Supporting Information
|
|
|
- Amos York
- 5 years ago
- Views:
Transcription
1 Supporting Information Conflict in the Mechanism and Kinetics of the Barrierless Reaction between SH and NO 2 Radicals Ramanpreet Kaur and Vikas * Quantum Chemistry Group, Department of Chemistry & Centre of Advanced Studies in Chemistry, Panjab University, Chandigarh INDIA. *Corresponding Author Phone: , qlabspu@pu.ac.in, qlabspu@yahoo.com S1
2 Table of Contents Contents Figure S1: Various EQs obtained through the FirstOnly GRRM search at BHandHLYP/ G(d,p) level of the theory. The numerical values depicted are relative energy (in kcal/mol) w.r.t. separated reactants. Figure S2 : The re-optimized geometries of the important stationary points at M06-2X/cc-pVTZ level of the theory. The bond distances depicted are in angstrom and bond angles are in degrees. Figure S3. Same as Figure S1 but using spin-unrestricted UBH&HLYP/6-31G method with GRRM search following two largest ADD. Figure S4. Structures of the key isomers (a) HSNO 2 (b) HSONO trans (c) HSONO cis obtained through the GRRM search at (1) UBHandHLYP/6-31G and (2) UM06-2X/cc-pVTZ levels of the theory. Table S1. Relative energies ( E) and standard Gibbs free energy change ( G), in kcal/mol, w.r.t. separated reactants (R1) of the relevant stationary points on the singlet potential energy surface of the reaction between HS and NO 2 radicals at the BHandLYP/ G(d,p), M06-2X/cc-pVTZ and CCSD(T)/cc-pVTZ//DFT/M06-2X/cc-pVTZ levels of the theory. The T1 diagnostic, harmonic frequency values (in cm -1 ) and rotational constants (in GHz) are given at M06-2X/cc-pVTZ level. Figure S5. Significant connections, both barrierless (depicted in dashed lines) and with barrier (depicted in solid lines) on the singlet potential energy surface of the HS + NO 2 reaction. Relative energies ( E) including ZPE and BSSE (in kcal/mol) depicted in parentheses were calculated at the CCSD(T)/cc-pVTZ//M06-2X/cc-pVTZ level of the theory. The bond distances and angles depicted are in angstroms and degrees, respectively. Figure S6. Same as Figure S5 but for the standard Gibbs free energy profile relative to the separated reactants at K. Figure S7. Relaxed scan of the potential energy as a function of S-N distance in the isomer HS-NO 2 calculated at spin-restricted (a) B3LYP/6-31G level of the theory, and (b) at the ZPE- & BSSE-corrected CCSD(T)/ccpVTZ//M06-2X/cc-pVTZ levels of the theory w.r.t. separated reactants R1 (depicted in Figure S5). Figure S8. Same as Fig. S7 but for the relaxed potential energy scan as a function of S-O distance in the isomer HS-ONO trans. Page No. S4-S5 S5-S6 S7 S8 S9 S10 S11 S12 S13 S2
3 Figure S9. Same as Fig. S7 but for the relaxed potential energy scan as a function of S-O distance in the isomer HS-ONO cis(a). Figure S10. Same as Fig. S7 but for the relaxed potential energy scan as a function of S-O distance in the isomer HS-ONO cis(b). Figure S11. Logarithm of vibrational frequencies (υ in cm -1 ) calculated at the M06-2X/cc-pvTZ level of theory as functions of (a) S N bond distance R S-N for the isomer HSNO 2 Figure S12. Arrhenius plots for the rate constants calculated at the BSSEcorrected CCSD(T)/cc-pVTZ//M06-2X/cc-pVTZ level of the theory in the temperature range of K at varying pressures, for mode 1 corresponding to S-N distances: (a) 3.38 Å, (b) 2.88 Å; and for mode 2 corresponding to S-O distances: (c) 3.89 Å, (d) 4.19 Å. Table S2. CVTST trial rate constants (in cm 3 molecule 1 s 1 ) for the barrierless reaction channel: HS + NO 2 HSNO 2 calculated using the spin-restricted method in the temperature range of K, at 1.01 bar, with varying S-N distance (r in Å).The values depicted in bold corresponds to experimentally observed range of rate constants at 298 K Table S3. Same as Table S2 but for the barrierless reaction channel: HS +NO 2 HSONO cis(a) or cis(b), in the temperature range of K, at 1.01 bar, with varying S-O distance (r in Å). Table S4. Same as Table S2 but for the barrierless reaction channel: HS +NO 2 HSONO trans, in the temperature range of K, at1.01 bar, with varying S-O distance (r in Å). Table S5. CVTST rate constants (in cm 3 molecule 1 s 1 ) for the barrierless reaction channel HS +NO 2 HS ---NO 2 in the temperature range of K and pressure ranging between bar. Table S6. RRKM unimolecular rate constants k (in sec -1 ) for the isomerization pathways with intrinsic energy barrier (TSs) located on the singlet PES of the reaction between HS and NO 2 radicals at 298 K. k rev are the values for the pathway in the reverse direction. Cartesian coordinates of the stationary points on the PES. S14 S15 S16 S17 S18 S18 S19 S19 S20 S20-S22 S3




")

 w.r.t.")
4 EQ1 = EQ2 = EQ3 = EQ4= EQ5= EQ6= EQ7= EQ8= EQ9= EQ10= EQ11= EQ12= Figure S1. Various EQs obtained through the FirstOnly GRRM search at BHandHLYP/ G(d,p) level of the theory. The numerical values depicted are relative energy (in kcal/mol) w.r.t. separated reactants. Figure S1 Continues S4


")




5 Figure S1 Continues EQ13= EQ14= EQ15= EQ16= NO 2 & HS radical HSNO 2 TS0 HSONO cis(a) TS1 HSONO trans Figure S2. The re-optimized geometries of the important stationary points at M06-2X/cc-pVTZ level of the theory. The bond lnghts depicted are in angstroms and the bond angles are in degrees. Figure S2 continues.. S5

 TS2")
H")


6 Figure S2 continues.. TS3 HSONO cis(b) TS2 HON(S)O cis TS4 SON(O)H TS5 HON(S)O trans HSO & NO HNO & SO SNO & OH S6


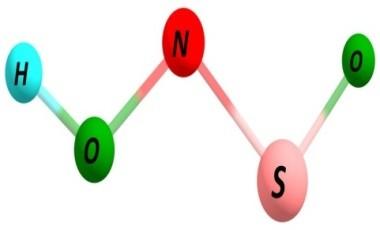

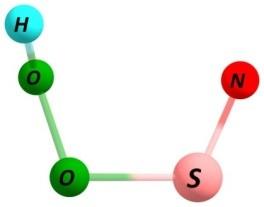
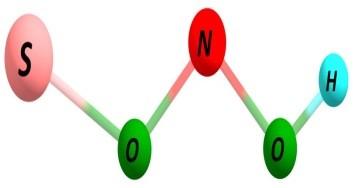
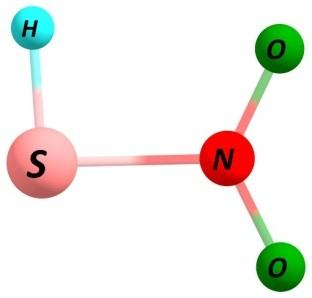

7 Figure S3. Same as Figure S1 but using spin-unrestricted UBH&HLYP/6-31G method with GRRM search following two largest ADDs. S7

 HSONO cis (c) HSONO trans")
 UBHandHLYP/6-31G and (ii)")
8 (i) (a) (b) (c) (ii) (a) (b) (c) Figure S4. Structures of the key isomers (a) HSNO 2 (b) HSONO cis (c) HSONO trans obtained through the GRRM search at (i) UBHandHLYP/6-31G and (ii) UM06-2X/cc-pVTZ levels of the theory. S8
9 Table S1. Relative energies ( E) and standard Gibbs free energy change ( G), in kcal/mol, w.r.t. separated reactants (R1) of the relevant stationary points on the singlet potential energy surface of the reaction between HS and NO 2 radicals at the BHandLYP/ G(d,p), M06-2X/cc-pVTZ and CCSD(T)/cc-pVTZ//DFT/M06-2X/ccpVTZ levels of the theory. The T1 diagnostic, harmonic frequency values (in cm -1 ) and rotational constants (in GHz) are given at M06-2X/cc-pVTZ level. Note that the stationary points are similar in nature and relative energy values to those on the spin-unrestricted singlet PES analyzed in Figure 1 and Table 1 of the article. Stationary point ΔE BHandHLYP/ G(d,p) +ZPE1 ΔE M06-2X/ccpVTZ (ZPE) +ZPE2 ΔE CCSD(T)/ cc-pvtz// M06-2X/ccpVTZ+ ZPE2 ΔG //M06-2X/ c-pvtz ΔG CCSD(T)/cc- pvtz//m06-2x/cc-pvtz T1 diagnostic values Harmonic Frequencies (M06-2X/cc-pVTZ) (cm -1 ) R1 (NO 2 + SH) NO 2 : 789, 1468, 1781 SH : 2758 Rotational Constants (M06-2X/cc-pVTZ) (GHz) NO 2 : ,13.28, HS : HSNO 2 HSONO trans HSONO cis(a) HSONO cis(b) SON(O)H HON(S)O cis HON(S)O trans TS0 i TS1 i TS2 i TS3 i TS4 i TS5 i P1 (HSO + NO) HSO P2 (HNO + SO) 14.12(0.01) HNO: 1593, 1758, 3005 SO: 1238 P3 (SNO + OH) SNO HSO HNO: , 43.67, SO: SNO a The total energy including (ZPE) of R1 at the default spin-unrestricted UBH&LYP/ G(d,p), UM06-2X/cc-pVTZ and UCCSD(T)/cc-pVTZ//DFT/M06-2X/cc-pVTZ levels of the theory are and a.u., respectively, and the total Gibbs free energy of R1 at UM06-2X/cc-pVTZ, UCCSD(T)/cc-pVTZ//DFT/UM06-2X/cc-pVTZ level are and a.u., respectively (1 a.u. = kcal/mol). S9
 HS + NO 2 R1 (0.0) TS4 (-7.53) TS3 (-9.68) TS5 (-20.90) NO + HSO (P1) (-20.")
 HSONO trans (-30.04) HSONO cis(a) (-30.72) Reaction coordinate Figure S5.")
 on the singlet potential energy surface of the HS + NO 2 reaction.")
/cc-pVTZ//M06-2X/cc-pVTZ level of the theory.")
10 ΔE(in kcal/mol) TS0 (19.82) SO + HNO (P2) (12.42) TS2 (11.99) SON(O)H (10.16) OH + SNO (P3) (12.28) HS + NO 2 R1 (0.0) TS4 (-7.53) TS3 (-9.68) TS5 (-20.90) NO + HSO (P1) (-20.13) HON(S)O cis (-27.78) TS1 (-22.56) HON(S)O trans (-26.96) HSNO 2 (-33.26) HSONO cis(b) (-29.33) HSONO trans (-30.04) HSONO cis(a) (-30.72) Reaction coordinate Figure S5. Significant connections, both barrierless (depicted in dashed lines) and with barrier (depicted in solid lines) on the singlet potential energy surface of the HS + NO 2 reaction. Relative energies ( E) including ZPE (in kcal/mol) depicted in parentheses were calculated at the CCSD(T)/cc-pVTZ//M06-2X/cc-pVTZ level of the theory. The bond distances and angles depicted are in angstroms and degrees, respectively. S10
11 TS0 (29.04) TS2 (21.31) SON(O)H (19.47) SO + HNO (P2) (12.27) HS + NO 2 R1 (0.0) TS4 (2.1) TS3 (--0.29) OH + SNO (P3) (12.14) TS5 (-11.42) TS1 (-13.27) HON(S)O cis (-18.35) HON(S)O trans (-17.53) NO + HSO (P1) (-21.20) HSNO 2 (-23.93) HSONO trans (-21.14) HSONO cis(a) (-21.53) HSONO cis(b) (-20.17) Reaction coordinates Figure S6. Same as Figure 1 but for the standard Gibbs free energy profile relative to the separated reactants at K. S11
12 (a) (b) 2.78 Å 2.98 Å 3.38 Å Figure S7. Relaxed scan of the potential energy as a function of S-N distance in the isomer HS- NO 2 calculated at spin-restricted (a) B3LYP/6-31G level of the theory, and (b) at the ZPE- & BSSE-corrected CCSD(T)/cc-pVTZ//M06-2X/cc-pVTZ levels of the theory w.r.t. separated reactants R1 (depicted in Figure S5). S12


13 (a) (b) Figure S8. Same as Fig. S7 but for the relaxed potential energy scan as a function of S-O distance in the isomer HS-ONO trans. S13







14 (a) (b) Figure S9. Same as Fig. S7 but for the relaxed potential energy scan as a function of S-O distance in the isomer HS-ONO cis(a). S14




15 Figure S10. Same as Fig. S7 but for the relaxed potential energy scan as a function of S-O distance in the isomer HS-ONO cis(b). S15
16 Figure S11. Logarithm of vibrational frequencies (υ in cm -1 ) calculated at the M06-2X/cc-pvTZ level of theory as functions of (a) S N bond distance R S-N for the isomer HSNO 2. S16
17 (a) (b) (d) (c) Figure S12. Arrhenius plots for the rate constants calculated at the BSSE-corrected CCSD(T)/cc-pVTZ//M06-2X/cc-pVTZ level of the theory in the temperature range of K at varying pressures, for mode 1 corresponding to S-N distances: (a) 3.38 Å, (b) 2.88 Å; and for mode 2 corresponding to S-O distances: (c) 3.89 Å, (d) 4.19 Å. S17
18 Table S2. CVTST trial rate constants (in cm 3 molecule 1 s 1 ) for the barrierless reaction channel: HS + NO 2 HSNO 2 using the spin-restricted method, in the temperature range of K, at 1.01 bar, with varying S-N distance (R in Å).The values depicted in bold corresponds to experimentally observed range of rate constants at 298 K R(Å) T(K) x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x Table S3. Same as Table S2 but for the barrierless reaction channel: HS +NO 2 HSONO cis(a) or cis(b), in the temperature range of K, at 1.01 bar, with varying S-O distance (R in Å). R(Å) T(K) x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x S18
19 Table S4. Same as Table S2 but for the barrierless reaction channel: HS +NO 2 bar, with varying S-O distance (R in Å). HSONO trans, in the temperature range of K, at1.01 RÅ) T(K) x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x Table S5. CVTST rate constants (in cm 3 molecule 1 s 1 ) for the barrierless reaction channel HS +NO 2 HS ---NO 2 in the temperature range of K and pressure ranging between bar. T(K) P(bar) x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x x S19
20 Table S6. RRKM unimolecular rate constants k (in sec -1 ) for the isomerization pathways with intrinsic energy barrier (TSs) located on the singlet PS of the reaction between HS and NO 2 radicals at 298 K. k rev are the values for the pathway in the reverse direction. Pathway k k rev HSNO 2 TS0 HSONO trans 7.52 x x HSONO trans TS1 HSONO cis(a) 1.26 x x 10 7 HSONO cis(a) TS3 HSONO cis(b) 1.95 x x 10-2 HSNO 2 TS4 HON(S)O cis 6.27 x x 10-3 HON(S)O cis TS4 HON(S)O trans 5.40 x x 10 8 HSONO trans TS2 SON(O)H 3.64 x x Cartesian coordinates (in angstroms) of the stationary points on the PES [depicted in Figure 1 (of main article) and SI Figure S5] at (U)M06-2X/ccpVTZ level X Y Z NO 2 N O O SH S H HSNO 2 N O O S H HSONO trans N O O S H S20
21 HSONO cis(a) N O O S H HSONO cis(b) N O O S H SON(O)H N O O S H HON(S)O cis N O O S H HON(S)O trans N O O S H TS0 N O O S H S21
22 TS1 N O O S H TS2 N O O S H TS3 N O O S H TS4 N O O S H TS5 N O O S H S22
New Reaction Classes in the Kinetic Modeling of Low Temperature Oxidation of n-alkanes
 Supplemental Material for paper New Reaction Classes in the Kinetic Modeling of Low Temperature Oxidation of n-alkanes Eliseo Ranzi, Carlo Cavallotti, Alberto Cuoci, Alessio Frassoldati, Matteo Pelucchi,
Supplemental Material for paper New Reaction Classes in the Kinetic Modeling of Low Temperature Oxidation of n-alkanes Eliseo Ranzi, Carlo Cavallotti, Alberto Cuoci, Alessio Frassoldati, Matteo Pelucchi,
Physical Chemistry Chemical Physics
 Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 2018 Physical Chemistry Chemical Physics Supporting Information Thermochemistry of the
Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 2018 Physical Chemistry Chemical Physics Supporting Information Thermochemistry of the
Theoretical study on reactions of ground state boron atom with ethylene(c 2 H 4 ), allene
, allene") Theoretical study on reactions of ground state boron atom with ethylene(c 2 H 4 ), allene (H 2 CCCH 2 ), and methylacetylen (CH 3 CCH) C. H. Huang 1, C. H. Kao 1, H. L. Sun 1, A. H. H. Chang 1, and R.
Theoretical study on reactions of ground state boron atom with ethylene(c 2 H 4 ), allene (H 2 CCCH 2 ), and methylacetylen (CH 3 CCH) C. H. Huang 1, C. H. Kao 1, H. L. Sun 1, A. H. H. Chang 1, and R.
Molecular tailoring: a possible synthetic route to hexasilabenzene
 SUPPORTING INFORMATION Molecular tailoring: a possible synthetic route to hexasilabenzene Zsolt Benedek, Tibor Szilvási and Tamás Veszprémi* Department of Inorganic and Analytical Chemistry, Budapest University
SUPPORTING INFORMATION Molecular tailoring: a possible synthetic route to hexasilabenzene Zsolt Benedek, Tibor Szilvási and Tamás Veszprémi* Department of Inorganic and Analytical Chemistry, Budapest University
Theoretical Study of Oxygen Isotope Exchange and Quenching in the O( 1 D) + CO 2 Reaction
 + CO 2 Reaction") Theoretical Study of Oxygen Isotope Exchange and Quenching in the O( 1 D) + CO 2 Reaction A. M. Mebel,*,, M. Hayashi, V. V. Kislov, and S. H. Lin*, Department of Chemistry and Biochemistry, Florida International
Theoretical Study of Oxygen Isotope Exchange and Quenching in the O( 1 D) + CO 2 Reaction A. M. Mebel,*,, M. Hayashi, V. V. Kislov, and S. H. Lin*, Department of Chemistry and Biochemistry, Florida International
Gas Phase Vibrational Spectroscopy of the Protonated Water Pentamer: The Role of Isomers and Nuclear Quantum Effects
 Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 2016 Supplementary Information Gas Phase Vibrational Spectroscopy of the Protonated
Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 2016 Supplementary Information Gas Phase Vibrational Spectroscopy of the Protonated
Research Article Theoretical Mechanism Study on the Reaction of FOO Radical with NO
 Chemistry Volume 2016, Article ID 5387853, 6 pages http://dx.doi.org/10.1155/2016/5387853 Research Article Theoretical Mechanism Study on the Reaction of Radical with ShunLi u Yang, 1 anan Wu, 1,2 JingYao
Chemistry Volume 2016, Article ID 5387853, 6 pages http://dx.doi.org/10.1155/2016/5387853 Research Article Theoretical Mechanism Study on the Reaction of Radical with ShunLi u Yang, 1 anan Wu, 1,2 JingYao
Supporting Information
 Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 2015 Supporting Information Are intramolecular frustrated Lewis pairs also intramolecular
Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 2015 Supporting Information Are intramolecular frustrated Lewis pairs also intramolecular
Thiourea Derivatives as Brønsted Acid Organocatalysts
 Supporting Information Thiourea Derivatives as Brønsted Acid Organocatalysts Ádám Madarász, Zsolt Dósa, Szilárd Varga, * Tibor Soós, Antal Csámpai, Imre Pápai * Institute of Organic Chemistry, Research
Supporting Information Thiourea Derivatives as Brønsted Acid Organocatalysts Ádám Madarász, Zsolt Dósa, Szilárd Varga, * Tibor Soós, Antal Csámpai, Imre Pápai * Institute of Organic Chemistry, Research
Supporting Information for Atmospheric Hydroxyl Radical Source: Reaction of Triplet SO 2 and Water
 Supporting Information for Atmospheric Hydroxyl Radical Source: Reaction of Triplet SO 2 and Water Authors: Jay A. Kroll 1,2,#, Benjamin N. Frandsen 3,#, Henrik G. Kjaergaard 3,*, and Veronica Vaida 1,2,*
Supporting Information for Atmospheric Hydroxyl Radical Source: Reaction of Triplet SO 2 and Water Authors: Jay A. Kroll 1,2,#, Benjamin N. Frandsen 3,#, Henrik G. Kjaergaard 3,*, and Veronica Vaida 1,2,*
Theoretical study of the BF 3-promoted rearrangement of oxiranyl N-methyliminodiacetic acid boronates
 Theoretical study of the BF 3-promoted rearrangement of oxiranyl N-methyliminodiacetic acid boronates Margarita M. Vallejos a* and Silvina C. Pellegrinet b* a Laboratorio de Química Orgánica, IQUIBA-NEA,
Theoretical study of the BF 3-promoted rearrangement of oxiranyl N-methyliminodiacetic acid boronates Margarita M. Vallejos a* and Silvina C. Pellegrinet b* a Laboratorio de Química Orgánica, IQUIBA-NEA,
List of Figures Page Figure No. Figure Caption No. Figure 1.1.
 List of Figures Figure No. Figure Caption Page No. Figure 1.1. Cation- interactions and their modulations. 4 Figure 1.2. Three conformations of benzene dimer, S is not a minimum on the potential energy
List of Figures Figure No. Figure Caption Page No. Figure 1.1. Cation- interactions and their modulations. 4 Figure 1.2. Three conformations of benzene dimer, S is not a minimum on the potential energy
Cethrene: The Chameleon of Woodward Hoffmann Rules
 Supporting Information Cethrene: The Chameleon of Woodward Hoffmann Rules Tomáš Šolomek,*, Prince Ravat,, Zhongyu Mou, Miklos Kertesz, and Michal Juríček*,, Department of Chemistry, University of Basel,
Supporting Information Cethrene: The Chameleon of Woodward Hoffmann Rules Tomáš Šolomek,*, Prince Ravat,, Zhongyu Mou, Miklos Kertesz, and Michal Juríček*,, Department of Chemistry, University of Basel,
A Theoretical Study of Oxidation of Phenoxy and Benzyl Radicals by HO 2
 A Theoretical Study of xidation of Phenoxy and Benzyl adicals by 2 S. Skokov, A. Kazakov, and F. L. Dryer Department of Mechanical and Aerospace Engineering Princeton University, Princeton, NJ 08544 We
A Theoretical Study of xidation of Phenoxy and Benzyl adicals by 2 S. Skokov, A. Kazakov, and F. L. Dryer Department of Mechanical and Aerospace Engineering Princeton University, Princeton, NJ 08544 We
Rate Constant and Branching Fraction for the NH 2 +NO 2 Reaction
 pubs.acs.org/jpca Rate Constant and Branching Fraction for the NH 2 +NO 2 Reaction Stephen J. Klippenstein, Lawrence B. Harding, Peter Glarborg,*, Yide Gao, Huanzhen Hu, and Paul Marshall Chemical Sciences
pubs.acs.org/jpca Rate Constant and Branching Fraction for the NH 2 +NO 2 Reaction Stephen J. Klippenstein, Lawrence B. Harding, Peter Glarborg,*, Yide Gao, Huanzhen Hu, and Paul Marshall Chemical Sciences
The Silacyclobutene Ring: An Indicator of Triplet State Baird-Aromaticity
 The Silacyclobutene Ring: An Indicator of Triplet State Baird-Aromaticity Rabia Ayub, 1,2 Kjell Jorner, 1,2 and Henrik Ottosson 1,2 * 1 Department of Chemistry - BMC, Uppsala University, Box 576, 751 23
The Silacyclobutene Ring: An Indicator of Triplet State Baird-Aromaticity Rabia Ayub, 1,2 Kjell Jorner, 1,2 and Henrik Ottosson 1,2 * 1 Department of Chemistry - BMC, Uppsala University, Box 576, 751 23
A Two Transition State Model for Radical-Molecule Reactions: A Case Study of the Addition of OH to C 2 H 4
 J. Phys. Chem. A 2005, 109, 6031-6044 6031 A Two Transition State Model for Radical-Molecule Reactions: A Case Study of the Addition of OH to C 2 H 4 Erin E. Greenwald and Simon W. North Department of
J. Phys. Chem. A 2005, 109, 6031-6044 6031 A Two Transition State Model for Radical-Molecule Reactions: A Case Study of the Addition of OH to C 2 H 4 Erin E. Greenwald and Simon W. North Department of
Atmospheric Fate of Methyl Vinyl Ketone: Peroxy. Radical Reactions with NO and HO 2. Supporting Information
 Atmospheric Fate of Methyl Vinyl Ketone: Peroxy Radical Reactions with NO and HO 2 Supporting Information Eric Praske, John D. Crounse*, Kelvin H. Bates, Theo Kurtén, Henrik G. Kjaergaard, Paul O. Wennberg
Atmospheric Fate of Methyl Vinyl Ketone: Peroxy Radical Reactions with NO and HO 2 Supporting Information Eric Praske, John D. Crounse*, Kelvin H. Bates, Theo Kurtén, Henrik G. Kjaergaard, Paul O. Wennberg
From Roaming Atoms to Hopping Surfaces: Mapping Out Global Reaction Routes in Photochemistry
 This is an open access article published under an ACS AuthorChoice License, which permits copying and redistribution of the article or any adaptations for non-commercial purposes. pubs.acs.org/jacs From
This is an open access article published under an ACS AuthorChoice License, which permits copying and redistribution of the article or any adaptations for non-commercial purposes. pubs.acs.org/jacs From
Supporting information for: On the atmospheric fate of methacrolein: 1. Peroxy. radical isomerization following addition of OH and O 2
 Supporting information for: On the atmospheric fate of methacrolein: 1. Peroxy radical isomerization following addition of OH and O 2 John D. Crounse,, Hasse C. Knap, Kristian B. Ørnsø, Solvejg Jørgensen,
Supporting information for: On the atmospheric fate of methacrolein: 1. Peroxy radical isomerization following addition of OH and O 2 John D. Crounse,, Hasse C. Knap, Kristian B. Ørnsø, Solvejg Jørgensen,
Figure 1: Transition State, Saddle Point, Reaction Pathway
 Computational Chemistry Workshops West Ridge Research Building-UAF Campus 9:00am-4:00pm, Room 009 Electronic Structure - July 19-21, 2016 Molecular Dynamics - July 26-28, 2016 Potential Energy Surfaces
Computational Chemistry Workshops West Ridge Research Building-UAF Campus 9:00am-4:00pm, Room 009 Electronic Structure - July 19-21, 2016 Molecular Dynamics - July 26-28, 2016 Potential Energy Surfaces
Supporting Information. P,N Ligands. General Information:
 Supporting Information A Dynamic Kinetic C Cross Coupling for the Asymmetric Synthesis of Axially Chiral,N Ligands edro Ramírez-López, Abel Ros, *, Beatriz Estepa, Rosario Fernández, *, Béla Fiser, Enrique
Supporting Information A Dynamic Kinetic C Cross Coupling for the Asymmetric Synthesis of Axially Chiral,N Ligands edro Ramírez-López, Abel Ros, *, Beatriz Estepa, Rosario Fernández, *, Béla Fiser, Enrique
Computational details, X-ray datas and spectral copies of 1 H, 13 C NMR of compounds obtained in this study
 Stereo, Regio-, and Chemoselective [3+2]-Cycloaddition of (2E,4E)-Ethyl 5-(Phenylsulfonyl)penta-2,4-dienoate with Various Azomethine Ylides, Nitrones, and Nitrile Oxides: Synthesis of Pyrrolidine, Isoxazolidine,
Stereo, Regio-, and Chemoselective [3+2]-Cycloaddition of (2E,4E)-Ethyl 5-(Phenylsulfonyl)penta-2,4-dienoate with Various Azomethine Ylides, Nitrones, and Nitrile Oxides: Synthesis of Pyrrolidine, Isoxazolidine,
Theoretical study of the OH-initiated atmospheric oxidation mechanism. of perfluoro methyl vinyl ether, CF 3 OCF=CF 2
 Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 2015 Theoretical study of the OH-initiated atmospheric oxidation mechanism of perfluoro
Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 2015 Theoretical study of the OH-initiated atmospheric oxidation mechanism of perfluoro
Theoretical Studies of Reaction Mechanisms Relevant to Hydrocarbon Growth in Titan s Atmosphere
 Theoretical Studies of Reaction Mechanisms Relevant to Hydrocarbon Growth in Titan s Atmosphere Alexander M. Mebel, Adeel Jamal, Alexander Landera, and Ralf I. Kaiser Department of Chemistry and Biochemistry,
Theoretical Studies of Reaction Mechanisms Relevant to Hydrocarbon Growth in Titan s Atmosphere Alexander M. Mebel, Adeel Jamal, Alexander Landera, and Ralf I. Kaiser Department of Chemistry and Biochemistry,
- 1 - Institute of Organic Chemistry and Biochemistry AS CR, v.v.i., Flemingovo náměstí 2, CZ, Praha, Czech Republic
 Electronic Supplementary Material (ESI) for RSC Advances. This journal is The Royal Society of Chemistry 2014-1 - The activation of N-glycosidic bond cleavage operated by base-excision repair enzyme hogg1;
Electronic Supplementary Material (ESI) for RSC Advances. This journal is The Royal Society of Chemistry 2014-1 - The activation of N-glycosidic bond cleavage operated by base-excision repair enzyme hogg1;
Pyramidal Fe(CO) 5. P. Aiswaryalakshmi, Devendra Mani and E. Arunan* Department of Inorganic and Physical Chemistry, Indian Institute of Science,
 5. P. Aiswaryalakshmi, Devendra Mani and E. Arunan* Department of Inorganic and Physical Chemistry, Indian Institute of Science,") Fe as Hydrogen/Halogen Bond Acceptor in Square Pyramidal Fe(CO) 5 Supporting Information P. Aiswaryalakshmi, Devendra Mani and E. Arunan* Department of Inorganic and Physical Chemistry, Indian Institute
Fe as Hydrogen/Halogen Bond Acceptor in Square Pyramidal Fe(CO) 5 Supporting Information P. Aiswaryalakshmi, Devendra Mani and E. Arunan* Department of Inorganic and Physical Chemistry, Indian Institute
Electronic Supplementary Information. for
 Electronic Supplementary Material (ESI) for Chemical Science. This journal is The Royal Society of Chemistry 2018 Electronic Supplementary Information for Two Chiral Catalysts in Action: Insights on Cooperativity
Electronic Supplementary Material (ESI) for Chemical Science. This journal is The Royal Society of Chemistry 2018 Electronic Supplementary Information for Two Chiral Catalysts in Action: Insights on Cooperativity
Supporting Information
 Supporting Information Copyright Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, 2013 Photochemistry of N-Methylformamide: Matrix Isolation and Nonadiabatic Dynamics Rachel Crespo-Otero, [a] Artur Mardyukov,
Supporting Information Copyright Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, 2013 Photochemistry of N-Methylformamide: Matrix Isolation and Nonadiabatic Dynamics Rachel Crespo-Otero, [a] Artur Mardyukov,
Protonation of N 2 O and NO 2 in a solid phase
 Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 2017 SUPPORTING INFORMATION Protonation of N 2 O and NO 2 in a solid phase Evgenii S.
Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 2017 SUPPORTING INFORMATION Protonation of N 2 O and NO 2 in a solid phase Evgenii S.
SUPPORTING INFORMATION. Elucidation of the role of betaine hydrochloride in glycerol esterification: towards bio-based ionic building blocks
 Electronic Supplementary Material (ESI) for Green Chemistry. This journal is The Royal Society of Chemistry 2017 SUPPORTING INFORMATION Elucidation of the role of betaine hydrochloride in glycerol esterification:
Electronic Supplementary Material (ESI) for Green Chemistry. This journal is The Royal Society of Chemistry 2017 SUPPORTING INFORMATION Elucidation of the role of betaine hydrochloride in glycerol esterification:
Size-extensive wave functions for QMC A linear-scaling GVB approach
 Size-extensive wave functions for QMC A linear-scaling GVB approach Claudia Filippi, University of Twente, The Netherlands Francesco Fracchia, University of Pisa, Italy Claudio Amovilli, University of
Size-extensive wave functions for QMC A linear-scaling GVB approach Claudia Filippi, University of Twente, The Netherlands Francesco Fracchia, University of Pisa, Italy Claudio Amovilli, University of
Supporting Information (SI) For. Reaction of Aniline with Singlet Oxygen (O 2 1 g )
 For. Reaction of Aniline with Singlet Oxygen (O 2 1 g )") Supporting Information (SI) For Reaction of Aniline with Singlet Oxygen (O 2 1 g ) Jomana Al-Nu airat 1, Mohammednoor Altarawneh* 1, Xiangpeng Gao 1, Phillip R. Westmoreland 2 and Bogdan Z. Dlugogorski
Supporting Information (SI) For Reaction of Aniline with Singlet Oxygen (O 2 1 g ) Jomana Al-Nu airat 1, Mohammednoor Altarawneh* 1, Xiangpeng Gao 1, Phillip R. Westmoreland 2 and Bogdan Z. Dlugogorski
Supporting Information
 Supporting Information Tuning of Second-Order Nonlinear Optical Response Properties of Aryl-Substituted Boron-Dipyrromethene Dyes: Unidirectional Charge Transfer Coupled with Structural Tailoring Ramprasad
Supporting Information Tuning of Second-Order Nonlinear Optical Response Properties of Aryl-Substituted Boron-Dipyrromethene Dyes: Unidirectional Charge Transfer Coupled with Structural Tailoring Ramprasad
Density Functional Theory Study on Mechanism of Forming Spiro-Geheterocyclic Ring Compound from Me 2 Ge=Ge: and Acetaldehyde
 CHINESE JURNAL F CHEMICAL PHYSICS VLUME 26, NUMBER 1 FEBRUARY 27, 2013 ARTICLE Density Functional Theory Study on Mechanism of Forming Spiro-Geheterocyclic Ring Compound from Me 2 Ge=Ge: and Acetaldehyde
CHINESE JURNAL F CHEMICAL PHYSICS VLUME 26, NUMBER 1 FEBRUARY 27, 2013 ARTICLE Density Functional Theory Study on Mechanism of Forming Spiro-Geheterocyclic Ring Compound from Me 2 Ge=Ge: and Acetaldehyde
Temperature and pressure dependent rate coefficients for the reaction of vinyl radical with molecular oxygen
 Paper # 070RK-0274 Topic: Reaction Kinetics 8 th U. S. National Combustion Meeting rganized by the Western States Section of the Combustion Institute and hosted by the University of Utah May 19-22, 2013
Paper # 070RK-0274 Topic: Reaction Kinetics 8 th U. S. National Combustion Meeting rganized by the Western States Section of the Combustion Institute and hosted by the University of Utah May 19-22, 2013
Annual Report Combustion Energy Frontier Research Center Princeton, NJ Sept. 23, Truhlar group University of Minnesota
 Annual Report Combustion Energy Frontier Research Center Princeton, NJ Sept. 23, 2010 Truhlar group University of Minnesota John Steven Ewa Xuefei Tao Jingjing Alecu Mielke Papajak Xu Yu Zheng Practical
Annual Report Combustion Energy Frontier Research Center Princeton, NJ Sept. 23, 2010 Truhlar group University of Minnesota John Steven Ewa Xuefei Tao Jingjing Alecu Mielke Papajak Xu Yu Zheng Practical
Ab Initio MO and TST Calculations for the Rate Constant of the HNO NO : HONO NO
 Ab Initio M and TST Calculations for the Rate Constant of the N N : N N 2 Reaction A. M. MEBEL,* M. C. LIN, K. MRKUMA Cherry L. Emerson Center for Scientific Computation and Department of Chemistry, Emory
Ab Initio M and TST Calculations for the Rate Constant of the N N : N N 2 Reaction A. M. MEBEL,* M. C. LIN, K. MRKUMA Cherry L. Emerson Center for Scientific Computation and Department of Chemistry, Emory
NH 3 inversion: Potential energy surfaces and transition states CH342L March 28, 2016
 N 3 inversion: Potential energy surfaces and transition states C342L March 28, 2016 Last week, we used the IR spectrum of ammonia to determine the splitting of energy levels due to inversion of the umbrella
N 3 inversion: Potential energy surfaces and transition states C342L March 28, 2016 Last week, we used the IR spectrum of ammonia to determine the splitting of energy levels due to inversion of the umbrella
A. MP2 - Inclusion of counterpoise in the optimisation step
 A. MP2 - Inclusion of counterpoise in the optimisation step Figure S1. Top and side views of the M_FS_SF_A and M_FS_SF_R IP-dimer structures computed at the MP2 level with (orange) and without (blue) counterpoise
A. MP2 - Inclusion of counterpoise in the optimisation step Figure S1. Top and side views of the M_FS_SF_A and M_FS_SF_R IP-dimer structures computed at the MP2 level with (orange) and without (blue) counterpoise
The Potential Energy Surface (PES) Preamble to the Basic Force Field Chem 4021/8021 Video II.i
 Preamble to the Basic Force Field Chem 4021/8021 Video II.i") The Potential Energy Surface (PES) Preamble to the Basic Force Field Chem 4021/8021 Video II.i The Potential Energy Surface Captures the idea that each structure that is, geometry has associated with it
The Potential Energy Surface (PES) Preamble to the Basic Force Field Chem 4021/8021 Video II.i The Potential Energy Surface Captures the idea that each structure that is, geometry has associated with it
Optically Triggered Stepwise Double Proton Transfer in an Intramolecular Proton Relay: A Case Study of 1,8-Dihydroxy-2-naphthaldehyde (DHNA)
") Supporting Information Optically Triggered Stepwise Double Proton Transfer in an Intramolecular Proton Relay: A Case Study of 1,8-Dihydroxy-2-naphthaldehyde (DHNA) Chia-Yu Peng,, Jiun-Yi Shen,, Yi-Ting
Supporting Information Optically Triggered Stepwise Double Proton Transfer in an Intramolecular Proton Relay: A Case Study of 1,8-Dihydroxy-2-naphthaldehyde (DHNA) Chia-Yu Peng,, Jiun-Yi Shen,, Yi-Ting
Ab initio calculations for potential energy surfaces. D. Talbi GRAAL- Montpellier
 Ab initio calculations for potential energy surfaces D. Talbi GRAAL- Montpellier A theoretical study of a reaction is a two step process I-Electronic calculations : techniques of quantum chemistry potential
Ab initio calculations for potential energy surfaces D. Talbi GRAAL- Montpellier A theoretical study of a reaction is a two step process I-Electronic calculations : techniques of quantum chemistry potential
Quantum chemistry and vibrational spectra
 Chapter 3 Quantum chemistry and vibrational spectra This chapter presents the quantum chemical results for the systems studied in this work, FHF (Section 3.) and OHF (Section 3.3). These triatomic anions
Chapter 3 Quantum chemistry and vibrational spectra This chapter presents the quantum chemical results for the systems studied in this work, FHF (Section 3.) and OHF (Section 3.3). These triatomic anions
Express the transition state equilibrium constant in terms of the partition functions of the transition state and the
 Module 7 : Theories of Reaction Rates Lecture 33 : Transition State Theory Objectives After studying this Lecture you will be able to do the following. Distinguish between collision theory and transition
Module 7 : Theories of Reaction Rates Lecture 33 : Transition State Theory Objectives After studying this Lecture you will be able to do the following. Distinguish between collision theory and transition
Modelling of Reaction Mechanisms KJE-3102
 Modelling of Reaction Mechanisms KJE-3102 Kathrin H. Hopmann Kathrin.hopmann@uit.no Outline Potential energy surfaces Transition state optimization Reaction coordinates Imaginary frequencies Verification
Modelling of Reaction Mechanisms KJE-3102 Kathrin H. Hopmann Kathrin.hopmann@uit.no Outline Potential energy surfaces Transition state optimization Reaction coordinates Imaginary frequencies Verification
The Mechanism of Directed Ni(II)-Catalyzed C H Iodination with Molecular Iodine
-Catalyzed C H Iodination with Molecular Iodine") Electronic Supplementary Material (ESI) for Chemical Science. This journal is The Royal Society of Chemistry 2017 Supporting Information The Mechanism of Directed Ni(II)-Catalyzed C H Iodination with Molecular
Electronic Supplementary Material (ESI) for Chemical Science. This journal is The Royal Society of Chemistry 2017 Supporting Information The Mechanism of Directed Ni(II)-Catalyzed C H Iodination with Molecular
SUPPORTING INFORMATION Control Elements in Dynamically-Determined Selectivity on a Bifurcating Surface
 S1 SUPPORTING INFORMATION Control Elements in Dynamically-Determined Selectivity on a Bifurcating Surface Jacqueline B. Thomas, Jack R. Waas, Michael Harmata,*, and Daniel A. Singleton*, Department of
S1 SUPPORTING INFORMATION Control Elements in Dynamically-Determined Selectivity on a Bifurcating Surface Jacqueline B. Thomas, Jack R. Waas, Michael Harmata,*, and Daniel A. Singleton*, Department of
Supplementary Figure S1 Stable structures of I, I', II, and II' optimized at the
 H C Si 2.492 (2.348) 1.867 (2.005) Fe O 2.106 (2.077) 2.360 (2.497) N 1.126 (1.115) 2.105 (2.175) I quintet (triplet) 0.0 (+4.1) kcal/mol II triplet (quintet) 0.0 (+4.3) kcal/mol 1.857 (2.076) 2.536 (2.351)
H C Si 2.492 (2.348) 1.867 (2.005) Fe O 2.106 (2.077) 2.360 (2.497) N 1.126 (1.115) 2.105 (2.175) I quintet (triplet) 0.0 (+4.1) kcal/mol II triplet (quintet) 0.0 (+4.3) kcal/mol 1.857 (2.076) 2.536 (2.351)
Uptake of OH radical to aqueous aerosol: a computational study
 Uptake of OH radical to aqueous aerosol: a computational study Grigory Andreev Karpov Institute of Physical Chemistry 10 Vorontsovo pole, Moscow, 105064, Russia Institute of Physical Chemistry and Electrochemistry
Uptake of OH radical to aqueous aerosol: a computational study Grigory Andreev Karpov Institute of Physical Chemistry 10 Vorontsovo pole, Moscow, 105064, Russia Institute of Physical Chemistry and Electrochemistry
Mechanism of Iron Carbonyl-Catalyzed Hydrogenation of Ethylene. 1. Theoretical Exploration of Molecular Pathways
 pubs.acs.org/jpca Mechanism of Iron Carbonyl-Catalyzed Hydrogenation of Ethylene. 1. Theoretical Exploration of Molecular Pathways Rubik Asatryan* and Eli Ruckenstein Department of Chemical and Biological
pubs.acs.org/jpca Mechanism of Iron Carbonyl-Catalyzed Hydrogenation of Ethylene. 1. Theoretical Exploration of Molecular Pathways Rubik Asatryan* and Eli Ruckenstein Department of Chemical and Biological
Supporting Information Computational Part
 Supporting Information Computational Part Ruthenium-Catalyzed Alkyne trans-hydrometalation: Mechanistic Insights and Preparative Implications Dragoş Adrian Roşca, Karin Radkowski, Larry M. Wolf, Minal
Supporting Information Computational Part Ruthenium-Catalyzed Alkyne trans-hydrometalation: Mechanistic Insights and Preparative Implications Dragoş Adrian Roşca, Karin Radkowski, Larry M. Wolf, Minal
OH CH 3 D H 2 O CH 2 D and HDO CH 3, R2
 JOURNAL OF CHEMICAL PHYSICS VOLUME 114, NUMBER 5 1 FEBRUARY 2001 The reactions CH n D 4Àn OH\P and CH 4 OD\CH 3 HOD as a test of current direct dynamics computational methods to determine variational transition-state
JOURNAL OF CHEMICAL PHYSICS VOLUME 114, NUMBER 5 1 FEBRUARY 2001 The reactions CH n D 4Àn OH\P and CH 4 OD\CH 3 HOD as a test of current direct dynamics computational methods to determine variational transition-state
An Introduction to Quantum Chemistry and Potential Energy Surfaces. Benjamin G. Levine
 An Introduction to Quantum Chemistry and Potential Energy Surfaces Benjamin G. Levine This Week s Lecture Potential energy surfaces What are they? What are they good for? How do we use them to solve chemical
An Introduction to Quantum Chemistry and Potential Energy Surfaces Benjamin G. Levine This Week s Lecture Potential energy surfaces What are they? What are they good for? How do we use them to solve chemical
Rate Constant for the NH 3 NO 2. HONO Reaction: Comparison of Kinetically Modeled and Predicted Results
 Rate Constant for the NH 3 HONO Reaction: Comparison of Kinetically Modeled and Predicted Results A. GRANT THAXTON, C.-C. HSU, M. C. LIN Department of Chemistry, Emory University, Atlanta, Georgia 30322
Rate Constant for the NH 3 HONO Reaction: Comparison of Kinetically Modeled and Predicted Results A. GRANT THAXTON, C.-C. HSU, M. C. LIN Department of Chemistry, Emory University, Atlanta, Georgia 30322
Ab initio CASPT2ÕÕCASSCF study of the O 1 D H 2 O X 1 A 1 reaction
 JOURNAL OF CHEMICAL PHYSICS VOLUME 115, NUMBER 19 15 NOVEMBER 2001 Ab initio CASPT2ÕÕCASSCF study of the O 1 D H 2 O X 1 A 1 reaction R. Sayós, a) Carolina Oliva, and Miguel González a) Departament de
JOURNAL OF CHEMICAL PHYSICS VOLUME 115, NUMBER 19 15 NOVEMBER 2001 Ab initio CASPT2ÕÕCASSCF study of the O 1 D H 2 O X 1 A 1 reaction R. Sayós, a) Carolina Oliva, and Miguel González a) Departament de
Chemical Science EDGE ARTICLE
 Chemical Science View Online Dynamic Article Links C < Cite this: Chem. Sci., 2011, 2, 2199 www.rsc.org/chemicalscience Multi-structural variational transition state theory. Kinetics of the 1,4-hydrogen
Chemical Science View Online Dynamic Article Links C < Cite this: Chem. Sci., 2011, 2, 2199 www.rsc.org/chemicalscience Multi-structural variational transition state theory. Kinetics of the 1,4-hydrogen
# ( 1) j=1. 1 Computer Experiment 5: Computational Thermochemistry. 1.1 Background:
 j=1. 1 Computer Experiment 5: Computational Thermochemistry. 1.1 Background:") Computational Thermochemistry 1 1 Computer Experiment 5: Computational Thermochemistry 1.1 Background: Within the frame of this experiment you will employ methods of statistical thermodynamics in order
Computational Thermochemistry 1 1 Computer Experiment 5: Computational Thermochemistry 1.1 Background: Within the frame of this experiment you will employ methods of statistical thermodynamics in order
Supplementary Information
 Electronic Supplementary Material (ESI) for New Journal of Chemistry. This journal is The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2018 Supplementary Information
Electronic Supplementary Material (ESI) for New Journal of Chemistry. This journal is The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2018 Supplementary Information
Roaming and Spectroscopy
 Roaming and Spectroscopy D. Townsend, S. A. Lahankar, S. K. Lee, S. D. Chambreau, A. G. Suits, X. Zhang, and J. Rheinecker, L. B. Harding, J. M. Bowman, Science. 306, 1158 (2004). J. M. Bowman, X. Zhang,
Roaming and Spectroscopy D. Townsend, S. A. Lahankar, S. K. Lee, S. D. Chambreau, A. G. Suits, X. Zhang, and J. Rheinecker, L. B. Harding, J. M. Bowman, Science. 306, 1158 (2004). J. M. Bowman, X. Zhang,
Chemistry 4021/8021 Computational Chemistry 3/4 Credits Spring Semester 2007 Key PS3
 Chemistry 4021/8021 Computational Chemistry 3/4 Credits Spring Semester 2007 Key PS3 1. Below are two isomeric geometries that we previously examined in Problem Sets 1 and 2 as both C 10 H 16 and Si 10
Chemistry 4021/8021 Computational Chemistry 3/4 Credits Spring Semester 2007 Key PS3 1. Below are two isomeric geometries that we previously examined in Problem Sets 1 and 2 as both C 10 H 16 and Si 10
Introduction to Theories of Chemical Reactions. Graduate Course Seminar Beate Flemmig FHI
 Introduction to Theories of Chemical Reactions Graduate Course Seminar Beate Flemmig FHI I. Overview What kind of reactions? gas phase / surface unimolecular / bimolecular thermal / photochemical What
Introduction to Theories of Chemical Reactions Graduate Course Seminar Beate Flemmig FHI I. Overview What kind of reactions? gas phase / surface unimolecular / bimolecular thermal / photochemical What
5.62 Physical Chemistry II Spring 2008
 MIT OpenCourseWare http://ocw.mit.edu 5.62 Physical Chemistry II Spring 2008 For information about citing these materials or our Terms of Use, visit: http://ocw.mit.edu/terms. 5.62 Spring 2007 Lecture
MIT OpenCourseWare http://ocw.mit.edu 5.62 Physical Chemistry II Spring 2008 For information about citing these materials or our Terms of Use, visit: http://ocw.mit.edu/terms. 5.62 Spring 2007 Lecture
Energy Barriers and Rates - Transition State Theory for Physicists
 Energy Barriers and Rates - Transition State Theory for Physicists Daniel C. Elton October 12, 2013 Useful relations 1 cal = 4.184 J 1 kcal mole 1 = 0.0434 ev per particle 1 kj mole 1 = 0.0104 ev per particle
Energy Barriers and Rates - Transition State Theory for Physicists Daniel C. Elton October 12, 2013 Useful relations 1 cal = 4.184 J 1 kcal mole 1 = 0.0434 ev per particle 1 kj mole 1 = 0.0104 ev per particle
Ab initio study on the paths of oxygen abstraction of hydrogen trioxide (HO 3 ) molecule in the HO 3 + SO 2 reaction
 molecule in the HO 3 + SO 2 reaction") J. Chem. Sci. Vol. 125, No. 4, July 2013, pp. 927 932. c Indian Academy of Sciences. Ab initio study on the paths of oxygen abstraction of hydrogen trioxide (HO 3 ) molecule in the HO 3 + SO 2 reaction
J. Chem. Sci. Vol. 125, No. 4, July 2013, pp. 927 932. c Indian Academy of Sciences. Ab initio study on the paths of oxygen abstraction of hydrogen trioxide (HO 3 ) molecule in the HO 3 + SO 2 reaction
Energetics and Mechanism of the Hydrogenation of XH n for Group IV to Group VII Elements X
 pubs.acs.org/jctc Energetics and Mechanism of the Hydrogenation of XH n for Group IV to Group VII Elements X Elfi Kraka,* Wenli Zou, Marek Freindorf, and Dieter Cremer* CATCO Group, Department of Chemistry,
pubs.acs.org/jctc Energetics and Mechanism of the Hydrogenation of XH n for Group IV to Group VII Elements X Elfi Kraka,* Wenli Zou, Marek Freindorf, and Dieter Cremer* CATCO Group, Department of Chemistry,
Supporting Information. Substitutent Rate Effects
 Supporting Information Gosteli Claisen Rearrangement: DFT Study of Substitutent Rate Effects Julia Rehbein* and Martin Hiersemann* Fakultät Chemie, Technische Universität Dortmund, 44227 Dortmund, Germany
Supporting Information Gosteli Claisen Rearrangement: DFT Study of Substitutent Rate Effects Julia Rehbein* and Martin Hiersemann* Fakultät Chemie, Technische Universität Dortmund, 44227 Dortmund, Germany
eight-valence electron species
 DOI: 0.038/NCHEM.263 Quadruple bonding in C 2 and analogous eight-valence electron species Sason Shaik*, David Danovich, Wei Wu 2, Peifeng Su 2, Henry Rzepa 3, Philippe C. Hiberty 4. Institute of Chemistry
DOI: 0.038/NCHEM.263 Quadruple bonding in C 2 and analogous eight-valence electron species Sason Shaik*, David Danovich, Wei Wu 2, Peifeng Su 2, Henry Rzepa 3, Philippe C. Hiberty 4. Institute of Chemistry
THE PERFORMANCE OF DENSITY FUNCTIONALS WITH RESPECT TO THE CORRELATION CONSISTENT BASIS SETS. Xuelin Wang, B.S., M.S
 THE PERFORMANCE OF DENSITY FUNCTIONALS WITH RESPECT TO THE CORRELATION CONSISTENT BASIS SETS Xuelin Wang, B.S., M.S Dissertation Prepared for the Degree of DOCTOR OF PHILOSOPHY UNIVERSITY OF NORTH TEXAS
THE PERFORMANCE OF DENSITY FUNCTIONALS WITH RESPECT TO THE CORRELATION CONSISTENT BASIS SETS Xuelin Wang, B.S., M.S Dissertation Prepared for the Degree of DOCTOR OF PHILOSOPHY UNIVERSITY OF NORTH TEXAS
Supporting Information
 Supporting Information Formation of Ruthenium Carbenes by gem-hydrogen Transfer to Internal Alkynes: Implications for Alkyne trans-hydrogenation Markus Leutzsch, Larry M. Wolf, Puneet Gupta, Michael Fuchs,
Supporting Information Formation of Ruthenium Carbenes by gem-hydrogen Transfer to Internal Alkynes: Implications for Alkyne trans-hydrogenation Markus Leutzsch, Larry M. Wolf, Puneet Gupta, Michael Fuchs,
Supplementary Material. Computational Study of Hydrido Boronium Dications and Comparison with the Isoelectronic Carbon Analogs
 Supplementary Material Computational Study of Hydrido Boronium Dications and Comparison with the Isoelectronic Carbon Analogs Golam Rasul, George A. Olah, and G. K. Surya Prakash* Loker Hydrocarbon Research
Supplementary Material Computational Study of Hydrido Boronium Dications and Comparison with the Isoelectronic Carbon Analogs Golam Rasul, George A. Olah, and G. K. Surya Prakash* Loker Hydrocarbon Research
Excited State Intramolecular Proton Transfer in Julolidine Derivatives: an ab initio Study Electronic Supplementary Information (ESI)
") Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 2018 Excited State Intramolecular Proton Transfer in Julolidine Derivatives: an ab initio
Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 2018 Excited State Intramolecular Proton Transfer in Julolidine Derivatives: an ab initio
QUANTUM CHEMISTRY PROJECT 3: PARTS B AND C
 Chemistry 460 Fall 2017 Dr. Jean M. Standard November 6, 2017 QUANTUM CHEMISTRY PROJECT 3: PARTS B AND C PART B: POTENTIAL CURVE, SPECTROSCOPIC CONSTANTS, AND DISSOCIATION ENERGY OF DIATOMIC HYDROGEN (20
Chemistry 460 Fall 2017 Dr. Jean M. Standard November 6, 2017 QUANTUM CHEMISTRY PROJECT 3: PARTS B AND C PART B: POTENTIAL CURVE, SPECTROSCOPIC CONSTANTS, AND DISSOCIATION ENERGY OF DIATOMIC HYDROGEN (20
DFT Study on the Reaction of Molybdenum with Acetaldehyde in Gas Phase
 Asian Journal of Chemistry; Vol. 25, No. 1 (2013), 89-94 http://dx.doi.org/10.14233/ajchem.2013.12753 DFT Study on the Reaction of Molybdenum with Acetaldehyde in Gas Phase YONG WANG 1,2 and GUO-LIANG
Asian Journal of Chemistry; Vol. 25, No. 1 (2013), 89-94 http://dx.doi.org/10.14233/ajchem.2013.12753 DFT Study on the Reaction of Molybdenum with Acetaldehyde in Gas Phase YONG WANG 1,2 and GUO-LIANG
Direct ab initio dynamics studies of N H 2^NH H reaction
 JOURNAL OF CHEMICAL PHYSICS VOLUME 113, NUMBER 15 15 OCTOBER 2000 Direct ab initio dynamics studies of N H 2^NH H reaction Shaowen Zhang and Thanh N. Truong a) Henry Eyring Center for Theoretical Chemistry,
JOURNAL OF CHEMICAL PHYSICS VOLUME 113, NUMBER 15 15 OCTOBER 2000 Direct ab initio dynamics studies of N H 2^NH H reaction Shaowen Zhang and Thanh N. Truong a) Henry Eyring Center for Theoretical Chemistry,
Mapping the Excited State Potential Energy Surface of a Retinal Chromophore Model with Multireference and Equation-of-Motion Coupled-Cluster Methods
 pubs.acs.org/jctc Mapping the Excited State Potential Energy Surface of a Retinal Chromophore Model with Multireference and Equation-of-Motion Coupled-Cluster Methods Samer Gozem, Federico Melaccio, Roland
pubs.acs.org/jctc Mapping the Excited State Potential Energy Surface of a Retinal Chromophore Model with Multireference and Equation-of-Motion Coupled-Cluster Methods Samer Gozem, Federico Melaccio, Roland
The Abstraction of Iodine from Aromatic Iodides by Alkyl Radicals. Steric and Electronic Effects 1
 Supporting Information: The Abstraction of Iodine from Aromatic Iodides by Alkyl Radicals. Steric and Electronic Effects 1 Darko Dolenc* and Božo Plesničar University of Ljubljana, Faculty of Chemistry
Supporting Information: The Abstraction of Iodine from Aromatic Iodides by Alkyl Radicals. Steric and Electronic Effects 1 Darko Dolenc* and Božo Plesničar University of Ljubljana, Faculty of Chemistry
Electronic Supplementary Information (ESI): First Principles Study of Photo-oxidation Degradation Mechanisms in P3HT for Organic Solar Cells
: First Principles Study of Photo-oxidation Degradation Mechanisms in P3HT for Organic Solar Cells") Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is The Royal Society of Chemistry 2014 Electronic Supplementary Information (ESI): First Principles Study of
Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is The Royal Society of Chemistry 2014 Electronic Supplementary Information (ESI): First Principles Study of
A Two Transition State Model for Radical-Molecule Reactions: Applications to Isomeric Branching in the OH-Isoprene Reaction
 5582 J. Phys. Chem. A 2007, 111, 5582-5592 A Two Transition State Model for Radical-Molecule Reactions: Applications to Isomeric Branching in the OH-Isoprene Reaction Erin E. Greenwald, Simon W. North,*,
5582 J. Phys. Chem. A 2007, 111, 5582-5592 A Two Transition State Model for Radical-Molecule Reactions: Applications to Isomeric Branching in the OH-Isoprene Reaction Erin E. Greenwald, Simon W. North,*,
Supporting Information
 Supporting Information Roles of Zeolite Confinement and Cu O Cu Angle on the Direct Conversion of Methane to Methanol by [Cu 2 (µ-o)] 2+ -exchanged AEI, CHA, AFX, and MFI Zeolites M. Haris Mahyuddin,,
Supporting Information Roles of Zeolite Confinement and Cu O Cu Angle on the Direct Conversion of Methane to Methanol by [Cu 2 (µ-o)] 2+ -exchanged AEI, CHA, AFX, and MFI Zeolites M. Haris Mahyuddin,,
Nonadiabatic Decomposition of Gas-Phase RDX through Conical Intersections: An ONIOM-CASSCF Study
 pubs.acs.org/jpca Nonadiabatic Decomposition of Gas-Phase RDX through Conical Intersections: An ONIOM-CASSCF Study A. Bhattacharya and E. R. Bernstein* Department of Chemistry, Colorado State University,
pubs.acs.org/jpca Nonadiabatic Decomposition of Gas-Phase RDX through Conical Intersections: An ONIOM-CASSCF Study A. Bhattacharya and E. R. Bernstein* Department of Chemistry, Colorado State University,
Supporting Information
 Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 2018 Supporting Information Beryllium-Beryllium only double-π bonds in the octahedral
Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 2018 Supporting Information Beryllium-Beryllium only double-π bonds in the octahedral
Photoinduced Water Oxidation at the Aqueous. GaN Interface: Deprotonation Kinetics of. the First Proton-Coupled Electron-Transfer Step
 Supporting Information Photoinduced Water Oxidation at the Aqueous Interface: Deprotonation Kinetics of the First Proton-Coupled Electron-Transfer Step Mehmed Z. Ertem,,,* eerav Kharche,,* Victor S. Batista,
Supporting Information Photoinduced Water Oxidation at the Aqueous Interface: Deprotonation Kinetics of the First Proton-Coupled Electron-Transfer Step Mehmed Z. Ertem,,,* eerav Kharche,,* Victor S. Batista,
I. INTRODUCTION JOURNAL OF CHEMICAL PHYSICS VOLUME 110, NUMBER 21 1 JUNE 1999
 JOURNAL OF CHEMICAL PHYSICS VOLUME 110, NUMBER 21 1 JUNE 1999 Crossed-beam reaction of carbon atoms with hydrocarbon molecules. V. Chemical dynamics of n-c 4 H 3 formation from reaction of C 3 P j with
JOURNAL OF CHEMICAL PHYSICS VOLUME 110, NUMBER 21 1 JUNE 1999 Crossed-beam reaction of carbon atoms with hydrocarbon molecules. V. Chemical dynamics of n-c 4 H 3 formation from reaction of C 3 P j with
Theorical study of the thermal decomposition mechanism of phenylperoxy radical.
 Theorical study of the thermal decomposition mechanism of phenylperoxy radical. B. Sirjean (1), M.F. Ruiz-Lopez (1)*, P.A. Glaude (2), F. Battin-Leclerc (2), R. Fournet (2) (1) Equipe de Chimie et Biochimie
Theorical study of the thermal decomposition mechanism of phenylperoxy radical. B. Sirjean (1), M.F. Ruiz-Lopez (1)*, P.A. Glaude (2), F. Battin-Leclerc (2), R. Fournet (2) (1) Equipe de Chimie et Biochimie
Potential Energy Surface and Binding Energy in External Electric Field: Modulation of Anion π Interactions for Graphene Based Receptors
 Potential Energy Surface and Binding Energy in External Electric Field: Modulation of Anion π Interactions for Graphene Based Receptors Cina Foroutan Nejad a and Radek Marek a,b a National Center for Biomolecular
Potential Energy Surface and Binding Energy in External Electric Field: Modulation of Anion π Interactions for Graphene Based Receptors Cina Foroutan Nejad a and Radek Marek a,b a National Center for Biomolecular
Autoxidation of α-pinene at High Oxygen Pressure SUPPORTING INFORMATION
 Autoxidation of α-pinene at High Oxygen Pressure Ulrich Neuenschwander and Ive Hermans* Institute for Chemical and Bioengineering, Department of Chemistry and Applied Biosciences, Swiss Federal Institute
Autoxidation of α-pinene at High Oxygen Pressure Ulrich Neuenschwander and Ive Hermans* Institute for Chemical and Bioengineering, Department of Chemistry and Applied Biosciences, Swiss Federal Institute
Current address: Department of Chemistry, Hong Kong Baptist University, Kowloon Tong, Hong Kong,
 Hydrolysis of Cisplatin - A Metadynamics Study Supporting Information Justin Kai-Chi Lau a and Bernd Ensing* b Department of Chemistry and Applied Bioscience, ETH Zurich, USI Campus, Computational Science,
Hydrolysis of Cisplatin - A Metadynamics Study Supporting Information Justin Kai-Chi Lau a and Bernd Ensing* b Department of Chemistry and Applied Bioscience, ETH Zurich, USI Campus, Computational Science,
Theoretical studies on the bifunctionality of chiral thiourea-based organocatalysts: Competing routes to C C bond formation
 Supporting Information Theoretical studies on the bifunctionality of chiral thiourea-based organocatalysts: Competing routes to C C bond formation Andrea Hamza, a Gábor Schubert, a Tibor Soós b and Imre
Supporting Information Theoretical studies on the bifunctionality of chiral thiourea-based organocatalysts: Competing routes to C C bond formation Andrea Hamza, a Gábor Schubert, a Tibor Soós b and Imre
Activation of CH 4 by Th + as Studied by Guided Ion Beam Mass Spectrometry and Quantum Chemistry
 pubs.acs.org/ic Activation of CH 4 by Th + as Studied by Guided Ion Beam Mass Spectrometry and Quantum Chemistry Richard M Cox, P. B. Armentrout,*, and Wibe A. de Jong Department of Chemistry, University
pubs.acs.org/ic Activation of CH 4 by Th + as Studied by Guided Ion Beam Mass Spectrometry and Quantum Chemistry Richard M Cox, P. B. Armentrout,*, and Wibe A. de Jong Department of Chemistry, University
Chemistry 1B, Fall 2016 Topic 23
 Chemistry 1B, Fall 016 Topic 3 Chemistry 1B Fall 016 Topic 3 [more] Chemical Kinetics goals for topic 3 inetics and mechanism of chemical reaction energy profile and reaction coordinate activation energy
Chemistry 1B, Fall 016 Topic 3 Chemistry 1B Fall 016 Topic 3 [more] Chemical Kinetics goals for topic 3 inetics and mechanism of chemical reaction energy profile and reaction coordinate activation energy
Photo-Dissociation Resonances of Jet-Cooled NO 2 by CW-CRDS
 Photo-Dissociation Resonances of Jet-Cooled NO 2 by CW-CRDS Patrick DUPRÉ Laboratoire de Physico-Chimie de l Atmosphère, Université du Littoral, Côte d Opale Dunkerque, France ISMS 22-26 June 2015 Patrick
Photo-Dissociation Resonances of Jet-Cooled NO 2 by CW-CRDS Patrick DUPRÉ Laboratoire de Physico-Chimie de l Atmosphère, Université du Littoral, Côte d Opale Dunkerque, France ISMS 22-26 June 2015 Patrick
Ab initio studies of ClO x reactions. IV. Kinetics and mechanism for the self-reaction of ClO radicals
 Ab initio studies of ClO x reactions. IV. Kinetics and mechanism for the self-reaction of ClO radicals R. S. Zhu and M. C. Lin Citation: The Journal of Chemical Physics 118, 4094 (2003); doi: 10.1063/1.1540623
Ab initio studies of ClO x reactions. IV. Kinetics and mechanism for the self-reaction of ClO radicals R. S. Zhu and M. C. Lin Citation: The Journal of Chemical Physics 118, 4094 (2003); doi: 10.1063/1.1540623
Chemistry 1B, Fall 2012 Lecture 23
 Chemistry 1B Fall 01 [more] Chemical Kinetics 1 kinetics and mechanism of reaction NO (g) + CO(g) ô NO(g) + CO (g) at T< 500K if the reaction was a collision between a NO molecule and a CO molecule one
Chemistry 1B Fall 01 [more] Chemical Kinetics 1 kinetics and mechanism of reaction NO (g) + CO(g) ô NO(g) + CO (g) at T< 500K if the reaction was a collision between a NO molecule and a CO molecule one
Chemistry 334 Part 2: Computational Quantum Chemistry
 Chemistry 334 Part 2: Computational Quantum Chemistry 1. Definition Louis Scudiero, Ben Shepler and Kirk Peterson Washington State University January 2006 Computational chemistry is an area of theoretical
Chemistry 334 Part 2: Computational Quantum Chemistry 1. Definition Louis Scudiero, Ben Shepler and Kirk Peterson Washington State University January 2006 Computational chemistry is an area of theoretical
Theoretical investigation of mechanism for the gas-phase reaction of OH radical and ethane
 J. At. Mol. Sci. doi: 10.4208/jams.122810.011811a Vol. 2, No. 3, pp. 225-233 August 2011 Theoretical investigation of mechanism for the gas-phase reaction of OH radical and ethane Xiao-Ping Hu a, Bing-Xing
J. At. Mol. Sci. doi: 10.4208/jams.122810.011811a Vol. 2, No. 3, pp. 225-233 August 2011 Theoretical investigation of mechanism for the gas-phase reaction of OH radical and ethane Xiao-Ping Hu a, Bing-Xing
DE-MAN HAN, GUO-LIANG DAI*, ZHEN-ZHONG YAN, CHUAN-FENG WANG, AI-GUO ZHONG
 J. Chil. Chem. Soc., 55, Nº 1 (010) CARBON DIOXIDE ACTIVATION BY Y ATOM AND Y + CATION IN THE GAS PHASE: A DENSITY FUNCTIONAL THEORETICAL STUDY DE-MAN HAN, GUO-LIANG DAI*, ZHEN-ZHONG YAN, CHUAN-FENG WANG,
J. Chil. Chem. Soc., 55, Nº 1 (010) CARBON DIOXIDE ACTIVATION BY Y ATOM AND Y + CATION IN THE GAS PHASE: A DENSITY FUNCTIONAL THEORETICAL STUDY DE-MAN HAN, GUO-LIANG DAI*, ZHEN-ZHONG YAN, CHUAN-FENG WANG,
CD Basis Set of Spectra that is used is that derived from comparing the spectra of globular proteins whose secondary structures are known from X-ray
 CD Basis Set of Spectra that is used is that derived from comparing the spectra of globular proteins whose secondary structures are known from X-ray crystallography An example of the use of CD Modeling
CD Basis Set of Spectra that is used is that derived from comparing the spectra of globular proteins whose secondary structures are known from X-ray crystallography An example of the use of CD Modeling
The role of NNH in NO formation and control
 Downloaded from orbit.dtu.dk on: Oct 05, 2018 The role of NNH in NO formation and control Klippenstein, Stephen J.; Harding, Lawrence B.; Glarborg, Peter; Miller, James A. Published in: Combustion and
Downloaded from orbit.dtu.dk on: Oct 05, 2018 The role of NNH in NO formation and control Klippenstein, Stephen J.; Harding, Lawrence B.; Glarborg, Peter; Miller, James A. Published in: Combustion and
Appendix D Simulating Spectroscopic Bands Using Gaussian and PGopher
 429 Appendix D Simulating Spectroscopic Bands Using Gaussian and PGopher This appendix contains methods for using Gaussian 09 121 and PGopher 120 to simulate vibrational and electronic bands of molecules.
429 Appendix D Simulating Spectroscopic Bands Using Gaussian and PGopher This appendix contains methods for using Gaussian 09 121 and PGopher 120 to simulate vibrational and electronic bands of molecules.