MO s in BeH 2. MO diagram for BeH 2
 Be-H σ bonds. The two σ bonding MO s in BeH 2. MO diagram for BeH 2") The Delocalized Approach to Bonding: The localized models for bonding we have examined (Lewis and VBT) assume that all electrons are restricted to specific bonds between atoms or in lone pairs. In contrast,
The Delocalized Approach to Bonding: The localized models for bonding we have examined (Lewis and VBT) assume that all electrons are restricted to specific bonds between atoms or in lone pairs. In contrast,
The symmetry properties & relative energies of atomic orbitals determine how they react to form molecular orbitals. These molecular orbitals are then
 1 The symmetry properties & relative energies of atomic orbitals determine how they react to form molecular orbitals. These molecular orbitals are then filled with the available electrons according to
1 The symmetry properties & relative energies of atomic orbitals determine how they react to form molecular orbitals. These molecular orbitals are then filled with the available electrons according to
Lecture 18, March 2, 2015 graphene, bucky balls, bucky tubes
 Lecture 18, March 2, 2015 graphene, bucky balls, bucky tubes Elements of Quantum Chemistry with Applications to Chemical Bonding and Properties of Molecules and Solids Course number: Ch125a; Room 115 BI
Lecture 18, March 2, 2015 graphene, bucky balls, bucky tubes Elements of Quantum Chemistry with Applications to Chemical Bonding and Properties of Molecules and Solids Course number: Ch125a; Room 115 BI
(1/2) M α 2 α, ˆTe = i. 1 r i r j, ˆV NN = α>β
 M α 2 α, ˆTe = i. 1 r i r j, ˆV NN = α>β") Chemistry 26 Spectroscopy Week # The Born-Oppenheimer Approximation, H + 2. Born-Oppenheimer approximation As for atoms, all information about a molecule is contained in the wave function Ψ, which is the
Chemistry 26 Spectroscopy Week # The Born-Oppenheimer Approximation, H + 2. Born-Oppenheimer approximation As for atoms, all information about a molecule is contained in the wave function Ψ, which is the
Molecular orbitals, potential energy surfaces and symmetry
 Molecular orbitals, potential energy surfaces and symmetry mathematical presentation of molecular symmetry group theory spectroscopy valence theory molecular orbitals Wave functions Hamiltonian: electronic,
Molecular orbitals, potential energy surfaces and symmetry mathematical presentation of molecular symmetry group theory spectroscopy valence theory molecular orbitals Wave functions Hamiltonian: electronic,
Quantum Analogs Chapter 3 Student Manual
 Quantum Analogs Chapter 3 Student Manual Broken Symmetry in the Spherical Resonator and Modeling a Molecule Professor Rene Matzdorf Universitaet Kassel 3. Broken symmetry in the spherical resonator and
Quantum Analogs Chapter 3 Student Manual Broken Symmetry in the Spherical Resonator and Modeling a Molecule Professor Rene Matzdorf Universitaet Kassel 3. Broken symmetry in the spherical resonator and
CHEMISTRY Topic #1: Bonding What Holds Atoms Together? Spring 2012 Dr. Susan Lait
 CHEMISTRY 2000 Topic #1: Bonding What Holds Atoms Together? Spring 2012 Dr. Susan Lait Why Do Bonds Form? An energy diagram shows that a bond forms between two atoms if the overall energy of the system
CHEMISTRY 2000 Topic #1: Bonding What Holds Atoms Together? Spring 2012 Dr. Susan Lait Why Do Bonds Form? An energy diagram shows that a bond forms between two atoms if the overall energy of the system
Chemistry 6 (9 am section) Spring Covalent Bonding
 Spring Covalent Bonding") Chemistry 6 (9 am section) Spring 000 Covalent Bonding The stability of the bond in molecules such as H, O, N and F is associated with a sharing (equal) of the VALENCE ELECTRONS between the BONDED ATOMS.
Chemistry 6 (9 am section) Spring 000 Covalent Bonding The stability of the bond in molecules such as H, O, N and F is associated with a sharing (equal) of the VALENCE ELECTRONS between the BONDED ATOMS.
Molecular Physics. Attraction between the ions causes the chemical bond.
 Molecular Physics A molecule is a stable configuration of electron(s) and more than one nucleus. Two types of bonds: covalent and ionic (two extremes of same process) Covalent Bond Electron is in a molecular
Molecular Physics A molecule is a stable configuration of electron(s) and more than one nucleus. Two types of bonds: covalent and ionic (two extremes of same process) Covalent Bond Electron is in a molecular
PHYSICAL CHEMISTRY I. Chemical Bonds
 PHYSICAL CHEMISTRY I Chemical Bonds Review The QM description of bonds is quite good Capable of correctly calculating bond energies and reaction enthalpies However it is quite complicated and sometime
PHYSICAL CHEMISTRY I Chemical Bonds Review The QM description of bonds is quite good Capable of correctly calculating bond energies and reaction enthalpies However it is quite complicated and sometime
5.111 Principles of Chemical Science
 MIT OpenCourseWare http://ocw.mit.edu 5.111 Principles of Chemical Science Fall 2008 For information about citing these materials or our Terms of Use, visit: http://ocw.mit.edu/terms. 5.111 Lecture Summary
MIT OpenCourseWare http://ocw.mit.edu 5.111 Principles of Chemical Science Fall 2008 For information about citing these materials or our Terms of Use, visit: http://ocw.mit.edu/terms. 5.111 Lecture Summary
Structure of diatomic molecules
 Structure of diatomic molecules January 8, 00 1 Nature of molecules; energies of molecular motions Molecules are of course atoms that are held together by shared valence electrons. That is, most of each
Structure of diatomic molecules January 8, 00 1 Nature of molecules; energies of molecular motions Molecules are of course atoms that are held together by shared valence electrons. That is, most of each
Chemistry 2. Lecture 1 Quantum Mechanics in Chemistry
 Chemistry 2 Lecture 1 Quantum Mechanics in Chemistry Your lecturers 8am Assoc. Prof Timothy Schmidt Room 315 timothy.schmidt@sydney.edu.au 93512781 12pm Assoc. Prof. Adam J Bridgeman Room 222 adam.bridgeman@sydney.edu.au
Chemistry 2 Lecture 1 Quantum Mechanics in Chemistry Your lecturers 8am Assoc. Prof Timothy Schmidt Room 315 timothy.schmidt@sydney.edu.au 93512781 12pm Assoc. Prof. Adam J Bridgeman Room 222 adam.bridgeman@sydney.edu.au
Lecture 19: Building Atoms and Molecules
 Lecture 19: Building Atoms and Molecules +e r n = 3 n = 2 n = 1 +e +e r y even Lecture 19, p 1 Today Nuclear Magnetic Resonance Using RF photons to drive transitions between nuclear spin orientations in
Lecture 19: Building Atoms and Molecules +e r n = 3 n = 2 n = 1 +e +e r y even Lecture 19, p 1 Today Nuclear Magnetic Resonance Using RF photons to drive transitions between nuclear spin orientations in
Lecture 9 Electronic Spectroscopy
 Lecture 9 Electronic Spectroscopy Molecular Orbital Theory: A Review - LCAO approximaton & AO overlap - Variation Principle & Secular Determinant - Homonuclear Diatomic MOs - Energy Levels, Bond Order
Lecture 9 Electronic Spectroscopy Molecular Orbital Theory: A Review - LCAO approximaton & AO overlap - Variation Principle & Secular Determinant - Homonuclear Diatomic MOs - Energy Levels, Bond Order
In this lecture we will understand how the molecular orbitals are formed from the interaction of atomic orbitals.
 Lecture 7 Title: Understanding of Molecular Orbital Page-1 In this lecture we will understand how the molecular orbitals are formed from the interaction of atomic orbitals. We will see how the electrons
Lecture 7 Title: Understanding of Molecular Orbital Page-1 In this lecture we will understand how the molecular orbitals are formed from the interaction of atomic orbitals. We will see how the electrons
The Postulates of Quantum Mechanics Common operators in QM: Potential Energy. Often depends on position operator: Kinetic Energy 1-D case: 3-D case
 The Postulates of Quantum Mechanics Common operators in QM: Potential Energy Often depends on position operator: Kinetic Energy 1-D case: 3-D case Time Total energy = Hamiltonian To find out about the
The Postulates of Quantum Mechanics Common operators in QM: Potential Energy Often depends on position operator: Kinetic Energy 1-D case: 3-D case Time Total energy = Hamiltonian To find out about the
H 2 in the minimal basis
 H 2 in the minimal basis Alston J. Misquitta Centre for Condensed Matter and Materials Physics Queen Mary, University of London January 27, 2016 Overview H 2 : The 1-electron basis. The two-electron basis
H 2 in the minimal basis Alston J. Misquitta Centre for Condensed Matter and Materials Physics Queen Mary, University of London January 27, 2016 Overview H 2 : The 1-electron basis. The two-electron basis
5.111 Lecture Summary #13 Monday, October 6, 2014
 5.111 Lecture Summary #13 Monday, October 6, 2014 Readings for today: Section 3.8 3.11 Molecular Orbital Theory (Same in 5 th and 4 th ed.) Read for Lecture #14: Sections 3.4, 3.5, 3.6 and 3.7 Valence
5.111 Lecture Summary #13 Monday, October 6, 2014 Readings for today: Section 3.8 3.11 Molecular Orbital Theory (Same in 5 th and 4 th ed.) Read for Lecture #14: Sections 3.4, 3.5, 3.6 and 3.7 Valence
Quantum Chemistry. NC State University. Lecture 5. The electronic structure of molecules Absorption spectroscopy Fluorescence spectroscopy
 Quantum Chemistry Lecture 5 The electronic structure of molecules Absorption spectroscopy Fluorescence spectroscopy NC State University 3.5 Selective absorption and emission by atmospheric gases (source:
Quantum Chemistry Lecture 5 The electronic structure of molecules Absorption spectroscopy Fluorescence spectroscopy NC State University 3.5 Selective absorption and emission by atmospheric gases (source:
Brief review of Quantum Mechanics (QM)
") Brief review of Quantum Mechanics (QM) Note: This is a collection of several formulae and facts that we will use throughout the course. It is by no means a complete discussion of QM, nor will I attempt
Brief review of Quantum Mechanics (QM) Note: This is a collection of several formulae and facts that we will use throughout the course. It is by no means a complete discussion of QM, nor will I attempt
Quantum mechanics can be used to calculate any property of a molecule. The energy E of a wavefunction Ψ evaluated for the Hamiltonian H is,
 Chapter : Molecules Quantum mechanics can be used to calculate any property of a molecule The energy E of a wavefunction Ψ evaluated for the Hamiltonian H is, E = Ψ H Ψ Ψ Ψ 1) At first this seems like
Chapter : Molecules Quantum mechanics can be used to calculate any property of a molecule The energy E of a wavefunction Ψ evaluated for the Hamiltonian H is, E = Ψ H Ψ Ψ Ψ 1) At first this seems like
Lecture 9: Molecular Orbital theory for hydrogen molecule ion
 Lecture 9: Molecular Orbital theory for hydrogen molecule ion Molecular Orbital Theory for Hydrogen Molecule Ion We have seen that the Schrödinger equation cannot be solved for many electron systems. The
Lecture 9: Molecular Orbital theory for hydrogen molecule ion Molecular Orbital Theory for Hydrogen Molecule Ion We have seen that the Schrödinger equation cannot be solved for many electron systems. The
Chapter 4 Symmetry and Chemical Bonding
 Chapter 4 Symmetry and Chemical Bonding 4.1 Orbital Symmetries and Overlap 4.2 Valence Bond Theory and Hybrid Orbitals 4.3 Localized and Delocalized Molecular Orbitals 4.4 MX n Molecules with Pi-Bonding
Chapter 4 Symmetry and Chemical Bonding 4.1 Orbital Symmetries and Overlap 4.2 Valence Bond Theory and Hybrid Orbitals 4.3 Localized and Delocalized Molecular Orbitals 4.4 MX n Molecules with Pi-Bonding
we have to deal simultaneously with the motion of the two heavy particles, the nuclei
 157 Lecture 6 We now turn to the structure of molecules. Our first cases will be the e- quantum mechanics of the two simplest molecules, the hydrogen molecular ion, H +, a r A r B one electron molecule,
157 Lecture 6 We now turn to the structure of molecules. Our first cases will be the e- quantum mechanics of the two simplest molecules, the hydrogen molecular ion, H +, a r A r B one electron molecule,
Symmetry and Molecular Orbitals (I)
") Symmetry and Molecular Orbitals (I) Simple Bonding Model http://chiuserv.ac.nctu.edu.tw/~htchiu/chemistry/fall-2005/chemical-bonds.htm Lewis Structures Octet Rule Resonance Formal Charge Oxidation Number
Symmetry and Molecular Orbitals (I) Simple Bonding Model http://chiuserv.ac.nctu.edu.tw/~htchiu/chemistry/fall-2005/chemical-bonds.htm Lewis Structures Octet Rule Resonance Formal Charge Oxidation Number
An Introduction to Quantum Chemistry and Potential Energy Surfaces. Benjamin G. Levine
 An Introduction to Quantum Chemistry and Potential Energy Surfaces Benjamin G. Levine This Week s Lecture Potential energy surfaces What are they? What are they good for? How do we use them to solve chemical
An Introduction to Quantum Chemistry and Potential Energy Surfaces Benjamin G. Levine This Week s Lecture Potential energy surfaces What are they? What are they good for? How do we use them to solve chemical
Bonding in Molecules Prof John McGrady Michaelmas Term 2009
 Bonding in Molecules Prof John McGrady Michaelmas Term 2009 6 lectures building on material presented in Introduction to Molecular Orbitals (HT Year 1). Provides a basis for analysing the shapes, properties,
Bonding in Molecules Prof John McGrady Michaelmas Term 2009 6 lectures building on material presented in Introduction to Molecular Orbitals (HT Year 1). Provides a basis for analysing the shapes, properties,
CHEMISTRY 333 Fall 2006 Exam 3, in class
 CHEMISTRY 333 Fall 006 Exam 3, in class Name SHOW YOUR WORK. Mathematica & calculators. You may use Mathematica or a calculator to do arithmetic. Do not use Mathematica for any higher-level manipulations
CHEMISTRY 333 Fall 006 Exam 3, in class Name SHOW YOUR WORK. Mathematica & calculators. You may use Mathematica or a calculator to do arithmetic. Do not use Mathematica for any higher-level manipulations
Lecture 19: Building Atoms and Molecules
 Lecture 19: Building Atoms and Molecules +e r n = 3 n = 2 n = 1 +e +e r ψ even Lecture 19, p 1 Today Nuclear Magnetic Resonance Using RF photons to drive transitions between nuclear spin orientations in
Lecture 19: Building Atoms and Molecules +e r n = 3 n = 2 n = 1 +e +e r ψ even Lecture 19, p 1 Today Nuclear Magnetic Resonance Using RF photons to drive transitions between nuclear spin orientations in
MOLECULAR STRUCTURE. The general molecular Schrödinger equation, apart from electron spin effects, is. nn ee en
 MOLECULAR STRUCTURE The Born-Oppenheimer Approximation The general molecular Schrödinger equation, apart from electron spin effects, is ( ) + + V + V + V =E nn ee en T T ψ ψ n e where the operators in
MOLECULAR STRUCTURE The Born-Oppenheimer Approximation The general molecular Schrödinger equation, apart from electron spin effects, is ( ) + + V + V + V =E nn ee en T T ψ ψ n e where the operators in
CHEM3023: Spins, Atoms and Molecules
 CHEM3023: Spins, Atoms and Molecules Lecture 3 The Born-Oppenheimer approximation C.-K. Skylaris Learning outcomes Separate molecular Hamiltonians to electronic and nuclear parts according to the Born-Oppenheimer
CHEM3023: Spins, Atoms and Molecules Lecture 3 The Born-Oppenheimer approximation C.-K. Skylaris Learning outcomes Separate molecular Hamiltonians to electronic and nuclear parts according to the Born-Oppenheimer
Diatomic Molecules. 14th May Chemical Bonds in Diatomic Molecules: Overlaps and Delocalization of Electrons
 Diatomic Molecules 14th May 2009 1 Chemical Bonds in Diatomic Molecules: Overlaps and Delocalization of Electrons 1.1 H + 2 Molecule Consider the process where 2 atomic nuclei and associated electron (1
Diatomic Molecules 14th May 2009 1 Chemical Bonds in Diatomic Molecules: Overlaps and Delocalization of Electrons 1.1 H + 2 Molecule Consider the process where 2 atomic nuclei and associated electron (1
wbt Λ = 0, 1, 2, 3, Eq. (7.63)
") 7.2.2 Classification of Electronic States For all diatomic molecules the coupling approximation which best describes electronic states is analogous to the Russell- Saunders approximation in atoms The orbital
7.2.2 Classification of Electronic States For all diatomic molecules the coupling approximation which best describes electronic states is analogous to the Russell- Saunders approximation in atoms The orbital
Rethinking Hybridization
 Rethinking Hybridization For more than 60 years, one of the most used concepts to come out of the valence bond model developed by Pauling was that of hybrid orbitals. The ideas of hybridization seemed
Rethinking Hybridization For more than 60 years, one of the most used concepts to come out of the valence bond model developed by Pauling was that of hybrid orbitals. The ideas of hybridization seemed
Symmetry III: Molecular Orbital Theory. Reading: Shriver and Atkins and , 6.10
 Lecture 9 Symmetry III: Molecular Orbital Theory Reading: Shriver and Atkins 2.7-2.9 and g 6.6-6.7, 6.10 The orbitals of molecules H H The electron energy in each H atom is -13.6 ev below vacuum. What
Lecture 9 Symmetry III: Molecular Orbital Theory Reading: Shriver and Atkins 2.7-2.9 and g 6.6-6.7, 6.10 The orbitals of molecules H H The electron energy in each H atom is -13.6 ev below vacuum. What
Lecture 13 February 1, 2011 Pd and Pt, MH + bonding, metathesis
 Lecture 13 February 1, 2011 Pd and Pt, MH + bonding, metathesis Nature of the Chemical Bond with applications to catalysis, materials science, nanotechnology, surface science, bioinorganic chemistry, and
Lecture 13 February 1, 2011 Pd and Pt, MH + bonding, metathesis Nature of the Chemical Bond with applications to catalysis, materials science, nanotechnology, surface science, bioinorganic chemistry, and
Lecture 15 February 15, 2013 Transition metals
 Lecture 15 February 15, 2013 Transition metals Nature of the Chemical Bond with applications to catalysis, materials science, nanotechnology, surface science, bioinorganic chemistry, and energy Course
Lecture 15 February 15, 2013 Transition metals Nature of the Chemical Bond with applications to catalysis, materials science, nanotechnology, surface science, bioinorganic chemistry, and energy Course
Be H. Delocalized Bonding. Localized Bonding. σ 2. σ 1. Two (sp-1s) Be-H σ bonds. The two σ bonding MO s in BeH 2. MO diagram for BeH 2
 Be-H σ bonds. The two σ bonding MO s in BeH 2. MO diagram for BeH 2") The Delocalized Approach to Bonding: The localized models for bonding we have examined (Lewis and VBT) assume that all electrons are restricted to specific bonds between atoms or in lone pairs. In contrast,
The Delocalized Approach to Bonding: The localized models for bonding we have examined (Lewis and VBT) assume that all electrons are restricted to specific bonds between atoms or in lone pairs. In contrast,
Lecture 10. Born-Oppenheimer approximation LCAO-MO application to H + The potential energy surface MOs for diatomic molecules. NC State University
 Chemistry 431 Lecture 10 Diatomic molecules Born-Oppenheimer approximation LCAO-MO application to H + 2 The potential energy surface MOs for diatomic molecules NC State University Born-Oppenheimer approximation
Chemistry 431 Lecture 10 Diatomic molecules Born-Oppenheimer approximation LCAO-MO application to H + 2 The potential energy surface MOs for diatomic molecules NC State University Born-Oppenheimer approximation
Time part of the equation can be separated by substituting independent equation
 Lecture 9 Schrödinger Equation in 3D and Angular Momentum Operator In this section we will construct 3D Schrödinger equation and we give some simple examples. In this course we will consider problems where
Lecture 9 Schrödinger Equation in 3D and Angular Momentum Operator In this section we will construct 3D Schrödinger equation and we give some simple examples. In this course we will consider problems where
Lecture 14 February 3, 2014 Rules for Chem. React. - Woodward-Hoffmann
 Lecture 14 February 3, 2014 Rules for Chem. React. - Woodward-Hoffmann Nature of the Chemical Bond with applications to catalysis, materials science, nanotechnology, surface science, bioinorganic chemistry,
Lecture 14 February 3, 2014 Rules for Chem. React. - Woodward-Hoffmann Nature of the Chemical Bond with applications to catalysis, materials science, nanotechnology, surface science, bioinorganic chemistry,
Molecular Orbitals. Based on Inorganic Chemistry, Miessler and Tarr, 4 th edition, 2011, Pearson Prentice Hall
 Molecular Orbitals Based on Inorganic Chemistry, Miessler and Tarr, 4 th edition, 2011, Pearson Prentice Hall Images from Miessler and Tarr Inorganic Chemistry 2011 obtained from Pearson Education, Inc.
Molecular Orbitals Based on Inorganic Chemistry, Miessler and Tarr, 4 th edition, 2011, Pearson Prentice Hall Images from Miessler and Tarr Inorganic Chemistry 2011 obtained from Pearson Education, Inc.
Molecular Bonding. Molecular Schrödinger equation. r - nuclei s - electrons. M j = mass of j th nucleus m 0 = mass of electron
 Molecular onding Molecular Schrödinger equation r - nuclei s - electrons 1 1 W V r s j i j1 M j m i1 M j = mass of j th nucleus m = mass of electron j i Laplace operator for nuclei Laplace operator for
Molecular onding Molecular Schrödinger equation r - nuclei s - electrons 1 1 W V r s j i j1 M j m i1 M j = mass of j th nucleus m = mass of electron j i Laplace operator for nuclei Laplace operator for
Chapter 4 Symmetry and Chemical Bonding
 Chapter 4 Symmetry and Chemical Bonding 4.1 Orbital Symmetries and Overlap 4.2 Valence Bond Theory and Hybrid Orbitals 4.3 Localized and Delocalized Molecular Orbitals 4.4 MX n Molecules with Pi-Bonding
Chapter 4 Symmetry and Chemical Bonding 4.1 Orbital Symmetries and Overlap 4.2 Valence Bond Theory and Hybrid Orbitals 4.3 Localized and Delocalized Molecular Orbitals 4.4 MX n Molecules with Pi-Bonding
Chem 442 Review for Exam 2. Exact separation of the Hamiltonian of a hydrogenic atom into center-of-mass (3D) and relative (3D) components.
 and relative (3D) components.") Chem 44 Review for Exam Hydrogenic atoms: The Coulomb energy between two point charges Ze and e: V r Ze r Exact separation of the Hamiltonian of a hydrogenic atom into center-of-mass (3D) and relative
Chem 44 Review for Exam Hydrogenic atoms: The Coulomb energy between two point charges Ze and e: V r Ze r Exact separation of the Hamiltonian of a hydrogenic atom into center-of-mass (3D) and relative
Lecture 8 January 24, 2013 GaAs crystal surfaces, n-p dopants Si
 Lecture 8 January 24, 2013 Ga crystal surfaces, n-p dopants Si Nature of the Chemical Bond with applications to catalysis, materials science, nanotechnology, surface science, bioinornic chemistry, and
Lecture 8 January 24, 2013 Ga crystal surfaces, n-p dopants Si Nature of the Chemical Bond with applications to catalysis, materials science, nanotechnology, surface science, bioinornic chemistry, and
Chapter IV: Electronic Spectroscopy of diatomic molecules
 Chapter IV: Electronic Spectroscopy of diatomic molecules IV.2.1 Molecular orbitals IV.2.1.1. Homonuclear diatomic molecules The molecular orbital (MO) approach to the electronic structure of diatomic
Chapter IV: Electronic Spectroscopy of diatomic molecules IV.2.1 Molecular orbitals IV.2.1.1. Homonuclear diatomic molecules The molecular orbital (MO) approach to the electronic structure of diatomic
ECE440 Nanoelectronics. Lecture 07 Atomic Orbitals
 ECE44 Nanoelectronics Lecture 7 Atomic Orbitals Atoms and atomic orbitals It is instructive to compare the simple model of a spherically symmetrical potential for r R V ( r) for r R and the simplest hydrogen
ECE44 Nanoelectronics Lecture 7 Atomic Orbitals Atoms and atomic orbitals It is instructive to compare the simple model of a spherically symmetrical potential for r R V ( r) for r R and the simplest hydrogen
Same idea for polyatomics, keep track of identical atom e.g. NH 3 consider only valence electrons F(2s,2p) H(1s)
 H(1s)") XIII 63 Polyatomic bonding -09 -mod, Notes (13) Engel 16-17 Balance: nuclear repulsion, positive e-n attraction, neg. united atom AO ε i applies to all bonding, just more nuclei repulsion biggest at low
XIII 63 Polyatomic bonding -09 -mod, Notes (13) Engel 16-17 Balance: nuclear repulsion, positive e-n attraction, neg. united atom AO ε i applies to all bonding, just more nuclei repulsion biggest at low
Theoretical Chemistry - Level II - Practical Class Molecular Orbitals in Diatomics
 Theoretical Chemistry - Level II - Practical Class Molecular Orbitals in Diatomics Problem 1 Draw molecular orbital diagrams for O 2 and O 2 +. E / ev dioxygen molecule, O 2 dioxygenyl cation, O 2 + 25
Theoretical Chemistry - Level II - Practical Class Molecular Orbitals in Diatomics Problem 1 Draw molecular orbital diagrams for O 2 and O 2 +. E / ev dioxygen molecule, O 2 dioxygenyl cation, O 2 + 25
( R) MOLECULES: VALENCE BOND DESCRIPTION VALENCE BOND APPROXIMATION METHOD. Example: H 2. at some fixed R, repeat for many different values
 MOLECULES: VALENCE BOND DESCRIPTION VALENCE BOND APPROXIMATION METHOD. Example: H 2. at some fixed R, repeat for many different values") 5.6 004 Lecture #9 page MOLECULES: VLENCE OND DESCRIPTION VLENCE OND PPROXIMTION METHOD Example: H H = + + r r r r r R Determine E ( R) at some fixed R, repeat for many different values Electronic Schrodinger
5.6 004 Lecture #9 page MOLECULES: VLENCE OND DESCRIPTION VLENCE OND PPROXIMTION METHOD Example: H H = + + r r r r r R Determine E ( R) at some fixed R, repeat for many different values Electronic Schrodinger
NH 3 H 2 O N 2. Why do they make chemical bonds? Molecular Orbitals
 N 2 NH 3 H 2 O Why do they make chemical bonds? 5 Molecular Orbitals Why do they make chemical bonds? Stabilization Bond energy Types of Chemical Bonds Metallic Bond Ionic Bond Covalent Bond Covalent Bond
N 2 NH 3 H 2 O Why do they make chemical bonds? 5 Molecular Orbitals Why do they make chemical bonds? Stabilization Bond energy Types of Chemical Bonds Metallic Bond Ionic Bond Covalent Bond Covalent Bond
Section 10: Many Particle Quantum Mechanics Solutions
 Physics 143a: Quantum Mechanics I Section 10: Many Particle Quantum Mechanics Solutions Spring 015, Harvard Here is a summary of the most important points from this week (with a few of my own tidbits),
Physics 143a: Quantum Mechanics I Section 10: Many Particle Quantum Mechanics Solutions Spring 015, Harvard Here is a summary of the most important points from this week (with a few of my own tidbits),
26 Group Theory Basics
 26 Group Theory Basics 1. Reference: Group Theory and Quantum Mechanics by Michael Tinkham. 2. We said earlier that we will go looking for the set of operators that commute with the molecular Hamiltonian.
26 Group Theory Basics 1. Reference: Group Theory and Quantum Mechanics by Michael Tinkham. 2. We said earlier that we will go looking for the set of operators that commute with the molecular Hamiltonian.
Chemistry 483 Lecture Topics Fall 2009
 Chemistry 483 Lecture Topics Fall 2009 Text PHYSICAL CHEMISTRY A Molecular Approach McQuarrie and Simon A. Background (M&S,Chapter 1) Blackbody Radiation Photoelectric effect DeBroglie Wavelength Atomic
Chemistry 483 Lecture Topics Fall 2009 Text PHYSICAL CHEMISTRY A Molecular Approach McQuarrie and Simon A. Background (M&S,Chapter 1) Blackbody Radiation Photoelectric effect DeBroglie Wavelength Atomic
Lecture 6. Tight-binding model
 Lecture 6 Tight-binding model In Lecture 3 we discussed the Krönig-Penny model and we have seen that, depending on the strength of the potential barrier inside the unit cell, the electrons can behave like
Lecture 6 Tight-binding model In Lecture 3 we discussed the Krönig-Penny model and we have seen that, depending on the strength of the potential barrier inside the unit cell, the electrons can behave like
Chancellor Phyllis Wise invites you to a birthday party!
 Chancellor Phyllis Wise invites you to a birthday party! 50 years ago, Illinois alumnus Nick Holonyak Jr. demonstrated the first visible light-emitting diode (LED) while working at GE. Holonyak returned
Chancellor Phyllis Wise invites you to a birthday party! 50 years ago, Illinois alumnus Nick Holonyak Jr. demonstrated the first visible light-emitting diode (LED) while working at GE. Holonyak returned
Chemistry 120A 2nd Midterm. 1. (36 pts) For this question, recall the energy levels of the Hydrogenic Hamiltonian (1-electron):
 For this question, recall the energy levels of the Hydrogenic Hamiltonian (1-electron):") April 6th, 24 Chemistry 2A 2nd Midterm. (36 pts) For this question, recall the energy levels of the Hydrogenic Hamiltonian (-electron): E n = m e Z 2 e 4 /2 2 n 2 = E Z 2 /n 2, n =, 2, 3,... where Ze is
April 6th, 24 Chemistry 2A 2nd Midterm. (36 pts) For this question, recall the energy levels of the Hydrogenic Hamiltonian (-electron): E n = m e Z 2 e 4 /2 2 n 2 = E Z 2 /n 2, n =, 2, 3,... where Ze is
Lecture 6 January 18, 2012 CC Bonds diamond, ΔHf, Group additivity
 Lecture 6 January 18, 2012 CC Bonds diamond, ΔHf, Group additivity Nature of the Chemical Bond with applications to catalysis, materials science, nanotechnology, surface science, bioinorganic chemistry,
Lecture 6 January 18, 2012 CC Bonds diamond, ΔHf, Group additivity Nature of the Chemical Bond with applications to catalysis, materials science, nanotechnology, surface science, bioinorganic chemistry,
MOLECULES. ENERGY LEVELS electronic vibrational rotational
 MOLECULES BONDS Ionic: closed shell (+) or open shell (-) Covalent: both open shells neutral ( share e) Other (skip): van der Waals (He-He) Hydrogen bonds (in DNA, proteins, etc) ENERGY LEVELS electronic
MOLECULES BONDS Ionic: closed shell (+) or open shell (-) Covalent: both open shells neutral ( share e) Other (skip): van der Waals (He-He) Hydrogen bonds (in DNA, proteins, etc) ENERGY LEVELS electronic
Chemistry 2000 Lecture 2: LCAO-MO theory for homonuclear diatomic molecules
 Chemistry 2000 Lecture 2: LCAO-MO theory for homonuclear diatomic molecules Marc R. Roussel January 5, 2018 Marc R. Roussel Homonuclear diatomics January 5, 2018 1 / 17 MO theory for homonuclear diatomic
Chemistry 2000 Lecture 2: LCAO-MO theory for homonuclear diatomic molecules Marc R. Roussel January 5, 2018 Marc R. Roussel Homonuclear diatomics January 5, 2018 1 / 17 MO theory for homonuclear diatomic
Lecture February 13-15, Silicon crystal surfaces
 Lecture 18-19 February 13-15, 2012 Nature of the Chemical Bond with applications to catalysis, materials science, nanotechnology, surface science, bioinorganic chemistry, and energy Course number: Ch120a
Lecture 18-19 February 13-15, 2012 Nature of the Chemical Bond with applications to catalysis, materials science, nanotechnology, surface science, bioinorganic chemistry, and energy Course number: Ch120a
Lecture #13 1. Incorporating a vector potential into the Hamiltonian 2. Spin postulates 3. Description of spin states 4. Identical particles in
 Lecture #3. Incorporating a vector potential into the Hamiltonian. Spin postulates 3. Description of spin states 4. Identical particles in classical and QM 5. Exchange degeneracy - the fundamental problem
Lecture #3. Incorporating a vector potential into the Hamiltonian. Spin postulates 3. Description of spin states 4. Identical particles in classical and QM 5. Exchange degeneracy - the fundamental problem
Yingwei Wang Computational Quantum Chemistry 1 Hartree energy 2. 2 Many-body system 2. 3 Born-Oppenheimer approximation 2
 Purdue University CHM 67300 Computational Quantum Chemistry REVIEW Yingwei Wang October 10, 2013 Review: Prof Slipchenko s class, Fall 2013 Contents 1 Hartree energy 2 2 Many-body system 2 3 Born-Oppenheimer
Purdue University CHM 67300 Computational Quantum Chemistry REVIEW Yingwei Wang October 10, 2013 Review: Prof Slipchenko s class, Fall 2013 Contents 1 Hartree energy 2 2 Many-body system 2 3 Born-Oppenheimer
The Hydrogen Molecule-Ion
 Sign In Forgot Password Register ashwenchan username password Sign In If you like us, please share us on social media. The latest UCD Hyperlibrary newsletter is now complete, check it out. ChemWiki BioWiki
Sign In Forgot Password Register ashwenchan username password Sign In If you like us, please share us on social media. The latest UCD Hyperlibrary newsletter is now complete, check it out. ChemWiki BioWiki
On the Uniqueness of Molecular Orbitals and limitations of the MO-model.
 On the Uniqueness of Molecular Orbitals and limitations of the MO-model. The purpose of these notes is to make clear that molecular orbitals are a particular way to represent many-electron wave functions.
On the Uniqueness of Molecular Orbitals and limitations of the MO-model. The purpose of these notes is to make clear that molecular orbitals are a particular way to represent many-electron wave functions.
Valence bond theory accounts, at least qualitatively, for the stability of the covalent bond in terms of overlapping atomic orbitals.
 Molecular Orbital Theory Valence bond theory accounts, at least qualitatively, for the stability of the covalent bond in terms of overlapping atomic orbitals. Using the concept of hybridization, valence
Molecular Orbital Theory Valence bond theory accounts, at least qualitatively, for the stability of the covalent bond in terms of overlapping atomic orbitals. Using the concept of hybridization, valence
CHEM-UA 127: Advanced General Chemistry I
 CHEM-UA 7: Advanced General Chemistry I I. LINEAR COMBINATION OF ATOMIC ORBITALS Linear combination of atomic orbitals (LCAO) is a simple method of quantum chemistry that yields a qualitative picture of
CHEM-UA 7: Advanced General Chemistry I I. LINEAR COMBINATION OF ATOMIC ORBITALS Linear combination of atomic orbitals (LCAO) is a simple method of quantum chemistry that yields a qualitative picture of
Chapter 8 Problem Solutions
 Chapter 8 Problem Solutions 1. The energy needed to detach the electron from a hydrogen atom is 13.6 ev, but the energy needed to detach an electron from a hydrogen molecule is 15.7 ev. Why do you think
Chapter 8 Problem Solutions 1. The energy needed to detach the electron from a hydrogen atom is 13.6 ev, but the energy needed to detach an electron from a hydrogen molecule is 15.7 ev. Why do you think
The Schrodinger Equation and Postulates Common operators in QM: Potential Energy. Often depends on position operator: Kinetic Energy 1-D case:
 The Schrodinger Equation and Postulates Common operators in QM: Potential Energy Often depends on position operator: Kinetic Energy 1-D case: 3-D case Time Total energy = Hamiltonian To find out about
The Schrodinger Equation and Postulates Common operators in QM: Potential Energy Often depends on position operator: Kinetic Energy 1-D case: 3-D case Time Total energy = Hamiltonian To find out about
Problem Set 5 Solutions
 Chemistry 362 Dr Jean M Standard Problem Set 5 Solutions ow many vibrational modes do the following molecules or ions possess? [int: Drawing Lewis structures may be useful in some cases] In all of the
Chemistry 362 Dr Jean M Standard Problem Set 5 Solutions ow many vibrational modes do the following molecules or ions possess? [int: Drawing Lewis structures may be useful in some cases] In all of the
Electronic structure theory: Fundamentals to frontiers. 1. Hartree-Fock theory
 Electronic structure theory: Fundamentals to frontiers. 1. Hartree-Fock theory MARTIN HEAD-GORDON, Department of Chemistry, University of California, and Chemical Sciences Division, Lawrence Berkeley National
Electronic structure theory: Fundamentals to frontiers. 1. Hartree-Fock theory MARTIN HEAD-GORDON, Department of Chemistry, University of California, and Chemical Sciences Division, Lawrence Berkeley National
Chapter 10 Chemical Bonding II
 Chapter 10 Chemical Bonding II Valence Bond Theory Valence Bond Theory: A quantum mechanical model which shows how electron pairs are shared in a covalent bond. Bond forms between two atoms when the following
Chapter 10 Chemical Bonding II Valence Bond Theory Valence Bond Theory: A quantum mechanical model which shows how electron pairs are shared in a covalent bond. Bond forms between two atoms when the following
221B Lecture Notes Many-Body Problems II Molecular Physics
 1 Molecules 221B Lecture Notes Many-Body Problems II Molecular Physics In this lecture note, we discuss molecules. I cannot go into much details given I myself am not familiar enough with chemistry. But
1 Molecules 221B Lecture Notes Many-Body Problems II Molecular Physics In this lecture note, we discuss molecules. I cannot go into much details given I myself am not familiar enough with chemistry. But